 Massimo Baudo1,2
Massimo Baudo1,2 Alessandro Varrica1Matteo Reali1Antonio Saracino3
Alessandro Varrica1Matteo Reali1Antonio Saracino3 Mario Carminati3
Mario Carminati3 Alessandro Frigiola1Alessandro Giamberti1
Alessandro Frigiola1Alessandro Giamberti1 Mauro Lo Rito1*
Mauro Lo Rito1*
- 1Department of Congenital Cardiac Surgery, IRCCS Policlinico San Donato, San Donato Milanese, Italy
- 2Department of Cardiac Surgery, ASST Spedali Civili di Brescia, University of Brescia, Brescia, Italy
- 3Department of Pediatric and Adult Congenital Cardiology, IRCCS Policlinico San Donato, San Donato Milanese, Italy
Background: This is the first meta-analysis to analyze all reports of published pediatric cases of cervical aortic arch (CAA) by highlighting the clinical characteristics and treatment outcomes using the reported individual data of the patients. The aim of the study is to investigate the clinical features and surgical outcomes of such a rare disease in the pediatric population.
Methods: A comprehensive search was conducted in various academic databases, including PubMed, ScienceDirect, SciELO, DOAJ, and Cochrane Library, until June 2022 for case reports describing the presence of cervical aortic arch in the pediatric age. Case reports and series were included if the following criteria were met: (1) description of the cervical aortic arch; (2) patient of pediatric age; and (3) published in the English language. All other types of publications that lacked patient-specific information were excluded from the analysis. This systematic review was conducted in accordance with the PRISMA guidelines. The primary outcome measure of the analysis was early and late mortality.
Results: The literature search identified 2,272 potentially eligible articles, 72 of which met our inclusion criteria with 96 patients including the author's institutional case. At a median of 365 (90–730) days, the overall cohort registered a 7.3% (7/96) mortality rate. In the subset of patients who underwent surgery, the mortality rate was also 7.3% (4/55), and the mortality rate following surgery to treat only CAA was 2.4% (1/42). Dyspnea was identified as an independent determinant of mortality by employing the univariable Firth bias-reduced logistic regression method.
Conclusion: Cervical aortic arch is a rare congenital heart disease that poses treatment challenges due to the high anatomical variability, diverse clinical presentations, and the presence of other concomitant diseases. The surgical treatment appears to be a safe and effective approach for resolving the symptoms, although it needs to be tailored individually for each patient.
Systematic Review Registration: https://www.crd.york.ac.uk/prospero/display_record.php?RecordID=346826, Identifier: CRD42022346826.
Introduction
The cervical aortic arch (CAA) is a relatively rare congenital anomaly of the aorta development in which the aortic arch is located above the superior aspect of the clavicle, occasionally protruding high into the neck. CAA was initially introduced by Reid in 1914 (1), and the number of recorded cases in literature remains relatively low. CAA may be associated with other structural anomalies (such as kinking, coarctation, or aneurysm) or congenital heart disease (CHD), or it can occur as an isolated cardiovascular anomaly (2). Aortic arch anomalies exhibit a higher prevalence among patients with chromosome 22q11 deletion, whether associated with heart malformation or not (3).
Embryologically, the arch normally arises from the fourth branchial arch. It is theorized that in cases of CAA, the arch arises in a more cephalad location from the second or third branchial arch, resulting in an elevated final position of the arch (4).
Most often asymptomatic, CAA can manifest as a swelling pulsatile mass at the base of the neck where a murmur can be heard or a thrill can be felt. In cases when it is observed, signs and symptoms are associated with the presence of a vascular ring that compresses the trachea or esophagus (i.e., stridor, dyspnea, recurrent pulmonary infections, or dysphagia) (5).
Historically, Haughton et al. (6) proposed the first classification of CAA in 1975, encompassing five morphological types (A–E) based on their own observations and a review of the available literature cases. More recently, Zhong et al. (7) proposed a revised classification of CAA in an attempt to provide a more intuitive classification that could be used for surgical decision making. The classification consists of two types and six subtypes on the basis of the presence of a vascular ring (i.e., retroesophageal aortic segment and/or aberrant subclavian artery) and the relationship of the descending thoracic aorta to the side of the aortic arch (Figure 1).

Figure 1. Haughton and Zhong cervical aortic arch classification diagram.
The existing reports on CAA are currently constrained to studies involving small cohorts and case reports. In the present study, we seek to provide the most comprehensive review of pediatric cases and evaluate the demographics, clinical presentation, and surgical procedures of patients of pediatric age with CAA.
Materials and methods
Literature search strategy
This systematic review was conducted in accordance with the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines (see the Supplementary Material) (8). The PRISMA flow diagram is presented in Figure 2. PubMed, ScienceDirect, SciELO, DOAJ, and Cochrane Library databases were searched until June 2022 for case reports and series describing the presence of cervical aortic arch in the pediatric age. The complete search strategy is shown in Supplementary Table S1. Furthermore, the references of all studies and meta-analyses were examined to identify additional articles (i.e., “backward snowballing”). The study selection process involved the following steps: (1) identification of titles of records through database search; (2) removal of duplicates; (3) screening and selection of abstracts; (4) assessment for eligibility through full-text articles; and (5) final inclusion in the study. Two authors (MB and MLR) independently screened the studies for inclusion. Discrepancies were arbitrated by a third author (AG) to achieve consensus.
