 Jiaying Luo
Jiaying Luo Zhiwei He
Zhiwei He Qingwen Li2
Qingwen Li2 Yuli Cai
Yuli Cai Xuan Niu
Xuan Niu Zhaohui Zhang
Zhaohui Zhang- 1Department of Neurology, Renmin Hospital of Wuhan University, Wuhan, China
- 2Department of Anesthesiology, Renmin Hospital of Wuhan University, Wuhan, China
- 3Department of Endocrinology, Renmin Hospital of Wuhan University, Wuhan, China
Adipokines are biologically active factors secreted by adipose tissue that act on local and distant tissues through autocrine, paracrine, and endocrine mechanisms. However, adipokines are believed to be involved in an increased risk of atherosclerosis. Classical adipokines include leptin, adiponectin, and ceramide, while newly identified adipokines include visceral adipose tissue-derived serpin, omentin, and asprosin. New evidence suggests that adipokines can play an essential role in atherosclerosis progression and regression. Here, we summarize the complex roles of various adipokines in atherosclerosis lesions. Representative protective adipokines include adiponectin and neuregulin 4; deteriorating adipokines include leptin, resistin, thrombospondin-1, and C1q/tumor necrosis factor-related protein 5; and adipokines with dual protective and deteriorating effects include C1q/tumor necrosis factor-related protein 1 and C1q/tumor necrosis factor-related protein 3; and adipose tissue-derived bioactive materials include sphingosine-1-phosphate, ceramide, and adipose tissue-derived exosomes. However, the role of a newly discovered adipokine, asprosin, in atherosclerosis remains unclear. This article reviews progress in the research on the effects of adipokines in atherosclerosis and how they may be regulated to halt its progression.
1. Introduction
Atherosclerosis (AS) is the leading cause of death from cardiovascular disease (CVD) in Western countries (1). It is characterized by vascular lesion formation, involving dysfunction of vessel wall cells and lipid deposition due to dyslipidemia (2, 3). Obesity, diabetes, and hypertension, which are major cardiovascular risk factors, induce endothelial injury leading to various proatherogenic effects (4). These effects include increased platelet adhesion and aggregation, monocyte adhesion/infiltration, accumulation of oxidatively modified lipoproteins, and vasoconstriction (5). In humans, the initial stage of de novo atherosclerotic plaque formation is known as adaptive intimal thickening. It is primarily characterized by the migration and proliferation of vascular smooth muscle cells (VSMCs) (5), during which VSMC transitions from a contracted to a proliferative state. At the late atheroma, VSMCs could migrate to the surface, forming a “fibrous cap” that protects the lesion from rupture (6). Early pathological development is facilitated by VSMC proliferation and migration, while VSMC apoptosis, cellular senescence, and the presence of macrophage-like cells derived from VSMCs may contribute to inflammation (7). The hallmark of AS lesions is the formation of foam cells. Differentiated macrophages express scavenger receptors that recognize and take up oxidized low-density lipoprotein (ox-LDL) (8). Macrophages that accumulate excessive lipids from ox-LDL, transforming into foam cells. In addition to the well-established involvement of lipid accumulation in AS pathogenesis, emerging evidence suggests that adipose tissue, once perceived as a passive energy storage depot, plays an active role in AS by secreting bioactive proteins or products called adipokines (9) (Table 1).
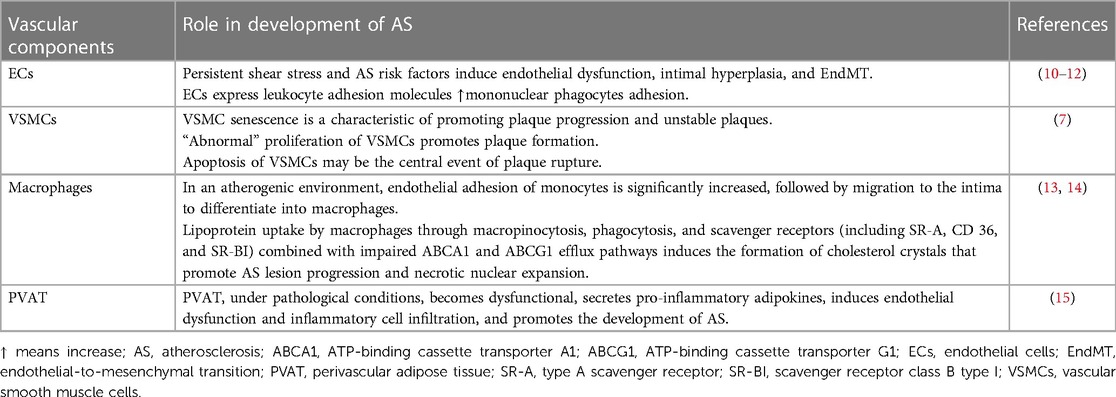
Table 1. Vascular components’ role in the development of AS.
As one of the largest organs in the body, white adipose tissue (WAT) is mainly composed of adipocytes, blood vessels, lymphocytes, and stem cells, and occupies a central position in energy regulation and metabolism. In the human body, WAT is mainly distributed in subcutaneous, visceral, and gonadal areas. The percentage of WAT in body weight and cell size varies greatly among individuals of different body sizes, from approximately 9%–28% in lean adults to 40%–70% in obese people (16). This disparity primarily arises from the expansion of subcutaneous and visceral WAT depots (17). Notably, there exists considerable heterogeneity in fat cell size, both within an individual and across different individuals. Generally, during periods of weight gain, fat cell size tends to increase, whereas weight loss is associated with a reduction in fat cell size (18). White adipocytes contain large single-compartment lipid droplets with fewer mitochondria, which store excess energy in the body and respond to the body's energy and nutrient needs at all times. Macrophages are the most abundant immune cells in the adipose tissue of obese individuals, and their recruitment and proliferation during high-caloric feeding are usually associated with adipose tissue inflammation and insulin resistance (19, 20). Adipose tissue macrophages in lean organisms tend to be inflammation-ameliorating phenotype, whereas the phenotype of adipose tissue macrophages in obese individuals is more pro-inflammatory (21). The activation of CD8+ effector T cells and the recruitment of monocytes and macrophages in obese adipose tissue contribute to the accumulation of adipose tissue macrophages and the promotion of inflammation within the adipose tissue (22). WAT has an endocrine function and secretes characteristic adipokines such as leptin, adiponectin, omentin, and visceral adipose tissue-derived serpin (vaspin). In the obesity model, adipocyte hypertrophy occurred before hyperplasia, with increased secretion of leptin, ceramide, and vaspin and decreased secretion of adiponectin and omentin in hypertrophied adipocytes. One distinguishing feature of hypertrophic WAT is the presence of enlarged adipocytes, which occurs due to lipid accumulation within the cells. In contrast, hyperplastic WAT is characterized by a higher abundance of smaller adipocytes compared to normal or hypertrophic WAT (18). Inducing a brown adipose tissue (BAT) phenotype in WAT is called “browning,” that is, beige adipose tissue (BeAT).
BAT is named for its brown appearance. Color reflects the number of iron-containing mitochondria in adipocytes. The darker the color, the more mitochondria there are (23). In the human body, BAT is mainly found in the interscapular region, back of the neck, and mediastinum. BAT is most abundant in newborns, whereas adults have less than 2% of their body weight in BAT. BAT function decreases with age. Nevertheless, BAT can be restored to its young state when necessary in response to the body's needs (24). Unlike white adipocytes, which are microscopically visible as tiny multi-compartmented lipid droplets, brown adipocytes partly originate from myogenic factor 5 positive progenitor cells and contain more mitochondria than white adipocytes (25). The abundance of mitochondria allows brown adipocytes to achieve adaptive thermogenesis via fatty acid uncoupling and oxidative phosphorylation (26). In addition to this mechanism, alternative pathways for thermogenesis include the succinate cycle, creatine cycle, calcium cycle, fatty acid cycle, and ATP/ADP carrier-mediated thermogenesis (27, 28). Apart from its thermogenic function, BAT and BeAT play a role in metabolic regulation through the secretion of adipokines. Notable adipokines secreted by BAT include neuregulin 4 (NRG4), growth differentiation factor 15 (GDF-15), and fibroblast growth factor 21 (FGF21). While the precise impact of these adipokines on AS remains to be fully elucidated, they hold potential for further exploration in AS research.
Adipose tissue is divided into WAT, BAT and BeAT, the adipocytes of which exhibit different morphological and functional characteristics. BeAT is a reversible state between WAT and BAT and is homologous with WAT,which is considered “browned”WAT (29). Perivascular adipose tissue (PVAT), a connective tissue that envelopes the adventitia of the blood vessels and provide mechanical support, serves as a distinctive form of adipose tissue (30). Perivascular adipose tissue could undergo phenotype change and participate in vascular inflammation and remodeling during atherosclerosis (31). Adipose tissue is now recognized as the largest endocrine organ in the body and a metabolically active organ that plays a vital role in regulating the balance of the systemic energy environment (32). The dysfunction of adipose tissue is directly related to various metabolic diseases, including obesity, CVD, and type 2 diabetes (33, 34). Adipokines are bioactive proteins or products secreted from adipose tissues, and they could exert their effect on local and distant tissues through autocrine, paracrine, and endocrine mechanisms (19, 35). Adipokines could also bind to surface receptors on endothelial cells (ECs), VSMCs, and macrophages, influencing their behavior and modulating AS lesion progression. While adipokines such as adiponectin, NRG4, and C1q/tumor necrosis factor-related protein 9 (CTRP9) have been found to play a protective role in AS, others like leptin, resistin, and thrombospondin-1 (TSP-1) can exacerbate disease progression. Adipokines exert a complex regulatory role in the development of AS. Understanding the beneficial and detrimental effects of specific adipokines can not only serve as biomarkers to predict AS outcomes but also aid in identifying potential therapeutic targets for AS treatment.
Adipokines produced by adipose tissue are biologically active. Obese adipose tissue alters the balance of these adipokines and is associated with accelerated CVD. In this review, we classified the role of adipokines in AS progression into five categories: protective, deteriorating, dual-acting, indeterminate and adipose tissue-derived bioactive materials, and summarized the role of these adipokines on AS in Table 2 and Figure 1.
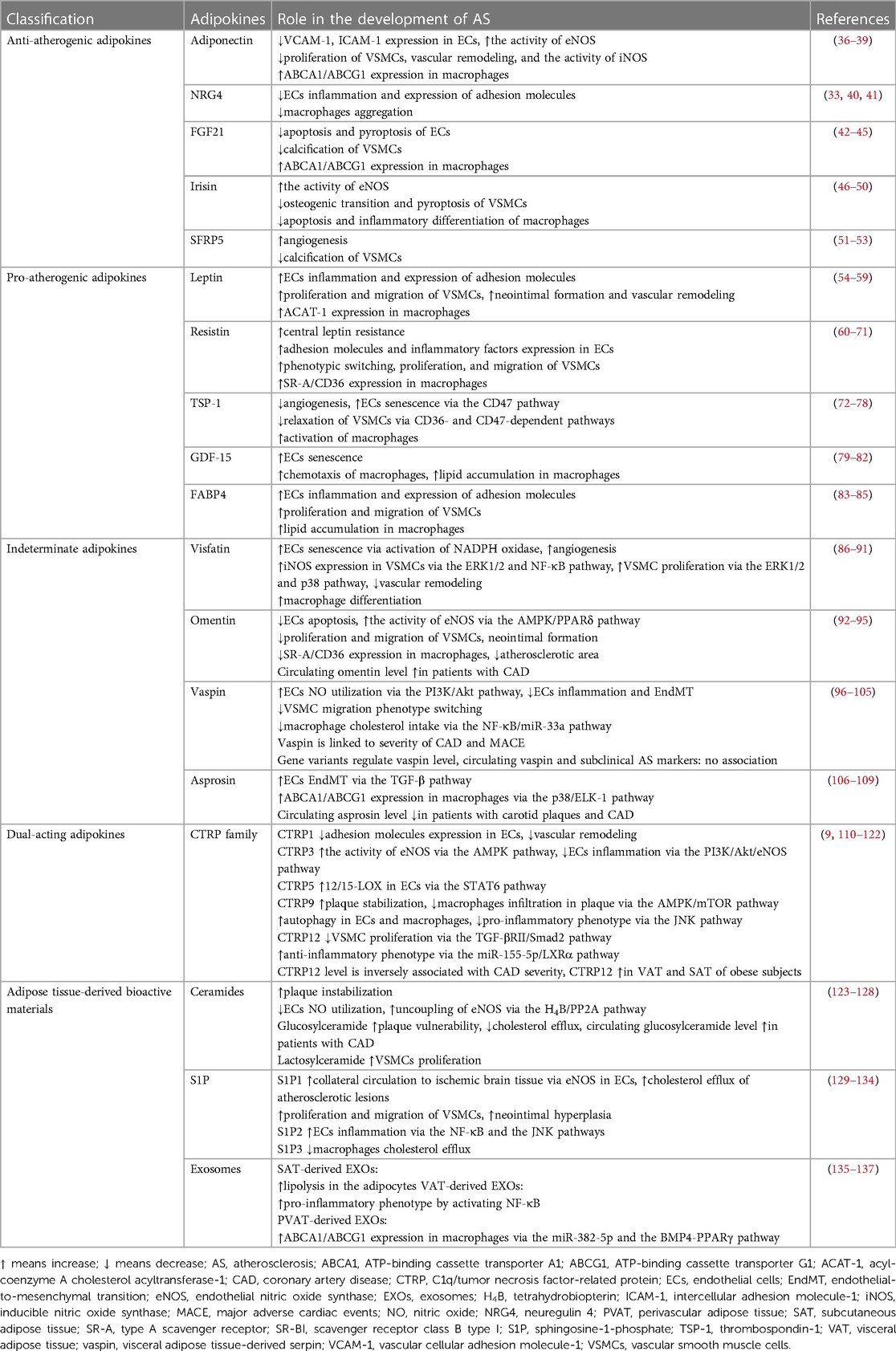
Table 2. Adipokines for atherosclerosis.
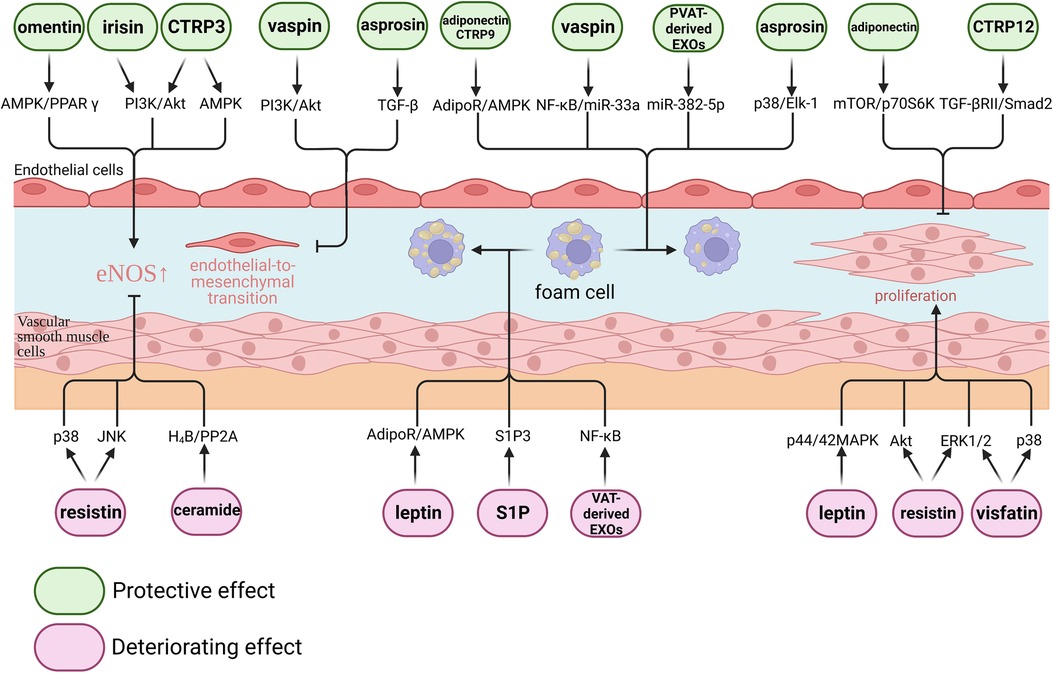
Figure 1. Effect of adipokines on atherosclerosis. Adipokines play complex regulatory roles in atherosclerosis. From a molecular perspective, certain adipokines such as omentin, irisin, CTRP3, vaspin, and asprosin have been shown to exert protective effects on endothelial cells. Conversely, resistin and ceramide have been found to have detrimental effects on endothelial cells. Adiponectin, CTRP9, vaspin, asprosin, and perivascular adipose tissue-derived EXOs have demonstrated the ability to inhibit macrophage foam cell formation. On the other hand, leptin, S1P, and visceral adipose tissue-derived EXOs have been found to promote macrophage foam cell formation. Additionally, adiponectin and CTRP12 have been shown to inhibit vascular smooth muscle cell proliferation, while leptin, resistin, and visfatin have been found to promote vascular smooth muscle cell proliferation.
2. Protective adipokines for AS
2.1. Adiponectin
Adiponectin is a protein-like adipokine secreted by adipocytes (138). It is secreted as multimers of different molecular weights: hexameric, trimeric, and globular. Adiponectin binds to receptors to regulate lipid metabolism and insulin sensitivity. Adiponectin is composed of a globular domain and a collagen-like domain. It interacts with three types of receptors: adiponectin receptors (AdipoRs), calreticulin, and T-cadherin (139). Notably, T-cadherin exhibits high expression levels in the cardiovascular system (140). The binding of the globular domain of adiponectin to T-cadherin has been shown to have beneficial effects on intimal hyperplasia and AS (141). Additionally, the adiponectin/T-cadherin system plays a role in promoting the synthesis of exosomes, small extracellular vesicles, while simultaneously reducing the release of cellular ceramide (142). This dual mechanism contributes to the modulation of insulin sensitivity, thereby potentially mitigating the development of insulin resistance (143). There are two AdipoR types, AdipoR1 and AdipoR2 (144). AdipoR1 and AdipoR2 may have opposite effects on obesity resistance and glucose clearance by activating 5′ AMP-activated protein kinase (AMPK)α1 and AMPKα2, respectively.
