 Yan Zhong
Yan Zhong YingWen Chen2,†
YingWen Chen2,† XinYue Zhang
XinYue Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med., 12 October 2023
Sec. Cardiovascular Epidemiology and Prevention
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1243867
Objective: Observational studies have suggested an increased risk of cardiovascular disease in individuals with ankylosing spondylitis. However, these studies are prone to confounding factors and reverse causality. To address these limitations, we conducted a Mendelian randomization study to assess the causal relationship between AS and CVD.
Methods: The study population comprises 9,069 individuals with ankylosing spondylitis and 509,093 individuals with either of six common cardiovascular diseases and a related indicator. Causal analysis using summary effect estimates and inverse variance weighting were employed as the main methods.
Results: The CAUSE analysis showed no evidence of a causal relationship between AS and CVD. The odds ratios for total CVD, heart failure, myocardial infarction, valvular heart disease, ischemic heart disease, and venous thromboembolism, Arterial stiffness index, were as follows: OR, 1.01; 95% confidence interval, 0.96–1.05; P = 0.91; OR, 1.03; 95% CI, 0.99–1.08; P = 0.50; OR, 0.94; 95% CI, 0.86–1.03; P = 0.53; OR, 0.99; 95% CI, 0.94–1.04; P = 0.99; OR, 0.98; 95% CI, 0.91–1.04; P = 0.94; OR, 0.98; 95% CI, 0.91–1.04; P = 0.99; β, −0.0019; 95% CI, 0.97–1.01; P = 0.99. The IVW and weighted median methods also yielded consistent results, and no heterogeneity or pleiotropy was found. Likewise, a reverse Mendelian randomization analysis did not uncover a heritable causal relationship between AS and CVD.
Conclusion: This Mendelian randomization study does not support a causal relationship between AS and CVD. Further research is needed to confirm this association.
Cardiovascular disease (CVD) is a condition that impacts the heart and blood vessels, and it stands out as having the highest mortality and morbidity rates among non-infectious diseases (1). Over the past decade, there has been a notable 12.5% increase in CVD-related deaths, primarily attributed to the aging population (2). The escalating burden of CVD presents a significant challenge to both society and families, underscoring the crucial need to identify risk factors associated with CVD and implement effective preventive measures.
Ankylosing Spondylitis (AS) is a chronic inflammatory disease classified as a form of axial spondyloarthritis (3). In the United States, it affects approximately 0.9% to 1.4% of adults, which is comparable to the prevalence of rheumatoid arthritis (4). AS primarily manifests as chronic inflammatory low back pain, peripheral arthritis, and sacroiliac arthritis (5). Patients with AS commonly experience restricted spinal mobility as a result of back pain and stiffness, and over time, they may develop progressive hyperkyphosis (6), leading to compromised cardiopulmonary function (7).
Observational studies have demonstrated an association between AS and increased morbidity and mortality related to CVD (8, 9). However, the causal relationship between AS and CVD remains contentious. Cohort studies have reported an elevated risk of ischemic heart disease (IHD) and myocardial infarction (MI) in AS patients, yet multiple studies have failed to reproduce these findings (10). Furthermore, there is ongoing controversy surrounding the association between AS and atherosclerosis (11, 12). It is worth noting that comorbidities commonly observed in AS, such as hypertension (HTN) and diabetes mellitus (DM) (13), along with well-established risk factors like smoking, obesity, and reduced physical activity, are strongly associated with CVD morbidity and mortality (14, 15). Consequently, it remains uncertain whether the development of CVD is directly attributed to AS itself or indirectly influenced by the comorbidities associated with AS. This uncertainty arises from inherent limitations in previous studies, including small sample sizes, issues of reverse causality, and potential confounding factors.
Mendelian randomization (MR) is a statistical method that uses genetic variation as an instrumental variable (IV) to assess causal relationships between exposures and outcomes, particularly in situations where randomized controlled trials (RCTs) are not feasible or practical (16). One of the key advantages of MR is its ability to overcome issues of confounding, as genetic variants are typically not influenced by environmental factors or disease status (17). Another advantage of MR is the potential for larger sample sizes. This is particularly useful when traditional observational studies may not have enough statistical power to detect a causal relationship (18). Therefore, our goal is to explore the causal relationship between AS and CVD through two sample MR analysis.
In MR analysis, genetic variations associated with the exposure are employed as instrumental variables (IVs) to explore the causation between exposure and outcome. The design of a Mendelian randomization study must adhere to three fundamental assumptions: First, the instrumental variables and exposure should exhibit a strong association. Second, the instrumental variables should be independent of confounding factors. And third, the instrumental variables should solely impact the outcome through their influence on the exposure (Figure 1).
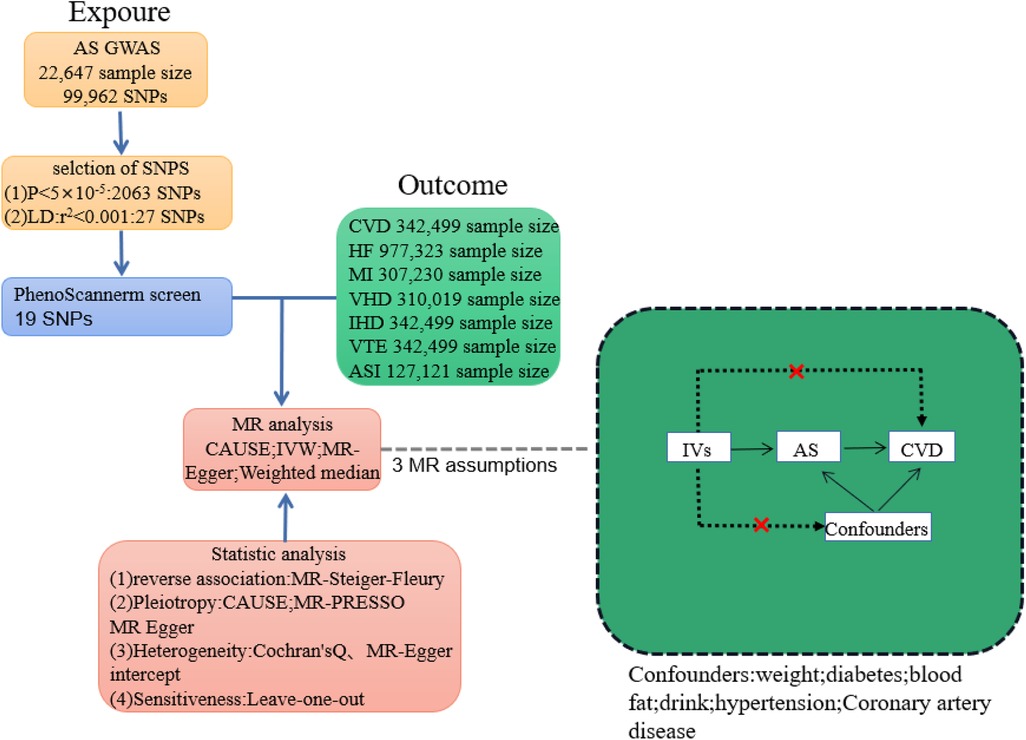
Figure 1. Plot of Mendelian randomization study between ankylosing spondylitis and cardiovascular disease. IVs, instrumental variable; CVD, total cardiovascular disease; HF, heart failure; MI, myocardial infarction; VHD, heart valve disease; CHD, coronary heart disease; IHD; ischaemic heart disease; VTE, venous embolism; ASI, arterial stiffness index.
