Abstract
Vascular fibrosis is a widespread pathologic condition that arises during vascular remodeling in cardiovascular dysfunctions. According to previous studies, vascular fibrosis is characterized by endothelial matrix deposition and vascular wall thickening. The RAAS and TGF-β/Smad signaling pathways have been frequently highlighted. It is, however, far from explicit in terms of understanding the cause and progression of vascular fibrosis. In this review, we collected and categorized a large number of molecules which influence the fibrosing process, in order to acquire a better understanding of vascular fibrosis, particularly of pathologic dysfunction. Furthermore, several mediators that prevent vascular fibrosis are discussed in depth in this review, with the aim that this will contribute to the future prevention and treatment of related conditions.
Highlights
- •
Endothelial-to-mesenchymal mesenchymal transition, smooth muscle cell proliferation, and extracellular matrix deposition are all symptoms of vascular fibrosis.
- •
Progressive vascular fibrosis is tied closely to multisystem diseases including hypertension, atherosclerosis, and diabetes.
- •
Multiple newly discovered variables, including the renin–angiotensin–aldosterone system and transforming growth factors-β, contribute to vascular fibrosis.
- •
Inflammation and mitochondrial damage can both contribute to vascular fibrosis.
1. Introduction
The vascular system is made up of numerous arteries, capillaries, and veins, and is found throughout the body, from organs to tissues. Its major role is to keep the blood flowing, which is required for cell growth, nutrition absorption, metabolite transfer, and hence intracellular stability (1). However, changes in the vascular anatomy can bring crucial disruption to the homeostasis in vascular function. Vascular fibrosis is one of the most prevalent biochemical abnormalities in the cardiovascular system. More than 60% of individuals over the age of 70 have arterial stiffness (2), which can be aggravated by vascular fibrosis (3). Initially, vascular fibrosis begins as a reversible compensatory adaptation process. Persistent stimulation of the renin–angiotensin–aldosterone system (RAAS), inflammation, oxidative stress (OS), and other factors might result in irreversible vascular fibrosis due to myofibroblast activation and extracellular matrix (ECM) deposition (1). Their harsh impacts would spread from blood vessels into parenchymal tissue, triggering reactions in a variety of systems and organs, for instance, the heart, brain, stomach, kidneys (3). In this review, we outline the structure and function of the vascular system, lay stress on the processes and mediators of vascular fibrosis, and highlight some therapeutic strategies for associated conditions. We also analyze the most recent research methodologies and advances in vascular fibrosis, meanwhile bringing different theoretical foundations and approaches for future clinical trials.
Box 1. Pathogenic shifts during vascular fibrosis
Vascular fibrosis is the overgrowth, hardening, scarring of blood vessels, which is attributed to excess deposition of ECM. Many scholars believe there are four major pathogenic variables that cause vascular fibrosis: (i) Fibroblasts as the primary cell involving in the production and deposition of ECM proteins in the vascular wall. Changes in genes that express distinct collagen subtypes in fibroblasts effect fibrotic process (4). (ii) ECM proteins accumulation is secondary to fibroblast changes, and can be mediated by plenty of molecules or events, such as TNF-α, IL-6, IL-1, and OSM. (iii) TGF-β action appears to be dictated by its biological source, with macrophage originated TGF-β typically demonstrating wound-healing and profibrotic activity, while Treg cells produced TGF-β operates as an anti-inflammatory and antifibrotic mediator (5). (iv) Endothelial cells release cytokines and adhesion molecules which elevate the inflammatory and potential fibrotic response, in response to wounded tissues and activated immune cells (6).
2. Vascular structure and function: from physiology to fibrosis
Under physiological conditions, blood vessels behave as more than simply a pipeline for blood transport, but as an organ capable of adjusting their diameter to regulate blood pressure elastically or muscle-dependently. In the circulatory system, blood flows from the left ventricle to the artery, which then branches into arterioles and connects with venules through capillaries, before returning to the right atrium.
Notwithstanding the circulatory system's heterogeneity and mutability, the histological arrangement is indeed very comparable. Except for capillaries, blood vessels are composed of three layers: tunica intima, tunica medium, and tunica adventitia (7), in which ECM is their mutual internal environment for cell survival. This critical component is filled with numerous proteins including elastin fibers, collagen (Col), fibronectin (FN), proteoglycans, and others (8), making it indispensable in transducing cellular signaling and cellular interactions, as well as connecting all the cells in vascular wall.
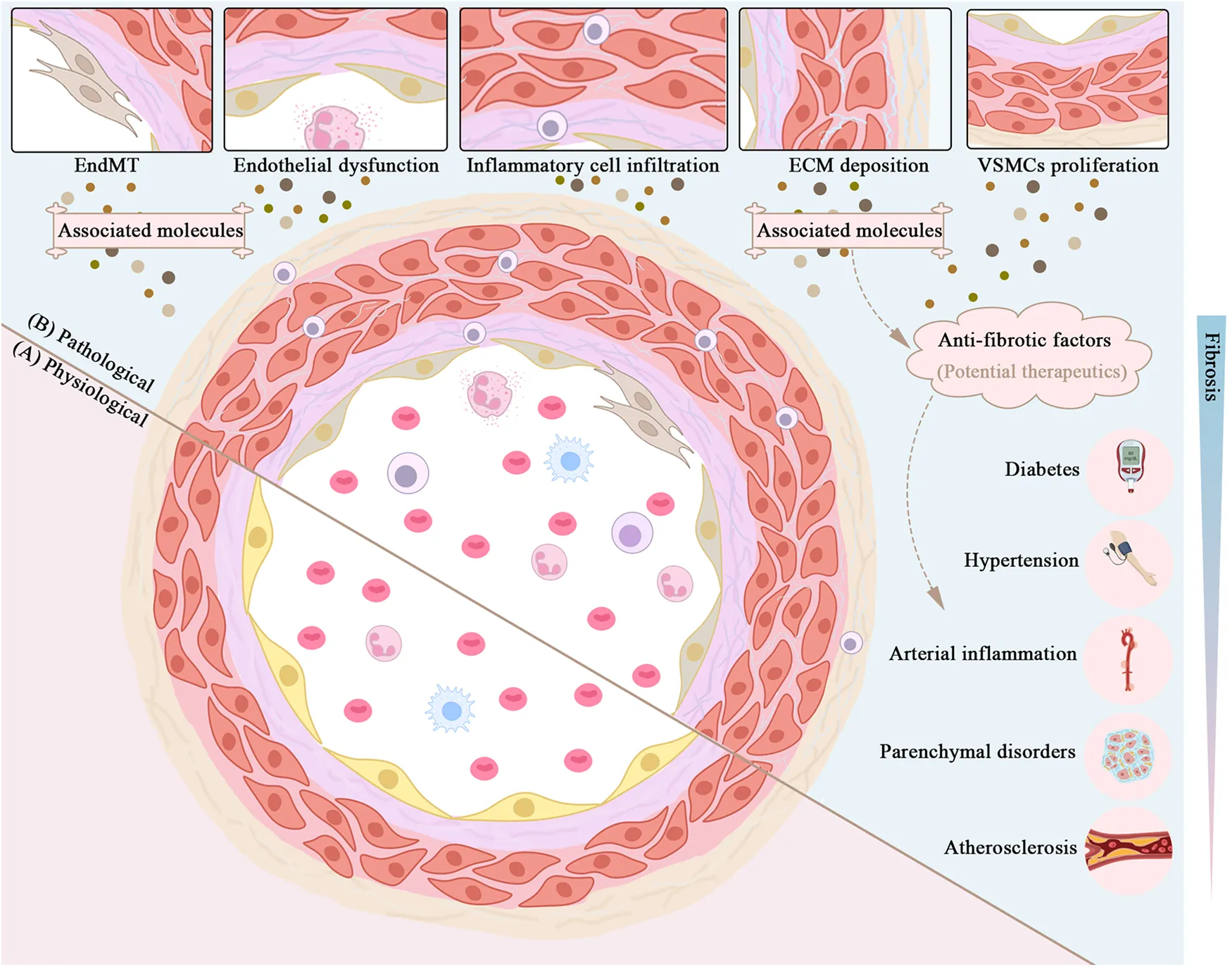
The intima, the thinnest of the three layers, is consisted of a single layer of endothelial cells (ECs) that make direct contact with the lumen. Therefore, EC is taking on a crucial role in cellular homeostasis, such as vascular tone modulation, coagulation and anticoagulation, and immune response (7). Antigen presentation characteristics in EC were discovered due to a coating of glycocalyx on its surface (9). However, since ECs have such high biological activity, plasma antigens or chemicals may trigger specific clinical disorders. A sub-endothelial layer packed with fibro elastic connective tissue lies beneath the ECs, stabilizing them yet enabling them to be flexible (7). Another type of cell that should not be overlooked is Pericyte. They can differentiate into ECs, fibroblasts, vascular smooth muscle cells (VSMCs), adipocytes, and osteoblasts and eventually contribute in blood flow dynamics modulation (10, 11). In most cases, vascular injury begins with intima damage induced by hemodynamics or morbific materials in the circulation (Figure 1).
Figure 1
The media is thicker than the intima and adventitia. It is composed of a significant number of VSMCs and well-ordered elastin fibers that act as vessel dimension regulators (12). The adventitia is the most complicated part of the arterial wall since it comprises not only a considerable amount of fibro-elastic connective tissue, but also a diversity of cell phenotypes such as fibroblasts, pericytes, and immune cells (13) (Figure 1).
Unfortunately, once fibrosis worsens, substantial abnormalities in the structure of blood vessels emerge, leading to perfusion dysfunction in systemic circulation and organs. ECM deposition and media hypertrophy lead to thickening of the vascular wall and reduction in elasticity, which terminally contribute to hemorheological abnormalities (2). During vascular fibrosis, the media augmented VSMC contraction and calcification (1). Meanwhile the adventitia experiences remodeling (14), and the intima undergoes endothelial dysfunction due to decreased endothelial relaxation, increased permeability, and inflammation (15). These alterations in endothelium are associated with vascular fibrosis and sclerosis, illustrating its importance as a physiologic barrier against circulating immune cells and inflammatory cells. However, recent research has revealed that cytokines can initiate EC to secrete pro-inflammatory and profibrotic molecules, thereby facilitating inflammation and fibrosis in endothelial dysfunction (16). EC to mesenchymal transition (EndMT) also represents multiple cardiovascular disorders (CVD), fibrosis included (16), once more emphasizing the role of endothelial cells (Figure 1). So far, we have elaborated a histological and functional feature in vascular fibrosing process, which is of great significance in understanding what we discuss later.
3. Molecular mechanisms of vascular fibrosis
Vascular fibrosis is caused mainly by EC dysregulation, VSMC proliferation, fibroblast activation, ECM remodeling, and immune cell infiltration. In this section, we highlighted some molecules of concern in cardiovascular pathophysiology and categorized them in Table 1. Propelling the understanding on underlying processes and associated molecules will provide a number of strategies for treating or alleviating sickness.