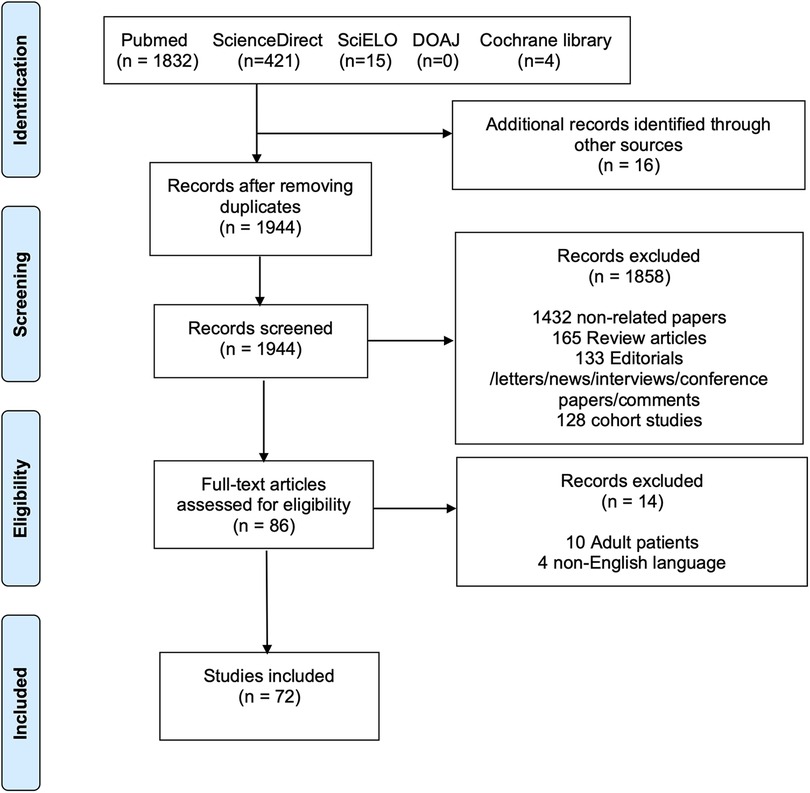
Figure 2. PRISMA flow diagram of the included studies. The following steps were taken for study selection: (1) identification of titles of records through database search; (2) removal of duplicates; (3) screening and selection of abstracts; (4) assessment for eligibility through full-text articles; and (5) final inclusion in the study.
This review was registered with the PROSPERO register of systematic reviews (ID: CRD42022346826). For the systematic review, data were obtained from published papers. As such, the approval of the research ethics board or the consent of the patient was not required. Regarding the institutional case report, the consent to use personal data for scientific research purposes was obtained through the signing of the surgical consent form. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Selection criteria
Using the Population, Interventions, Comparison, Outcome, and Study design (PICOS) strategy, the case reports and series were included if the following criteria were met: (1) description of the cervical aortic arch; (2) patient of pediatric age; (3) published in the English language. Exclusion criteria for analysis were all other forms of publications that lacked patient-specific information.
Data extraction and critical appraisal
Microsoft Office 365 Excel software (Microsoft, Redmond, WA, USA) was used for data extraction. The following patient characteristics were extracted: age, sex, cervical arch laterality, presence of other aortic defects, concomitant CHD, signs, symptoms, and surgical procedure performed.
The expected differences in the information reported from the cases were observed, and to a certain degree, each article presented distinct variables that were not found in other reports. As a result, the absence of data for certain variables necessitated subjective interpretation, Significant relevant complications that were not reported were assumed not to have occurred. Denominators were determined in the data analysis based on either explicit indications of the presence or absence of a variable, or through suitable inferences of their existence.
The Joanna Briggs Institute Critical Appraisal tool was used for the critical appraisal of the quality of the included case reports (9).
Statistical analysis
The primary outcome measure of the analysis was early and late mortality, while the secondary outcome measure was to analyze the clinical presentation of CAA. Categorical variables were presented as frequency counts and percentages and compared between groups using the Chi-square test or Fisher's exact test, as required. After assessing the normality of continuous variables using Kolmogorov–Smirnov test, data were presented as means and standard deviations if normally distributed and were compared between groups using Student's t-test or analysis of variance (ANOVA). The data were presented as medians and interquartile ranges (IQR) if not normally distributed and compared between groups using Mann–Whitney U test or Kruskal–Wallis test, accordingly. Symptom and mortality predictors were identified using univariable Firth bias-reduced logistic regression. The independent predictors of late mortality were evaluated using univariable Cox regression with Firth's correction method. The Firth bias-reduced correction method has become a standard approach for analyzing binary outcomes with small samples and reduces the bias in maximum likelihood estimates of coefficients. All tests were two-sided, and the alpha level was set at 0.05 for statistical significance.
All analyses were performed using R version 4.2.1 (R Project for Statistical Computing, Vienna, Austria) and RStudio version 2022.07.1 Built 554.
Results
The literature review was conducted adding the authors' institutional case. In brief, a 6-year-old girl was diagnosed with isolated aortic arch aneurysm with surgical indication. The pre-natal cardiologic evaluation revealed the presence of a dilated aortic arch. Marfan syndrome and other collagenopathies were excluded at the genetic evaluation after birth. At the cardiologic follow-up visit, a contrast-enhanced CT scan was performed, revealing the presence of a left CAA aneurysm (max diameter 29 mm) with a branching left subclavian artery (Figure 3). The patient underwent resection of the aneurysm, followed by a termino-lateral anastomosis with an anterior autologous pericardium patch. In addition, the subclavian artery was reimplanted into the ascending aorta using a 6 mm vascular conduit through a median sternotomy. The postoperative hospital stay was uneventful, and the patient was discharged on the sixth postoperative day. At the last follow-up visit, she was found to be alive and in good health.

Figure 3. CT scan 3D reconstruction of the author's institutional cervical aortic arch case. 3D reconstructed CT scan showing the left-sided cervical aortic arch anatomy of the institutional patient included in the analysis.