Instrumental variables for AS were used from the GWAS genome-wide data of Crotes et al. (19). The total sample size was 22,647 Europeans (9,069 ncase and 13,578 ncontrol). To fulfill the first hypothesis of the MR analysis, we employed three selection methods. Firstly, SNPs with a significance threshold of P < 5 × 10−8 were chosen. Secondly, any screened genes displaying linkage disequilibrium (R2 < 0.001; distance <10,000 kb) were excluded to ensure the independence of genetic variations. Thirdly, to prevent weak instrumental variables from biasing our findings, we used the F-value statistic (F = beta2/se2). If the calculated F-statistic was greater than 10, we concluded that the relationship between IV and exposure was sufficiently strong and would not affect the results of our MR analysis (20). In cases where the requested SNPs were unavailable, we supplemented them using SNPs from the 1,000 Genomes Project proxy (LD; r2 > 0.8) (21).
To mitigate potential confounding effects, we consulted the PhenoScanner database (http://www.phenoscanner.medschl.cam.ac.uk/) to assess whether the selected SNPs violated hypotheses (2) and (3). As a result, six traits (i.e., weight, cardiovascular disease, alcohol, blood glucose, total cholesterol and hypertension) were found to be potential confounders. Thus, we further excluded SNPs strongly associated with these six potential confounders as well as CVD to ensure that the resulting instrumental variables influenced CVD only through AS, with no confounding factors affecting the final results (22). After examining the potential synergistic effects between exposure and the outcome, we excluded SNPs with palindromic properties. Ultimately, we identified 19 SNPs as genetic variables for the instrumental variable (IV) analysis (Table 1).
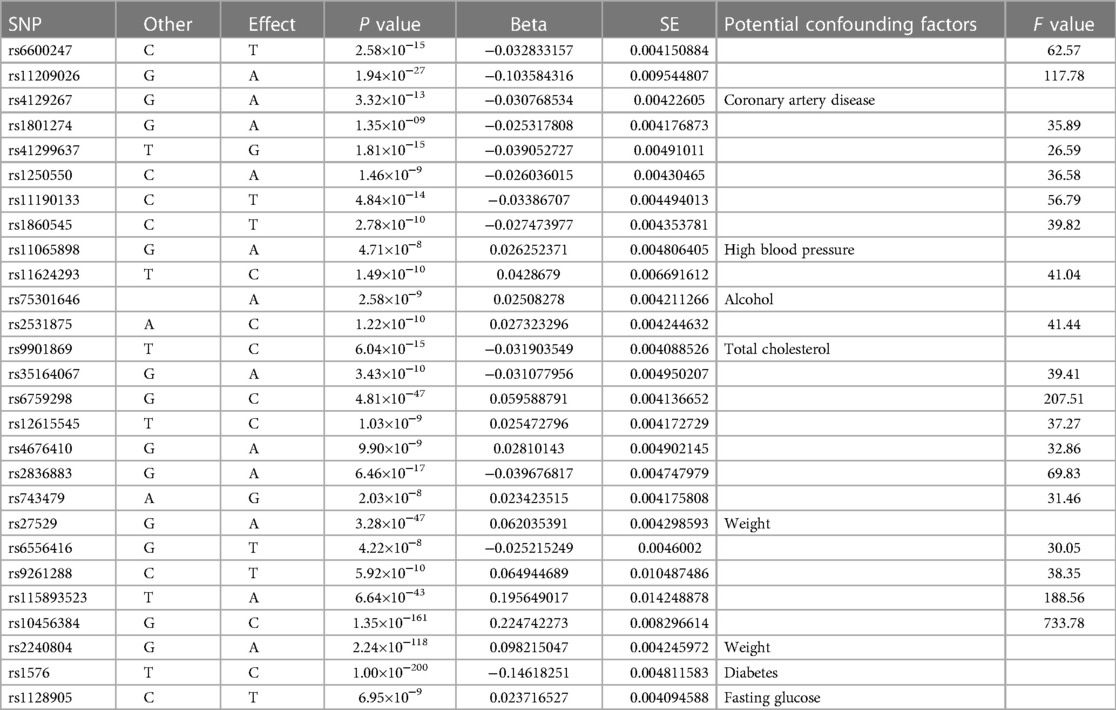
Table 1. As instrumental variables.
Data for total cardio-vascular disease (CVD) (n = 174,499 cases, n = 168,000 controls), myocardial infarction (MI) (n = 21,609 cases, n = 285,621 controls), valvular heart disease (VHD) (n = 72,756 cases, n = 237,263 controls), ischemic heart disease (IHD) (n = 56,730 cases, n = 285,769 controls), and venous thromboembolism (VTE) (n = 17,048 cases, n = 325,451 controls) were extracted from the Finnish database. The FinnGen research project aggregated the final genetic data for various diseases from the Finnish Biobank and the Finnish Health Registry (23). Arterial stiffness, which offers a different perspective on hemodynamic alterations (24), is closely associated with the development of CVD (25). Therefore, we retrieved summary-level data on the arterial stiffness index (ASI) for 127,121 participants from the UKB database (26). The data were obtained by measuring finger pulse waveforms of the participants (25). Statistics regarding heart failure (HF) were sourced from the Heart Failure Molecular Therapeutic Target Epidemiology (HERMES) Consortium, which includes 47,309 cases and 930,014 individuals of European ancestry (27).
The data used in this study were obtained from publicly available GWAS databases that previously obtained ethical approval, eliminating the need for further ethical approval. To minimize data bias caused by ethnic differences, the study population was limited to individuals of European descent, and information on the exposure and outcomes is presented in (Table 2).
