 Sepiso K. Masenga
Sepiso K. Masenga Annet Kirabo
Annet Kirabo- 1HAND Research Group, School of Medicine and Health Sciences, Mulungushi University, Livingstone Cam-Pus, Livingstone, Zambia
- 2School of Medicine, University of Zambia, Lusaka, Zambia
- 3Department of Medicine, Vanderbilt University Medical Centre, Nashville, TN, United States
Hypertensive heart disease constitutes functional and structural dysfunction and pathogenesis occurring primarily in the left ventricle, the left atrium and the coronary arteries due to chronic uncontrolled hypertension. Hypertensive heart disease is underreported and the mechanisms underlying its correlates and complications are not well elaborated. In this review, we summarize the current understanding of hypertensive heart disease, we discuss in detail the mechanisms associated with development and complications of hypertensive heart disease especially left ventricular hypertrophy, atrial fibrillation, heart failure and coronary artery disease. We also briefly highlight the role of dietary salt, immunity and genetic predisposition in hypertensive heart disease pathogenesis.
Introduction
Hypertensive heart disease is a term applied to abnormalities of the heart, involving structure and function of the left ventricle, the left atrium and intramural coronary arteries due to sustained elevated blood pressure (1). Although the blood pressure cut-off criteria for the diagnosis of hypertension differs based on the American (2) and European (3, 4) guidelines, most of the recommendations are similar (5). Moreover, the complications of chronic hypertension remain the same. Consensus on the criteria for hypertensive heart disease is not yet universal. However the European criteria as proposed by Gonzalez-Maqueda et al. from the Spanish Society of Cardiology define and classify hypertensive heart disease based on the acronym “VIA” referring to alterations of function and structure occurring in the left ventricle (V), myocardial ischaemia (I), and atrial fibrillation (6). In general, both the European and American hypertension guidelines or other international working groups/societies agree that hypertensive heart disease may involve left ventricular hypertrophy (LVH), left atrial dilatation, systolic and diastolic dysfunction including some clinical symptoms or manifestations such as arrhythmias, myocardial ischemia and heart failure (1, 6). LVH is one of the earliest manifestations of hypertensive heart disease and is thought to be a compensatory mechanism to minimize the increase in ventricular wall stress and an intermediate pathological change in the advancement of hypertensive heart disease (7). However, LVH may progress to complications such as heart failure, arrhythmias, sudden cardiac arrest, ischemic stroke, end stage renal disease (ESRD) and death (8–10).
Globally, the age-standardized prevalence of hypertension in women and in men was 32% and 34%, respectively (11). The prevalence has been increasing with time (12), by more than 138% between 1990 and 2019, affecting a total of 20 million (13). Among persons with hypertension, LVH prevalence is about 40% (14) and black persons tend to have increased left ventricular mass and more severe diastolic dysfunction compared to white persons (1, 15, 16).
Risk factors of hypertensive heart disease
Hypertension is the most common risk factor for development of hypertensive heart disease (1). Additional risk factors include older age, ethnicity, being overweight, physical inactivity, excess dietary salt intake, smoking, alcohol intake, concomitant diseases such as diabetes mellitus (17–19). All these factors contribute to an increased hemodynamic stress on the heart and with chronicity, the left ventricle of the heart hypertrophies to compensate for the load, but in the long run this can lead to heart failure (Figure 1). Obesity is an important risk factor for the development of hypertensive heart disease due to the associated increase in renin secretion that is mediated by leptin production via adipose cells, resulting in blood pressure elevation and exacerbating already existing hypertension (20). Moreover, obesity-related increase in sympathetic tone counteracts the body's compensatory mechanisms for the abnormally elevated renin and aldosterone level that induce cardiac fibrosis and endothelial dysfunction and contribute to the development and complications of hypertensive heart disease (20, 21). Beside altering hemodynamics, obesity also contributes to hypertensive heart disease by inducing inflammation, lipid accumulation in tissue and dysregulating several intracellular pathways (22–24). Obesity and hypertension synergize through overlapping neurohormonal pathways and contribute to hypertensive heart disease and its complications such as LVH and heart failure (25). However, the mechanisms are still complex.
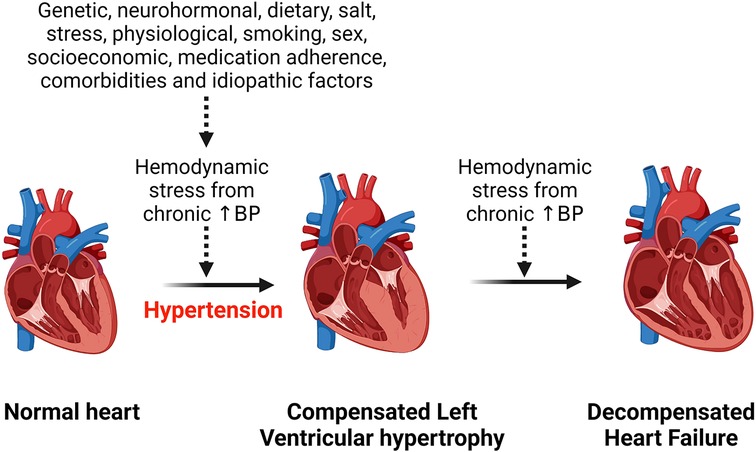
Figure 1. Effect of chronic hemodynamic stress on the heart. Chronic hypertension resulting from various stimuli leads to left ventricular hypertrophy as a compensation for the hemodynamic stress and metabolic demand on the heart. However, heart failure results when the heart can no longer withstand the persistent hemodynamic stress. BP, blood pressure.
Mechanisms of hypertensive heart disease and its complications
The common complication of hypertensive heart disease is either diastolic heart failure, systolic failure, or a combination of the two and individuals with hypertensive heart disease have a higher risk for development of atrial fibrillation (AFib), perioperative ischaemia, coronary artery disease, rehospitalization, kidney disease, heart valve diseases, aortic dissection, intramural hematoma, and aortic ulcer (26–28).
Left ventricular hypertrophy mechanisms in hypertensive heart disease
LVH is an early compensatory response to hemodynamic overload in chronic hypertension (29, 30). LVH can predispose to cardiovascular events in individuals with hypertension who have coronary artery calcifications but without symptoms (31) or in individuals with hypertension independent of treatment and other existing cardiovascular risk factors (32). In addition, malignant LVH characterized by elevated biomarkers of cardiac injury and hemodynamic stress such as N-terminal prohormone of brain natriuretic peptide (NT-proBNP) and troponins is associated with severe adverse outcomes such as heart failure with reduced ejection fraction, left ventricular dysfunction and cardiovascular death (33). Thus, high-sensitive cardiac troponin T (hs-cTnT) and NT-proBNP levels can be used to identify patients who are more likely to develop adverse outcomes (34). It is also important to note that malignant LVH and related adverse events may be more pronounced in black persons compared to white persons (35). However, intensive therapy to lower blood pressure can prevent malignant LVH and reduce the risk for adverse events (36). The pathogenesis and severity of LVH is different by sex. For example, aortic characteristic impedance, systemic vascular resistance, augmentation index, and carotid-femoral pulse wave velocity and proximal aortic compliance are independently associated with relative wall thickness in women but not in men (37). LVH is therefore, more often and more pronounced in women compared to men (38, 39). LVH can be pathological and physiological in pattern (40, 41). The distinguishing pathological changes include increased extracellular connective tissue relative to myocytes without commensurate capillary growth, and myocardial fibrosis that often manifests as diastolic dysfunction (1, 30, 42). In Physiological LVH, extracellular matrix and micro-vessel increase is proportional to the myocyte hypertrophy without deleterious effects on left ventricular function (42). This physiological pattern is seen as a normal response to physical exercise (42).
The mechanisms by which chronic hypertension leads to LVH may involve gap junction lateralization and overexpression of the fatty acid transporter cluster of differentiation 36 (CD36), redox-sensitive Protein Kinase C (PKC), increased oxidative stress, increased matrix metalloproteinase-2 (MMP-2) activity, abnormal Ca2+ homeostasis, increased activation of the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway, apoptosis, and abnormal regulation of junctional proteins (7, 43). All these compensatory changes participate in an orchestrated manner to compensate for increased stress, metabolic and functional demand but are likely to also induce decompensated heart failure when the pathological insult is persistent (7, 44, 45). At cellular level, LVH is a consequence of an increase in cardiac myocyte size due to functional demand but chronic blood pressure elevations may lead to death of cardiomyocytes progressing to dilated cardiomyopathy, a feature described as a transition from compensated to decompensated heart failure and mediated by neurohormonal signaling (45). The whole mark and characteristic feature of LVH is an increase in cardiomyocyte with fibrotic changes including medial hypertrophy and perivascular fibrosis (1). The structural features of LVH can either take concentric or an eccentric pattern depending on volume load, genetic factors, specific alterations of the extracellular matrix, neurohormonal milieu, pressure load severity, duration, rapidity of onset, concomitant medical conditions such as cardiovascular disease, metabolic disease such as diabetes mellitus and demographic factors such as age, race/ethnicity, gender (1, 46). The ultimate consequence of LVH with continued hemodynamic stress is progression to heart failure with either a preserved or a reduced left ventricular ejection fraction (47, 48). There is evidence of LVH regression with use of different classes of antihypertensive medication, however, there is considerable variability in individual responses including sex differences (49) owing to variability of pathological changes per individual and a number of studies have reported failure to regress LVH even when blood pressure is controlled (49–54).
Several proteins, mediators and cellular responses such as angiotensin II, cardiac myosin-binding protein C, endothelin-1, S-thiolated protein, norepinephrine, Rho and Ras proteins, thiols, oxidative stress, heat shock proteins, Fractalkine/C-X3-C Motif Chemokine Ligand 1 (CX3CL1), leukotriene-A4 hydrolase, calcineurin, and some kinases have been implicated in LVH which is associated with chronic elevated blood pressure (42, 55–57). The enzymatic cleavage of angiotensinogen by renin converts angiotensinogen to Angiotensin I and then angiotensin converting enzyme (ACE) converts Angiotensin I to Angiotensin II (58, 59). Angiotensin II is the main effector molecule of the renin angiotensin aldosterone system (RAAS) that serves to control blood pressure (58). Angiotensin II increases blood pressure by inducing water and sodium reabsorption, inducing vasoconstriction and exerting proliferative, pro-inflammatory and pro-fibrotic activities by binding to angiotensin type 1 and 2 receptors (59, 60) (Figure 2).
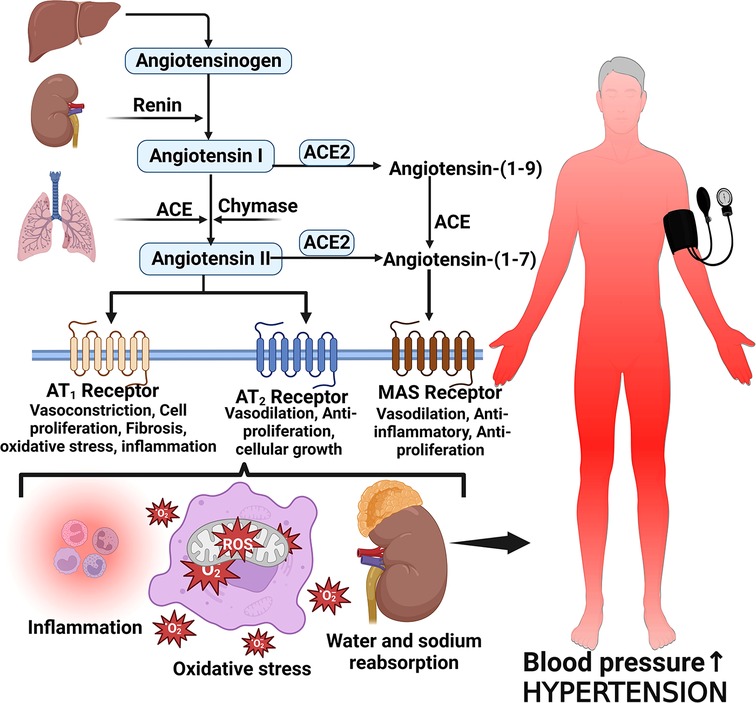
Figure 2. Angiotensin II formation and effects on blood pressure. Angiotensinogen is produced by the liver and converted to angiotensin I by the action of renin produced by the juxtaglomerular cells of the kidney. Angiotensin I is then converted to angiotensin II via angiotensin-converting enzyme (ACE). Angiotensin II binds to its receptors to induce several activities such as inflammation, vasoconstriction oxidative stress and reabsorption of water and sodium leading to elevated blood pressure and hypertension; MAS, marker assisted selected.
Angiotensin II increases blood pressure and induces pathological features characteristic of hypertensive heart disease by activating angiotensin II receptors, regulating cardiac contractility, cardiac remodeling, growth, inflammation, apoptosis and impulse propagation (59, 61). Angiotensin II activates extracellular signal-regulated kinase (ERK)/ mitogen-activated protein kinases (MAPKs) pathways via G protein-coupled receptors (GPCRs) or growth factor-stimulated tyrosine kinase receptors leading to an increase in protein synthesis, extracellular matrix proteins and activation of endothelin-1 effects which result in induction of cardiac fibrosis (42). Heat shock proteins 90 (HSP90) mediate cardiac hypertrophy that is induced by angiotensin II through the stabilization of IкB kinase (IKK) complex (62). Activation of the Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) during hemodynamic stress, inflammation and reactive oxygen species (ROS) production in cardiac myocytes also contributes to cardiac hypertrophy in hypertensive heart disease (42).
Another mechanism associated with LVH is activation of calcineurin and calmodulin kinase II (CaMKII) due to enhanced sensitivity to calcium resulting in calcineurin binding to and dephosphorylating nuclear factor of activated T cells (NFAT)(42, 63). This step increases hypertrophic gene expression through mechanisms that are not yet clear (63). Intergrins have also been implicated in LVH mediated by RAAS and MAPKs pathways (42).