Study selection and characteristics
The literature search identified 2,272 potentially eligible articles. Additional 16 articles were identified using the backward snowballing method. After removing the duplicates, 1,944 papers were screened. A total of 86 full-text articles were assessed for eligibility, and 72 publications (6, 10–80) were found to meet our inclusion criteria (Figure 2) with 95 patients. The authors’ institutional case was added to the study population. Thus, a total of 96 patients were finally included. The publication year ranged from 1947 to 2021. The critical assessment of the included articles is shown in Supplementary Table S2.
Meta-analysis
The median age of the general population was 6 years (IQR: 0.83–11) with a female prevalence of 62.8% (54/86). Two-thirds of the CAA were right-sided (64/96, 66.7%), and 86.5% (83/96) of the descending aorta were left-sided. Concomitant arch-associated anomalies and concomitant CHD were present in 69.8% (67/96) and 41.7% (40/96) of the patients, respectively. The most prevalent Haughton class was type B (41/92, 44.6%), while the most prevalent Zhong class was type B (67/92, 72.8%) with B2 being the most prevalent subclass (48/92, 52.2%, Table 1). In total, 46 out of 88 patients (52.3%) were asymptomatic, while the most prevailing symptom reported was dyspnea (21/88, 23.9%, Table 2). In total, 55 out of 93 (59.1%) patients underwent surgery with or without CAA correction, while 42 out of 93 (45.2%) patients underwent surgery to treat CAA. Thus, 13 out of 93 patients (14.0%) did not undergo CAA surgical correction. Of note, there were 27 patients in total that underwent surgery to treat an isolated CAA (including all diameter variations, i.e., aneurysm, coarctation, arch hypoplasia, and double aortic arch). A sternotomy was chosen in 57.9% (11/19) of the cases, while a thoracotomy was preferred in 42.1% (8/19) of the patients. None of these patients died. At a median of 365 (90–730) days, the overall cohort registered a 7.3% (7/96) mortality rate. Three of these cases were related to surgery for treating CHD without the CAA (14, 27, 39), while only one case focused on surgery for isolated CAA (2). The three non-operated patients experienced different causes of death: one patient died due to acute cerebral hemorrhage (6), another patient died due to sepsis with deranged coagulation, renal failure, and cardiopulmonary failure (50), and the third patient experienced unexpected death 24 h after undergoing cerebral arteriography, with no identifiable cause (17). After surgery, only four patients (among the symptomatic), all with a vascular ring, did not completely resolve their symptoms, which persisted in a milder form than prior to intervention (6, 19, 39, 54). More details can be seen in Table 3.
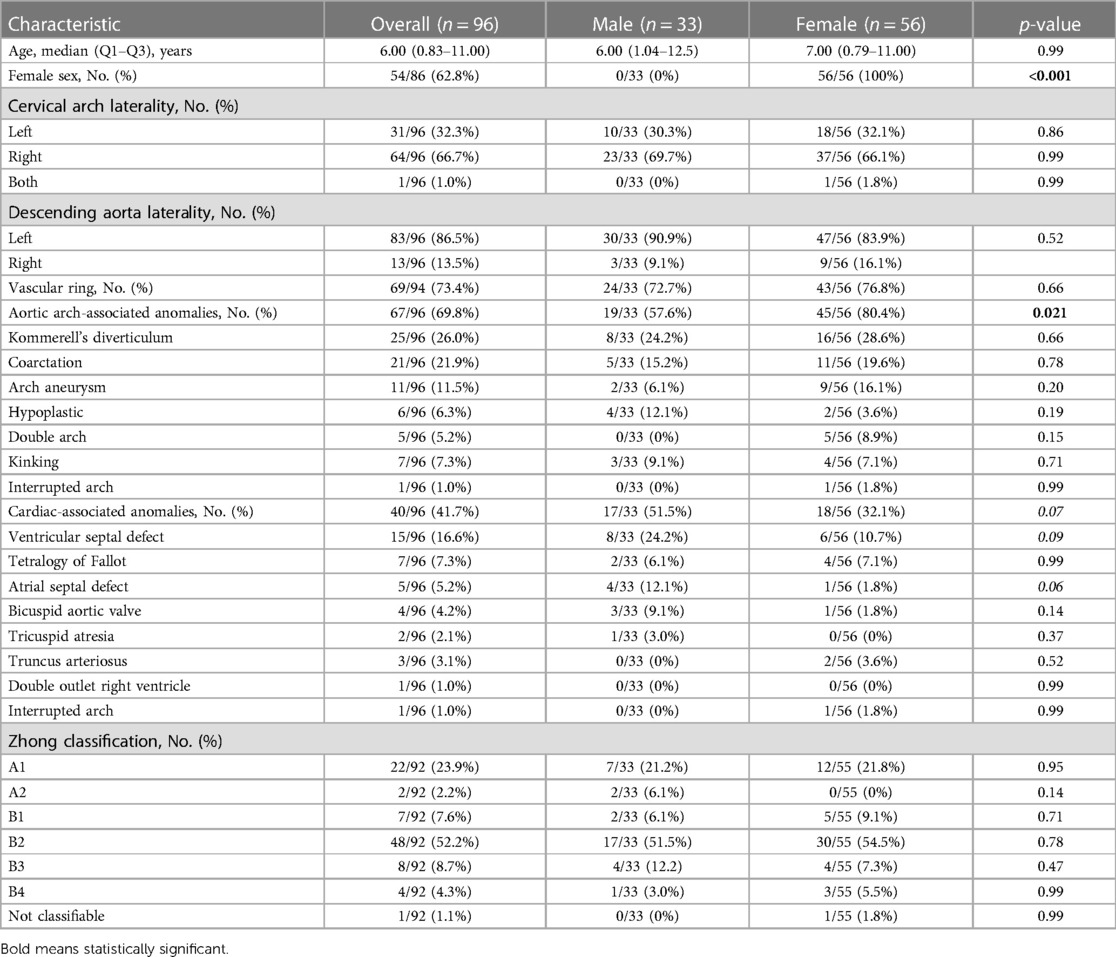
Table 1. Baseline characteristics of the included patients overall and divided by sex.