Table 1
| Category | Mediator | Pro-fibrotic or Anti-fibrotic | Influence | Notes | References |
|---|---|---|---|---|---|
| Canonical | Ang II | Pro-fibrotic | ECM deposition (collagen & fibronectin overexpression, elastin reduction) and VSMC proliferation | via AT-1R | (1, 2, 17) |
| Anti-fibrotic | Vasodilation, anti-inflammation and growth inhibition | via AT-2R | (18, 19) | ||
| Aldo | Pro-fibrotic | ECM deposition (collagen & fibronectin overexpression, elastin reduction) and VSMC proliferation | via MR | (17, 20, 21) | |
| Ang-(1-7) | Anti-fibrotic | Vasodilation, anti-inflammation, and Ang II inhibition | ACE2-Ang-(1-7)/MasR axis | (22–24) | |
| TGF-β | Pro-fibrotic | ECM deposition (collagen & fibronectin overexpression, elastin reduction) and VSMC proliferation | via Smad2/3/4, RhoA/ERK, p38/MAPK, JAK/STAT, PI3K/AKT, Galectin-3, IL-11, etc. | (25–29) | |
| MMP | Pro-fibrotic | Degrading collagen & elastin | Imbalance in MMP family is the cause of ECM deposition | (1, 30, 31) | |
| TIMP | Anti-fibrotic | Inhibiting MMP | / | (30) | |
| TG 2 | Pro-fibrotic | ECM deposition (collagen secretion & scaffolding ECM proteins) | / | (17, 32, 33) | |
| CTGF | Pro-fibrotic | ECM deposition | / | (2, 30) | |
| FGF | Pro-fibrotic | ECM deposition | / | (2, 30) | |
| Non-canonical | Fetuin-A | Anti-fibrotic | Suppressing TGF-β signaling | Inhibiting Smad3 phosphorylation | (34) |
| Salusin-β | Pro-fibrotic | Promoting TGF-β signaling, increasing Col-1/3, FN, TGF-β and CTGF expression | Stimulating Smad2/3 phosphorylation | (35) | |
| Alamandine | Anti-fibrotic | Attenuating Ang II-induced col-1, TGF-β and CTGF overexpression | via stimulating MrgD expression and inhibiting p38 phosphorylation | (36) | |
| Galectin-3 | Pro-fibrotic | Increasing Col-1 deposition, MMP-9 decrease, VSMC proliferation | Increased by multiple factors (inflammation, ROS, TGF-β, aging) | (27, 37, 38) | |
| IL-11 | Pro-fibrotic | Increasing collagen & MMP (via ERK) | Downstream molecule of TGF-β; X203 alleviates vascular fibrosis | (28, 29) | |
| StAR | Pro-fibrotic | Increasing Aldo | Downstream signal of c-Fos/c-Jun | (39) | |
| BMS | Anti-fibrotic | Decreasing collagen deposition & media thickness | ET-1 antagonist | (40) | |
| Energy metabolism in mitochondria | Mitochondria OS | Pro-fibrotic | Increasing Col-1/3, CTGF, TGF-β expression & media thickness | Along with ER stress | (41–43) |
| HFD | Pro-fibrotic | ECM deposition (Col-1/3, CTGF expression) and vascular thickness | / | (44) | |
| MitoQ | Anti-fibrotic | Inhibiting mitochondria OS | Alleviating Ang II-induced fibrosis | (44–46) | |
| FABP3 | Pro-fibrotic | ECM deposition (Col-1, FN-1, MMP3/9 overexpression) | FABP3 deficiency is also unfavorable | (47, 48) | |
| EndMT | EndMT | Pro-fibrotic | Increasing collagen expression; NO shortage; ROS accumulation; inflammation | Increased by multiple factors (IL-1, IL-6, TNF-α, IFN-γ, ET-1, etc.) | (49–52) |
| let-7 | Anti-fibrotic | Reversing EndMT | Forming TGF-βR1/let-7i double-negative feedback pathway | (53) | |
| H2S | Anti-fibrotic | Suppressing TGF signal & RAAS; increasing NO; inhibiting CD11b + leukocytes; inhibiting OS | Blocker of EndMT | (54–57) | |
| Immune cells and inflammation | IL-17 | Pro-fibrotic | Increasing gal-3, collagen deposition, ROS accumulation, fibroblasts | Increased by Aldo via MR | |
| Caspase-1 | Pro-fibrotic | Increasing Col-1 & FN expression | Pyroptosis related | (58–60) | |
| Macrophage autophagy | Anti-fibrotic | Inhibiting M1 polarization; suppressing EndMT; | / | (61–63) | |
| Macrophage polarization | Pro-fibrotic | Dual stimulation between pro-inflammatory (IL-1β, IL-6, TNF-α, ROS, NO) molecules and anti-inflammatory (IL-4, IL-10, IL-13) molecules | Imbalanced M1/M2 ratio cause chronic inflammation in fibrosis | (62–65) | |
| IL-17A | Pro-fibrotic | Increasing ROS, endothelial dysfunction, fibroblasts proliferation; NGAL&PYK2 expression upregulation | Can be expressed in response to Aldo signal | (66, 67) | |
| PTEN | Anti-fibrotic | Decreasing TGF-β, Col-1, FN-1, MMP-2 expression | Preventing pyroptosis and apoptosis through PTEN/PI3K/AKT axis | (68–70) | |
| Galectin-3 | Pro-fibrotic | Increasing IL-1 & IL-18 expression and inflammatory cell extravasation | Inducing NF-κB pathway | (71–73) | |
| EVs | Pro-fibrotic | EVs form different sources can carry different pro-fibrotic cargos (miR-23a-3p, lncRNA GAS5, circUbe3, miR-142-3p, etc.) | The pro-fibrotic feature is shown by molecules within EVs, not EVs themselves | (74–77) | |
| miR-21 | Pro-fibrotic | Regulating inflammation via PPAR-α; TGFβ1/Smad3 pathway | Has anti-inflammatory effect | (78, 79) | |
| miR-122 | Pro-fibrotic | Inhibiting PTEN; inducing OS, apoptosis, and inflammatory response | / | (80) | |
| Others | MBG | Pro-fibrotic | Promoting TGF-β signaling; suppressing Fli-1 | Being an endogenous cardiotonic steroid | (81, 82) |
| Fli-1 | Anti-fibrotic | Inhibiting Col-1 expression | Being a nuclear inhibitory transcription factor | (81, 82) | |
| spironolactone | Anti-fibrotic | Suppressing MBG's effects | / | (83) | |
| ROCK | Pro-fibrotic | Promoting VSMC activation, inflammation, OS, NO production and EndMT | Can be suppressed by statins | (84–86) | |
| PAI-1 | Pro-fibrotic | Increasing Col-1 expression; inducing Rho kinase activation | Increased by Ang II; can be suppressed by apelin | (87, 88) | |
| apelin | Anti-fibrotic | Inhibiting Ang II-induced PAI-1, collagen and MMP-2 expression | Can be suppressed by miR-122 | (88) |
Summary of pro-fibrotic and anti-fibrotic molecules discussed in this review.
3.1. Renin–angiotensin–aldosterone system and TGF-β induce profibrotic changes in ECM
RAAS and TGF-β are the canonical regulators in vascular fibrosis. Although they are not new to public attention, it is still necessary to be elaborated for the better understanding of newfound evidences. Initially, studies on RAAS concentrated on hypertension and heart failure, but later shifted to vascular fibrosis. TGF-β has been identified as an essential mediator in fibrotic parenchymal organs. In the case of vascular fibrosis, however, a linkage between RAAS and TGF-β is revealed.
3.1.1. RAAS
Renin, also known as angiotensinogenase, is a kidney-secreted enzyme that catalyzes angiotensin II (Ang II) synthesis on its substrate via angiotensin-converting enzyme (ACE), meanwhile Ang II can further stimulate aldosterone (Aldo) secretion (20). RAAS is essential to water-sodium retention and vasopressor function, therefore being one of the most highly considered mediator in cardiovascular system. Intriguingly, previous studies indicated that Ang II and Aldo have an influence on developing vascular fibrosis, respectively via Ang II receptor 1 (AT-1R) and mineralocorticoid receptor (MR). These signals subsequently upregulates TGF-β expression in ECs, VSMCs, fibroblasts, and macrophages (17) (Figure 2). Additionally, activated AT-1R and MR is also responsible to EC/VSMC dysfunction, OS, macrophage activation, dysregulation of autophagy, during fibrosis in vascular pathology (17, 21, 89, 90). It is noteworthy that AT-1R triggers vasoconstriction, VSMC proliferation, inflammation, and extracellular matrix remodeling, whereas AT-2R activation have completely contradictory effects and often represents protection in blood vessels (18). In fact, scholars have found AT-2R exhibits vasodilation through regulating production of bradykinin, NO, and cGMP (19). Meanwhile, decreased TNF-α-mediated IL-6 secretion via inhibiting NF-κB activation and reducing MAPK activity are also the ways how AT-2R respectively manage anti-inflammation and growth inhibition (19). Additionally, research found AT-2R can significantly attenuate the expression of TGF-β type I receptor in VSMCs of WKY rat (91). Recent researches claim that AT-2R agonists have astonishing potential in clinical treatment, for instance, Compound 21 are now in phase III clinical trial (92, 93). Still, few studies have focused their significance into vascular fibrosis.
Figure 2
However, there are two opposing arms in RAAS, and above-discussed ACE/Ang II/AT1R axis refers to a pro-fibrotic, pro-inflammatory “classical arm”. The “protective arm”, though not dominant, have essential role in counteracting negative effects in “classical arm” via ACE2/Ang-(1-7)/MasR axis (22). ACE2 as a homolog of ACE, converts Ang II to Ang-(1-7) through a carboxypeptidase feature, therefore inhibits Ang II by degradation (94). Furthermore, Ang-(1-7) can be formed in an Ang II alternative way, by firstly synthesizing Ang-(1-9) from Ang I via ACE2, then transforming into Ang-(1-7) via ACE (23). Ang-(1-7) then activates its specific Mas receptor (MasR) in EC, which increases the production of vasodilators such as nitric oxide (NO) and prostacyclin (24). In addition to vasodilation, MasR is also involved in many vascular protective functions, including anti-fibrotsis, anti-inflammation, and anti-proliferation (22). Meanwhile, Ang-(1-7) has been reported to downregulate TGF-β1/Smad2 level in rabbits after angioplasty (95). Therefore, multiple researches have been attracted to the therapeutic potential that ACE2/Ang-(1-7)/MasR axis possesses in cardiovascular field.
Accumulating research found that treatment with human recombinant ACE2 (hrACE2) constrains the cardiac fibrosis, OS, and inflammation that Ang II induced (96). A small molecule ACE2 activator, XNT, reduced hydroxyproline accumulation in kidney and myocardium (97). Furthermore, diminazene aceturate (DIZE) as a ACE2 activator and AT1R inhibitor, shows alleviation to collagen accumulation in cardiac infraction mice (98). Besides ACE2 activators, researchers recently found a nonpeptide, Ang-1(1-7)-like compound named AVE 0991 that can selectively activates MasR and is of great potential as cardiovascular medicine (99). However, more evidences are needed when it comes to vascular fibrosis.
3.1.2. TGF-β
TGF-β literally act as an intersection, mediating a series of downstream pro-fibrotic signaling pathway that eventually promote ECM protein reconfiguration and VSMC proliferation, while receiving multiple upstream mediators that modulate the preceding process. Through decades of researching, evidences suggest TGF-β induces Smad (small mothers against decapentaplegic)-2/3/4 phosphorylation, which results in further profibrotic gene expression (25). Yet, not all Smad are pro-fibrotic, Smad-7 has been proved to decrease Smad-2/3 phosphorylation (25). Intriguingly, scholars have explicated that Smad signal can be triggered independently to TGF-β (100). Study have shown that Ang II-mediated ECM synthesis can be managed by activation of PKC and p38 MAPK, triggering NF-κB and Smad2/3 cross-talk pathway, which is independent to TGF-β (100). It is supported by further study that Ang II-induced Smad2/3 nuclear translocation and DNA-binding activity via AT-1R have not been dissolved by blockade of endogenous TGF-β (101). Besides TGF-β/Smad, many studies have suggested several other pathways that are as well involved in TGF-β signaling, including galectin-3, IL-11, RhoA/ERK, p38/MAPK, JAK/STAT and PI3K/AKT (26) as depicted in Figure 2.
Noteworthy, there are plenty of TGF-β interacting molecules with great importance. To begin with, fetuin-A is a serum glycoprotein produced by liver, and can suppress TGF-β1 signaling by upregulating pseudoreceptor of TGF-β1 (34). A study among 106 participants found a negative connection between serum fetuin-A levels and peripheral artery disease in atherosclerosis (102). In vitro, fetuin-A stimulation can significantly relieve TGF-β1-induced overexpression of TGF-β1 and Col-1 (34). Secondly, salusin-β is a bioactive peptide that participates in CVD. Recent study indicates their part in promoting vascular fibrosis through TGF-β1/Smad and VSMCs proliferating (35), as well as Col-1/3 overexpression, CTGF mRNA upregulation and Smad2/3 phosphorylation (35). Additionally, a RAAS component named alamandine can attenuate Ang II-induced elevation of Col-1, TGF-β and CTGF, alleviating vascular fibrosis via MrgD (Mas-related G protein-coupled receptor member D) expression (36). Galectin-3 (gal-3) is another TGF-β interactive molecule that worth special noticing. It has lately been explored intensively throughout many studies, for it contributes to multiple cardiovascular fibrotic pathways (37). Gal-3 expresses in fibroblasts, ECs, and immune cells, causing Col-1a deposition and MMP (matrix metalloproteinases) reduction, in response to TGF-β1 signaling via STAT-3 (27). This process can be reversed by gal-3 siRNA (38). Meanwhile, a univariate analysis found gal-3 being correlated with carotid intima-media thickness, arterial stiffness, cardiac output, indicating gal-3 is a favorable marker of cardiovascular function and fibrosis (103). Last but not least, recent studies have demonstrated that IL-11 (Interleukin 11), as the downstream molecule of TGF-β, triggers fibrosis along with ET-1 and PDGF in an ERK-dependent way (28, 29). It can also activate fibroblasts via IL-11RA (28). Meanwhile, a study on the IL-11 antibody, X203, revealed its efficacy in MMP and collagen downregulation, as well as VSMCs reduction in vivo (29). It is worth emphasizing that IL-11 intervention may be more advantageous than TGF-β, for TGF-β has pleiotropic roles, suppressing which may result in certain physiological dysfunction (28).
3.1.3. ECM accumulation
ECM deposition is the ultimate lesion in vascular fibrosis, in response to all molecular signals that we discuss. Collagen and elastin fibers are the two most important ECM proteins. In particular, their balance and their absolute amount determines the structure and mechanical properties of the blood vessel. It is of great significance to stress that ECM proteins can be regulated by an endopeptidases family named matrix metalloproteinases (MMPs). There lies multiple factors that can regulate MMP expression, including RAAS related molecules (Ang II, TGF-β, Aldo, ET-1, etc.), pro-inflammatory cytokines (IL-6, TNF-α, IL-1, etc.), and reactive oxygen species (ROS) (1, 17, 30). However, it is believed that different MMP isoforms function diversely on ECM proteins, resulting in different fibrotic outcomes. For instance, MMP-2/9 participated in TGF-β1/Smad-induced myofibroblasts activation and monocytes/macrophages infiltration (31), yet MMP-1 overexpression alleviates fibrosing (104). Upregulation of MMP-2/9 causes collagen accumulation, whereas MMP-8/13 degrades collagen, which is hazardous in arterial plaque rupture (105, 106). Apart from MMPs, an endogenous inhibitor called TIMP (tissue inhibitor of metalloproteinase) is also essential in ECM remodeling. TIMP are inhibitive to MMPs, which echoes the fact that imbalance in MMP/TIMP frequently accompanies with vascular fibrosis (30).
Transglutaminase 2 (TG2), another ECM associated proteinases that acts as a crosslinking protein as well as an ECM scaffold enzyme, promotes fibrosis by acting on fibronectin, collagen, and laminin (17, 32). As research deepens, some researchers have revealed that TG2 participates in TGF-β1/Smad signaling for collagen synthesis and secretion, causing VSMCs proliferation and ECM remodeling in vascular fibrosis (33). According to earlier research, preceding ECM changes are as well related to the overexpression of connective tissue growth factor (CTGF), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF) (2, 30) (Figure 2). Furthermore, there are additional molecules that worth notice. BMS, an ET-1 antagonist, is beneficial in reducing collagen deposition and medium thickness (40). StAR (steroidogenic acute regulatory protein) is a downstream factor of the steroidogenic acute regulatory protein c-Fos/c-Jun, which leads to hypoxia-induced upregulation of ECM proteins including CTGF, Col-3, MMP-2/9 (39).
From the above experimental evidence, we can conclude that RAAS and access TGF-β play an important role in the induction and promotion of extracellular matrix. Meanwhile, there are complex and extensive universal links between them, therefore they are important targets for therapeutic intervention in fibrosis.
3.2. Energy metabolism dysfunction and oxidative stress in mitochondria as prerequisite in vascular fibrosis
According to research findings, mitochondrial oxidative stress is increased in cardiac remodeling and failure (107), and is highly linked to inflammation and fibrosis (41). It is well acknowledged that OS is characterized by an overabundance of ROS, which can be generated by aberrant mitochondrial metabolism and lead to mitochondrial malfunction. Mitochondrial DNA is extremely vulnerable since oxidative damage may set off a vicious cycle of ROS generation (42). Ang II has also been demonstrated to promote mitochondrial dysfunction in vascular endothelial cells, through increasing ROS production (43). Besides, mitochondrial dysfunction interacts with TGF-β on various levels to promote or inhibit tissue fibrosis (108). Histological and western-blotting studies show that high-fat diet (HFD) animals had an increase in cardiac and aortic fibrosis biomarker genes such as Col-1/3, CTGF, and TGF-β, as well as aortic media and carotid intima media thickness (44, 109). It is noteworthy that aforementioned abnormalities can be reversed by MitoQ, a mitochondrial antioxidant (44, 45), which partly contributes to the of OS induces fibrosis.
