 Siqi Gao1,†
Siqi Gao1,† Alan T. Tang1,†
Alan T. Tang1,† Min Wang1David W. Buchholz2Brian Imbiakha2Jisheng Yang1Xiaowen Chen1
Min Wang1David W. Buchholz2Brian Imbiakha2Jisheng Yang1Xiaowen Chen1 Peter Hewins3Patricia Mericko-Ishizuka1N. Adrian Leu4Stephanie Sterling4
Peter Hewins3Patricia Mericko-Ishizuka1N. Adrian Leu4Stephanie Sterling4 Avery August2
Avery August2 Kellie A. Jurado3Edward E. Morrisey1,5
Kellie A. Jurado3Edward E. Morrisey1,5 Hector Aguilar-Carreno2
Hector Aguilar-Carreno2 Mark L. Kahn1*
Mark L. Kahn1*
- 1Department of Medicine and Cardiovascular Institute, University of Pennsylvania, Philadelphia, PA, United States
- 2Department of Microbiology and Immunology, College of Veterinary Medicine, Cornell University, Ithaca, NY, United States
- 3Department of Microbiology, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, United States
- 4Department of Biomedical Sciences, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 5Penn-CHOP Lung Biology Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
Endothelial damage and vascular pathology have been recognized as major features of COVID-19 since the beginning of the pandemic. Two main theories regarding how severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) damages endothelial cells and causes vascular pathology have been proposed: direct viral infection of endothelial cells or indirect damage mediated by circulating inflammatory molecules and immune mechanisms. However, these proposed mechanisms remain largely untested in vivo. In the present study, we utilized a set of new mouse genetic tools developed in our lab to test both the necessity and sufficiency of endothelial human angiotensin-converting enzyme 2 (hACE2) in COVID-19 pathogenesis. Our results demonstrate that endothelial ACE2 and direct infection of vascular endothelial cells do not contribute significantly to the diverse vascular pathology associated with COVID-19.
Introduction
The most common clinical feature reported in patients with COVID-19 is respiratory symptoms (1, 2). In addition to primarily causing pulmonary symptoms, COVID-19 disease is accompanied by vascular pathology, endothelial damage, and vascular coagulopathy (3–5). Reports emerged around the world confirming a disproportionate prevalence of abnormal thrombotic events and vascular pathology in patients with COVID-19, even in those not in intensive care units (6–14). Theories regarding the mechanism of vascular disease observed in patients with COVID-19 have been proposed, including direct infection of endothelial cells and systemic inflammatory responses (15–22). However, these hypotheses remain largely untested, and the cellular basis of vascular pathology remains controversial. In this study, we used a set of new mouse genetic tools (23) to rigorously test endothelial contribution to COVID-19-associated vascular pathology.
Materials and methods
Mice
hACE2fl/y mice and LSL-hACE2+/0 mice have been generated through CRISPR/Cas9-assisted mouse embryonic stem cell targeting and have been described (23). Briefly, for loss of function mouse line, the human ACE2 cDNA sequence was inserted after the ATG-start codon of mouse Ace2 in exon2, flanking the polyA cassette with loxP sites, to achieve Cre-mediated cell type-specific deletion of ACE2. For gain of function mouse line, the human ACE2 cDNA sequence was targeted to Rosa26 locus with Lox-STOP-Lox cassette to permit tissue-specific gain of expression of ACE2. Tie2-Cre transgenic mice have been used for tissue-specific drivers as previously described (24). All mice were maintained on a mixed genetic background, including C57BL/8 and other strains, at the University of Pennsylvania animal facility. Mice were genotyped by PCR as described (23). The number of male mice used in each experiment ranged from three to nine.
Viral inoculation and tissue harvest
Viral inoculations were performed as described previously (23). Briefly, mice were anesthetized with isoflurane and then intranasally infected with SARS-CoV-2 (Isolate USA-WA1/2020; BEI resources: NR-52281) that was obtained from BEI Resource. Mice were monitored and weighed daily, then euthanized at a humane endpoint when they lost 20% of their starting weights. Mice studies were combined results from Penn ABSL3 laboratory and Cornell ABSL3 laboratory in accordance with protocols approved by the IACUC at the University of Pennsylvania and Cornell University. For tissue harvest, mice were euthanized with ketamine/xylazine. Lungs were gently inflated with PBS infusion via trachea cannulation. Then lungs were fixed in 4% paraformaldehyde with a minimum of 72 h to ensure viral inactivation. Tissues were removed from the animal BSL3 facility, followed by ethanol dehydration and embedding in paraffin blocks for histology. Hematoxylin and eosin staining was performed on paraffin sections.