Decreased plasma adiponectin levels are associated with an increased risk of intima-media thickness (145) and intracranial AS stenosis (146). Gasbarrino et al. analyzed plasma adiponectin in patients with severe carotid AS undergoing carotid endarterectomy (147). They found that high circulating adiponectin levels were associated with a lower risk of atherosclerotic cardiovascular events. Adiponectin levels are lower in unstable plaques than in stable plaques (148), suggesting that adiponectin levels are negatively associated with the risk of CVD events. Smoking is an essential trigger of AS, and nicotine in tobacco accelerates the progression of AS by reducing adiponectin expression in adipocytes via ATP-dependent potassium channels (149). Physiological concentrations of adiponectin can inhibit the expression of cell adhesion molecules vascular cellular adhesion molecule-1 (VCAM-1), E-selectin, and intercellular adhesion molecule-1 (ICAM-1) on EC (36). Inducible nitric oxide synthase (iNOS) is a nitric oxide (NO) and peroxynitrite-forming enzyme that is overproduced in vascular diseases such as AS or diabetes-related vasculopathy and promotes vascular inflammation and endothelial dysfunction. The globular domain treatment of adiponectin significantly increased endothelial nitric oxide synthase (eNOS) activity but decreased iNOS activity in hyperlipidemic vessels (37), which suggested that adiponectin protects the endothelium from hyperlipidemia through multiple mechanisms. Adiponectin inhibits the mammalian target of rapamycin (mTOR)/p70S6K signaling-mediated proliferation of VSMCs in a receptor-activation-AMPK-dependent (38) or AMPK-independent (39) manner. In addition, adiponectin attenuates angiotensin II–induced vascular remodeling through NO–dependent inhibition of the RhoA/Rho-associated protein kinase pathway and reactive oxygen species production in vascular smooth muscle (150). Adiponectin also regulates lipid efflux in macrophages. Adiponectin reduces lipid accumulation by promoting ATP-binding cassette transporter A1 (ABCA1)- and ATP-binding cassette transporter G1 (ABCG1)-dependent cholesterol efflux through activation of the peroxisome proliferator-activated receptor (PPAR)γ/liver × receptor α signaling pathway (151, 152). Activation of the AdipoR1/AMPK pathway in macrophages reduces apoptosis and improves cholesterol efflux from foam cells, thereby reducing foam cell cholesterol and triglyceride accumulation (153, 154).
2.2. Neuregulin 4
NRG4, a novel adipokine, is a member of the NRG family of neuromodulatory proteins. It is mainly expressed in specific peripheral tissues, with the highest expression levels observed in BAT. Li et al. reported that NRG4 could regulate glucose metabolism and improve insulin resistance (155); however, NRG4 has been less studied in AS. NRG4 expression is upregulated in PVAT after vascular injury (33), which mediates ECs to attenuate the expression of inflammatory factors, tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β), and adhesion molecules VCAM-1 and ICAM-1 through the protein Kinase B (Akt)/nuclear factor kappa-B (NF-κB) pathway (40) and inhibits leukocyte migration to the subintima and macrophage accumulation within AS plaques (41). NRG4 also attenuates the levels of inflammatory cytokines in classically activated macrophages (33). The current study suggests that NRG4 inhibits AS development and exerts atheroprotective effects on AS lesions.
2.3. FGF21
FGF21 is a signaling protein synthesized in WAT and BAT (156, 157). Exercise can increase plasma levels of FGF21, primarily due to hepatic secretion (158). FGF21 binds to FGF receptors, with the highest affinity for FGF receptors 1 subtype, and β-klotho serves as an essential coreceptor (159). FGF21 has the capacity to reduce fat mass, decrease insulin resistance, and lower plasma glucose and triglyceride levels. Research on FGF21 in adipocytes has primarily focused on human and mouse adipocytes. FGF21 can stimulate glucose uptake in adipocytes by inducing the expression of glucose transporter 1. This process operates independently of insulin and relies on the extracellular regulated protein kinases 1/2 (ERK1/2) signaling pathway and the activation of the serum response factor Ets-like protein 1 (160, 161). FGF21 can stimulate adipocytes to produce and secrete adiponectin through the induction of adiponectin gene expression and PPARγ-dependent mechanisms (162). Additionally, FGF21 can induce browning and adaptive thermogenesis in adipose tissue through various mechanisms, including the induction of PPARγ coactivator 1α and the chemokine C–C motif chemokine ligand 11 (163).
FGF21 exhibits a protective effect on AS. It can inhibit EC apoptosis by suppressing the Fas signaling pathway (42) and inhibit the activation of the NLRP3 inflammasome, thereby preventing EC pyroptosis (43). Aerobic exercise can promote this process by increasing FGF21 levels and downregulating NLRP3 expression (164). Furthermore, FGF21 can promote cholesterol efflux by inducing the expression of ABCA1 and ABCG1 in foam cells and reduce cholesterol accumulation in foam cells through AMPK-mediated autophagy (44, 165). FGF21 also inhibits the calcification of VSMCs (45). These findings suggest that FGF21 may be a potential therapeutic target for preventing and treating AS by modulating EC function, cholesterol metabolism, and vascular calcification.
2.4. Irisin
Irisin, an adipomyokine synthesized by skeletal muscle and adipose tissue (166, 167), results from the proteolytic cleavage of membrane-bound FNDC5 (168). It plays a crucial role in regulating energy metabolism and improving insulin resistance by binding to various receptors, including fibroblast growth factor receptors and hemojuvelin (169). Additionally, irisin promotes mitochondrial synthesis and induces browning of WAT (168). Its expression in adipose tissue is decreased in obese individuals (170). Serum irisin levels are significantly lower in patients with coronary artery disease (CAD) and ischemic stroke (171–173), making it a potential predictive marker for early CVDs. Irisin enhances lipid metabolism by facilitating the transport of biliary cholesterol and fecal cholesterol excretion (174). In the experimental model, Irisin-ApoE−/− mice are a strain obtained by crossing Irisin transgenic mice with ApoE−/− mice. Compared to ApoE−/− mice, an improvement in hyperlipidemia was observed in Irisin-ApoE−/− mice, and the irisin levels were negatively correlated with high-density lipoprotein cholesterol (175). Irisin exerts a protective effect against vascular injury and ECs inflammation induced by ox-LDL (176, 177). The protective effect of irisin on ECs is mediated through the activation of the AMPK-PI3K-Akt-eNOS signaling pathway (46). Moreover, irisin reduces macrophage apoptosis induced by ox-LDL, potentially through the inhibition of endoplasmic reticulum stress signaling pathways (47). Irisin also promotes the anti-inflammatory differentiation of macrophages by activating JAK2-STAT6-dependent signaling (48). Additionally, irisin mitigates vascular calcification by suppressing the osteogenic transition and pyroptosis of vascular smooth muscle cells (49, 50). In conclusion, irisin, an adipomyokine with diverse physiological functions, demonstrates significant potential as a biomarker and therapeutic target in various metabolic and CVDs. However, further research is needed to fully elucidate the underlying mechanisms and explore the clinical implications of irisin in human health and disease.
2.5. Secreted frizzled-related protein 5
Secreted frizzled-related protein 5 (SFRP5) is an adipokine synthesized by white adipocytes (178). Its expression level is disrupted in obesity under metabolic stress conditions. Studies by Ouchi et al. have demonstrated that SFRP5 expression in adipose tissue is reduced in obesity (179). Functionally, SFRP5 binds to Wnt ligands, thereby interfering with Wnt signaling pathway transduction, which is crucial for promoting adipogenesis (180). Emerging research has consistently shown decreased circulating levels of SFRP5 and increased levels of Wnt5a in patients with CAD and obesity when compared to healthy controls (181, 182). The decrease in circulating SFRP5 levels may serve as an indication that the clearance function of SFRP5 during the obesity stage can still be compensated. By binding to the Wnt ligand Wnt5a, SFRP5 inhibits the activation of the Wnt non-canonical pathway, subsequently promoting inflammation in macrophages and adipose tissue (179).
In the context of vascular health, SFRP5 plays a notable role in inhibiting high phosphate-induced calcification of VSMCs. It achieves this through suppression of the Wnt/β-Catenin pathway and the Wnt3a-mediated signaling (51, 52). In addition to its impact on vascular health, SFRP5 also exhibits angiogenic properties in human umbilical vein ECs by inhibiting the Wnt5a/c-Jun N-terminal kinase (JNK) signaling pathway (53). Furthermore, SFRP5 has been observed to mitigate apoptosis induced by oxidative stress in human aortic ECs (183). The aforementioned findings shed light on the complex involvement of SFRP5 in adipose tissue regulation, metabolic disorders, vascular calcification, and EC function, contributing to our understanding of the intricate interplay between adipokines and physiological processes.
3. Deteriorating adipokines for AS
3.1. Leptin
Leptin is a pleiotropic hormone secreted by adipocytes, involved in various biological processes, and including inflammatory responses, immune function, and regulation of biological behavior and metabolism. Bäckdahl et al. classified human WAT into three types of mature adipocytes based on distinct transcriptional profiles and spatial arrangement: AdipoLEP, AdipoPLIN, and AdipoSAA. Among these, adipocytes expressing the marker gene LEP, which is associated with the synthesis of the adipokine leptin, were categorized as AdipoLEP (184). Leptin synthesis is regulated by the lipid content in adipocytes, the Lepob gene, and adipocyte size (185, 186). There are six subtypes of leptin receptors (LepRa to LepRf) (187), and leptin acts on receptors in the hypothalamus to regulate biological behavior, food intake, and indirectly regulate glucolipid metabolism. Thus, ob/ob mice (leptin deficient) and db/db mice (leptin receptor deficient) are commonly used models of hyperglycemia and obesity. The same receptors that respond to leptin are also present in the vasculature. In adipose metabolism disorders with reduced serum leptin levels, leptin treatment ameliorates the endothelial-to-mesenchymal transition (188), suggesting an endothelial protective effect of leptin at physiological concentrations.
Central leptin resistance and preservation of peripheral vascular leptin responsiveness allow high leptin concentrations to induce vascular dysfunction (186). A high leptin concentration increases ERK1/2 phosphorylation and NF-κB activation in ECs (54), which leads to increased secretion of the inflammatory factor TNF-α, expression of the cell adhesion molecule VCAM-1 (55), and endothelial leptin resistance (186), disrupting endothelial barrier function. It also exacerbates neointimal growth and vascular remodeling by promoting VSMCs proliferation and metalloproteinase-9 expression that induces migration (56, 57). However, in apolipoprotein E deficient (ApoE−/−) mice, leptin-induced neointima formation was entirely blocked (58), indicating that ApoE mediates leptin-induced neointima formation. Additionally, leptin promotes foam cell formation. High leptin concentrations upregulate the expression of acyl-coenzyme A cholesterol acyltransferase-1 in acetylated LDL-induced macrophages, increasing intracellular cholesteryl ester accumulation and promoting foam cell formation (59).
One study demonstrated that the decreased expression of type A scavenger receptor (SR-A) and platelet glycoprotein 4 (CD36) in macrophages of ob/ob mice resulted in reduced macrophage cholesterol accumulation and decreased foam cell formation (189). However, another study reported that CD36 expression was upregulated in macrophages of ob/ob mice in the presence of defective insulin signaling, leading to increased macrophage foaminess (190). This discrepancy may be because different cellular microenvironments interfere with the macrophage phenotype. In conclusion, high leptin levels can disrupt ECs barrier function, promote VSMCs proliferation and migration, and accelerate AS progression by exacerbating lipid accumulation in foam cells.
3.2. Resistin
Resistin belongs to a family of cysteine-rich secreted proteins that are almost exclusively derived from adipose tissue in rodents, with elevated adipose expression and serum levels in models of obesity and insulin resistance (191). Resistin is primarily released by visceral WAT macrophage, exacerbating WAT inflammation (192, 193). Additionally, elevated resistin triggers central leptin resistance (194), exacerbating impaired glucose and lipid metabolism. High resistin expression levels in circulating (60) and unstable plaques (195) in carotid plaque subjects suggest that resistin correlates with carotid disease severity and may serve as a potential marker of plaque instability. Resistin was positively correlated with the degree of thoracic aortic calcification (196) and coronary artery calcification (191), all suggesting a significant association between resistin and the severity of CVD.
Resistin directly contributes to EC activation by promoting the release of the endothelin-1 (197). The major cardiovascular risks contribute to an increase in vascular reactive oxygen species production, which in turn promotes the oxidative degradation of tetrahydrobiopterin, a critical cofactor for eNOS. This process leads to eNOS “uncoupling” and reduced NO production (198). Resistin directly induces eNOS downregulation by excessive ROS production and activation of p38 and JNK in human coronary artery ECs (61). Resistin exacerbates monocyte/macrophage adhesion by stimulating EC upregulation of adhesion molecules VCAM-1 and ICAM-1 via the NF-κB pathway and p38 mitogen-activated protein kinase (MAPK) pathway (62, 63, 199). Additionally, resistin enhances inflammatory factors TNF-α and IL-1β expression via the NF-κB signaling pathway in response to stimulation of human coronary artery ECs (64). PVAT-derived resistin-cultured VSMCs upregulate osteopontin, a hallmark of the phenotype of proliferative VSMCs, via the transcription factor AP-1 (200). Resistin acts on VSMCs in a paracrine or endocrine manner to promote VSMC migration via the protein kinase C protein ε pathway (65, 66) and induce human aortic smooth muscle cell proliferation via the ERK 1/2 and Akt signaling pathways (67), respectively. Additionally, protein kinase C protein ε-mediated Nox activation induces ROS production, causing VSMC dysfunction and endothelial proliferation (68). In the absence of natural or modified lipoproteins, resistin induces an increase in cholesterol and triglyceride cell mass in human macrophages (201). In contrast, ox-LDL-treated macrophages significantly increased the expression of resistin mRNA (69). Resistin induces a pro-inflammatory phenotype in macrophages via an NF-κB-dependent pathway, increasing macrophage inflammatory factors IL-1, IL-6, IL-12, and TNF-α, as well as VCAM-1, exacerbating vascular inflammation and promoting AS progression (62, 70, 71). Resistin upregulates macrophage SR-A and CD36 via transcription factors AP-1 and PPARγ (69), respectively. Resistin enhances proteasome-mediated degradation of ABCA1, exacerbating cholesterol uptake and facilitating foam cell formation (202). In summary, resistin directly stimulates VSMC proliferation and migration and promotes the inflammatory phenotype of ECs and macrophages.
3.3. TSP-1
TSP-1 is a multifunctional glycoprotein secreted by platelets (203), macrophages (204), and adipocytes (205). TSP-1 exerts multiple biological effects by binding to extracellular matrix proteins and cell surface receptors to regulate cell and cell-matrix interactions (206). TSP-1 expression was increased in plasma and visceral adipose tissue (VAT) in both diabetic and obese patients and animals (207). Moreover, TSP-1 expression was greater in VAT than in subcutaneous adipose tissue (SAT) in obese subjects (205). Varma et al. found that it was adipose tissue-derived macrophages rather than THP-1-derived macrophages that expressed TSP-1 (205). Hence, TSP-1 is suggested to be a candidate gene for visceral obesity (208). As an essential ligand of TSP-1, CD36 is also a fatty acid translocase, suggesting that TSP-1 may control the metabolism of fatty acids in adipocytes. Loss of TSP-1 reduced macrophage infiltration in adipose tissue, suggesting that TSP-1 may also regulate inflammatory cells infiltrating expanded adipose tissue.
Not only do CVD risk factors promote TSP-1 expression in adipose tissue, but leptin also promotes TSP-1 synthesis in VSMCs, thus hastening AS progression. High leptin concentrations upregulate TSP-1 expression in VSMCs via JAK2 and MAPK-dependent pathways (209). Furthermore, leptin promotes a synergistic interaction between the transcription factor interferon regulatory factor-1 and the cAMP response element binding protein at the promoter of the TSP-1 gene that drives TSP-1 transcription in VSMCs (210). Moreover, TSP-1 deficiency inhibited leptin-induced VSMC dedifferentiation, lipid loading, and increased plaque area (207), suggesting that the pro-AS effect of leptin is mediated via the TSP-1 pathway.
Diabetes upregulates TSP-1-CD47 signaling in ECs to induce senescence and impair angiogenesis (72). Increased TSP-1 can also interfere with ECs chemotaxis, inhibit ECs proliferation, and capillary formation, thereby inhibiting angiogenesis in vitro (73–75). Anti-TSP-1 antibody C6.7 blocks the inhibitory effect of TSP-1 on ECs growth and reendothelialization (211). NO/cGMP signaling enhances intracellular calcium ions and relaxes VSMCs via cGMP-dependent protein kinase (212), while exogenous TSP-1 blocks this diastolic effect of NO via a CD36-dependent pathway (76). Endogenous TSP-1 has the same effect via CD36- and CD47-dependent pathways and also blocks NO-driven ECs adhesion via the CD47 pathway (76, 77). In their study, Moura et al. found that TSP-1 expression increased after carotid ligation and was able to activate VSMCs to induce de novo neointima formation (213). In addition, Li et al. found that TSP-1 activates macrophages through a toll-like receptor 4-dependent pathway (78). All of the above suggest that TSP-1 may be a target of AS lesions and play an aggressive role in AS progression.
3.4. Growth differentiation factor 15
Growth differentiation factor 15 (GDF-15), a member of the TGFβ superfamily, is expressed in various tissues, including the placenta (214), liver (215), kidney, and brown adipose tissue (216). GDF-15 may play a role in the metabolic dysregulation associated with obesity. Studies have shown that serum levels of GDF-15 are elevated in obese and type 2 diabetes patients compared to lean control groups (217). Similarly, circulating levels of GDF-15 are higher in both obese humans and rodents (218). In the latest research by Sjøberg et al., it was discovered that GDF-15 increases local insulin-stimulated glucose uptake in BAT and WAT through β-adrenergic signaling (219). Notably, GDF-15 expression is strongly induced in BAT in response to cold exposure, while circulating levels of GDF-15 remain unchanged (216). The released GDF-15 from brown adipocytes can target macrophages and downregulate the expression of pro-inflammatory genes through paracrine effects (216). Research has also revealed that mice with GDF-15 deficiency exhibit elevated levels of cholesterol and triglycerides in circulation, and this effect is independent of the ApoE gene (220). Moreover, GDF-15 induces anorexia and weight loss through its interaction with the glial-derived neurotrophic factor receptor alpha-like in the central nervous system (221). However, an analysis of the relationship between GDF-15 and weight in non-obese monozygotic twins found a negative correlation between serum levels of GDF-15 and body mass index (217). These findings suggest that the observed increase in GDF-15 in obesity may be a consequence rather than a cause of obesity.