On the other hand, ER stress is associated to mitochondrial OS. ER stress protein markers, BiP and PDIA6, was also found increased in HFD mice and positively correlated with cardiac interstitial fibrosis (44). Luckily, MitoQ can relieve those variations. Further experiments on fibroblasts in vitro suggest the same, for that MitoQ and ER stress inhibitor (4-phenyl butyric acid) alleviates Ang II induced fibrotic alternations (44). Besides, research on human umbilical vein endothelial cells have also shown that Aldo suppresses the expression of SOD2 protein, mitochondria DNA and protein through MR. Intriguingly, another mitochondrial antioxidant (Mito-TEMPO) can also twist this attenuation (46). These evidences further indicate OS is taking part in fibrosis in an ER stress-dependent manner, and mitochondrial OS antagonists like MitoQ and Mito-TEMPO possess their therapeutic benefits. Besides, scholars discovered that particulate matter 2.5 (PM2.5) exposure triggers aortic fibrosis via mitochondrial OS with the mechanisms mentioned above (45).
Mitochondria serve as the cell's energy factory, in which fatty acid binding protein 3 (FABP3) subtly modulates the process of fatty acid oxidation (FAO) (110). FAO generates ATPs as an energy source during fibrosis, for VSMC proliferation, ECM protein secretion, and immune cell functions (110). Interestingly, recent research reveals that FABP3 elevation induces aortic adventitia fibroblasts growth and ECM deposition (47), hence contributing to vascular fibrosis. Immunohistochemistry data demonstrate a substantial relationship between the expression of FABP3 and adventitia ECM proteins, including Col-1, FN-1, MMP-3/9 (47). However, another research on FABP3 suggests the opposite. FABP3−/− mice exhibited severer cardiac hypertrophy than wildtype mice owing to FAO dysfunction, which enhances glycolysis and toxic lipid accumulation (48). As a conclusion, neither elevation nor deficiency in FABP3 is beneficial, meaning that mitochondrial energy metabolism has deeper mechanism that merits further investigation.
Last but not least, emerging studies have corroborated that mitophagy effects mitochondrial dysfunction in organs and tissues fibrosing (111). Consequently, there emerges plenty of potential therapeutic targets, such as targeting mitophagy, miR-30a, mTORC1/PGC-1 axis, and targeting mitochondrial quality control (108, 112, 113). These targets have the potential to improve mitochondrial function and reduce oxidative stress, thereby slowing or reversing the accumulation of ECM. Besides, employing wide tactics of genetic engineering, small compounds targeting mitochondria, and mito-therapy are all therapeutic options.
3.3. Endothelial to mesenchymal transition in vessel as initial fibrosis
EndMT, as mentioned above, is pathological condition of endothelium that results in vascular fibrosis. It occurs when ECs cease to secrete their particular biomarkers, but instead products of mesenchymal and myofibroblasts like Col-1 and α-SMA (49). The problem is that EndMT not only manifests a dysfunction of endothelium integrity and altering to a migratory form, but also turns ECs into invasive and proliferative phenotype, all of which are contributive to fibrosing. Multiple signaling pathways are involved in the formation of EndMT, for instance, IL-1β, IL-6 and TNF-α can be triggers of TGF-β pathways via NF-κB (50). Meanwhile, IFN-γ acting through JAK/STAT pathway and ET-1 participation in TGF-β/Smad signaling as well promotes EndMT process (51, 52). Unfortunately, EndMT causes certain dysfunctions in vascular system. For instance, NO production in ECs was essential to maintaining integrative structures and functions of vessels, whereas growing EndMT causes NO shortage. Besides, EndMT also leads to vascular fibrosis related ROS accumulation, vascular inflammation and ECM deposition. Furthermore, evidence manifests that ischemia-reperfusion injury in particular, can be the direct trigger of EndMT, which in turn promotes atherosclerosis and arteriosclerosis, especially in ischemic stroke in brain (53). However, a circulating human microRNA family named let-7 has been identified to play crucial role in remodeling the blood-brain barrier and reversing EndMT caused by ischemic stroke via a TGF-βR1/let-7i double-negative feedback pathway (53).
Recently, hydrogen sulfide (H2S), an essential mediator, generates from L-cysteine through enzymatic reactions, has been established as a blocker of EndMT and vascular fibrosis process. There are currently four major approaches. Initially, H2S acts as a vasodilator by inhibiting the TGF-β/cGMP-PKG pathway and lowering RAAS (54, 114), especially in chronic hypertension-induced vascular alteration. Secondly, H2S can boost endothelial nitric oxide synthase (eNOS) for more sufficient NO synthesizing in guarding against EC dysfunctions and reducing blood pressure (55). Additionally, H2S also has an anti-inflammatory impact by blocking the migration of CD11b+ leukocytes (56). Finally, H2S has been demonstrated to be a negative regulator of OS, which is thought to be connected to EndMT (57). The foregoing facts have surely pushed H2S's function into the future treatments for vascular fibrosis.
3.4. Inflammation associated cellular and molecular changes in fibrosis
Inflammation is a key factor in the development and progression of fibrosis. Inflammatory cells and mediators are playing a crucial role in myofibroblast activation and ECM deposition. Understanding the cellular and molecular mechanisms of fibrosis may lead to the development of new therapeutic interventions.
3.4.1. Inflammation related cellular events
There are certain cellular activities that are highly related to fibrosis. First and foremost is pyroptosis, a pro-inflammatory apoptosis event that lately drew attention to vascular fibrosis associated diseases (58). The creation of cell membrane holes, membrane rupture, and nuclear condensation are the primary characteristics of pyroptosis (115). These processes can be initiated by three separate signals, including inflammasome, death receptor, and mitochondrial dysfunctions (116). The molecular mechanism of pyroptosis is complicated, but the caspase family is generally considered to be involved. Caspase-1 activation and recruitment is responsive to the first signal pathway——inflammasome, which NLRP3 regulates (59). Through activation, caspase-1 cleaves pyroptosis-related molecules to their mature form, such as IL-1β, IL-18, and N-terminal fragment of gasdermin D (GSDMD) (59). Meanwhile, LPS induced caspase-4/5/11 signal as well catalyzes GSDMD (117). In vitro, those factors have been proved to be attenuated by NLRP3 inhibitor MCC950, in coronary artery inflammation model mice (59). Immunohistochemistry and western blot analysis shows that caspase-1 suppression reduces Col-1 and fibronectin expression, implying caspase-1 related pyroptosis is playing a critical role in fibrosis (58). In addition to inflammasomes, death receptor and mitochondrial dysfunctions drives caspase-3 activation, resulting in gasdermin E (GSDME) cleavage (118). The N-terminal fragment of both GSDMD and GSDME induces pore formation, which leads to pyroptosis (115). Eventually, myofibroblasts are activated by caspase-induced inflammatory cytokines release and the direct production of pyroptosis (119). This is how pyroptosis helps the progression of fibrosis. It is worth noting that some research claims SARS-CoV-2 infection also involves caspase-1-induced excessive inflammation (60). However, another research demonstrates that SARS-COV-2 has an inhibitory effect in pyroptosis (120). This inconsistency suggests pyroptosis in COVID-19 are not legible enough to be concluded unilaterally.
Furthermore, autophagy has been linked to fibrotic disorder by its regulatory influence on macrophage polarization and EndMT (61, 121). Autophagy is an essential self-degrading system in normal eukaryotic cells, for it maintains cell homeostasis by converting cellular wastes into reusable matters (62, 122). Studies have shown the inhibition of macrophage autophagy promotes M1 polarization (61, 62), which leads to the secretion of pro-inflammatory cytokines that starts ECM remodeling (63). Intriguingly, in vitro data shows that endothelial-specific ATG5 (autophagy-related protein 5) knockout mice exhibited EndMT-related organ fibrosis compared to littermate controls, which can be alleviated by IL-6 specific antibody neutralization (61). Although these findings were not targeted on vascular, there is no doubt that they indicated the importance of autophagy in macrophage polarization related fibrosis and in IL-6-driven EndMT. They could be the potential breach of vascular fibrosis.
3.4.2. Imbalanced macrophage polarization cause chronic inflammation in vascular fibrosis
Inflammation is accompanied by both destroying and mending, and maintaining their balance is critical (62). Excessive inflammatory damage or uncontrolled tissue regeneration can both be detrimental to the healing process, while also promoting a pathological fibrotic process. By polarization and activation, macrophage is particularly connected with the above-mentioned equilibrium as well as fibrosis among all inflammation-associated cells (62). Polarization is the process by which macrophages phenotypically differentiate into two major subtypes (M1 and M2) in response to environmental signals (64). It is widely accepted that M1 is pro-inflammatory while M2 is anti-inflammatory during inflammation, echoing the previous balance of destroying and mending. In order to show an intuitionistic understanding, we highlighted the difference between M1 and M2 in Table 2.
Table 2
| M1 macrophages | M2 macrophages | References | |
|---|---|---|---|
| Inflammation | Pro-inflammation | Anti-inflammation | (123, 124) |
| Fibrosis | Initiating inflammatory environment that begins the progression of fibrotic diseases | Excessive matrix deposition and remodeling replace the functional tissue into nonfunctional fibrotic tissue | (123) |
| Myofibroblasts | Activation and proliferation | Differentiation and Proliferation | (121, 124) |
| Fibrotic mediators | MMP-2, IL-6, ROS, CCL2 | IL-10, TGF-β, Ang II, Wnt, CTGF, PDGF, galectin-3, MMP-9 | (121, 125) |
| Fibrocyte | Recruitment of fibrocyte | Drive EndMT and FMT | (126, 127) |
Summary macrophages in different roles in formation of fibrosis.
In early stage of inflammation, classically activated M1 phenotype initiates the tissue inflammation, and manage the removal of pathogens. To do so, stimuli such as TNF-α, IFN-γ, and LPS orient the polarization of M0 to M1, and the later further secretes pro-inflammatory molecules including IL-1β, IL-6, TNF-α, ROS, iNOS (63). These molecules can directly trigger myofibroblasts activation, meanwhile, M1 also produces chemokines (CCL2, CCL3, CXCL1, etc.) that assist fibrocytes recruitment (123, 126). In response to M1 signal, fibrocytes are recruited and migrates by the help of several chemokine receptors (CCR2, CCR3, CXCR2, etc.) (121, 123). Myofibroblasts are as well indirectly activated and proliferates (124). Furthermore, M1 produces an amount of MMP-2, which exacerbates fibrosis process through EndMT and direct degradation of ECM proteins (125).
Conversely, M2 is alternatively polarized to inhibit persistent inflammatory response (65). M2 polarization is governed by Th2 cytokines (IL-4, IL-10, IL-13, etc.) and results in fibrotic responses such as tissue remodeling (M2a), Th2 activation (M2b), and angiogenesis (M2d) (63). In order to create an environment for healing and regenerating, anti-inflammatory molecules are produced (TGF-β, IL-10, arginase, etc.) (128). However, if initial lesion prolonged, M2 exhibits a pro-fibrotic feature. As a result, multiple fibrotic molecules are produced by M2, including TGF-β, galectin-3, Ang II, Wnt, CTGF, and PDGF (63, 121, 125). The pro-fibrotic mechanisms of M2 signal involving EndMT, fibroblast-myofibroblast (FMT), cellular differentiation towards myofibroblasts, and fibrocytes proliferation (127). In addition, MMP expression was found diversely featured in M2, in comparison with M1. Scholars found that MMP-9 is overexpressed in M2, while M2 polarization results in MMP-12 elevation (129). Meanwhile, TIMP secretion accompanies. The above molecules and reactions support the fact that M2 contribute greatly to fibrosis. Nevertheless, blindly suppressing M2 polarization does not fully stand for alleviating fibrosis. Instead, persistent inflammation is why tissue repair being misled into fibrosis, therefore M1 and M2 are both heavily implicated in this process.
3.4.3. Potential molecules may mediate fibrosis as inflammatory regulator
While acute inflammation facilitates healing, chronic inflammation typically implies a failure to resolve itself. While inflammation prolonged, several molecules displayed their fibrotic associated roles. Aldo, for example, can activate Th17 cells via MRs, increasing IL-17A release in adventitia (66). As a result, transcription of neutrophil gelatinase-associated lipocalin (NGAL) in fibroblasts is promoted, facilitating the elevation of gal-3, triggering collagen synthesizing and depositing (66). To further assess the fibrotic process of IL-17A, researchers conducted a study on CD4-IL-17Aind/+ mice, which reflect the chronic overexpression of IL-17A in vivo. The results are explicit, since CD4-IL-17Aind/+ mice exhibit an increase of serum ROS, endothelial dysfunction, and aggravated fibroblasts proliferation (67). Additionally, in CD4-IL-17Aind/+ mice, NO/cGMP downregulation and T-cell associated protein tyrosine kinase 2 (PYK2) upregulation were observed in contrast to control mice (67). In general, IL-17A as a critical molecule in initiating myeloid cells infiltration into the arterial wall, suppressing which has considerable potential in avoiding vascular fibrosis.
An increase in PTEN (phosphatase and tensin homolog) has been shown to be protective against Ang II-induced immune cell infiltration and vascular fibrosis. A study on sPTEN mice (transgenic mice carrying additional Pten gene) discovered its beneficial impact in reducing pro-fibrotic genes expression (Tgfb1, Col1a1, Fn1, Mmp2, etc.) triggered by Ang II (68). Evidence suggests that PTEN negatively modulates PI3K/AKT, establishing a PTEN/PI3K/AKT axis to prevent pyroptosis and apoptosis (69, 70).
Gal-3, as mentioned before, exhibit additional effect in immunoregulation (130). Although we have described its more direct influence on fibrosis, there are other mechanisms by which gal-3 affects inflammation, such as inflammatory cell extravasation regulation via binding on CD11b (Macrophages) and CD66 (Neutrophils) (71, 72). Furthermore, gal-3 enhances IL-1 and IL-18 expression by serving as a PRR and DAMP, thereby boosting the NF-κB pathway and pyroptosis (73). Nonetheless, there is currently limited evidence on gal-3 indirectly promoting fibrosis through inflammation.