Immunofluorescence staining and analysis
Immunohistochemistry staining was performed as previously described (23) with control and experimental samples on the same slide and under identical staining conditions. Primary antibodies were as follows: pan-ACE2 (goat, 1:1,000, R&D AF933), hACE2 (rabbit, 1:200, Abcam ab108209), SARS-CoV-2 nucleocapsid (rabbit, 1:500, Rockland 200-401-A50), ICAM-1 (rabbit, 1:500, Abcam ab179707), vWF (rabbit 1:1,000, Novus Biologicals NB600-586), and PECAM (goat 1:500, R&D AF3628). Fluorescence-conjugated Alexa Fluor secondary antibodies were used (1:500, Invitrogen) according to the primary antibody species and counterstained with DAPI (1:1,000). ICAM1 and vWF fluorescence intensity were calculated by integrated fluorescence intensity. All images were analyzed using ImageJ/FIJI software.
Statistics
Mice were inoculated with SARS-CoV-2 in a blinded fashion without knowledge of genotypes, and infections were performed in two different ABSL-3 facilities with independent experimenters. Statistical tests used to determine significance are described in the figure legends. GraphPad Prism 9.5.1 was used to generate graphs and statistical analyses. Survival curve statistics were performed with log-rank Mantel-Cox tests. All t-tests performed were two-tailed.
Results and discussion
Cellular expression of ACE2 is indispensable for SARS-CoV-2 infection in pneumocytes (25, 26), but SARS-CoV-2 is unable to bind mouse ACE2. To determine if endothelial cells directly contribute to lethal infection, we generated animals that express human ACE2 (hACE2) from the mouse Ace2 locus in a manner that enables cell-specific loss of hACE2 using Cre recombinase (hACE2fl/y mice) (23). We crossed hACE2fl/y mice onto a Tie2-Cre transgenic mouse line that drives Cre expression in endothelial cells (ECs) to generate mice that express hACE2 in all cells except vascular ECs. hACE2fl/y; Tie2-Cre+ mice and control littermates were exposed to 105 PFU of SARS-CoV-2 virus via nasal inhalation. hACE2fl/y; Tie2-Cre+ mice showed no significant difference in survival after exposure to SARS-CoV-2 compared with the littermate controls (Figure 1A). Histological analysis revealed the presence of alveolar infiltrates and pulmonary vascular thrombi in the lungs of infected hACE2fl/y; Tie2-Cre+ mice that were indistinguishable from findings observed in control hACE2fl/y mice (Figure 1B). Histological analysis using hematoxylin-eosin staining of tissue sections from the small intestine, kidney, liver, and heart also failed to identify any vascular pathology (Supplementary Figure S1). Next, we evaluated the expression of inflammation-induced protein intracellular adhesion marker 1 (ICAM1) and the pro-coagulant, inflammation-induced protein Von Willebrand factor (vWF) in the mice following the SARS-CoV-2 infection, given both ICAM1 and vWF have been closely associated with COVID-19 induced vascular damage (27, 28). Expression of ICAM1 and vWF were also similar in the lung capillary endothelial cells of SARS-CoV-2-infected hACE2fl/y and hACE2fl/y; Tie2-Cre+ mice (Figures 1C,D).