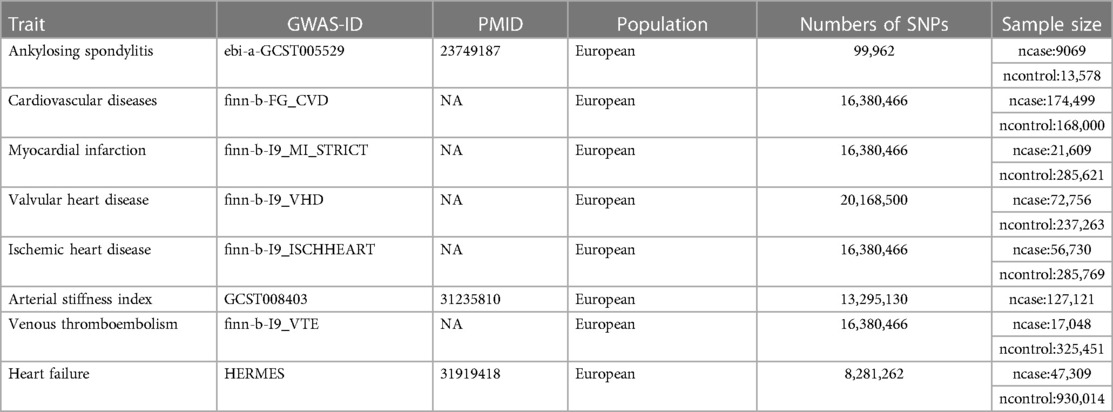
Table 2. Mendelian randomization analysis data details.
In this study, we used the “TwoSampleMR”, “MRPRESSO”, and “CAUSE” packages in R (4.2.3) for data analysis. The study adhered to the STROBE-MR statement (28). To address the potential impact of reverse association (i.e., CVD for exposure and AS for outcome), we applied the MR-Steiger-Fleury method and reverse MR methods. These two methods helped us ensure that all IVs were correctly oriented in our inferences. We assessed the directionality of the association between them and employed bidirectional MR to investigate their potential causal relationship.
We employed the Causal Analysis Using Summary Effect estimates (CASUSE) approach for causal analysis (29). The issue of horizontal pleiotropy has been a challenge in MR studies, wherein it can be categorized as “correlated horizontal pleiotropy” and “uncorrelated horizontal pleiotropy” (30). While previous studies have addressed the impact of “uncorrelated horizontal pleiotropy” using MR-PRESSO (31), ignoring the effect of “correlated horizontal pleiotropy” can result in a high false-positive rate in MR studies (32). Thus, we used CAUSE, which is superior to other established methods in detecting causality in the presence of pleiotropy (29). CAUSE leverages genome-wide summary statistics that distinguish causality from correlated pleiotropy. Additionally, Bayesian modeling is employed to further evaluate uncorrelated horizontal pleiotropy to ensure the rigor of the final conclusions.
The primary method for detecting causality in MR analyses is inverse variance weighting (IVW), provided there is no multiplicity of effects and all instrumental variables are valid (33). We used weighted median (WM) analysis as a complementary measure (34). For identifying fixed-point pleiotropy, we employed MR-Egger and used MR-PRESSO analysis for detecting unrelated pleiotropy (31). SNPs with pleiotropy were excluded when it occurred. We assessed SNP homogeneity using both the Cochran Q test and MR-Egger intercept test (35). Sensitivity analyses via the “leave-one-out” methods were conducted to investigate the effects of individual SNPs on MR.
The F-value statistics for selected IVs in MR analysis ranged from 26 to 733, showing a strong prediction of AS by IVs and validating the MR hypothesis (1). The MR-Steiger-Fleury test did not reveal the presence of reverse causality (Table 3). Reverse MR analysis also did not reveal a causal relationship between CVD and AS (Supplementary Tables S1, S2).
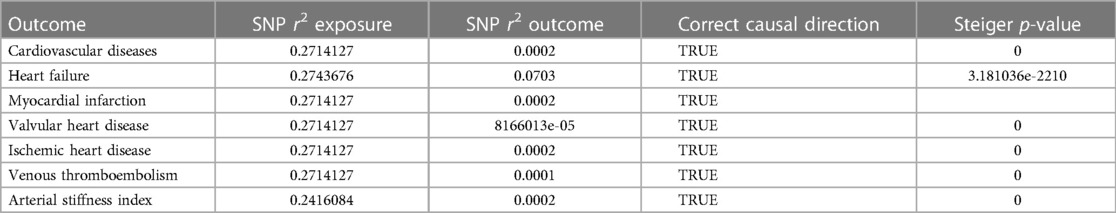
Table 3. A directional test for Mendelian randomization about the effect of AS on CVD.
SNP r2 exposure: r-squared values of the exposed SNP; SNP r2 outcome: r-squared values of the outcome SNP; correct causal direction: AS to CVD directional correctness; Steiger p-value: the p-value of the MR-Steiger-Fleury test.
CAUSE analysis revealed no association between AS and CVD (total CVD: OR, 1.01; 95% CI, 0.96–1.05; P = 0.91; HF: OR, 1.03; 95% CI, 0.99–1.08; P = 0.50; MI: OR, 0.94; 95% CI, 0.86–1.03; P = 0.53; VHD: OR, 0.99; 95% CI, 0.94–1.04; P = 0.99; IHD: OR, 0.98; 95% CI, 0.91–1.04; P = 0.94; VTE: OR, 0.98; 95% CI, 0.91-1.04; P = 0.99; ASI: β, −0.0019; 95% CI, 0.97–1.01; P = 0.99;). Similarly, the trends of IVW and WM were consistent (Figures 2, 3).