Table 2. Signs and symptoms of the included patients overall and divided by sex.
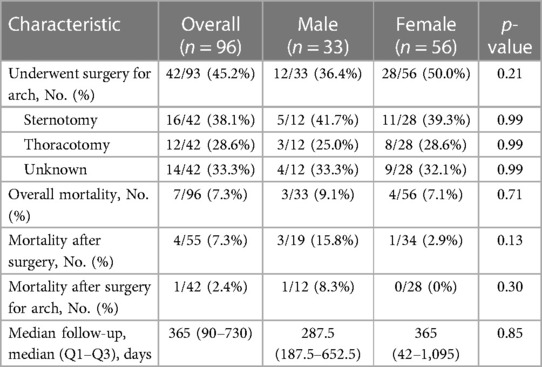
Table 3. Outcomes of included studies divided by sex.
When analyzing sex differences, it was observed that females exhibited a significantly higher prevalence of concomitant arch-associated anomalies (80.4% vs. 57.6%, p = 0.021), while experiencing a lower incidence of dyspnea (12.0% vs. 32.3%, p = 0.026) compared with males. Females demonstrated a lower prevalence of concomitant CHD (32.1% vs. 51.5%, p = 0.071), specifically a reduced occurrence of atrial septal defects (1.8% vs. 12.1%, p = 0.061). More details are shown in Table 1.
The patients undergoing surgery were younger when compared with the non-surgical subgroup [4.5 (0.47–10) years vs. 8.0 (2.56–11.25) years, p = 0.036]. Furthermore, the surgical patients were more symptomatic compared with the non-operated patients (60.4% vs. 31.6%, p = 0.008), particularly those who suffered more from dyspnea (35.4% vs. 2.6%, p < 0.001). The operated patients presented significantly more concomitant arch-associated anomalies (80.0% vs. 57.9%, p = 0.021), particularly of arch aneurism (18.2% vs. 2.6%, p = 0.025), when compared with the non-surgical subgroup. Of note, the overall mortality rate was similar between the two groups (7.3% vs. 7.9%, p = 0.999). All details can be seen in Supplementary Table S3.
The cervical aortic arches classified as Zhong type A exhibited a higher tendency for the presence of a CAA on the left side compared with those classified as Zhong type B (84.0% vs. 13.4%, p < 0.001). Interestingly, Zhong type A was more prone to show a concomitant arch aneurysm or kinking of the aorta when compared with Zhong type B (36.0% vs. 3.0%, p < 0.001 and 20.0% vs. 3.0%, p = 0.015, respectively), but with a lower prevalence of Kommerell's diverticulum (8.0% vs. 34.3%, p = 0.016) (Supplementary Table S4).
Regression analysis
The univariable Firth bias-reduced logistic regression analysis revealed that interrupted arch (p = 0.017), tricuspid atresia (p = 0.049), and dyspnea (p = 0.034) were identified as independent determinants of mortality. A higher mortality rate was noted in individuals with right-sided CAA (p = 0.057) (Supplementary Table S5).
Kinking of the arch was negatively associated with symptoms (p = 0.007), while surgery (p = 0.008) was positively associated with symptoms. A positive association between symptoms and concomitant CHD (p = 0.062), double aortic arch (p = 0.087), and coarctation (p = 0.082) was observed. In addition, a negative association was seen between age (p = 0.057) and symptoms (Supplementary Table S6).
The results of the univariable Cox regression analysis with Firth's correction revealed that interrupted arch was found to be an independent risk factor for mortality (p = 0.014). A lower mortality rate was observed when surgery was performed (p = 0.065) (Supplementary Table S7).
Discussion
To our knowledge, this is the first meta-analysis to analyze all reports of published pediatric cases of CAA by highlighting the demographics, clinical characteristics, and treatment outcomes using the individual data of the patients. Since all the papers were documented as either case reports or case series, they provided a comprehensive of the most relevant data needed.
CAA is a rare congenital anomaly whose embryological origin remains unsolved. Normally, the aortic arch develops from the fourth of the six aortic arches during embryonic development. There are currently two competing theories to explain the CAA variation (4). The first one hypothesizes that the cervical arch arises from the persistence of the third embryonic arch, while the ipsilateral fourth arch regresses. The other theory assumes an anomalous positioned fourth arch that did not undergo the typical descent process.
Two main CAA classifications have been proposed by Haughton et al. (6) and Zhong et al. (7). The classification proposed by Haughton is considered to be a predominantly historical categorization, as it was developed based on a limited number of cases. Zhong's more recent classification can be viewed as a revised classification that was established with a greater emphasis on potential surgical approaches and anatomical traits, i.e., the presence of a vascular ring. Nevertheless, the population used to construct the sample comprised a cohort of young adult patients. The present review of the pediatric population showed how Zhong's classification identifies additional anatomical characteristics apart from the vascular ring in the pediatric population. Zhong type A is associated to more left-sided aortic arches, while type B is associated to more right-sided variants. Moreover, arch aneurysms and kinking are more related to Zhong type A than type B.