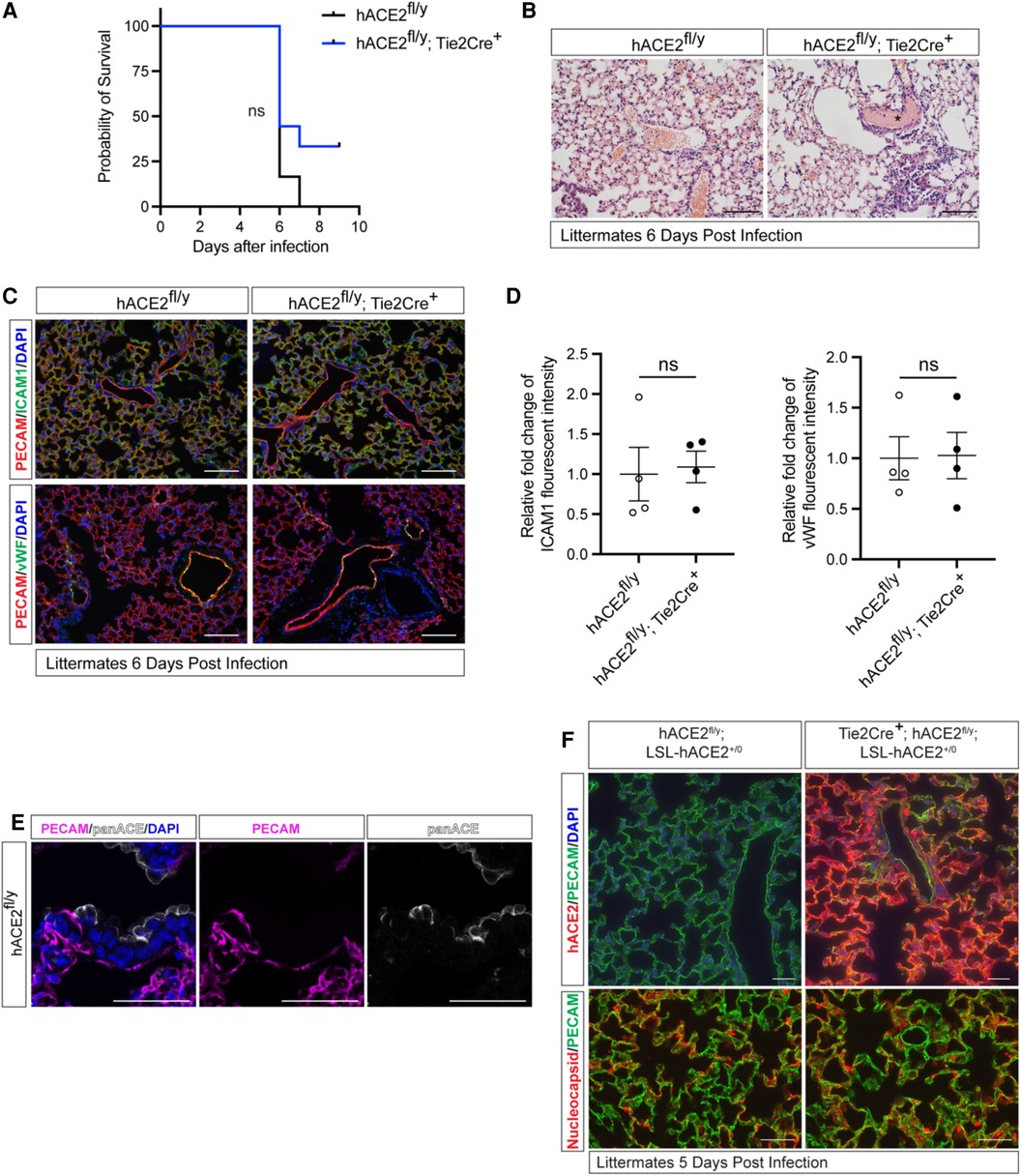
Figure 1. Loss or gain of endothelial hACE2 does not alter SARS-CoV2 infection. (A) Survival of hACE2fl/y and hACE2fl/y; Tie2-Cre+ male mice (12 to 16-week-old males) after infection with 105 PFU of SARS-CoV-2 via intranasal administration. This viral inoculation method was used in all experiments. n = 6 (hACE2fl/y) and 9 (hACE2fl/y; Tie2-Cre+); ns, non-significant; data are from two independent experiments. (B) H&E staining of hACE2fl/y and hACE2fl/y; Tie2-Cre+ lung tissue 6 days after infection. The asterisk indicates intravascular thrombosis. Scale bars: 100 μm. (C) Immunofluorescent staining of the lung from hACE2fl/y and hACE2fl/y; Tie2-Cre+ mice with antibodies against ICAM1 or vWF (green), and PECAM (red). Images are representative of four animals per genotype. Scale bars: 100 μm. (D) Quantification of ICAM1 and vWF fluorescent intensity. The error bars represent mean ± s.d; statistical analyses were performed using an unpaired two-tailed t-test; ns, non-significant. (E) Immunofluorescent staining of hACE2fl/y lung tissue using pan-ACE2 antibodies (grey) that recognize both hACE2 and mACE2 proteins and co-stained with PECAM (magenta). Images are representative of three animals. Scale bars 50 μm. (F) Immunofluorescent staining of the lung from hACE2fl/y; LSL-hACE2+/0 and Tie2Cre+; hACE2fl/y; LSL-hACE2+/0 mice is performed using anti-hACE2 antibody or anti-SARS-CoV-2 nucleocapsid (red) and costained with PECAM (green) 5 days after infection with SARS-CoV-2. The hACE2fl/y allele enables these mice to be productively infected intranasally. Representative of three animals per genotype. Scale bars 100 μm.
