 Georgia Atkinson
Georgia Atkinson Rosaria Bianco
Rosaria Bianco Karina Di Gregoli
Karina Di Gregoli Jason L. Johnson
Jason L. Johnson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 19 September 2023
Sec. General Cardiovascular Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1248561
This article is part of the Research Topic Abdominal Aortic Aneurysms: Advancement in Diagnose, Biomarkers, Drug Therapeutics, Surgical, and Endovascular Treatment Vol. II View all 4 articles
Abdominal aortic aneurysms (AAAs) account for up to 8% of deaths in men aged 65 years and over and 2.2% of women. Patients with AAAs often have atherosclerosis, and intimal atherosclerosis is generally present in AAAs. Accordingly, AAAs are considered a form of atherosclerosis and are frequently referred to as atherosclerotic aneurysms. Pathological observations advocate inflammatory cell infiltration alongside adverse extracellular matrix degradation as key contributing factors to the formation of human atherosclerotic AAAs. Therefore, macrophage production of proteolytic enzymes is deemed responsible for the damaging loss of ECM proteins, especially elastin and fibrillar collagens, which characterise AAA progression and rupture. Matrix metalloproteinases (MMPs) and their regulation by tissue inhibitors metalloproteinases (TIMPs) can orchestrate not only ECM remodelling, but also moderate the proliferation, migration, and apoptosis of resident aortic cells, alongside the recruitment and subsequent behaviour of inflammatory cells. Accordingly, MMPs are thought to play a central regulatory role in the development, progression, and eventual rupture of abdominal aortic aneurysms (AAAs). Together, clinical and animal studies have shed light on the complex and often diverse effects MMPs and TIMPs impart during the development of AAAs. This dichotomy is underlined from evidence utilising broad-spectrum MMP inhibition in animal models and clinical trials which have failed to provide consistent protection from AAA progression, although more encouraging results have been observed through deployment of selective inhibitors. This review provides a summary of the supporting evidence connecting the contribution of individual MMPs to AAA development, progression, and eventual rupture. Topics discussed include structural, functional, and cell-specific diversity of MMP members; evidence from animal models of AAA and comparisons with findings in humans; the dual role of MMPs and the requirement to selectively target individual MMPs; and the advances in identifying aberrant MMP activity. As evidenced, our developing understanding of the multifaceted roles individual MMPs perform during the progression and rupture of AAAs, should motivate clinical trials assessing the therapeutic potential of selective MMP inhibitors, which could restrict AAA-related morbidity and mortality worldwide.
Abdominal aortic aneurysms (AAA) demonstrate a major, life-threatening cardiovascular condition as demonstrated through mortality rates associated with ruptured AAAs estimated at 90% (1). Progress has been made in elucidating the multifactorial process of AAA pathogenesis, in particular the components of chronic inflammation which has led to promising candidates for conservative treatments currently being tested in several prospective clinical trials. Yet to date, surgical repair remains the only curative approach, but is associated with considerable morbidity and mortality rates.
Matrix metalloproteinases (MMPs) are a family of enzymes which degrade major components of the vessel wall. Additionally, resident and recruited cells can utilise MMPs directly and indirectly to facilitate their behaviour, including proliferation, migration, and survival. As a result, MMPs intimately contribute to the development and progression of multiple cardiovascular diseases, including AAA. An imbalance in the ratio between MMPs and the endogenous tissue inhibitors of MMPs (TIMPs) is believed to be a critical part of the dysregulated extracellular matrix degradation seen in AAAs, with numerous studies observing increased MMP activity in AAA patients (2). This review provides an overview of the role of MMPs and TIMPs in AAA development, progression, and rupture.
Aortic aneurysms are principally found within the abdominal and thoracic aorta and are respectively referred to as AAA and TAA. AAAs are the most common as data revealed a prevalence of 5%–6% for men and 1%–2% for women, in people older than 65 years, in comparison to a prevalence of five-fold lower for TAA (3). TAAs can be observed in different aortic segments including the aortic root, ascending aorta, aortic arch, or descending aorta. Sixty percent of TAAs develop in the aortic root and ascending aorta, forty percent are attributed to the descending aorta, whereas only ten percent involve the arch and the thoracic-abdominal area (4). AAAs are characterised as suprarenal when located within the suprarenal region of the abdominal aorta where the visceral arteries are involved. They are defined as juxtarenal (also termed pararenal) if the aneurysm extends up to but does not involve the renal arteries, or as infrarenal if they begin 10 mm below the renal arteries (5, 6). The pathogenesis of AAAs and TAAs is distinct.
AAAs have a complex multifactorial pathogenesis in which genetic and environmental risk factors play a prominent role (5). Clinically, there is no universal definition of AAA, however, it is widely accepted to be a maximum infrarenal abdominal aortic diameter of >30 mm, measured using ultrasonography or computed tomography angiogram (CTA). Although the exact cause of AAAs is unknown, several factors are considered to contribute to the focal weakening of the aortic wall, increasing an individual's risk of developing an aneurysm.
Males are around four-times more likely to be diagnosed with an aneurysm during their lifetime compared to females, with this risk further increased by 40% every five years after the age of 65 (7). Moreover, AAA incidence is higher among the Caucasian population compared to those of African-American, Asian and Hispanic descent (8). Smoking has been highlighted as a major risk factor for aneurysm initiation and progression (9). The deleterious effects of smoking have been attributed to alterations in the inflammatory response of the aortic wall, alongside increased matrix degradation due to an imbalance between proteases (such as matrix metalloproteinases) and protease inhibitors. Moreover, there is an association between smoking duration and number of cigarettes smoked with AAA risk. Other risk factors include a family history of aneurysms. In particular, it has been shown that individuals with a first-degree relative with an AAA have a 30% higher risk of developing an AAA themselves as well as a higher risk of aneurysm rupture and increased likelihood of developing an aneurysm at a younger age (10). Another study revealed that the growth rate of aneurysms is doubled in patients with familial history when compared to patients with no evidence of previous familial disease (11).
AAAs often grow slowly and are typically asymptomatic until they rupture, making them difficult to detect. This is the main complication of AAAs and underlies their high mortality rates of approximately 60%–80%. However, when symptoms do arise, these usually display as constant abdominal or back tenderness or pain, which can last for hours up to days. Some patients report a palpable abdominal mass. At times, AAAs may cause symptoms because of local compression, leading to early satiety, nausea, vomiting, urinary symptoms, or venous thrombosis due to venous compression. Aneurysms that produce symptoms are at an increased risk for rupture, leading to further increased mortality rates. Due to the asymptomatic nature of most AAAs, diagnosis commonly results from incidental abdominal screening or during a routine examination involving abdominal palpation (12). Nationwide screening programmes have been implemented in several countries with the aim to alleviate AAA-specific mortality. Ultrasonography and computed tomography angiography are first-line imaging tools to identify and manage AAAs (13). An ultrasound scan is an accurate method to measure aneurysm size, with the benefit of being inexpensive and non-invasive. Unfortunately, many AAAs remain undetected and a ruptured aortic aneurysm can lead to life-threatening internal bleeding, with larger aneurysms exhibiting a greater risk (14, 15).
Therapies for AAAs depend on a variety of factors such as location and size, as well as patient-specific factors like age, risk factors, or other existing conditions that may increase the risk during surgical intervention. Small aneurysms (less than 5.5 cm in diameter) are deemed at a low risk of rupture and are typically treated with pharmacological interventions to control blood pressure, aiming to reduce shear stress within the aorta (15). For example, beta-blockers may decrease the rate at which aneurysms grow by reducing blood pressure and contractility of the left ventricle (15). Statins have also been considered as a good therapy due to their pleiotropic anti-inflammatory properties, as well as their ability to reduce the expression of MMPs (16). Similarly, angiotensin-converting enzyme (ACE)-inhibitors and Ang II receptor blockers (ARBs) have been proposed, and are already widely deployed for the treatment of other cardiovascular diseases (15, 16). Additionally, antibiotic therapy has been evaluated as a method to reduce aneurysm expansion by reducing elastin degradation and inflammation (17). Finally, lifestyle changes are recommended to improve outcomes and prevent aneurysm growth and rupture. As mentioned above, smoking is a risk factor of aneurysm growth and rupture, therefore the American College of Cardiology and American Heart Association have advised those with a positive familial history of AAA to stop smoking (18). However, there is currently no pharmacological therapy established to slow or prevent AAA growth and rupture directly.
Accordingly, two main methods of elective intervention are used as surgical options and are the only available curative treatment for AAA: open surgical repair and endovascular aneurysm repair (EVAR). Usually, surgical treatment is adopted only when the diameter of the aorta is greater than 5.5 cm, as risk of rupture outweighs the risk of surgical complications. Open surgical repair involves the removal of the damaged section of the aorta and replacement with a synthetic vascular graft. This will allow blood to flow through the graft, bypassing the aneurysmal area and therefore, reducing pressure on the damaged wall of the aorta. This procedure requires a 2–3-month recovery and is associated with a high risk of mortality (19). Alternatively, EVAR involves the minimally invasive insertion of a synthetic graft within the aneurysm via groin incisions to access the femoral artery. This is considered a less invasive procedure due to less interference with the circulation, resulting in a reduced recovery period. Considering outcomes, a study comparing both short-term and long-term survival of patients undergoing either procedure revealed that mortality rates are similar and influenced by age, co-morbidities, and pharmacological interventions (20).
As shown in Figure 1, human aneurysms are characterised by structural deterioration of the aortic wall, consequent progressive aortic dilatation and ultimately rupture. Multiple factors are involved in the pathogenesis of AAA which cause focal destructive remodelling of connective tissue through all layers of the aortic wall. A major pathobiological aspect of AAA includes loss of key extracellular matrix (ECM) proteins through the proteolytic degradation of collagen and elastin, accompanied by the infiltration and accumulation of inflammatory cells throughout all layers of the aortic wall, alongside resultant secretion of inflammatory growth factors and cytokines. Additionally, AAAs are commonly characterised by a reduction in VSMC content due to apoptosis, and the presence of neovascularisation (1, 21). MMPs and other proteases are produced by macrophages and VSMCs and are proposed to contribute to many of the above processes.
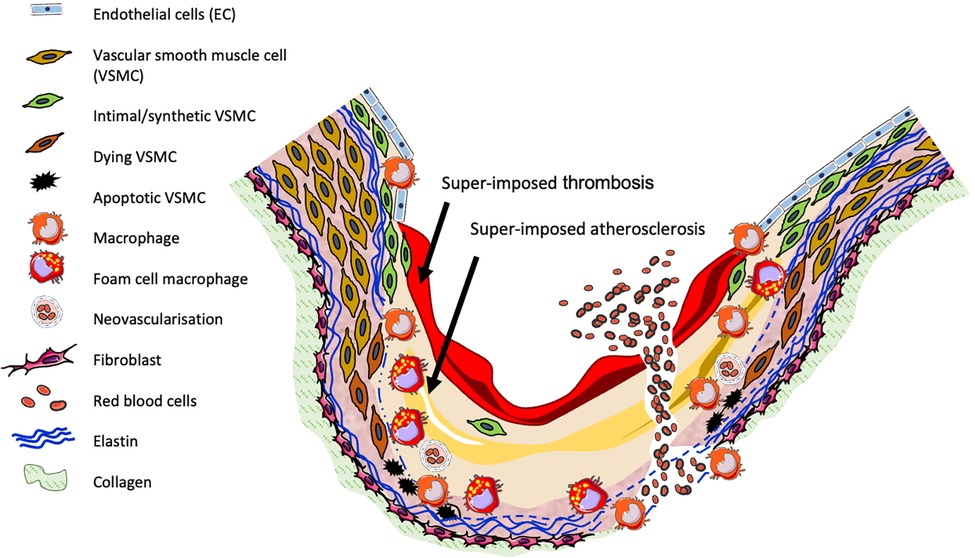
Figure 1. Diagram of human AAA pathogenesis.
Collagen and elastin are the most abundant extracellular matrix proteins found within large and mid-sized arteries, imparting the elastic properties, structural strength and mechanical resistance to the vessel wall (22). Collagen fibrils are stable structures formed from the aggregation of several subunits, called tropocollagen, which play an essential role in the artery wall providing the necessary strength and stability. Twenty-nine types of collagen have been identified in the human body, although 80%–90% of collagen present in the body is of type I, II, and III (1, 23). Within the aortic wall, type I collagen accounts for 70% of the total collagen content, and is located predominantly in the intima and adventitia, whereas type III collagen is expressed within the media and along the elastic lamina (24). Several studies have identified collagen loss within aneurysmal aortae, contributing to the instability of the aortic wall. ECM degradation is prevalent during aneurysm progression, because of increased expression and activity of proteases, especially MMPs, leading to increased collagen degradation (25, 26). Accordingly, the combination of decreased vascular smooth muscle cell (VSMC) collagen synthesis (25) alongside enhanced degradation highlights the major contribution attributed to collagen turnover during aneurysm progression. Accordingly, products released by collagen catabolism have been indicated as potential aneurysm biomarkers, especially the amino terminal pro-peptide of type III procollagen (PIIIN). In AAA patients, serum levels of PIIIN were significantly elevated compared to control subjects in two separate studies, however the serum level of the carboxyterminal pro-peptide of type I procollagen (PICP) was similar between groups within multiple studies (27).
Elastin is organised as fibres within the medial portion of the aortic wall. The interaction of these fibres with VSMCs and collagen provide a solid structure which is necessary for the biomechanical and elastic properties of the aorta (28). Alterations in elastin fibre quantity alongside variations in collagen structure underlie the mechanical and functional changes related to aortic diseases. Specifically, elastin degradation occurs during aneurysm progression, releasing elastin-derived peptides into the circulation. A screening study revealed that patients with a ruptured AAA greater than 6 cm display increased levels of elastin-derived peptide, compared to those with small aneurysms that have not ruptured (29). The primary catabolic enzyme responsible of elastin degradation is elastase, a family of eight human genes including chymotrypsin (CTRC), neutrophil elastase (ELANE), and MMP-12 (also known as macrophage metalloelastase) (30), whose activity is inhibited by endogenous protease inhibitors such as alpha-1 anti-trypsin (A1AT) (31) and TIMP-3 (32). It has previously been shown that serum levels of A1AT are higher in AAA patients than those with aortic-occlusive disease, highlighting the role of elastin degradation during aneurysm progression and the response to alleviate increased elastase activity (1). As discussed in greater detail later, researchers have exploited the proposed central role of elastase in AAA development and progression through deploying intra-aortic elastase perfusion to induce AAA formation in mice, rats, rabbits, dogs, and pigs (33). The utility of this experimental approach demonstrated that elastase activity contributed to aneurysm formation and corroborated quantitative correlation between elastase concentration and aneurysm size (34). In summary, alterations in the structure of elastin and collagen contributes towards aneurysm formation and progression. Moreover, aortic dilation is linked with elastin loss, whereas aortic rupture is associated with loss of collagen (22, 35).