Due to the rarity of CAA and the associated complexity with concomitant CHD and arch abnormalities, the surgical management of CAA is technically challenging and remains unstandardized. The aims of surgery encompass the correction of concomitant CHD when present, along with decompression of the esophagus and trachea in the presence of a vascular ring. Therefore, surgical accesses and procedures are tailored individually according to all of these parameters. The selection of the thoracotomy incision side is determined by the CAA laterality, while a sternotomy is generally preferred for cases of higher complexity. By definition, the presence of a vascular ring in Zhong type B CAA implies a more complicated surgery. In fact, thoracotomies were performed twice as frequently in type A CAA as in type B CAA (38.5% vs. 18.5%, respectively), despite the fact that the difference was not statistically significant. However, based on the limited number of reported deaths, it is not feasible to draw conclusions whether distinctions exist among the various surgical strategies.
The regression analysis conducted in the present review provided insights into the prevailing trends regarding surgical interventions, indicating a preference for performing surgery on patients displaying symptoms as opposed to those who were asymptomatic. Within this context, symptomatology, particularly dyspnea, emerged as a prominent factor influencing the decision to proceed with surgical intervention. Moreover, factors indicative of a more severe pathological condition, such as interrupted aortic arch and tricuspid atresia, were identified as independent determinants of mortality. However, this last point warrants further discussion. In the current study population, only two cases of tricuspid atresia and one case of interrupted aortic arch were observed. The patient in the latter case did not undergo surgery and died a few weeks later. Both patients diagnosed with tricuspid atresia had surgical intervention: one patient survived, while the other patient died 1 year after surgery. On the basis of a comprehensive analysis of these statistics and the observation of extremely wide confidence intervals, it becomes evident that the clinical significance of the two identified risk factors is not firmly established. Rather, their influence appears to be primarily statistical in nature, highlighting the need for cautious interpretation.
Limitations
While the majority of individual data could be retrieved from the published papers, it is important to note that certain minor information was not always present, and this could be a possible source of bias. In addition, the exclusion of non-English studies may introduce a potential source of selection bias. Finally, the limited duration of the follow-up period restricts the scope of the evaluated outcome data.
Conclusion
CAA is a rare CHD that poses treatment challenges due to the high anatomical variability, diverse clinical presentations, and the presence of other concomitant CHD. The surgical treatment appears to be a safe and effective approach for resolving the symptoms, although it needs to be tailored individually for each patient. Finally, Zhong's classification offers anatomical associations that prove to be useful in informing surgical strategies.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
MB: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Data curation, Writing – Original draft, Writing – Review and editing, Visualization, Project administration. AV: Methodology, Validation, Writing – Review and editing, Supervision. MR: Investigation, Data curation, Writing – Original draft. AS: Writing – Review and editing, Resources, Supervision. MC: Resources, Supervision, Writing – Review and editing. AF: Writing – Review and editing, Resources, Supervision; AG: Writing – Review and editing, Resources, Supervision. MLR: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Writing – Original draft, Writing – Review and editing, Project administration.
Funding
This work was supported by IRCCS Policlinico San Donato, a Clinical Research Hospital partially funded by the Italian Ministry of Health.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1266956/full#supplementary-material
References
2. Baravelli M, Borghi A, Rogiani S, Preda L, Quattrociocchi M, Fantoni C, et al. Clinical, anatomopathological and genetic pattern of 10 patients with cervical aortic arch. Int J Cardiol. (2007) 114:236–40. doi: 10.1016/j.ijcard.2005.12.028
3. Momma K, Matsuoka R, Takao A. Aortic arch anomalies associated with chromosome 22q11 deletion (CATCH 22). Pediatr Cardiol. (1999) 20:97–102. doi: 10.1007/s002469900414
4. Rosen RD, Bordoni B. Embryology, aortic arch. Treasure Island, FL: StatPearls Publishing (2022). Available at: http://www.ncbi.nlm.nih.gov/books/NBK553173/ (Accessed October 11, 2022).
5. Zientara A, Schwegler I, Attigah N, Genoni M, Dzemali O. Anomaly of Haughton type D left cervical aortic arch in combination with type B dissection: case report and literature review. J Cardiothorac Surg. (2018) 13:79. doi: 10.1186/s13019-018-0768-8