The studies described above suggested that endothelial cell infection is not required for vascular COVID-19 pathology when hACE2 is expressed at endogenous levels. In fact, immunostaining of lung sections using anti-ACE2 antibodies was able to detect ACE2 expression in epithelial but not endothelial cells (23) (Figure 1E). To more rigorously test the role of endothelial hACE2, we next crossed Tie2-Cre onto a recently described Cre-activated gain of function hACE2 allele (loxP-stop-loxP-hACE2 or LSL-hACE2+/0) (23) to over-express hACE2 in vascular endothelial cells. Tie2-Cre;LSL-hACE2+/0 animals exhibited very high endothelial-specific expression of hACE2, assessed by immunostaining of tissue sections compared with hACE2fl/y mice (Figure 1F). To ensure that Tie2-Cre;LSL-hACE2+/0 animals would be productively infected following SARS-CoV-2 exposure, we generated Tie2-Cre;LSL-hACE2+/0;hACE2fl/y animals that support robust infection of the nasal and respiratory epithelium (23) (Figure 1F). Despite high levels of endothelial hACE2 expression, we failed to detect nucleocapsid protein that colocalized with PECAM+ endothelial cells following nasal SARS-CoV-2 infection (Figure 1F). In contrast, we have previously shown that this gain of function allele is sufficient to drive hACE2 expression and support SARS-CoV-2 infection in both neuronal cells and lung epithelial cells (23). These studies support the conclusion that SARS-CoV-2 does not confer endothelial cell damage and vascular thrombosis through direct viral infection of those cells. They further demonstrate that the levels of circulating virus are too low to infect even endothelial cells that express very high levels of hACE2, and therefore that most COVID-19 pathology arises due to aerosol infection of the nasal and pulmonary epithelium.
It has been debated whether direct viral infection of endothelial cells or indirect damage from systematic inflammation underlie COVID-19-associated vascular pathology (3). Our murine vascular endothelial loss and gain of function studies reported here provide strong in vivo evidence that endothelial ACE2 and direct infection of vascular endothelial cells do not contribute significantly to the diverse vascular pathology associated with COVID-19. These findings are consistent with previously reported in vitro studies that showed human endothelial cells are not readily infected by SARS-CoV-2 (21). Together with our recently reported studies, these findings strongly support a mechanism in which SARS-CoV-2 infection of nasal epithelial and neuronal cells stimulates a powerful inflammatory response that is the cause of COVID-19 vascular pathology.