VSMCs are the most abundant cell type within the blood vessel wall, and play an important role in the structure of aorta wall by maintaining production of elastin, collagen, matrix proteins, several proteases, and inhibitors (36). VSMCs regulate the contractile tone of the vessel alongside the secretion of ECM proteins which highlights the two different VSMC phenotypes, synthetic and contractile (37). In healthy blood vessels, the predominant phenotype present is the contractile form, due to the limited secretion of ECM proteins and profuse myofilament production to allow regulation of blood flow and vessel diameter. During pathological conditions, the switching of VSMC phenotype from contractile to synthetic is induced, which is characterised through a decrease in contractile protein expression alongside an increased production of proteases such as collagenolytic and elastinolytic MMPs, which can degrade elastin and collagen and promote vessel dilation, weakening, and eventual rupture (21, 38).
Together with VSMC phenotypic modulation, a reduced number of VSMCs is observed in AAAs (39), attributed to their enhanced susceptibility to apoptosis, as VSMCs undergoing apoptosis are observed within the medial portion of human AAAs (40). Multiple factors have been proposed to induce VSMC apoptosis including oxidised lipoproteins, cytokines, and increased reactive oxygen species (41), alongside extracellular matrix degradation (42) and mechanical stress (43). Angiotensin II can also trigger VSMC apoptosis through ligation of angiotensin receptor types 1 and 2, with angiotensin II infusion shown to induce AAA formation in mice (33) which is associated with medial VSMC apoptosis (44). It was also found that VSMCs within aneurysms showed increased production and accumulation of p53, a pro-apoptotic protein and a member of the Bcl-2 protein family (39), a mechanism corroborated in a mouse elastase-induced aneurysm model (45).
Abdominal aortic calcification (AAC) has been observed as a sign of a degenerative inflammatory process within the arterial wall, occurring at two distinct sites: the intima and the media. Mainly, intimal calcification is observed in advanced atherosclerosis, with many studies revealing an association between calcification and risk of cardiovascular events, supporting its potential as a biomarker (46, 47). Calcification is characterised by lipid deposition, macrophage infiltration and VSMC proliferation, whereas medial calcification can exist independently of atherosclerosis and is usually associated with elastic fibres (48). Arterial calcification causes a reduction in the elasticity of the vessel wall which increases the risk of rupture during aneurysm development and progression (49, 50). A proteomics study of calcified AAAs (CAAs) revealed type I collagen α-1 and type III collagen α-1 chains were increased in CAA, whereas type XIV collagen α-1 was decreased compared to healthy controls (51). Furthermore, levels of ECM proteins such as fibulin-5 were decreased in calcified TAA (CTA) patients (51). Fibulin-5 plays an important role in endothelial cell adhesion and elastin fibre integrity, and is therefore important in preserving the stability of the aorta wall, through its integrin-binding matricellular protein properties (52).
Angiogenesis, characterised by the formation of new blood vessels from pre-existing vessels, plays an essential role in many developmental and pathological processes, including embryogenesis, inflammation, tissue development and repair (53). The induction of angiogenesis begins with ECM degradation and activation of vascular endothelial cells, with their increased proliferation and migration essential for new blood vessel formation. In healthy individuals, the balance of pro- and anti-angiogenic factors is maintained, whereas in pathological conditions there is an imbalance which favours the accumulation of pro-angiogenic factors (54). MMPs have been recognised as a factor strongly involved in angiogenesis, and a histological study of human AAAs demonstrated prevalent angiogenesis (also termed neovascularisation) within the media of the aortic wall which associated with increased MMP expression and activity (55), especially ruptured AAAs (56). Accordingly, neovascularisation within AAAs co-localise with areas of elastin degradation and macrophage accumulation, which are mainly restricted to the outer medial aspect and remodelling adventitia (53, 57).
Inflammation has been postulated to play a pivotal role in the pathogenesis of aneurysm. Inflammatory cells including neutrophils, T cells, B cells, macrophages, mast cells and NK cells can permeate through the aortic wall and are associated with increased production of pro-inflammatory cytokines and chemokines (58). Furthermore, VSMCs, endothelial cells and monocyte/macrophage MMP expression can be regulated by chemokines and lead to ECM degradation, contributing to the weakening of the aortic wall during aneurysm progression (59, 60). Cytokines are intercellular messenger proteins involved in the regulation of inflammation, haematopoiesis, cellular and humoral responses, and wound healing (60). Prevalent inflammatory cells found within aneurysms include cluster of differentiation 4+ (CD4+) T cells, B cells and macrophages. CD4 + type 1 T helper cells (Th1) and CD8+ T-cytotoxic type-1 (Tc1) cells produce interferon gamma (IFNγ), interleukin-2 (IL-2) and tumour necrosis factor (TNFα), whereas type 2 T helper cells (Th2) and type 2 cytotoxic cells (Tc2) secrete IL-4, IL-5, IL-10 and IL-13, which are also produced by B cells, macrophages, NK cells and ECs.
TNFα and IFNγ-driven inflammatory processes are prominent in the pathogenesis of aneurysms due its role in activation and recruitment of immune cells to inflammatory sites, alongside secretion of pro-inflammatory cytokines and MMPs, and inducing VSMC death (59, 61). Elevated levels of TNFα mRNA and protein were reported in aneurysmal tissue of rodent models, and elastase levels reduced after delivery of a TNFα binding protein, which suggests that TNFα may play a role in the pathogenic mechanisms of AAA (62). Similarly, increased levels of IFNγ have been observed in human AAA compared to controls and has been shown to suppress VSMC collagen production (63). IFNγ also stimulates macrophage and VSMC MMP production, further contributing to aneurysm formation (59). However, there is conflicting evidence over the role of this cytokine in AAA pathogenesis, as conversely, studies have found that blocking the IFNγ receptor led to accelerated aneurysm formation (64). This may be due to the divergent roles of IFNγ, which can exert both anti- and pro-inflammatory effects, dependent on the pathology and cytokines involved.
Transforming growth factor-β (TGFβ) can modulate the structure and composition of the ECM and therefore considered an important regulator of vascular remodelling. TGFβ is produced by multiple cell types and participates in a wide array of cellular responses including proliferation, angiogenesis, differentiation, apoptosis, inflammation, and wound healing. Reduced TGFβ levels have been detected in AAA patients, which correlated with reduced cystatin C, a cysteine protease inhibitor (65). TGFβ can also retard MMP-12 expression and therefore abrogate aneurysm progression in several mouse models (66). However, similarly to IFNγ, divergent roles for TGFβ have been proposed, with increased TGFβ activity shown to associate with AAA formation, and subsequent functional blocking of TGFβ reduced aneurysm formation (67, 68).
In addition, other cytokines such as macrophage migration inhibitory factor (MIF) have been implicated in aneurysm pathogenesis. MIF functions as a pleiotropic protein, participating in inflammatory and immune responses, in particular the stimulation of angiogenesis and regulation of proliferation (69). MIF also regulates the production and activation of T and B cells as well as modulating proteolysis through the regulation of MMP expression, and activation of urokinase plasminogen activator (uPA) and tissue plasminogen activator (tPA) (61). Granulocyte/macrophage-colony stimulating factor (GM-CSF) regulates monocyte production and maturation, while also polarising macrophages towards a pro-inflammatory phenotype with heightened expression of MMPs (70). GM-CSF administration to hypercholesterolaemic mice was shown to accelerate AAA formation (71), while GM-CSF inhibition dampened AAA development in mice which was associated with decreased inflammation and related MMP expression/activity, and elevated GM-CSF expressing inflammatory cells were detected within human AAA samples compared to non-diseased aortic samples (72). These findings further support a direct role for inflammation in the pathogenesis of AAA and highlight GM-CSF as a potential therapeutic target.
Relatedly, C-reactive protein (CRP) is considered a biomarker of cardiovascular events and an indicator of systemic inflammation. A study by Vainas and colleagues found high levels of serum CRP in patients with an increased aortic diameter (73). Patients with symptomatic or ruptured AAA showed higher circulating levels of CRP and this correlated with an increased aneurysm size. This relationship between CRP and aneurysm size was further confirmed in a study that demonstrated that the CRP level was greater in patients with large AAAs, compared to small ones, and a marked difference between AAA patients and healthy controls (74), further qualifying a central role for inflammation in AAA development and progression.
Atherosclerosis is characterised by thickening of the arterial wall due to the accumulation of lipids, vascular and inflammatory cells, and the deposition of various ECM proteins within the tunica intima (75). Several studies have revealed a strong association with atherosclerosis and aneurysm formation, with atherosclerosis commonly being considered a risk factor for subsequent aneurysm formation (76). However, it is still unclear if these pathologies are linked due to common risk factors or exert direct causal effects (77). In the past, different concepts have been developed to explain the relationship between AAA and atherosclerosis, with the general consensus being that AAAs are caused by atherosclerosis and are therefore called atherosclerotic aneurysms typically occurring within the abdominal aorta (78). Indeed, the pathophysiology of AAA has been termed as a highly proteolytic from of atherothrombosis (79). It has been proposed that luminal narrowing of the aorta, typical of atherosclerosis formation, induces modification and remodelling of the vascular wall to compensate for the loss of lumen patency, involving ECM remodelling to try and correct the vessel diameter. However, this inadvertently induces medial thinning and therefore weakening of the vessel wall, typical of AAA development (80). It is also plausible that AAA and atherosclerosis are independent pathologies with different aetiologies but are strongly linked with common risk factors such as smoking, hypertension and hypercholesterolaemia, although as discussed above, human AAAs harbour overlying atherosclerotic lesions. A final concept suggests that AAA or aortic atherosclerosis can develop first and consequently encourage development of the other. A study conducted by Golledge et al. stated that several independent mechanisms are responsible for AAA and atherosclerotic plaque formation which are different from patient to patient, concluding that it is important to avoid broad standardised therapeutics that would not be successful (80).
Aortic dissection is a lethal condition in which the inner layer of the aorta tears causing blood to push through the tear, separating the middle layer of the wall from the outer layer, creating a new lumen (false lumen). In some cases, the dissection will cross all three layers of the aortic wall and cause immediate rupture and almost certain death (81, 82). Several conditions can alter and weaken the aorta wall, subsequently predisposing individuals to an aortic dissection. These include hypertension, atherosclerosis, disorders of the connective tissue (Marfan Syndrome, Ehlers-Danlos Syndrome), genetic disorders or trauma, which can cause damage to the aorta wall, so as to cause an aortic dissection (83). Conversely, an aneurysm involves focal dilatation of a blood vessel, which may become weakened and subsequently rupture and cause death (84). Common risk factors for both aneurysm and dissection include ageing, atherosclerosis, and hypertension (85). Moreover, if an individual develops an AAA, this becomes a strong risk factor for future aortic dissection. In addition, once an aortic dissection occurs, the aorta becomes weak and may enlarge over time, which may ultimately lead to the development of an aneurysm. Therefore, although aneurysms and dissections have separate pathologies and aetiologies, they can be associated.
AAA is a common degenerative disorder associated with aortic rupture and consequently sudden death, with current therapies being limited. Accordingly, several animal models have been developed with the purpose of further elucidating the pathogenesis of AAA and illuminate the biochemical and cellular mechanisms underlying this disease (86). A wide range of animal models have been developed across a variety of species including pig, sheep, dogs, rabbits, rodents, and primates. Like humans, certain dog breeds have comparable artery size as well as ability to endure prolonged anaesthesia. Pigs also display a high level of similarity in the morphology of the arterial system compared to humans, and the coagulation process is comparable between sheep and humans. Finally, primates have a high level of similarity with humans, especially the fibrinolytic system. However, costs and ethical concerns related to the use of these large animal species restrict their utilisation in aneurysm studies.
Accordingly, the mouse is a desirable species to model AAA due to their small size, relatively low costs and their capacity to overexpress or delete specific genes (87). There are several classes of experimental AAA which can be divided in three categories: genetically predisposed animal models, chemical and physical models (33). The most commonly used approach is chemical induction and includes methods such as the localised perfusion of elastase, application of calcium chloride to the adventitia of the aorta, or systemic infusion of angiotensin-II (88). Most published studies utilised the angiotensin II (Ang II)-infusion mouse model which involves the subcutaneous implantation of osmotic mini-pumps in male and/or female apolipoprotein E-deficient (Apoe-/-) mice to deliver Ang II (500–1000 ng/kg/min). This induces suprarenal aneurysm formation, approximately 3–15 days after implantation with a reported mortality rate of 15%–30% attributed to aortic rupture (89, 90). Although rarely reported in mouse AAA studies, early atherosclerotic plaque formation can be induced in the Ang II-infusion mouse model when combined with high-fat feeding. The aneurysms which from within the suprarenal abdominal aortae of Ang II-infused Apoe-/-mice are characterised by medial degeneration, inflammation, and intra-mural thrombosis (89). Furthermore, although Ang II is a hypertension-inducing molecule, blood pressure assessment in Apoe-/-mice revealed that aneurysm formation in this model was independent of blood pressure changes (88). However, it should be noted that ultra-high-resolution imaging studies have suggested that Ang II-infusion induces rupture of the aortic medial layer which results in formation of an intra-mural haematoma/thrombus and subsequent partial dissection of the adventitia within the supra-renal region of the abdominal aorta (91). Accordingly, it has been proposed that the Ang II-infusion Apoe-/- mouse may model aortic dissection rather than fusiform AAA-related rupture, with mice displaying intra-mural rather than intra-luminal thrombus, which is prevalent in most human AAAs (92); these disparities are highlighted in Figure 2.