Compared to the normal control group, patients with CAD exhibit significantly elevated circulating levels of GDF-15, suggesting that GDF-15 may serve as an independent predictor of CAD mortality (222). Furthermore, elevated plasma levels of GDF-15 have been identified as an independent predictor of long-term adverse cardiovascular events in patients with moderate CAD (223). These findings indicate that GDF-15 may play a crucial role in the occurrence and progression of CAD. In addition, increased expression of GDF-15 has been observed in atherosclerotic vessels (220), while systemic deficiency of GDF-15 in mice has shown improvements in luminal narrowing in affected vessels (224). Treatment of macrophages with GDF-15 leads to increased levels of autophagy and intracellular lipid accumulation, thereby affecting lipid homeostasis (79). Furthermore, GDF-15 has been shown to exert a pro-inflammatory effect in the progression of AS by mediating the chemotaxis of macrophages through the CCR2 pathway (80). The expression of GDF-15 in ECs is upregulated in response to inflammatory reactions and ROS-mediated cellular senescence (81, 225), and it exerts paracrine effects that impact neighboring non-senescent cells (82). These findings suggest that during vascular stress, the expression of GDF-15 is elevated, and it plays a detrimental role in the progression of AS.
3.5. Fatty acid-binding protein 4
Fatty acid-binding protein 4 (FABP4), primarily expressed in adipocytes and macrophages (226). The expression of FABP4 can be reduced through administration of metformin (227) and atorvastatin (228). Under normal conditions, FABP4 is expressed in capillary and venous ECs, but not in arterial ECs (229). However, damaged arteries can induce ectopic expression of FABP4, although the specific receptors involved in this process remain unclear (230). FABP4 has been implicated in AS, and studies using small-molecule inhibitors of aP2 have demonstrated effective treatment for severe AS and type 2 diabetes in mouse models (231).
FABP4 is closely associated with lipid accumulation in monocytes and macrophages. Inhibition of FABP4 can reduce ox-LDL-induced monocyte adhesion by downregulating the expression of integrin β2, integrin α4, and P-selectin glycoprotein ligand 1 (83). Treatment with FABP4 inhibitors in macrophages significantly reduces cholesterol accumulation by increasing the expression of ABCA1 (231). FABP4 promotes inflammatory responses by inhibiting the PPARγ-LXRα-ABCA1 pathway, leading to cholesterol ester accumulation and foam cell formation, and by activating the JNK-AP-1 signaling pathway (84). Metformin reduces lipid accumulation in macrophages by decreasing FOXO1-mediated FABP4 transcription (227). Regarding FABP4 and VSMCs, it has been found that FABP4 can promote migration and proliferation of coronary artery smooth muscle cells through a MAPK-dependent pathway (85). In the context of ECs, inhibition of FABP4 can reduce ox-LDL-induced adhesion of coronary artery ECs by decreasing the expression of ICAM-1, VCAM-1, and P-selectin (83).
Overall, FABP4 plays a significant role in various cellular processes associated with AS, including monocyte adhesion, cholesterol accumulation, foam cell formation, inflammatory responses, and the behavior of smooth muscle and ECs. Inhibition of FABP4 has shown promise as a potential therapeutic strategy for AS and related complications.
4. Dual-acting adipokines
4.1. CTRP family
C1q/tumor necrosis factor-related proteins (CTRPs) are paralogous homologs of adiponectin. To date, 15 members of this family (CTRP1 to CTRP15) have been identified, exhibiting different or even opposite physiological functions (232). In contrast to adiponectin, which is expressed only in adipocytes, CTRPs are widely distributed in vivo and are expressed in the heart, liver, and kidney (232). CTRP1 synthesized in adipose tissue induces proinflammatory and pro-foam cell formation, accelerating AS progression. However, CTRP1 expressed in the vessels has an antithrombotic effect after AS plaque rupture. CTRP1 upregulates ECs adhesion molecule expression via the p38 MAPK/NF-κB pathway (9).
TNF-induced adhesion molecule and cytokine expressions are reduced in ECs and macrophages of CTRP1-deficient mice (9), indicating that CTRP1 induces adhesion molecule expression, inflammatory cytokine production, and promotes leukocyte adhesion to ECs. Moreover, CTRP1 exerts antithrombotic effects by inhibiting collagen-induced platelet agglutination (233). CTRP1 enhances endothelial adhesion molecule-mediated leukocyte homing and accelerates AS progression. CTRP1 overexpression promotes blood monocyte adherence to the vessel wall and their differentiation into macrophages (9). After entering the subendothelium, ox-LDL can induce macrophage CTRP1 expression, leading to enhanced expression of pro-atherogenic inflammatory factors, and PPARγ regulates this effect (234). Systemic administration of an adenoviral vector encoding CTRP1 reduces the growth of human VSMCs via a cAMP-dependent pathway attenuates intimal thickening after vascular injury (110), and prevents pathological vascular remodeling.
In rodent adipose tissue (235), CTRP3 expression is the highest in the mesentery, followed by the epididymis and subcutaneous tissue, and is relatively low in the thorax, perirenal, and BAT. The sources of CTRP3 in adipose tissue are adipocytes, monocytes (236), and fibroblasts (237). CTRP3 is an adipokine with vascular endothelial–protective effects. Decreased CTRP3 expression in epicardial adipose tissue increases the risk of AS in patients with CAD (238). CTRP3 activates the PI3K/Akt/eNOS pathway to attenuate ox-LDL–induced inflammatory responses in mouse aortic ECs (111). The C1q-like globular domain of CTRP3 has been shown to enhance diastolic function in ECs by activating the AMPK/eNOS/NO signaling pathway, thereby preserving vascular endothelial function (112).
CTRP5 is a pro-atherogenic glycoprotein secreted by adipocytes. Adipocyte CTRP5 expression levels in SAT are positively correlated with the degree of obesity in children (239). Serum CTRP5 levels were increased in rodent models of obesity (232). Upregulation of 12/15-lipoxygenase, a key enzyme mediating LDL transport and oxidation in ECs via the signal transducer and activator of transcription 6 signaling pathway, promotes LDL transport across the endothelium and oxidative modifications (113). The globular form of CTRP5 is responsible for diabetic vascular EC dysfunction via Nox1-mediated mitochondrial apoptosis (240). Moreover, CTRP5 can inhibit the expression of uncoupling protein 1, a negative regulator of WAT browning, and cold exposure decreases CTRP5 expression in SAT (241).
The human CTRP9 gene is located on chromosome 13q12.12 (242) and encodes two isoforms, CTRP9A and CTRP9B, whereas mice lack CTRP9B (243). While CTRP9A is secreted as a multimeric protein, CTRP9B requires physical association with CTRP9A or adiponectin for secretion (242). CTRP9 stabilizes plaques and is atheroprotective. Reduced CTRP9 levels are an independent risk factor for CAD in AS patients with thin fibrous caps (114). Defects in the CTRP9 gene alter the gut microbial composition of mice, increase serum cholesterol and LDL levels, and promote AS progression (244). Transplantation of wild-type mice into the intestinal microflora can reverse this effect. Zhang et al. found that CTRP9 exerts atheroprotective effects via the CTRP9-AMPK-NLRP3 inflammatory vesicle pathway (245). CTRP9 promotes EC function and ischemia-induced revascularization through an eNOS-dependent mechanism (246). CTRP9 inhibits EC senescence by promoting autophagy and autophagic flow by activating the AMPK and AMPKα/KLF4 signaling pathways (115, 116). VSMC apoptosis is closely associated with the stability and progression of AS plaques. CTRP9 induces macrophage polarization to the pro-inflammatory phenotype and promotes the apoptosis of VSMCs by activating the JNK pathway (117). CTRP9 overexpression significantly attenuated AS lesion size and reduced the accumulation of macrophages and VSMCs in ApoE−/− mice, which was associated with the activation of the AMPK/mTOR signaling pathway by CTRP9 to induce autophagy (118). CTRP9 also downregulates the inflammatory response of macrophages through the AdipoR1/AMPK pathway, attenuates apoptosis, improves cholesterol efflux from foam cells (153, 245), and improves AS.
CTRP12, also known as adipolin, was found to be a conserved paralog of adiponectin, with the lowest homology to adiponectin among other CTRPs (247). CTRP12 improves insulin sensitivity and glycemic control in obese and diabetic mice (248), with increased expression of CTRP12 in obese VAT and SAT (119). In CAD subjects, serum CTRP12 levels were negatively correlated with the extent of stenosis (120), suggesting that CTRP12 may be an independent protective factor for CAD. CTRP12 reduces the proliferation of VSMCs and inhibits macrophage inflammatory mediator gene expression through the TGF-β receptor II/Smad2-dependent pathway (121, 249). In a study of the effects of CTRP12 on macrophages, it was found that CTRP12 increases ABCA1 and ABCG1-dependent cholesterol efflux and promotes macrophage polarization to the inflammation-ameliorating phenotype via the miR-155-5p/LXRα pathway (122). The above findings suggest that CTRP12, a paralog of adiponectin, may have the same protective role as adiponectin in CVD.
5. Indeterminate adipokines
5.1. Visfatin
Visfatin is also known as pre-B-cell colony-enhancing factor because it was originally isolated as a secreted factor for synthesizing IL-7 and stem cell factors that promote the growth of B cell precursors. Moreover, visfatin is known as nicotinamide phosphoribosyltransferase (NAMPT), which has enzymatic activity, and intracellular NAMPT is the rate-limiting enzyme that catalyzes the salvage pathway of nicotinamide adenine dinucleotide (NAD+) biosynthesis (250). Extracellular NAMPT, such as adipocyte-derived NAMPT and plasma NAMPT, is enzymatically active and can affect vascular function in an autocrine and paracrine manner (251). Visfatin is mainly released by visceral WAT macrophages (192), while in unstable atherosclerotic lesions, visfatin is expressed by foam cells and macrophages (252). The levels are even higher in subjects with symptomatic carotid plaques, suggesting that visfatin may play a role in unstable plaques.
Visfatin expression is highest in PVAT compared to SAT and VAT. PVAT-derived visfatin stimulates VSMC proliferation through ERK1/2 and p38 signaling pathways (86). iNOS is a NO and peroxynitrite-forming enzyme overproduced in vascular diseases such as AS or diabetes-related vasculopathy and promotes vascular inflammation and endothelial dysfunction. Visfatin induces iNOS expression in VSMCs via ERK1/2- and NF-κB-dependent mechanisms, an effect that can be blocked by the NAMPT inhibitor APO866 (87). Visfatin enhances IL-1β-dependent induction of IL-6 and CD36 production through distinct signaling pathways mediated by JNK and NF-κB, respectively, which leads to accelerated monocyte/macrophage differentiation (88). Activation of NADPH oxidase by visfatin mediates the induction of senescence in human ECs (89). The above findings suggest that visfatin is a cytokine promoting vascular inflammation and AS. However, different from these results, exogenous visfatin ameliorates Ang II-induced endothelial dysfunction and vascular remodeling by targeting NAD/SIRT1 signaling in the study by Zhou et al. (90). Additionally, visfatin can also activate eNOS and improve EC function and angiogenesis in vitro and in vivo through Akt and MAPK pathways (91). However, it is important to note that these experiments were conducted under conditions of intact endothelial function. Given that endothelial dysfunction is a characteristic feature of AS, further investigations are required to explore the potential beneficial effects of visfatin in the presence of endothelial dysfunction. These inconsistencies between in vivo and in vitro results still require further studies to explore the underlying mechanisms.
5.2. Omentin
Omentin is a relatively new adipokine primarily expressed in VAT (253, 254). There are two highly homologous omentins: omentin-1 and omentin-2. Omentin-1 is the predominant circulating form in human plasma (254). Expression is decreased in metabolic syndromes, such as obesity (255). Elevated levels of omentin-1 in advanced coronary plaques and the circulatory system in patients with acute coronary syndromes may be related to high counteracting AS reactivity (256). Omentin facilitates vasodilation, promotes inflammatory resolution, and inhibits foam cells and neoplastic intima. Compared with ApoE−/− mice, the atherosclerotic area in the aortic sinus of mice expressing the human omentin gene in adipose tissue was significantly reduced, suggesting that omentin may have a protective role in AS (92). At low serum concentrations, omentin promotes the differentiation of human umbilical vein ECs into vascular-like structures to reduce apoptotic activity, activates the AMPK/PPARδ pathway to increase NO production (93), and stimulates ECs to exert vasodilatory physiological effects via an eNOS-dependent mechanism (94), which suggests that circulating omentin concentrations can be a valuable indicator of endothelial function. In addition, omentin-1 regulates macrophage function. Omentin-1 can promote the phosphorylation of Akt in macrophages to exert anti-inflammatory effects and, in turn, promote the conversion of monocytes to anti-inflammatory macrophages in vitro and in vivo (256), retarding AS development. Omentin inhibits the migration of VSMCs induced by Ang II and platelet-derived growth factor BB (94), decreases matrix metalloproteinase 2 expression after TNF-α stimulation, and significantly inhibits carotid intimal hyperplasia. However, Saely et al. analyzed plasma omentin in patients with coronary angiography and found that increased plasma omentin was a predictor of cardiovascular events in patients with CAD (95). This study's contradictory findings compared to previous conclusions may be attributed to potential racial differences, highlighting the need for further research and investigation. Watanabe et al. found that patients with CAD have decreased omentin levels in the coronary endothelium (256), which may involve a negative feedback regulation mechanism. This opposite result suggests that the role of omentin in the process of AS cannot be fully characterized.
5.3. Vaspin
Vaspin is a newly identified adipokine belonging to the serine protease inhibitor family. It is derived from the VAT, and circulating vaspin concentrations are significantly higher in obese vs. lean children (257, 258). Elevated circulating vaspin levels target WAT to exert insulin sensitization and improve glucose tolerance in high-fat diet-induced obese (DIO) rats (259). The long-term injection of vaspin into ApoE−/− mice significantly inhibits the development of AS aortic lesions and increases plaque stability (96). Vaspin is a novel ligand for the GRP78/voltage-dependent negative ion channel complex on the ECs surface (260) and inhibits ECs inflammation by binding to the receptor. Vaspin improves ECs NO utilization through the PI3K/Akt signaling pathway (97, 98) and improves hypoxia-stimulated cell injury and glucose tolerance. Vaspin inhibits acetylcholinesterase in mesenteric arteries to increase acetylcholine-induced eNOS phosphorylation (99). Vaspin inhibits ECs activation induced by the glucose reactive metabolite methylglyoxal (261) and ECs inflammation mediated by proinflammatory factors (100). Serum vaspin levels decrease in patients upon restenosis after coronary stenting. In vitro experiments have shown that vaspin inhibits the migration of human coronary artery smooth muscle cells (101), suggesting that it may improve vascular remodeling and prevent stenosis. The osteogenic phenotype switching of VSMCs facilitated vascular calcification. Vaspin eliminates lncRNA LEF1-AS1-mediated VSMCs osteogenic phenotype switching (102) and inhibits the progression of vascular calcification, thereby exacerbating AS. Vaspin inhibits the expression of the receptor for ox-LDL uptake by macrophages via the NF-κB/miR-33a pathway (96), increases cholesterol transport protein expression, and promotes cholesterol efflux, thereby inhibiting the foaming phenotype of macrophages. Decreased circulating vaspin concentrations appear to be associated with CAD severity and a higher incidence of major adverse cardiac events (103, 104). However, Rueda-Gotor et al. did not observe a statistically significant association between vaspin and subclinical AS markers in the study of the relationship between CVD risk and vaspin in patients with axial spondyloarthritis (105). Moreover, they found that serum vaspin concentration was regulated by gene variants (105). Such contradictory results suggest that the role of vaspin in the AS process still requires further investigation.
5.4. Asprosin
Asprosin is a newly identified adipokine and a C-terminal cleavage product of pro-fibronectin, primarily expressed in WAT (262). Placental cells, hepatocytes, and cardiomyocytes can also produce this adipokine (263, 264). It plays a vital role in glucose metabolism, appetite regulation, and inflammation; however, little is known about its role in AS (265). Serum asprosin levels are considerably higher in patients with carotid plaques and multiple coronary lesions than in healthy and asymptomatic patients (106, 107, 266). These results suggest that asprosin may be used as a marker for detecting CVD and determining its severity. The expression levels of subcutaneous WAT asprosin are significantly downregulated in mice fed a high-fat diet (265, 267).
Asprosin directly induces the endothelial-to-mesenchymal transition in a TGF-β-dependent manner (108). Asprosin upregulates ABCA1 and ABCG1 expression through activation of the p38/ELK-1 signaling pathway, inhibites lipid expression in THP-1 macrophages during lipid deposition, and reduces AS load in ApoE−/− mice (109). In addition, asprosin negatively regulates WAT browning and enhances lipid accumulation in adipose tissue (267). Based on the previous research findings, the elevated levels of asprosin in the patient's circulation may be a result of negative feedback regulation. However, this conclusion requires further investigation to explore the underlying mechanisms involved.
6. Adipose tissue-derived bioactive materials
6.1. Ceramides
Ceramide is the precursor of most sphingolipids and the central molecule involved in sphingolipid metabolism. It is also an essential component of cell membranes and a second messenger in critical signaling pathways in vivo. Ceramide synthesis occurs in the endoplasmic reticulum (123). There are three pathways for ceramide synthesis in vivo (268): de novo, sphingolipase, and remedial. In adipocytes, free fatty acids enter the cell and combine with coenzyme A to form acyl-CoAs. In obesity, triglyceride stores in adipocytes become saturated. Excess acyl-CoAs enter the ceramide synthesis pathway, and the concentration of ceramide in the cell increases with free fatty acid accumulation. The adiponectin receptor (AdipoR) has ceramidase activity (269) and works with ceramidase to maintain the dynamic balance between ceramide levels in cells and circulation. Under physiological conditions, ceramide contributes to the regulation of lipid metabolic homeostasis. However, in impaired lipid metabolism, ceramide levels are increased due to the activation of sphingomyelinase, accumulation of fatty acids, and reduced stimulation of AdipoR by downregulating circulating adiponectin levels (270).