Mechanisms of heart failure associated with hypertensive heart disease
LVH progresses to heart failure when the compensatory mechanisms have failed to meet the functional and metabolic demands of the myocardium (64). Heart failure is a progressive clinical syndrome characterized by reduced ability of the heart to pump blood to meet the body's metabolic demand (65, 66). Symptoms include dyspnoea, fatigue, peripheral oedema or distended jugular veins (65, 66). The symptoms/signs are caused by abnormality in cardiac function and structure and the clinical syndrome is characterized by an increase in natriuretic peptide levels and pulmonary or systemic congestion (67). Heart failure can either be acute or chronic (68). The classification of heart failure is based on left ventricular ejection fraction (LVEF) and can present either with reduced ejection fraction (HFrEF)) or preserved ejection fraction (HFpEF) (65, 67). The clinical stages of heart failure based on United states (US) guidelines fall into four categories (67, 69) as shown in Table 1. The pathogenesis, risk factors and therapeutic response in heart failure is sex dependent. For example, women are more susceptible to traditional risk factors for heart failure and have more severe symptoms especially with higher left ventricular ejection fraction compared to men (70, 71). However, in terms of the adverse outcomes such as hospitalization and mortality, the prognosis regardless of the ejection fraction state, appears to be better for women than men (72). In general, specific data on sex differences in heart failure is still limited due to the fact that women are underrepresented in most studies (72). Also, while women may have more disease severity on some outcomes, this is not the case with other outcomes or symptoms such as plaque rupture which is more common in men (71, 73).

Table 1. Clinical stages of heart failure based on symptoms according to AHA/ACC/HFSA guidelines.
The transition from hypertrophy to heart failure in hypertensive heart disease is driven by several cellular mediators many of which are progressive changes already explained under LVH and these include oxidant stress, apoptosis, insufficient angiogenesis, mitochondria dysfunction, metabolic derangements and fetal gene program induction (74, 75). The underlying pathways and cellular mediators that are activated to mediate the pathology of heart failure in hypertensive heart disease include peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) and PGC-1β (64), GPCRs, p38, ERK1/2, JNK, CAMKII, protein kinases (type C, G, and A), Growth factor-mediated stimulation of mechanistic target of rapamycin (mTOR) (74), epigenetic modulators (such as NFAT, MEF2) and transient receptor potentials (74, 76). Most of these pathways have been reviewed elsewhere (74). PGC-1α and PGC-1β both play a role in the maintenance of cardiac function during pressure overload such that in the progression to heart failure, a deficiency of PGC-1β is shown to accelerate the transition (64). The hormone ligands that mediate the activation of these pathways include angiotensin II, endothelin 1, α-adrenergic receptors and β- adrenergic receptors (74). At organ level, multiple cardiac, vascular, and non-cardiac abnormalities associated with hypertensive heart disease underly the pathophysiology of heart failure (77, 78). These include impaired structural and functional changes of the left ventricle, myocardial ischemia, autonomic deregulation, endothelial dysfunction and vascular stiffening (77, 79). The RAAS is an important contributor that plays a central role in the transition from LVH to heart failure in hypertensive heart disease and has been explained above.
Mechanisms of conduction arrhythmias associated with hypertensive heart disease
The most common manifestation or complication of hypertensive heart disease is cardiac arrhythmias, and the most common among these is AFib (80). AFib is an irregular and very rapid heart rhythm associated with increased risk for blood clots in the heart, stroke, and heart failure (81–84). AFib can be detected on an electrocardiography (ECG) (Figure 3). During normal heart conduction, electrical signals from the sinoatrial node travel through the atria to the atrioventricular node, passes through the ventricles causing them to contract (Figures 3A,B). In AFib, the electrical signals conduct in a chaotic manner firing from multiple locations leading to faster and irregular heartbeats and characterized by lack of a P-wave and irregular QRS complexes on an ECG (85) (Figure 3C).
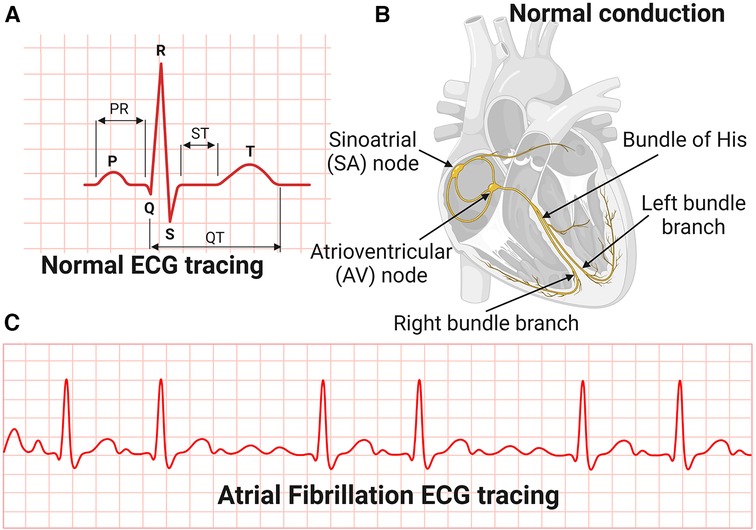
Figure 3. Electrocardiography and conduction system of the heart. (A) Normal Electrocardiography (ECG). (B) Conduction system of the heart. (C) Atrial Fibrillation ECG tracing.
Although effective control of blood pressure prevents AFib, some antihypertensive drugs such as thiazide diuretics used to control blood pressure can contribute to AFib risk by inducing hypokalaemia and hypomagnesemia (80). The mechanisms associated with AFib are not yet clear but include modulation of L-type Ca2+ and K + currents and gap junction function, cardiac structural remodeling and autonomic remodeling which is characterized by altered sympathovagal activity and hyperinnervation (85). One explanation for the chaotic rhythms in AFib is that structural remodeling characterized by atrial fibrosis occurring in hypertensive heart disease is associated with reentry of a self-sustaining cardiac rhythm abnormality (86). Ectopic conduction activities originating from the pulmonary veins is one of the common triggers of AFib due to specific action potential properties of the pulmonary vein cardiomyocytes (86). Overall, the mechanisms of conduction arrhythmias associated with hypertensive heart disease are not yet clear. In addition, sex differences in the risk, pathogenesis and outcomes of AFib also exist but data is limited (87).
Mechanisms of coronary artery disease associated with hypertensive heart disease
Coronary artery disease in hypertensive heart disease is accelerated by chronic elevation of blood pressure that induces endothelial dysfunction and exacerbates atherosclerotic processes (88). LVH exacerbates coronary artery disease by promoting myocardial ischemia mediated by a decreased coronary reserve and increased myocardial oxygen demand (88). Atherosclerosis remains the main cause of cardiovascular diseases and hypertensive heart disease accelerates complications of atherosclerotic diseases (89). Coronary arteries are considered the most susceptible blood vessels to atherosclerosis in the entire cardiovascular system due to their structurally higher curvature and torsion that plays a role in the localization of early coronary artery thickening (90) (Figure 4A). Blood pressure disturbances and irregularities and cardiac remodeling associated with hypertensive heart disease increase the risk of coronary artery disease and related complications such as myocardial infarction, angina, heart failure and AFib (88, 91, 92) (Figure 4B). The risk factors for coronary artery disease are similar with those associated with hypertension (93, 94). Sex differences in the burden, pathogenesis or severity of coronary artery disease do exist. For example incidental finding of coronary microvascular dysfunction is more common in women than men (95–98). In addition, coronary microvascular dysfunction occurs more in women than in men (99).
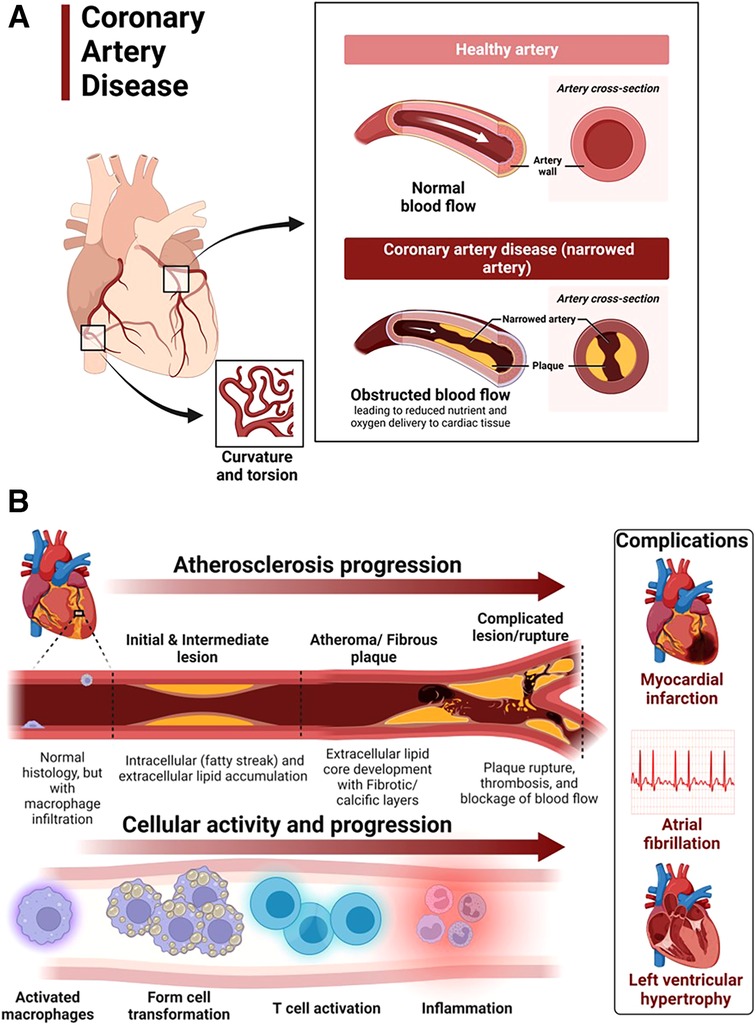
Figure 4. Atherosclerosis progression in coronary artery disease. (A) Coronary artery disease is characterized by narrowed coronary arteries due to advanced atherosclerotic fatty plaque. (B) Atherosclerosis in coronary artery disease and complications. Atherosclerosis is initiated by fatty streak and macrophage infiltration to rid of cholesterol deposits in the coronary arteries. Macrophages that have engulfed cholesterol deposits transform into lipid laden foam cells, enter the subendothelial layer and also activate the immune system leading to inflammation. Increased deposition of fat, calcium and persistent inflammation leads to formation of fibrotic and calcic changes that can result in plaque rupture, thrombosis and blockage of blood flow in the vasculature. The resulting complications of advanced atherosclerotic lesions include myocardial infarction, atrial fibrillation and left ventricular hypertrophy among others. Activated macrophages and T cells are the main players.
The mechanisms underlying the pathophysiology of coronary artery disease include fatty streak formation that activates macrophages to take up these lipids and deposit in the sub endothelium (100, 101). Immune cells including T cells are activated and recruited, secreting inflammatory cytokines in the process that results in the deposition of oxidized low-density lipoprotein (LDL) particles and collagen to form a stable subendothelial plaque that grows with time predisposing to vessel occlusion and atherothrombotic activity in chronic hypertension (101, 102) (Figure 4B). The immune system is intricately involved in atherosclerotic processes. Studies using apolipoprotein E–deficient mice have reported that when chronic inflammation does not resolve, tertiary lymphoid organs emerge in tissues and the adventitia of the aorta which become infiltrated with activated dendritic cells, B cells, and T cells of varying types (103, 104). These cells seem to target unknown antigens released from injured tissue and contribute to advanced atherosclerosis. The whole mark of disease progression is mediated by autoimmune B and T cells that become overly activated as a result of failure of anti-inflammatory effects to remove the continuous discharge of antigens from injured atherosclerotic tissue (104). Further, evidence of formation of neuroimmune cardiovascular interfaces characterized by expanded axon networks and activated artery-brain circuit activity in the adventitia is another proposed mechanism contributing to the progression of atherosclerosis in coronary artery disease (105). The whole process of atherosclerosis development has been described elsewhere (106). Shear stress associated with hypertensive heart disease and atherosclerotic changes leading to plaque progression and remodeling activates PKC epsilon, c-Jun N-terminal Kinase (JNK) MAP kinase, and p53 that worsen endothelial remodeling in the vasculature (107). High shear stress also activates matrix metalloproteinases (MMPs) resulting in thinning of artery wall and eccentric remodeling (107).
Although there are many proposed models of atherosclerosis based on animal studies and a few focused on humans, the challenge remains in translating our understanding to clinical practice (108).
Dietary salt in hypertensive heart disease and its complications
Excess dietary salt is associated with the development of hypertension and increases the risk for cardiovascular disease, stroke and death (109, 110). Through several mechanisms, excess dietary salt modulates endothelial function and structure, increases systemic peripheral resistance, modulates nervous system function and activates cells of the immune system (109, 111, 112) and accelerates the complications of hypertensive heart disease. The adverse effects of salt also affect normotensive individuals (113–115). Reduction in salt intake of less than 5 grams per day has been shown to lower the risk of developing hypertension and ameliorate cardiovascular diseases (110, 116–125). However, programs aimed at reducing salt intake at population level face a lot of compliance challenges (126).