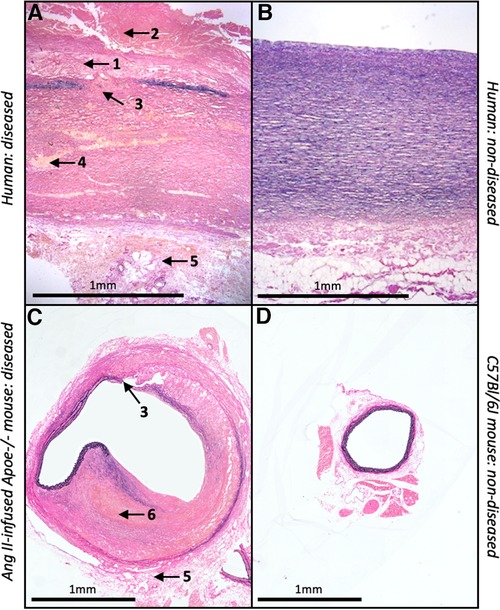
Figure 2. Similarities and differences between human and mouse AAAs. Histological images of EVG-stained abdominal aortae from (A) diseased and (B) non-diseased humans alongside (C) Ang II-infused Apoe-/- mouse and (D) C57Bl/6J wild-type mouse. Numbers and associated arrows indicate the following histological characteristics; 1 = atherosclerosis; 2 = intra-luminal thrombus; 3 = medial degeneration; 4 = VSMC loss; 5 = adventitial remodelling; 6 = intra-mural thrombus.
Adventitial application of calcium chloride solution is another method adopted to induce aortic aneurysm formation. This method was first applied to rabbit aortas, and subsequently mice, involving the application of 0.25–1 molar calcium chloride to the adventitia of the infra-renal abdominal aorta, using either a soaked swab or directly in solution (93). Results illustrated an increase in the aorta diameter by 64% after two weeks of application and by 110% after three weeks, alongside a marked inflammatory response (94). Further features of this model that correlate with human AAAs include medial calcification, elastin loss, VSMC apoptosis, and increased proteolytic activity (33). The elastase infusion model is further model deployed in AAA studies. This method requires the introduction of a catheter into the infra-renal aorta and isolation of a segment of the abdominal aorta by distal suture before perfusion with elastase, commonly porcine pancreatic elastase (34). This leads to medial destruction, dilatation of the aorta and consequently, aneurysm formation within 2–4 weeks after the procedure in rats and mice (86).
Matrix metalloproteinases (MMPs), also known as matrixins, are a family of proteolytic enzymes capable of degrading many components of the extracellular matrix (ECM) (2). MMPs are involved in a multitude of physiological processes such as morphogenesis, tissue repair, and remodelling. However, they also play a crucial role in pathological events including many cardiovascular diseases such as atherosclerosis, post MI remodelling, and aneurysm formation, progression, and rupture (26). In addition to degradation of ECM components, MMPs have the ability to target and process non-ECM molecules directly affecting cell behaviour (95). Consequently, MMP activity has been linked to migration/invasion, proliferation, and apoptosis of several component cells of the blood vessels, VSMCs, endothelial cells, and monocyte/macrophages (96). The MMP family consists of 23 members which share structural similarities and are included in the Metzincin family together with ADAMs (a disintegrin and metalloproteinase family) and ADAMTs (ADAM with thrombospondin motifs) proteases (97). The structure and classification of MMPs are summarised in Figure 3. MMPs are produced as latent enzymes (pro-forms) and therefore they require activation through pro-domain cleavage. Stepwise activation of secreted MMPs require the destabilisation, followed by full cleavage of the pro-domain by either plasma, bacterial protease and/or, in some cases, by other active MMPs (97). Membrane type-1 and type-2 MMPs (MT-MMPs) are fully activated intracellularly by furin or other pro-protein convertases before being expressed on the cell membrane as active enzymes. Similarly, MMP-11 and -28 are intracellularly activated by furin and secreted as active enzymes into the extracellular space.
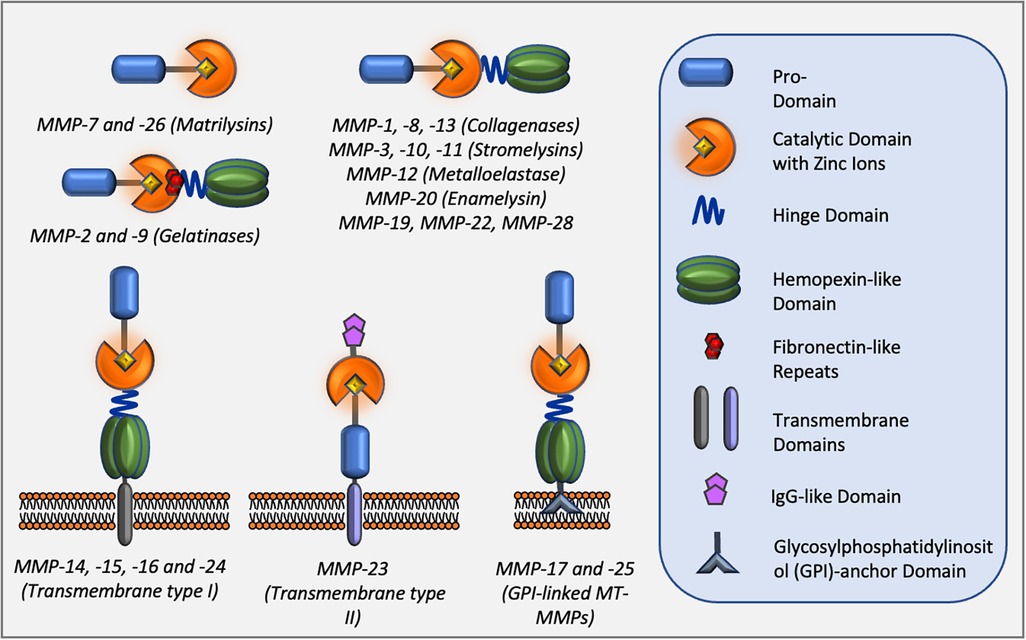
Figure 3. Classes of MMPs and their domain structure.
Due to the potential damaging actions of uncontrolled MMP expression, their activity is tightly regulated by multiple endogenous protease inhibitors; these include α2-macroglobulin, the reversion-inducing cysteine-rich protein with Kazal motifs (RECK), tissue factor pathway inhibitor-2 (TFPI-2), and the pro-collagen C-terminal proteinase enhancer (PCPE) (97). However, tissue inhibitor of MMPs (TIMPs) are the most potent endogenous inhibitors of MMPs, playing a major role in regulating MMP activity during physiological and pathobiological processes (98). The balance between MMPs and TIMPs is crucial for homeostasis and dysregulation can result in pathological dysfunctions associated with an aberrant turnover of the extracellular matrix and/or alterations in cell behaviour. Four different TIMPs have been identified in vertebrates (TIMP-1, -2, -3, and -4); they are secreted proteins able to form tight complexes with MMP catalytic domains due to inhibitory residues existing within their N-terminal domain. Each distinct TIMP exhibits diverse inhibitory efficacy against different members of the MMP family (i.e., TIMP-1 has a poor inhibitory effect on MMP-9, -14, -15, -16, and -24) (96). Moreover, TIMPs also retain the ability to inhibit members of both the ADAM and ADAMTS family of proteinases (96) which, however, will not be discussed in the present review. Finally, TIMP-3 is distinct to the other TIMPs as it displays high-affinity for extracellular matrix proteins through interactions with its C-terminal domain, facilitating TIMP-3 accumulation within the matrix and subsequently extending its half-life (98).
MMPs share structural homology and are classified into six classes based upon their structure. All members contain a pro-domain (blue) which requires removal during activation. A linker hinge domain separates the catalytic domain (orange) from the hemopexin-like domain (green) for members of the collagenase and stromelysin families. Three fibronectin-like repeats in the catalytic domain (red) are present within members of the gelatinase sub-family. Membrane-type MMPs also contain a transmembrane domain and a cytoplasmic tail which is linked to the cell membrane. Two members of membrane-type MMPs are glycosylphosphatidylinositol (GPI)-anchored MMPs.
MMPs have been proposed to play a significant role during inflammatory responses, as they can promote accumulation of inflammatory cells within areas of injury/damage through enabling their invasion, proliferation, and susceptibility to apoptosis. For instance, MMP-12 contributes to the invasion of monocyte/macrophages and their subsequent accrual at sites of inflammation including atherosclerotic plaques, attributed to the potent elastinolytic activity of MMP-12 (99, 100, 101). Similarly, MMP-14 has been shown to promote monocyte invasion through a synthetic matrix, and monocyte/macrophage accumulation at sites of sterile inflammation (mouse model of granuloma formation), as well as within atherosclerotic lesions (102, 103). Additionally, it has been indicated that TIMP-2 and TIMP-3, but not TIMP-1, can inhibit macrophage apoptosis by inhibiting MMP-14 and MMP-12 dependent cleavage of N-Cadherin, respectively (101, 103). Moreover, MMPs can modulate the bioavailability of inflammatory mediators either by modifying chemokines, cytokines, and growth factors, or by shedding membrane receptors (104). In particular, the MT-MMPs (such as MMP-14) are considered effective sheddases due to their pericellular location, and are afforded the capacity to control cell behaviour through modifying membrane protein localisation, including the cleavage of growth-factor and cytokine receptors, integrins, and proteins that regulate cell–cell contacts (105). These effects can be mediated by direct targeting of inflammatory molecules as well as indirect control of their activity through the modulation of other substrates, such as ECM proteins or co-receptors which are able to bind and maintain pro-inflammatory mediators in specific locations during inflammatory responses (106).
In addition to a prominent role in inflammation, MMP activity is also required for vascular remodelling, where degradation of ECM components is fundamental alongside substantial evidence indicating MMPs as modulators of VSMC migration, proliferation, and apoptosis. Heightened MMP-2 and MMP-9 expression/activity promotes VSMC migration and subsequent neointima formation in an in vivo model of carotid ligation-induced vascular injury (107, 108, 109), with a similar role proposed for MMP-3, ascribed to its regulation of MMP-9 activation (110). Regarding VSMC proliferation, conflicting results indicate a more complicated role of MMPs. Several MMPs are able to disrupt VSMC cell-cell contact through shedding of N-cadherin and subsequent activation of intracellular β-catenin signalling, successively promoting a pro-proliferative VSMC phenotype, particularly MMP-9 and MMP-12 (111). However, although Mmp9 deficiency resulted in retarded VSMC proliferation in response to arterial injury, but not in carotid ligation experiments (108, 109). Similarly, several studies utilising pharmacological broad-spectrum MMP inhibitors revealed ambiguous findings on VSMC proliferation (26). Whereas adenoviral-based gene transfer studies have shown that over-expression of individual TIMPs can abrogate adverse vascular remodelling and neointima formation, but with limited effects on VSMC proliferation (112, 113, 114, 115). MMP-9 has also been demonstrated to facilitate reorganisation of the ECM alongside its degradation, and therefore participated in adverse arterial remodelling in response to haemodynamic changes (116), with clear connotations for vessel expansion during AAA development. MMP activity can also mediate mobilisation of growth factors, such as fibroblast growth factor (FGF)-1 and FGF-2, potentially promoting VSMC proliferation (106). Together with migration and proliferation, VSMC apoptosis also contributes to vascular remodelling, and alongside MMP-12 and MMP-14, MMP-7 proteolytic activity has been linked to cleavage of N-cadherin in VSMCs promoting their apoptosis (117). Finally, dysregulated MMP activity can also modulate apoptosis through the processing death ligands (i.e., TNFα and Fas ligand) and/or their receptors, prompting apoptosis in an autocrine as well as paracrine manner (42).
Histological studies assessing the expression and activity of MMP family members and TIMPs in AAA tissues has demonstrated their spatial and temporal profile, including cell-type expression (Table 1). Most of the MMPs evaluated within AAAs are highly expressed by inflammatory cell infiltrates alongside VSMCs and endothelial cells. Inflammatory cells—which comprise of neutrophils, monocytes/macrophages, mast cells, B- and T-cells—may serve as a prominent source of MMPs in AAA, as their numbers increase during aneurysm progression through amplified cell recruitment and proliferation (135). The collagenase family members, MMP-1, -8, and -13 have been reported to be increased in macrophages and VSMCs within AAA tissues in comparison to healthy aortic tissue (136, 137), with MMP-2, -3, -9, -12, and -14 also displaying elevated expression (26). Additionally, genetic studies examining relationships between gene polymorphisms and AAA provide support for MMP-3 as an important contributory factor (138, 139). Spatially, MMP-2 expression and activity are co-localised primarily with VSMCs in human AAAs (140). Conversely, MMP-9 is predominantly expressed by macrophages, and its activity as assessed by zymography, is increased in ruptured aneurysms when matched to similar sized intact AAAs (140, 141). MMP-12 (also known as macrophage metalloelastase), is expressed by infiltrating-macrophages within the atherosclerotic portions and medial aspect of AAAs, which co-localise with areas of elastin fragmentation and loss (142). Furthermore, proteomics analysis of human AAAs confirmed increased accumulation of MMP-12 along with degradation of collagen XII, fibronectin, periostin, tenascin, and thrombospondin 2, and suggested MMP-12 as the dominant protease within human AAAs (143). Accordingly, MMP-12 is considered to have a direct role in the pathogenesis of the AAAs through its ability to preferentially degrade elastin, and facilitate macrophage invasion, two key characteristics of ruptured human AAAs. MMP-14, one of the most predominant membrane-bound MMPs in the vasculature, has also been detected in VSMCs within human AAAs (144). Lastly, transcriptomic analysis of monocyte-derived macrophages isolated from AAA and peripheral arterial occlusion patients revealed heightened MMP-27 levels in AAA individuals (145).