6. Haughton VM, Fellows KE, Rosenbaum AE. The cervical aortic arches. Radiology. (1975) 114:675–81. doi: 10.1148/114.3.675
7. Zhong Y-L, Ma W-G, Zhu J-M, Qiao Z-Y, Zheng J, Liu Y-M, et al. Surgical repair of cervical aortic arch: an alternative classification scheme based on experience in 35 patients. J Thorac Cardiovasc Surg. (2020) 159:2202–13.e4. doi: 10.1016/j.jtcvs.2019.03.143
8. Page MJ, Moher D, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. PRISMA 2020 explanation and elaboration: updated guidance and exemplars for reporting systematic reviews. Br Med J. (2021) 372:n160. doi: 10.1136/bmj.n160
9. Moola S, Munn Z, Tufanaru C, Aromataris E, Sears K, Sfetcu R, et al. Chapter 7: systematic reviews of etiology and risk – JBI manual for evidence synthesis. JBI Man Evid Synth. (2020). doi: 10.46658/JBIMES-20-08
10. Adaletli I, Kurugoglu S, Davutoglu V, Ozer H, Besirli K, Sayin AG. Pseudocoarctation. Can J Cardiol. (2007) 23:675–6. doi: 10.1016/s0828-282x(07)70232-5
11. Ahluwalia GS, Rashid AG, Griselli M, Szczeklik M, Rigby ML, Mohiaddin RH, et al. Hypoplastic circumflex retroesophageal right-sided cervical aortic arch with unusual vascular arrangement and severe coarctation. Ann Thorac Surg. (2007) 84:1014–6. doi: 10.1016/j.athoracsur.2007.04.070
12. Almeida R, Alvares S, Fortuna A, Moreira J, Vieira A. Cervical aortic arch and 22q11 deletion–the role of MRI in diagnosis. Rev Port Cardiol. (2003) 22:1241–8.14708337
13. Baker KS, Bezirdjian DR, Tisnado J, Cho SR. Cervical aortic arch: case report with a 12-year follow-up. Can Assoc Radiol J. (1987) 38:302–4.2961766
14. Beavan TED, Fatti L. Ligature of aortic arch in the neck. Br J Surg. (1947) 34:414–6. doi: 10.1002/bjs.18003413615
15. Binsalamah ZM, Zea-Vera R, Fraser CD. Cervical left aortic arch with distal tortuosity causing coarctation and aneurysmal formation in a child. J Card Surg. (2018) 33:466–8. doi: 10.1111/jocs.13750
16. Bisset GS, Towbin RB, Strife JL, Gole D. Pediatric case of the day. Cervical aortic arch. Radiographics. (1987) 7:186–9. doi: 10.1148/radiographics.7.1.3448630
17. Bourdon JL, Hoeffel JC, Worms AM, Picard L, Pernot C. The cervical aortic arch: a case with diffuse arterial dysplasia and neurocutaneous angiomatosis. A review of the subject. Pediatr Radiol. (1981) 10:143–50. doi: 10.1007/BF00975188
18. Caputo S, Villanacci R, Ciampi Q, Villari B. Cervical aortic arch: echocardiographic and three-dimensional computed tomography view. Echocardiography. (2010) 27:E44–5. doi: 10.1111/j.1540-8175.2009.01139.x
19. Chang LW, Kaplan EL, Baum D, Figley MM. Aortic arch in the neck: a case report. J Pediatr. (1971) 79:788–93. doi: 10.1016/s0022-3476(71)80392-x
20. Chen F-L, Vick GW, Ge S. Left cervical aortic arch with right ligamentum arteriosum forming a vascular ring. Tex Heart Inst J. (2008) 35:78–9.18427661
21. Chen HY, Chen LK, Su CT, Chen SJ, Lin CH, Tsai YF, et al. Left cervical aortic arch with aneurysm and obstruction: three-dimensional computed tomographic angiography and magnetic resonance angiographic appearance. Int J Cardiovasc Imaging. (2002) 18:463–8. doi: 10.1023/a:1021155625397
22. Chibane S, Bouzid A, Abderahmane RAO. Cervical arch with aortic kinking. World J Pediatr Congenit Heart Surg. (2013) 4:453–4. doi: 10.1177/2150135113484501
23. Cornali M, Reginato E, Azzolina G. Cervical aortic arch and a new type of double aortic arch. Report of a case. Br Heart J. (1976) 38:993–6. doi: 10.1136/hrt.38.9.993
24. Costanzo L, Caruso E, Agati S, Guccione P. Double aortic arch with hypoplastic right aortic arch and type C atresia of left aortic arch. Interact Cardiovasc Thorac Surg. (2014) 19:331–3. doi: 10.1093/icvts/ivu147
25. D’Cruz IA, Stanley A, Vitullo D, Desai P, Chiemmongkoltip P. Noninvasive diagnosis of right cervical aortic arch. Chest. (1983) 83:820–2. doi: 10.1378/chest.83.5.820
26. Doorenbos BM, Mooyaart EL, Hoorntje JC. MR diagnosis of a right cervical aortic arch. J Comput Assist Tomogr. (1991) 15:864–6.1885816
27. Duke C, Chan KC. Isolated innominate artery in 22q11 microdeletion. Pediatr Cardiol. (2001) 22:80–2. doi: 10.1007/s002460010163
28. Fernández-Doblas J, Tauron M, Blasco A, Abella RF. Right corkscrew cervical aortic arch. Eur J Cardiothorac Surg. (2012) 42:903. doi: 10.1093/ejcts/ezs340
29. Floemer F, Ulmer HE, Brockmeier K. Images in congenital heart disease. Use of 3D volume rendered magnetic resonance angiography to demonstrate a cervical aortic arch. Cardiol Young. (2000) 10:423–4. doi: 10.1017/s104795110000977x
30. Gagnier JJ, Kienle G, Altman DG, Moher D, Sox H, Riley D, et al. The CARE guidelines: consensus-based clinical case reporting guideline development. Headache. (2013) 53:1541–7. doi: 10.1111/head.12246
31. Gerrah R, Shah A, Langley SM, Quaegebeur JM. Hypoplastic right cervical aortic arch. Ann Thorac Surg. (2012) 94:2127–9. doi: 10.1016/j.athoracsur.2012.04.136