Limitations of the study
In the present study, we utilized both loss of function and gain of function hACE2 mouse lines and demonstrated that direct endothelial viral infection does not contribute to COVID-19-associated vascular pathology. Future studies are needed to define the cytokines that likely drive secondary vascular inflammation and thrombosis and to understand the molecular mechanism by which systemic inflammation damages endothelium following SARS-CoV-2 infection. We used the original isolate SARS-CoV-2 USA-WA1/2020 strain in our study because that isolate is the best characterized regarding vascular complication. Omicron BA.1 variant failed to confer lethal disease and associated vascular phenotypes in our mouse models (23). Future studies testing the impact of other variants on the vascular system will be needed. We performed our studies on male mice due to the Ace2 allele being located on the X chromosome, enabling a straightforward comparison. Mouse models are not humans, and our mouse model hACE2fl/y expresses a higher level of hACE2 as previously reported (23). Thus there are likely to be differences in pathogenic mechanisms identified using our model compared to human studies. However, this difference should bias toward rather than against a direct endothelial infection mechanism and it does not weaken our negative conclusions. Future studies looking at longer-term vascular events in mice with lower levels of hACE2 expression will be needed to address non-acute mechanisms of COVID-19-related cardiovascular disease.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by University of Pennsylvania IACUC. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
MK: Conceptualization, Formal analysis, Funding acquisition, Project administration, Writing – original draft. SG: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Writing – original draft. AT: Conceptualization, Formal analysis, Writing – original draft. MW: Conceptualization, Data curation, Formal analysis, Methodology, Writing – original draft. DB: Conceptualization, Data curation, Writing – original draft. BI: Data curation, Formal analysis, Conceptualization, Writing – original draft. JY: Data curation, Formal analysis, Writing – original draft. XC: Conceptualization, Data curation, Formal analysis, Writing – original draft. PM: Methodology, Project administration, Writing – original draft. NL: Data curation, Formal analysis, Methodology, Writing – original draft. SS: Methodology, Project administration, Writing – original draft. AA: Project administration, Data curation, Methodology, Writing – original draft. KJ: Data curation, Methodology, Project administration, Investigation, Writing – original draft. EM: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Writing – original draft, Conceptualization. HA: Conceptualization, Data curation, Formal analysis, Funding acquisition, Project administration, Writing – original draft. PH: Formal analysis, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
This work was supported by National Institute of Health grants R01HL39552-04S1 (MLK), R01HL164929 (MLK and EEM), AHA 963048 (MLK), R01AI109022 and R21AI156731 (HAC), and T32EB023860 (DWB), an AHA Postdoctoral Fellowship 906488 (SG), and a Penn CVI Dream Team grant (MK).
Acknowledgments
We thank Kahn lab members for their helpful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1266276/full#supplementary-material
References
1. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
2. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
3. Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol. (2020) 20:389–91. doi: 10.1038/s41577-020-0343-0
4. Smadja DM, Mentzer SJ, Fontenay M, Laffan MA, Ackermann M, Helms J, et al. COVID-19 is a systemic vascular hemopathy: insight for mechanistic and clinical aspects. Angiogenesis. (2021) 24:755–88. doi: 10.1007/s10456-021-09805-6
5. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. (2020) 135:2033–40. doi: 10.1182/blood.2020006000
6. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
7. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. (2020) 18:844–7. doi: 10.1111/jth.14768
8. Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, et al. Large-vessel stroke as a presenting feature of COVID-19 in the young. N Engl J Med. (2020) 382:e60. doi: 10.1056/NEJMc2009787
9. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb Res. (2020) 191:148–50. doi: 10.1016/j.thromres.2020.04.041