3.4.4. Extracellular vesicles and non-coding RNAs
Accumulating evidence suggesting non-coding RNA (ncRNA), including microRNA (miR) and lncRNA, have essential role in inflammatory and fibrosis related diseases. They can function as decoys, guides, or scaffolds in endothelial cells, contributing to the development and progression of vascular disorders (131). Meanwhile, extracellular vesicles (EVs) have been studied broadly in cardiovascular diseases, especially for their ncRNA-shipping-role in inflammatory induced fibrosis. EVs can be found in nearly every type of cell, and are composed of a lipid bilayer enclosing cargo of bioactive molecules which has significant meaning to intercellular communication (132). During atherogenesis, atherosclerosis plaque release EVs that cargos miR-23a-3p into serum, which later captured by ECs and accelerates endothelial inflammation via enhanced expression of ICAM-1, VCAM-1, E-selectin (74). Beyond that, EVs from CD4+ T cell and macrophage also facilitate a major part in pro-fibrosis. For example, macrophage derived EVs carrying with lncRNA GAS5 can lead to EC apoptosis, in response to excessive lipoproteins accumulation (75). Through targeting miR-138-5p/RhoC axis, EVs from macrophages show pro-fibrotic effect by transmitting circUbe3 (76). Furthermore, activated CD4+ T cells secretes EVs loading miR-142-3p, to contribute to fibrotic remodeling after myocardial infraction, via activation of myofibroblasts (77).
However, the significance of ncRNA goes beyond. miR-21 is abundantly expressed in mammalian cells, and is known for their anti-inflammatory effect. Novel findings show that miR-21 have essential role in regulating inflammation in ECs and VSMCs, targeting PPAR-α (78). miR-21 is also involved in cardiac fibrosis formation via TGFβ1/Smad3 pathway (79). Overexpressed miR-122 are found suppressive to PTEN, leaving an indication that miR-122 participates in pyroptosis. Similarly, studies found miR-122 knockdown alleviates apoptosis, inflammation, and OS (80), which suggests a viable target against fibrosis. miR-126 has special role in angiogenesis via VEGF signal activation. In addition, miR-126 displays certain anti-inflammatory feature, since its deficiency in EC results in TNF-α-induced VCAM1 expression, which mediates leukocyte adherence to EC (133). lincRNA-p21 is downregulated in atherosclerosis mice and patients with coronary artery disease, silencing which leads to VSMC and macrophage proliferation, yet apoptosis repression (78). LEENE represents for lncRNA that enhances endothelial nitric oxide synthase (eNOS) expression. LEENE works as an enhancer to mRNA promoter, which facilitates EC inflammation suppression (134). The lncRNA myocardial infarction associated transcript (MIAT) is upregulated in diabetic patient monocytes, and decreased in senescent fibroblasts. MIAT overexpression has shown relevance in high-glucose induced apoptosis, and diabetic pathological angiogenesis, reversing inhibition to VEGF by decoying miR-150 and miR-200a (131).
In summary, inflammation, in its diverse aspects, has enormous influence and closely related to vascular fibrosis. Deepening its understanding would be a major breakthrough in related pathological conditions.
3.5. Other mediators in vascular fibrosis
We have so far elaborated the molecular mechanisms of vascular fibrosis from several different aspects including RAAS and TGF-β, mitochondria, EndMT, as well as inflammation. However, there are several more factors closely related to vascular fibrosis that cannot be overlooked.
Marinobufagenin (MBG), an endogenous cardiotonic steroid being identified in mammalian extracellular fluids, promotes vascular fibrosis by inhibiting friend leukemia virus integration 1 (Fli-1) along with activating TGF-β1/Smad pathway (81). Since Fli-1 function as a nuclear inhibitory transcription factor to Col-1 gene, MBG elevation results in Col-1 overexpression (82). Interestingly, spironolactone has shown alleviation to the profibrotic effect of MBG (83). ROCK (Rho-associated coiled-coil forming protein kinases), which expresses in most vascular cells (ECs, VSMCs, fibroblasts, macrophages, and lymphocytes), has been shown to mediate cellular migration, adhesion, polarity, and cytokinesis via phosphorylating downstream cytoskeleton mediator proteins (84). ROCK suppresses eNOS (NO generation), promotes EndMT, switches VSMCs to a migratory, proliferative phenotype, and regulates M1/2 alternation in macrophages (84). Moreover, ROCK promotes inflammation and OS, since the upregulation of IL-6, TNF-α, IFN-γ, and SOD is accompanied with ROCK overexpression in sepsis models (85). Nonetheless, several drugs have been reported to be ROCK-resistant. Statins, for instance, can suppress RhoA/ROCK via competing TGF-β1/Smad activation, as well as p38-MAPK (86). A study has shown that PAI-1 (plasminogen activator inhibitor-1) mutant mice have higher levels of Col-1 expression and coronary perivascular fibrosis (87). Intriguingly, apelin, an endogenous peptide expressed by ECs, has the capacity to block Ang II induced collagen and MMP-2 expression as well as PAI-1-induced Rho kinase activation, indicating a role in anti-fibrosis (88).
4. Age and sex brought heterogeneousness to diverse fibrotic responses
Before putting clinical disorders into discussion, it is important to know that age and sex play a vital role in individual responses. When it comes to vascular fibrosis, age and sex exhibits unique characteristics in immune system and inflammation, collagen deposition, and eventually, fibrosis development (135, 136). The concept of those certain aspects being influenced by aging and sex will be generally explored in this section.
4.1. Age and sex in immune system and inflammation
Gender and age perform substantially in the immune response, influencing susceptibility to infections, vaccine response, and function in immune system (137). Aging is linked to the occurrence of long-lasting inflammation as well as a general decrease in immunological function (138).
First of all, there are certain variance in immune cells: NK cells are observed to be higher in male, while plasma cell levels are higher in female (139). The mechanisms within could be related to the prohibit effect of estrogen and progesterone on NK cells (140). Meanwhile, female exerts more activated T cells and B cells than male (141). These differences are claimed to be more significantly impacted through aging. Meanwhile, some research suggests aging to be linked with the increase in monocytes, decrease in T cells, and expression of PBMCs inflammatory markers (142).
Secondly, gene and molecular expression impacts the bias of gender and age difference in immune system. Aging influences pro-inflammatory genes expression, which is severer in male (138). According to recent studies, male experience more IFN-γ receptor activation and MAPK signal induced inflammations (139). They also discovered ZFP36 and DUSP2 are two genes that have the highest level in elder male, which represents weaker adaptive immunity, due to T cell inhibition (139). In addition, we have mentioned a more active T and B cell in female, which could be related to upregulated BAFF/APRIL system in women. While BAFF manages the viability of B cells, APRIL controls cellular proliferation which are essential in humoral immunity (139). Unfortunately, BAFF and APRIL levels lead to their increased risk in autoimmune diseases (143).
Lastly, male and female, young and elder, have distinct sensitivity in inflammation or infection. Aging is linked to the development of chronic inflammation as well as a general decrease in immunological function. Some research suggests that the innate immune system of elderly females is more prone to inflammation than that of elderly males (137). However, women have an enhanced immune response to viral infections than men, resulting in quicker pathogen clearance and increased vaccination effectiveness in females (144). For instance, men have a higher susceptibility to several infectious illnesses, as well as a higher fatality rate (139). After the age of 65, men and women's immune system cells differed the most (141). Older female functions slower in acute reaction or sickness yet shows resistance to certain long-term infections (141). Older men, on the other hand, exhibited higher levels of activity in the innate immune system, the body's generic but faster-reacting defense force (145).
4.2. Age and sex in collagen deposition
Age as well as gender may additionally impact collagen deposition in different tissues. Increased collagen and fibrin deposition occurred in elderly normal controls when compared to non-elderly controls, and this was even more pronounced in elderly patients with nasal polyps (146), which was further confirmed by studying model organisms such as mice and C. elegans (147). The deposition of collagen fibers after surgery is significantly reduced in older men but not in women (148). These results imply that age and sex can have a major effect on collagen deposition throughout different tissues and that analyzing these differences is vital for developing potent therapies for age-related diseases.
4.3. Age and sex in fibrosis development
It is eventually our consideration of fibrotic diseases for any heterogeneousness that gender and age may cause. Age-related alterations to the cardiac ECM differ by gender, with elderly male hearts being more fibrotic than female hearts (149). Gender-dependent cardiac fibrosis occurs with age in rats, but the underlying processes remain unknown (150). In those with atrial fibrillation, increasing age and female sex are correlated with a higher incidence of atrial fibrosis (151). We should additionally pay close attention to the fact that the occurrence of greater fibrosis in adult skeletal muscle cells increases dramatically with age, which is associated with TGF-β signaling (152). Similarly, it has been presented that female hormones can ameliorate the advancement of cystic fibrosis in the lung, and there is substantial evidence that estrogen plays an essential role in the development of cystic fibrosis (153). One issue that cannot be overlooked is that aging and diabetes are substantial risk factors for the development of penile fibrosis (154). The above findings emphasize the intricate relationships between age, gender, and the genesis of fibrosis in diverse tissues and disorders. These distinctions can aid the development of tailored and effective fibrotic treatments.
5. Vascular fibrosis correlated diseases and possible interventions: bench to bed
Fibrosis is a widely identified vascular condition that develops progressively and is linked to aging and hypertension. Understanding the processes behind vascular fibrosis may lead to the development of new treatment strategies, as vascular involvement in fibrosis pathophysiology is an area of ongoing research. However, there is currently no cure for vascular fibrosis, except there are some interventions that can manage the condition. In this section, we just highlight the associated disorders as well as provide our evaluation in order to establish a novel therapeutic approach for future clinical practice (Figure 3 for details).
Figure 3
5.1. Hypertension
Hypertension is the leading cause of morbidity and mortality worldwide, and is associated with an increased risk of cardiovascular disease. Normally, the heart ejects blood, and the arcus aortae dilate and buffer the systolic and diastolic pressures as they pass to provide appropriate tissue perfusion (155). This mechanism is commonly accepted to be strongly dependent on artery elasticity and compliance, whereas vascular fibrosis-induced arterial stiffness lacks this feature (1). It is especially unsettling since hypertension being a direct outcome of arterial remodeling and fibrosing, may be easily obtained through collagen deposition (36). The expression and proliferation of VSMCs can also lead to the thickening of the vessel wall, which enhances transverse aortic contraction and blood pressure (48).
There are several medications that alleviate hypertension through various methods, some of which grabbed our attention due to their anti-fibrosis properties. Firstly, alamandine can reduce blood pressure by preventing Ang II-induced arterial remodeling (36). Secondly, statins, as the inhibitors of the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, have been found to be pleiotropically beneficial against fibrosis and hypertension through inhibiting TGF-β/Smad (86), in addition to its well-known cholesterol-lowering activity. Their anti-fibrotic role leaves us a novel target for resistant hypertension.
Pulmonary arterial hypertension (PAH) is one particular hypertension in the lungs, defined as an ≥20 mmHg resting pulmonary arterial pressure in mean (156). Pulmonary illness and dyspnea are inseparable from PAH, partly due to pulmonary artery smooth muscle cell (PASMC) proliferation, inflammation, and severe fibrosis (73). Scholars show special interest in their latest discovery in a cluster of PAH-related mediators, including gal-3, StAR, caspase-1, and programmed death ligand-1 (PDL-1). PDL-1, along with caspase-1, is implicated in pyroptosis, which is associated with the production of fibrosis and PAH (58). Furthermore, the research found that NEDD9, a TGF-β independent mediator, can trigger fibrosis in PAH via oxidative modification at Cys 18 (157).
5.2. Vascular disorders
Atherosclerosis, one of the leading causes of CVD, is characterized by impaired degradation of ECM (collagen and fibronectin), VSMCs proliferation, as well as calcification (49), therefore lies connection with fibrosis. EndMT has been widely discovered in atherosclerosis due to its promotive effect in plaque formation. EndMT generates mesenchymal cells that produce pro-inflammatory cytokines, ECM proteins, and MMPs (158). As research deepens, certain chemicals strongly linked to atherosclerosis have been discovered. For example, gal-3 overexpresses in plaque, salusin-β TGF-β-dependently promotes Col-1/3 expression, and ncRNAs (miR-125b, miR-21, Let-7c, Let-7g) generally expresses in the thickened vascular wall (35, 49, 103). Intriguingly, a study on a natural substance paeonol revealed that it has a role in atherosclerosis suppression by enhancing gut microorganisms' short-chain fatty acids (SCFAs) synthesis to modulate Treg/Th17 balance in the spleen, hence preventing vascular fibrosis (159). Statins also alleviates atherosclerosis by down-regulating the expression of Ang II-induced profibrotic factors and ECM proteins (86). Furthermore, H2S and specific microRNAs (miR-483, miR-126, miR-155) have a certain role in alleviating vascular fibrosis and atherosclerosis. The experimental evidence shown above illustrates that the occurrence, progression, and therapy of atherosclerosis is a tremendously complicated process involving different routes and cytokines or active substances.
As mentioned above, vascular inflammation is highly associated with fibrosis. Takayasu's arteritis (TAK) is a chronic inflammatory disease characterized by granulomatous vasculitis and fibrosis in the aorta and its branches (47). Perfusion is hampered by persistent inflammation and fibrosis, which leads to organ failure and death (160). Multiple fibrotic biomarkers, including TGF-β, Col-1/3, fibronectin, and α-SMA, have been found elevated in TAK arteries than in normal arteries (161). Unfortunately, conventional therapies with glucocorticoids and immunosuppressants can only stabilize inflammation, but behaved badly against existing fibrosis-induced structural damage. Research has revealed that vascular fibrosis in TAK is promoted by IL-6 related autophagy via JAK1/STAT3 pathway, implying impeding autophagy, as well as IL-6 inhibitor, may be beneficial in the treatment of TAK (161, 162). Leflunomide (LEF), an anti-inflammatory medication, has been demonstrated to diminish TAK by blocking and lowering macrophage subtype M2 rather than M1 (160). Another study found that Curcumin and Etomoxir exhibit properties of anti-fibrosing, targeting FABP3 to modulate FAO in AAFs as a possible treatment for TAK and other vascular fibrosing conditions.