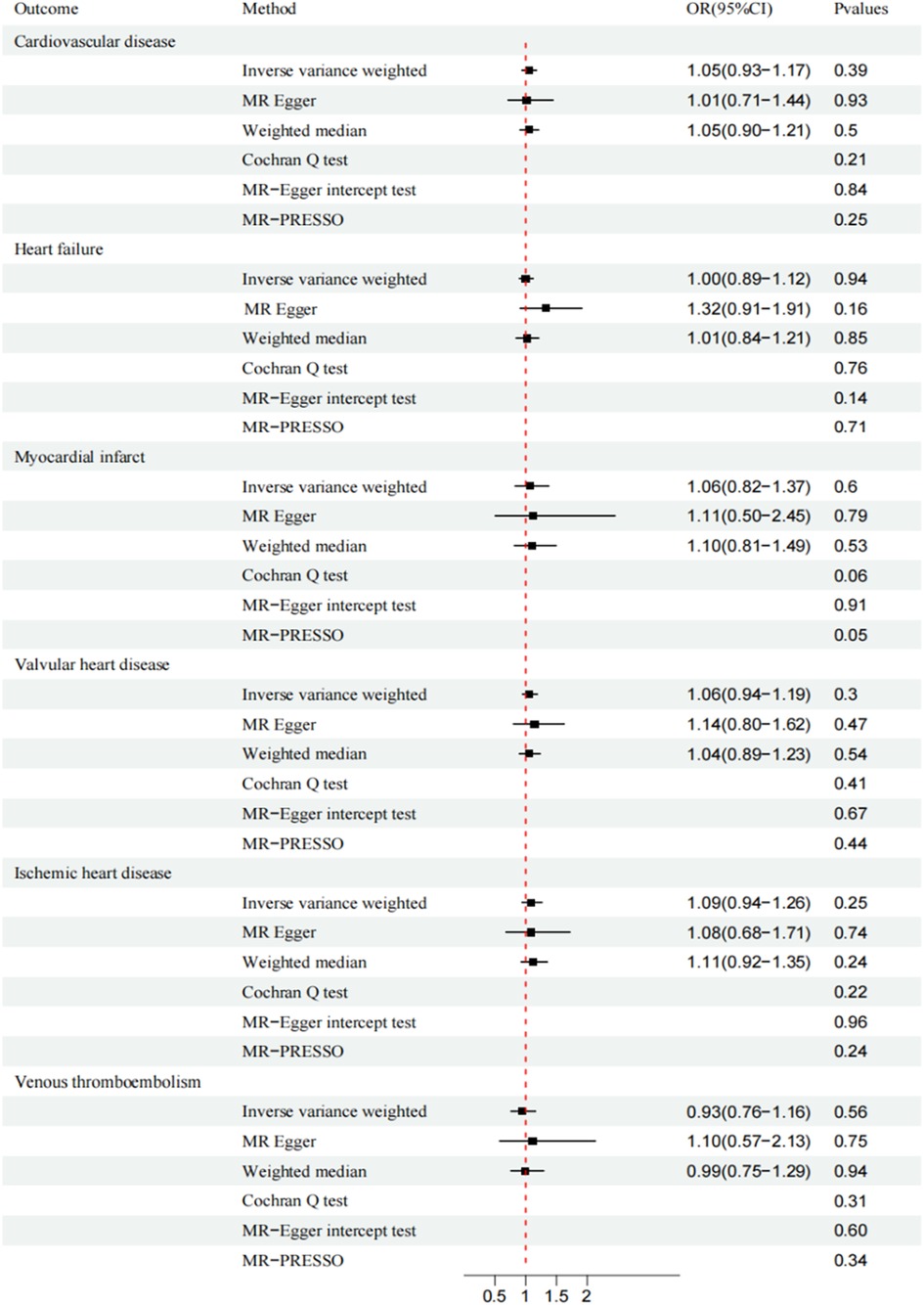
Figure 2. Mendelian randomization analysis of ankylosing spondylitis and cardiovascular disease. CI, confidence interval; OR, odds ratio.
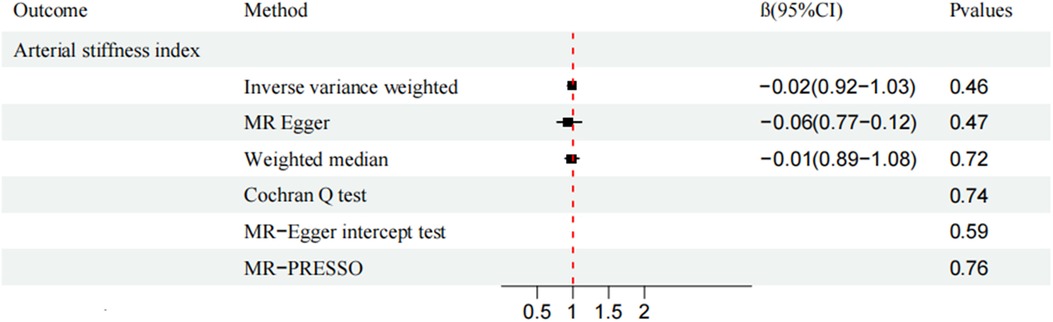
Figure 3. Mendelian randomization analysis of ankylosing spondylitis and arterial stiffness index.
MR-Egger and MR-PRESSO secondary analyses found no fixed-point or horizontal pleiotropy in the MR analysis, and both analyses revealed identical trends. The Cochran Q test and MR-Egger intercept test indicated low or no heterogeneity (Figures 2, 3). The leave-one-out method found no evidence of overall instability or sensitivity abnormalities when eliminating one SNP. The absence of potentially confounding SNPs, the low or no heterogeneity, and the results of the multipotency robustness analysis confirmed the validity of MR hypotheses (2) and (3). For more detailed information, please refer to (Supplementary Table S3).
In our current study, we explored the causal relationship between AS and CVD using multiple large open databases. The MR analyses conducted did not demonstrate any effect of AS on CVD, with consistent outcomes across all analyses. The findings contradicted several observational studies that have implied a causal connection between AS and CVD. As the evidence for the causal relationship between AS and CVD remains inconclusive (36), the results of our MR analysis are of great interest.
Atherosclerosis is well known as the basis of cardiovascular disease (37). Research has demonstrated differences in extracardiac pulse wave velocity (PWV) between individuals with AS and control groups (38), and two observational studies have corroborated that AS patients are at higher risk for atherosclerosis (39, 40). Two recent meta-analyses have also suggested that AS patients may have an increased of developing atherosclerosis (41, 42). However, some disagreement persists around this issue; studies have found that atherosclerosis does not accelerate when controlling for inflammatory manifestations and disease progression in AS patients (43, 44). Evidence also indicates that individuals with AS who have a shorter disease duration do not exhibit increased carotid or femoral artery plaque thickness (43). It should be noted that although observational studies have made efforts to avoid confounding factors, issues such as selection bias and reverse causality inherent in design have the potential to skew results. As a result, the causal relationship between AS and atherosclerosis remains inconclusive.
The heightened risk of CVD in individuals with AS may be attributed to the significant overlap between conventional CVD risk factors and the comorbidities of AS, such as hypertension, dyslipidemia, diabetes, smoking, and obesity (45). This is corroborated by a recent meta-analysis which reported hypertension and hyperlipidemia to be the most prevalent comorbidities among AS patients (46). Findings from another cross-sectional study also indicated that AS patients have a higher prevalence of diabetes and smoking compared to the general population (47), and that obesity is a notable issue due to various reasons (48). Being overweight or obese can have a direct impact on AS activity (49), with evidence suggesting that it can lead to reduced efficacy of biologic medications (50), and an increased likelihood of exercise-induced hypertensive (51). Matters are further complicated by the role of smoking, and in 2,021, the European League Against Rheumatism (EULAR) issued guidelines for managing cardiovascular risks, which includes cigarette cessation as a key strategy (52). The reason is that, in addition to increasing the risk of CVD (53), smoking also undermines the effectiveness of drug therapy (54). Therefore, smoking assumes a dual role in individuals with AS: on one hand, it embodies a traditional CVD risk factor; on the other hand, it influences and worsens disability and the development of AS, which ultimately leads to a more sedentary lifestyle—an additional conventional CVD risk factor (55, 56).