Elevated circulating ceramide levels contribute to AS development, insulin resistance, and diabetes mellitus by modulating insulin sensitivity (270), glucose metabolism (271), and lipid metabolism. Increased levels of C16:0, C22:0, and C24:0 ceramides exacerbate the risk of CVDs such as carotid plaque and stroke (272). Reducing ceramide synthesis in adipocytes slows AS development. Ceramide induces uncoupling of eNOS in ECs via the tetrahydrobiopterin/protein phosphatase 2 pathway, decreases NO utilization (124), blocks vasodilation, and increases the risk of CVD. Simultaneously, ceramide promotes apoptosis of VSMCs (123) and the development of unstable plaques. The expression and secretion of glucosylceramide and lactosylceramide increase in adipocytes when adipose tissue metabolism is impaired. Human aortic AS plaque analysis revealed high glucosylceramide and lactosylceramide levels (125). Fiorelli et al. found increased expression of lactosylceramide in monocytes from patients with acute myocardial infarction (126). In a plasma lipid profile analysis of 200 patients with carotid plaques, high circulating glucosylceramide levels increased plaque vulnerability (273). Lactosylceramide also stimulates VSMC proliferation by stimulating nuclear antigen expression, promoting aortic VSMC proliferation (127), and facilitating monocyte migration (126). In ApoE−/− mice, inhibition of glucosylceramide synthesis promotes cholesterol excretion (274) and reduces AS plaque load.
6.2. S1P
S1P is a bioactive sphingolipid produced by the phosphorylation of sphingosine and is catalyzed by two sphingosine kinase isozymes (SphK1 and SphK2). S1P triggers inflammation by interacting with five different receptor types, S1P1–5, which belong to the G-protein-coupled receptor family. The major S1P receptors in the vascular system are S1P2, S1P1, and S1P3. The major carrier proteins of S1P are apolipoprotein M (ApoM) and albumin. The effect of S1P on the inflammatory response depends on its carrier protein. Most plasma S1P binds to ApoM to form the ApoM-S1P complex, which preferentially binds to high-density lipoprotein (HDL) and activates S1P1 (270). Although it is a product of the same sphingomyelin as ceramide, S1P has a different physiological function. Unlike S1P2, which aggravates endothelial impairment (129), S1P1 and S1P3 safeguard ECs barrier function and promote vasodilation to improve the tissue blood supply (130, 275, 276). S1P1 is expressed in cerebrovascular ECs and, upon activation, maintains responsiveness to vasodilatory stimuli, and may ensure collateral circulation to ischemic brain tissue via eNOS (130). Apolipoprotein M and S1P1 promote transcytosis of HDL across endothelial monolayers and induce cholesterol efflux from AS lesions to reduce the plaque lipid load (131). S1P2 promotes ECs expression of inflammatory factors and cell adhesion molecules by activating NF-κB (132) and JNK phosphorylation pathways (129). S1P2 also induces senescence-associated damage in young ECs (277). Under hypoxic conditions, S1P3 is activated in ECs, resulting in NO-dependent vasodilation (278).
S1P1 and S1P2 play different roles in VSMCs proliferation and migration. After ligation of the carotid arteries in mice, S1P1 expression is upregulated in the carotid arteries. After vascular injury, the S1P-S1P1 signaling pathway promotes VSMCs proliferation and migration and promotes neointimal hyperplasia (133). In contrast, S1P2 can inhibit S1P-induced migration of human coronary artery smooth muscle cells via HDL (279). S1P1 can also promote the conversion of macrophages to an anti-inflammatory phenotype (280). High endogenous S1P levels activate S1P3, impair macrophage cholesterol efflux, and cause plaque rupture (134). S1P4 is expressed in primary brain microvascular ECs, and S1P4 expression decreases after stroke (281); S1P5 reduces activation of the transcription factor NF-κB and maintains low expression levels of leukocyte adhesion molecules, inflammatory chemokines, and cytokines (282). Both S1P4 and S1P5 in brain ECs have strong protective barriers.
6.3. Exosomes
After the fusion of the intracellular body of multivesicular vesicles with the plasma membrane of the parent cell, the inner vesicles inside are expelled from the cell, forming exosomes (EXOs) (283). EXOs can affect the composition of the extracellular matrix and mediate intercellular communication and information transfer after cellular excretion (284). EXOs differ by secretory tissue, resulting in heterogeneity in size, composition, and cellular or organ uptake (285). WAT-derived EXOs are pro-atherogenic in obesity. In high-fat DIO mice, EXOs from SAT caused changes in lipid profiles (135). EXOs extracted from SAT of DIO mice aggravated obesity by affecting fatty acid metabolism in mice, while EXOs from VAT promoted macrophage foaminess and converted them to the pro-inflammatory phenotype by activating NF-κB (136). PVAT-derived EXOs can reduce macrophage foaminess through the miR-382-5p- and bone morphogenetic protein 4-PPARγ-mediated upregulation of the cholesterol efflux transporters ABCA1 and ABCG1. In contrast, this effect is weakened in CAD patients due to the downregulation of miR-382-5p expression (137). EXOs secreted by adipose-derived mesenchymal stem cells reduce reactive oxygen species production in ECs, promote angiogenesis (286, 287), and induce inflammation-ameliorating phenotype polarization of macrophages (288). In addition, ECs-derived (289) and VSMCs-derived (290) EXOs interact with each other, forming a new mode of cell-cell communication. Cytokine-stimulated VSMCs-derived EXOs cause VSMCs self-malfunction/proliferation and induce ECs malfunction, which can be attenuated by the miR548ai inhibitor (290). ECs-derived EXOs, in turn, enhance leukocyte adhesion to VSMCs and induce VSMCs protein synthesis and senescence (291). Exosomal lncRNA LIPCAR derived from THP-1 cells modified by ox-LDL significantly increases the expression levels of cyclin dependent kinase 2 and proliferating cell nuclear antigen in human vascular VSMCs to promote AS progression (292). Activated platelet-derived EXOs (293) decreases macrophage CD36 content, attenuates the CD36-dependent lipid loading capacity of macrophages, and inhibites platelet aggregation and thrombosis.
7. Conclusions
The increasing number of overweight and obese individuals has sparked interest in the role of adipose tissue in inducing associated comorbidities. Adipokines have been extensively studied due to their multifactorial properties in regulating physiological functions. Leptin, adiponectin, resistin, and visfatin have been extensively studied and play important roles in regulating glucose metabolism and cardiovascular homeostasis. Additionally, deteriorative adipokines contribute to metabolic dysregulation, endothelial dysfunction, vascular remodeling, and foam cell formation, leading to AS and an increased risk of CVD. The role of some adipokines in the pathogenesis of AS has been established. However, adipokines with unclear roles, such as omentin, vaspin, etc., still require larger prospective studies in the general population and patients with CVD to determine whether measuring circulating levels of adipokines improves AS prediction. Nevertheless, the functions of adipokines are coordinated, and changes in one adipokine may affect others. Adipokines may represent a novel clinical approach to reduce CVD-related mortality and disability, but only positive and highly effective results from well-designed clinical trials will allow broad therapeutic intervention targeting circulating adipokines.
In recent years, various adipokines have been gradually recognized has research progressed, and their role in the mechanism of AS has been explored. On the one hand, the representative protective adipokines, such as adiponectin, omentin, CTRP9, and vaspin, mainly prevent AS by protecting ECs function, inhibiting VSMCs proliferation and migration, and reducing macrophage inflammation and foam cell formation. However, their specific regulatory mechanisms differ from each other. For example, adiponectin inhibits mTOR/p70S6K signaling (38, 39) to attenuate VSMCs proliferation, whereas CTRP9 promotes VSMCs apoptosis by activating the JNK pathway (117). Asprosin ameliorates macrophage cholesterol efflux by activating the p38/ELK-1 signaling pathway to upregulate ABCA1 and ABCG1 expression (109), whereas CTRP9 improves macrophage cholesterol efflux via the AdipoR1/AMPK pathway (153). Similarly, to increase NO utilization by ECs, omentin activates the AMPK/PPARδ pathway (93), whereas vaspin activates the PI3K/Akt signaling pathway (98).
In contrast, ceramide, leptin, and CTRP5 are risk factors for AS, inducing eNOS uncoupling in ECs, promoting neointima formation, and accelerating macrophage foam cell formation. Leptin and CTRP5 have different regulatory mechanisms in ECs. High concentrations of leptin increase phosphorylation of ERK1/2 and activate NF-κB in ECs, leading to increased expression of inflammatory factors and cell adhesion molecules, and worsening endothelial injury (54). CTRP5 promotes LDL transcytosis transport and oxidative damage by activating the signal transducer and activator of transcription 6 signaling pathway to upregulate 12/15-lipoxygenase (113), a key enzyme mediating LDL transport and oxidation in ECs.
Some adipokines and adipose tissue-derived bioactive materials, such as S1P, CTRP1, and CTRP3, have both positive and negative effects on AS lesions. The inconsistency in the roles played by different cell types or ligands in the progression of AS after activation has revealed various roles played by these adipokines in AS. The roles of EXOs secreted by different adipose tissues vary, resulting in different effects on the same cells, forming the basis for the diverse functions of EXOs. Finally, the role of adipokine asprosin in AS lesions is uncertain. Due to the limited research on asprosin in vascular lesions, no conclusions can be drawn, and further investigation is warranted.
In conclusion, adipokines play a complex regulatory role in the development of AS. Increasing our understanding of which adipokines are beneficial or detrimental will help predict atherogenesis biomarkers and help identify potential therapeutic targets for AS.
Author contributions
JL and ZH wrote the draft; QL, ML, YC, and WK performed the data analysis and data presentation; XN and ZZ reviewed, edited and proofed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 82071183 to ZZ and No. 82001245 to XN).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Townsend N, Kazakiewicz D, Lucy Wright F, Timmis A, Huculeci R, Torbica A, et al. Epidemiology of cardiovascular disease in Europe. Nat Rev Cardiol. (2022) 19:133–43. doi: 10.1038/s41569-021-00607-3
3. Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
4. Wang D, Wang Z, Zhang L, Wang Y. Roles of cells from the arterial vessel wall in atherosclerosis. Mediators Inflamm. (2017) 2017:8135934. doi: 10.1155/2017/8135934
5. Johnson JL. Emerging regulators of vascular smooth muscle cell function in the development and progression of atherosclerosis. Cardiovasc Res. (2014) 103:452–60. doi: 10.1093/cvr/cvu171
6. Björkegren JLM, Lusis AJ. Atherosclerosis: recent developments. Cell. (2022) 185:1630–45. doi: 10.1016/j.cell.2022.04.004
7. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
8. Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. (2002) 8:1235–42. doi: 10.1038/nm1102-1235
9. Lu L, Zhang RY, Wang XQ, Liu ZH, Shen Y, Ding FH, et al. C1q/TNF-related protein-1: an adipokine marking and promoting atherosclerosis. Eur Heart J. (2016) 37:1762–71. doi: 10.1093/eurheartj/ehv649
10. He L, Zhang C-L, Chen Q, Wang L, Huang Y. Endothelial shear stress signal transduction and atherogenesis: from mechanisms to therapeutics. Pharmacol Ther. (2022) 235:108152. doi: 10.1016/j.pharmthera.2022.108152
11. Souilhol C, Harmsen MC, Evans PC, Krenning G. Endothelial–mesenchymal transition in atherosclerosis. Cardiovasc Res. (2018) 114:565–77. doi: 10.1093/cvr/cvx253
12. Libby P. The changing landscape of atherosclerosis. Nature. (2021) 592:524–33. doi: 10.1038/s41586-021-03392-8
13. Kuznetsova T, Prange KHM, Glass CK, De Winther MPJ. Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nat Rev Cardiol. (2020) 17:216–28. doi: 10.1038/s41569-019-0265-3
14. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. (2016) 118:653–67. doi: 10.1161/CIRCRESAHA.115.306256
15. Qi X-Y, Qu S-L, Xiong W-H, Rom O, Chang L, Jiang Z-S. Perivascular adipose tissue (PVAT) in atherosclerosis: a double-edged sword. Cardiovasc Diabetol. (2018) 17:134. doi: 10.1186/s12933-018-0777-x
16. Hausman DB, DiGirolamo M, Bartness TJ, Hausman GJ, Martin RJ. The biology of white adipocyte proliferation. Obes Rev. (2001) 2:239–54. doi: 10.1046/j.1467-789X.2001.00042.x
17. Maniyadath B, Zhang Q, Gupta RK, Mandrup S. Adipose tissue at single-cell resolution. Cell Metab. (2023) 35:386–413. doi: 10.1016/j.cmet.2023.02.002
18. Morigny P, Boucher J, Arner P, Langin D. Lipid and glucose metabolism in white adipocytes: pathways, dysfunction and therapeutics. Nat Rev Endocrinol. (2021) 17:276. doi: 10.1038/s41574-021-00471-8
19. Fuster JJ, Ouchi N, Gokce N, Walsh K. Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ Res. (2016) 118:1786–807. doi: 10.1161/CIRCRESAHA.115.306885
20. AlZaim I, Hammoud SH, Al-Koussa H, Ghazi A, Eid AH, El-Yazbi AF. Adipose tissue immunomodulation: a novel therapeutic approach in cardiovascular and metabolic diseases. Front Cardiovasc Med. (2020) 7:602088. doi: 10.3389/fcvm.2020.602088
21. Jiang E, Perrard XD, Yang D, Khan IM, Perrard JL, Smith CW, et al. Essential role of CD11a in CD8+ T-cell accumulation and activation in adipose tissue. Arterioscler Thromb Vasc Biol. (2014) 34:34–43. doi: 10.1161/ATVBAHA.113.302077
22. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. (2009) 15:914–20. doi: 10.1038/nm.1964
23. Li X, Ma Z, Zhu YZ. Regional heterogeneity of perivascular adipose tissue: morphology, origin, and secretome. Front Pharmacol. (2021) 12:697720. doi: 10.3389/fphar.2021.697720
24. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. (2004) 84:277–359. doi: 10.1152/physrev.00015.2003
25. Seale P, Bjork B, Yang W, Kajimura S, Kuang S, Scime A, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. (2008) 454:961–7. doi: 10.1038/nature07182
26. Matthias A, Ohlson KBE, Fredriksson JM, Jacobsson A, Nedergaard J, Cannon B. Thermogenic responses in brown fat cells are fully UCP1-dependent. J Biol Chem. (2000) 275:25073–81. doi: 10.1074/jbc.M000547200
27. Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S, et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature. (2018) 560:102–6. doi: 10.1038/s41586-018-0353-2
28. Roesler A, Kazak L. UCP1-independent thermogenesis. Biochem J. (2020) 477:709–25. doi: 10.1042/BCJ20190463
29. Adachi Y, Ueda K, Nomura S, Ito K, Katoh M, Katagiri M, et al. Beiging of perivascular adipose tissue regulates its inflammation and vascular remodeling. Nat Commun. (2022) 13:5117. doi: 10.1038/s41467-022-32658-6
30. Chen Y, Qin Z, Wang Y, Li X, Zheng Y, Liu Y. Role of inflammation in vascular disease-related perivascular adipose tissuedysfunction. Front Endocrinol (Lausanne). (2021) 12:710842. doi: 10.3389/fendo.2021.710842
31. Adachi Y, Ueda K, Nomura S, Ito K, Katoh M, Katagiri M, et al. Beiging of perivascular adipose tissue regulates its inflammation and vascular remodeling. Nat. Commun. (2022) 13(1):5117. doi: 10.1038/s41467-022-32658-6