Furthermore, vascular fibrosis is found in Kawasaki disease (KD), a well-documented but poorly understood systemic pediatric vasculitis characterized by multisystem inflammatory syndrome (MIS), severe toxic shock syndrome, and coronary artery aneurysm (163, 164). In KD model mice, coronary artery inflammation pyroptosis-related factors (NLRP3, caspase-1, IL-1β, IL-18) exhibited a significant upregulation in ECs (59), showing EC pyroptosis participates in KD. Intriguingly, SARS-CoV-2 was identified to coincide with KD in MIS-C degree (163, 165), hence understanding the mechanisms of SARS-CoV-2 may be the potential key to unmasking KD. In addition, studies have demonstrated that gal-3 and TGF-β/Smad levels are much higher in KD patients, making them appealing therapeutic targets for KD and fibrotic dysfunction (166, 167).
5.3. Diabetes and endocrine disorders
Evidence has confirmed that there is a linkage between vascular fibrosis and endocrine disturbance. Diabetes, particularly type 2 diabetic mellitus (DM2), that shares consistency with vascular fibrosis according to multiple studies. On one hand, fibrosis directs DM: Experiencing ECM deposition, cytoarchitectural abnormalities, microvascular dysfunction, and impaired endocrine cell-blood exchanges in pancreatic islets lead to aberrant insulin secretion (168). Cell type modifications, such as the conversion of pericytes to fibroblasts and EndMT, are critical in triggering DM2 vascular fibrosis (49, 168). DM is also linked to autonomous cortisol secretion (ACS) in individuals with primary aldosteronism (PA) (169). PA patients with ACS had a considerably greater diabetes incidence and a larger fibrosis area in blood vessels, indicating that an ACS show-up implies poor vascular remodeling (169). On the other hand, DM2 lures fibrosis in aortic or other cardiovascular tissue: developing DM boosts myofibroblasts activation, inhibits macrophage M2 polarization, induces a profibrotic phenotype in immune cells, increases ROS, and initiates EndMT (81, 170, 171). Interestingly, various fibrotic-associated molecular alterations were observed in salt-loaded DM2 mice, including increased TGF-β, Col-1, and fibronectin, as well as decreased Fli-1, which could be reversed by MBG (81, 83). In salt-loaded DM2 patients, experiencing a Na+–K+–ATPase malfunction might be the cause of vascular injury and remodeling. MBG, as its inhibitor, contributes to this pathological condition by suppressing it, therefore benefiting DM2 (81). In addition, rosiglitazone as a classic drug on insulin resistance DM2 patient, have attached repressive impact on Ang II-induced AT-1R, IL-6, and TNF-α elevation (172), as well as VSMCs and myofibroblasts proliferation (173, 174). These changes vastly benefit vascular fibrosis. Another study indicates rosiglitazone, as a PPAR-γ activator, benefits in DM2 by lowering CTGF expression and ECM deposition in Ang II-induced fibrosis (175). However, along with other thiazolidinediones (TZDs), rosiglitazone may be associated with severe side effects including heart failure and pulmonary edema (176), therefore its clinical application is restricted and must be paid particular attention. Nevertheless, many research suggest rosiglitazone attenuative to serum level of fibronectin, TGF-β, PAI-1, and inflammatory marker (MMP-2, MMP-9, COX-2) in DM2 patients (177, 178). Based on the evidence presented above, we may deduce that there is a pertinent association between diabetes mellitus and vascular fibrosis, and that this link impacts the prognosis and progression of the disease, while also providing a novel target for vascular fibrosis treatment.
5.4. Parenchymal organ lesions
Since myocardial fibrosis is frequently coupled with vascular fibrosis, both molecular mechanisms and pathological processes are intertwined. Chronic hypertension and arteriosclerosis increase cardiac afterload, leading to myocardial hypertrophy, coronary heart disease, heart failure, and other complications. In the aging kidney, T and B cells, together with resident fibroblasts, form tertiary lymphoid tissues that cause uncontrollable inflammation and delay tissue repair (179). Furthermore, severe fibrosis of small blood vessels can cause parenchymal tissues and cells to grow, leading to fatal diseases including liver cirrhosis and brain fibrosis (58). However, fibrosis is not a one-way road. To slow the process of organ fibrosis, ECM protein degradation, chronic inflammation regression, tissue injury resolution, and myofibroblasts deactivation are all required (180). Currently, a number of anti-fibrosis medications are being tested in clinical trials. Nintedanib is an antifibrotic drug licensed for patients with idiopathic pulmonary fibrosis (IPF) by blocking several tyrosine kinases implicated in fibrosis development (181). Another anti-fibrotic medicine, Pirfenidone, has been licensed for use in individuals with IPF to decrease disease development by reducing the production of pro-fibrotic cytokines and growth factors (182). It should be emphasized that lowering oxidative stress can also help slow or stop fibrosis progression (183). As previously stated, some immune modulation is involved in the genesis of fibrosis (184).
6. Limitation and challenge
Despite numerous experimental research on vascular fibrosis, it remains a limitation when it comes to clinical trials and treatments (185). To begin with, although several medications have been explored for fibrosis therapy, there are presently no viable therapeutics for vascular fibrosis. This emphasizes the importance of additional research to uncover novel treatment targets (186). The second and most essential reason is that the processes of vascular sclerosis are still poorly understood. Although the processes of vascular fibrosis have been established, the fundamental mechanisms are still poorly understood (186). The third issue that can't be overlooked is the lack of a reliable biomarker for vascular fibrosis diagnosis and monitoring (187). Finally, despite several clinical cohort studies that have been conducted, there is currently a paucity of viable treatments for vascular fibrosis (188). Many anti-fibrosis medicines have been tested exclusively on animals and have shown promising results, but the transition from bench to bed remains a lengthy time. Based on the facts stated above, we have reason to believe that there are still particular challenges that will necessitate collaboration between researchers, doctors, and clinical trials in the treatment of vascular fibrosis.
7. Conclusion
Fibrosis affects almost all tissues in the body. Although fibrosis is frequently associated with strong inflammatory response, growing studies indicate that there are particular mediators and pathways contributing to the pathophysiology of fibrosis that are differ from the inflammatory processes. We must first recognize this distinction before developing effective therapies for fibrotic illness. In this review, we emphasized the underlying changes in structure and function in vascular fibrosis, concluded some recently discovered or solid mediators, and illustrated a few related ailments with prospective interventions. Vascular fibrosis, characterized by ECM deposition, VSMC proliferation, hindered matrix breakdown, and EC dysfunction, causes numerous dysfunctions in cardiovascular and other systems via RAAS, TGF-β, MMPs, and other linked mediators. Targeting on these mediators may be of valuable means in discovering related treatments. Despite decades of research in vascular fibrosis, therapeutic drugs have always been few or ineffective, especially to pre-existing fibrosis. Due to the complexity and diversity of the vascular system, it is difficult for a single element to be successful and target-specific in the overall rehabilitation of vascular fibrosis. This is why we believe the understanding presented in this review would be especially beneficial to researchers in developing practical and therapeutic medicines or methods for vascular fibrosis-related disorders.
Statements
Author contributions
RH: Formal analysis, Writing – original draft, Investigation, Writing – review & editing. HG: Conceptualization, Formal analysis, Writing – original draft. CH: Formal analysis, Resources, Writing – original draft. SX: Conceptualization, Formal analysis, Writing – original draft. BW: Formal analysis, Validation, Writing – original draft. SZ: Conceptualization, Supervision, Writing – review & editing. LG: Software, Writing – review & editing. QT: Supervision, Visualization, Writing – review & editing. WW: Investigation, Visualization, Writing – review & editing. FS: Formal analysis, Validation, Writing – review & editing. JX: Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by the National Natural Science Foundation of China Grant (No. 82174056 JDX).
Acknowledgments
We are especially grateful to Dr. Tao Xin, Department of Applied Linguistics, Capital University of Medical Sciences, for polishing the language of this manuscript and making it more readable.
Conflict of interest
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
HarveyAMontezanoACLopesRARiosFTouyzRM. Vascular fibrosis in aging and hypertension: molecular mechanisms and clinical implications. Can J Cardiol. (2016) 32(5):659–68. 10.1016/j.cjca.2016.02.070
2.
LanTHHuangXQTanHM. Vascular fibrosis in atherosclerosis. Cardiovasc Pathol. (2013) 22(5):401–7. 10.1016/j.carpath.2013.01.003
3.
SelvinENajjarSSCornishTCHalushkaMK. A comprehensive histopathological evaluation of vascular medial fibrosis: insights into the pathophysiology of arterial stiffening. Atherosclerosis. (2010) 208(1):69–74. 10.1016/j.atherosclerosis.2009.06.025
4.
KendallRTFeghali-BostwickCA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. (2014) 5(123):1–13. 10.3389/fphar.2014.00123
5.
KitaniAFussINakamuraKKumakiFUsuiTStroberW. Transforming growth factor (TGF)-beta1-producing regulatory T cells induce Smad-mediated interleukin 10 secretion that facilitates coordinated immunoregulatory activity and amelioration of TGF-beta1-mediated fibrosis. J Exp Med. (2003) 198(8):1179–88. 10.1084/jem.20030917
6.
TabitCEChungWBHamburgNMVitaJA. Endothelial dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev Endocr Metab Disord. (2010) 11(1):61–74. 10.1007/s11154-010-9134-4
7.
PugsleyMKTabrizchiR. The vascular system: an overview of structure and function. J Pharmacol Toxicol Methods. (2000) 44(2):333–40. 10.1016/s1056-8719(00)00125-8
8.
PatilMSCartlandSPKavurmaMM. TRAIL signals, extracellular matrix and vessel remodelling. Vasc Biol. (2020) 2(1):R73–84. 10.1530/VB-20-0005
9.
SimionescuMSimionescuNPaladeGE. Differentiated microdomains on the luminal surface of capillary endothelium: distribution of lectin receptors. J Cell Biol. (1982) 94(2):406–13. 10.1083/jcb.94.2.406
10.
SimsDE. The pericyte—a review. Tissue Cell. (1986) 18(2):153–74. 10.1016/0040-8166(86)90026-1
11.
KelleyCD'AmorePHechtmanHBSheproD. Microvascular pericyte contractility in vitro: comparison with other cells of the vascular wall. J Cell Biol. (1987) 104(3):483–90. 10.1083/jcb.104.3.483
12.
MilutinovicASuputDZorc-PleskovicR. Pathogenesis of atherosclerosis in the tunica intima, media, and adventitia of coronary arteries: an updated review. Bosn J Basic Med Sci. (2020) 20(1):21–30. 10.17305/bjbms.2019.4320
13.
StenmarkKRYeagerMEEl KasmiKCNozik-GrayckEGerasimovskayaEVLiMet alThe adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol. (2013) 75:23–47. 10.1146/annurev-physiol-030212-183802
14.
LakattaEG. The reality of aging viewed from the arterial wall. Artery Res. (2013) 7(2):73–80. 10.1016/j.artres.2013.01.003
15.
QianCSunYJiangJ. Diagnostic values of epicardial adipose tissue thickness with right common carotid artery elasticity and intima-media thickness for middle-aged and elderly patients with coronary heart disease. Int J Gen Med. (2021) 14:633–9. 10.2147/IJGM.S292426
16.
HsuTNguyen-TranHHTrojanowskaM. Active roles of dysfunctional vascular endothelium in fibrosis and cancer. J Biomed Sci. (2019) 26(1):86. 10.1186/s12929-019-0580-3
17.
JiaGAroorARHillMASowersJR. Role of renin–angiotensin–aldosterone system activation in promoting cardiovascular fibrosis and stiffness. Hypertension. (2018) 72(3):537–48. 10.1161/HYPERTENSIONAHA.118.11065
18.
HoriuchiMAkishitaMDzauVJ. Recent progress in angiotensin II type 2 receptor research in the cardiovascular system. Hypertension. (1999) 33(2):613–21. 10.1161/01.hyp.33.2.613
19.
ZhangYYYuYYuC. Antifibrotic roles of RAAS blockers: update. Adv Exp Med Biol. (2019) 1165:671–91. 10.1007/978-981-13-8871-2_33
20.
PatelSRaufAKhanHAbu-IzneidT. Renin–angiotensin–aldosterone (RAAS): the ubiquitous system for homeostasis and pathologies. Biomed Pharmacother. (2017) 94:317–25. 10.1016/j.biopha.2017.07.091
21.
WuJSalehMAKiraboAItaniHAMontanielKRXiaoLet alImmune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. (2016) 126(1):50–67. 10.1172/JCI80761
22.
NiWYangXYangDBaoJLiRXiaoYet alRole of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care. (2020) 24(1):422. 10.1186/s13054-020-03120-0
23.
SantosRASSampaioWOAlzamoraACMotta-SantosDAleninaNBaderMet alThe ACE2/angiotensin-(1-7)/MAS axis of the renin–angiotensin system: focus on angiotensin-(1-7). Physiol Rev. (2018) 98(1):505–53. 10.1152/physrev.00023.2016
24.
KuriakoseJMontezanoACTouyzRM. ACE2/Ang-(1-7)/Mas1 Axis and the vascular system: vasoprotection to COVID-19-associated vascular disease. Clin Sci (Lond). (2021) 135(2):387–407. 10.1042/CS20200480
25.
HuHHChenDQWangYNFengYLCaoGVaziriNDet alNew insights into TGF-beta/smad signaling in tissue fibrosis. Chem Biol Interact. (2018) 292:76–83. 10.1016/j.cbi.2018.07.008
26.
TzavlakiKMoustakasA. TGF-beta signaling. Biomolecules. (2020) 10(3):487. 10.3390/biom10030487
27.
WangXWangYZhangJGuanXChenMLiYet alGalectin-3 contributes to vascular fibrosis in monocrotaline-induced pulmonary arterial hypertension rat model. J Biochem Mol Toxicol. (2017) 31(5):e21879. 10.1002/jbt.21879
28.
SchaferSViswanathanSWidjajaAALimWWMoreno-MoralADeLaughterDMet alIL-11 is a crucial determinant of cardiovascular fibrosis. Nature. (2017) 552(7683):110–5. 10.1038/nature24676
29.
SchumacherDLiehnEANilchamPMayanDCRattanasopaCAnandKet alA neutralizing IL-11 antibody reduces vessel hyperplasia in a mouse carotid artery wire injury model. Sci Rep. (2021) 11(1):20674. 10.1038/s41598-021-99880-y
30.
GiannandreaMParksWC. Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech. (2014) 7(2):193–203. 10.1242/dmm.012062
31.