AS triggers a systemic inflammatory response that plays a significant role in the development of CVD (57). Cardiovascular inflammation is closely associated with elevated inflammatory cytokines, particularly interleukin-6 (IL-6) (58, 59). In patients with AS, total cholesterol and high-density lipoprotein (HDL) levels are lower than in the general population, which is closely linked to their own inflammatory activity (60). When IL-6 is increased, total cholesterol and HDL levels decrease significantly (61). IL-6 also interacts with the hypothalamic-pituitary-adrenal axis, resulting in increased insulin sensitivity that triggers diabetes, as well as increased BMI and blood pressure that lead to hypertension (62). Non-steroidal anti-inflammatory drugs (NSAIDs) are recommended as a first-line treatment to alleviate inflammation and pain (63). However, although NSAIDs have good anti-inflammatory and analgesic effects, their cardiovascular side effects are a concern for physicians, and the US Food and Drug Administration (FDA) has recently issued a warning (64). A meta-analysis found that almost all NSAIDs increase the risk of MI (65). Even short-term NSAID use can lead to an increase in thrombotic cardiovascular events (66). Real-world studies conducted in four European countries discovered that NSAIDs increased the risk of heart failure death in patients (67), and high doses of diclofenac or ibuprofen can more than double this risk. The administration of NSAIDs can also lead to adverse effects on thrombosis. Several studies suggest that even short-term concurrent use of NSAIDs and antithrombotics increases the risk of bleeding, with no specific window period (68, 69).
Observational studies have indicated a potential association between ankylosing spondylitis and the development of VHD (70). But our MR analysis did not reveal any evidence supporting a genetic causative link between ankylosing spondylitis and VHD. Nevertheless, patients with AS frequently exhibit clinical manifestations linked to VHD, such as aortic valve insufficiency and aortic inflammation (71). Notably, individuals positive for the HLA-B27 marker often exhibit an increased aortic root index, which correlates with a heightened risk of complications like aortic valve regurgitation (AVR) (72). In the real-world scenario, HLA-B27 positivity is associated with sustained activation of IL-23 + T cells, leading to an inflammatory response within the heart (73). A comprehensive cohort study involving AS patients from an East Asian population suggested that chronic systemic inflammation may contribute to the early development of VHD (74). Furthermore, a related study has solidified the connections among aortic stiffness, myocardial function, and the activity of AS (75). These findings align with autopsy results, which reveal aortic valve fibrosis in AS patients (76). However, the relationship between AS and cardiovascular disease is profoundly intricate. Given the existence of different AS subtypes with varying manifestations across diverse racial and gender groups, it is imperative to conduct more extensive research to thoroughly explore the associations between different AS subtypes, races, and CVD (77).
Recent genetic and MR studies have demonstrated the role of IL-6 in atherosclerosis and CVD (78, 79). Although our MR analysis did not reveal a causal link between AS and CVD, we identified the IL-6R SNP rs4129267 as a pleiotropic SNP that was removed from our analysis. Given the critical role of IL-6 in CVD, we postulated that specific loci such as IL-6 or local genomic correlations might be responsible for the relationship between AS and CVD. Further analysis using a co-localization approach would help elucidate this relationship. However, we must note that the usage of rs4129267 as a potential therapeutic target for CVD would require in-depth demonstration via clinical studies.
Our study provides a unique perspective on the prevention and treatment of CVD in patients with AS. Although our MR analysis does not find a genetic causal relationship between AS and CVD, we acknowledge the need for caution while interpreting our findings. Given the comparatively lower appreciation for the prevention and management of CVD comorbidities in patients with AS, we believe additional experimental studies and clinical research are necessary to inform evidence-based interventions.
Our study has several strengths. Firstly, the utilization of the MR approach facilitated the avoidance of interference from environmental factors, inherent bias, and reverse causality into the study's outcomes. Secondly, through a rigorous screening process, we minimized confounding factors that could affect the final results. Lastly, we employed multiple analytical techniques such as CAUSE, IVW, and WM to ensure the rigorousness of the study's findings regarding the causal relationship between AS and CVD.
Our study has several limitations. Firstly, our population was restricted to European populations, which could limit the generalization of the findings to other races. Secondly, AS is a progressive disease with different comorbidities at each stage, which could influence the relationship between AS and CVD in various ways. Although we made efforts to include as many cardiovascular diseases associated with AS as possible, some cardiovascular events that were not considered in our study may also influence the relationship between AS and CVDs. Thirdly, we have five GWAS datasets derived from the FinnGen database which introduces certain limitations and the possibility of selection bias. In future studies we will incorporate more diverse data for more in-depth research. Further studies that address these limitations may provide a more comprehensive understanding of the relationship between AS and CVDs.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
YZ and YWC: performed data collection and collation, as well as for the initial drafting and revision of the thesis. XYZ and WJC: conducted data visualization and verified the accuracy of the data. CWZ and WHZ: performed substantive review and revised the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Science and Technology Department of Jilin Province (YDZJ202201ZYTS216).
We extend our sincere appreciation to previous researchers, particularly the HERMES consortium, the OPENGWAS database, the Finnish database, and the UKB database, whose extensive work has provided us with vital genetic data.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1243867/full#supplementary-material
1. Omran AR. The epidemiologic transition—a theory of the epidemiology of population change (reprinted from the milbank memorial fund quarterly. Bul World Health Organ. (2001) 79(2):161–70.
2. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the global burden of disease study 2015. Lancet (2016) 388(10053):1459–544. doi: 10.1016/s0140-6736(16)31012-1
3. Chen CW, Wei JC, Gu J, Yu D. Editorial: advances in pathogenesis, etiology, and therapies for ankylosing spondylitis. Front Immunol. (2021) 12:822582. doi: 10.3389/fimmu.2021.822582
4. Reveille JD, Witter JP, Weisman MH. Prevalence of axial spondylarthritis in the United States: estimates from a cross-sectional survey. Arthritis Care Res (Hoboken). (2012) 64(6):905–10. doi: 10.1002/acr.21621
5. Duba AS, Mathew SD. The seronegative spondyloarthropathies. Prim Care. (2018) 45(2):271–87. doi: 10.1016/j.pop.2018.02.005
6. Ladd JR, Cassidy JT, Martel W. Juvenile ankylosing spondylitis. Arthritis Rheum. (1971) 14(5):579–90. doi: 10.1002/art.1780140505
7. Klingberg E, Sveälv BG, Täng MS, Bech-Hanssen O, Forsblad-d'Elia H, Bergfeldt L. Aortic regurgitation is common in ankylosing spondylitis: time for routine echocardiography evaluation? Am J Med. (2015) 128(11):1244–50. doi: 10.1016/j.amjmed.2015.04.032
8. Wysham KD, Murray SG, Hills N, Yelin E, Gensler LS. Cervical spinal fracture and other diagnoses associated with mortality in hospitalized ankylosing spondylitis patients. Arthritis Care Res (Hoboken). (2017) 69(2):271–7. doi: 10.1002/acr.22934
9. Fernández-Carballido C, Martín-Martínez MA, García-Gómez C, Castañeda S, González-Juanatey C, Sánchez-Alonso F, et al. Impact of comorbidity on physical function in patients with ankylosing spondylitis and psoriatic arthritis attending rheumatology clinics: results from a cross-sectional study. Arthritis Care Res (Hoboken). (2020) 72(6):822–8. doi: 10.1002/acr.23910
10. Essers I, Stolwijk C, Boonen A, De Bruin ML, Bazelier MT, de Vries F, et al. Ankylosing spondylitis and risk of ischaemic heart disease: a population-based cohort study. Ann Rheum Dis. (2016) 75(1):203–9. doi: 10.1136/annrheumdis-2014-206147
11. Stanek A, Cholewka A, Wielkoszyński T, Romuk E, Sieroń K, Sieroń A. Increased levels of oxidative stress markers, soluble Cd40 ligand, and carotid intima-media thickness reflect acceleration of atherosclerosis in male patients with ankylosing spondylitis in active phase and without the classical cardiovascular risk factors. Oxid Med Cell Longevity. (2017) 2017:9712536. doi: 10.1155/2017/9712536
12. Serdaroğlu Beyazal M, Erdoğan T, Türkyılmaz AK, Devrimsel G, Cüre MC, Beyazal M, et al. Relationship of serum osteoprotegerin with arterial stiffness, preclinical atherosclerosis, and disease activity in patients with ankylosing spondylitis. Clin Rheumatol. (2016) 35(9):2235–41. doi: 10.1007/s10067-016-3198-9
13. Ionescu M, Ionescu P, Suceveanu AP, Stoian AP, Motofei I, Ardeleanu V, et al. Cardiovascular risk estimation in young patients with ankylosing spondylitis: a new model based on a prospective study in constanta county, Romania. Exp Ther Med. (2021) 21(5):529. doi: 10.3892/etm.2021.9961
14. Bremander A, Petersson IF, Bergman S, Englund M. Population-based estimates of common comorbidities and cardiovascular disease in ankylosing spondylitis. Arthritis Care Res (Hoboken). (2011) 63(4):550–6. doi: 10.1002/acr.20408
15. Kelty E, Ognjenovic M, Raymond WD, Inderjeeth CA, Keen HI, Preen DB, et al. Mortality rates in patients with ankylosing spondylitis with and without extraarticular manifestations and comorbidities: a retrospective cohort study. J Rheumatol. (2022) 49(7):688–93. doi: 10.3899/jrheum.210909
16. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. (2008) 27(8):1133–63. doi: 10.1002/sim.3034
17. Rosoff DB, Davey Smith G, Mehta N, Clarke TK, Lohoff FW. Evaluating the relationship between alcohol consumption, tobacco use, and cardiovascular disease: a multivariable Mendelian randomization study. PLoS Med. (2020) 17(12):e1003410. doi: 10.1371/journal.pmed.1003410
18. Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The Mr-base platform supports systematic causal inference across the human phenome. Elife. (2018) 7. doi: 10.7554/eLife.34408
19. Cortes A, Hadler J, Pointon JP, Robinson PC, Karaderi T, Leo P, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet. (2013) 45(7):730–8. doi: 10.1038/ng.2667
20. Feng R, Lu M, Xu J, Zhang F, Yang M, Luo P, et al. Pulmonary embolism and 529 human blood metabolites: genetic correlation and two-sample Mendelian randomization study. BMC Genomic Data. (2022) 23(1):69. doi: 10.1186/s12863-022-01082-6
21. Hu S, Xing H, Wang X, Zhang N, Xu Q. Causal relationships between total physical activity and ankylosing spondylitis: a Mendelian randomization study. Front Immunol. (2022) 13:887326. doi: 10.3389/fimmu.2022.887326
22. Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, et al. Phenoscanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. (2019) 35(22):4851–3. doi: 10.1093/bioinformatics/btz469
23. Chen Y, Shen J, Wu Y, Ni M, Deng Y, Sun X, et al. Tea consumption and risk of lower respiratory tract infections: a two-sample Mendelian randomization study. Eur J Nutr. (2023) 62(1):385–93. doi: 10.1007/s00394-022-02994-w
24. Hamilton PK, Lockhart CJ, Quinn CE, McVeigh GE. Arterial stiffness: clinical relevance, measurement and treatment. Clin. Sci. (London, England: 1979). (2007) 113(4):157–70. doi: 10.1042/cs20070080
25. Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol. (2010) 55(13):1318–27. doi: 10.1016/j.jacc.2009.10.061
26. Fung K, Ramírez J, Warren HR, Aung N, Lee AM, Tzanis E, et al. Genome-wide association study identifies loci for arterial stiffness index in 127,121 UK biobank participants. Sci Rep. (2019) 9(1):9143. doi: 10.1038/s41598-019-45703-0
27. Shah S, Henry A, Roselli C, Lin H, Sveinbjörnsson G, Fatemifar G, et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. (2020) 11(1):163. doi: 10.1038/s41467-019-13690-5
28. Skrivankova VW, Richmond RC, Woolf BAR, Davies NM, Swanson SA, VanderWeele TJ, et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomisation (strobe-Mr): explanation and elaboration. BMJ (Clin Res) (2021) 375:n2233. doi: 10.1136/bmj.n2233
29. Morrison J, Knoblauch N, Marcus JH, Stephens M, He X. Mendelian Randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat Genet. (2020) 52(7):740–7. doi: 10.1038/s41588-020-0631-4
30. Zhang G, Feenstra B, Bacelis J, Liu X, Muglia LM, Juodakis J, et al. Genetic associations with gestational duration and spontaneous preterm birth. N Engl J Med. (2017) 377(12):1156–67. doi: 10.1056/NEJMoa1612665
31. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. (2018) 50(5):693–8. doi: 10.1038/s41588-018-0099-7
32. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int J Epidemiol. (2015) 44(2):512–25. doi: 10.1093/ije/dyv080
33. Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat Med. (2017) 36(11):1783–802. doi: 10.1002/sim.7221
34. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. (2016) 40(4):304–14. doi: 10.1002/gepi.21965
35. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the Mr-egger method. Eur J Epidemiol. (2017) 32(5):377–89. doi: 10.1007/s10654-017-0255-x