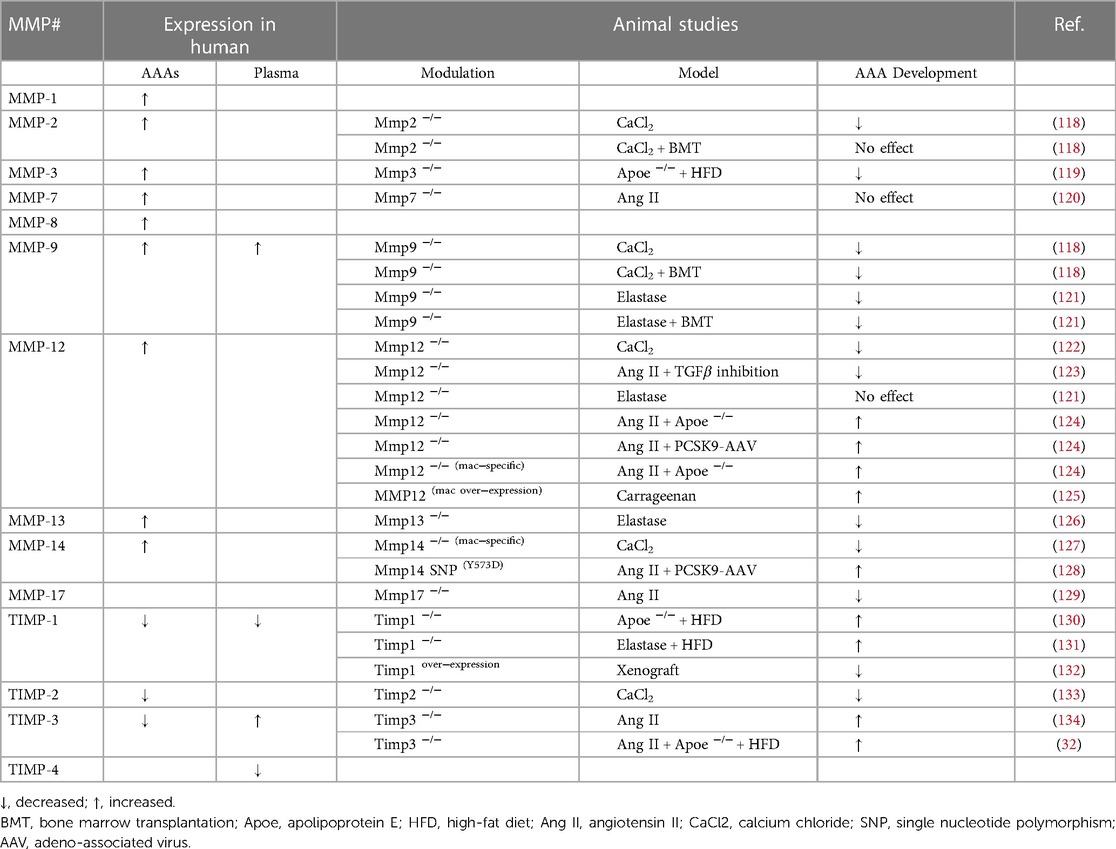
Table 1. MMP expression in human AAA tissues and plasma, alongside results of in vivo animal studies evaluating the effects of genetic deficiency for select matrix metalloproteinases (MMP) or tissue inhibitors of metalloproteinases (TIMPs) on AAA formation.
At the mRNA level, TIMP-1 (146) and TIMP-3 (32) were unchanged between AAA and non-aneurysmal tissues, although TIMP-2 levels were modestly increased within AAAs (146). When comparing human AAA samples to healthy aortae, although protein expression of TIMP-2 was decreased while TIMP-1 and TIMP-3 were increased, heightened activity of MMP-1, MMP-9, MMP-12, and MMP-14 were observed (147), and therefore credited with the excess degradation of collagen and elastin, and driving aneurysmal dilatation. Further supporting a protective role for TIMP-2, a promoter polymorphism (-418 G/C; rs8179090) that diminishes TIMP2 gene expression rate, has been identified as a risk factor of AAA (148). Although TIMP-3 expression is elevated in the plasma from AAA patients, it is considered a potential positive feed-back mechanism to negate increased MMP activity (145, 149, 150). Interestingly, macrophage TIMP-3 expression was decreased in human AAAs, independent of changes in mRNA levels, and associated with increased MMP activity (32). Although reduced plasma and tissue levels of TIMP-4 were reported in patients with ascending aortic aneurysmal patients compared to controls (151), expression in AAAs has not been examined.
In line with elevated tissue levels of select MMPs, several studies have ascertained MMP plasma levels in AAA patients alongside healthy controls or patients with aortic obstructive diseases, to determine if they may serve as biomarkers of AAA presence and progression (Table 1). Specifically, elevated MMP-9 plasma levels were detected in AAA patients compared to controls and, additionally, a reduction of plasma MMP-9 was observed after surgical repair of AAA (152, 153, 154). Moreover, MMP-9 plasma levels have also been associated with evidence of aortic medial remodelling and increased luminal diameter (155). Furthermore, MMP-1 alongside MMP-9 were identified as elevated in the plasma of patients with ruptured AAAs when compared with individuals harbouring intact AAAs, with MMP-9 levels also associating with 30-day mortality (156). However, no correlation between MMP-9 plasma levels and the yearly expansion of small diameter AAAs (35–49 mm) was observed, questioning the utility of MMP-9 as a plasma biomarker for AAA progression (157).
Single-cell transcriptomics have been deployed to examine the cell phenotype expression of genes during the development and progression of human and mouse aortic aneurysms including AAAs (158), with certain studies identifying changes in the expression of select MMPs and TIMPs. Focussing upon VSMC phenotypes, single-cell sequencing data analysis combined from two independent studies demonstrated that a “fibroblast-like” VSMC phenotype (characterised through expression of ACTA2, MYH11, COL1A1, COL1A2, and PDGFRA) was increased in human aortic aneurysm samples compared to normal aorta, and displayed augmented MMP2 expression compared to contractile VSMCs (159). In mice, two standalone studies both deploying the Ang II-infusion mouse model of AAA revealed a pro-inflammatory macrophage subpopulation (Netrin1 + ve) and VSMC Mmp3 levels are increased and contribute to AAA formation (160, 161). A further study using the Ang II-infusion approach in Apoe−/− mice identified a “fibrocyte” cell cluster (Cd34+ and Col1a2+) which was more prevalent compared to control aorta, and enriched for Mmp2, Mmp3, Mmp14, and Mmp23 (162). Utilising the elastase-induced mouse AAA model and examining the infrarenal abdominal aorta, Mmp9 and Mmp4 were identified as enriched genes across multiple monocyte/macrophage subpopulations, with Mmp9 highly expressed in a specific subpopulation termed “aortic-resident” and characterised through expression of Cx3cr1 and Flt3, which decreased proportionally in elastase-treated mice (163). No differentially expressed MMPs were identified in single-cell RNA sequencing on mouse AAA tissues from the perivascular CaCl2 application mouse model (164).
Multiple proof-of-principle animal studies have been conducted to determine the contribution of specific MMP and TIMP family members to AAA formation (Table 1). A deleterious role for MMP-2 in AAA formation was proposed based upon reduced aortic diameter in mice with global Mmp2-deficiency using the CaCl2-application model (118). The adverse involvement of MMP-2 was attributed to heightened VSMC production as intravenous administration of peritoneal macrophages from wild-type mice failed to reverse the phenotype (118). Assessment of spontaneous AAA formation in long-term high fat-fed Apoe−/− mice with and without Mmp3-deficiency revealed increased presence of aneurysms in Mmp3 wild-type mice, as characterised by elastin fragmentation, aortic wall thinning, and rupture of medial elastin fibres (119). Mice harbouring deletion of Mmp-7 displayed reduced VSMC proliferation and apoptosis numbers within Ang II-induced AAAs compared to wild-type controls, however this did not translate to a difference in AAA expansion or severity (120). Two independent studies demonstrated absence of MMP-9 suppressed aneurysm development in the CaCl2-application model (118) and elastase-induced AAAs (121). In addition, both studies utilised a bone marrow transplant technique to suggest that macrophage-derived MMP-9 underlies the induction of AAA in both models (118, 121).
Multiple reports utilising an array of mouse and rabbit models have yielded conflicting findings regarding the role of MMP-12 in AAA development. Mmp12 deficiency attenuated AAA development allied with reduced macrophage recruitment in the peri-aortic CaCl2-application model (122), and a mouse model of combined Ang II-infusion and TGF-β neutralising antibody treatment (66). Further supporting the proposition that MMP-12 promotes AAA formation, medial elastin fragmentation and aortic dilation were increased in transgenic rabbits over-expressing MMP-12 in macrophages and subjected to carrageenan-induced AAA, compared to wild-type rabbits (125). However, no effect of Mmp12 knockout on AAA formation was observed in the elastase-perfusion mouse model (121). Conversely, the development of AAAs was reported to have accelerated in two different hypercholesterolaemic mouse models (Apoe-deficient mice or mice over-expressing PCSK9) (124). Additionally, it was proposed that the protective effects were through macrophage expression of MMP-12 suppressing complement activation and subsequent neutrophil infiltration, as macrophage-restricted Mmp12-deficeincy mirrored global Mmp12-deficeincy effects on AAA (124). Aortic dilation was reduced in Mmp13-deficient mice subjected to elastase infusion compared to wild-type animals, suggesting MMP-13 promotes AAA formation (126). An adverse role for MMP-14 in aneurysm progression has been suggested given bone marrow transplantation of Mmp14-deficient macrophages into wild-type mice prevented AAA development in the peri-aortic CaCl2-application model (127). Although in opposition, a transgenic mouse harbouring a point mutation in the cytoplasmic domain MMP-14 (Y573D) which abrogates its signalling function without affecting its proteolytic activity, protected from Ang II-induced AAA development (128). Finally, a beneficial effect for MMP-17 was proposed given that lack of Mmp17 resulted in increased susceptibility to ang II-induced AAA expansion, although incidence was unaffected (129).
Focussing on the endogenous MMP inhibitors, TIMPs are largely considered as protective during aneurysm formation, development, and rupture through combatting MMP proteolytic activity. Indeed, deletion of TIMP-1, in both wild type and Apoe−/− mice increased AAA development (130). Moreover, Timp1 knockout, in an elastase-induced model of AAA, resulted in increased aortic diameter and augmented loss of medial elastin (131). Hence, overexpression of TIMP-1 in a rat model of guinea-pig xenograft-induced AAA reduced elastin degradation together with aneurysm formation and rupture (132). On the contrary, the genetic deletion of Timp2 in in the peri-aortic CaCl2-application model, suppressed aortic expansion compared to wild type controls with no effect upon medial elastin fragmentation, despite a reduction in MMP-2 activity in Timp2-deficient mice (133). The role of TIMP-3 in the formation of AAA has been explored in the non-atherosclerotic and atherosclerotic Ang II-induced AAA mouse model, with both approaches demonstrating Timp3-deficiency adversely affected vascular remodelling through increased inflammation and proteolytic activity, consequently contributing to reduced collagen and elastin content (32, 134), highlighting the protective role of TIMP-3 on AAAs. No animal study addressing the role of TIMP-4 in AAA development and rupture has been conducted at present.
The accumulating human histological analysis alongside complimentary animal studies provide robust proof-of-principle evidence for dysregulated MMP expression and activity actively contributing to AAA formation and progression. Broad-spectrum targeting of aberrant MMP activity has been explored as a therapeutic strategy to limit the development of experimental AAAs in numerous rodent models (Table 2). Researchers have deployed tetracycline derivatives as non-specific MMP inhibitors alongside other pleiotropic compounds with established MMP-inhibitory capacity. Doxycycline, a broad-spectrum antibiotic, and tetracycline analogue, harbours the ability to non-specifically inhibit MMP activity and expression, as demonstrated in AAA explants and cultured VSMCs (181). In the rat elastase-perfusion AAA model, doxycycline administration was shown to suppress aortic dilation, alongside decreased incidence of AAA formation and medial elastin degradation, and inhibited MMP-9 expression/activity (165, 166, 167). Comparable beneficial effects of doxycycline treatment were detected in mice with established elastase-induced AAAs (168) or during their development (121). Doxycycline treatment induced equivalent effects within a rat aortic ligature model of AAA (176), and in rats where AAAs were stimulated through application of thioglycolate and plasmin (175). Aortic dilation was equally dampened in the mouse peri-aortic CaCl2-application AAA model, which was associated with decreased MMP activity (169, 170). Doxycycline was also shown to diminish aortic expansion and inflammation within Ang II-induced AAAs from wild-type mice (171), with reduced AAA development also observed in Apoe−/− mice but with mixed effects upon MMP expression and activity (172, 173). Lastly, AAA incidence and severity were reduced in Ldlr−/− mice infused with Ang II alongside prolonged high fat feeding, although atherosclerotic burden within the aorta was unaffected (174).
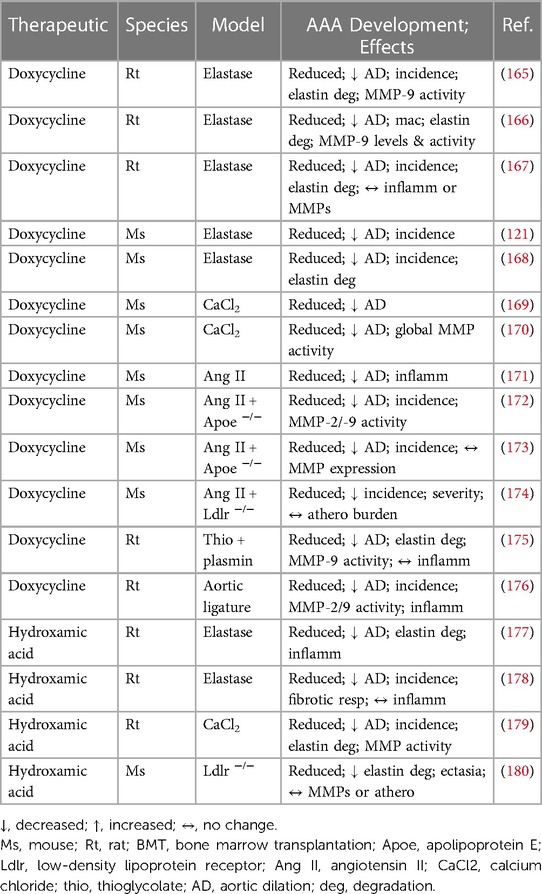
Table 2. Results of in vivo animal studies evaluating the effects of modulating non-specific MMP activity on abdominal aortic aneurysm formation and cellular composition, using broad-spectrum pharmacological inhibitors of MMPs.