Salt sensitivity of blood pressure
Although excess dietary salt raises blood pressure, the effect of salt on blood pressure is variable in the population (127). While significantly elevating blood pressure in some individuals (salt sensitivity), excess salt has no effect on blood pressure in others (salt resistant) and there is a group of individuals (about 15%) whose blood pressure increases with low salt intake (inverse salt sensitivity) (127–129). Salt sensitivity of blood pressure (SSBP) results in part from genetic polymorphisms in genes regulating sodium handling and those not related to sodium handling such as the Protein Kinase CGMP-Dependent 1 (PRKG1), cytochrome b-245 alpha (CYBA) chain (also known as p22-phox), branched chain amino acid transaminase 1 (BCAT1), Solute Carrier Family 8 Member A1 (SLC8A1), SLC4A5, Angiotensin II Receptor Type 1 (AGTR1), Selectin E (SELE), cytochrome P450 family 4 subfamily A member 11 (CYP4A11), Neuronal precursor cell expressed developmentally down-regulated 4-like (NEDD4l) and Visinin Like 1 (VSNL1) (129–133). As explained above, RAAS activation leads to vasoconstriction, increased systemic vascular resistance (SVR) and elevation in blood pressure (134). In individuals with SSBP, RAAS is altered in that renin stimulation is reduced in salt depletion and the mechanisms are not adequate to suppress renin in high salt intake hence worsening the adverse effects of salt on blood pressure (135–138).
Salt induced immune-activation in the skin in hypertensive heart disease
The handling of salt by the kidney and how salt contributes to water retention and elevated blood pressure is well known. The current dogma that sodium in the interstitial space equilibrates with plasma has been challenged in emerging studies that have now identified extrarenal handling of sodium that contributes to hypertension and sustenance of blood pressure in hypertensive heart disease (139, 140). It is now known that sodium can accumulate in tissues and skin without commensurate volume retention and activate innate and adaptive immunity leading to or sustaining hypertension (139, 141). Accumulation of salt in the skin is associated with autoimmune disease severity and heightening of inflammation in several diseases such as lipedema, diffuse cutaneous systemic sclerosis, multiple sclerosis, psoriasis and systemic lupus erythematosus (142–146). Several studies have demonstrated similar findings of increased sodium accumulation in the skin in hypertension using sodium magnetic resonance imaging (23Na MRI) (147–150). For dietary sodium to reach the skin from the intestinal lumen, it is first absorbed across the apical membrane of enterocytes through sodium-hydrogen exchangers (NHE), sodium glucose cotransporter 1 (SGLT1), sodium-dependent phosphate transporter 2b (NaPi2b), glucose transporters (GLUT) and endothelial sodium channels (ENaC) and pumped across the basal membrane of the intestine into the interstitium by Na-K ATPases (151–154). From the insterstitium sodium diffuses into the intestinal capillaries for transport. In the vasculature, excess dietary salt diminishes the buffering capacity of the negatively charged glycocalyx lining the endothelium and the red blood cells leading to extravasation of sodium and accumulation of salt in the interstitial tissues (140, 155, 156). Accumulation of salt in the skin increases the density and hyperplasia of the lymph-capillary network and this effect is mediated by activation of tonicity-responsive enhancer binding protein (TonEBP) in mononuclear phagocyte system (MPS) cells (140). TonEBP binds to and activates the promoter of the gene encoding vascular endothelial growth factor-C (VEGF-C) resulting in VEGF-C secretion and trapping by macrophages, augmenting interstitial hypertonic volume retention, decreasing endothelial nitric oxide synthase expression and elevating blood pressure in response to excess dietary salt (140). In addition, the hypertonic milieu contributed by the accumulation of sodium in the skin that induces the expression of VEGF-C increases lymphangiogenesis as a compensatory mechanism to eliminate sodium from the skin but this process is usually disrupted in hypertension, exacerbating hypertensive heart disease (147, 148). Low salt diet has shown to improve dermal capillary density and blood pressure in hypertension (157).
A group by Laffer et al. investigated hemodynamic changes in individuals with SSBP and found that compared to salt resistant individuals, individuals with SSBP had higher total peripheral resistance after salt loading which did not change after salt depletion and further, they also gained weight during salt loading but lost more weight during salt depletion that reflected failure to correct fluid retention (158). This study suggests that individuals with SSBP are unable to maintain and modulate a proper hemodynamic balance that reflects a dysfunction in the storage of salt in the interstitial compartment probably due to vascular dysfunction (156, 159).
Resident macrophages and dendritic cells in the interstitium of the skin are activated in the presence of excess dietary salt and via increased activity of the ROS producing reduced nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase, the ROS oxidize arachidonic acid leading to formation of Isolevuglandins (IsoLGs) (160, 161). IsoLGs adduct to lysine residues and alter intracellular protein structure and function and the resulting IsoLG-protein adducts act as neoantigens presented to and activating T cells (160). The activated T cells produce interferon-gamma (IFN-γ), interleukin 17A (IL-17A) and tumor necrosis factor-alpha (TNF-α) which causes vascular damage and lead to hypertension (160, 162). The activated macrophages and dendritic cells produce inflammatory cytokines IL-1β, IL-6 and IL-23 which induce T cell proliferation and production of inflammatory cytokines implicated in hypertension (160) (Figure 5). It has been demonstrated in many studies that T cells infiltrate the kidney causing vascular injury via inflammatory cytokines and increased oxidative stress and contributing to salt sensitive hypertension (163–165).
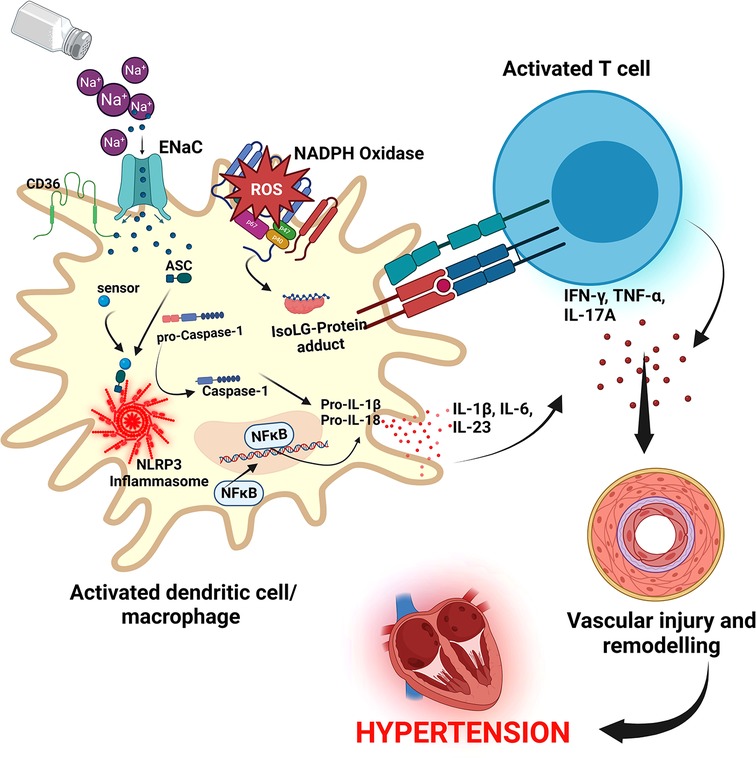
Figure 5. Salt induced hypertension. In high salt environments, dendritic cells or macrophages become activated through increased intracellular sodium that enter via the epithelial sodium channel (ENaC). Sodium activates NADPH oxidase and the inflammasome leading to formation of Isolevuglandins(IsoLGs)-protein adducts that are processed in major histocompatibility molecules and presented to T cells, activating them. Activated T cells produce inflammatory cytokines IFN-γ, TNF-α and IL-17A that lead to hypertension. NF-Kb, Nuclear factor kappa-light-chain-enhancer of activated B cells; IFN-γ, interferon gamma; TNF-α, Tumor necrosis factor alpha; ROS, reactive oxygen species; NLRP3, NACHT, LRR, and PYD domains-containing protein 3; ASC, Apoptosis-associated speck-like protein containing a CARD.
Cellular pathways activated by excess dietary salt
Several cellular pathways have been implicated in salt sensitive hypertension. The NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome is an oligomeric complex containing the NOD-like receptor NLRP3, the adaptor Apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and caspase-1 implicated in salt sensitive hypertension (166). The inflammasome is activated when NF-κB upregulates the inflammasome components and pro-IL-1β leading to the assembly of components to form the NLRP3 inflammasome signaling complex (134, 166) (Figure 5). Activation of the NLRP3 inflammasome leads to the release of pro-inflammatory cytokines IL-1β and IL-18 via pyroptosis that involves the cleavage of gasdermin D and development of pores in the membrane of cells through which the cytokines and other cellular contents are released (134, 166). It has been demonstrated that the NLRP3 inflammasome can be activated in high salt environments in an ENaC-dependent manner leading to IsoLG-protein adduct formation in dendritic cells and macrophages and antigen presentation to activate cells of the adaptive immune system and leading to hypertension as explained above (167).
In high salt diets, small guanosine triphosphatases (GTP)ases Rho and Rac kinases are activated and lead to activation of sympathetic nerve outflow that results in blood pressure elevation (168). In vascular smooth muscles, Rho kinases facilitate vasoconstriction though GPCRs and Wnt pathways, and in vascular endothelium, Rho/Rho kinase inhibits nitric oxide (NO) leading to increased vascular resistance and vascular tone and salt sensitive hypertension (168).
A study by Chu et al., in 329 subjects looked at growth factors that are produced in relation to the activation of the phosphoinositide 3-kinase/ Ak strain transforming (PI3K-Akt) pathway, which is activated through serine and/or threonine phosphorylation of a range of downstream substrates (169). They found that individuals with SSBP had elevated levels of several growth factors compared to salt resistant group (169). The signal transduction PI3K-Akt Pathway regulates metabolism, proliferation, cell survival, growth and angiogenesis (170). The PI3K-Akt Pathway activation has been implicated to contribute to the progression of atherosclerotic plaque formation and pathological changes in the vasculature leading to hypertension and cardiovascular disorders in many studies (171–175). Several other cellular pathways associated with salt sensitive hypertension such as the WNK signaling pathway (176), Kelch-like 3/Cullin 3 ubiquitin ligase complex (177), brain Gαi2-proteins of GPCR (178, 179) in the central nervous system, MAPK/extracellular signal regulated kinase [ERK] mediated by angiotensin II in vascular smooth muscles (180), and redox signaling (181) have been well elucidated.
Genetic predisposition in hypertensive heart disease complications
There is substantial evidence for genetic involvement in hypertensive heart disease and its complications (heart failure, AFib, and coronary artery disease). Evidence from observational, sibling and longitudinal twin studies reported that LVH phenotypes are highly heritable (182–184). Specific variants have been associated with abnormalities in cardiac structure and function related to hypertensive heart disease using gene association and genome-wide association studies (185, 186). Genome wide association studies and international collaborative metanalysis studies have also reported more than 30 gene loci associated with AFib (187–191). Several studies have demonstrated that most polymorphisms associated with blood pressure also increases the risk for coronary artery disease (192), incident hypertension and cardiovascular diseases (193). We found several studies that have reported genes associated with cardiomyopathies and heart failure such as myosin heavy chain 7 (MYH7), troponin T (TNNT2), troponin I (TNNI3), cardiac myosin binding protein 3 (MYBPC3), tropomyosin alpha-1 (TPM1), Lamin A/C (LMNA/C), plakophilin 2 (PKP2), desmocollin 2 (DSC2), desmoglein 2 (DSG2), desmoplakin (DSP), plakoglobin (JUP) and titin (194). We also know that the genetic component requires interaction with environmental factors for the effect or risk for hypertension and cardiovascular disease to be heightened (195). The genetic predisposition to hypertensive heart disease has been extensively reviewed and studied elsewhere (185, 192, 195–200).
Current diagnostic techniques
Echocardiography, carotid ultrasound and cardiac magnetic resonance imaging are important diagnostic techniques used in the clinic to detect functional and structural changes in the heart such as occurs in LVH (1, 50, 201). Echocardiography is cheap, readily available and more preferred to the high cost and limited availability of the gold standard, cardiac magnetic resonance imaging (42). Cardiac magnetic resonance imaging is a noninvasive, tomographic, nonionizing technique used to detect structural changes in the heart and therefore important for the diagnosis of hypertrophic cardiomyopathy, coronary heart disease, congenital heart disease, heart failure and other cardiac abnormalities (202).
Therapy for hypertensive heart disease and its complications
Controlling hypertension with current medication reduces the risk for complications and adverse cardiovascular events. The current US and European guidelines have extensively discussed therapy for hypertension and all related cardiovascular complications (2, 203). Thus, we will briefly focus on recent clinical studies reporting potential therapies that are especially used in combination for the treatment of LVH, AFib, coronary artery disease, and heart failure.
Left ventricular hypertrophy therapies
Patients with LVH benefit remarkably from intensive blood pressure lowering (<120 mmHg) to prevent complications (36). In clinical trials, several therapies have been reported to reduce LVH and its complications. Use of the neprilysin inhibitor sacubitril used for treatment of heart failure and the angiotensin receptor blocker valsartan was associated with reduced left ventricular mass index when compared to the angiotensin receptor blocker (ARB) Olmesartan, in participants with hypertension (204). Another clinical trial reported that combination of the ARB telmisartan and simvastatin did not only significantly reduce blood pressure but was able to reverse LVH and improve left ventricular systolic function (205). Similar findings were reported for a triple fixed dose combination of perindopril/indapamide/amlodipine (angiotensin-converting enzyme inhibitor (ACEI)/diuretic/calcium channel blocker (CCB)) in patients with essential hypertension followed for 14 months (206). Another interesting finding is from a clinical trial by Lal et al. where they used allopurinol, a xanthine oxidase inhibitor commonly used to reduce plasma uric acid in patients with gout, to determine its efficacy in reducing LVH (207). High dose allopurinol was more effective in reducing left ventricular mass and LVH when compared to febuxostatin (207) but caution must be exercised in using allopurinol in normouricemic individuals with controlled blood pressure as it can increase oxidative stress (208). In general, it appears that significant reversal of LVH is greater when both RAAS and sympathetic nervous system (SNS) inhibitors are used compared to drugs that just target blood pressure reduction (209). Other drugs as well as natural compounds or interventions used in combination have also been reported in clinical trials to ameliorate progression of LVH, examples include a nutraceutical combination of berberine, red yeast rice extract and policosanol (210), azelnidipine (211), losartan (212), low-dose eplerenone (213), metformin in patients with coronary artery disease without diabetes (214), diets low in fat and carbohydrate and regular consumption of green tea (215, 216).