32. Guha S, Grover V, Aiyer P, Dhull J. A unique case of right cervical aortic arch with anomalous left common carotid artery and absent right common carotid artery. Ann Med Surg (Lond). (2016) 9:58–60. doi: 10.1016/j.amsu.2016.06.013
33. Haliloglu M, Karcaaltincaba M, Oguz B, Celiker A. MR angiography of left-sided cervical aortic arch with aberrant right subclavian artery. Br J Radiol. (2007) 80:e260–4. doi: 10.1259/bjr/30659566
34. Harley HR. The development and anomalies of the aortic arch and its branches with the report of a case of right cervical aortic arch and intrathoracic vascular ring. Br J Surg. (1959) 46:561–73. doi: 10.1002/bjs.18004620003
35. Hastreiter AR, D’Cruz IA, Cantez T, Namin EP, Licata R. Right-sided aorta. I. Occurrence of right aortic arch in various types of congenital heart disease. II. Right aortic arch, right descending aorta, and associated anomalies. Br Heart J. (1966) 28:722–39. doi: 10.1136/hrt.28.6.722
36. Hellenbrand WE, Kelley MJ, Talner NS, Stansel HC, Berman MA. Cervical aortic arch with retroesophageal aortic obstruction: report of a case with successful surgical intervention. Ann Thorac Surg. (1978) 26:86–92. doi: 10.1016/s0003-4975(10)63638-7
37. Higuchi K, Koseni K, Takamoto S. Left-sided cervical aortic arch aneurysm: case report. J Thorac Cardiovasc Surg. (2003) 126:2098–100. doi: 10.1016/s0022-5223(03)01225-x
38. Huang S-C, Wang C-J, Su W-J, Chu J-J, Hwang M-S. The rare association of truncus arteriosus with a cervical double aortic arch presenting with left main bronchial compression. Cardiology. (2008) 111:16–20. doi: 10.1159/000113421
39. Hunter JG, Jeter OL, Keats TE. Right cervical aortic arch associated with ventricular septal defect: a case report. J Can Assoc Radiol. (1973) 24:331–3.4773910
40. Hyman RA, Stein HL. The cervical aortic arch anomaly. Angiology. (1975) 26:749–58. doi: 10.1177/000331977502601007
41. Jain S, Kleiner B, Moon-Grady A, Hornberger LK. Prenatal diagnosis of vascular rings. J Ultrasound Med. (2010) 29:287–94. doi: 10.7863/jum.2010.29.2.287
42. Jhaveri S, Komarlu R. Prenatal diagnosis of right-sided cervical aortic arch with aberrant left subclavian artery and absent ductus arteriosus in tetralogy of Fallot. Ultrasound Obstet Gynecol. (2019) 54:414–6. doi: 10.1002/uog.20151
43. Karakurt C, Dogan M, Baysal T, Erdil N, Kocak G, Elkiran O. Right cervical aortic arch with unusual origin of left carotid artery. Tex Heart Inst J. (2011) 38:94–5.21423483
44. Kasar T, Kafalı HC, Türkvatan A, Ergül Y. A rare case of right corkscrew cervical aortic arch associated with retrotracheal aberrant left brachiocephalic vein. Kardiol Pol. (2018) 76:812. doi: 10.5603/KP.2018.0086
45. Kazuma N, Murakami M, Suzuki Y, Umezu R, Murata M. Cervical aortic arch associated with 22q11.2 deletion. Pediatr Cardiol. (1997) 18:149–51. doi: 10.1007/s002469900138
46. Kumar A, McCombs JL, Sapire DW. Deletions in chromosome 22q11 region in cervical aortic arch. Am J Cardiol. (1997) 79:388–90. doi: 10.1016/s0002-9149(96)00772-2
47. Kumar S, Mandalam KR, Unni M, Roy S, Gupta AK, Rao VR. Left cervical arch and associated abnormalities. Cardiovasc Intervent Radiol. (1989) 12:88–91. doi: 10.1007/BF02577395
48. Lipchik EO, Young LW. Unusual symptomatic arch anomalies. Radiology. (1967) 89:85–90. doi: 10.1148/89.1.85
49. Lu JC, Shah SS, Owens ST, Dorfman AL, Vedre A, Goble MM, et al. Successful staged neonatal repair of tetralogy of Fallot with long-segment hypoplasia of the aorta. Pediatr Cardiol. (2010) 31:124–7. doi: 10.1007/s00246-009-9547-6
50. Dey M, Garg N, Kumar K. An unusual case of interrupted cervical aortic arch associated with long segment coarctation of the descending thoracic aorta. Cardiol Young. (2018) 28:592–4. doi: 10.1017/S1047951117001913
51. Nagashima M, Shikata F, Higaki Kawachi K. Cervical aortic arch and Kommerell’s diverticulum associated with the anomalous subaortic left brachiocephalic vein in a patient with chromosome 22q11.2 deletion. Interact Cardiovasc Thorac Surg (2010) 11:202–3. doi: 10.1510/icvts.2010.235416
52. Mahoney EB, Manning JA. Congenital abnormalities of the aortic arch. Surgery. (1964) 55:1–14.14114264
53. Makani S, Mitchell J, Metton O, Di Filippo S, Henaine R, Ninet J. Surgical repair of a pseudocoarctation with cervical aortic arch complicated by multiple aneurysms of the aorta: a case report. Pan Afr Med J. (2017) 26:236. doi: 10.11604/pamj.2017.26.236.11800
54. Massumi R, Wiener L, Charif P. The syndrome of cervical aorta. Report of a case and review of the previous cases. Am J Cardiol. (1963) 11:678–85. doi: 10.1016/0002-9149(63)90089-4
55. McCue CM, Mauck HP, Tingelstad JB, Kellett GN. Cervical aortic arch. Am J Dis Child. (1973) 125:738–42. doi: 10.1001/archpedi.1973.04160050082017
56. McElhinney DB, Thompson LD, Weinberg PM, Jue KL, Hanley FL. Surgical approach to complicated cervical aortic arch: anatomic, developmental, and surgical considerations. Cardiol Young. (2000) 10:212–9. doi: 10.1017/s1047951100009136
57. Moncada R, Shannon M, Miller R, White H, Friedman J, Shuford WH. The cervical aortic arch. Am J Roentgenol Radium Ther Nucl Med. (1975) 125:591–601. doi: 10.2214/ajr.125.3.591