Batimastat, a synthetic peptide also known as BB-94, is a hydroxamic acid-based broad-spectrum inhibitor of MMPs, and it was demonstrated that systemic administration of BB-94 reduced aortic dilatation and medial elastin fragmentation in elastase-induced AAAs in rats, and also suppressed inflammatory cell infiltration (177). Moreover, targeted delivery and inhibition achieved through delivery of elastin antibody-conjugated BB-94-loaded nanoparticles retarded AAA MMP activity, elastin degradation, calcification, and aneurysmal development in a peri-aortic CaCl2-application rat model (179). An alternative hydroxamate-based MMP antagonist, RS 132908, also suppressed aortic dilatation in a rat intraluminal elastase-perfusion AAA model, preserving medial elastin integrity and exerting a medial pro-fibrotic response, although no effect on inflammation was observed (178). Furthermore, CGS 27023A, a similar hydroxamic acid MMP inhibitor, reduced elastin degradation and medial degeneration within the atherosclerotic abdominal aortae of hypercholesterolaemic Ldlr−/− mice (180).
The positive response to doxycycline treatment in animal models of AAA resulted in the undertaking of several clinical trials being carried out globally. Doxycycline treatment for 3-months in patients with small AAAs (diameter <5.5 cm) significantly reduced AAA expansion at 6- to -12 and 12- to 18-month follow-up periods, in a double-blind, placebo-controlled, randomised clinical trial (182). Moreover, longer-term (6-month) doxycycline treatment reduced MMP-9 plasma levels as well as maximum aortic diameter in EVAR patients within a randomised, placebo-controlled study (183). Short-term doxycycline treatment (2-weeks) increased TIMP-1 and reduced MMP-8, MMP-9, MMP-3, and MMP-25 expression, resulting in a decrease of inflammatory cell accumulation within diseased aorta of AAA patients (184, 185). Nevertheless, conflicting results in other studies have raised some concerns. A phase II study indicated no changes in AAA diameter after 6-months of doxycycline treatment, despite an observed reduction in MMP-9 plasma levels (186). Additionally, in a randomised, placebo-controlled, and double-blind study, doxycycline treatment did not affect expression or activity of any of the MMPs and TIMPs assessed (187). A further randomised, placebo-controlled, double-blind study utilising a larger patient cohort, demonstrated that long-term doxycycline treatment (18-months) associated with increased AAA growth, and did not impact the requirement for AAA repair or time to surgical repair (188). Lastly, a parallel, two-group randomised clinical trial across 22 US clinical centres demonstrated doxycycline had no effect upon the growth of small infrarenal AAAs at two-years (189). Accordingly, the ambivalent outcomes have not fostered the therapeutic adoption of doxycycline as a treatment for reducing the expansion of AAAs in humans, with ambiguous outcomes with broad-spectrum MMP inhibitors suggesting the selective targeting of individual MMPs is desirable.
Although an important role for MMPs in AAAs is evident from the published literature, a major limitation of targeting these proteases clinically rests in the difficulty in designing highly selective MMP inhibitors that remain effective in vivo. MMPs govern a variety of biological processes that can exert beneficial and detrimental effects upon the pathophysiology of AAAs alongside physiological processes throughout the body, hence the ongoing attempts to pharmacologically target select MMPs using small organic inhibitors. However, due to the high structural homology of MMP family members, early-stage MMP inhibition has commonly failed as a therapeutic strategy in clinical trials. Such failures have been attributed to the poor specificity of the compounds, in part due and their broad inhibitory profile, resulting in effects on multiple MMPs which exert divergent properties on differing matrix proteins and disparate cell types. Additionally, shared substrates and cross-reactivity between MMP family members (alongside ADAMs and ADAMTSs) remains a continual obstacle, that requires novel strategies which can distinguish between the activities of separate MMPs. Next generation MMP inhibitors must be highly selective, specific, and preferably inhibit a single MMP function, and would benefit from cell-type directed delivery advances to negate off-target effects.
In summary, there is considerable evidence supporting a direct role for dysregulated MMP expression and subsequent activity to the formation and progression of AAAs. Evidence demonstrates that destruction of elastin and collagen can be limited or reversed with MMP inhibition, while animal studies have confirmed the anomalous role and function of individual MMPs during AAA development. Accordingly, modifications to restore or over-express TIMP family members have proven to effectively suppress MMP activity and prevent the development and progression of AAAs in animal models. However, the inability of broad-spectrum MMP inhibition to successfully provide clinical translation has led to renewed interest in the elaboration of MMP inhibitors that exhibit narrow specificity, as has been successfully demonstrated in atherosclerosis studies utilising inhibitors restricted to the targeting of MMP-12 (101) or MMP-13 (190). Indeed, the direct association of heightened macrophage infiltration and elastin degradation to the pathogenesis of AAAs, in marriage with the central role of MMP-12 to both processes suggests that perturbing the resulting medial wall inflammation and destruction of elastin could be achieved through inhibition of MMP-12 activity. Therefore, future studies focussing on the development of MMP-12 specific inhibitors as a potential therapeutic for AAA would appear desirable. However, evaluating the efficacy of MMP inhibition while also identifying patients who will benefit most from associated therapeutics would also be advantageous. Accordingly, there is a keen interest to develop imaging approaches to identify MMP activity within AAAs. Two recent studies demonstrating how MMP inhibitors or substrates can be engineered to permit visualisation of MMP activity within AAAs (191, 192) may therefore facilitate prognosis and potential treatment stratification. So, notwithstanding the limitations associated with translating findings from animal models to human AAA, the reviewed studies should encourage renewed motivation for future clinical trials of selective MMP inhibitors for the treatment of existing AAAs. Furthermore, new insight into the spatial and temporal expression and activity of MMPs and TIMPs, alongside cell-type expression patterns, may aid the design of future therapeutics.
All authors contributed to the construction, writing, and editing of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hellenthal F, Buurman WA, Wodzig W, Schurink GWH. Biomarkers of AAA progression. Part 1: extracellular matrix degeneration. Nat Rev Cardiol. (2009) 6(7):464–74. doi: 10.1038/nrcardio.2009.80
2. Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. (2006) 69(3):562–73. doi: 10.1016/j.cardiores.2005.12.002
3. Norman PE, Powell JT. Site specificity of aneurysmal disease. Circulation. (2010) 121(4):560–8. doi: 10.1161/CIRCULATIONAHA.109.880724
4. Isselbacher EM. Trends in thoracic aortic aneurysms and dissection: out of the shadows and into the light. Circulation. (2014) 130(25):2267–8. doi: 10.1161/CIRCULATIONAHA.114.013603
5. Kuivaniemi H, Platsoucas CD, Tilson MD III. Aortic aneurysms: an immune disease with a strong genetic component. Circulation. (2008) 117(2):242–52. doi: 10.1161/CIRCULATIONAHA.107.690982
6. Wanhainen A, Verzini F, Van Herzeele I, Allaire E, Bown M, Cohnert T, et al. European society for vascular surgery (ESVS) 2019 clinical practice guidelines on the management of abdominal aorto-iliac artery aneurysms. Eur J Vasc Endovasc Surg. (2019) 57(1):8–93. doi: 10.1016/j.ejvs.2018.09.020
7. Lederle FA, Johnson GR, Wilson SE. Abdominal aortic aneurysm in women. J Vasc Surg. (2001) 34(1):122–6. doi: 10.1067/mva.2001.115275
8. Kent KC. Abdominal aortic aneurysms. N Engl J Med. (2014) 371(22):2101–8. doi: 10.1056/NEJMcp1401430
9. Forsdahl Signe H, Singh K, Solberg S, Jacobsen Bjarne K. Risk factors for abdominal aortic aneurysms. Circulation. (2009) 119(16):2202–8. doi: 10.1161/CIRCULATIONAHA.108.817619
10. Sakalihasan N, Defraigne JO, Kerstenne MA, Cheramy-Bien JP, Smelser DT, Tromp G, et al. Family members of patients with abdominal aortic aneurysms are at increased risk for aneurysms: analysis of 618 probands and their families from the liège AAA family study. Ann Vasc Surg. (2014) 28(4):787–97. doi: 10.1016/j.avsg.2013.11.005
11. Akai A, Watanabe Y, Hoshina K, Obitsu Y, Deguchi J, Sato O, et al. Family history of aortic aneurysm is an independent risk factor for more rapid growth of small abdominal aortic aneurysms in Japan. J Vasc Surg. (2015) 61(2):287–90. doi: 10.1016/j.jvs.2014.07.007
12. Fink HA, Lederle FA, Roth CS, Bowles CA, Nelson DB, Haas MA. The accuracy of physical examination to detect abdominal aortic aneurysm. Arch Intern Med. (2000) 160(6):833–6. doi: 10.1001/archinte.160.6.833
13. Kontopodis N, Lioudaki S, Pantidis D, Papadopoulos G, Georgakarakos E, Ioannou CV. Advances in determining abdominal aortic aneurysm size and growth. World J Radiol. (2016) 8(2):148–58. doi: 10.4329/wjr.v8.i2.148
14. Kuzmik GA, Sang AX, Elefteriades JA. Natural history of thoracic aortic aneurysms. J Vasc Surg. (2012) 56(2):565–71. doi: 10.1016/j.jvs.2012.04.053
15. Danyi P, Elefteriades J, Jovin I S. Medical therapy of thoracic aortic aneurysms are we there yet? Circulation. (2011) 124:1469–76. doi: 10.1161/CIRCULATIONAHA.110.006486
16. Miyake T, Morishita R. Pharmacological treatment of abdominal aortic aneurysm. Cardiovasc Res. (2009) 83(3):436–43. doi: 10.1093/cvr/cvp155
17. Aggarwal S, Qamar A, Sharma V, Sharma A. Abdominal aortic aneurysm: a comprehensive review. Exp Clin Cardiol. (2011) 16(1):11–5. PMID: 21523201, PMCID: PMC3076160
18. Hirsch Alan T, Haskal Ziv J, Hertzer Norman R, Bakal Curtis W, Creager Mark A, Halperin Jonathan L, et al. ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic). Circulation. (2006) 113(11):e463–654. doi: 10.1161/CIRCULATIONAHA.106.174526
19. Epstein D, Sculpher MJ, Powell JT, Thompson SG, Brown LC, Greenhalgh RM. Long-term cost-effectiveness analysis of endovascular versus open repair for abdominal aortic aneurysm based on four randomized clinical trials. Br J Surg. (2014) 101(6):623–31. doi: 10.1002/bjs.9464
20. Lee HG, Clair DG, Ouriel K. Ten-year comparison of all-cause mortality after endovascular or open repair of abdominal aortic aneurysms: a propensity score analysis. World J Surg. (2013) 37(3):680–7. doi: 10.1007/s00268-012-1863-y
21. Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL, et al. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg. (2009) 138(6):1392–9. doi: 10.1016/j.jtcvs.2009.07.075
22. Tsamis A, Krawiec JT, Vorp DA. Elastin and collagen fibre microstructure of the human aorta in ageing and disease: a review. J R Soc Interface. (2013) 10(83):20121004. doi: 10.1098/rsif.2012.1004
23. Shoulders MD, Raines RT. Collagen structure and stability. Annu Rev Biochem. (2009) 78:929–58. doi: 10.1146/annurev.biochem.77.032207.120833
24. Jana S, Hu M, Shen M, Kassiri Z. Extracellular matrix, regional heterogeneity of the aorta, and aortic aneurysm. Exp Mol Med. (2019) 51(12):1–15. doi: 10.1038/s12276-019-0286-3
25. Abdul-Hussien H, Soekhoe RGV, Weber E, von der Thusen JH, Kleemann R, Mulder A, et al. Collagen degradation in the abdominal aneurysm—a conspiracy of matrix metalloproteinase and cysteine collagenases. Am J Pathol. (2007) 170(3):809–17. doi: 10.2353/ajpath.2007.060522
26. Johnson JL. Matrix metalloproteinases and their inhibitors in cardiovascular pathologies: current knowledge and clinical potential. Metalloproteinases Med. (2014) 1:21–36. doi: 10.2147/MNM.S50999
27. Golledge J, Tsao PS, Dalman RL, Norman PE. Circulating markers of abdominal aortic aneurysm presence and progression. Circulation. (2008) 118(23):2382–92. doi: 10.1161/CIRCULATIONAHA.108.802074
28. Wagenseil JE, Mecham RP. Elastin in large artery stiffness and hypertension. J Cardiovasc Transl Res. (2012) 5(3):264–73. doi: 10.1007/s12265-012-9349-8