Therapies for atrial fibrillation
Several clinical trials have reported beneficial therapies in the management of AFib. A few are discussed below.
When AFib is controlled, patients remain at risk for cardiovascular events, however, early rhythm control achieved by using antiarrhythmic drugs or atrial fibrillation ablation was effective in treating AFib and reducing the risk for cardiovascular events (217). In clinical practice, patients are first prescribed drugs such as beta blockers or a CCB in patients with asymptomatic AFib but a few clinical trials found that cryoballoon ablation was more effective compared to drug therapy as initial therapy for AFib (218, 219). Thus, rhythm control may be beneficial in both asymptomatic and symptomatic AFib (220). Further, radiofrequency ablation may delay or prevent paroxysmal AFib from progressing into persistent AFib (221). Despite its beneficial effect, caution should be exercised, as catheter ablation may also increase left atrial stiffness and worsen post-ablation diastolic function (222). Additional interventions for the management of AFib and its complications have been reported in other clinical trials elsewhere (223–227).
Therapies for coronary artery disease and heart failure
Patients with coronary artery disease also benefit from several interventional strategies including dietary interventions (228, 229), rivaroxaban monotherapy and other drugs (230–232), and physical exercise (233, 234). Further, lifestyle modifications have also been reported to be beneficial (235).
To alleviate heart failure and reduce its complications, several interventions are available (236). For example, in a clinical trial by Hieda et al. they found that physical exercise training for one year reversed left ventricular myocardial stiffness in patients with stage B heart failure with preserved ejection fraction that is characterized by LVH and N-terminal pro-B-type natriuretic peptide or high-sensitivity troponin (237). Therapeutic interventions for patients with heart failure also exist. Empagliflozin, dapagliflozin and spironolactone improves and ameliorates adverse outcomes of heart failure with persevered ejection fraction (238–240). In addition, individualized nutritional support as well as treatment with vericiguat for hospitalized patients with heart failure is also beneficial in reducing the risk for death and morbidity (241–243). Further, in patients with acute decompensated heart failure, usage of levosimendan in combination with Shenfu injection was effective in improving hemodynamics and enhance myocardial contractility (244). In severe heart failure where therapy is limited, use of omecamtiv mecarbil therapy is reported to have beneficial effects in reducing adverse outcomes (245). Management of heart failure is discussed in detail in the US and European guidelines.
Future directions
Future studies should focus on clinical studies (especially prospective) to understand the pathogenesis and complications of hypertensive heart disease as there are few studies in this area. Understanding the implications of physiological and pathological LVH and the potential for regression will be important for clinical application.
Conclusions
Hypertensive heart disease progresses through several mechanisms that amplify and increase the risk for adverse complications. Excess dietary salt is one of the modifiable factors that contribute enormously to the pathogenesis of hypertensive heart disease. Reduction of dietary salt has potential to reduce blood pressure and the risk for development of hypertensive heart disease.
Author contributions
SM: conceptualized the study and wrote the draft manuscript. SM and AK: wrote and edited different sections of the manuscript. SM: created all the figures. AK: conceptualized the frame-work and finalized the manuscript as well as obtained funding for the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Fogarty International Center of the National Institutes of Health grants R03HL155041, R01HL147818 and R01HL144941 (AK) and 2D43TW009744 (SM). The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Drazner MH. The progression of hypertensive heart disease. Circulation. (2011) 123:327–34. doi: 10.1161/CIRCULATIONAHA.108.845792
2. Whelton PK, Carey RM, Aronow WS, Casey DE, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Hypertension. (2018) 71:1269–324. doi: 10.1161/HYP.0000000000000066
3. Stergiou GS, Palatini P, Parati G, O’Brien E, Januszewicz A, Lurbe E, et al. 2021 European society of hypertension practice guidelines for office and out-of-office blood pressure measurement. J Hypertens. (2021) 39:1293. doi: 10.1097/HJH.0000000000002843
4. Unger T, Borghi C, Charchar F, Khan NA, Poulter NR, Prabhakaran D, et al. 2020 International society of hypertension global hypertension practice guidelines. Hypertension. (2020) 75:1334–57. doi: 10.1161/HYPERTENSIONAHA.120.15026
5. de la Sierra A. New American and European hypertension guidelines, reconciling the differences. Cardiol Ther. (2019) 8:157–66. doi: 10.1007/s40119-019-0144-3
6. Gonzalez-Maqueda I, Alegria-Ezquerra E, Gonzalez-Juanatey JR. Hypertensive heart disease: a new clinical classification (VIA). e-Journal of Cardiology Practice. (2009) 7. https://www.escardio.org/Journals/E-Journal-of-Cardiology-Practice/Volume-7/Hypertensive-heart-disease-a-new-clinical-classification-VIA, https://www.escardio.org/Journals/E-Journal-of-Cardiology-Practice/Volume-7/Hypertensive-heart-disease-a-new-clinical-classification-VIA (Accessed May 17, 2023).
7. Dumitrescu M, Constantin A, Nemecz AM, Drăgan E, Popov LD, Tanko G. Hypertension induces compensatory left ventricular hypertrophy by a mechanism involving gap junction lateralization and overexpression of CD36, PKC and MMP-2. Rom J Morphol Embryol. (2021) 62:713–21. doi: 10.47162/RJME.62.3.08
8. Boner G, Cooper ME, McCarroll K, Brenner BM, de Zeeuw D, Kowey PR, et al. Adverse effects of left ventricular hypertrophy in the reduction of endpoints in NIDDM with the angiotensin II antagonist losartan (RENAAL) study. Diabetologia. (2005) 48:1980–7. doi: 10.1007/s00125-005-1893-1
9. Liao Y, Cooper RS, Mensah GA, McGee DL. Left ventricular hypertrophy has a greater impact on survival in women than in men. Circulation. (1995) 92:805–10. doi: 10.1161/01.CIR.92.4.805
10. Kim YH, Her A-Y, Choi BG, Choi SY, Byun JK, Baek MJ, et al. Impact of left ventricular hypertrophy on long-term clinical outcomes in hypertensive patients who underwent successful percutaneous coronary intervention with drug-eluting stents. Medicine. (2018) 97:e12067. doi: 10.1097/MD.0000000000012067
11. Zhou B, Carrillo-Larco RM, Danaei G, Riley LM, Paciorek CJ, Stevens GA, et al. Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. Lancet. (2021) 398:957–80. doi: 10.1016/S0140-6736(21)01330-1
12. Mills KT, Stefanescu A, He J. The global epidemiology of hypertension. Nat Rev Nephrol. (2020) 16:223–37. doi: 10.1038/s41581-019-0244-2
13. Lu Y, Lan T. Global, regional, and national burden of hypertensive heart disease during 1990–2019: an analysis of the global burden of disease study 2019. BMC Public Health. (2022) 22:841. doi: 10.1186/s12889-022-13271-0
14. Dai H, Bragazzi NL, Younis A, Zhong W, Liu X, Wu J, et al. Worldwide trends in prevalence, mortality, and disability-adjusted life years for hypertensive heart disease from 1990 to 2017. Hypertension. (2021) 77:1223–33. doi: 10.1161/HYPERTENSIONAHA.120.16483
15. Kizer JR, Arnett DK, Bella JN, Paranicas M, Rao DC, Province MA, et al. Differences in left ventricular structure between black and white hypertensive adults. Hypertension. (2004) 43:1182–8. doi: 10.1161/01.HYP.0000128738.94190.9f
16. Sharp A, Tapp R, Francis DP, Thom SA, Hughes AD, Stanton AV, et al. Ethnicity and left ventricular diastolic function in hypertension: an ASCOT (anglo-scandinavian cardiac outcomes trial) substudy. J Am Coll Cardiol. (2008) 52:1015–21. doi: 10.1016/j.jacc.2008.04.065
17. Lawson CA, Zaccardi F, Squire I, Okhai H, Davies M, Huang W, et al. Risk factors for heart failure. Circ Heart Fail. (2020) 13:e006472. doi: 10.1161/CIRCHEARTFAILURE.119.006472
18. Messerli FH, Rimoldi SF, Bangalore S. The transition from hypertension to heart failure: contemporary update. JACC Heart Fail. (2017) 5:543–51. doi: 10.1016/j.jchf.2017.04.012
19. Roumie CL, Hung AM, Russell GB, Basile J, Kreider KE, Nord J, et al. Blood pressure control and the association with diabetes incidence: results from the SPRINT randomized trial. Hypertension. (2020) 75:331–8. doi: 10.1161/HYPERTENSIONAHA.118.12572
20. Saliba LJ, Maffett S. Hypertensive heart disease and obesity: a review. Heart Fail Clin. (2019) 15:509–17. doi: 10.1016/j.hfc.2019.06.003
21. Murdolo G, Angeli F, Reboldi G, Di Giacomo L, Aita A, Bartolini C, et al. Left ventricular hypertrophy and obesity: only a matter of fat? High Blood Press Cardiovasc Prev. (2015) 22:29–41. doi: 10.1007/s40292-014-0068-x
22. Venteclef N, Guglielmi V, Balse E, Gaborit B, Cotillard A, Atassi F, et al. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur Heart J. (2015) 36:795–805. doi: 10.1093/eurheartj/eht099
23. Szczepaniak LS, Dobbins RL, Metzger GJ, Sartoni-D’Ambrosia G, Arbique D, Vongpatanasin W, et al. Myocardial triglycerides and systolic function in humans: in vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn Reson Med. (2003) 49:417–23. doi: 10.1002/mrm.10372
24. Garcia JN, Wanjalla CN, Mashayekhi M, Hasty AH. Immune cell activation in obesity and cardiovascular disease. Curr Hypertens Rep. (2022) 24:627–37. doi: 10.1007/s11906-022-01222-4
25. daSilva-deAbreu A, Alhafez BA, Lavie CJ, Milani RV, Ventura HO. Interactions of hypertension, obesity, left ventricular hypertrophy, and heart failure. Curr Opin Cardiol. (2021) 36:453–60. doi: 10.1097/HCO.0000000000000868
26. Fuchs FD, Whelton PK. High blood pressure and cardiovascular disease. Hypertension. (2020) 75:285–92. doi: 10.1161/HYPERTENSIONAHA.119.14240
27. Howell SJ, Sear JW, Foëx P. Hypertension, hypertensive heart disease and perioperative cardiac risk†. BJA: British Journal of Anaesthesia. (2004) 92:570–83. doi: 10.1093/bja/aeh091
28. Rosenfeld EB, Graham HL, Brandt JS, Ananth CV. Patients with chronic hypertension are at increased risk for postpartum cardiovascular disease complications. Am J Obstetr Gynecol. (2022) 226:S352. doi: 10.1016/j.ajog.2021.11.592
29. Lorell BH, Carabello BA. Left ventricular hypertrophy. Circulation. (2000) 102:470–9. doi: 10.1161/01.CIR.102.4.470
30. Généreux P, Pibarot P, Redfors B, Mack MJ, Makkar RR, Jaber WA, et al. Staging classification of aortic stenosis based on the extent of cardiac damage. Eur Heart J. (2017) 38:3351–8. doi: 10.1093/eurheartj/ehx381
31. Grossman C, Levin M, Koren-Morag N, Bornstein G, Leibowitz A, Ben-Zvi I, et al. Left ventricular hypertrophy predicts cardiovascular events in hypertensive patients with coronary artery calcifications. Am J Hypertens. (2018) 31:313–20. doi: 10.1093/ajh/hpx181
32. Bang CN, Soliman EZ, Simpson LM, Davis BR, Devereux RB, Okin PM. Electrocardiographic left ventricular hypertrophy predicts cardiovascular morbidity and mortality in hypertensive patients: the ALLHAT study. Am J Hypertens. (2017) 30:914–22. doi: 10.1093/ajh/hpx067
33. Peters MN, Seliger SL, Christenson RH, Hong-Zohlman SN, Daniels LB, Lima JAC, et al. “Malignant” left ventricular hypertrophy identifies subjects at high risk for progression to asymptomatic left ventricular dysfunction, heart failure, and death: mESA (multi-ethnic study of atherosclerosis). J Am Heart Assoc. (2018) 7:e006619. doi: 10.1161/JAHA.117.006619
34. Seliger SL, de Lemos J, Neeland IJ, Christenson R, Gottdiener J, Drazner MH, et al. Older adults, “malignant” left ventricular hypertrophy, and associated cardiac-specific biomarker phenotypes to identify the differential risk of new-onset reduced versus preserved ejection fraction heart failure: cHS (cardiovascular health study). JACC Heart Fail. (2015) 3:445–55. doi: 10.1016/j.jchf.2014.12.018