OkadaMKosakaNHoshinoYYamawakiHHaraY. Effects of captopril and telmisartan on matrix metalloproteinase-2 and -9 expressions and development of left ventricular fibrosis induced by isoprenaline in rats. Biol Pharm Bull. (2010) 33(9):1517–21. 10.1248/bpb.33.1517
32.
WangZGriffinM. TG2, A novel extracellular protein with multiple functions. Amino Acids. (2012) 42(2–3):939–49. 10.1007/s00726-011-1008-x
33.
SteppanJBergmanYViegasKArmstrongDTanSWangHet alTissue transglutaminase modulates vascular stiffness and function through crosslinking-dependent and crosslinking-independent functions. J Am Heart Assoc. (2017) 6(2):e004161. 10.1161/JAHA.116.004161
34.
SatoMKamadaYTakedaYKidaSOharaYFujiiHet alFetuin-A negatively correlates with liver and vascular fibrosis in nonalcoholic fatty liver disease subjects. Liver Int. (2015) 35(3):925–35. 10.1111/liv.12478
35.
SunHJLiuTYZhangFXiongXQWangJJChenQet alSalusin-beta contributes to vascular remodeling associated with hypertension via promoting vascular smooth muscle cell proliferation and vascular fibrosis. Biochim Biophys Acta. (2015) 1852(9):1709–18. 10.1016/j.bbadis.2015.05.008
36.
YangCWuXShenYLiuCKongXLiP. Alamandine attenuates angiotensin II-induced vascular fibrosis via inhibiting p38 MAPK pathway. Eur J Pharmacol. (2020) 883:173384. 10.1016/j.ejphar.2020.173384
37.
BarmanSALiXHaighSKondrikovDMahboubiKBordanZet alGalectin-3 is expressed in vascular smooth muscle cells and promotes pulmonary hypertension through changes in proliferation, apoptosis, and fibrosis. Am J Physiol Lung Cell Mol Physiol. (2019) 316(5):L784–97. 10.1152/ajplung.00186.2018
38.
CalvierLMianaMReboulPCachofeiroVMartinez-MartinezEde BoerRAet alGalectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler, Thromb, Vasc Biol. (2013) 33(1):67–75. 10.1161/ATVBAHA.112.300569
39.
MaronBAOldhamWMChanSYVargasSOAronsEZhangYYet alUpregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation. (2014) 130(2):168–79. 10.1161/CIRCULATIONAHA.113.007690
40.
ParkJBSchiffrinEL. Cardiac and vascular fibrosis and hypertrophy in aldosterone-infused rats: role of endothelin-1. Am J Hypertens. (2002) 15(2 Pt 1):164–9. 10.1016/s0895-7061(01)02291-9
41.
PanduriVLiuGSurapureddiSKondapalliJSoberanesSde Souza-PintoNCet alRole of mitochondrial hOGG1 and aconitase in oxidant-induced lung epithelial cell apoptosis. Free Radic Biol Med. (2009) 47(6):750–9. 10.1016/j.freeradbiomed.2009.06.010
42.
PeoplesJNSarafAGhazalNPhamTTKwongJQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. (2019) 51(12):1–13. 10.1038/s12276-019-0355-7
43.
BhattiJSBhattiGKReddyPH. Mitochondrial dysfunction and oxidative stress in metabolic disorders—a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. (2017) 1863(5):1066–77. 10.1016/j.bbadis.2016.11.010
44.
Souza-NetoFVJimenez-GonzalezSDelgado-ValeroBJurado-LopezRGentyMRomero-MirandaAet alThe interplay of mitochondrial oxidative stress and endoplasmic Reticulum stress in cardiovascular fibrosis in obese rats. Antioxidants. (2021) 10(8):1274. 10.3390/antiox10081274
45.
NingRLiYDuZLiTSunQLinLet alThe mitochondria-targeted antioxidant MitoQ attenuated PM2.5-induced vascular fibrosis via regulating mitophagy. Redox Biol. (2021) 46:102113. 10.1016/j.redox.2021.102113
46.
PengSYTsaiCHWuXMHuangHHChenZWLeeBCet alAldosterone suppresses endothelial mitochondria through mineralocorticoid receptor/mitochondrial reactive oxygen Species pathway. Biomedicines. (2022) 10(5):1119. 10.3390/biomedicines10051119
47.
WuSKongXSunYDaiXYuWChenRet alFABP3 Overexpression promotes vascular fibrosis in Takayasu’s arteritis by enhancing fatty acid oxidation in aorta adventitial fibroblasts. Rheumatology. (2022) 61(7):3071–81. 10.1093/rheumatology/keab788
48.
ZhuangLMaoYLiuZLiCJinQLuLet alFABP3 Deficiency exacerbates metabolic derangement in cardiac hypertrophy and heart failure via PPARalpha pathway. Front Cardiovasc Med. (2021) 8:722908. 10.3389/fcvm.2021.722908
49.
TestaiLBrancaleoneVFloriLMontanaroRCalderoneV. Modulation of EndMT by hydrogen sulfide in the prevention of cardiovascular fibrosis. Antioxidants. (2021) 10(6):910. 10.3390/antiox10060910
50.
OkayamaKAzumaJDosakaNIekushiKSanadaFKusunokiHet alHepatocyte growth factor reduces cardiac fibrosis by inhibiting endothelial-mesenchymal transition. Hypertension. (2012) 59(5):958–65. 10.1161/HYPERTENSIONAHA.111.183905
51.
ChrobakILennaSStawskiLTrojanowskaM. Interferon-gamma promotes vascular remodeling in human microvascular endothelial cells by upregulating endothelin (ET)-1 and transforming growth factor (TGF) beta2. J Cell Physiol. (2013) 228(8):1774–83. 10.1002/jcp.24337
52.
CiprianiPDi BenedettoPRuscittiPCapeceDZazzeroniFLiakouliVet alThe endothelial-mesenchymal transition in systemic sclerosis is induced by endothelin-1 and transforming growth factor-beta and may be blocked by macitentan, a dual endothelin-1 receptor antagonist. J Rheumatol. (2015) 42(10):1808–16. 10.3899/jrheum.150088
53.
ChenDLiLWangYXuRPengSZhouLet alIschemia-reperfusion injury of brain induces endothelial-mesenchymal transition and vascular fibrosis via activating let-7i/TGF-betaR1 double-negative feedback loop. FASEB J. (2020) 34(5):7178–91. 10.1096/fj.202000201R
54.
XueHYuanPNiJLiCShaoDLiuJet alH(2)S inhibits hyperglycemia-induced intrarenal renin–angiotensin system activation via attenuation of reactive oxygen species generation. PloS One. (2013) 8(9):e74366. 10.1371/journal.pone.0074366
55.
LinFYangYWeiSHuangXPengZKeXet alHydrogen sulfide protects against high glucose-induced human umbilical vein endothelial cell injury through activating PI3K/Akt/eNOS pathway. Drug Des Devel Ther. (2020) 14:621–33. 10.2147/DDDT.S242521
56.
WuTLiHWuBZhangLWuSWWangJNet alHydrogen sulfide reduces recruitment of CD11b(+)Gr-1(+) cells in mice with myocardial infarction. Cell Transplant. (2017) 26(5):753–64. 10.3727/096368917X695029
57.
GeroDTorregrossaRPerryAWatersALe-TrionnaireSWhatmoreJLet alThe novel mitochondria-targeted hydrogen sulfide (H2S) donors AP123 and AP39 protect against hyperglycemic injury in microvascular endothelial cells in vitro. Pharmacol Res. (2016) 113(Pt A):186–98. 10.1016/j.phrs.2016.08.019
58.
ZhangMXinWYuYYangXMaCZhangHet alProgrammed death-ligand 1 triggers PASMCs pyroptosis and pulmonary vascular fibrosis in pulmonary hypertension. J Mol Cell Cardiol. (2020) 138:23–33. 10.1016/j.yjmcc.2019.10.008
59.
JiaCZhangJChenHZhugeYChenHQianFet alEndothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. (2019) 10(10):778. 10.1038/s41419-019-2021-3
60.
PanPShenMYuZGeWChenKTianMet alSARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun. (2021) 12(1):4664. 10.1038/s41467-021-25015-6
61.
TakagakiYLeeSMDongqingZKitadaMKanasakiKKoyaD. Endothelial autophagy deficiency induces IL6—dependent endothelial mesenchymal transition and organ fibrosis. Autophagy. (2020) 16(10):1905–14. 10.1080/15548627.2020.1713641
62.
WenJHLiDYLiangSYangCTangJXLiuHF. Macrophage autophagy in macrophage polarization, chronic inflammation and organ fibrosis. Front Immunol. (2022) 13:946832. 10.3389/fimmu.2022.946832
63.
Shapouri-MoghaddamAMohammadianSVaziniHTaghadosiMEsmaeiliSAMardaniFet alMacrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233(9):6425–40. 10.1002/jcp.26429
64.
SicaAMantovaniA. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122(3):787–95. 10.1172/JCI59643
65.
BashirSSharmaYElahiAKhanF. Macrophage polarization: the link between inflammation and related diseases. Inflamm Res. (2016) 65(1):1–11. 10.1007/s00011-015-0874-1
66.
LeopoldJA. The central role of neutrophil gelatinase-associated lipocalin in cardiovascular fibrosis. Hypertension. (2015) 66(1):20–2. 10.1161/HYPERTENSIONAHA.115.05479
67.
SchulerREfentakisPWildJLagrangeJGarlapatiVMolitorMet alT cell-derived IL-17A induces vascular dysfunction via perivascular fibrosis formation and dysregulation of (.)NO/cGMP signaling. Oxid Med Cell Longev. (2019) 2019:6721531. 10.1155/2019/6721531
68.
LuSStrandKAMutrynMFTuckerRMJollyAJFurgesonSBet alPTEN (phosphatase and tensin homolog) protects against Ang II (angiotensin II)-induced pathological vascular fibrosis and remodeling-brief report. Arterioscler Thromb Vasc Biol. (2020) 40(2):394–403. 10.1161/ATVBAHA.119.313757
69.
LiuBHTuYNiGXYanJYueLLiZLet alTotal flavones of Abelmoschus manihot ameliorates podocyte pyroptosis and injury in high glucose conditions by targeting METTL3-dependent m(6)A modification-mediated NLRP3-inflammasome activation and PTEN/PI3K/Akt signaling. Front Pharmacol. (2021) 12:667644. 10.3389/fphar.2021.667644
70.
LiuHYZhangYYZhuBLFengFZYanHZhangHYet almiR-21 regulates the proliferation and apoptosis of ovarian cancer cells through PTEN/PI3K/AKT. Eur Rev Med Pharmacol Sci. (2019) 23(10):4149–55. 10.26355/eurrev_201905_17917
71.
DongSHughesRC. Macrophage surface glycoproteins binding to galectin-3 (Mac-2-antigen). Glycoconj J. (1997) 14(2):267–74. 10.1023/a:1018554124545
72.
SatoSOuelletNPelletierISimardMRancourtABergeronMG. Role of galectin-3 as an adhesion molecule for neutrophil extravasation during streptococcal pneumonia. J Immunol. (2002) 168(4):1813–22. 10.4049/jimmunol.168.4.1813
73.
BarmanSABordanZBatoriRHaighSFultonDJR. Galectin-3 promotes ROS, inflammation, and vascular fibrosis in pulmonary arterial hypertension. Adv Exp Med Biol. (2021) 1303:13–32. 10.1007/978-3-030-63046-1_2
74.
PengMSunRHongYWangJXieYZhangXet alExtracellular vesicles carrying proinflammatory factors may spread atherosclerosis to remote locations. Cell Mol Life Sci. (2022) 79(8):430. 10.1007/s00018-022-04464-2
75.
WangYZhaoMLiuSGuoJLuYChengJet alMacrophage-derived extracellular vesicles: diverse mediators of pathology and therapeutics in multiple diseases. Cell Death Dis. (2020) 11(10):924. 10.1038/s41419-020-03127-z
76.
ZhuJWangSYangDXuWQianH. Extracellular vesicles: emerging roles, biomarkers and therapeutic strategies in fibrotic diseases. J Nanobiotechnol. (2023) 21(1):164. 10.1186/s12951-023-01921-3
77.
CaiLChaoGLiWZhuJLiFQiBet alActivated CD4(+) T cells-derived exosomal miR-142-3p boosts post-ischemic ventricular remodeling by activating myofibroblast. Aging. (2020) 12(8):7380–96. 10.18632/aging.103084
78.
OnoKHorieTBabaOKimuraMTsujiSRodriguezRRet alFunctional non-coding RNAs in vascular diseases. FEBS J. (2021) 288(22):6315–30. 10.1111/febs.15678
79.
ZhangYYuanBXuYZhouNZhangRLuLet alMiR-208b/miR-21 promotes the progression of cardiac fibrosis through the activation of the TGF-beta1/Smad-3 signaling pathway: an in vitro and in vivo study. Front Cardiovasc Med. (2022) 9:924629. 10.3389/fcvm.2022.924629
80.
LiuYSongJWLinJYMiaoRZhongJC. Roles of MicroRNA-122 in cardiovascular fibrosis and related diseases. Cardiovasc Toxicol. (2020) 20(5):463–73. 10.1007/s12012-020-09603-4
81.
FedorovaOVFadeevAVGrigorovaYNMarshallCAZernetkinaVKolodkinNIet alCardiotonic steroids induce vascular fibrosis via pressure-independent mechanism in NaCl-loaded diabetic rats. J Cardiovasc Pharmacol. (2019) 74(5):436–42. 10.1097/FJC.0000000000000730
82.
NikitinaERMikhailovAVNikandrovaESFrolovaEVFadeevAVShmanVVet alIn preeclampsia endogenous cardiotonic steroids induce vascular fibrosis and impair relaxation of umbilical arteries. J Hypertens. (2011) 29(4):769–76. 10.1097/HJH.0b013e32834436a7
83.
FedorovaOVEmelianovIVBagrovKAGrigorovaYNWeiWJuhaszOet alMarinobufagenin-induced vascular fibrosis is a likely target for mineralocorticoid antagonists. J Hypertens. (2015) 33(8):1602–10. 10.1097/HJH.0000000000000591
84.
YuBSladojevicNBlairJELiaoJK. Targeting Rho-associated coiled-coil forming protein kinase (ROCK) in cardiovascular fibrosis and stiffening. Expert Opin Ther Targets. (2020) 24(1):47–62. 10.1080/14728222.2020.1712593
85.