58. Mullins CE, Gillette PC, McNamara DG. The complex of cervical aortic arch. Pediatrics. (1973) 51:210–5. doi: 10.1542/peds.51.2.210
59. Ojha V, Vadher A, Chandrashekhara SH, Malhi AS, Nayak SK, Kumar S. A unique case of separate origins of left internal and external carotid arteries from high aortic arch with aberrant right subclavian artery – an unreported association in tetralogy of Fallot. J Cardiovasc Comput Tomogr. (2020) 14:e71–2. doi: 10.1016/j.jcct.2019.04.004
60. Oztunç F, Babaoĝlu K, Türkoglu H. Left cervical aortic arch with proximal obstruction. Cardiol Young. (2004) 14:453–5. doi: 10.1017/S1047951104004184
61. Patel SS, Morriss JH. Cervical aortic arch with a new twist. Pediatr Cardiol. (2009) 30:566–7. doi: 10.1007/s00246-009-9430-5
62. Pearson GD, Kan JS, Neill CA, Midgley FM, Gardner TJ, Hougen TJ. Cervical aortic arch with aneurysm formation. Am J Cardiol. (1997) 79:112–4. doi: 10.1016/s0002-9149(96)00695-9
63. Priya S, Nagpal P. Virtual modeling and interactive virtual reality display of unusual high-riding cervical aortic arch. Ann Pediatr Cardiol. (2021) 14:122–4. doi: 10.4103/apc.APC_188_19
64. Rajbanshi BG, Gautam NC, Pradhan S, Sharma A, Ghimire RK, Joyce LD. Complex cervical aortic arch with hypoplasia: a simple solution to a complex problem. Ann Thorac Surg. (2016) 102:e27–9. doi: 10.1016/j.athoracsur.2015.11.030
65. Ramaswamy P, Harrington JK. Fetal echocardiographic diagnosis of a triad with common embryological origins: cervical aortic arch, retro-aortic left innominate vein and coarctation of the aorta. Echocardiography. (2021) 38:1657–61. doi: 10.1111/echo.15179
67. Schleman MM, Kory LA, Gootman N, Silbert D. Right cervical aortic arch associated with a ventricular septal defect. Chest. (1975) 68:601–3. doi: 10.1378/chest.68.4.601
68. Shakerian B, Mandegar MH, Moradi B, Roshanali F. Right-sided cervical aortic arch in Loeys–Dietz syndrome. J Cardiol Cases. (2015) 11:60–1. doi: 10.1016/j.jccase.2014.10.004
69. Shepherd RM, Kerth WJ, Rosenthal JH. Right cervical aortic arch with left descending aorta. Case report and review of the literature. Am J Dis Child (1969) 118:642–8. doi: 10.1001/archpedi.1969.02100040644021
70. Shuford WH, Sybers RG, Schlant RC. Right aortic arch with isolation of the left subclavian artery. Am J Roentgenol Radium Ther Nucl Med. (1970) 109:75–83. doi: 10.2214/ajr.109.1.75
71. Takao R, Imamura H, Koga Y, Baba H, Amamoto Y, Takao A. Right-sided cervical aortic arch associated with tetralogy of Fallot and peculiar tortuosity of the descending aorta. Cardiovasc Radiol. (1979) 2:51–4. doi: 10.1007/BF02552018
72. Tiraboschi R, Crupi G, Locatelli G, Ho SY, Parenzan L. Cervical aortic arch with aortic obstruction: report of two cases. Thorax. (1980) 35:26–30. doi: 10.1136/thx.35.1.26
73. Tjang YS, Aramendi JI, Crespo A, Hamzeh G, Voces R, Rodríguez MA. Right cervical aortic arch with aberrant left subclavian artery. Asian Cardiovasc Thorac Ann. (2008) 16:e37–9. doi: 10.1177/021849230801600425
74. Vaillant L, Lorette G, Chantepie A, Marchand M, Alison D, Vaillant MC, et al. Multiple cutaneous hemangiomas and coarctation of the aorta with right aortic arch. Pediatrics. (1988) 81:707–10. doi: 10.1542/peds.81.5.707
75. Van Nooten G, Deuvaert F, De Paepe J, Primo G. Left-sided cervical aortic arch. Acta Chir Belg. (1986) 86:248–50.3766025
76. van Son JA, Bossert T, Mohr FW. Surgical treatment of vascular ring including right cervical aortic arch. J Card Surg. (1999) 14:98–102. doi: 10.1111/j.1540-8191.1999.tb00957.x
77. Walker T, Heinemann M-K, Nagy S, Steil E, Ziemer G. Right-sided cervical aortic arch with stenosis – treatment with an extra-anatomic bypass graft. Thorac Cardiovasc Surg. (2002) 50:306–7. doi: 10.1055/s-2002-34576
78. Whitman G, Stephenson LW, Weinberg P. Vascular ring: left cervical aortic arch, right descending aorta, and right ligamentum arteriosum. J Thorac Cardiovasc Surg. (1982) 83:311–5. doi: 10.1016/S0022-5223(19)37313-1
79. Yeager SB, Balian AA, Neal WA. Cervical aortic arch: an unusual cause of a suprasternal notch thrill and diminished left arm pulse. W V Med J. (1984) 80:141–2.6589875
Keywords: cervical aortic arch, congenital heart disease, cardiac surgery, pediatrics, meta-analysis
Citation: Baudo M, Varrica A, Reali M, Saracino A, Carminati M, Frigiola A, Giamberti A and Lo Rito M (2023) Cervical aortic arch in the pediatric population: a meta-analysis of individual patient's data. Front. Cardiovasc. Med. 10:1266956. doi: 10.3389/fcvm.2023.1266956
Received: 25 July 2023; Accepted: 11 September 2023;
Published: 28 September 2023.
Edited by:
Arpit Kumar Agarwal, Baylor College of Medicine, United StatesReviewed by:
Cosimo Marco Campanale, Bambino Gesù Children's Hospital (IRCCS), ItalyMaruti Haranal, U N Mehta Institute of Cardiology and Research, India
© 2023 Baudo, Varrica, Reali, Saracino, Carminati, Frigiola, Giamberti and Lo Rito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauro Lo Rito mauro.lorito@gmail.com