36. Schieir O, Tosevski C, Glazier RH, Hogg-Johnson S, Badley EM. Incident myocardial infarction associated with major types of arthritis in the general population: a systematic review and meta-analysis. Ann Rheum Dis. (2017) 76(8):1396–404. doi: 10.1136/annrheumdis-2016-210275
37. Golub IS, Termeie OG, Kristo S, Schroeder LP, Lakshmanan S, Shafter AM, et al. Major global coronary artery calcium guidelines. JACC Cardiovasc Imaging. (2023) 16(1):98–117. doi: 10.1016/j.jcmg.2022.06.018
38. Demir K, Avcı A, Ergulu Esmen S, Tuncez A, Yalcın MU, Yılmaz A, et al. Assessment of arterial stiffness and epicardial adipose tissue thickness in predicting the subclinical atherosclerosis in patients with ankylosing spondylitis. Clin Exp Hypertension (New York, NY: 1993). (2021) 43(2):169–74. doi: 10.1080/10641963.2020.1833025
39. Ben Tekaya A, Boukriba S, Fendri A, Rouached L, Saidane O, Bouden S, et al. Endothelial dysfunction and increased carotid intima-media thickness in patients with spondyloarthritis without traditional cardiovascular risk factors. RMD Open. (2022) 8(2). doi: 10.1136/rmdopen-2022-002270
40. Ozdowska P, Wardziak Ł, Kruk M, Kępka C, Kowalik I, Szwed H, et al. Increased prevalence of subclinical coronary atherosclerosis in young patients with ankylosing spondylitis. Polish Arch Int Med. (2018) 128(7-8):455–61. doi: 10.20452/pamw.4300
41. Bai R, Zhang Y, Liu W, Ma C, Chen X, Yang J, et al. The relationship of ankylosing spondylitis and subclinical atherosclerosis: a systemic review and meta-analysis. Angiology. (2019) 70(6):492–500. doi: 10.1177/0003319718814309
42. Yuan Y, Yang J, Zhang X, Han R, Chen M, Hu X, et al. Carotid intima-media thickness in patients with ankylosing spondylitis: a systematic review and updated meta-analysis. J Atheroscler Thromb. (2019) 26(3):260–71. doi: 10.5551/jat.45294
43. Arida A, Protogerou AD, Konstantonis G, Konsta M, Delicha EM, Kitas GD, et al. Subclinical atherosclerosis is not accelerated in patients with ankylosing spondylitis with low disease activity: new data and metaanalysis of published studies. J Rheumatol. (2015) 42(11):2098–105. doi: 10.3899/jrheum.150316
44. Dzieża-Grudnik A, Sulicka J, Strach M, Siga O, Klimek E, Korkosz M, et al. Arterial stiffness is not increased in patients with short duration rheumatoid arthritis and ankylosing spondylitis. Blood Press. (2017) 26(2):115–21. doi: 10.1080/08037051.2016.1232586
45. Toussirot E. The risk of cardiovascular diseases in axial spondyloarthritis. current insights. Front Med (Lausanne). (2021) 8:782150. doi: 10.3389/fmed.2021.782150
46. Zhao SS, Robertson S, Reich T, Harrison NL, Moots RJ, Goodson NJ. Prevalence and impact of comorbidities in axial spondyloarthritis: systematic review and meta-analysis. Rheumatology (Oxford, England). (2020) 59(Suppl4):iv47–57. doi: 10.1093/rheumatology/keaa246
47. Liew JW, Reveille JD, Castillo M, Sawhney H, Naovarat BS, Heckbert SR, et al. Cardiovascular risk scores in axial spondyloarthritis versus the general population: a cross-sectional study. J Rheumatol. (2021) 48(3):361–6. doi: 10.3899/jrheum.200188
48. Ortolan A, Lorenzin M, Felicetti M, Ramonda R. Do obesity and overweight influence disease activity measures in axial spondyloarthritis? A systematic review and meta-analysis. Arthritis Care Res (Hoboken). (2021) 73(12):1815–25. doi: 10.1002/acr.24416
49. Liew JW, Gianfrancesco MA, Heckbert SR, Gensler LS. Relationship between body mass index, disease activity, and exercise in ankylosing spondylitis. Arthritis Care Res (Hoboken). (2022) 74(8):1287–93. doi: 10.1002/acr.24565
50. Shan J, Zhang J. Impact of obesity on the efficacy of different biologic agents in inflammatory diseases: a systematic review and meta-analysis. Joint Bone Spine. (2019) 86(2):173–83. doi: 10.1016/j.jbspin.2018.03.007
51. Klemz BN, Reis-Neto ET, Jennings F, Siqueira US, Klemz FK, Pinheiro HH, et al. The relevance of performing exercise test before starting supervised physical exercise in asymptomatic cardiovascular patients with rheumatic diseases. Rheumatology (Oxford, England). (2016) 55(11):1978–86. doi: 10.1093/rheumatology/kew277
52. Nikiphorou E, Santos EJF, Marques A, Böhm P, Bijlsma JW, Daien CI, et al. 2021 Eular recommendations for the implementation of self-management strategies in patients with inflammatory arthritis. Ann Rheum Dis. (2021) 80(10):1278–85. doi: 10.1136/annrheumdis-2021-220249
53. Roelsgaard IK, Esbensen BA, Østergaard M, Rollefstad S, Semb AG, Christensen R, et al. Smoking cessation intervention for reducing disease activity in chronic autoimmune inflammatory joint diseases. Cochrane Database Syst Rev. (2019) 9(9):Cd012958. doi: 10.1002/14651858.CD012958.pub2
54. Jones GT, Ratz T, Dean LE, Macfarlane GJ, Atzeni F. Disease severity in never smokers, ex-smokers, and current smokers with axial spondyloarthritis: results from the Scotland registry for ankylosing spondylitis. Arthritis Care Res (Hoboken). (2017) 69(9):1407–13. doi: 10.1002/acr.23157
55. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. (2014) 11(5):255–65. doi: 10.1038/nrcardio.2014.28
56. Neumann FJ, Ott I, Gawaz M, Richardt G, Holzapfel H, Jochum M, et al. Cardiac release of cytokines and inflammatory responses in acute myocardial infarction. Circulation. (1995) 92(4):748–55. doi: 10.1161/01.cir.92.4.748
57. Nakano A, Matsumori A, Kawamoto S, Tahara H, Yamato E, Sasayama S, et al. Cytokine gene therapy for myocarditis by in vivo electroporation. Hum Gene Ther. (2001) 12(10):1289–97. doi: 10.1089/104303401750270940
58. Esteve E, Castro A, López-Bermejo A, Vendrell J, Ricart W, Fernández-Real JM. Serum interleukin-6 correlates with endothelial dysfunction in healthy men independently of insulin sensitivity. Diabetes Care. (2007) 30(4):939–45. doi: 10.2337/dc06-1793
59. Azevedo VF, Pecoits-Filho R. Atherosclerosis and endothelial dysfunction in patients with ankylosing spondylitis. Rheumatol Int. (2010) 30(11):1411–6. doi: 10.1007/s00296-010-1416-3
60. Divecha H, Sattar N, Rumley A, Cherry L, Lowe GD, Sturrock R. Cardiovascular risk parameters in men with ankylosing spondylitis in comparison with non-inflammatory control subjects: relevance of systemic inflammation. Clin Sci. (London, England: 1979). (2005) 109(2):171–6. doi: 10.1042/cs20040326
61. Masi AT, Fessler SL, Brezka ML, Wang Y, Donohue SE. Systematic review and meta-analysis of individual serum lipids and analysis of lipid ratios in ankylosing spondylitis and healthy control cohorts: significantly lower mean hdl-cholesterol level in ankylosing spondylitis cohorts. Clin Exp Rheumatol. (2023) 41(9):1862–74. doi: 10.55563/clinexprheumatol/gtcard
62. Regel A, Sepriano A, Baraliakos X, van der Heijde D, Braun J, Landewé R, et al. Efficacy and safety of non-pharmacological and non-biological pharmacological treatment: a systematic literature review informing the 2016 update of the asas/eular recommendations for the management of axial spondyloarthritis. RMD Open. (2017) 3(1):e000397. doi: 10.1136/rmdopen-2016-000397
63. Evaluation CFD. Drug safety and availability—Fda drug safety communication: Fda strengthens warning that non-aspirin nonsteroidal anti-inflammatory drugs (Nsaids) can Cause Heart attacks or strokes. (2023).