29. Lindholt JS, Ashton HA, Heickendorff L, Scott RAP. Serum elastin peptides in the preoperative evaluation of abdominal aortic aneurysms. Eur J Vasc Endovasc Surg. (2001) 22(6):546–50. doi: 10.1053/ejvs.2001.1516
30. Heinz A. Elastases and elastokines: elastin degradation and its significance in health and disease. Crit Rev Biochem Mol Biol. (2020) 55(3):252–73. doi: 10.1080/10409238.2020.1768208
31. Pini L, Peroni M, Zanotti C, Pini A, Bossoni E, Giordani J, et al. Investigating the link between alpha-1 antitrypsin deficiency and abdominal aortic aneurysms. Ann Vasc Surg. (2021) 77:195–201. doi: 10.1016/j.avsg.2021.05.064
32. Di Gregoli K, Mohamad Anuar NN, Bianco R, White SJ, Newby AC, George SJ, et al. MicroRNA-181b controls atherosclerosis and aneurysms through regulation of TIMP-3 and elastin. Circ Res. (2017) 120(1):49–65. doi: 10.1161/CIRCRESAHA.116.309321
33. Sénémaud J, Caligiuri G, Etienne H, Delbosc S, Michel J-B, Coscas R. Translational relevance and recent advances of animal models of abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. (2017) 37(3):401–10. doi: 10.1161/ATVBAHA.116.308534
34. Anidjar S, Salzmann JL, Gentric D, Lagneau P, Camilleri JP, Michel JB. Elastase-induced experimental aneurysms in rats. Circulation. (1990) 82(3):973–81. doi: 10.1161/01.CIR.82.3.973
35. Carmo M, Colombo L, Bruno A, Corsi FR, Roncoroni L, Cuttin MS, et al. Alteration of elastin, collagen and their cross-links in abdominal aortic aneurysms. Eur J Vasc Endovasc Surg. (2002) 23(6):543–9. doi: 10.1053/ejvs.2002.1620
36. Milewicz DM, Guo DC, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, et al. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. (2008) 9:283–302. doi: 10.1146/annurev.genom.8.080706.092303
37. Beamish JA, He P, Kottke-Marchant K, Marchant RE. Molecular regulation of contractile smooth muscle cell phenotype: implications for vascular tissue engineering. Tissue Eng Part B Rev. (2010) 16(5):467–91. doi: 10.1089/ten.teb.2009.0630
38. Patel PD, Arora RR. Pathophysiology, diagnosis, and management of aortic dissection. Ther Adv Cardiovasc Dis. (2008) 2(6):439–68. doi: 10.1177/1753944708090830
39. López-Candales A, Holmes DR, Liao S, Scott MJ, Wickline SA, Thompson RW. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am J Pathol. (1997) 150(3):993–1007. PMID: 9060837, PMCID: PMC1857880
40. Rowe VL, Stevens SL, Reddick TT, Freeman MB, Donnell R, Carroll RC, et al. Vascular smooth muscle cell apoptosis in aneurysmal, occlusive, and normal human aortas. J Vasc Surg. (2000) 31(3):567–76. doi: 10.1067/mva.2000.102847
41. Hsieh C-C, Yen M-H, Yen C-H, Lau Y-T. Oxidized low density lipoprotein induces apoptosis via generation of reactive oxygen species in vascular smooth muscle cells. Cardiovasc Res. (2001) 49(1):135–45. doi: 10.1016/S0008-6363(00)00218-2
42. Johnson JL. Emerging regulators of vascular smooth muscle cell function in the development and progression of atherosclerosis. Cardiovasc Res. (2014) 103(4):452–60. doi: 10.1093/cvr/cvu171
43. Jia LX, Zhang WM, Li TT, Liu Y, Piao CM, Ma YC, et al. ER stress dependent microparticles derived from smooth muscle cells promote endothelial dysfunction during thoracic aortic aneurysm and dissection. Clin Sci (Lond). (2017) 131(12):1287–99. doi: 10.1042/CS20170252
44. Yamanouchi D, Morgan S, Kato K, Lengfeld J, Zhang F, Liu B. Effects of caspase inhibitor on angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (2010) 30(4):702–7. doi: 10.1161/ATVBAHA.109.200527
45. Leeper NJ, Raiesdana A, Kojima Y, Kundu RK, Cheng H, Maegdefessel L, et al. Loss of CDKN2B promotes p53-dependent smooth muscle cell apoptosis and aneurysm formation. Arterioscler Thromb Vasc Biol. (2013) 33(1):e1–e10. doi: 10.1161/ATVBAHA.112.300399
46. Jayalath RW, Mangan SH, Golledge J. Aortic calcification. Eur J Vasc Endovasc Surg. (2005) 30(5):476–88. doi: 10.1016/j.ejvs.2005.04.030
47. Chowdhury MM, Zieliński LP, Sun JJ, Lambracos S, Boyle JR, Harrison SC, et al. Calcification of thoracic and abdominal aneurysms is associated with mortality and morbidity. Eur J Vasc Endovasc Surg. (2018) 55(1):101–8. doi: 10.1016/j.ejvs.2017.11.007
48. McCarty MF, DiNicolantonio JJ. The molecular biology and pathophysiology of vascular calcification. J Postgrad Med. (2014) 126(2):54–64. doi: 10.3810/pgm.2014.03.2740
49. Ladich E, Yahagi K, Romero ME, Virmani R. Vascular diseases: aortitis, aortic aneurysms, and vascular calcification. Cardiovasc Pathol. (2016) 25(5):432–41. doi: 10.1016/j.carpath.2016.07.002
50. Buijs RV, Willems TP, Tio RA, Boersma HH, Tielliu IF, Slart RH, et al. Calcification as a risk factor for rupture of abdominal aortic aneurysm. Eur J Vasc Endovasc Surg. (2013) 46(5):542–8. doi: 10.1016/j.ejvs.2013.09.006
51. Matsumoto K, Satoh K, Maniwa T, Tanaka T, Okunishi H, Oda T. Proteomic comparison between abdominal and thoracic aortic aneurysms. Int J Mol Med. (2014) 33(4):1035–47. doi: 10.3892/ijmm.2014.1627
52. Orriols M, Varona S, Martí-Pàmies I, Galán M, Guadall A, Escudero JR, et al. Down-regulation of fibulin-5 is associated with aortic dilation: role of inflammation and epigenetics. Cardiovasc Res. (2016) 110(3):431–42. doi: 10.1093/cvr/cvw082
53. Thompson MM, Jones L, Nasim A, Sayers RD, Bell PR. Angiogenesis in abdominal aortic aneurysms. Eur J Vasc Endovasc Surg. (1996) 11(4):464–9. doi: 10.1016/S1078-5884(96)80183-3
54. Kubis N, Levy BI. Vasculogenesis and angiogenesis: molecular and cellular controls. Part 2: interactions between cell and extracellular environment. Interv Neuroradiol. (2003) 9(3):239–48. doi: 10.1177/159101990300900302
55. Choke E, Cockerill GW, Dawson J, Wilson RW, Jones A, Loftus IM, et al. Increased angiogenesis at the site of abdominal aortic aneurysm rupture. Ann NY Acad Sci. (2006) 1085(1):315–9. doi: 10.1196/annals.1383.007
56. Defawe OD, Colige A, Lambert CA, Delvenne P, Lapière CM, Limet R, et al. Gradient of proteolytic enzymes, their inhibitors and matrix proteins expression in a ruptured abdominal aortic aneurysm. Eur J Clin Invest. (2004) 34(7):513–4. doi: 10.1111/j.1365-2362.2004.01371.x
57. Sano M, Sasaki T, Hirakawa S, Sakabe J, Ogawa M, Baba S, et al. Lymphangiogenesis and angiogenesis in abdominal aortic aneurysm. PLoS One. (2014) 9(3):e89830. doi: 10.1371/journal.pone.0089830
58. Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. (2011) 8(2):92–102. doi: 10.1038/nrcardio.2010.180
59. Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. (2006) 26(5):987–94. doi: 10.1161/01.ATV.0000214999.12921.4f
60. Middleton RK, Lloyd GM, Bown MJ, Cooper NJ, London NJ, Sayers RD. The pro-inflammatory and chemotactic cytokine microenvironment of the abdominal aortic aneurysm wall: a protein array study. J Vasc Surg. (2007) 45(3):574–80. doi: 10.1016/j.jvs.2006.11.020
61. Hellenthal F, Buurman WA, Wodzig W, Schurink GWH. Biomarkers of abdominal aortic aneurysm progression. Part 2: inflammation. Nat Rev Cardiol. (2009) 6(8):543–52. doi: 10.1038/nrcardio.2009.102
62. Xiong W, MacTaggart J, Knispel R, Worth J, Persidsky Y, Baxter BT. Blocking TNF-alpha attenuates aneurysm formation in a murine model. J Immunol. (2009) 183(4):2741–6. doi: 10.4049/jimmunol.0803164
63. Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. (2009) 78(6):539–52. doi: 10.1016/j.bcp.2009.04.029
64. King VL, Lin AY, Kristo F, Anderson TJT, Ahluwalia N, Hardy GJ, et al. Interferon-γ and the interferon-inducible chemokine CXCL10 protect against aneurysm formation and rupture. Circulation. (2009) 119(3):426–35. doi: 10.1161/CIRCULATIONAHA.108.785949
65. Shi G-P, Sukhova GK, Grubb A, Ducharme A, Rhode LH, Lee RT, et al. Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J Clin Invest. (1999) 104(9):1191–7. doi: 10.1172/JCI7709
66. Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, et al. TGF-B activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest. (2010) 120(2):422–32. doi: 10.1172/JCI38136
67. Matt P, Schoenhoff F, Habashi J, Holm T, Van Erp C, Loch D, et al. Circulating transforming growth factor-beta in marfan syndrome. Circulation. (2009) 120(6):526–32. doi: 10.1161/CIRCULATIONAHA.108.841981
68. Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of marfan syndrome. Science. (2006) 312(5770):117–21. doi: 10.1126/science.1124287
69. Pan JH, Lindholt JS, Sukhova GK, Baugh JA, Henneberg EW, Bucala R, et al. Macrophage migration inhibitory factor is associated with aneurysmal expansion. J Vasc Surg. (2003) 37(3):628–35. doi: 10.1067/mva.2003.74
70. Di Gregoli K, Johnson JL. Role of colony-stimulating factors in atherosclerosis. Curr Opin Lipidol. (2012) 23(5):412–21. doi: 10.1097/MOL.0b013e328357ca6e
71. Haghighat A, Weiss D, Whalin MK, Cowan DP, Taylor WR. Granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor exacerbate atherosclerosis in apolipoprotein E-deficient mice. Circulation. (2007) 115(15):2049–54. doi: 10.1161/CIRCULATIONAHA.106.665570
72. Ye P, Chen W, Wu J, Huang X, Li J, Wang S, et al. GM-CSF contributes to aortic aneurysms resulting from SMAD3 deficiency. J Clin Invest. (2013) 123(5):2317–31. doi: 10.1172/JCI67356
73. Vainas T, Lubbers T, Stassen Frank RM, Herngreen Selma B, van Dieijen-Visser Marja P, Bruggeman Cathrien A, et al. Serum C-reactive protein level is associated with abdominal aortic aneurysm size and may be produced by aneurysmal tissue. Circulation. (2003) 107(8):1103–5. doi: 10.1161/01.CIR.0000059938.95404.92
74. Badger SA, Soong CV, O'Donnell ME, Mercer C, Young IS, Hughes AE. C-reactive protein (CRP) elevation in patients with abdominal aortic aneurysm is independent of the most important CRP genetic polymorphism. J Vasc Surg. (2009) 49(1):178–84. doi: 10.1016/j.jvs.2008.07.081
75. Insull W Jr. The pathology of atherosclerosis: plaque development and plaque responses to medical treatment. Am J Med. (2009) 122(1 Suppl):S3–S14. doi: 10.1016/j.amjmed.2008.10.013
76. Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet. (2005) 365(9470):1577–89. doi: 10.1016/S0140-6736(05)66459-8
77. Lee AJ, Fowkes FG, Carson MN, Leng GC, Allan PL. Smoking, atherosclerosis and risk of abdominal aortic aneurysm. Eur Heart J. (1997) 18(4):671–6. doi: 10.1093/oxfordjournals.eurheartj.a015314
78. Reed D, Reed C, Stemmermann G, Hayashi T. Are aortic aneurysms caused by atherosclerosis? Circulation. (1992) 85(1):205–11. doi: 10.1161/01.CIR.85.1.205
79. Michel J-B, Martin-Ventura J-L, Egido J, Sakalihasan N, Treska V, Lindholt J, et al. Novel aspects of the pathogenesis of aneurysms of the abdominal aorta in humans. Cardiovasc Res. (2011) 90(1):18–27. doi: 10.1093/cvr/cvq337
80. Golledge J, Norman PE. Atherosclerosis and abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. (2010) 30(6):1075–7. doi: 10.1161/ATVBAHA.110.206573
81. Gawinecka J, Schönrath F, von Eckardstein A. Acute aortic dissection: pathogenesis, risk factors and diagnosis. Swiss Med Wkly. (2017) 147:w14489. doi: 10.4414/smw.2017.14489
82. Tong J, Cheng Y, Holzapfel GA. Mechanical assessment of arterial dissection in health and disease: advancements and challenges. J Biomech. (2016) 49(12):2366–73. doi: 10.1016/j.jbiomech.2016.02.009
83. Juang D, Braverman AC, Eagle K. Aortic dissection. Circulation. (2008) 118(14):e507–e10. doi: 10.1161/CIRCULATIONAHA.108.799908
84. Simsek C, Onuma Y, Magro M, de Boer S, Battes L, van Domburg RT, et al. Four-year clinical outcome of sirolimus- and paclitaxel-eluting stents compared to bare-metal stents for the percutaneous treatment of stable coronary artery disease. Catheter Cardiovasc Interv. (2010) 76(1):41–9. doi: 10.1002/ccd.22533
85. Vianello E, Dozio E, Rigolini R, Marrocco-Trischitta MM, Tacchini L, Trimarchi S, et al. Acute phase of aortic dissection: a pilot study on CD40l, MPO, and MMP-1, -2, 9 and TIMP-1 circulating levels in elderly patients. Immun Ageing. (2016) 13:9. doi: 10.1186/s12979-016-0063-2
86. Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. (2004) 24(3):429–34. doi: 10.1161/01.ATV.0000118013.72016.ea
87. Trollope A, Moxon JV, Moran CS, Golledge J. Animal models of abdominal aortic aneurysm and their role in furthering management of human disease. Cardiovasc Pathol. (2011) 20(2):114–23. doi: 10.1016/j.carpath.2010.01.001
88. Tsui JC. Experimental models of abdominal aortic aneurysms. Open Cardiovasc Med J. (2010) 4:221–30. doi: 10.2174/1874192401004010221
89. Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E deficient mice. J Clin Invest. (2000) 105(11):1605–12. doi: 10.1172/JCI7818