35. Lewis AA, Ayers CR, Selvin E, Neeland I, Ballantyne CM, Nambi V, et al. Racial differences in malignant left ventricular hypertrophy and incidence of heart failure: a multicohort study. Circulation. (2020) 141:957–67. doi: 10.1161/CIRCULATIONAHA.119.043628
36. Ascher SB, de Lemos JA, Lee M, Wu E, Soliman EZ, Neeland IJ, et al. Intensive blood pressure lowering in patients with malignant left ventricular hypertrophy. J Am Coll Cardiol. (2022) 80:1516–25. doi: 10.1016/j.jacc.2022.08.735
37. Coutinho T, Pellikka PA, Bailey KR, Turner ST, Kullo IJ. Sex differences in the associations of hemodynamic load with left ventricular hypertrophy and concentric remodeling. Am J Hypertens. (2016) 29:73–80. doi: 10.1093/ajh/hpv071
38. Kuch B, Muscholl M, Luchner A, Döring A, Riegger GA, Schunkert H, et al. Sex differences in the correlation between obesity and hypertension with left ventricular mass and hypertrophy. Z Kardiol. (1996) 85:334–42. PMID: 8711946.8711946
39. Gerdts E, Izzo R, Mancusi C, Losi MA, Manzi MV, Canciello G, et al. Left ventricular hypertrophy offsets the sex difference in cardiovascular risk (the campania salute network). Int J Cardiol. (2018) 258:257–61. doi: 10.1016/j.ijcard.2017.12.086
40. Dorn GW. The fuzzy logic of physiological cardiac hypertrophy. Hypertension. (2007) 49:962–70. doi: 10.1161/HYPERTENSIONAHA.106.079426
41. Inagaki M, Yokota M, Izawa H, Ishiki R, Nagata K, Iwase M, et al. Impaired force-frequency relations in patients with hypertensive left ventricular hypertrophy. A possible physiological marker of the transition from physiological to pathological hypertrophy. Circulation. (1999) 99:1822–30. doi: 10.1161/01.cir.99.14.1822
42. Cacciapuoti F. Molecular mechanisms of left ventricular hypertrophy (LVH) in systemic hypertension (SH)—possible therapeutic perspectives. J Am Soc Hypertens. (2011) 5:449–55. doi: 10.1016/j.jash.2011.08.006
43. Oldfield CJ, Duhamel TA, Dhalla NS. Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can J Physiol Pharmacol. (2020) 98:74–84. doi: 10.1139/cjpp-2019-0566
44. Gradman AH, Alfayoumi F. From left ventricular hypertrophy to congestive heart failure: management of hypertensive heart disease. Prog Cardiovasc Dis. (2006) 48:326–41. doi: 10.1016/j.pcad.2006.02.001
45. Diwan A, Dorn GW. Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology. (2007) 22:56–64. doi: 10.1152/physiol.00033.2006
46. Bang CN, Gerdts E, Aurigemma GP, Boman K, de Simone G, Dahlöf B, et al. Four-group classification of left ventricular hypertrophy based on ventricular concentricity and dilatation identifies a low-risk subset of eccentric hypertrophy in hypertensive patients. Circ Cardiovasc Imaging. (2014) 7:422–9. doi: 10.1161/CIRCIMAGING.113.001275
47. Heinzel FR, Hohendanner F, Jin G, Sedej S, Edelmann F. Myocardial hypertrophy and its role in heart failure with preserved ejection fraction. J Appl Physiol (1985). (2015) 119:1233–42. doi: 10.1152/japplphysiol.00374.2015
48. Shah AM, Cikes M, Prasad N, Li G, Getchevski S, Claggett B, et al. Echocardiographic features of patients with heart failure and preserved left ventricular ejection fraction. J Am Coll Cardiol. (2019) 74:2858–73. doi: 10.1016/j.jacc.2019.09.063
49. Gerdts E, Okin PM, de Simone G, Cramariuc D, Wachtell K, Boman K, et al. Gender differences in left ventricular structure and function during antihypertensive treatment. Hypertension. (2008) 51:1109–14. doi: 10.1161/HYPERTENSIONAHA.107.107474
50. Lønnebakken MT, Izzo R, Mancusi C, Gerdts E, Losi MA, Canciello G, et al. Left ventricular hypertrophy regression during antihypertensive treatment in an outpatient clinic (the campania salute network). J Am Heart Assoc. (2017) 6:e004152. doi: 10.1161/JAHA.116.004152
51. Devereux RB, Dahlöf B, Gerdts E, Boman K, Nieminen MS, Papademetriou V, et al. Regression of hypertensive left ventricular hypertrophy by losartan compared with atenolol. Circulation. (2004) 110:1456–62. doi: 10.1161/01.CIR.0000141573.44737.5A
52. Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med. (2003) 115:41–6. doi: 10.1016/S0002-9343(03)00158-X
53. de Simone G, Okin PM, Gerdts E, Olsen MH, Wachtell K, Hille DA, et al. Clustered metabolic abnormalities blunt regression of hypertensive left ventricular hypertrophy: the LIFE study. Nutr Metab Cardiovasc Dis. (2009) 19:634–40. doi: 10.1016/j.numecd.2008.12.012
54. de Simone G, Devereux RB, Izzo R, Girfoglio D, Lee ET, Howard BV, et al. Lack of reduction of left ventricular mass in treated hypertension: the strong heart study. J Am Heart Assoc. (2013) 2:e000144. doi: 10.1161/JAHA.113.000144
55. Maron BJ, Niimura H, Casey SA, Soper MK, Wright GB, Seidman JG, et al. Development of left ventricular hypertrophy in adults in hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations. J Am Coll Cardiol. (2001) 38:315–21. doi: 10.1016/s0735-1097(01)01386-9
56. Quintana-Villamandos B, del Pozo IG, Pazó-Sayós L, Bellón JM, Pedraz-Prieto Á, Pinto ÁG, et al. Plasma protein thiolation index (PTI) as a potential biomarker for left ventricular hypertrophy in humans. PLoS One. (2019) 14:e0216359. doi: 10.1371/journal.pone.0216359
57. Katz DH, Tahir UA, Ngo D, Benson MD, Gao Y, Shi X, et al. Multiomic profiling in black and white populations reveals novel candidate pathways in left ventricular hypertrophy and incident heart failure specific to black adults. Circ Genom Precis Med. (2021) 14:e003191. doi: 10.1161/CIRCGEN.120.003191
58. Muñoz-Durango N, Fuentes CA, Castillo AE, González-Gómez LM, Vecchiola A, Fardella CE, et al. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: molecular and cellular mechanisms involved in End-organ damage during arterial hypertension. Int J Mol Sci. (2016) 17:797. doi: 10.3390/ijms17070797
59. Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. (2010) 2:247–57. doi: 10.1002/emmm.201000080
60. Saino A, Pomidossi G, Perondi R, Valentini R, Rimini A, Di Francesco L, et al. Intracoronary angiotensin II potentiates coronary sympathetic vasoconstriction in humans. Circulation. (1997) 96:148–53. doi: 10.1161/01.cir.96.1.148
61. De Mello WC, Danser AHJ. Angiotensin II and the heart. Hypertension. (2000) 35:1183–8. doi: 10.1161/01.HYP.35.6.1183
62. Lee KH, Jang Y, Chung JH. Heat shock protein 90 regulates IκB kinase complex and NF-κB activation in angiotensin II-induced cardiac cell hypertrophy. Exp Mol Med. (2010) 42:703–11. doi: 10.3858/emm.2010.42.10.069
63. van Rooij E, Doevendans PA, de Theije CC, Babiker FA, Molkentin JD, de Windt LJ. Requirement of nuclear factor of activated T-cells in calcineurin-mediated cardiomyocyte hypertrophy. J Biol Chem. (2002) 277:48617–26. doi: 10.1074/jbc.M206532200
64. Saheera S, Krishnamurthy P. Cardiovascular changes associated with hypertensive heart disease and aging. Cell Transplant. (2020) 29:0963689720920830. doi: 10.1177/0963689720920830
65. Arrigo M, Jessup M, Mullens W, Reza N, Shah AM, Sliwa K, et al. Acute heart failure. Nat Rev Dis Primers. (2020) 6:16. doi: 10.1038/s41572-020-0151-7
66. Tomasoni D, Adamo M, Lombardi CM, Metra M. Highlights in heart failure. ESC Heart Fail. (2020) 6:1105–27. doi: 10.1002/ehf2.12555
67. Bozkurt B, Coats AJS, Tsutsui H, Abdelhamid CM, Adamopoulos S, Albert N, et al. Universal definition and classification of heart failure: a report of the heart failure society of America, heart failure association of the European society of cardiology, Japanese heart failure society and writing committee of the universal definition of heart failure. Eur J Heart Fail. (2021) 23:352–80. doi: 10.1002/ejhf.2115
68. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. (2021) 42:3599–726. doi: 10.1093/eurheartj/ehab368
69. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: executive summary: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. Circulation. (2022) 145:e876–94. doi: 10.1161/CIR.0000000000001062
70. Lala A, Tayal U, Hamo CE, Youmans Q, Al-Khatib SM, Bozkurt B, et al. Sex differences in heart failure. J Card Fail. (2022) 28:477–98. doi: 10.1016/j.cardfail.2021.10.006
71. Shufelt C, Pacheco C, Tweet MS, Miller VM. Sex-specific physiology and cardiovascular disease. Adv Exp Med Biol. (2018) 1065:433–54. doi: 10.1007/978-3-319-77932-4_27
72. Savarese G, D’Amario D. Sex differences in heart Failure. Adv Exp Med Biol. (2018) 1065:529–44. doi: 10.1007/978-3-319-77932-4_32
73. Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R. Update on acute coronary syndromes: the pathologists’ view. Eur Heart J. (2013) 34:719–28. doi: 10.1093/eurheartj/ehs411
74. Mishra S, Kass DA. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. (2021) 18:400–23. doi: 10.1038/s41569-020-00480-6
75. Toyoda S, Haruyama A, Inami S, Arikawa T, Saito F, Watanabe R, et al. Effects of carvedilol vs bisoprolol on inflammation and oxidative stress in patients with chronic heart failure. J Cardiol. (2020) 75:140–7. doi: 10.1016/j.jjcc.2019.07.011
76. Goyal N, Skrdla P, Schroyer R, Kumar S, Fernando D, Oughton A, et al. Clinical pharmacokinetics, safety, and tolerability of a novel, first-in-class TRPV4 Ion channel inhibitor, GSK2798745, in healthy and heart failure subjects. Am J Cardiovasc Drugs. (2019) 19:335–42. doi: 10.1007/s40256-018-00320-6
77. Omote K, Verbrugge FH, Borlaug BA. Heart failure with preserved ejection fraction: mechanisms and treatment strategies. Annu Rev Med. (2022) 73:321–37. doi: 10.1146/annurev-med-042220-022745
78. Zheng Y-L, Yan BP, Zhang Y-T, Poon CCY. Noninvasive characterization of vascular tone by model-based system identification in healthy and heart failure patients. Ann Biomed Eng. (2015) 43:2242–52. doi: 10.1007/s10439-015-1266-y
79. Vasu S, Morgan TM, Kitzman DW, Bertoni A, Stacey RB, Hamilton C, et al. Abnormal stress-related measures of arterial stiffness in middle-aged and elderly men and women with impaired fasting glucose at risk for a first episode of symptomatic heart failure. J Am Heart Assoc. (2015) 4:e000991. doi: 10.1161/JAHA.114.000991
80. Lip GYH, Coca A, Kahan T, Boriani G, Manolis AS, Olsen MH, et al. Hypertension and cardiac arrhythmias: a consensus document from the European heart rhythm association (EHRA) and ESC council on hypertension, endorsed by the heart rhythm society (HRS), Asia-pacific heart rhythm society (APHRS) and sociedad latinoamericana de estimulación cardíaca y electrofisiología (SOLEACE). Europace. (2017) 19:891–911. doi: 10.1093/europace/eux091
81. Hald EM, Løchen M, Mathiesen EB, Wilsgaard T, Njølstad I, Brækkan SK, et al. Atrial fibrillation, venous thromboembolism, ischemic stroke, and all-cause mortality: the tromsø study. Res Pract Thromb Haemost. (2020) 4:1004–12. doi: 10.1002/rth2.12351
82. Hald EM, Rinde LB, Løchen M, Mathiesen EB, Wilsgaard T, Njølstad I, et al. Atrial fibrillation and cause-specific risks of pulmonary embolism and ischemic stroke. J Am Heart Assoc. (2018) 7:e006502. doi: 10.1161/JAHA.117.006502
83. Kotecha D, Banerjee A, Lip GYH. Increased stroke risk in atrial fibrillation patients with heart failure. Stroke. (2015) 46:608–9. doi: 10.1161/STROKEAHA.114.008421
84. Haywood LJ, Davis BR, Piller LB, Simpson LM, Ghosh A, Einhorn PT, et al. Risk factors influencing outcomes of atrial fibrillation in ALLHAT. J Natl Med Assoc. (2018) 110:343–51. doi: 10.1016/j.jnma.2017.07.003
85. Pellman J, Sheikh F. Atrial fibrillation: mechanisms, therapeutics, and future directions. Compr Physiol. (2015) 5:649–65. doi: 10.1002/cphy.c140047
86. Cheniti G, Vlachos K, Pambrun T, Hooks D, Frontera A, Takigawa M, et al. Atrial fibrillation mechanisms and implications for catheter ablation. Front Physiol. (2018) 9. doi: 10.3389/fphys.2018.01458
87. Westerman S, Wenger N. Gender differences in atrial fibrillation: a review of epidemiology, management, and outcomes. Curr Cardiol Rev. (2019) 15:136–44. doi: 10.2174/1573403X15666181205110624
88. Escobar E. Hypertension and coronary heart disease. J Hum Hypertens. (2002) 16 Suppl 1:S61–63. doi: 10.1038/sj.jhh.1001345