64. Jüni P, Nartey L, Reichenbach S, Sterchi R, Dieppe PA, Egger M. Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet. (2004) 364(9450):2021–9. doi: 10.1016/s0140-6736(04)17514-4
65. Roubille C, Richer V, Starnino T, McCourt C, McFarlane A, Fleming P, et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann Rheum Dis. (2015) 74(3):480–9. doi: 10.1136/annrheumdis-2014-206624
66. Schjerning Olsen AM, Fosbøl EL, Lindhardsen J, Folke F, Charlot M, Selmer C, et al. Duration of treatment with nonsteroidal anti-inflammatory drugs and impact on risk of death and recurrent myocardial infarction in patients with prior myocardial infarction: a nationwide cohort study. Circulation. (2011) 123(20):2226–35. doi: 10.1161/circulationaha.110.004671
67. Arfè A, Scotti L, Varas-Lorenzo C, Nicotra F, Zambon A, Kollhorst B, et al. Non-steroidal anti-inflammatory drugs and risk of heart failure in four European countries: nested case-control study. BMJ (Clin Res). (2016) 354:i4857. doi: 10.1136/bmj.i4857
68. Sørensen R, Hansen ML, Abildstrom SZ, Hvelplund A, Andersson C, Jørgensen C, et al. Risk of bleeding in patients with acute myocardial infarction treated with different combinations of aspirin, clopidogrel, and vitamin K antagonists in Denmark: a retrospective analysis of nationwide registry data. Lancet. (2009) 374(9706):1967–74. doi: 10.1016/s0140-6736(09)61751-7
69. Randomised A. Blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (caprie). caprie steering committee. Lancet. (1996) 348(9038):1329–39. doi: 10.1016/s0140-6736(96)09457-3
70. Ward MM. Lifetime risks of valvular heart disease and pacemaker use in patients with ankylosing spondylitis. J Am Heart Assoc. (2018) 7(20):e010016. doi: 10.1161/jaha.118.010016
71. O'Neill TW, Bresnihan B. The heart in ankylosing spondylitis. Ann Rheum Dis. (1992) 51(6):705–6. doi: 10.1136/ard.51.6.705
72. Baniaamam M, Heslinga SC, Konings TC, Handoko ML, Kamp O, van Halm VP, et al. Aortic root diameter is associated with hla-B27: identifying the patient with ankylosing spondylitis at risk for aortic valve regurgitation. Rheumatol Int. (2022) 42(4):683–8. doi: 10.1007/s00296-021-05040-w
73. Reinhardt A, Yevsa T, Worbs T, Lienenklaus S, Sandrock I, Oberdorfer L, et al. Interleukin-23-Dependent/T cells produce interleukin-17 and accumulate in the enthesis. Aortic valve, and ciliary body in mice. Arthritis Rheumatol. (2016) 68(10):2476–86. doi: 10.1002/art.39732
74. Siao WZ, Liu CH, Wang YH, Wei JCC, Jong GP. Increased risk of valvular heart disease in patients with ankylosing spondylitis: a nationwide population-based longitudinal cohort study. Ther Adv Musculoskelet Dis. (2021) 13:1759720x211021676. doi: 10.1177/1759720(211021676
75. Moyssakis I, Gialafos E, Vassiliou VA, Boki K, Votteas V, Sfikakis PP, et al. Myocardial performance and aortic elasticity are impaired in patients with ankylosing spondylitis. Scand J Rheumatol. (2009) 38(3):216–21. doi: 10.1080/03009740802474672
76. Bulkley BH, Roberts WC. Ankylosing spondylitis and aortic regurgitation. Description of the characteristic cardiovascular lesion from study of eight necropsy patients. Circulation. (1973) 48(5):1014–27. doi: 10.1161/01.Cir.48.5.1014
77. Hong C, Kwan YH, Leung YY, Lui NL, Fong W. Comparison of ankylosing spondylitis and non-radiographic axial spondyloarthritis in a multi-ethnic Asian population of Singapore. Int J Rheum Dis. (2019) 22(8):1506–11. doi: 10.1111/1756-185x.13603
78. Thériault S, Dina C, Messika-Zeitoun D, Le Scouarnec S, Capoulade R, Gaudreault N, et al. Genetic association analyses highlight Il6, Alpl, and Nav1 as 3 new susceptibility genes underlying calcific aortic valve stenosis. Circ Genomic Precision Med. (2019) 12(10):e002617. doi: 10.1161/circgen.119.002617
Keywords: epidemiology, Mendelian randomization, cardiovascular disease, ankylosing spondylitis, genome-wide association study
Citation: Zhong Y, Chen Y, Zhang X, Cai W, Zhao C and Zhao W (2023) No evidence of a causal relationship between ankylosing spondylitis and cardiovascular disease: a two-sample Mendelian randomization study. Front. Cardiovasc. Med. 10:1243867. doi: 10.3389/fcvm.2023.1243867
Received: 22 June 2023; Accepted: 2 October 2023;
Published: 12 October 2023.
Edited by:
James Cheng-Chung Wei, Chung Shan Medical University Hospital, TaiwanReviewed by:
Wun Zhih Siao, Chung Shan Medical University Hospital, Taiwan© 2023 Zhong, Chen, Zhang, Cai, Zhao and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: ChangWei Zhao emhhb2N3QGNjdWNtLmVkdS5jbg== WenHai Zhao endoOTg5OUAxNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.