10. Song WC, FitzGerald GA. COVID-19, microangiopathy, hemostatic activation, and complement. J Clin Invest. (2020) 130:3950–3. doi: 10.1172/JCI140183
11. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. (2020) 7:e575–82. doi: 10.1016/S2352-3026(20)30216-7
12. Al-Samkari H, Karp Leaf RS, Dzik WH, Carlson JCT, Fogerty AE, Waheed A, et al. COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood. (2020) 136:489–500. doi: 10.1182/blood.2020006520
13. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
14. Jimenez D, Garcia-Sanchez A, Rali P, Muriel A, Bikdeli B, Ruiz-Artacho P, et al. Incidence of VTE and bleeding among hospitalized patients with coronavirus disease 2019: a systematic review and meta-analysis. Chest. (2021) 159:1182–96. doi: 10.1016/j.chest.2020.11.005
15. Merrill JT, Erkan D, Winakur J, James JA. Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications. Nat Rev Rheumatol. (2020) 16:581–9. doi: 10.1038/s41584-020-0474-5
16. He L, Mäe MA, Muhl L, Sun Y, Pietilä R, Nahar K, et al. Pericyte-specific vascular expression of SARS-CoV-2 receptor ACE2—implications for microvascular inflammation and hypercoagulopathy in COVID-19. bioRxiv. 2020:2020.2005.2011.088500. doi: 10.1101/2020.05.11.088500
17. Henry BM, Vikse J, Benoit S, Favaloro EJ, Lippi G. Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: a novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin Chim Acta. (2020) 507:167–73. doi: 10.1016/j.cca.2020.04.027
18. Gu SX, Tyagi T, Jain K, Gu VW, Lee SH, Hwa JM, et al. Thrombocytopathy and endotheliopathy: crucial contributors to COVID-19 thromboinflammation. Nat Rev Cardiol. (2020) 18(3):194–209. doi: 10.1038/s41569-020-00469-1
19. Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat Rev Nephrol. (2021) 17:46–64. doi: 10.1038/s41581-020-00357-4
20. Schimmel L, Chew KY, Stocks CJ, Yordanov TE, Essebier P, Kulasinghe A, et al. Endothelial cells are not productively infected by SARS-CoV-2. Clin Transl Immunology. (2021) 10:e1350. doi: 10.1002/cti2.1350
21. McCracken IR, Saginc G, He L, Huseynov A, Daniels A, Fletcher S, et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation. (2021) 143:865–8. doi: 10.1161/CIRCULATIONAHA.120.052824
22. Nicosia RF, Ligresti G, Caporarello N, Akilesh S, Ribatti D. COVID-19 vasculopathy: mounting evidence for an indirect mechanism of endothelial injury. Am J Pathol. (2021) 191:1374–84. doi: 10.1016/j.ajpath.2021.05.007
23. Tang AT, Buchholz DW, Szigety KM, Imbiakha B, Gao S, Frankfurter M, et al. Cell-autonomous requirement for ACE2 across organs in lethal mouse SARS-CoV-2 infection. PLoS Biol. (2023) 21:e3001989. doi: 10.1371/journal.pbio.3001989
24. Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. (2001) 230:230–42. doi: 10.1006/dbio.2000.0106
25. Oudit GY, Wang K, Viveiros A, Kellner MJ, Penninger JM. Angiotensin-converting enzyme 2-at the heart of the COVID-19 pandemic. Cell. (2023) 186:906–22. doi: 10.1016/j.cell.2023.01.039
26. Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe A, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. (2021) 595:107–13. doi: 10.1038/s41586-021-03570-8
27. Nagashima S, Mendes MC, Camargo Martins AP, Borges NH, Godoy TM, Miggiolaro A, et al. Endothelial dysfunction and thrombosis in patients with COVID-19-brief report. Arterioscler Thromb Vasc Biol. (2020) 40:2404–7. doi: 10.1161/ATVBAHA.120.314860
Keywords: SARS-CoV-2, endothelial cell, COVID-19, vascular, hACE2
Citation: Gao S, Tang AT, Wang M, Buchholz DW, Imbiakha B, Yang J, Chen X, Hewins P, Mericko-Ishizuka P, Leu N.A, Sterling S, August A, Jurado KA, Morrisey EE, Aguilar-Carreno H and Kahn ML (2023) Endothelial SARS-CoV-2 infection is not the underlying cause of COVID-19-associated vascular pathology in mice. Front. Cardiovasc. Med. 10:1266276. doi: 10.3389/fcvm.2023.1266276
Received: 24 July 2023; Accepted: 5 September 2023;
Published: 26 September 2023.
Edited by:
Masanori Aikawa, Harvard Medical School, United StatesReviewed by:
Antonin Trimaille, Hôpitaux Universitaires de Strasbourg, FranceNicolas Gendron, Hôpital Européen Georges Pompidou, France
© 2023 Gao, Tang, Wang, Buchholz, Imbiakha, Yang, Chen, Hewins, Mericko-Ishizuka, Leu, Sterling, August, Jurado, Morrisey, Aguilar-Carreno and Kahn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark L. Kahn bWFya2thaG5AcGVubm1lZGljaW5lLnVwZW5uLmVkdQ==
†These authors share first authorship