32. Chen Q, Huang L, Pan D, Hu K, Li R, Friedline RH, et al. A brown fat-enriched adipokine adissp controls adipose thermogenesis and glucose homeostasis. Nat Commun. (2022) 13:7633. doi: 10.1038/s41467-022-35335-w
33. Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat RevDrug Discovery. (2016) 15:639–60. doi: 10.1038/nrd.2016.75
34. Koliaki C, Liatis S, Kokkinos A. Obesityand cardiovascular disease:revisiting an old relationship. Metab Clin Exp. (2019) 92:98–107. doi: 10.1016/j.metabol.2018.10.011
35. Kim HW, de Chantemèle EJB, Weintraub NL. Perivascular adipocytes in vascular disease. Arterioscler Thromb Vasc Biol. (2019) 39:2220–7. doi: 10.1161/ATVBAHA.119.312304
36. Ouchi N, Kihara S, Arita Y, Maeda K, Kuriyama H, Okamoto Y, et al. Novel modulator for endothelial adhesion molecules. Circulation. (1999) 100:2473–6. doi: 10.1161/01.CIR.100.25.2473
37. Li R, Wang W-Q, Zhang H, Yang X, Fan Q, Christopher TA, et al. Adiponectin improves endothelial function in hyperlipidemic rats by reducing oxidative/nitrative stress and differential regulation of eNOS/iNOS activity. Am J Physiol Endocrinol Metab. (2007) 293:E1703–8. doi: 10.1152/ajpendo.00462.2007
38. Osman I, Segar L. Pioglitazone, a PPARγ agonist, attenuates PDGF-induced vascular smooth muscle cell proliferation through AMPK-dependent and AMPK-independent inhibition of mTOR/p70S6K and ERK signaling. Biochem Pharmacol. (2016) 101:54–70. doi: 10.1016/j.bcp.2015.11.026
39. Kubota T, Kubota N, Sato H, Inoue M, Kumagai H, Iwamura T, et al. Pioglitazone ameliorates smooth muscle cell proliferation in cuff-induced neointimal formation by both adiponectin-dependent and -independent pathways. Sci Rep. (2016) 6:34707. doi: 10.1038/srep34707
40. Shi L, Li Y, Xu X, Cheng Y, Meng B, Xu J, et al. Brown adipose tissue-derived Nrg4 alleviates endothelial inflammation and atherosclerosis in male mice. Nat Metab. (2022) 4:1573–90. doi: 10.1038/s42255-022-00671-0
41. Schumacher MA, Hedl M, Abraham C, Bernard JK, Lozano PR, Hsieh JJ, et al. Erbb4 signaling stimulates pro-inflammatory macrophage apoptosis and limits colonic inflammation. Cell Death Dis. (2017) 8:e2622. doi: 10.1038/cddis.2017.42
42. Yan X, Gou Z, Li Y, Wang Y, Zhu J, Xu G, et al. Fibroblast growth factor 21 inhibits atherosclerosis in apoE−/− mice by ameliorating fas-mediated apoptosis. Lipids Health Dis. (2018) 17:203. doi: 10.1186/s12944-018-0846-x
43. Zeng Z, Zheng Q, Chen J, Tan X, Li Q, Ding L, et al. FGF21 mitigates atherosclerosis via inhibition of NLRP3 inflammasome-mediated vascular endothelial cells pyroptosis. Exp Cell Res. (2020) 393:112108. doi: 10.1016/j.yexcr.2020.112108
44. Shang W, Yu X, Wang H, Chen T, Fang Y, Yang X, et al. Fibroblast growth factor 21 enhances cholesterol efflux in THP-1 macrophage-derived foam cells. Mol Med Rep. (2015) 11:503–8. doi: 10.3892/mmr.2014.2731
45. Cao F, Wang S, Cao X, Liu X, Fu K, Hao P, et al. Fibroblast growth factor 21 attenuates calcification of vascular smooth muscle cells in vitro. J Pharm Pharmacol. (2017) 69:1802–16. doi: 10.1111/jphp.12826
46. Lu J, Xiang G, Liu M, Mei W, Xiang L, Dong J. Irisin protects against endothelial injury and ameliorates atherosclerosis in apolipoprotein E-null diabetic mice. Atherosclerosis. (2015) 243:438–48. doi: 10.1016/j.atherosclerosis.2015.10.020
47. Zheng G, Li H, Zhang T, Yang L, Yao S, Chen S, et al. Irisin protects macrophages from oxidized low density lipoprotein-induced apoptosis by inhibiting the endoplasmic reticulum stress pathway. Saudi J Biol Sci. (2018) 25:849–57. doi: 10.1016/j.sjbs.2017.08.018
48. Tu Y, Liu J, Kong D, Guo X, Li J, Long Z, et al. Irisin drives macrophage anti-inflammatory differentiation via JAK2-STAT6-dependent activation of PPARγ and Nrf2 signaling. Free Radic Biol Med. (2023) 201:98–110. doi: 10.1016/j.freeradbiomed.2023.03.014
49. Wang P-W, Pang Q, Zhou T, Song X-Y, Pan Y-J, Jia L-P, et al. Irisin alleviates vascular calcification by inhibiting VSMC osteoblastic transformation and mitochondria dysfunction via AMPK/Drp1 signaling pathway in chronic kidney disease. Atherosclerosis. (2022) 346:36–45. doi: 10.1016/j.atherosclerosis.2022.02.007
50. Pang Q, Wang P, Pan Y, Dong X, Zhou T, Song X, et al. Irisin protects against vascular calcification by activating autophagy and inhibiting NLRP3-mediated vascular smooth muscle cell pyroptosis in chronic kidney disease. Cell Death Dis. (2022) 13:283. doi: 10.1038/s41419-022-04735-7
51. Deng D, Diao Z, Han X, Liu W. Secreted frizzled-related protein 5 attenuates high phosphate-induced calcification in vascular smooth muscle cells by inhibiting the Wnt/ß-catenin pathway. Calcif Tissue Int. (2016) 99:66–75. doi: 10.1007/s00223-016-0117-7
52. Oh YJ, Kim H, Kim AJ, Ro H, Chang JH, Lee HH, et al. Reduction of secreted frizzled-related protein 5 drives vascular calcification through Wnt3a-mediated rho/ROCK/JNK signaling in chronic kidney disease. Int J Mol Sci. (2020) 21:3539. doi: 10.3390/ijms21103539
53. Ding N, Zheng C. Secreted frizzled-related protein 5 promotes angiogenesis of human umbilical vein endothelial cells and alleviates myocardial injury in diabetic mice with myocardial infarction by inhibiting Wnt5a/JNK signaling. Bioengineered. (2022) 13:11656–67. doi: 10.1080/21655979.2022.2070964
54. Indra MR, Karyono S, Ratnawati R, Malik SG. Quercetin suppresses inflammation by reducing ERK1/2 phosphorylation and NF kappa B activation in leptin-induced human umbilical vein endothelial cells (HUVECs). BMC Res Notes. (2013) 6:275. doi: 10.1186/1756-0500-6-275
55. Teixeira TM, da Costa DC, Resende AC, Soulage CO, Bezerra FF, Daleprane JB. Activation of Nrf2-antioxidant signaling by 1,25-dihydroxycholecalciferol prevents leptin-induced oxidative stress and inflammation in human endothelial cells. J Nutr. (2017) 147:506–13. doi: 10.3945/jn.116.239475
56. Huang F, Xiong X, Wang H, You S, Zeng H. Leptin-induced vascular smooth muscle cell proliferation via regulating cell cycle, activating ERK1/2 and NF-kappaB. Acta Biochim Biophys Sin (Shanghai). (2010) 42:325–31. doi: 10.1093/abbs/gmq025
57. Tsai Y-C, Lee Y-M, Hsu C-H, Leu S-Y, Chiang H-Y, Yen M-H, et al. The effect of ferulic acid ethyl ester on leptin-induced proliferation and migration of aortic smooth muscle cells. Exp Mol Med. (2015) 47:e180. doi: 10.1038/emm.2015.56
58. Schroeter MR, Leifheit-Nestler M, Hubert A, Schumann B, Glückermann R, Eschholz N, et al. Leptin promotes neointima formation and smooth muscle cell proliferation via NADPH oxidase activation and signalling in caveolin-rich microdomains. Cardiovasc Res. (2013) 99:555–65. doi: 10.1093/cvr/cvt126
59. Hongo S, Watanabe T, Arita S, Kanome T, Kageyama H, Shioda S, et al. Leptin modulates ACAT1 expression and cholesterol efflux from human macrophages. Am J Physiol Endocrinol Metab. (2009) 297:E474–82. doi: 10.1152/ajpendo.90369.2008
60. Gasbarrino K, Mantzoros C, Gorgui J, Veinot JP, Lai C, Daskalopoulou SS. Circulating chemerin is associated with carotid plaque instability, whereas resistin is related to cerebrovascular symptomatology. Arterioscler Thromb Vasc Biol. (2016) 36:1670–8. doi: 10.1161/ATVBAHA.115.306741
61. Chen C, Jiang J, Lü J-M, Chai H, Wang X, Lin PH, et al. Resistin decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Am J Physiol Heart Circ Physiol. (2010) 299:H193–201. doi: 10.1152/ajpheart.00431.2009
62. Cho Y, Lee S-E, Lee H-C, Hur J, Lee S, Youn S-W, et al. Adipokine resistin is a key player to modulate monocytes, endothelial cells, and smooth muscle cells, leading to progression of atherosclerosis in rabbit carotid artery. J Am Coll Cardiol. (2011) 57:99–109. doi: 10.1016/j.jacc.2010.07.035
63. Kawanami D, Maemura K, Takeda N, Harada T, Nojiri T, Imai Y, et al. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: a new insight into adipocytokine–endothelial cell interactions. Biochem Biophys Res Commun. (2004) 314:415–9. doi: 10.1016/j.bbrc.2003.12.104
64. Gao F, Si F, Feng S, Yi Q, Liu R. Resistin enhances inflammatory cytokine production in coronary artery tissues by activating the NF-κB signaling. Biomed Res Int. (2016) 2016:3296437. doi: 10.1155/2016/3296437
65. Jiang C, Zhang H, Zhang W, Kong W, Zhu Y, Zhang H, et al. Homocysteine promotes vascular smooth muscle cell migration by induction of the adipokine resistin. Am J Physiol Cell Physiol. (2009) 297:C1466–76. doi: 10.1152/ajpcell.00304.2009
66. Ding Q, Chai H, Mahmood N, Tsao J, Mochly-Rosen D, Zhou W. Matrix metalloproteinases modulated by PKCε mediate resistin-induced migration of human coronary artery smooth muscle cells. J Vasc Surg. (2011) 53:1044–51. doi: 10.1016/j.jvs.2010.10.117
67. Calabro P, Samudio I, Willerson JT, Yeh ETH. Resistin promotes smooth muscle cell proliferation through activation of extracellular signal–regulated kinase 1/2 and phosphatidylinositol 3-kinase pathways. Circulation. (2004) 110:3335–40. doi: 10.1161/01.CIR.0000147825.97879.E7
68. Raghuraman G, Zuniga MC, Yuan H, Zhou W. PKCε-mediates resistin-induced NADPH oxidase activation and inflammation leading to smooth muscle cell dysfunction and intimal hyperplasia. Atherosclerosis. (2016) 253:29–37. doi: 10.1016/j.atherosclerosis.2016.08.015
69. Xu W, Yu L, Zhou W, Luo M. Resistin increases lipid accumulation and CD36 expression in human macrophages. Biochem Biophys Res Commun. (2006) 351:376–82. doi: 10.1016/j.bbrc.2006.10.051
70. Zuniga MC, Raghuraman G, Hitchner E, Weyand C, Robinson W, Zhou W. PKC-epsilon and TLR4 synergistically regulate resistin-mediated inflammation in human macrophages. Atherosclerosis. (2017) 259:51–9. doi: 10.1016/j.atherosclerosis.2017.02.021
71. Silswal N, Singh AK, Aruna B, Mukhopadhyay S, Ghosh S, Ehtesham NZ. Human resistin stimulates the pro-inflammatory cytokines TNF-α and IL-12 in macrophages by NF-κB-dependent pathway. Biochem Biophys Res Commun. (2005) 334:1092–101. doi: 10.1016/j.bbrc.2005.06.202
72. Bitar MS. Diabetes impairs angiogenesis and induces endothelial cell senescence by up-regulating thrombospondin-CD47-dependent signaling. Int J Mol Sci. (2019) 20:673. doi: 10.3390/ijms20030673
73. DiPietro LA, Nebgen DR, Polverini PJ. Downregulation of endothelial cell thrombospondin 1 enhances in vitro angiogenesis. J Vasc Res. (1994) 31:178–85. doi: 10.1159/000319585
74. Iruela-Arispe ML, Bornstein P, Sage H. Thrombospondin exerts an antiangiogenic effect on cord formation by endothelial cells in vitro. Proc Natl Acad Sci USA. (1991) 88:5026–30. doi: 10.1073/pnas.88.11.5026
75. Panetti TS, Chen H, Misenheimer TM, Getzler SB, Mosher DF. Endothelial cell mitogenesis induced by LPA: inhibition by thrombospondin-1 and thrombospondin-2. J Lab Clin Med. (1997) 129:208–16. doi: 10.1016/S0022-2143(97)90141-4
76. Isenberg JS, Wink DA, Roberts DD. Thrombospondin-1 antagonizes nitric oxide-stimulated vascular smooth muscle cell responses. Cardiovasc Res. (2006) 71:785–93. doi: 10.1016/j.cardiores.2006.05.024
77. Isenberg JS, Ridnour LA, Dimitry J, Frazier WA, Wink DA, Roberts DD. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1*. J Biol Chem. (2006) 281:26069–80. doi: 10.1074/jbc.M605040200
78. Li Y, Qi X, Tong X, Wang S. Thrombospondin 1 activates the macrophage toll-like receptor 4 pathway. Cell Mol Immunol. (2013) 10:506–12. doi: 10.1038/cmi.2013.32
79. Ackermann K, Bonaterra GA, Kinscherf R, Schwarz A. Growth differentiation factor-15 regulates oxLDL-induced lipid homeostasis and autophagy in human macrophages. Atherosclerosis. (2019) 281:128–36. doi: 10.1016/j.atherosclerosis.2018.12.009
80. de Jager SCA, Bermúdez B, Bot I, Koenen RR, Bot M, Kavelaars A, et al. Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J Exp Med. (2011) 208:217–25. doi: 10.1084/jem.20100370
81. Park H, Kim C-H, Jeong J-H, Park M, Kim KS. GDF15 contributes to radiation-induced senescence through the ROS-mediated p16 pathway in human endothelial cells. Oncotarget. (2016) 7:9634–44. doi: 10.18632/oncotarget.7457
82. Ha G, De Torres F, Arouche N, Benzoubir N, Ferratge S, Hatem E, et al. GDF15 secreted by senescent endothelial cells improves vascular progenitor cell functions. PLoS One. (2019) 14:e0216602. doi: 10.1371/journal.pone.0216602
83. Wu Y-W, Chang T-T, Chang C-C, Chen J-W. Fatty-acid-binding protein 4 as a novel contributor to mononuclear cell activation and endothelial cell dysfunction in atherosclerosis. IJMS. (2020) 21:9245. doi: 10.3390/ijms21239245
84. Hui X, Li H, Zhou Z, Lam KSL, Xiao Y, Wu D, et al. Adipocyte fatty acid-binding protein modulates inflammatory responses in macrophages through a positive feedback loop involving c-jun NH2-terminal kinases and activator protein-1. J Biol Chem. (2010) 285:10273–80. doi: 10.1074/jbc.M109.097907
85. Girona J, Rosales R, Plana N, Saavedra P, Masana L, Vallvé J-C. FABP4 induces vascular smooth muscle cell proliferation and migration through a MAPK-dependent pathway. PLoS One. (2013) 8:e81914. doi: 10.1371/journal.pone.0081914
86. Wang P, Xu T-Y, Guan Y-F, Su D-F, Fan G-R, Miao C-Y. Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovasc Res. (2009) 81:370–80. doi: 10.1093/cvr/cvn288
87. Romacho T, Azcutia V, Vázquez-Bella M, Matesanz N, Cercas E, Nevado J, et al. Extracellular PBEF/NAMPT/visfatin activates pro-inflammatory signalling in human vascular smooth muscle cells through nicotinamide phosphoribosyltransferase activity. Diabetologia. (2009) 52:2455–63. doi: 10.1007/s00125-009-1509-2
88. Yun MR, Seo JM, Park HY. Visfatin contributes to the differentiation of monocytes into macrophages through the differential regulation of inflammatory cytokines in THP-1 cells. Cell Signal. (2014) 26:705–15. doi: 10.1016/j.cellsig.2013.12.010
89. Villalobos LA, Uryga A, Romacho T, Leivas A, Sánchez-Ferrer CF, Erusalimsky JD, et al. Visfatin/nampt induces telomere damage and senescence in human endothelial cells. Int J Cardiol. (2014) 175:573–5. doi: 10.1016/j.ijcard.2014.05.028
90. Zhou L, Zhang S, Bolor-Erdene E, Wang L, Tian D, Mei Y. NAMPT/SIRT1 attenuate ang II-induced vascular remodeling and vulnerability to hypertension by inhibiting the ROS/MAPK pathway. Oxid Med Cell Longev. (2020) 2020:1974265. doi: 10.1155/2020/1974265
91. Yamawaki H, Hara N, Okada M, Hara Y. Visfatin causes endothelium-dependent relaxation in isolated blood vessels. Biochem Biophys Res Commun. (2009) 383:503–8. doi: 10.1016/j.bbrc.2009.04.074
92. Hiramatsu-Ito M, Shibata R, Ohashi K, Uemura Y, Kanemura N, Kambara T, et al. Omentin attenuates atherosclerotic lesion formation in apolipoprotein E-deficient mice. Cardiovasc Res. (2016) 110:107–17. doi: 10.1093/cvr/cvv282
93. Liu F, Fang S, Liu X, Li J, Wang X, Cui J, et al. Omentin-1 protects against high glucose-induced endothelial dysfunction via the AMPK/PPARδ signaling pathway. Biochem Pharmacol. (2020) 174:113830. doi: 10.1016/j.bcp.2020.113830
94. Kazama K, Okada M, Hara Y, Yamawaki H. A novel adipocytokine, omentin, inhibits agonists-induced increases of blood pressure in rats. J Vet Med Sci. (2013) 75:1029–34. doi: 10.1292/jvms.12-0537
95. Saely CH, Leiherer A, Muendlein A, Vonbank A, Rein P, Geiger K, et al. High plasma omentin predicts cardiovascular events independently from the presence and extent of angiographically determined atherosclerosis. Atherosclerosis. (2016) 244:38–43. doi: 10.1016/j.atherosclerosis.2015.10.100
96. Sato K, Shirai R, Yamaguchi M, Yamashita T, Shibata K, Okano T, et al. Anti-atherogenic effects of vaspin on human aortic smooth muscle cell/macrophage responses and hyperlipidemic mouse plaque phenotype. Int J Mol Sci. (2018) 19:E1732. doi: 10.3390/ijms19061732
97. Ke X, Chen X, Yan L, Zhang Y. Vaspin contributes to autophagy and endothelial-to-mesenchymal transition via PI3K-/AKT-mTOR pathway. Acta Histochem. (2022) 124:151881. doi: 10.1016/j.acthis.2022.151881