90. Phillips EH, Yrineo AA, Schroeder HD, Wilson KE, Cheng JX, Goergen CJ. Morphological and biomechanical differences in the elastase and AngII apoE(-/-) rodent models of abdominal aortic aneurysms. Biomed Res Int. (2015) 2015:413189. doi: 10.1155/2015/413189
91. Trachet B, Fraga-Silva RA, Piersigilli A, Tedgui A, Sordet-Dessimoz J, Astolfo A, et al. Dissecting abdominal aortic aneurysm in ang II-infused mice: suprarenal branch ruptures and apparent luminal dilatation. Cardiovasc Res. (2015) 105(2):213–22. doi: 10.1093/cvr/cvu257
92. Krishna SM, Morton SK, Li J, Golledge J. Risk factors and mouse models of abdominal aortic aneurysm rupture. Int J Mol Sci. (2020) 21(19):7250. doi: 10.3390/ijms21197250
93. Wang Y, Krishna S, Golledge J. The calcium chloride-induced rodent model of abdominal aortic aneurysm. Atherosclerosis. (2013) 226(1):29–39. doi: 10.1016/j.atherosclerosis.2012.09.010
94. Chiou AC, Chiu B, Pearce WH. Murine aortic aneurysm produced by periarterial application of calcium chloride. J Surg Res. (2001) 99(2):371–6. doi: 10.1006/jsre.2001.6207
95. Somerville RPT, Oblander SA, Apte SS. Matrix metalloproteinases: old dogs with new tricks. Genome Biol. (2003) 4(216):1–11. doi: 10.1186/gb-2003-4-6-216
96. Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol. (2013) 13(9):649–65. doi: 10.1038/nri3499
97. Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases. Circ Res. (2003) 93:827–39. doi: 10.1161/01.RES.0000070112.80711.3D
98. Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. (2002) 115:3719–27. doi: 10.1242/jcs.00063
99. Shipley JM, Wesselschmidt RL, Kobayashi DK, Ley TJ, Shapiro SD. Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci USA. (1996) 93:3942–6. doi: 10.1073/pnas.93.9.3942
100. Warner RL, Lukacs NW, Shapiro SD, Bhagarvathula N, Nerusu KC, Varani J, et al. Role of metalloelastase in a model of allergic lung responses induced by cockroach allergen. Am J Pathol. (2004) 165(6):1921–30. doi: 10.1016/S0002-9440(10)63244-0
101. Johnson JL, Devel L, Czarny B, George SJ, Jackson CL, Rogakos V, et al. A selective matrix metalloproteinase-12 inhibitor retards atherosclerotic plaque development in apolipoprotein E-knockout mice. Arterioscler Thromb Vasc Biol. (2011) 31(3):528–35. doi: 10.1161/ATVBAHA.110.219147
102. Di Gregoli K, Jenkins N, Salter R, White S, Newby AC, Johnson JL. MicroRNA-24 regulates macrophage behavior and retards atherosclerosis. Arterioscler Thromb Vasc Biol. (2014) 34(9):1990–2000. doi: 10.1161/ATVBAHA.114.304088
103. Di Gregoli K, George SJ, Jackson CL, Newby AC, Johnson JL. Differential effects of tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2 on atherosclerosis and monocyte/macrophage invasion. Cardiovasc Res. (2016) 109(2):318–30. doi: 10.1093/cvr/cvv268
104. Parks WC, Wilson CL, López-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. (2004) 4(8):617–29. doi: 10.1038/nri1418
105. Murphy G. Fell-Muir lecture: metalloproteinases: from demolition squad to master regulators. Int J Exp Path. (2010) 91(4):303–13. doi: 10.1111/j.1365-2613.2010.00736.x
106. Newby AC. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res. (2006) 69(3):614–24. doi: 10.1016/j.cardiores.2005.08.002
107. Kuzuya M, Kanda S, Sasaki T, Tamaya-Mori N, Cheng XW, Itoh T, et al. Deficiency of gelatinase a suppresses smooth muscle cell invasion and development of experimental intimal hyperplasia. Circulation. (2003) 108(11):1375–81. doi: 10.1161/01.CIR.0000086463.15540.3C
108. Cho A, Reidy MA. Matrix metalloproteinase-9 is necessary for the regulation of smooth muscle cell replication and migration after arterial injury. Circ Res. (2002) 91:845–51. doi: 10.1161/01.RES.0000040420.17366.2E
109. Galis ZS, Johnson C, Godin D, Magid R, Shipley JM, Senior RM, et al. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Circ Res. (2002) 91:852–9. doi: 10.1161/01.RES.0000041036.86977.14
110. Johnson JL, Dwivedi A, Somerville M, George SJ, Newby AC. Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler Thromb Vasc Biol. (2011) 31(9):e35–44. doi: 10.1161/ATVBAHA.111.225623
111. Dwivedi A, Slater SC, George SJ. MMP-9 and -12 cause N-cadherin shedding and thereby {beta}-catenin signalling and vascular smooth muscle cell proliferation. Cardiovasc Res. (2009) 81(1):178–86. doi: 10.1093/cvr/cvn278
112. George SJ, Johnson JL, Angelini GD, Newby AC, Baker AH. Adenovirus-mediated gene transfer of the human TIMP-1 gene inhibits SMC migration and neointima formation in human saphenous vein. Hum Gene Ther. (1998) 9:867–77. doi: 10.1089/hum.1998.9.6-867
113. George SJ, Baker AH, Angelini GD, Newby AC. Gene transfer of tissue inhibitor of metalloproteinase-2 inhibits metalloproteinase activity and neointima formation in human saphenous veins. Gene Ther. (1998) 5:1552–60. doi: 10.1038/sj.gt.3300764
114. George SJ, Lloyd CT, Angelini GD, Newby AC, Baker AH. Inhibition of late vein graft neointima formation in human and porcine models by adenovirus-mediated overexpression of tissue inhibitor of metalloproteinase-3. Circulation. (2000) 101(3):296–304. doi: 10.1161/01.CIR.101.3.296
115. Dollery CM, Humphries SE, McClelland A, Latchman DS, McEwan JR. Expression of tissue inhibitor of matrix metalloproteinases 1 by use of an adenoviral vector inhibits smooth muscle cell migration and reduces neointimal hyperplasia in the rat model of vascular balloon injury. Circulation. (1999) 99(24):3199–205. doi: 10.1161/01.CIR.99.24.3199
116. Johnson C, Galis ZS. Matrix metalloproteinase-2 and-9 differentially regulate smooth muscle cell migration and cell-mediated collagen organization. Arterioscler Thromb Vasc Biol. (2004) 24(1):54–60. doi: 10.1161/01.ATV.0000100402.69997.C3
117. Williams H, Johnson JL, Jackson CL, White SJ, George SJ. MMP-7 mediates cleavage of N-cadherin and promotes smooth muscle cell apoptosis. Cardiovasc Res. (2010) 87(1):137–46. doi: 10.1093/cvr/cvq042
118. Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinase 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. (2002) 110:625–32. doi: 10.1172/JCI0215334
119. Silence J, Lupu F, Collen D, Lijnen HR. Persistence of atherosclerotic plaque but reduced aneurysm formation in mice with stromelysin-1 (MMP-3) gene inactivation. Arterioscler Thromb Vasc Biol. (2001) 21:1440–5. doi: 10.1161/hq0901.097004
120. Lyon CA, Williams H, Bianco R, Simmonds SJ, Brown BA, Wadey KS, et al. Aneurysm severity is increased by combined mmp-7 deletion and N-cadherin mimetic (EC4-fc) over-expression. Sci Rep. (2017) 7(1):17342. doi: 10.1038/s41598-017-17700-8
121. Pyo R, Lee JK, Shipley JM, Curci JA, Mao DL, Ziporin SJ, et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. (2000) 105(11):1641–9. doi: 10.1172/JCI8931
122. Longo GM, Buda SJ, Fiotta N, Xiong W, Griener T, Shapiro S, et al. MMP-12 has a role in abdominal aortic aneurysms in mice. Surgery. (2005) 137:457–62. doi: 10.1016/j.surg.2004.12.004
123. Wang B, Hsu SH, Majumder S, Kutay H, Huang W, Jacob ST, et al. TGF[Beta]-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene. (2010) 29(12):1787–97. doi: 10.1038/onc.2009.468
124. Salarian M, Ghim M, Toczek J, Han J, Weiss D, Spronck B, et al. Homeostatic, non-canonical role of macrophage elastase in vascular integrity. Circ Res. (2023) 132(4):432–48. doi: 10.1161/CIRCRESAHA.122.322096
125. Chen Y, Yang X, Kitajima S, Quan L, Wang Y, Zhu M, et al. Macrophage elastase derived from adventitial macrophages modulates aortic remodeling. Front Cell Dev Biol. (2022) 10:1097137. doi: 10.3389/fcell.2022.1097137
126. Lizarbe TR, Tarín C, Gómez M, Lavin B, Aracil E, Orte LM, et al. Nitric oxide induces the progression of abdominal aortic aneurysms through the matrix metalloproteinase inducer EMMPRIN. Am J Pathol. (2009) 175(4):1421–30. doi: 10.2353/ajpath.2009.080845
127. Xiong W, Knispel R, MacTaggart J, Greiner TC, Weiss SJ, Baxter BT. Membrane-type 1 matrix metalloproteinase regulates macrophage-dependent elastolytic activity and aneurysm formation in vivo. J Biol Chem. (2009) 284(3):1765–71. doi: 10.1074/jbc.M806239200
128. Silvestro M, Rivera CF, Alebrahim D, Vlahos J, Pratama MY, Lu C, et al. The nonproteolytic intracellular domain of membrane-type 1 matrix metalloproteinase coordinately modulates abdominal aortic aneurysm and atherosclerosis in mice—brief report. Arterioscler Thromb Vasc Biol. (2022) 42(10):1244–53. doi: 10.1161/ATVBAHA.122.317686
129. Martín-Alonso M, García-Redondo AB, Guo D, Camafeita E, Martínez F, Alfranca A, et al. Deficiency of MMP17/MT4-MMP proteolytic activity predisposes to aortic aneurysm in mice. Circ Res. (2015) 117(2):e13–26. doi: 10.1161/CIRCRESAHA.117.305108
130. Silence J, Collen D, Lijnen HR. Reduced atherosclerotic plaque but enhanced aneurysm formation in mice with inactivation of the tissue inhibitor of metalloproteinase-1 (TIMP-1) gene. Circ Res. (2002) 90:897–903. doi: 10.1161/01.RES.0000016501.56641.83
131. Eskandari MK, Vijungco JD, Flores A, Borensztajn J, Shively V, Pearce WH. Enhanced abdominal aortic aneurysm in TIMP-1-deficient mice. J Surg Res. (2005) 123(2):289–93. doi: 10.1016/j.jss.2004.07.247
132. Allaire E, Forough R, Clowes M, Starcher B, Clowes AW. Local overexpression of TIMP-1 prevents aortic aneurysm degeneration and rupture in a rat model. J Clin Invest. (1998) 102(7):1413–20. doi: 10.1172/JCI2909
133. Xiong WF, Knispel R, Mactaggart J, Baxter BT. Effects of tissue inhibitor of metalloproteinase 2 deficiency on aneurysm formation. J Vasc Surg. (2006) 44(5):1061–6. doi: 10.1016/j.jvs.2006.06.036
134. Basu R, Fan D, Kandalam V, Lee J, Das SK, Wang X, et al. Loss of Timp3 gene leads to abdominal aortic aneurysm formation in response to angiotensin II. J Biol Chem. (2012) 287(53):44083–96. doi: 10.1074/jbc.M112.425652
135. Rizas KD, Ippagunta N, Tilson MD 3rd. Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol Rev. (2009) 17(5):201–10. doi: 10.1097/CRD.0b013e3181b04698
136. Wilson WR, Schwalbe EC, Jones JL, Bell PR, Thompson MM. Matrix metalloproteinase 8 (neutrophil collagenase) in the pathogenesis of abdominal aortic aneurysm. Br J Surg. (2005) 92(7):828–33. doi: 10.1002/bjs.4993
137. Tromp G, Gatalica Z, Skunca M, Berguer R, Siegel T, Kline RA, et al. Elevated expression of matrix metalloproteinase-13 in abdominal aortic aneurysms. Ann Vasc Surg. (2004) 18(4):414–20. doi: 10.1007/s10016-004-0050-5
138. Morris DR, Biros E, Cronin O, Kuivaniemi H, Golledge J. The association of genetic variants of matrix metalloproteinases with abdominal aortic aneurysm: a systematic review and meta-analysis. Heart. (2014) 100(4):295–302. doi: 10.1136/heartjnl-2013-304129
139. Saratzis A, Bown MJ, Wild B, Nightingale P, Smith J, Johnson C, et al. Association between seven single nucleotide polymorphisms involved in inflammation and proteolysis and abdominal aortic aneurysm. J Vasc Surg. (2015) 61(5):1120–8. doi: 10.1016/j.jvs.2013.11.099
140. Davis V, Persidskaia R, Baca-Regen L, Itoh Y, Nagase H, Persidsky Y, et al. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. (1998) 18(10):1625–33. doi: 10.1161/01.ATV.18.10.1625
141. Petersen E, Gineitis A, Wågberg F, Ängquist KA. Activity of matrix metalloproteinase-2 and -9 in abdominal aortic aneurysms. Relation to size and rupture. Eur J Vasc Endovasc Surg. (2000) 20(5):457–61. doi: 10.1053/ejvs.2000.1211
142. Curci JA, Liao S, Huffman MD, Shapiro SD, Thompson RW. Expression and localization of macrophage elastase (matrix metalloproteinase-12) in abdominal aortic aneurysms. J Clin Invest. (1998) 102:1900–10. doi: 10.1172/JCI2182
143. Didangelos A, Yin X, Mandal K, Saje A, Smith A, Xu Q, et al. Extracellular matrix composition and remodeling in human abdominal aortic aneurysms: a proteomics approach. Mol Cell Proteomics. (2011) 10(8):1–15. doi: 10.1074/mcp.M111.008128
144. Nollendorfs A, Greiner TC, Nagase H, Baxter BT. The expression and localization of membrane type-1 matrix metalloproteinase in human abdominal aortic aneurysms. J Vasc Surg. (2001) 34(2):316–22. doi: 10.1067/mva.2001.115962
145. Lamblin N, Ratajczak P, Hot D, Dubois E, Chwastyniak M, Beseme O, et al. Profile of macrophages in human abdominal aortic aneurysms: a transcriptomic, proteomic, and antibody protein array study. J Proteome Res. (2010) 9(7):3720–9. doi: 10.1021/pr100250s
146. Elmore JR, Keister BF, Franklin DP, Youkey JR, Carey DJ. Expression of matrix metalloproteinases and TIMPs in human abdominal aortic aneurysms. Ann Vasc Surg. (1998) 12(3):221–8. doi: 10.1007/s100169900144