WangYWangXYangWZhaoXZhangR. Effect of simvastatin on the intestinal Rho/ROCK signaling pathway in rats with sepsis. J Surg Res. (2018) 232:531–8. 10.1016/j.jss.2018.07.016
86.
Rodrigues DiezRRodrigues-DiezRLavozCRayego-MateosSCivantosERodriguez-VitaJet alStatins inhibit Angiotensin II/Smad pathway and related vascular fibrosis, by a TGF-beta-independent process. PloS One. (2010) 5(11):e14145. 10.1371/journal.pone.0014145
87.
KaikitaKFogoABMaLSchoenhardJABrownNJVaughanDE. Plasminogen activator inhibitor-1 deficiency prevents hypertension and vascular fibrosis in response to long-term nitric oxide synthase inhibition. Circulation. (2001) 104(7):839–44. 10.1161/hc3301.092803
88.
SiddiqueeKHamptonJKhanSZadoryDGleavesLVaughanDEet alApelin protects against Angiotensin II-induced cardiovascular fibrosis and decreases plasminogen activator inhibitor type-1 production. J Hypertens. (2011) 29(4):724–31. 10.1097/HJH.0b013e32834347de
89.
GhatageTGoyalSGDharABhatA. Novel therapeutics for the treatment of hypertension and its associated complications: peptide- and nonpeptide-based strategies. Hypertens Res. (2021) 44(7):740–55. 10.1038/s41440-021-00643-z
90.
DongRQWangZFZhaoCGuHRHuZWXieJet alToll-like receptor 4 knockout protects against isoproterenol-induced cardiac fibrosis: the role of autophagy. J Cardiovasc Pharmacol Ther. (2015) 20(1):84–92. 10.1177/1074248414539564
91.
SuJZFukudaNJinXQLaiYMSuzukiRTahiraYet alEffect of AT2 receptor on expression of AT1 and TGF-beta receptors in VSMCs from SHR. Hypertension. (2002) 40(6):853–8. 10.1161/01.hyp.0000042096.17141.b1
92.
BaloghMAguilarCNguyenNTShepherdAJ. Angiotensin receptors and neuropathic pain. Pain Rep. (2021) 6(1):e869. 10.1097/PR9.0000000000000869
93.
RiceASCDworkinRHMcCarthyTDAnandPBountraCMcCloudPIet alEMA401, An orally administered highly selective Angiotensin II type 2 receptor antagonist, as a novel treatment for postherpetic neuralgia: a randomised, double-blind, placebo-controlled phase 2 clinical trial. Lancet. (2014) 383(9929):1637–47. 10.1016/S0140-6736(13)62337-5
94.
KhajehpourSAghazadeh-HabashiA. Targeting the protective arm of the renin–angiotensin system: focused on Angiotensin-(1-7). J Pharmacol Exp Ther. (2021) 377(1):64–74. 10.1124/jpet.120.000397
95.
ZengWChenWLengXHeJGMaH. Chronic Angiotensin-(1-7) administration improves vascular remodeling after angioplasty through the regulation of the TGF-beta/Smad signaling pathway in rabbits. Biochem Biophys Res Commun. (2009) 389(1):138–44. 10.1016/j.bbrc.2009.08.112
96.
PatelVBPutkoBWangZZhongJCOuditGY. Manipulating angiotensin metabolism with angiotensin converting enzyme 2 (ACE2) in heart failure. Drug Discov Today Ther Strateg. (2012) 9(4):e141–148. 10.1016/j.ddstr.2013.11.001
97.
PaulisLRajkovicovaRSimkoF. New developments in the pharmacological treatment of hypertension: dead-end or a glimmer at the horizon?Curr Hypertens Rep. (2015) 17(6):557. 10.1007/s11906-015-0557-x
98.
CastardeliCSartorioCLPimentelEBForechiLMillJG. The ACE 2 activator diminazene aceturate (DIZE) improves left ventricular diastolic dysfunction following myocardial infarction in rats. Biomed Pharmacother. (2018) 107:212–8. 10.1016/j.biopha.2018.07.170
99.
DengYDingWPengQWangWDuanRZhangY. Advancement in beneficial effects of AVE 0991: a brief review. Mini Rev Med Chem. (2023) 24(2):139–58. 10.2174/1389557523666230328134932
100.
LiAZhangJZhangXWangJWangSXiaoXet alAngiotensin II induces connective tissue growth factor expression in human hepatic stellate cells by a transforming growth factor beta-independent mechanism. Sci Rep. (2017) 7(1):7841. 10.1038/s41598-017-08334-x
101.
Rodriguez-VitaJSanchez-LopezEEstebanVRuperezMEgidoJRuiz-OrtegaM. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation. (2005) 111(19):2509–17. 10.1161/01.CIR.0000165133.84978.E2
102.
JirakPMirnaMWernlyBPaarVThiemeMBetgeSet alAnalysis of novel cardiovascular biomarkers in patients with peripheral artery disease. Minerva Med. (2018) 109(6):443–50. 10.23736/S0026-4806.18.05628-8
103.
AnyfantiPGkaliagkousiEGavriilakiETriantafyllouADolgyrasPGalanopoulouVet alAssociation of galectin-3 with markers of myocardial function, atherosclerosis, and vascular fibrosis in patients with rheumatoid arthritis. Clin Cardiol. (2019) 42(1):62–8. 10.1002/clc.23105
104.
GeervlietEMorenoSBaiamonteLBooijinkRBoyeSWangPet alMatrix metalloproteinase-1 decorated polymersomes, a surface-active extracellular matrix therapeutic, potentiates collagen degradation and attenuates early liver fibrosis. J Control Release. (2021) 332:594–607. 10.1016/j.jconrel.2021.03.016
105.
NewbyAC. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev. (2005) 85(1):1–31. 10.1152/physrev.00048.2003
106.
WangMKimSHMonticoneRELakattaEG. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension. (2015) 65(4):698–703. 10.1161/HYPERTENSIONAHA.114.03618
107.
Souza-NetoFVIslasFJimenez-GonzalezSLuacesMRamchandaniBRomero-MirandaAet alMitochondrial oxidative stress promotes cardiac remodeling in myocardial infarction through the activation of endoplasmic Reticulum stress. Antioxidants. (2022) 11(7):1232. 10.3390/antiox11071232
108.
BuenoMCalyecaJRojasMMoraAL. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. (2020) 33:101509. 10.1016/j.redox.2020.101509
109.
NingRShiYJiangJLiangSXuQDuanJet alMitochondrial dysfunction drives persistent vascular fibrosis in rats after short-term exposure of PM2.5. Sci Total Environ. (2020) 733:139135. 10.1016/j.scitotenv.2020.139135
110.
LiMXianHCTangYJLiangXHTangYL. Fatty acid oxidation: driver of lymph node metastasis. Cancer Cell Int. (2021) 21(1):339. 10.1186/s12935-021-02057-w
111.
Larson-CaseyJLHeCCarterAB. Mitochondrial quality control in pulmonary fibrosis. Redox Biol. (2020) 33:101426. 10.1016/j.redox.2020.101426
112.
LiXZhangWCaoQWangZZhaoMXuLet alMitochondrial dysfunction in fibrotic diseases. Cell Death Discov. (2020) 6:80. 10.1038/s41420-020-00316-9
113.
RangarajanSBernardKThannickalVJ. Mitochondrial dysfunction in pulmonary fibrosis. Ann Am Thorac Soc. (2017) 14(Supplement_5):S383–388. 10.1513/AnnalsATS.201705-370AW
114.
MengGMaYXieLFerroAJiY. Emerging role of hydrogen sulfide in hypertension and related cardiovascular diseases. Br J Pharmacol. (2015) 172(23):5501–11. 10.1111/bph.12900
115.
YuPZhangXLiuNTangLPengCChenX. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. (2021) 6(1):128. 10.1038/s41392-021-00507-5
116.
WangYYLiuXLZhaoR. Induction of pyroptosis and its implications in cancer management. Front Oncol. (2019) 9:971. 10.3389/fonc.2019.00971
117.
DevantPCaoAKaganJC. Evolution-inspired redesign of the LPS receptor caspase-4 into an interleukin-1beta converting enzyme. Sci Immunol. (2021) 6(62):eabh3567. 10.1126/sciimmunol.abh3567
118.
JiangMQiLLiLLiY. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. (2020) 6:112. 10.1038/s41420-020-00349-0
119.
SongZGongQGuoJ. Pyroptosis: mechanisms and links with fibrosis. Cells. (2021) 10(12):3509. 10.3390/cells10123509
120.
MaJZhuFZhaoMShaoFYuDMaJet alSARS-CoV-2 nucleocapsid suppresses host pyroptosis by blocking gasdermin D cleavage. EMBO J. (2021) 40(18):e108249. 10.15252/embj.2021108249
121.
KishoreAPetrekM. Roles of macrophage polarization and macrophage-derived miRNAs in pulmonary fibrosis. Front Immunol. (2021) 12:678457. 10.3389/fimmu.2021.678457
122.
LiangSWuYSLiDYTangJXLiuHF. Autophagy in viral infection and pathogenesis. Front Cell Dev Biol. (2021) 9:766142. 10.3389/fcell.2021.766142
123.
BragaTTAgudeloJSCamaraNO. Macrophages during the fibrotic process: M2 as friend and foe. Front Immunol. (2015) 6:602. 10.3389/fimmu.2015.00602
124.
HouJShiJChenLLvZChenXCaoHet alM2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun Signal. (2018) 16(1):89. 10.1186/s12964-018-0300-8
125.
Lis-LopezLBausetCSeco-CerveraMCosin-RogerJ. Is the macrophage phenotype determinant for fibrosis development?Biomedicines. (2021) 9(12):1747. 10.3390/biomedicines9121747
126.
StrizovaZBenesovaIBartoliniRNovysedlakRCecrdlovaEFoleyLKet alM1/M2 macrophages and their overlaps—myth or reality?Clin Sci (Lond). (2023) 137(15):1067–93. 10.1042/CS20220531
127.
HuMYaoZXuLPengMDengGLiuLet alM2 macrophage polarization in systemic sclerosis fibrosis: pathogenic mechanisms and therapeutic effects. Heliyon. (2023) 9(5):e16206. 10.1016/j.heliyon.2023.e16206
128.
CaoQHarrisDCWangY. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda). (2015) 30(3):183–94. 10.1152/physiol.00046.2014
129.
JagerNAWallis de VriesBMHillebrandsJLHarlaarNJTioRASlartRHet alDistribution of matrix metalloproteinases in human atherosclerotic carotid plaques and their production by smooth muscle cells and macrophage subsets. Mol Imaging Biol. (2016) 18(2):283–91. 10.1007/s11307-015-0882-0
130.
LiuFTHsuDKZuberiRIKuwabaraIChiEYHendersonWRJr. Expression and function of galectin-3, a beta-galactoside-binding lectin, in human monocytes and macrophages. Am J Pathol. (1995) 147(4):1016–28. PMCID: PMC1871012
131.
BinkDILozano-VidalNBoonRA. Long non-coding RNA in vascular disease and aging. Noncoding RNA. (2019) 5(1):26. 10.3390/ncrna5010026
132.
Berumen SanchezGBunnKEPuaHHRafatM. Extracellular vesicles: mediators of intercellular communication in tissue injury and disease. Cell Commun Signal. (2021) 19(1):104. 10.1186/s12964-021-00787-y
133.
HarrisTAYamakuchiMFerlitoMMendellJTLowensteinCJ. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci U S A. (2008) 105(5):1516–21. 10.1073/pnas.0707493105
134.
MiaoYAjamiNEHuangTSLinFMLouCHWangYTet alEnhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat Commun. (2018) 9(1):292. 10.1038/s41467-017-02113-y
135.
Garate-CarrilloAGonzalezJCeballosGRamirez-SanchezIVillarrealF. Sex related differences in the pathogenesis of organ fibrosis. Transl Res. (2020) 222:41–55. 10.1016/j.trsl.2020.03.008
136.
MacneeW. Premature vascular ageing in cystic fibrosis. Eur Respir J. (2009) 34(6):1217–8. 10.1183/09031936.00155209
137.
Gubbels BuppMRPotluriTFinkALKleinSL. The confluence of sex hormones and aging on immunity. Front Immunol. (2018) 9:1269. 10.3389/fimmu.2018.01269
138.
LiXLiCZhangWWangYQianPHuangH. Inflammation and aging: signaling pathways and intervention therapies. Signal Transduct Target Ther. (2023) 8(1):239. 10.1038/s41392-023-01502-8
139.
HuangZChenBLiuXLiHXieLGaoYet alEffects of sex and aging on the immune cell landscape as assessed by single-cell transcriptomic analysis. Proc Natl Acad Sci U S A. (2021) 118(33)::e2023216118. 10.1073/pnas.2023216118
140.
HaoSZhaoJZhouJZhaoSHuYHouY. Modulation of 17beta-estradiol on the number and cytotoxicity of NK cells in vivo related to MCM and activating receptors. Int Immunopharmacol. (2007) 7(13):1765–75. 10.1016/j.intimp.2007.09.017
141.
KleinSLFlanaganKL. Sex differences in immune responses. Nat Rev Immunol. (2016) 16(10):626–38. 10.1038/nri.2016.90
142.
ZhengYLiuXLeWXieLLiHWenWet alA human circulating immune cell landscape in aging and COVID-19. Protein Cell. (2020) 11(10):740–70. 10.1007/s13238-020-00762-2
143.
KhorramOGarthwaiteMGolosT. The influence of aging and sex hormones on expression of growth hormone-releasing hormone in the human immune system. J Clin Endocrinol Metab. (2001) 86(7):3157–61. 10.1210/jcem.86.7.7652
144.
Giefing-KrollCBergerPLepperdingerGGrubeck-LoebensteinB. How sex and age affect immune responses, susceptibility to infections, and response to vaccination. Aging Cell. (2015) 14(3):309–21. 10.1111/acel.12326
145.
GranataLGildawieKRIsmailNBrenhouseHCKopecAM. Immune signaling as a node of interaction between systems that sex-specifically develop during puberty and adolescence. Dev Cogn Neurosci. (2022) 57:101143. 10.1016/j.dcn.2022.101143
146.
JoAChoiTGHanJYTaborMHKolliputiNLockeyRFet alAge-related increase of collagen/fibrin deposition and high PAI-1 production in human nasal polyps. Front Pharmacol. (2022) 13:845324. 10.3389/fphar.2022.845324
147.