98. Jung CH, Lee WJ, Hwang JY, Lee MJ, Seol SM, Kim YM, et al. Vaspin increases nitric oxide bioavailability through the reduction of asymmetric dimethylarginine in vascular endothelial cells. PLoS One. (2012) 7:e52346. doi: 10.1371/journal.pone.0052346
99. Kameshima S, Yamada K, Morita T, Okada M, Yamawaki H. Visceral adipose tissue-derived serine protease inhibitor augments acetylcholine-induced relaxation via the inhibition of acetylcholine esterase activity in rat isolated mesenteric artery. Acta Physiol (Oxf). (2016) 216:203–10. doi: 10.1111/apha.12563
100. Liu S, Dong Y, Wang T, Zhao S, Yang K, Chen X, et al. Vaspin inhibited proinflammatory cytokine induced activation of nuclear factor-kappa B and its downstream molecules in human endothelial EA.hy926 cells. Diabetes Res Clin Pract. (2014) 103:482–8. doi: 10.1016/j.diabres.2013.12.002
101. Kastl SP, Katsaros KM, Krychtiuk KA, Jägersberger G, Kaun C, Huber K, et al. The adipokine vaspin is associated with decreased coronary in-stent restenosis in vivo and inhibits migration of human coronary smooth muscle cells in vitro. PLoS One. (2020) 15:e0232483. doi: 10.1371/journal.pone.0232483
102. Ma X, Wang Y, Liu Q, Han B, Wang G, Zhang R, et al. Vaspin alleviates the lncRNA LEF1-AS1-induced osteogenic differentiation of vascular smooth muscle cells via the hippo/YAP signaling pathway. Exp Cell Res. (2022) 421:113407. doi: 10.1016/j.yexcr.2022.113407
103. Kadoglou NPE, Gkontopoulos A, Kapelouzou A, Fotiadis G, Theofilogiannakos EK, Kottas G, et al. Serum levels of vaspin and visfatin in patients with coronary artery disease—Kozani study. Clin Chim Acta. (2011) 412:48–52. doi: 10.1016/j.cca.2010.09.012
104. Zhou X, Chen Y, Tao Y, Zhang W, Xu W, Lu X. Serum vaspin as a predictor of adverse cardiac events in acute myocardial infarction. J Am Heart Assoc. (2019) 8:e010934. doi: 10.1161/JAHA.118.010934
105. Rueda-Gotor J, López-Mejías R, Remuzgo-Martínez S, Pulito-Cueto V, Corrales A, Lera-Gómez L, et al. Vaspin in atherosclerotic disease and cardiovascular risk in axial spondyloarthritis: a genetic and serological study. Arthritis Res Ther. (2021) 23:111. doi: 10.1186/s13075-021-02499-7
106. Moradi N, Fouani FZ, Vatannejad A, Bakhti Arani A, Shahrzad S, Fadaei R. Serum levels of asprosin in patients diagnosed with coronary artery disease (CAD): a case-control study. Lipids Health Dis. (2021) 20:88. doi: 10.1186/s12944-021-01514-9
107. Deng X, Zhao Z, Zhao L, Wang C, Li Y, Cai Z, et al. Association between circulating asprosin levels and carotid atherosclerotic plaque in patients with type 2 diabetes. Clin Biochem. (2022) 109–110:44–50. doi: 10.1016/j.clinbiochem.2022.04.018
108. You M, Liu Y, Wang B, Li L, Zhang H, He H, et al. Asprosin induces vascular endothelial-to-mesenchymal transition in diabetic lower extremity peripheral artery disease. Cardiovasc Diabetol. (2022) 21:25. doi: 10.1186/s12933-022-01457-0
109. Zou J, Xu C, Zhao Z-W, Yin S-H, Wang G. Asprosin inhibits macrophage lipid accumulation and reduces atherosclerotic burden by up-regulating ABCA1 and ABCG1 expression via the p38/elk-1 pathway. J Transl Med. (2022) 20:337. doi: 10.1186/s12967-022-03542-0
110. Kanemura N, Shibata R, Ohashi K, Ogawa H, Hiramatsu-Ito M, Enomoto T, et al. C1q/TNF-related protein 1 prevents neointimal formation after arterial injury. Atherosclerosis. (2017) 257:138–45. doi: 10.1016/j.atherosclerosis.2017.01.014
111. Chen L, Qin L, Liu X, Meng X. CTRP3 alleviates ox-LDL–induced inflammatory response and endothelial dysfunction in mouse aortic endothelial cells by activating the PI3K/Akt/eNOS pathway. Inflammation. (2019) 42:1350–9. doi: 10.1007/s10753-019-00996-1
112. Yan Z, Cao X, Wang C, Liu S, Li Y, Lu G, et al. C1q/tumor necrosis factor-related protein-3 improves microvascular endothelial function in diabetes through the AMPK/eNOS/NO· signaling pathway. Biochem Pharmacol. (2022) 195:114745. doi: 10.1016/j.bcp.2021.114745
113. Li C, Chen JW, Liu ZH, Shen Y, Ding FH, Gu G, et al. CTRP5 promotes transcytosis and oxidative modification of low-density lipoprotein and the development of atherosclerosis. Atherosclerosis. (2018) 278:197–209. doi: 10.1016/j.atherosclerosis.2018.09.037
114. Liu Y, Wang X, Wang T, Zhang W, Chang Y, Dai W, et al. Relationship between coronary VH-IVUS plaque characteristics and CTRP9, SAA, and hcy in patients with coronary heart disease. J Healthc Eng. (2022) 2022:1635446. doi: 10.1155/2022/1635446
115. Lee J, Yoo JH, Kim HS, Cho YK, Lee YL, Lee WJ, et al. C1q/TNF-related protein-9 attenuates palmitic acid-induced endothelial cell senescence via increasing autophagy. Mol Cell Endocrinol. (2021) 521:111114. doi: 10.1016/j.mce.2020.111114
116. Wang G, Han B, Zhang R, Liu Q, Wang X, Huang X, et al. C1q/TNF-related protein 9 attenuates atherosclerosis by inhibiting hyperglycemia-induced endothelial cell senescence through the AMPKα/KLF4 signaling pathway. Front Pharmacol. (2021) 12:758792. doi: 10.3389/fphar.2021.758792
117. Chen J-Y, Song C-X, Lei S-Y, Li J, Zuo A-J, Xu D, et al. CTRP9 induces macrophages polarization into m1 phenotype through activating JNK pathway and enhances VSMCs apoptosis in macrophages and VSMCs co-culture system. Exp Cell Res. (2020) 395:112194. doi: 10.1016/j.yexcr.2020.112194
118. Huang C, Zhang P, Li T, Li J, Liu T, Zuo A, et al. Overexpression of CTRP9 attenuates the development of atherosclerosis in apolipoprotein E-deficient mice. Mol Cell Biochem. (2019) 455:99–108. doi: 10.1007/s11010-018-3473-y
119. Omidifar A, Toolabi K, Rahimipour A, Emamgholipour S, Shanaki M. The gene expression of CTRP12 but not CTRP13 is upregulated in both visceral and subcutaneous adipose tissue of obese subjects. Diabetes Metab Syndr. (2019) 13:2593–9. doi: 10.1016/j.dsx.2019.07.027
120. Nadimi Shahraki Z, Azimi H, Ilchi N, Rohani Borj M, Pourghadamyari H, Mosallanejad S, et al. Circulating C1q/TNF-related protein-12 levels are associated with the severity of coronary artery disease. Cytokine. (2021) 144:155545. doi: 10.1016/j.cyto.2021.155545
121. Ogawa H, Ohashi K, Ito M, Shibata R, Kanemura N, Yuasa D, et al. Adipolin/CTRP12 protects against pathological vascular remodelling through suppression of smooth muscle cell growth and macrophage inflammatory response. Cardiovasc Res. (2020) 116:237–49. doi: 10.1093/cvr/cvz074
122. Wang G, Chen J-J, Deng W-Y, Ren K, Yin S-H, Yu X-H. CTRP12 ameliorates atherosclerosis by promoting cholesterol efflux and inhibiting inflammatory response via the miR-155-5p/LXRα pathway. Cell Death Dis. (2021) 12:254. doi: 10.1038/s41419-021-03544-8
123. Okrzeja J, Karwowska A, Błachnio-Zabielska A. The role of obesity, inflammation and sphingolipids in the development of an abdominal aortic aneurysm. Nutrients. (2022) 14:2438. doi: 10.3390/nu14122438
124. Zhang Q-J, Holland WL, Wilson L, Tanner JM, Kearns D, Cahoon JM, et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes. (2012) 61:1848–59. doi: 10.2337/db11-1399
125. Akawi N, Checa A, Antonopoulos AS, Akoumianakis I, Daskalaki E, Kotanidis CP, et al. Fat-secreted ceramides regulate vascular redox state and influence outcomes in patients with cardiovascular disease. J Am Coll Cardiol. (2021) 77:2494–513. doi: 10.1016/j.jacc.2021.03.314
126. Fiorelli S, Anesi A, Porro B, Cosentino N, Werba JP, Di Minno A, et al. Lipidomics analysis of monocytes from patients with acute myocardial infarction reveals lactosylceramide as a new player in monocyte migration. FASEB J. (2021) 35:e21494. doi: 10.1096/fj.202001872RRR
127. Chatterjee S. Oxidized low density lipoproteins and lactosylceramide both stimulate the expression of proliferating cell nuclear antigen and the proliferation of aortic smooth muscle cells. Indian J Biochem Biophys. (1997) 34:56–60.9343929
128. Meikle PJ, Wong G, Tsorotes D, Barlow CK, Weir JM, Christopher MJ, et al. Plasma lipidomic analysis of stable and unstable coronary artery disease. Arterioscler Thromb Vasc Biol. (2011) 31:2723–32. doi: 10.1161/ATVBAHA.111.234096
129. Ganbaatar B, Fukuda D, Shinohara M, Yagi S, Kusunose K, Yamada H, et al. Inhibition of S1P receptor 2 attenuates endothelial dysfunction and inhibits atherogenesis in apolipoprotein E-deficient mice. J Atheroscler Thromb. (2021) 28:630–42. doi: 10.5551/jat.54916
130. Nitzsche A, Poittevin M, Benarab A, Bonnin P, Faraco G, Uchida H, et al. Endothelial S1P1 signaling counteracts infarct expansion in ischemic stroke. Circ Res. (2021) 128:363–82. doi: 10.1161/CIRCRESAHA.120.316711
131. Velagapudi S, Rohrer L, Poti F, Feuerborn R, Perisa D, Wang D, et al. Apolipoprotein M and sphingosine-1-phosphate receptor 1 promote the transendothelial transport of HDL. Arterioscler Thromb Vasc Biol. (2021) 41:e468–79. doi: 10.1161/ATVBAHA.121.316725
132. Zhang W, An J, Jawadi H, Siow DL, Lee J-F, Zhao J, et al. Sphingosine-1-phosphate receptor-2 mediated NFκB activation contributes to tumor necrosis factor-α induced VCAM-1 and ICAM-1 expression in endothelial cells. Prostag Oth Lipid M. (2013) 106:62–71. doi: 10.1016/j.prostaglandins.2013.06.001
133. Kitano T, Usui S, Takashima S, Inoue O, Goten C, Nomura A, et al. Sphigosine-1-phosphate receptor 1 promotes neointimal hyperplasia in a mouse model of carotid artery injury. Biochem Biophys Res Commun. (2019) 511:179–84. doi: 10.1016/j.bbrc.2019.02.047
134. Keul P, Peters S, von Wnuck Lipinski K, Schröder NH, Nowak MK, Duse DA, et al. Sphingosine-1-Phosphate (S1P) lyase inhibition aggravates atherosclerosis and induces plaque rupture in ApoE−/− mice. Int J Mol Sci. (2022) 23:9606. doi: 10.3390/ijms23179606
135. Chen M, Zhang F, Chen B, Lau C, Xu K, Tong T, et al. Omics approach to reveal the effects of obesity on the protein profiles of the exosomes derived from different adipose depots. Cell Mol Life Sci. (2022) 79:570. doi: 10.1007/s00018-022-04597-4
136. Xie Z, Wang X, Liu X, Du H, Sun C, Shao X, et al. Adipose-derived exosomes exert proatherogenic effects by regulating macrophage foam cell formation and polarization. J Am Heart Assoc. (2018) 7:e007442. doi: 10.1161/JAHA.117.007442
137. Liu Y, Sun Y, Lin X, Zhang D, Hu C, Liu J, et al. Perivascular adipose-derived exosomes reduce macrophage foam cell formation through miR-382-5p and the BMP4-PPARγ-ABCA1/ABCG1 pathways. Vasc Pharmacol. (2022) 143:106968. doi: 10.1016/j.vph.2022.106968
138. Saxton SN, Clark BJ, Withers SB, Eringa EC, Heagerty AM. Mechanistic links between obesity, diabetes, and blood pressure: role of perivascular adipose tissue. Physiol Rev. (2019) 99:1701–63. doi: 10.1152/physrev.00034.2018
139. Kita S, Maeda N, Shimomura I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J Clin Invest (2019) 129(10):4041–9. doi: 10.1172/JCI129193
140. Ivanov D, Philippova M, Antropova J, Gubaeva F, Iljinskaya O, Tararak E, et al. Expression of cell adhesion molecule T-cadherin in the human vasculature. Histochem Cell Biol. (2001) 115:231–42. doi: 10.1007/s004180100252
141. Fujishima Y, Maeda N, Matsuda K, Masuda S, Mori T, Fukuda S, et al. Adiponectin association with T-cadherin protects against neointima proliferation and atherosclerosis. FASEB J. (2017) 31:1571–83. doi: 10.1096/fj.201601064R
142. Obata Y, Kita S, Koyama Y, Fukuda S, Takeda H, Takahashi M, et al. Adiponectin/T-cadherin system enhances exosome biogenesis and decreases cellular ceramides by exosomal release. JCI Insight (2018) 3(8):e99680. doi: 10.1172/jci.insight.99680
143. Chaurasia B, Tippetts TS, Monibas RM, Liu J, Li Y, Wang L, et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science. (2019) 365:386–92. doi: 10.1126/science.aav3722
144. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. (2003) 423:762–9. doi: 10.1038/nature01705
145. Liu L, Shi Z, Ji X, Zhang W, Luan J, Zahr T, et al. Adipokines, adiposity, and atherosclerosis. Cell Mol Life Sci. (2022) 79:272. doi: 10.1007/s00018-022-04286-2
146. Wang F-H, Meng L-Y, Yu T-Y, Tan Y, Quan H, Hu J-Y, et al. Associations of abdominal visceral fat content and plasma adiponectin level with intracranial atherosclerotic stenosis: a cross-sectional study. Front Neurol. (2022) 13:893401. doi: 10.3389/fneur.2022.893401
147. Gasbarrino K, Hafiane A, Gianopoulos I, Zheng H, Mantzoros CS, Daskalopoulou SS. Relationship between circulating adipokines and cholesterol efflux in subjects with severe carotid atherosclerosis. Metab Clin Exp. (2023) 140:155381. doi: 10.1016/j.metabol.2022.155381
148. Gasbarrino K, Zheng H, Hafiane A, Veinot JP, Lai C, Daskalopoulou SS. Decreased adiponectin-mediated signaling through the AdipoR2 pathway is associated with carotid plaque instability. Stroke. (2017) 48:915–24. doi: 10.1161/STROKEAHA.116.015145
149. Fan LH, He Y, Xu W, Tian HY, Zhou Y, Liang Q, et al. Adiponectin may be a biomarker of early atherosclerosis of smokers and decreased by nicotine through KATP channel in adipocytes. Nutrition. (2015) 31:955–8. doi: 10.1016/j.nut.2015.01.010
150. Nour-Eldine W, Ghantous CM, Zibara K, Dib L, Issaa H, Itani HA, et al. Adiponectin attenuates angiotensin II-induced vascular smooth muscle cell remodeling through nitric oxide and the RhoA/ROCK pathway. Front Pharmacol. (2016) 7:86. doi: 10.3389/fphar.2016.00086
151. Hafiane A, Gasbarrino K, Daskalopoulou SS. The role of adiponectin in cholesterol efflux and HDL biogenesis and metabolism. Metabolis. (2019) 100:153953. doi: 10.1016/j.metabol.2019.153953
152. Hafiane A, Daskalopoulou SS. Adiponectin's mechanisms in high-density lipoprotein biogenesis and cholesterol efflux. Metab Clin Exp. (2020) 113:154393. doi: 10.1016/j.metabol.2020.154393
153. Lei S, Chen J, Song C, Li J, Zuo A, Xu D, et al. CTRP9 alleviates foam cells apoptosis by enhancing cholesterol efflux. Mol Cell Endocrinol. (2021) 522:111138. doi: 10.1016/j.mce.2020.111138
154. Luo N, Chung BH, Wang X, Klein RL, Tang C-K, Garvey WT, et al. Enhanced adiponectin actions by overexpression of adiponectin receptor 1 in macrophages. Atherosclerosis. (2013) 228:124–35. doi: 10.1016/j.atherosclerosis.2013.02.026
155. Li Y, Jin L, Jiang F, Yan J, Lu Y, Yang Q, et al. Mutations of NRG4 contribute to the pathogenesis of nonalcoholic fatty liver disease and related metabolic disorders. Diabetes. (2021) 70:2213–24. doi: 10.2337/db21-0064
156. Fisher FM, Maratos-Flier E. Understanding the physiology of FGF21. Annu Rev Physiol. (2016) 78:223–41. doi: 10.1146/annurev-physiol-021115-105339
157. Hondares E, Iglesias R, Giralt A, Gonzalez FJ, Giralt M, Mampel T, et al. Thermogenic activation induces FGF21 expression and release in brown adipose tissue. J Biol Chem. (2011) 286:12983–90. doi: 10.1074/jbc.M110.215889
158. Gao Y, Zhang W, Zeng L-Q, Bai H, Li J, Zhou J, et al. Exercise and dietary intervention ameliorate high-fat diet-induced NAFLD and liver aging by inducing lipophagy. Redox Biol. (2020) 36:101635. doi: 10.1016/j.redox.2020.101635
159. Kurosu H, Choi M, Ogawa Y, Dickson AS, Goetz R, Eliseenkova AV, et al. Tissue-specific expression of βKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem. (2007) 282:26687–95. doi: 10.1074/jbc.M704165200
160. Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. (2005) 115:1627–35. doi: 10.1172/JCI23606
161. Ge X, Chen C, Hui X, Wang Y, Lam KSL, Xu A. Fibroblast growth factor 21 induces glucose transporter-1 expression through activation of the Serum response factor/ets-like protein-1 in adipocytes. J Biol Chem. (2011) 286:34533–41. doi: 10.1074/jbc.M111.248591
162. Lin Z, Tian H, Lam KSL, Lin S, Hoo RCL, Konishi M, et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. (2013) 17:779–89. doi: 10.1016/j.cmet.2013.04.005