147. Annabi B, Shedid D, Ghosn P, Kenigsberg RL, Desrosiers RR, Bojanowski MW, et al. Differential regulation of matrix metalloproteinase activities in abdominal aortic aneurysms. J Vasc Surg. (2002) 35(3):539–46. doi: 10.1067/mva.2002.121124
148. Mikołajczyk-Stecyna J, Korcz A, Gabriel M, Pawlaczyk K, Oszkinis G, Słomski R. Gene polymorphism -418 G/C of tissue inhibitor of metalloproteinases 2 is associated with abdominal aortic aneurysm. J Vasc Surg. (2015) 61(5):1114–9. doi: 10.1016/j.jvs.2013.12.045
149. Lipp C, Lohoefer F, Reeps C, Rudelius M, Baummann M, Heemann U, et al. Expression of a disintegrin and metalloprotease in human abdominal aortic aneurysms. J Vasc Res. (2012) 49(3):198–206. doi: 10.1159/000332959
150. Tsarouhas K, Soufla G, Apostolakis S, Zaravinos A, Panagiotou M, Khoury M, et al. Transcriptional regulation of TIMPs in ascending aorta aneurysms. Thromb Res. (2010) 126(5):399–405. doi: 10.1016/j.thromres.2010.08.015
151. Ikonomidis JS, Ivey CR, Wheeler JB, Akerman AW, Rice A, Patel RK, et al. Plasma biomarkers for distinguishing etiologic subtypes of thoracic aortic aneurysm disease. J Thorac Cardiovasc Surg. (2013) 145(5):1326–33. doi: 10.1016/j.jtcvs.2012.12.027
152. McMillan WD, Pearce WH. Increased plasma levels of metalloproteinase-9 are associated with abdominal aortic aneurysms. J Vasc Surg. (1999) 29(1):122–7; discussion 7–9. doi: 10.1016/S0741-5214(99)70363-0
153. Watanabe T, Sato A, Sawai T, Uzuki M, Goto H, Yamashita H, et al. The elevated level of circulating matrix metalloproteinase-9 in patients with abdominal aortic aneurysms decreased to levels equal to those of healthy controls after an aortic repair. Ann Vasc Surg. (2006) 20(3):317–21. doi: 10.1007/s10016-006-9038-7
154. Hovsepian DM, Ziporin SJ, Sakurai MK, Lee JK, Curci JA, Thompson RW. Elevated plasma levels of matrix metalloproteinase-9 in patients with abdominal aortic aneurysms: a circulating marker of degenerative aneurysm disease. J Vasc Interv Radiol. (2000) 11(10):1345–52. doi: 10.1016/S1051-0443(07)61315-3
155. Grodin JL, Powell-Wiley TM, Ayers CR, Kumar DS, Rohatgi A, Khera A, et al. Circulating levels of matrix metalloproteinase-9 and abdominal aortic pathology: from the Dallas heart study. Vasc Med. (2011) 16(5):339–45. doi: 10.1177/1358863X11422110
156. Wilson WR, Anderton M, Choke EC, Dawson J, Loftus IM, Thompson MM. Elevated plasma MMP1 and MMP9 are associated with abdominal aortic aneurysm rupture. Eur J Vasc Endovasc Surg. (2008) 35(5):580–4. doi: 10.1016/j.ejvs.2007.12.004
157. Karlsson L, Bergqvist D, Lindbäck J, Pärsson H. Expansion of small-diameter abdominal aortic aneurysms is not reflected by the release of inflammatory mediators IL-6, MMP-9 and CRP in plasma. Eur J Vasc Endovasc Surg. (2009) 37(4):420–4. doi: 10.1016/j.ejvs.2008.11.027
158. Bontekoe J, Liu B. Single-cell RNA sequencing provides novel insights to pathologic pathways in abdominal aortic aneurysm. Front Cardiovasc Med. (2023) 10:1172080. doi: 10.3389/fcvm.2023.1172080
159. Cao G, Xuan X, Li Y, Hu J, Zhang R, Jin H, et al. Single-cell RNA sequencing reveals the vascular smooth muscle cell phenotypic landscape in aortic aneurysm. Cell Commun Signal. (2023) 21(1):113. doi: 10.1186/s12964-023-01120-5
160. Hadi T, Boytard L, Silvestro M, Alebrahim D, Jacob S, Feinstein J, et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nature Commun. (2018) 9(1):5022. doi: 10.1038/s41467-018-07495-1
161. Qian W, Hadi T, Silvestro M, Ma X, Rivera CF, Bajpai A, et al. Microskeletal stiffness promotes aortic aneurysm by sustaining pathological vascular smooth muscle cell mechanosensation via Piezo1. Nature Commun. (2022) 13(1):512. doi: 10.1038/s41467-021-27874-5
162. Li B, Song X, Guo W, Hou Y, Hu H, Ge W, et al. Single-Cell transcriptome profiles reveal fibrocytes as potential targets of cell therapies for abdominal aortic aneurysm. Front Cardiovasc Med. (2021) 8:753711. doi: 10.3389/fcvm.2021.753711
163. Zhao G, Lu H, Chang Z, Zhao Y, Zhu T, Chang L, et al. Single-cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiovasc Res. (2020) 117(5):1402–16. doi: 10.1093/cvr/cvaa214
164. Yang H, Zhou T, Stranz A, DeRoo E, Liu B. Single-Cell RNA sequencing reveals heterogeneity of vascular cells in early stage murine abdominal aortic aneurysm—brief report. Arterioscler Thromb Vasc Biol. (2021) 41(3):1158–66. doi: 10.1161/ATVBAHA.120.315607
165. Petrinec D, Liao S, Holmes DR, Reilly JM, Parks WC, Thompson RW. Doxycycline inhibition of aneurysmal degeneration in an elastase-induced rat model of abdominal aortic aneurysm: preservation of aortic elastin associated with suppressed production of 92 kD gelatinase. J Vasc Surg. (1996) 23(2):336–46. doi: 10.1016/S0741-5214(96)70279-3
166. Sho E, Chu J, Sho M, Fernandes B, Judd D, Ganesan P, et al. Continuous periaortic infusion improves doxycycline efficacy in experimental aortic aneurysms. J Vasc Surg. (2004) 39(6):1312–21. doi: 10.1016/j.jvs.2004.01.036
167. Curci JA, Petrinec D, Liao S, Golub LM, Thompson RW. Pharmacologic suppression of experimental abdominal aortic aneurysms: a comparison of doxycycline and four chemically modified tetracyclines. J Vasc Surg. (1998) 28(6):1082–93. doi: 10.1016/S0741-5214(98)70035-7
168. Yu M, Chen C, Cao Y, Qi R. Inhibitory effects of doxycycline on the onset and progression of abdominal aortic aneurysm and its related mechanisms. Eur J Pharmacol. (2017) 811:101–9. doi: 10.1016/j.ejphar.2017.05.041
169. Prall AK, Longo GM, Mayhan WG, Waltke EA, Fleckten B, Thompson RW, et al. Doxycycline in patients with abdominal aortic aneurysms and in mice: comparison of serum levels and effect on aneurysm growth in mice. J Vasc Surg. (2002) 35(5):923–9. doi: 10.1067/mva.2002.123757
170. Sheth RA, Maricevich M, Mahmood U. In vivo optical molecular imaging of matrix metalloproteinase activity in abdominal aortic aneurysms correlates with treatment effects on growth rate. Atherosclerosis. (2010) 212(1):181–7. doi: 10.1016/j.atherosclerosis.2010.05.012
171. Tedesco MM, Terashima M, Blankenberg FG, Levashova Z, Spin JM, Backer MV, et al. Analysis of in situ and ex vivo vascular endothelial growth factor receptor expression during experimental aortic aneurysm progression. Arterioscler Thromb Vasc Biol. (2009) 29(10):1452–7. doi: 10.1161/ATVBAHA.109.187757
172. Turner GH, Olzinski AR, Bernard RE, Aravindhan K, Karr HW, Mirabile RC, et al. In vivo serial assessment of aortic aneurysm formation in apolipoprotein E–deficient mice via MRI. Circ Cardiovasc Imaging. (2008) 1(3):220–6. doi: 10.1161/CIRCIMAGING.108.787358
173. Vinh A, Gaspari TA, Liu HB, Dousha LF, Widdop RE, Dear AE. A novel histone deacetylase inhibitor reduces abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. J Vasc Res. (2008) 45(2):143–52. doi: 10.1159/000110041
174. Manning MW, Cassis LA, Daugherty A. Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. (2003) 23:483–8. doi: 10.1161/01.ATV.0000058404.92759.32
175. Kaito K, Urayama H, Watanabe G. Doxycycline treatment in a model of early abdominal aortic aneurysm. Surg Today. (2003) 33(6):426–33. doi: 10.1007/s10595-002-2513-0
176. Mata KM, Tefé-Silva C, Floriano EM, Fernandes CR, Rizzi E, Gerlach RF, et al. Interference of doxycycline pretreatment in a model of abdominal aortic aneurysms. Cardiovasc Pathol. (2015) 24(2):110–20. doi: 10.1016/j.carpath.2014.10.004
177. Bigatel DA, Elmore JR, Carey DJ, Cizmeci-Smith G, Franklin DP, Youkey JR. The matrix metalloproteinase inhibitor BB-94 limits expansion of experimental abdominal aortic aneurysms. J Vasc Surg. (1999) 29(1):130–9. doi: 10.1016/S0741-5214(99)70354-X
178. Moore G, Liao S, Curci JA, Starcher BC, Martin RL, Hendricks RT, et al. Suppression of experimental abdominal aortic aneurysms by systemic treatment with a hydroxamate-based matrix metalloproteinase inhibitor (RS 132908). J Vasc Surg. (1999) 29(3):522–32. doi: 10.1016/S0741-5214(99)70281-8
179. Nosoudi N, Nahar-Gohad P, Sinha A, Chowdhury A, Gerard P, Carsten CG, et al. Prevention of abdominal aortic aneurysm progression by targeted inhibition of matrix metalloproteinase activity with batimastat-loaded nanoparticles. Circ Res. (2015) 117(11):e80–e9. doi: 10.1161/CIRCRESAHA.115.307207
180. Prescott MF, Sawyer WK, Von Linden-Reed J, Jeune M, Chou M, Caplan SL, et al. Effect of matrix metalloproteinase inhibition on progression of atherosclerosis and aneurysm in LDL receptor-deficient mice overexpressing MMP3, MMP-12, and MMP-13 and on restenosis in rats after balloon injury. Ann NY Acad Sci. (1999) 878:179–90. doi: 10.1111/j.1749-6632.1999.tb07683.x
181. Liu J, Xiong W, Baca-Regen L, Nagase H, Baxter BT. Mechanism of inhibition of matrix metalloproteinase-2 expression by doxycycline in human aortic smooth muscle cells. J Vasc Surg. (2003) 38(6):1376–83. doi: 10.1016/S0741-5214(03)01022-X
182. Mosorin M, Juvonen J, Biancari F, Satta J, Surcel H-M, Leinonen M, et al. Use of doxycycline to decrease the growth rate of abdominal aortic aneurysms: a randomized, double-blind, placebo-controlled pilot study. J Vasc Surg. (2001) 34(4):606–10. doi: 10.1067/mva.2001.117891
183. Hackmann AE, Rubin BG, Sanchez LA, Geraghty PA, Thompson RW, Curci JA. A randomized, placebo-controlled trial of doxycycline after endoluminal aneurysm repair. J Vasc Surg. (2008) 48(3):519–26. doi: 10.1016/j.jvs.2008.03.064
184. Lindeman JHN, Abdul-Hussien H, van Bockel JH, Wolterbeek R, Kleemann R. Clinical trial of doxycycline for matrix metalloproteinase-9 inhibition in patients with an abdominal aneurysm: doxycycline selectively depletes aortic wall neutrophils and cytotoxic T cells. Circulation. (2009) 119(16):2209–16. doi: 10.1161/CIRCULATIONAHA.108.806505
185. Abdul-Hussien H, Hanemaaijer R, Verheijen JH, van Bockel JH, Geelkerken RH, Lindeman JH. Doxycycline therapy for abdominal aneurysm: improved proteolytic balance through reduced neutrophil content. J Vasc Surg. (2009) 49(3):741–9. doi: 10.1016/j.jvs.2008.09.055
186. Baxter BT, Pearce WH, Waltke EA, Littooy FN, Hallett Jr JW, Kent KC, et al. Prolonged administration of doxycycline in patients with small asymptomatic abdominal aortic aneurysms: report of a prospective (phase II) multicenter study. J Vasc Surg. (2002) 36(1):1–12. doi: 10.1067/mva.2002.125018
187. Ding R, McGuinness CL, Burnand KG, Sullivan E, Smith A. Matrix metalloproteinases in the aneurysm wall of patients treated with low-dose doxycycline. Vascular. (2005) 13(5):290–7. doi: 10.1258/rsmvasc.13.5.290
188. Meijer CA, Stijnen T, Wasser MNJM, Hamming JF, van Bockel JH, Lindeman JHN. Doxycycline for stabilization of abdominal aortic aneurysms: a randomized trial. Ann Intern Med. (2013) 159(12):815–23. doi: 10.7326/0003-4819-159-12-201312170-00007
189. Baxter BT, Matsumura J, Curci JA, McBride R, Larson L, Blackwelder W, et al. Effect of doxycycline on aneurysm growth among patients with small infrarenal abdominal aortic aneurysms: a randomized clinical trial. JAMA. (2020) 323(20):2029–38. doi: 10.1001/jama.2020.5230
190. Quillard T, Tesmenitsky Y, Croce K, Travers R, Shvartz E, Koskinas KC, et al. Selective inhibition of matrix metalloproteinase-13 increases collagen content of established mouse atherosclerosis. Arterioscler Thromb Vasc Biol. (2011) 31(11):2464–72. doi: 10.1161/ATVBAHA.111.231563
191. Toczek J, Gona K, Liu Y, Ahmad A, Ghim M, Ojha D, et al. Positron emission tomography imaging of vessel wall matrix metalloproteinase activity in abdominal aortic aneurysm. Circ Cardiovasc Imaging. (2023) 16(1):e014615. doi: 10.1161/CIRCIMAGING.122.014615
Keywords: aortic aneurysm (AA), matrix metalloproteinase (MMP), animal models, TIMP (tissue inhibitors of metallproteinase), VSMC = vascular smooth muscle cell, macrophage
Citation: Atkinson G, Bianco R, Di Gregoli K and Johnson JL (2023) The contribution of matrix metalloproteinases and their inhibitors to the development, progression, and rupture of abdominal aortic aneurysms. Front. Cardiovasc. Med. 10:1248561. doi: 10.3389/fcvm.2023.1248561
Received: 27 June 2023; Accepted: 7 September 2023;
Published: 19 September 2023.
Edited by:
Ho Pei, National University of Singapore, SingaporeReviewed by:
Renata Gomes, Bravo Victor, United Kingdom© 2023 Atkinson, Bianco, Di Gregoli and Johnson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason L. Johnson amFzb24ubC5qb2huc29uQGJyaXN0b2wuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.