TeuscherACStatzerCPantasisSBordoliMREwaldCY. Assessing collagen deposition during aging in mammalian tissue and in caenorhabditis elegans. Methods Mol Biol. (2019) 1944:169–88. 10.1007/978-1-4939-9095-5_13
148.
LenhardtRHopfHWMarkerEAkcaOKurzAScheuenstuhlHet alPerioperative collagen deposition in elderly and young men and women. Arch Surg. (2000) 135(1):71–4. 10.1001/archsurg.135.1.71
149.
YusifovAWoulfeKCBrunsDR. Mechanisms and implications of sex differences in cardiac aging. J Cardiovasc Aging. (2022) 2:20. 10.20517/jca.2022.01
150.
AchkarASalibaYFaresN. Differential gender-dependent patterns of cardiac fibrosis and fibroblast phenotypes in aging mice. Oxid Med Cell Longev. (2020) 2020:8282157. 10.1155/2020/8282157
151.
AkoumNMahnkopfCKholmovskiEGBrachmannJMarroucheNF. Age and sex differences in atrial fibrosis among patients with atrial fibrillation. Europace. (2018) 20(7):1086–92. 10.1093/europace/eux260
152.
ParkerLCaldowMKWattsRLevingerPCameron-SmithDLevingerI. Age and sex differences in human skeletal muscle fibrosis markers and transforming growth factor-beta signaling. Eur J Appl Physiol. (2017) 117(7):1463–72. 10.1007/s00421-017-3639-4
153.
LamGYGoodwinJWilcoxPGQuonBS. Sex disparities in cystic fibrosis: review on the effect of female sex hormones on lung pathophysiology and outcomes. ERJ Open Res. (2021) 7(1):00475-2020. 10.1183/23120541.00475-2020
154.
Gonzalez-CadavidNF. Mechanisms of penile fibrosis. J Sex Med. (2009) 6(Suppl 3):353–62. 10.1111/j.1743-6109.2008.01195.x
155.
MagderS. The meaning of blood pressure. Crit Care. (2018) 22(1):257. 10.1186/s13054-018-2171-1
156.
ThenappanTOrmistonMLRyanJJArcherSL. Pulmonary arterial hypertension: pathogenesis and clinical management. Br Med J. (2018) 360:j5492. 10.1136/bmj.j5492
157.
SamokhinAOStephensTWertheimBMWangRSVargasSOYungLMet alNEDD9 Targets COL3A1 to promote endothelial fibrosis and pulmonary arterial hypertension. Sci Transl Med. (2018) 10(445):eaap7294. 10.1126/scitranslmed.aap7294
158.
SouilholCHarmsenMCEvansPCKrenningG. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc Res. (2018) 114(4):565–77. 10.1093/cvr/cvx253
159.
ShiXHuangHZhouMLiuYWuHDaiM. Paeonol attenuated vascular fibrosis through regulating treg/Th17 balance in a gut Microbiota-dependent manner. Front Pharmacol. (2021) 12:765482. 10.3389/fphar.2021.765482
160.
CuiXKongXChenRMaLJiangL. The potential role of leflunomide in inhibiting vascular fibrosis by down-regulating type-II macrophages in Takayasu’s arteritis. Clin Exp Rheumatol. (2020) 38(2):69–78. PMID: 31969231.
161.
KongXMaLJiZDongZZhangZHouJet alPro-fibrotic effect of IL-6 via aortic adventitial fibroblasts indicates IL-6 as a treatment target in Takayasu arteritis. Clin Exp Rheumatol. (2018) 36(1):62–72. PMID: 28770707.
162.
ChenRSunYCuiXJiZKongXWuSet alAutophagy promotes aortic adventitial fibrosis via the IL-6/Jak1 signaling pathway in Takayasu’s arteritis. J Autoimmun. (2019) 99:39–47. 10.1016/j.jaut.2019.01.010
163.
KabeerdossJPilaniaRKKarkheleRKumarTSDandaDSinghS. Severe COVID-19, multisystem inflammatory syndrome in children, and Kawasaki disease: immunological mechanisms, clinical manifestations and management. Rheumatol Int. (2021) 41(1):19–32. 10.1007/s00296-020-04749-4
164.
MaruottiNCantatoreFPNicoBVaccaARibattiD. Angiogenesis in vasculitides. Clin Exp Rheumatol. (2008) 26(3):476–83. PMID: 18578974.
165.
Sancho-ShimizuVBrodinPCobatABiggsCMToubianaJLucasCLet alSARS-CoV-2-related MIS-C: a key to the viral and genetic causes of Kawasaki disease?J Exp Med. (2021) 218(6):e20210446. 10.1084/jem.20210446
166.
NumanoFShimizuCJimenez-FernandezSVejarMOharasekiTTakahashiKet alGalectin-3 is a marker of myocardial and vascular fibrosis in Kawasaki disease patients with giant aneurysms. Int J Cardiol. (2015) 201:429–37. 10.1016/j.ijcard.2015.07.063
167.
LeeAMShimizuCOharasekiTTakahashiKDanielsLBKahnAet alRole of TGF-beta signaling in remodeling of noncoronary artery aneurysms in Kawasaki disease. Pediatr Dev Pathol. (2015) 18(4):310–7. 10.2350/14-12-1588-OA.1
168.
Mateus GoncalvesLPereiraEWerneck de CastroJPBernal-MizrachiEAlmacaJ. Islet pericytes convert into profibrotic myofibroblasts in a mouse model of islet vascular fibrosis. Diabetologia. (2020) 63(8):1564–75. 10.1007/s00125-020-05168-7
169.
TsaiCHLiaoCWWuXMChenZWPanCTChangYYet alAutonomous cortisol secretion is associated with worse arterial stiffness and vascular fibrosis in primary aldosteronism: a cross-sectional study with follow-up data. Eur J Endocrinol. (2022) 187(1):197–208. 10.1530/EJE-21-1157
170.
TuletaIFrangogiannisNG. Diabetic fibrosis. Biochim Biophys Acta Mol Basis Dis. (2021) 1867(4):166044. 10.1016/j.bbadis.2020.166044
171.
LouiselleAENiemiecSMZgheibCLiechtyKW. Macrophage polarization and diabetic wound healing. Transl Res. (2021) 236:109–16. 10.1016/j.trsl.2021.05.006
172.
JonesADebRTorsneyEHoweFDunkleyMGnaneswaranYet alRosiglitazone reduces the development and rupture of experimental aortic aneurysms. Circulation. (2009) 119(24):3125–32. 10.1161/CIRCULATIONAHA.109.852467
173.
RenLLiuNZhiHLiYLiYTangRet alVasculoprotective effects of rosiglitazone through modulating renin-angiotensin system in vivo and vitro. Cardiovasc Diabetol. (2011) 10:10. 10.1186/1475-2840-10-10
174.
HouXZhangYShenYHLiuTSongSCuiLet alPPAR-gamma activation by rosiglitazone suppresses angiotensin II-mediated proliferation and phenotypictransition in cardiac fibroblasts via inhibition of activation of activator protein 1. Eur J Pharmacol. (2013) 715(1–3):196–203. 10.1016/j.ejphar.2013.05.021
175.
GaoDFNiuXLHaoGHPengNWeiJNingNet alRosiglitazone inhibits angiotensin II-induced CTGF expression in vascular smooth muscle cells—role of PPAR-gamma in vascular fibrosis. Biochem Pharmacol. (2007) 73(2):185–97. 10.1016/j.bcp.2006.09.019
176.
ArnoldSVInzucchiSEEchouffo-TcheuguiJBTangFLamCSPSperlingLSet alUnderstanding contemporary use of thiazolidinediones. Circ Heart Fail. (2019) 12(6):e005855. 10.1161/CIRCHEARTFAILURE.118.005855
177.
ShyuLYChenKMLuCYLaiSC. Regulation of proinflammatory enzymes by peroxisome proliferator-activated receptor gamma in astroglia infected with toxoplasma gondii. J Parasitol. (2020) 106(5):564–71. 10.1645/18-184
178.
TostesGCCunhaMRFukuiRTCorreiaMRRochaDMDos SantosRFet alEffects of nateglinide and rosiglitazone on pancreatic alpha- and beta-cells, GLP-1 secretion and inflammatory markers in patients with type 2 diabetes: randomized crossover clinical study. Diabetol Metab Syndr. (2016) 8:1. 10.1186/s13098-015-0120-6
179.
HuangRFuPMaL. Kidney fibrosis: from mechanisms to therapeutic medicines. Signal Transduct Target Ther. (2023) 8(1):129. 10.1038/s41392-023-01379-7
180.
WeiskirchenRWeiskirchenSTackeF. Organ and tissue fibrosis: molecular signals, cellular mechanisms and translational implications. Mol Aspects Med. (2019) 65:2–15. 10.1016/j.mam.2018.06.003
181.
MaherTMStrekME. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respir Res. (2019) 20(1):205. 10.1186/s12931-019-1161-4
182.
BehrJPrasseAKreuterMJohowJRabeKFBonellaFet alPirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med. (2021) 9(5):476–86. 10.1016/S2213-2600(20)30554-3
183.
RosenbloomJMendozaFAJimenezSA. Strategies for anti-fibrotic therapies. Biochim Biophys Acta. (2013) 1832(7):1088–103. 10.1016/j.bbadis.2012.12.007
184.
LinSNMaoRQianCBettenworthDWangJLiJet alDevelopment of antifibrotic therapy for stricturing Crohn’s disease: lessons from randomized trials in other fibrotic diseases. Physiol Rev. (2022) 102(2):605–52. 10.1152/physrev.00005.2021
185.
WhiteESThomasMStowasserSTetzlaffK. Challenges for clinical drug development in pulmonary fibrosis. Front Pharmacol. (2022) 13:823085. 10.3389/fphar.2022.823085
186.
O'DwyerDNAshleySLMooreBB. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am J Physiol Lung Cell Mol Physiol. (2016) 311(3):L590–601. 10.1152/ajplung.00221.2016
187.
MewtonNLiuCYCroisillePBluemkeDLimaJA. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol. (2011) 57(8):891–903. 10.1016/j.jacc.2010.11.013
188.
MaherTMBendstrupEDronLLangleyJSmithGKhalidJMet alGlobal incidence and prevalence of idiopathic pulmonary fibrosis. Respir Res. (2021) 22(1):197. 10.1186/s12931-021-01791-z
Summary
Keywords
vascular fibrosis, ECM, TGF-β, RAAS, arterial stiffness, CVD
Citation
Hua R, Gao H, He C, Xin S, Wang B, Zhang S, Gao L, Tao Q, Wu W, Sun F and Xu J (2023) An emerging view on vascular fibrosis molecular mediators and relevant disorders: from bench to bed. Front. Cardiovasc. Med. 10:1273502. doi: 10.3389/fcvm.2023.1273502
Received
06 August 2023
Accepted
27 November 2023
Published
21 December 2023
Volume
10 - 2023
Edited by
Muhammad Aslam, University of Giessen, Germany
Reviewed by
Shafeeq Ahmed Mohammed, University Hospital Zürich, Switzerland
Maria Luisa Barcena, Charité University Medicine Berlin, Germany
Updates
Copyright
© 2023 Hua, Gao, He, Xin, Wang, Zhang, Gao, Tao, Wu, Sun and Xu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingdong Xu xujingdong@ccmu.edu.cn
Abbreviations: ACE, angiotensin-converting enzyme; ACS, autonomous cortisol secretion; AKT(PKB), Protein kinase B; Aldo, aldosterone; Ang II, Angiotensin II; AT-1R, Ang II receptor 1; AT-2R, Ang II receptor 2; ATG5, autophagy-related protein 5; Col, collagen; CTGF, connective tissue growth factor; CVD, cardiovascular disease; DAMP, damage-associated molecular pattern; DIZE, diminazene aceturate; DM2, type 2 diabetes mellitus; ECM, extracellular matrix; ECs, endothelial cells; EndMT, endothelial to mesenchymal transition; eNOS, endothelial nitric oxide synthase; ER, endoplasmic reticulum; ERK, extracellular regulated protein kinases; ET-1, endothelin-1; FABP3, fatty acid binding protein 3; FAO, fatty acid oxidation; FGF, fibroblast growth factor; Fli-1, friend leukemia virus integration 1; FN, fibronectin; gal-3, galectin-3; GSDMD, gasdermin D; GSDME, gasdermin E; HFD, high-fat diet; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; IFN-γ, interferon-γ; IL, interleukin; IPF, idiopathic pulmonary fibrosis; KD, Kawasaki disease; LEF, leflunomide; LPS, lipopolysaccharide; MBG, marinobufagenin; MIAT, myocardial infarction associated transcript; MIS, multisystem inflammatory syndrome; MMP, matrix metalloproteinase; MR, mineralocorticoid receptor; MrgD, Mas-related G protein-coupled receptor member D; NEDD9, neural precursor cell expressed developmentally down-regulated 9; NF-κB, nuclear factor κB; NGAL, neutrophil gelatinase-associated lipocalin; OS, oxidative stress; OSM, oncostatin-M; PA, primary aldosteronism; PAH, pulmonary arterial hypertension; PAI, plasminogen activator inhibitor; PBMCs, peripheral blood mononuclear cells; PDGF, platelet-derived growth factor; PDIA6, protein disulfide isomerase family 6; PDL-1, programmed death ligand-1; PI3K, phosphatidylinositol 3 kinase; PPAR, peroxisome proliferator-activated receptor; PRR, pattern recognition receptor; PTEN, phosphatase and tensin homolog; PYK2, protein tyrosine kinase 2; RAAS, renin–angiotensin–aldosterone system; Rho, Ras homolog gene family member A; ROCK, Rho-associated coiled-coil forming protein kinases; ROS, reactive oxygen species; SCFAs, short-chain fatty acids; Smad, small mothers against decapentaplegic; SOD2, superoxide dismutase 2; StAR, steroidogenic acute regulatory protein; STAT-3, signal transducer and activator of transcription 3; TAK, Takayasu's arteritis; TG-2, transglutaminase-2; TGF-β, transforming growth factor-β; TIMP, tissue inhibitor of metalloproteinase; TNF, tumor necrosis factor; TZD, thiazolidinediones; VEGF, vascular endothelial growth factor; VSMCs, vascular smooth muscle cells; α-SMA, α-smooth muscle actin.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.