163. Huang Z, Zhong L, Lee JTH, Zhang J, Wu D, Geng L, et al. The FGF21-CCL11 axis mediates beiging of white adipose tissues by coupling sympathetic nervous system to type 2 immunity. Cell Metab. (2017) 26:493–508.e4. doi: 10.1016/j.cmet.2017.08.003
164. Li X-H, Liu L-Z, Chen L, Pan Q-N, Ouyang Z-Y, Fan D-J, et al. Aerobic exercise regulates FGF21 and NLRP3 inflammasome-mediated pyroptosis and inhibits atherosclerosis in mice. PLoS One. (2022) 17:e0273527. doi: 10.1371/journal.pone.0273527
165. Xiaolong L, Dongmin G, Liu M, Zuo W, Huijun H, Qiufen T, et al. FGF21 induces autophagy-mediated cholesterol efflux to inhibit atherogenesis via RACK1 up-regulation. J Cell Mol Med. (2020) 24:4992–5006. doi: 10.1111/jcmm.15118
166. Huh JY, Panagiotou G, Mougios V, Brinkoetter M, Vamvini MT, Schneider BE, et al. FNDC5 and irisin in humans: I. Predictors of circulating concentrations in serum and plasma and II. mRNA expression and circulating concentrations in response to weight loss and exercise. Metab Clin Exp. (2012) 61:1725–38. doi: 10.1016/j.metabol.2012.09.002
167. Roca-Rivada A, Castelao C, Senin LL, Landrove MO, Baltar J, Crujeiras AB, et al. FNDC5/irisin is not only a myokine but also an adipokine. PLoS One. (2013) 8:e60563. doi: 10.1371/journal.pone.0060563
168. Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1α-dependent myokine that drives browning of white fat and thermogenesis. Nature. (2012) 481:463–8. doi: 10.1038/nature10777
169. Kim H, Wrann CD, Jedrychowski M, Vidoni S, Kitase Y, Nagano K, et al. Irisin mediates effects on bone and fat via αV integrin receptors. Cell. (2018) 175:1756–68.e17. doi: 10.1016/j.cell.2018.10.025
170. Moreno-Navarrete JM, Ortega F, Serrano M, Guerra E, Pardo G, Tinahones F, et al. Irisin is expressed and produced by human muscle and adipose tissue in association with obesity and insulin resistance. J Clin Endocrinol Metab. (2013) 98:E769–78. doi: 10.1210/jc.2012-2749
171. Pan J, Zhang H, Yu Q, Zhang J, Wang C, Gu J, et al. Association of circulating irisin levels and the characteristics and prognosis of coronary artery disease. Am J Med Sci. (2021) 362:63–71. doi: 10.1016/j.amjms.2021.02.020
172. El-Lebedy DH, Ibrahim AA, Ashmawy IO. Novel adipokines vaspin and irisin as risk biomarkers for cardiovascular diseases in type 2 diabetes mellitus. Diabetes Metab Synd. (2018) 12:643–8. doi: 10.1016/j.dsx.2018.04.025
173. Wu H, Guo P, Jin Z, Li X, Yang X, Tang C, et al. Serum levels of irisin predict short-term outcomes in ischemic stroke. Cytokine. (2019) 122:154303. doi: 10.1016/j.cyto.2018.02.017
174. Li H, Shen J, Wu T, Kuang J, Liu Q, Cheng S, et al. Irisin is controlled by farnesoid X receptor and regulates cholesterol homeostasis. Front Pharmacol. (2019) 10:548. doi: 10.3389/fphar.2019.00548
175. Panagiotou G, Mu L, Na B, Mukamal KJ, Mantzoros CS. Circulating irisin, omentin-1, and lipoprotein subparticles in adults at higher cardiovascular risk. Metab Clin Exp. (2014) 63:1265–71. doi: 10.1016/j.metabol.2014.06.001
176. Zhang M, Xu Y, Jiang L. Irisin attenuates oxidized low-density lipoprotein impaired angiogenesis through AKT/mTOR/S6K1/Nrf2 pathway. J Cell Physiol. (2019) 234:18951–62. doi: 10.1002/jcp.28535
177. Deng X, Huang W, Peng J, Zhu T-T, Sun X-L, Zhou X-Y, et al. Irisin alleviates advanced glycation end products-induced inflammation and endothelial dysfunction via inhibiting ROS-NLRP3 inflammasome signaling. Inflammation. (2018) 41:260–75. doi: 10.1007/s10753-017-0685-3
178. Wang D, Zhang Y, Shen C. Research update on the association between SFRP5, an anti-inflammatory adipokine, with obesity, type 2 diabetes mellitus and coronary heart disease. J Cell Mol Med. (2020) 24:2730–5. doi: 10.1111/jcmm.15023
179. Ouchi N, Higuchi A, Ohashi K, Oshima Y, Gokce N, Shibata R, et al. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science. (2010) 329:454–7. doi: 10.1126/science.1188280
180. Mori H, Prestwich TC, Reid MA, Longo KA, Gerin I, Cawthorn WP, et al. Secreted frizzled-related protein 5 suppresses adipocyte mitochondrial metabolism through WNT inhibition. J Clin Invest. (2012) 122:2405–16. doi: 10.1172/JCI63604
181. Akoumianakis I, Sanna F, Margaritis M, Badi I, Akawi N, Herdman L, et al. Adipose tissue–derived WNT5A regulates vascular redox signaling in obesity via USP17/RAC1-mediated activation of NADPH oxidases. Sci Transl Med. (2019) 11:eaav5055. doi: 10.1126/scitranslmed.aav5055
182. Catalán V, Gómez-Ambrosi J, Rodríguez A, Pérez-Hernández AI, Gurbindo J, Ramírez B, et al. Activation of noncanonical Wnt signaling through WNT5A in visceral adipose tissue of obese subjects is related to inflammation. J Clin Endocrinol Metab. (2014) 99:E1407–17. doi: 10.1210/jc.2014-1191
183. Wang X, Peng Q, Jiang F, Xue L, Li J, Fan Z, et al. Secreted frizzled-related protein 5 protects against oxidative stress-induced apoptosis in human aortic endothelial cells via downregulation of bax. J Biochem Mol Toxicol. (2017) 31:e21978. doi: 10.1002/jbt.21978
184. Bäckdahl J, Franzén L, Massier L, Li Q, Jalkanen J, Gao H, et al. Spatial mapping reveals human adipocyte subpopulations with distinct sensitivities to insulin. Cell Metab. (2021) 33:1869–82.e6. doi: 10.1016/j.cmet.2021.07.018
185. Seoane-Collazo P, Martínez-Sánchez N, Milbank E, Contreras C. Incendiary leptin. Nutrients. (2020) 12:472. doi: 10.3390/nu12020472
186. Beltowski J. Leptin and atherosclerosis. Atherosclerosis. (2006) 189:47–60. doi: 10.1016/j.atherosclerosis.2006.03.003
187. Gorska E, Popko K, Stelmaszczyk-Emmel A, Ciepiela O, Kucharska A, Wasik M. Leptin receptors. Eur J Med Res. (2010) 15:50–4. doi: 10.1186/2047-783X-15-S2-50
188. Stürzebecher PE, Kralisch S, Schubert MR, Filipova V, Hoffmann A, Oliveira F, et al. Leptin treatment has vasculo-protective effects in lipodystrophic mice. Proc Natl Acad Sci U S A. (2022) 119:e2110374119. doi: 10.1073/pnas.2110374119
189. Kjerrulf M, Berke Z, Aspegren A, Umaerus M, Nilsson T, Svensson L, et al. Reduced cholesterol accumulation by leptin deficient (ob/ob) mouse macrophages. Inflamm Res. (2006) 55:300–9. doi: 10.1007/s00011-006-0087-8
190. Liang C-P, Han S, Okamoto H, Carnemolla R, Tabas I, Accili D, et al. Increased CD36 protein as a response to defective insulin signaling in macrophages. J Clin Invest. (2004) 113:764–73. doi: 10.1172/JCI19528
191. Reilly MP, Lehrke M, Wolfe ML, Rohatgi A, Lazar MA, Rader DJ. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. (2005) 111:932–9. doi: 10.1161/01.CIR.0000155620.10387.43
192. Curat CA, Wegner V, Sengenès C, Miranville A, Tonus C, Busse R, et al. Macrophages in human visceral adipose tissue: increased accumulation in obesity and a source of resistin and visfatin. Diabetologia. (2006) 49:744–7. doi: 10.1007/s00125-006-0173-z
193. Qatanani M, Szwergold NR, Greaves DR, Ahima RS, Lazar MA. Macrophage-derived human resistin exacerbates adipose tissue inflammation and insulin resistance in mice. J Clin Invest. (2009) 119:531–9. doi: 10.1172/JCI37273
194. Asterholm IW, Rutkowski JM, Fujikawa T, Cho Y-R, Fukuda M, Tao C, et al. Elevated resistin levels induce central leptin resistance and increased atherosclerotic progression in mice. Diabetologia. (2014) 57:1209–18. doi: 10.1007/s00125-014-3210-3
195. Yanofsky R, Sancho C, Gasbarrino K, Zheng H, Doonan RJ, Jaunet F, et al. Expression of resistin, chemerin, and chemerin’s receptor in the unstable carotid atherosclerotic plaque. Stroke. (2021) 52:2537–46. doi: 10.1161/STROKEAHA.120.030228
196. Sweeney T, Ogunmoroti O, Ndumele CE, Zhao D, Varma B, Allison MA, et al. Associations of adipokine levels with the prevalence and extent of valvular and thoracic aortic calcification: the multi-ethnic study of atherosclerosis (MESA). Atherosclerosis. (2021) 338:15–22. doi: 10.1016/j.atherosclerosis.2021.11.002
197. Verma S, Li S-H, Wang C-H, Fedak PWM, Li R-K, Weisel RD, et al. Resistin promotes endothelial cell activation. Circulation. (2003) 108:736–40. doi: 10.1161/01.CIR.0000084503.91330.49
198. Landmesser U, Hornig B, Drexler H. Endothelial function. Circulation. (2004) 109:II–27. doi: 10.1161/01.CIR.0000129501.88485.1f
199. Hsu W-Y, Chao Y-W, Tsai Y-L, Lien C-C, Chang C-F, Deng M-C, et al. Resistin induces monocyte–endothelial cell adhesion by increasing ICAM-1 and VCAM-1 expression in endothelial cells via p38MAPK-dependent pathway. J Cell Physiol. (2011) 226:2181–8. doi: 10.1002/jcp.22555
200. Park SY, Kim KH, Seo KW, Bae JU, Kim YH, Lee SJ, et al. Resistin derived from diabetic perivascular adipose tissue up-regulates vascular expression of osteopontin via the AP-1 signalling pathway. J Pathol. (2014) 232:87–97. doi: 10.1002/path.4286
201. Rae C, Graham A. Human resistin promotes macrophage lipid accumulation. Diabetologia. (2006) 49:1112–4. doi: 10.1007/s00125-006-0187-6
202. Lee T-S, Lin C-Y, Tsai J-Y, Wu Y-L, Su K-H, Lu K-Y, et al. Resistin increases lipid accumulation by affecting class A scavenger receptor, CD36 and ATP-binding cassette transporter-A1 in macrophages. Life Sci. (2009) 84:97–104. doi: 10.1016/j.lfs.2008.11.004
203. Baenziger NL, Brodie GN, Majerus PW. Isolation and properties of a thrombin-sensitive protein of human platelets. J Biol Chem. (1972) 247:2723–31. doi: 10.1016/S0021-9258(19)45271-X
204. Jaffe EA, Ruggiero JT, Falcone DJ. Monocytes and macrophages synthesize and secrete thrombospondin. Blood. (1985) 65:79–84. doi: 10.1182/blood.V65.1.79.79
205. Varma V, Yao-Borengasser A, Bodles AM, Rasouli N, Phanavanh B, Nolen GT, et al. Thrombospondin-1 is an adipokine associated with obesity, adipose inflammation, and insulin resistance. Diabetes. (2008) 57:432–9. doi: 10.2337/db07-0840
206. Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. (2001) 107:929–34. doi: 10.1172/JCI12749
207. Ganguly R, Khanal S, Mathias A, Gupta S, Lallo J, Sahu S, et al. TSP-1 (Thrombospondin-1) deficiency protects ApoE−/− mice against leptin-induced atherosclerosis. Arterioscler Thromb Vasc Biol. (2021) 41:e112–27. doi: 10.1161/ATVBAHA.120.314962
208. Ramis JM, Franssen-van Hal NLW, Kramer E, Llado I, Bouillaud F, Palou A, et al. Carboxypeptidase E and thrombospondin-1 are differently expressed in subcutaneous and visceral fat of obese subjects. Cell Mol Life Sci. (2002) 59:1960–71. doi: 10.1007/PL00012518
209. Chavez RJ, Haney RM, Cuadra RH, Ganguly R, Adapala RK, Thodeti CK, et al. Upregulation of thrombospondin-1 expression by leptin in vascular smooth muscle cells via JAK2- and MAPK-dependent pathways. Am J Physiol Cell Physiol. (2012) 303:C179–91. doi: 10.1152/ajpcell.00008.2012
210. Sahu S, Ganguly R, Raman P. Leptin augments recruitment of IRF-1 and CREB to thrombospondin-1 gene promoter in vascular smooth muscle cells in vitro. Am J Physiol Cell Physiol. (2016) 311(2):C212–C24. doi: 10.1152/ajpcell.00068.2016
211. Chen D, Asahara T, Krasinski K, Witzenbichler B, Yang J, Magner M, et al. Antibody blockade of thrombospondin accelerates reendothelialization and reduces neointima formation in balloon-injured rat carotid artery. Circulation. (1999) 100:849–54. doi: 10.1161/01.CIR.100.8.849
212. Bolz S-S, Vogel L, Sollinger D, Derwand R, de Wit C, Loirand G, et al. Nitric oxide-induced decrease in calcium sensitivity of resistance arteries is attributable to activation of the myosin light chain phosphatase and antagonized by the RhoA/Rho kinase pathway. Circulation. (2003) 107:3081–7. doi: 10.1161/01.CIR.0000074202.19612.8C
213. Moura R, Tjwa M, Vandervoort P, Cludts K, Hoylaerts MF. Thrombospondin-1 activates medial smooth muscle cells and triggers neointima formation upon mouse carotid artery ligation. Arterioscler Thromb Vasc Biol. (2007) 27:2163–9. doi: 10.1161/ATVBAHA.107.151282
214. Yokoyama-Kobayashi M, Saeki M, Sekine S, Kato S. Human cDNA encoding a novel TGF-β superfamily protein highly expressed in Placenta1. J Biochem. (1997) 122:622–6. doi: 10.1093/oxfordjournals.jbchem.a021798
215. Hsiao EC, Koniaris LG, Zimmers-Koniaris T, Sebald SM, Huynh TV, Lee S-J. Characterization of growth-differentiation factor 15, a transforming growth factor β superfamily member induced following liver injury. Mol Cell Biol. (2000) 20:3742–51. doi: 10.1128/MCB.20.10.3742-3751.2000
216. Campderrós L, Moure R, Cairó M, Gavaldà-Navarro A, Quesada-López T, Cereijo R, et al. Brown adipocytes secrete GDF15 in response to thermogenic activation. Obesity. (2019) 27:1606–16. doi: 10.1002/oby.22584
217. Dostálová I, Roubíček T, Bártlová M, Mráz M, Lacinová Z, Haluzíková D, et al. Increased serum concentrations of macrophage inhibitory cytokine-1 in patients with obesity and type 2 diabetes mellitus: the influence of very low calorie diet. Eur J Endocrinol. (2009) 161:397–404. doi: 10.1530/EJE-09-0417
218. Xiong Y, Walker K, Min X, Hale C, Tran T, Komorowski R, et al. Long-acting MIC-1/GDF15 molecules to treat obesity: evidence from mice to monkeys. Sci Transl Med. (2017) 9(412):eaan8732. eaan8732.doi: doi: 10.1126/scitranslmed.aan8732
219. Sjøberg KA, Sigvardsen CM, Alvarado-Diaz A, Andersen NR, Larance M, Seeley RJ, et al. GDF15 increases insulin action in the liver and adipose tissue via a β-adrenergic receptor-mediated mechanism. Cell Metab. (2023) S1550-4131(23)00226-7. doi: 10.1016/j.cmet.2023.06.016
220. Bonaterra GA, Zügel S, Thogersen J, Walter SA, Haberkorn U, Strelau J, et al. Growth differentiation factor-15 deficiency inhibits atherosclerosis progression by regulating interleukin-6–dependent inflammatory response to vascular injury. J Am Heart Assoc. (2012) 1:e002550. doi: 10.1161/JAHA.112.002550
221. Tsai VW-W, Zhang HP, Manandhar R, Schofield P, Christ D, Lee-Ng KKM, et al. GDF15 mediates adiposity resistance through actions on GFRAL neurons in the hindbrain AP/NTS. Int J Obes. (2019) 43:2370–80. doi: 10.1038/s41366-019-0365-5
222. Kempf T, Sinning J-M, Quint A, Bickel C, Sinning C, Wild PS, et al. Growth-differentiation factor-15 for risk stratification in patients with stable and unstable coronary heart disease: results from the AtheroGene study. Circ Cardiovasc Genet. (2009) 2:286–92. doi: 10.1161/CIRCGENETICS.108.824870
223. Wang W, Song X-T, Chen Y-D, Yuan F, Xu F, Zhang M, et al. Growth differentiation factor-15 is a prognostic marker in patients with intermediate coronary artery disease. J Geriatr Cardiol. (2020) 17:210–6. doi: 10.11909/j.issn.1671-5411.2020.04.004
224. Heduschke A, Ackermann K, Wilhelm B, Mey L, Bonaterra GA, Kinscherf R, et al. GDF-15 deficiency reduces autophagic activity in human macrophages in vitro and decreases p62-accumulation in atherosclerotic lesions in mice. Cells. (2021) 10:2346. doi: 10.3390/cells10092346
225. Kim Y, Noren Hooten N, Evans MK. CRP stimulates GDF15 expression in endothelial cells through p53. Mediators Inflamm. (2018) 2018:8278039. doi: 10.1155/2018/8278039
226. Guo D, Lin C, Lu Y, Guan H, Qi W, Zhang H, et al. FABP4 secreted by M1-polarized macrophages promotes synovitis and angiogenesis to exacerbate rheumatoid arthritis. Bone Res. (2022) 10:45. doi: 10.1038/s41413-022-00211-2
227. Song J, Ren P, Zhang L, Wang XL, Chen L, Shen YH. Metformin reduces lipid accumulation in macrophages by inhibiting FOXO1-mediated transcription of fatty acid-binding protein 4. Biochem Biophys Res Commun. (2010) 393:89–94. doi: 10.1016/j.bbrc.2010.01.086
228. Llaverias G, Noé V, Peñuelas S, Vázquez-Carrera M, Sánchez RM, Laguna JC, et al. Atorvastatin reduces CD68, FABP4, and HBP expression in oxLDL-treated human macrophages. Biochem Biophys Res Commun. (2004) 318:265–74. doi: 10.1016/j.bbrc.2004.04.021
229. Elmasri H, Karaaslan C, Teper Y, Ghelfi E, Weng M, Ince TA, et al. Fatty acid binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J. (2009) 23:3865–73. doi: 10.1096/fj.09-134882