89. Frostegård J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. (2013) 11:117. doi: 10.1186/1741-7015-11-117
90. Zhu H, Friedman MH. Relationship between the dynamic geometry and wall thickness of a human coronary artery. Arterioscler Thromb Vasc Biol. (2003) 23:2260–5. doi: 10.1161/01.ATV.0000095976.40874.E0
91. Olafiranye O, Zizi F, Brimah P, Jean-louis G, Makaryus AN, McFarlane S, et al. Management of hypertension among patients with coronary heart disease. Int J Hypertens. (2011) 2011:e653903. doi: 10.4061/2011/653903
92. Rosendorff C, Committee on behalf of the W. Hypertension and coronary artery disease: a summary of the American heart association scientific statement. J Clin Hypertens. (2007) 9:790–5. doi: 10.1111/j.1751-7176.2007.tb00006.x
93. Hajar R. Risk factors for coronary artery disease: historical perspectives. Heart Views. (2017) 18:109–14. doi: 10.4103/HEARTVIEWS.HEARTVIEWS_106_17
95. Leuzzi C, Modena MG. Coronary artery disease: clinical presentation, diagnosis and prognosis in women. Nutr Metab Cardiovasc Dis. (2010) 20:426–35. doi: 10.1016/j.numecd.2010.02.013
96. Shaw LJ, Bugiardini R, Merz CNB. Women and ischemic heart disease: evolving knowledge. J Am Coll Cardiol. (2009) 54:1561–75. doi: 10.1016/j.jacc.2009.04.098
97. Gulati M, Shaw LJ, Bairey Merz CN. Myocardial ischemia in women: lessons from the NHLBI WISE study. Clin Cardiol. (2012) 35:141–8. doi: 10.1002/clc.21966
98. Eisenberg E, Di Palo KE, Piña IL. Sex differences in heart failure. Clin Cardiol. (2018) 41:211–6. doi: 10.1002/clc.22917
99. Tweet MS, Hayes SN, Pitta SR, Simari RD, Lerman A, Lennon RJ, et al. Clinical features, management, and prognosis of spontaneous coronary artery dissection. Circulation. (2012) 126:579–88. doi: 10.1161/CIRCULATIONAHA.112.105718
100. Alexander RW. Hypertension and the pathogenesis of atherosclerosis. Hypertension. (1995) 25:155–61. doi: 10.1161/01.HYP.25.2.155
101. Poznyak AV, Sadykhov NK, Kartuesov AG, Borisov EE, Melnichenko AA, Grechko AV, et al. Hypertension as a risk factor for atherosclerosis: cardiovascular risk assessment. Front Cardiovasc Med. (2022) 9:959285. doi: 10.3389/fcvm.2022.959285
102. Alfarisi HAH, Mohamed ZBH, Ibrahim MB. Basic pathogenic mechanisms of atherosclerosis. Egypt J Basic Appl Sci. (2020) 7:116–25. doi: 10.1080/2314808X.2020.1769913
103. Mohanta SK, Yin C, Peng L, Srikakulapu P, Bontha V, Hu D, et al. Artery tertiary lymphoid organs contribute to innate and adaptive immune responses in advanced mouse atherosclerosis. Circ Res. (2014) 114:1772–87. doi: 10.1161/CIRCRESAHA.114.301137
104. King CG, Koehli S, Hausmann B, Schmaler M, Zehn D, Palmer E. T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity. (2012) 37:709–20. doi: 10.1016/j.immuni.2012.06.021
105. Mohanta SK, Peng L, Li Y, Lu S, Sun T, Carnevale L, et al. Neuroimmune cardiovascular interfaces control atherosclerosis. Nature. (2022) 605:152–9. doi: 10.1038/s41586-022-04673-6
106. Katakami N. Mechanism of development of atherosclerosis and cardiovascular disease in diabetes Mellitus. J Atheroscler Thromb. (2018) 25:27–39. doi: 10.5551/jat.RV17014
107. Kwak BR, Bäck M, Bochaton-Piallat M-L, Caligiuri G, Daemen MJAP, Davies PF, et al. Biomechanical factors in atherosclerosis: mechanisms and clinical implications†. Eur Heart J. (2014) 35:3013–20. doi: 10.1093/eurheartj/ehu353
108. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. (2011) 473:317–25. doi: 10.1038/nature10146
109. Grillo A, Salvi L, Coruzzi P, Salvi P, Parati G. Sodium intake and hypertension. Nutrients. (2019) 11:1970. doi: 10.3390/nu11091970
110. He FJ, MacGregor GA. Effect of modest salt reduction on blood pressure: a meta-analysis of randomized trials. Implications for public health. J Hum Hypertens. (2002) 16:761–70. doi: 10.1038/sj.jhh.1001459
111. Masenga SK, Hamooya BM, Nzala S, Kwenda G, Heimburger DC, Mutale W, et al. Patho-immune mechanisms of hypertension in HIV: a systematic and thematic review. Curr Hypertens Rep. (2019) 21:56. doi: 10.1007/s11906-019-0956-5
112. Masenga SK, Kirabo A, Hamooya BM, Nzala S, Kwenda G, Heimburger DC, et al. HIV-positive demonstrate more salt sensitivity and nocturnal non-dipping blood pressure than HIV-negative individuals. Clin Hypertens. (2021) 27. doi: 10.1186/s40885-020-00160-0
113. Masenga SK, Pilic L, Hamooya BM, Nzala S, Heimburger DC, Mutale W, et al. Immediate pressor response to oral salt and its assessment in the clinic: a time series clinical trial. Clin Hypertens. (2022) 28:25. doi: 10.1186/s40885-022-00209-2
114. Elias SO, Azinge EC, Umoren GA, Jaja SI, Sofola OA. Salt-sensitivity in normotensive and hypertensive Nigerians. Nig Q J Hosp Med. (2011) 21:85–91. PMID: 21913548.21913548
115. Marketou ME, Maragkoudakis S, Anastasiou I, Nakou H, Plataki M, Vardas PE, et al. Salt-induced effects on microvascular function: a critical factor in hypertension mediated organ damage. J Clin Hypertens. (2019) 21:749–57. doi: 10.1111/jch.13535
116. Law MR, Frost CD, Wald NJ. By how much does dietary salt reduction lower blood pressure? III–analysis of data from trials of salt reduction. Br Med J. (1991) 302:819–24. doi: 10.1136/bmj.302.6780.819
117. Grobbee DE, Hofman A. Does sodium restriction lower blood pressure? Br Med J (Clin Res Ed). (1986) 293:27–9. doi: 10.1136/bmj.293.6538.27
118. Hooper L, Bartlett C, Smith GD, Ebrahim S. Systematic review of long term effects of advice to reduce dietary salt in adults. Br Med J. (2002) 325:628. doi: 10.1136/bmj.325.7365.628
119. Geleijnse JM, Kok FJ, Grobbee DE. Blood pressure response to changes in sodium and potassium intake: a metaregression analysis of randomised trials. J Hum Hypertens. (2003) 17:471–80. doi: 10.1038/sj.jhh.1001575
120. Midgley JP, Matthew AG, Greenwood CMT, Logan AG. Effect of reduced dietary sodium on blood pressure: a meta-analysis of randomized controlled trials. JAMA. (1996) 275:1590–7. doi: 10.1001/jama.1996.03530440070039
121. Cutler J, Follmann D, Allender P. Randomized trials of sodium reduction: an overview. Am J Clin Nutr. (1997) 65:643S–51S. doi: 10.1093/ajcn/65.2.643S
122. Graudal NA, Hubeck-Graudal T, Jurgens G. Effects of low sodium diet versus high sodium diet on blood pressure, renin, aldosterone, catecholamines, cholesterol, and triglyceride. Cochrane Database Syst Rev. (2020. doi: 10.1002/14651858.CD004022.pub5
123. Aburto NJ, Ziolkovska A, Hooper L, Elliott P, Cappuccio FP, Meerpohl JJ. Effect of lower sodium intake on health: systematic review and meta-analyses. Br Med J. (2013) 346:f1326. doi: 10.1136/bmj.f1326
124. He FJ, Li J, MacGregor GA. Effect of longer term modest salt reduction on blood pressure: cochrane systematic review and meta-analysis of randomised trials. Br Med J. (2013) 346:f1325. doi: 10.1136/bmj.f1325
125. Antman EM, Appel LJ, Balentine D, Johnson RK, Steffen LM, Miller EA, et al. Stakeholder discussion to reduce population-wide sodium intake and decrease sodium in the food supply. Circulation. (2014) 129:e660–79. doi: 10.1161/CIR.0000000000000051
126. Wang J, Olendzki BC, Wedick NM, Persuitte GM, Culver AL, Li W, et al. Challenges in sodium intake reduction and meal consumption patterns among participants with metabolic syndrome in a dietary trial. Nutr J. (2013) 12:163. doi: 10.1186/1475-2891-12-163
127. Elijovich F, Weinberger MH, Anderson CAM, Appel LJ, Bursztyn CN, Dart RA, et al. Salt sensitivity of blood pressure. Hypertension. (2016) 68:e7–e46. doi: 10.1161/HYP.0000000000000047
128. Felder RA, Gildea JJ, Xu P, Yue W, Armando I, Carey RM, et al. Inverse salt sensitivity of blood pressure: mechanisms and potential relevance for prevention of cardiovascular disease. Curr Hypertens Rep. (2022) 24:361–74. doi: 10.1007/s11906-022-01201-9
129. Carey RM, Schoeffel CD, Gildea JJ, Jones JE, McGrath HE, Gordon LN, et al. Salt sensitivity of blood pressure is associated with polymorphisms in the sodium-bicarbonate cotransporter. Hypertension. (2012) 60:1359–66. doi: 10.1161/HYPERTENSIONAHA.112.196071
130. Xie Y, Liu Z, Liu K, Qi H, Peng W, Cao H, et al. Candidate gene polymorphisms influence the susceptibility to salt sensitivity of blood pressure in a han Chinese population: risk factors as mediators. Front Genet. (2021) 12. https://www.frontiersin.org/articles/10.3389/fgene.2021.675230 (Accessed April 10, 2023).
131. Georgieva T, Kuzova E, Arsova S, DImbareva D, Georgieva R, Duleva V. Study of rs10177833 polymorphism in SLC4A5 gene and salt sensitivity in Bulgarian population. Eur J Public Health. (2021) 31:ckab165. doi: 10.1093/eurpub/ckab165.290
132. Dahlberg J, Nilsson L-O, von Wowern F, Melander O. Polymorphism in NEDD4l is associated with increased salt sensitivity, reduced levels of P-renin and increased levels of nt-proANP. PLoS One. (2007) 2:e432. doi: 10.1371/journal.pone.0000432
133. Lupoli S, Salvi E, Barlassina C. Dietary salt intake, blood pressure, and genes. Curr Nutr Rep. (2013) 2:134–41. doi: 10.1007/s13668-013-0047-1
134. Maaliki D, Itani MM, Itani HA. Pathophysiology and genetics of salt-sensitive hypertension. Front Physiol. (2022) 13:1001434. doi: 10.3389/fphys.2022.1001434
135. Yatabe MS, Yatabe J, Yoneda M, Watanabe T, Otsuki M, Felder RA, et al. Salt sensitivity is associated with insulin resistance, sympathetic overactivity, and decreased suppression of circulating renin activity in lean patients with essential hypertension. Am J Clin Nutr. (2010) 92:77–82. doi: 10.3945/ajcn.2009.29028
136. Kobori H, Nishiyama A, Abe Y, Navar LG. Enhancement of intrarenal angiotensinogen in dahl salt-sensitive rats on high salt diet. Hypertension. (2003) 41:592–7. doi: 10.1161/01.HYP.0000056768.03657.B4
137. Fujita T. Mechanism of salt-sensitive hypertension: focus on adrenal and sympathetic nervous systems. J Am Soc Nephrol. (2014) 25:1148. doi: 10.1681/ASN.2013121258
138. Susic D, Frohlich ED, Kobori H, Shao W, Seth D, Navar LG. Salt-induced renal injury in SHRs is mediated by AT1 receptor activation. J Hypertens. (2011) 29:716. doi: 10.1097/HJH.0b013e3283440683
139. Kirabo A. A new paradigm of sodium regulation in inflammation and hypertension. Am J Physiol Regul Integr Comp Physiol. (2017) 313:R706–10. doi: 10.1152/ajpregu.00250.2017
140. Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C–dependent buffering mechanism. Nat Med. (2009) 15:545–52. doi: 10.1038/nm.1960
141. Jobin K, Müller DN, Jantsch J, Kurts C. Sodium and its manifold impact on our immune system. Trends Immunol. (2021) 42:469–79. doi: 10.1016/j.it.2021.04.002
142. Inglese M, Madelin G, Oesingmann N, Babb JS, Wu W, Stoeckel B, et al. Brain tissue sodium concentration in multiple sclerosis: a sodium imaging study at 3 tesla. Brain. (2010) 133:847–57. doi: 10.1093/brain/awp334
143. Kopp C, Beyer C, Linz P, Dahlmann A, Hammon M, Jantsch J, et al. Na+ deposition in the fibrotic skin of systemic sclerosis patients detected by 23Na-magnetic resonance imaging. Rheumatology. (2017) 56:556–60. doi: 10.1093/rheumatology/kew371
144. Crescenzi R, Marton A, Donahue PMC, Mahany HB, Lants SK, Wang P, et al. Tissue sodium content is elevated in the skin and subcutaneous adipose tissue in women with lipedema. Obesity. (2018) 26:310–7. doi: 10.1002/oby.22090
145. Maifeld A, Wild J, Karlsen TV, Rakova N, Wistorf E, Linz P, et al. Skin sodium accumulates in psoriasis and reflects disease severity. J Invest Dermatol. (2022) 142:166–178.e8. doi: 10.1016/j.jid.2021.06.013
146. Carranza-Leόn DA, Oeser A, Marton A, Wang P, Gore JC, Titze J, et al. Tissue sodium content in patients with systemic lupus erythematosus: association with disease activity and markers of inflammation. Lupus. (2020) 29:455–62. doi: 10.1177/0961203320908934
147. Machnik A, Dahlmann A, Kopp C, Goss J, Wagner H, van Rooijen N, et al. Mononuclear phagocyte system depletion blocks interstitial tonicity-responsive enhancer binding protein/vascular endothelial growth factor C expression and induces salt-sensitive hypertension in rats. Hypertension. (2010) 55:755–61. doi: 10.1161/HYPERTENSIONAHA.109.143339
148. Lankhorst S, Severs D, Markó L, Rakova N, Titze J, Müller DN, et al. Salt sensitivity of angiogenesis inhibition–induced blood pressure rise. Hypertension. (2017) 69:919–26. doi: 10.1161/HYPERTENSIONAHA.116.08565
149. Wiig H, Schröder A, Neuhofer W, Jantsch J, Kopp C, Karlsen TV, et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest. (2013) 123:2803–15. doi: 10.1172/JCI60113
150. Selvarajah V, Connolly K, McEniery C, Wilkinson I. Skin sodium and hypertension: a paradigm shift? Curr Hypertens Rep. (2018) 20:94. doi: 10.1007/s11906-018-0892-9
151. Xu H, Chen R, Ghishan FK. Subcloning, localization, and expression of the rat intestinal sodium-hydrogen exchanger isoform 8. Am J Physiol Gastrointest Liver Physiol. (2005) 289:G36–41. doi: 10.1152/ajpgi.00552.2004
152. Kiela PR, Ghishan FK. Recent advances in the renal–skeletal–gut axis that controls phosphate homeostasis. Lab Invest. (2009) 89:7–14. doi: 10.1038/labinvest.2008.114
153. Collins JF, Xu H, Kiela PR, Zeng J, Ghishan FK. Functional and molecular characterization of NHE3 expression during ontogeny in rat jejunal epithelium. Am J Physiol Cell Physiol. (1997) 273:C1937–46. doi: 10.1152/ajpcell.1997.273.6.C1937
154. Collins JF, Honda T, Knobel S, Bulus NM, Conary J, DuBois R, et al. Molecular cloning, sequencing, tissue distribution, and functional expression of a na+/H+ exchanger (NHE-2). Proc Natl Acad Sci USA. (1993) 90:3938–42. doi: 10.1073/pnas.90.9.3938
155. Oberleithner H. Sodium selective erythrocyte glycocalyx and salt sensitivity in man. Pflugers Arch. (2015) 467:1319–25. doi: 10.1007/s00424-014-1577-0
156. Masenga SK, Pilic L, Malumani M, Hamooya BM. Erythrocyte sodium buffering capacity status correlates with self-reported salt intake in a population from livingstone, Zambia. PLoS One. (2022) 17:e0264650. doi: 10.1371/journal.pone.0264650
157. He FJ, Marciniak M, Markandu ND, Antonios TF, MacGregor GA. Effect of modest salt reduction on skin capillary rarefaction in white, black, and Asian individuals with mild hypertension. Hypertension. (2010) 56:253–9. doi: 10.1161/HYPERTENSIONAHA.110.155747
158. Laffer CL, Scott RC, Titze JM, Luft FC, Elijovich F. Hemodynamics and salt-and-water balance link sodium storage and vascular dysfunction in salt-sensitive subjects. Hypertension. (2016) 68:195–203. doi: 10.1161/HYPERTENSIONAHA.116.07289
159. Oberleithner H, Peters W, Kusche-Vihrog K, Korte S, Schillers H, Kliche K, et al. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflugers Arch. (2011) 462:519–28. doi: 10.1007/s00424-011-0999-1
160. Xiao L, Patrick DM, Aden LA, Kirabo A. Mechanisms of isolevuglandin-protein adduct formation in inflammation and hypertension. Prostaglandins Other Lipid Mediators. (2018) 139:48–53. doi: 10.1016/j.prostaglandins.2018.09.008
161. Barbaro NR, Van Beusecum J, Xiao L, do Carmo L, Pitzer A, Loperena R, et al. Sodium activates human monocytes via the NADPH oxidase and isolevuglandin formation. Cardiovasc Res. (2020) 117. doi: 10.1093/cvr/cvaa207
162. Kirabo A, Fontana V, de Faria APC, Loperena R, Galindo CL, Wu J, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. (2014) 124:4642–56. doi: 10.1172/JCI74084
163. Mattson DL. Immune mechanisms of salt-sensitive hypertension and renal end-organ damage. Nat Rev Nephrol. (2019) 15:290–300. doi: 10.1038/s41581-019-0121-z
164. Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am J Physiol Renal Physiol. (2014) 307:F499–508. doi: 10.1152/ajprenal.00258.2014
165. Miguel CD, Guo C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol. (2011) 300:F734–42. doi: 10.1152/ajprenal.00454.2010
166. De Miguel C, Pelegrín P, Baroja-Mazo A, Cuevas S. Emerging role of the inflammasome and pyroptosis in hypertension. Int J Mol Sci. (2021) 22:1064. doi: 10.3390/ijms22031064
167. Pitzer A, Elijovich F, Laffer CL, Ertuglu LA, Sahinoz M, Saleem M, et al. DC ENaC-Dependent inflammasome activation contributes to salt-sensitive hypertension. Circ Res. (2022) 131:328–44. doi: 10.1161/CIRCRESAHA.122.320818
168. Kawarazaki W, Fujita T. Role of rho in salt-sensitive hypertension. Int J Mol Sci. (2021) 22:2958. doi: 10.3390/ijms22062958
169. Chu Y, Zhou Y, Lu S, Lu F, Hu Y. Pathogenesis of higher blood pressure and worse renal function in salt-sensitive hypertension. KBR. (2021) 46:236–44. doi: 10.1159/000515088
170. Xie Y, Shi X, Sheng K, Han G, Li W, Zhao Q, et al. PI3K/Akt Signaling transduction pathway, erythropoiesis and glycolysis in hypoxia. Mol Med Rep. (2019) 19:783–91. doi: 10.3892/mmr.2018.9713
171. Cheng W-H, Lu P-J, Hsiao M, Hsiao C-H, Ho W-Y, Cheng P-W, et al. Renin activates PI3K-akt-eNOS signalling through the angiotensin AT1 and Mas receptors to modulate central blood pressure control in the nucleus tractus solitarii. Br J Pharmacol. (2012) 166:2024–35. doi: 10.1111/j.1476-5381.2012.01832.x
172. Walkowski B, Kleibert M, Majka M, Wojciechowska M. Insight into the role of the PI3K/akt pathway in ischemic injury and post-infarct left ventricular remodeling in normal and diabetic heart. Cells. (2022) 11:1553. doi: 10.3390/cells11091553
173. Sata M, Nagai R. Phosphatidylinositol 3-kinase. Circ Res. (2002) 91:273–5. doi: 10.1161/01.RES.0000031956.29928.62
174. Ghafouri-Fard S, Khanbabapour Sasi A, Hussen BM, Shoorei H, Siddiq A, Taheri M, et al. Interplay between PI3K/AKT pathway and heart disorders. Mol Biol Rep. (2022) 49:9767–81. doi: 10.1007/s11033-022-07468-0
175. Tian H, Liu L, Wu Y, Wang R, Jiang Y, Hu R, et al. Resistin-like molecule β acts as a mitogenic factor in hypoxic pulmonary hypertension via the Ca2+-dependent PI3K/akt/mTOR and PKC/MAPK signaling pathways. Respir Res. (2021) 22:8. doi: 10.1186/s12931-020-01598-4
176. Furusho T, Uchida S, Sohara E. The WNK signaling pathway and salt-sensitive hypertension. Hypertens Res. (2020) 43:733–43. doi: 10.1038/s41440-020-0437-x
177. Sohara E, Uchida S. Kelch-like 3/cullin 3 ubiquitin ligase complex and WNK signaling in salt-sensitive hypertension and electrolyte disorder. Nephrol Dial Transplant. (2016) 31:1417–24. doi: 10.1093/ndt/gfv259
178. Carmichael C, Wainford R. Brain Gαi2-subunit proteins and the prevention of salt sensitive hypertension. Front Physiol. (2015) 6. doi: 10.3389/fphys.2015.00233
179. Wainford RD, Carmichael CY, Pascale CL, Kuwabara JT. Gαi2-protein–mediated signal transduction. Hypertension. (2015) 65:178–86. doi: 10.1161/HYPERTENSIONAHA.114.04463
180. Ohtsu H, Suzuki H, Nakashima H, Dhobale S, Frank GD, Motley ED, et al. Angiotensin II signal transduction through small GTP-binding proteins. Hypertension. (2006) 48:534–40. doi: 10.1161/01.HYP.0000237975.90870.eb
181. Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res. (2006) 71:247–58. doi: 10.1016/j.cardiores.2006.05.001
182. Kapuku GK, Ge D, Vemulapalli S, Harshfield GA, Treiber FA, Snieder H. Change of genetic determinants of left ventricular structure in adolescence: longitudinal evidence from the Georgia cardiovascular twin study. Am J Hypertens. (2008) 21:799–805. doi: 10.1038/ajh.2008.178
183. Arnett DK, Hong Y, Bella JN, Oberman A, Kitzman DW, Hopkins PN, et al. Sibling correlation of left ventricular mass and geometry in hypertensive African Americans and whites: the HyperGEN study*. Am J Hypertens. (2001) 14:1226–30. doi: 10.1016/S0895-7061(01)02200-2
184. Post WS, Larson MG, Myers RH, Galderisi M, Levy D. Heritability of left ventricular mass. Hypertension. (1997) 30:1025–8. doi: 10.1161/01.HYP.30.5.1025
185. Vasan RS, Glazer NL, Felix JF, Lieb W, Wild PS, Felix SB, et al. Genetic variants associated with cardiac structure and function: a meta-analysis and replication of genome-wide association data. JAMA. (2009) 302:168–78. doi: 10.1001/jama.2009.978-a
186. Petretto E, Sarwar R, Grieve I, Lu H, Kumaran MK, Muckett PJ, et al. Integrated genomic approaches implicate osteoglycin (ogn) in the regulation of left ventricular mass. Nat Genet. (2008) 40:546–52. doi: 10.1038/ng.134
187. Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, Sigurdsson A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. (2007) 448:353–7. doi: 10.1038/nature06007
188. Bapat A, Anderson CD, Ellinor PT, Lubitz SA. Genomic basis of atrial fibrillation. Heart. (2018) 104:201–6. doi: 10.1136/heartjnl-2016-311027
189. Sinner MF, Tucker NR, Lunetta KL, Ozaki K, Smith JG, Trompet S, et al. Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation. (2014) 130:1225–35. doi: 10.1161/CIRCULATIONAHA.114.009892
190. Iwasaki Y, Nishida K, Kato T, Nattel S. Atrial fibrillation pathophysiology. Circulation. (2011) 124:2264–74. doi: 10.1161/CIRCULATIONAHA.111.019893
191. Campbell HM, Wehrens XHT. Genetics of atrial fibrillation: an update. Curr Opin Cardiol. (2018) 33:304. doi: 10.1097/HCO.0000000000000505
192. Lieb W, Jansen H, Loley C, Pencina MJ, Nelson CP, Newton-Cheh C, et al. Genetic predisposition to higher blood pressure increases coronary artery disease risk. Hypertension. (2013) 61. doi: 10.1161/HYPERTENSIONAHA.111.00275
193. Lu X, Huang J, Wang L, Chen S, Yang X, Li J, et al. Genetic predisposition to higher blood pressure increases risk of incident hypertension and cardiovascular diseases in Chinese. Hypertension. (2015) 66:786–92. doi: 10.1161/HYPERTENSIONAHA.115.05961
194. Kaviarasan V, Mohammed V, Veerabathiran R. Genetic predisposition study of heart failure and its association with cardiomyopathy. Egypt Heart J. (2022) 74:5. doi: 10.1186/s43044-022-00240-6
195. Pazoki R, Dehghan A, Evangelou E, Warren H, Gao H, Caulfield M, et al. Genetic predisposition to high blood pressure and lifestyle factors. Circulation. (2018) 137:653–61. doi: 10.1161/CIRCULATIONAHA.117.030898
196. Ahn S-Y, Gupta C. Genetic programming of hypertension. Front Pediatr. (2018) 5. https://www.frontiersin.org/articles/10.3389/fped.2017.00285 (Accessed April 11, 2023).29404309
197. Roberts R, Chang CC, Hadley T. Genetic risk stratification. JACC Basic Transl Sci. (2021) 6:287–304. doi: 10.1016/j.jacbts.2020.09.004
198. Kraja AT, Hunt SC, Rao DC, Dávila-Román VG, Arnett DK, Province MA. Genetics of hypertension and cardiovascular disease and their interconnected pathways: lessons from large studies. Curr Hypertens Rep. (2011) 13:46–54. doi: 10.1007/s11906-010-0174-7
199. Patel RS, Masi S, Taddei S. Understanding the role of genetics in hypertension. Eur Heart J. (2017) 38:2309–12. doi: 10.1093/eurheartj/ehx273
200. Krogager ML, Skals RK, Appel EVR, Schnurr TM, Engelbrechtsen L, Have CT, et al. Hypertension genetic risk score is associated with burden of coronary heart disease among patients referred for coronary angiography. PLoS One. (2018) 13:e0208645. doi: 10.1371/journal.pone.0208645
201. Heckbert SR, Post W, Pearson GDN, Arnett DK, Gomes AS, Jerosch-Herold M, et al. Traditional cardiovascular risk factors in relation to left ventricular mass, volume, and systolic function by cardiac magnetic resonance imaging: the multiethnic study of atherosclerosis. J Am Coll Cardiol. (2006) 48:2285–92. doi: 10.1016/j.jacc.2006.03.072
202. Saeed M, Van TA, Krug R, Hetts SW, Wilson MW. Cardiac MR imaging: current status and future direction. Cardiovasc Diagn Ther. (2015) 5:290–310. doi: 10.3978/j.issn.2223-3652.2015.06.07
203. Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European society of cardiology and the European society of hypertension. J Hypertens. (2018) 36:1953. doi: 10.1097/HJH.0000000000001940
204. Schmieder RE, Wagner F, Mayr M, Delles C, Ott C, Keicher C, et al. The effect of sacubitril/valsartan compared to olmesartan on cardiovascular remodelling in subjects with essential hypertension: the results of a randomized, double-blind, active-controlled study. Eur Heart J. (2017) 38:3308–17. doi: 10.1093/eurheartj/ehx525
205. Pan X-D, Zeng Z-H, Liang L-Y, Luo J-D, Xiao A-Y, Lai Q, et al. The effects of simvastatin on left ventricular hypertrophy and left ventricular function in patients with essential hypertension. Clin Exp Hypertens. (2011) 33:558–64. doi: 10.3109/10641963.2011.577486
206. Mazza A, Townsend DM, Schiavon L, Torin G, Lenti S, Rossetti C, et al. Long-term effect of the perindopril/indapamide/amlodipine single-pill combination on left ventricular hypertrophy in outpatient hypertensive subjects. Biomed Pharmacother. (2019) 120:109539. doi: 10.1016/j.biopha.2019.109539
207. Lal B, Iqbal A, Butt NF, Randhawa FA, Rathore R, Waseem T. Efficacy of high dose allopurinol in reducing left ventricular mass in patients with left ventricular hypertrophy by comparing its efficacy with febuxostat—a randomized controlled trial. J Pak Med Assoc. (2018) 68:1446–50. PMID: 30317339.30317339
208. Gingles CR, Symon R, Gandy SJ, Struthers AD, Houston G, MacDonald TM, et al. Allopurinol treatment adversely impacts left ventricular mass regression in patients with well-controlled hypertension. J Hypertens. (2019) 37:2481–9. doi: 10.1097/HJH.0000000000002189
209. Burns J, Ball SG, Worthy G, Struthers AD, Mary DASG, Greenwood JP. Hypertensive left ventricular hypertrophy: a mechanistic approach to optimizing regression assessed by cardiovascular magnetic resonance. J Hypertens. (2012) 30:2039–46. doi: 10.1097/HJH.0b013e328356b850
210. Mercurio V, Pucci G, Bosso G, Fazio V, Battista F, Iannuzzi A, et al. A nutraceutical combination reduces left ventricular mass in subjects with metabolic syndrome and left ventricular hypertrophy: a multicenter, randomized, double-blind, placebo-controlled trial. Clin Nutr. (2020) 39:1379–84. doi: 10.1016/j.clnu.2019.06.026
211. Motoki H, Koyama J, Izawa A, Tomita T, Miyashita Y, Takahashi M, et al. Impact of azelnidipine and amlodipine on left ventricular mass and longitudinal function in hypertensive patients with left ventricular hypertrophy. Echocardiography. (2014) 31:1230–8. doi: 10.1111/echo.12548
212. Moroni C, Tolone S, Lopreiato F, Scrofani AR, Bossini A, Affricano C, et al. Effects of losartan on left ventricular mass: a three-year follow-up in elderly hypertensives with myocardial hypertrophy despite successful conventional antihypertensive treatment. Eur Rev Med Pharmacol Sci. (2017) 21:1323–8. PMID: 28387895.28387895
213. Schneider A, Schwab J, Karg MV, Kalizki T, Reinold A, Schneider MP, et al. Low-dose eplerenone decreases left ventricular mass in treatment-resistant hypertension. J Hypertens. (2017) 35:1086–92. doi: 10.1097/HJH.0000000000001264
214. Mohan M, Al-Talabany S, McKinnie A, Mordi IR, Singh JSS, Gandy SJ, et al. A randomized controlled trial of metformin on left ventricular hypertrophy in patients with coronary artery disease without diabetes: the MET-REMODEL trial. Eur Heart J. (2019) 40:3409–17. doi: 10.1093/eurheartj/ehz203
215. Haufe S, Utz W, Engeli S, Kast P, Böhnke J, Pofahl M, et al. Left ventricular mass and function with reduced-fat or reduced-carbohydrate hypocaloric diets in overweight and obese subjects. Hypertension. (2012) 59:70–5. doi: 10.1161/HYPERTENSIONAHA.111.178616
216. Al-Shafei AIM, El-Gendy OAA. Regular consumption of green tea improves pulse pressure and induces regression of left ventricular hypertrophy in hypertensive patients. Physiol Rep. (2019) 7:e14030. doi: 10.14814/phy2.14030
217. Kirchhof P, Camm AJ, Goette A, Brandes A, Eckardt L, Elvan A, et al. Early rhythm-control therapy in patients with atrial fibrillation. N Engl J Med. (2020) 383:1305–16. doi: 10.1056/NEJMoa2019422
218. Wazni OM, Dandamudi G, Sood N, Hoyt R, Tyler J, Durrani S, et al. Cryoballoon ablation as initial therapy for atrial fibrillation. N Engl J Med. (2021) 384:316–24. doi: 10.1056/NEJMoa2029554
219. Kuniss M, Pavlovic N, Velagic V, Hermida JS, Healey S, Arena G, et al. Cryoballoon ablation vs. Antiarrhythmic drugs: first-line therapy for patients with paroxysmal atrial fibrillation. Europace. (2021) 23:1033–41. doi: 10.1093/europace/euab029
220. Willems S, Borof K, Brandes A, Breithardt G, Camm AJ, Crijns HJGM, et al. Systematic, early rhythm control strategy for atrial fibrillation in patients with or without symptoms: the EAST-AFNET 4 trial. Eur Heart J. (2022) 43:1219–30. doi: 10.1093/eurheartj/ehab593
221. Kuck K-H, Lebedev DS, Mikhaylov EN, Romanov A, Gellér L, Kalējs O, et al. Catheter ablation or medical therapy to delay progression of atrial fibrillation: the randomized controlled atrial fibrillation progression trial (ATTEST). Europace. (2021) 23:362–9. doi: 10.1093/europace/euaa298
222. Park J-W, Yu HT, Kim T-H, Uhm J-S, Joung B, Lee M-H, et al. Atrial fibrillation catheter ablation increases the left atrial pressure. Circ Arrhythm Electrophysiol. (2019) 12:e007073. doi: 10.1161/CIRCEP.118.007073
223. Yang G, Zheng L, Jiang C, Fan J, Liu X, Zhan X, et al. Circumferential pulmonary vein isolation plus low-voltage area modification in persistent atrial fibrillation: the STABLE-SR-II trial. JACC Clin Electrophysiol. (2022) 8:882–91. doi: 10.1016/j.jacep.2022.03.012
224. Reed JL, Terada T, Vidal-Almela S, Tulloch HE, Mistura M, Birnie DH, et al. Effect of high-intensity interval training in patients with atrial fibrillation: a randomized clinical trial. JAMA Netw Open. (2022) 5:e2239380. doi: 10.1001/jamanetworkopen.2022.39380
225. Ezekowitz MD, Pollack CV, Halperin JL, England RD, VanPelt Nguyen S, Spahr J, et al. Apixaban compared to heparin/vitamin K antagonist in patients with atrial fibrillation scheduled for cardioversion: the EMANATE trial. Eur Heart J. (2018) 39:2959–71. doi: 10.1093/eurheartj/ehy148
226. Oldgren J, Steg PG, Hohnloser SH, Lip GYH, Kimura T, Nordaby M, et al. Dabigatran dual therapy with ticagrelor or clopidogrel after percutaneous coronary intervention in atrial fibrillation patients with or without acute coronary syndrome: a subgroup analysis from the RE-DUAL PCI trial. Eur Heart J. (2019) 40:1553–62. doi: 10.1093/eurheartj/ehz059
227. Rienstra M, Hobbelt AH, Alings M, Tijssen JGP, Smit MD, Brügemann J, et al. Targeted therapy of underlying conditions improves sinus rhythm maintenance in patients with persistent atrial fibrillation: results of the RACE 3 trial. Eur Heart J. (2018) 39:2987–96. doi: 10.1093/eurheartj/ehx739
228. Shah B, Newman JD, Woolf K, Ganguzza L, Guo Y, Allen N, et al. Anti-Inflammatory effects of a vegan diet versus the American heart association-recommended diet in coronary artery disease trial. J Am Heart Assoc. (2018) 7:e011367. doi: 10.1161/JAHA.118.011367
229. Jimenez-Torres J, Alcalá-Diaz JF, Torres-Peña JD, Gutierrez-Mariscal FM, Leon-Acuña A, Gómez-Luna P, et al. Mediterranean Diet reduces atherosclerosis progression in coronary heart disease: an analysis of the CORDIOPREV randomized controlled trial. Stroke. (2021) 52:3440–9. doi: 10.1161/STROKEAHA.120.033214
230. Naito R, Miyauchi K, Yasuda S, Kaikita K, Akao M, Ako J, et al. Rivaroxaban monotherapy vs combination therapy with antiplatelets on total thrombotic and bleeding events in atrial fibrillation with stable coronary artery disease: a post hoc secondary analysis of the AFIRE trial. JAMA Cardiol. (2022) 7:787–94. doi: 10.1001/jamacardio.2022.1561
231. Branch KR, Probstfield JL, Eikelboom JW, Bosch J, Maggioni AP, Cheng RK, et al. Rivaroxaban with or without aspirin in patients with heart failure and chronic coronary or peripheral artery disease. Circulation. (2019) 140:529–37. doi: 10.1161/CIRCULATIONAHA.119.039609
232. Opstal TSJ, van Broekhoven A, Fiolet ATL, Mosterd A, Eikelboom JW, Nidorf SM, et al. Long-term efficacy of colchicine in patients with chronic coronary disease: insights from LoDoCo2. Circulation. (2022) 145:626–8. doi: 10.1161/CIRCULATIONAHA.121.058233
233. Pedersen LR, Olsen RH, Anholm C, Astrup A, Eugen-Olsen J, Fenger M, et al. Effects of 1 year of exercise training versus combined exercise training and weight loss on body composition, low-grade inflammation and lipids in overweight patients with coronary artery disease: a randomized trial. Cardiovasc Diabetol. (2019) 18:127. doi: 10.1186/s12933-019-0934-x
234. Reed JL, Terada T, Cotie LM, Tulloch HE, Leenen FH, Mistura M, et al. The effects of high-intensity interval training, nordic walking and moderate-to-vigorous intensity continuous training on functional capacity, depression and quality of life in patients with coronary artery disease enrolled in cardiac rehabilitation: a randomized controlled trial (CRX study). Prog Cardiovasc Dis. (2022) 70:73–83. doi: 10.1016/j.pcad.2021.07.002
235. Jepma P, Jorstad HT, Snaterse M, Ter Riet G, Kragten H, Lachman S, et al. Lifestyle modification in older versus younger patients with coronary artery disease. Heart. (2020) 106:1066–72. doi: 10.1136/heartjnl-2019-316056
236. Sabatine MS, De Ferrari GM, Giugliano RP, Huber K, Lewis BS, Ferreira J, et al. Clinical benefit of evolocumab by severity and extent of coronary artery disease: analysis from FOURIER. Circulation. (2018) 138:756–66. doi: 10.1161/CIRCULATIONAHA.118.034309
237. Hieda M, Sarma S, Hearon CM, MacNamara JP, Dias KA, Samels M, et al. One-year committed exercise training reverses abnormal left ventricular myocardial stiffness in patients with stage B heart failure with preserved ejection fraction. Circulation. (2021) 144:934–46. doi: 10.1161/CIRCULATIONAHA.121.054117
238. Filippatos G, Butler J, Farmakis D, Zannad F, Ofstad AP, Ferreira JP, et al. Empagliflozin for heart failure with preserved left ventricular ejection fraction with and without diabetes. Circulation. (2022) 146:676–86. doi: 10.1161/CIRCULATIONAHA.122.059785
239. Peikert A, Martinez FA, Vaduganathan M, Claggett BL, Kulac IJ, Desai AS, et al. Efficacy and safety of dapagliflozin in heart failure with mildly reduced or preserved ejection fraction according to age: the DELIVER trial. Circ Heart Fail. (2022) 15:e010080. doi: 10.1161/CIRCHEARTFAILURE.122.010080
240. McDiarmid AK, Swoboda PP, Erhayiem B, Bounford KA, Bijsterveld P, Tyndall K, et al. Myocardial effects of aldosterone antagonism in heart failure with preserved ejection fraction. J Am Heart Assoc. (2020) 9:e011521. doi: 10.1161/JAHA.118.011521
241. Hersberger L, Dietz A, Bürgler H, Bargetzi A, Bargetzi L, Kägi-Braun N, et al. Individualized nutritional support for hospitalized patients with chronic heart failure. J Am Coll Cardiol. (2021) 77:2307–19. doi: 10.1016/j.jacc.2021.03.232
242. Butler J, Stebbins A, Melenovský V, Sweitzer NK, Cowie MR, Stehlik J, et al. Vericiguat and health-related quality of life in patients with heart failure with reduced ejection fraction: insights from the VICTORIA trial. Circ Heart Fail. (2022) 15:e009337. doi: 10.1161/CIRCHEARTFAILURE.121.009337
243. Bonilla-Palomas JL, Gámez-López AL, Castillo-Domínguez JC, Moreno-Conde M, López Ibáñez MC, Alhambra Expósito R, et al. Nutritional intervention in malnourished hospitalized patients with heart failure. Arch Med Res. (2016) 47:535–40. doi: 10.1016/j.arcmed.2016.11.005
244. Li M, Zhang Y, Wan Q, Li Y, Qu T, Yuan F. Use of levosimendan combined with shenfu injection to treat acute heart failure patients with hypotension: a prospective randomized controlled single-blind study. BMC Cardiovasc Disord. (2022) 22:130. doi: 10.1186/s12872-022-02572-2
245. Felker GM, Solomon SD, Claggett B, Diaz R, McMurray JJV, Metra M, et al. Assessment of omecamtiv mecarbil for the treatment of patients with severe heart failure: a post hoc analysis of data from the GALACTIC-HF randomized clinical trial. JAMA Cardiol. (2022) 7:26–34. doi: 10.1001/jamacardio.2021.4027
Keywords: hypertensive heart disease, heart failure, salt, hypertension, coronary artery disease, atrial fibrillation, immunity
Citation: Masenga SK and Kirabo A (2023) Hypertensive heart disease: risk factors, complications and mechanisms. Front. Cardiovasc. Med. 10:1205475. doi: 10.3389/fcvm.2023.1205475
Received: 13 April 2023; Accepted: 26 May 2023;
Published: 5 June 2023.
Edited by:
Ryszard Nosalski, University of Edinburgh, United KingdomReviewed by:
Piotr Szczepaniak, Jagiellonian University Medical College, PolandEvgeny Shlyakhto, Almazov National Medical Research Centre, Russia
© 2023 Masenga and Kirabo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annet Kirabo QW5uZXQuS2lyYWJvQHZ1bWMub3Jn