230. Fuseya T, Furuhashi M, Matsumoto M, Watanabe Y, Hoshina K, Mita T, et al. Ectopic fatty acid–binding protein 4 expression in the vascular endothelium is involved in neointima formation after vascular injury. JAHA. (2017) 6:e006377. doi: 10.1161/JAHA.117.006377
231. Furuhashi M, Tuncman G, Görgün CZ, Makowski L, Atsumi G, Vaillancourt E, et al. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. (2007) 447:959–65. doi: 10.1038/nature05844
232. Schäffler A, Buechler C. CTRP family: linking immunity to metabolism. Trends Endocrinol Metab. (2012) 23:194–204. doi: 10.1016/j.tem.2011.12.003
233. Lasser G, Guchhait P, Ellsworth JL, Sheppard P, Lewis K, Bishop P, et al. C1qTNF–related protein-1 (CTRP-1): a vascular wall protein that inhibits collagen-induced platelet aggregation by blocking VWF binding to collagen. Blood. (2006) 107:423–30. doi: 10.1182/blood-2005-04-1425
234. Wang XQ, Liu ZH, Xue L, Lu L, Gao J, Shen Y, et al. C1q/TNF-related protein 1 links macrophage lipid metabolism to inflammation and atherosclerosis. Atherosclerosis. (2016) 250:38–45. doi: 10.1016/j.atherosclerosis.2016.04.024
235. Schmid A, Vlacil A-K, Schuett J, Karrasch T, Schieffer B, Schäffler A, et al. Anti-inflammatory effects of C1q/tumor necrosis factor-related protein 3 (CTRP3) in endothelial cells. Cells. (2021) 10:2146. doi: 10.3390/cells10082146
236. Weigert J, Neumeier M, Schäffler A, Fleck M, Schölmerich J, Schütz C, et al. The adiponectin paralog CORS-26 has anti-inflammatory properties and is produced by human monocytic cells. FEBS Lett. (2005) 579:5565–70. doi: 10.1016/j.febslet.2005.09.022
237. Micallef P, Vujičić M, Wu Y, Peris E, Wang Y, Chanclón B, et al. C1QTNF3 is upregulated during subcutaneous adipose tissue remodeling and stimulates macrophage chemotaxis and M1-like polarization. Front Immunol. (2022) 13:914956. doi: 10.3389/fimmu.2022.914956
238. Matloch Z, Mraz M, Kasperova BJ, Kratochvilova H, Svoboda P, Pleyerova I, et al. Decreased epicardial CTRP3 mRNA levels in patients with type 2 diabetes Mellitus and coronary artery disease undergoing elective cardiac surgery: a possible association with coronary atherosclerosis. Int J Mol Sci. (2022) 23:9988. doi: 10.3390/ijms23179988
239. Schwartze JT, Landgraf K, Spielau U, Rockstroh D, Löffler D, Kratzsch J, et al. Adipocyte C1QTNF5 expression is BMI-dependently related to early adipose tissue dysfunction and systemic CTRP5 serum levels in obese children. Int J Obes (Loncd). (2017) 41:955–63. doi: 10.1038/ijo.2017.54
240. Liu J, Meng Z, Gan L, Guo R, Gao J, Liu C, et al. C1q/TNF-related protein 5 contributes to diabetic vascular endothelium dysfunction through promoting nox-1 signaling. Redox Biol. (2020) 34:101476. doi: 10.1016/j.redox.2020.101476
241. Rao C, Huang D, Mao X, Chen R, Huang D, Huang K. The novel adipokine CTRP5 is a negative regulator of white adipose tissue browning. Biochem Biophys Res Commun. (2019) 510:388–94. doi: 10.1016/j.bbrc.2019.01.111
242. Peterson JM, Wei Z, Wong GW. CTRP8 and CTRP9B are novel proteins that hetero-oligomerize with C1q/TNF family members. Biochem Biophys Res Commun. (2009) 388:360–5. doi: 10.1016/j.bbrc.2009.08.014
243. Zhang H, Zhang-Sun Z, Xue C, Li X, Ren J, Jiang Y, et al. CTRP family in diseases associated with inflammation and metabolism: molecular mechanisms and clinical implication. Acta Pharmacol Sin. (2022) 44(4):710–25. doi: 10.1038/s41401-022-00991-7
244. Es K, Bh Y, Sm L MC, Eh K, Bw L, Sy K, et al. Fecal microbiota transplantation ameliorates atherosclerosis in mice with C1q/TNF-related protein 9 genetic deficiency. Exp Mol Med. (2022) 54(2):103–14. doi: 10.1038/s12276-022-00728-w
245. Zhang L, Liu Q, Zhang H, Wang X-D, Chen S-Y, Yang Y, et al. C1q/TNF-related protein 9 inhibits THP-1 macrophage foam cell formation by enhancing autophagy. J Cardiovasc Pharmacol. (2018) 72:167–75. doi: 10.1097/FJC.0000000000000612
246. Yamaguchi S, Shibata R, Ohashi K, Enomoto T, Ogawa H, Otaka N, et al. C1q/TNF-related protein 9 promotes revascularization in response to ischemia via an eNOS-dependent manner. Front Pharmacol. (2020) 11:1313. doi: 10.3389/fphar.2020.01313
247. Enomoto T, Shibata R, Ohashi K, Kambara T, Kataoka Y, Uemura Y, et al. Regulation of adipolin/CTRP12 cleavage by obesity. Biochem Biophys Res Commun. (2012) 428:155–9. doi: 10.1016/j.bbrc.2012.10.031
248. Wei Z, Peterson JM, Lei X, Cebotaru L, Wolfgang MJ, Baldeviano GC, et al. C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J Biol Chem. (2012) 287:10301–15. doi: 10.1074/jbc.M111.303651
249. Enomoto T, Ohashi K, Shibata R, Higuchi A, Maruyama S, Izumiya Y, et al. Adipolin/C1qdc2/CTRP12 protein functions as an adipokine that improves glucose metabolism. J Biol Chem. (2011) 286:34552–8. doi: 10.1074/jbc.M111.277319
250. Revollo JR, Körner A, Mills KF, Satoh A, Wang T, Garten A, et al. Nampt/PBEF/visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab. (2007) 6:363–75. doi: 10.1016/j.cmet.2007.09.003
251. Wang P, Miao C-Y. NAMPT as a therapeutic target against stroke. Trends Pharmacol Sci. (2015) 36:891–905. doi: 10.1016/j.tips.2015.08.012
252. Dahl TB, Yndestad A, Skjelland M, Øie E, Dahl A, Michelsen A, et al. Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis. Circulation. (2007) 115:972–80. doi: 10.1161/CIRCULATIONAHA.106.665893
253. Schäffler A, Neumeier M, Herfarth H, Fürst A, Schölmerich J, Büchler C. Genomic structure of human omentin, a new adipocytokine expressed in omental adipose tissue. Biochim Biophys Acta Gene Struct Exp. (2005) 1732:96–102. doi: 10.1016/j.bbaexp.2005.11.005
254. Yang R-Z, Lee M-J, Hu H, Pray J, Wu H-B, Hansen BC, et al. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab. (2006) 290:E1253–61. doi: 10.1152/ajpendo.00572.2004
255. Batista CM DS, Yang R-Z, Lee M-J, Glynn NM, Yu D-Z, Pray J, et al. Omentin plasma levels and gene expression are decreased in obesity. Diabetes. (2007) 56:1655–61. doi: 10.2337/db06-1506
256. Watanabe K, Watanabe R, Konii H, Shirai R, Sato K, Matsuyama T-A, et al. Counteractive effects of omentin-1 against atherogenesis. Cardiovasc Res. (2016) 110:118–28. doi: 10.1093/cvr/cvw016
257. Yin C, Hu W, Wang M, Xiao Y. The role of the adipocytokines vaspin and visfatin in vascular endothelial function and insulin resistance in obese children. BMC Endocr Disord. (2019) 19:127. doi: 10.1186/s12902-019-0452-6
258. Suleymanoglu S, Tascilar E, Pirgon O, Tapan S, Meral C, Abaci A. Vaspin and its correlation with insulin sensitivity indices in obese children. Diabetes Res Clin Pract. (2009) 84:325–8. doi: 10.1016/j.diabres.2009.03.008
259. Hida K, Wada J, Eguchi J, Zhang H, Baba M, Seida A, et al. Visceral adipose tissue-derived serine protease inhibitor: a unique insulin-sensitizing adipocytokine in obesity. Proc Natl Acad Sci U S A. (2005) 102:10610–5. doi: 10.1073/pnas.0504703102
260. Gao J-H, Zeng M-Y, Yu X-H, Zeng G-F, He L-H, Zheng X-L, et al. Visceral adipose tissue-derived serine protease inhibitor accelerates cholesterol efflux by up-regulating ABCA1 expression via the NF-κB/miR-33a pathway in THP-1 macropahge-derived foam cells. Biochem Biophys Res Commun. (2018) 500:318–24. doi: 10.1016/j.bbrc.2018.04.066
261. Phalitakul S, Okada M, Hara Y, Yamawaki H. Vaspin prevents methylglyoxal-induced apoptosis in human vascular endothelial cells by inhibiting reactive oxygen species generation. Acta Physiol. (2013) 209:212–9. doi: 10.1111/apha.12139
262. Romere C, Duerrschmid C, Bournat J, Constable P, Jain M, Xia F, et al. Asprosin, a fasting-induced glucogenic protein hormone. Cell. (2016) 165:566–79. doi: 10.1016/j.cell.2016.02.063
263. Zhong L, Long Y, Wang S, Lian R, Deng L, Ye Z, et al. Continuous elevation of plasma asprosin in pregnant women complicated with gestational diabetes mellitus: a nested case-control study. Placenta. (2020) 93:17–22. doi: 10.1016/j.placenta.2020.02.004
264. Kocaman N, Kuloğlu T. Expression of asprosin in rat hepatic, renal, heart, gastric, testicular and brain tissues and its changes in a streptozotocin-induced diabetes mellitus model. Tissue Cell. (2020) 66:101397. doi: 10.1016/j.tice.2020.101397
265. Mishra I, Xie WR, Bournat JC, He Y, Wang C, Silva ES, et al. Protein tyrosine phosphatase receptor δ serves as the orexigenic asprosin receptor. Cell Metab. (2022) 34:549–63.e8. doi: 10.1016/j.cmet.2022.02.012
266. Güven C, Kafadar H. Evaluation of plasma asprosin concentration in patients with coronary artery disease. Braz J Cardiovasc Surg. (2022) 37:493–500. doi: 10.21470/1678-9741-2021-0003
267. Miao Y, Qin H, Zhong Y, Huang K, Rao C. Novel adipokine asprosin modulates browning and adipogenesis in white adipose tissue. J Endocrinol. (2021) 249:83–93. doi: 10.1530/JOE-20-0503
268. Summers SA, Chaurasia B, Holland WL. Metabolic messengers: ceramides. Nat Metab. (2019) 1:1051–8. doi: 10.1038/s42255-019-0134-8
269. Vasiliauskaité-Brooks I, Sounier R, Rochaix P, Bellot G, Fortier M, Hoh F, et al. Structural insights into adiponectin receptors suggest ceramidase activity. Nature. (2017) 544:120–3. doi: 10.1038/nature21714
270. Kojta I, Chacińska M, Błachnio-Zabielska A. Obesity, bioactive lipids, and adipose tissue inflammation in insulin resistance. Nutrients. (2020) 12:1305. doi: 10.3390/nu12051305
271. Chaurasia B, Summers SA. Ceramides in metabolism: key lipotoxic players. Annu Rev Physiol. (2021) 83:303–30. doi: 10.1146/annurev-physiol-031620-093815
272. Gui Y, Li Q, Liu L, Zeng P, Ren R-F, Guo Z-F, et al. Plasma levels of ceramides relate to ischemic stroke risk and clinical severity. Brain Res Bull. (2020) 158:122–7. doi: 10.1016/j.brainresbull.2020.03.009
273. Edsfeldt A, Dunér P, Ståhlman M, Mollet IG, Asciutto G, Grufman H, et al. Sphingolipids contribute to human atherosclerotic plaque inflammation. ATVB. (2016) 36:1132–40. doi: 10.1161/ATVBAHA.116.305675
274. Bietrix F, Lombardo E, van Roomen CPAA, Ottenhoff R, Vos M, Rensen PCN, et al. Inhibition of glycosphingolipid synthesis induces a profound reduction of plasma cholesterol and inhibits atherosclerosis development in APOE*3 Leiden and low-density lipoprotein receptor−/− mice. Arterioscler Thromb Vasc Biol. (2010) 30:931–7. doi: 10.1161/ATVBAHA.109.201673
275. Zhang Y, Wang L, Pan Q, Yang X, Cao Y, Yan J, et al. Selective sphingosine-1-phosphate receptor 1 modulator attenuates blood-brain barrier disruption following traumatic brain injury by inhibiting vesicular transcytosis. Fluids Barriers CNS. (2022) 19:57. doi: 10.1186/s12987-022-00356-6
276. Xu D, Gao Q, Wang F, Peng Q, Wang G, Wei Q, et al. Sphingosine-1-phosphate receptor 3 is implicated in BBB injury via the CCL2-CCR2 axis following acute intracerebral hemorrhage. CNS Neurosci Ther. (2021) 27:674–86. doi: 10.1111/cns.13626
277. Estrada R, Zeng Q, Lu H, Sarojini H, Lee J-F, Mathis SP, et al. Up-regulating sphingosine 1-phosphate receptor-2 signaling impairs chemotactic, wound-healing, and morphogenetic responses in senescent endothelial cells. J Biol Chem. (2008) 283:30363–75. doi: 10.1074/jbc.M804392200
278. Alganga H, Almabrouk TAM, Katwan OJ, Daly CJ, Pyne S, Pyne NJ, et al. Short periods of hypoxia upregulate sphingosine kinase 1 and increase vasodilation of arteries to sphingosine 1-phosphate (S1P) via S1P3. J Pharmacol Exp Ther. (2019) 371:63–74. doi: 10.1124/jpet.119.257931
279. Damirin A, Tomura H, Komachi M, Liu J-P, Mogi C, Tobo M, et al. Role of lipoprotein-associated lysophospholipids in migratory activity of coronary artery smooth muscle cells. Am J Physiol Heart Circ Physiol. (2007) 292:H2513–22. doi: 10.1152/ajpheart.00865.2006
280. Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, Hedrick CC. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ Res. (2008) 102:950–8. doi: 10.1161/CIRCRESAHA.107.170779
281. Hansen L, Lohfink N, Vutukuri R, Kestner R-I, Trautmann S, Hecht M, et al. Endothelial sphingosine-1-phosphate receptor 4 regulates blood-brain barrier permeability and promotes a homeostatic endothelial phenotype. J Neurosci. (2022) 42:1908–29. doi: 10.1523/JNEUROSCI.0188-21.2021
282. van Doorn R, Lopes Pinheiro MA, Kooij G, Lakeman K, van het Hof B, van der Pol SMA, et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J Neuroinflamm. (2012) 9:133. doi: 10.1186/1742-2094-9-133
283. Zhang H-G, Grizzle WE. Exosomes A novel pathway of local and distant intercellular communication that facilitates the growth and metastasis of neoplastic lesions. Am J Pathol. (2014) 184:28–41. doi: 10.1016/j.ajpath.2013.09.027
284. Wang H, Xie Y, Salvador AM, Zhang Z, Chen K, Li G, et al. Exosomes: multifaceted messengers in atherosclerosis. Curr Atheroscler Rep. (2020) 22:57. doi: 10.1007/s11883-020-00871-7
285. Mei R, Qin W, Zheng Y, Wan Z, Liu L. Role of adipose tissue derived exosomes in metabolic disease. Front Endocrinol (Lausanne). (2022) 13:873865. doi: 10.3389/fendo.2022.873865
286. Zhang Y, Bai X, Shen K, Luo L, Zhao M, Xu C, et al. Exosomes derived from adipose mesenchymal stem cells promote diabetic chronic wound healing through SIRT3/SOD2. Cells. (2022) 11:2568. doi: 10.3390/cells11162568
287. Sun Y, Ju Y, Fang B. Exosomes from human adipose-derived mesenchymal stromal/stem cells accelerate angiogenesis in wound healing: implication of the EGR-1/lncRNA-SENCR/DKC1/VEGF-A axis. Hum Cell. (2022) 35:1375–90. doi: 10.1007/s13577-022-00732-2
288. Shi R, Jin Y, Zhao S, Yuan H, Shi J, Zhao H. Hypoxic ADSC-derived exosomes enhance wound healing in diabetic mice via delivery of circ-Snhg11 and induction of M2-like macrophage polarization. Biomed Pharmacother. (2022) 153:113463. doi: 10.1016/j.biopha.2022.113463
289. Qin Z, Li Y, Li J, Jiang L, Zhang Z, Chang K, et al. Exosomal STAT1 derived from high phosphorus-stimulated vascular endothelial cells induces vascular smooth muscle cell calcification via the Wnt/β-catenin signaling pathway. Int J Mol Med. (2022) 50:1–13. doi: 10.3892/ijmm.2022.5195
290. Xie X, Guo L-W, Kent CK. Mir548ai antagonism attenuates exosome-induced endothelial cell dysfunction. Cell Death Discov. (2021) 7:318. doi: 10.1038/s41420-021-00720-9
291. Boyer MJ, Kimura Y, Akiyama T, Baggett AY, Preston KJ, Scalia R, et al. Endothelial cell-derived extracellular vesicles alter vascular smooth muscle cell phenotype through high-mobility group box proteins. J Extracell Vesicles. (2020) 9:1781427. doi: 10.1080/20013078.2020.1781427
292. Hu N, Zeng X, Tang F, Xiong S. Exosomal long non-coding RNA LIPCAR derived from oxLDL-treated THP-1 cells regulates the proliferation of human umbilical vein endothelial cells and human vascular smooth muscle cells. Biochem Biophys Res Commun. (2021) 575:65–72. doi: 10.1016/j.bbrc.2021.08.053
Keywords: adipokine, atherosclerosis, endothelial cell, vascular smooth muscle cell, macrophage
Citation: Luo J, He Z, Li Q, Lv M, Cai Y, Ke W, Niu X and Zhang Z (2023) Adipokines in atherosclerosis: unraveling complex roles. Front. Cardiovasc. Med. 10:1235953. doi: 10.3389/fcvm.2023.1235953
Received: 7 June 2023; Accepted: 2 August 2023;
Published: 14 August 2023.
Edited by:
Adriana Georgescu, Institute of Cellular Biology and Pathology (ICBP), RomaniaReviewed by:
Di Wang, Shanghai Jiao Tong University School of Medicine, ChinaChongxiu Sun, Nanjing Medical University, China
Ibrahim AlZaim, Aarhus University, Denmark
Zhaohua Cai, Shanghai Jiao Tong University, China
Sathish Babu Vasamsetti, University of Pittsburgh, United States
© 2023 Luo, He, Li, Lv, Cai, Ke, Niu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuan Niu cm1uaXV4dWFuQHdodS5lZHUuY24= Zhaohui Zhang emh6aHFpbmcxOTkwQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship