
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 13 July 2023
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1204483
This article is part of the Research TopicMitochondrial Maintenance and Cardiovascular AgingView all 3 articles
 Alessia Mongelli1
Alessia Mongelli1 Alessandro Mengozzi1Martin Geiger1
Alessandro Mengozzi1Martin Geiger1 Era Gorica1
Era Gorica1 Shafeeq Ahmed Mohammed1
Shafeeq Ahmed Mohammed1 Francesco Paneni1,2Frank Ruschitzka1,2
Francesco Paneni1,2Frank Ruschitzka1,2 Sarah Costantino1,2*
Sarah Costantino1,2*
Mitochondria are cellular organelles which generate adenosine triphosphate (ATP) molecules for the maintenance of cellular energy through the oxidative phosphorylation. They also regulate a variety of cellular processes including apoptosis and metabolism. Of interest, the inner part of mitochondria—the mitochondrial matrix—contains a circular molecule of DNA (mtDNA) characterised by its own transcriptional machinery. As with genomic DNA, mtDNA may also undergo nucleotide mutations that have been shown to be responsible for mitochondrial dysfunction. During physiological aging, the mitochondrial membrane potential declines and associates with enhanced mitophagy to avoid the accumulation of damaged organelles. Moreover, if the dysfunctional mitochondria are not properly cleared, this could lead to cellular dysfunction and subsequent development of several comorbidities such as cardiovascular diseases (CVDs), diabetes, respiratory and cardiovascular diseases as well as inflammatory disorders and psychiatric diseases. As reported for genomic DNA, mtDNA is also amenable to chemical modifications, namely DNA methylation. Changes in mtDNA methylation have shown to be associated with altered transcriptional programs and mitochondrial dysfunction during aging. In addition, other epigenetic signals have been observed in mitochondria, in particular the interaction between mtDNA methylation and non-coding RNAs. Mitoepigenetic modifications are also involved in the pathogenesis of CVDs where oxygen chain disruption, mitochondrial fission, and ROS formation alter cardiac energy metabolism leading to hypertrophy, hypertension, heart failure and ischemia/reperfusion injury. In the present review, we summarize current evidence on the growing importance of epigenetic changes as modulator of mitochondrial function in aging. A better understanding of the mitochondrial epigenetic landscape may pave the way for personalized therapies to prevent age-related diseases.
Mitochondria are double membrane organelles which are actively involved in a multitude of cellular activities such as energy production in the form of adenosine triphosphate (ATP), intracellular Ca2+ signalling, generation of reactive oxygen species (ROS), and catalysis of metabolites (1). Physiologically, the high plasticity of mitochondria makes them able to respond rapidly to cellular metabolic demands, such as during physical activity or fasting (1). Mitochondria are dynamic organelles that constantly alter their shape, oscillating between two opposing processes, fission and fusion, in response to different stimuli (1, 2).
In healthy individuals, these processes are well balanced. In fact, if the organism requires more energy, two mitochondria fuse together. To increase the amount of ATP, the cell transcribes the nuclear genes encoding mitochondrial proteins (NuGEMPs) which in turn promote the activity of the outer mitochondrial membrane (OMM) proteins, Mitofusin 1 and 2 (Mnf1/2) and the inner mitochondrial membrane (IMM) protein, Optic Atrophy Protein 1 (Opa1) (3).
On the other hand, under conditions of reduced energy demand or in the presence of dysfunctional mitochondria, such as during a sedentary lifestyle or aging, the fission mechanism (the removal of a mitochondria) is enhanced (1). This mechanism, triggered by the reduction of inner membrane potential or by high ROS production, induces the interaction among dynamin like 1 (Drp1), fission mitochondrial 1 (Fis1) and mitochondrial fission factor (Mff) (4). In physiological conditions, the unneeded or damaged mitochondrial fragments are cleared by the autophagosome (mitophagy process), while the accumulation of damaged mitochondria is strictly linked to the exacerbation of neurodegenerative and cardiovascular diseases, cancer and inflammation (5–8).
These mechanisms are also influenced by mitochondrial DNA (mtDNA). In fact, cells contain 100–10,000 copies of mtDNA in proportion to energy request (9). Despite some similarities with the genomic DNA, the mtDNA has specific features. First, the hereditary is not Mendelian because all mitochondria are transmitted by uniparental model (maternal hereditament). Second, the mtDNA is a circular covalently closed double stranded DNA with a length of 16.5 kbs in human (10). To distinguish the two strands, one is named “heavy” (the sense, which is purine rich), and the other is named “light” (the antisense, which contains high amounts of pyrimidine). The mtDNA has 37 genes, which encode 13 polypeptides involved into the oxidative phosphorylation, 2 rRNAs and 22 tRNAs (11). Third, similar to genomic DNA, the mtDNA assumes a secondary structure that regulates the genes transcription and the synthesis of new mtDNA molecules (12). This upper level of mitochondrial gene regulation has been named as “mitoepigenetics” (13–15).
Unlike genomic DNA, mtDNA is not packed into nucleosomes but is organized into protein complexes in which mtDNA is bound to mitochondrial transcription factor A (TFAM) to form nucleoids measuring 100 nm in diameter (16, 17). Despite its name, TFAM is not the main transcriptional player; in fact, mitochondrial RNA polymerase (POLRMT) and mitochondrial transcription factor B2 (TFB2M) are mainly involved in mitochondrial mRNA synthesis (18). However, it is essential for the maintenance, expression and transmission of mitochondrial DNA (mtDNA). Interestingly, post-translational modifications (PTMs) of TFAM play an important role in its affinity for mtDNA. In particular, acetylation in lysine (K62, K76, K111 and K118) or phosphorylation (S55 and/or S56) in serine can fine-tune TFAM-DNA binding affinity (19).
Specifically, acetylation of TFAM at lysine 76 (K76) mediated by GCN5L1 (General Control of Amino-Acid Synthesis, yeast homolog-like 1) inhibits the binding of TFAM to the mitochondrial transporter TOM70, resulting in reduced TFAM import into mitochondria and mitochondrial biogenesis (20). In contrast to these results, another work shows that increasing the acetylation levels of TFAM does not alter its binding to mtDNA, while significantly reducing TFAM-mediated DNA unwinding capacity (21). Although the role of TFAM acetylation is still debated, a growing body of evidence shows that PMTs in the C-tail of TFAM are critical for the recruitment and positioning of POLRMT (22) and TFBM2, leading to mitochondrial gene transcription (18).
Sirtuins (Sirts) are an evolutionarily conserved family of class III histone deacetylases that require NAD+ as a cofactor (23). Among these Sirt3, 4 and 5 have been found in mitochondria (24–26). However, only Sirt3 has been shown to have deacetylation activity, whereas the role of Sirt4 is that of ADP-ribosylase and lipoamidase, and Sirt5 is involved in succinylation, malonylation, and glutarylation (27). Interestingly, one of the targets of Sirt3 in the mitochondrion is TFAM (28) whose deacetylation results in increased binding to mtDNA, thus repressing gene transcription (28). Similarly, ERK2-mediated phosphorylation of TFAM in serine 177 (29) increases the binding of TFAM to mtDNA, resulting in suppression of transcription (19, 29). In addition, recent work shows that protein kinase A (PKA) regulates the phosphorylation of TFAM in serine 55 and promotes its degradation (30, 31).
In the nucleus, gene expression is not only modulated by PTMs of histones, but it is also regulated by the methylation in 5th position of cytosine (5 mC) in cytosine-guanine dinucleotide (CpG) at the level of regulatory sequences. Specifically, an increase in DNA methylation triggers the inhibition of transcription. The involvement and amount of methylated mtDNA are still debated. Some works report that methylation is a specific feature of genomic DNA that is not shared with mtDNA (32, 33), while other studies show the presence of methylation also at the level of mtDNA (34, 35, 36). Interestingly, in mtDNA, methylation of gene promoters has been observed at non-CpG sites (34). Although DNA methyltransferases DNMT1, DNMT3a and DNMT3b were shown to be active in mitochondria, their repression does not affect the mtDNA methylome (34). In mitochondrial DNA, the main methylation found is at the level of adenine (6 mA) at adenine-thymine dinucleotides (ApT) (37). In addition, accumulation of eukaryotic methyltransferase 4 MTA70 (METTL4) has been observed in the mitochondrial matrix (38), and knock-out of METTL4 results in a 6 mA decrease in mtDNA (38). Similar to nuclear DNA, in mitochondria 6 mA attenuates the binding of TFAM to transcription factors (38), suggesting the homology of the role of methylation in gene expression. In the nucleus, demethylation of 6 mA is driven by AlkB homolog 1 (ALKBH1) and ALKBH4 (39). However, there is currently no information on the mechanisms of 6 mA demethylation in mtDNA.
Another branch of epigenetic regulation is the role of noncoding RNAs (ncRNAs), a class of ribonucleic acid sequences that do not carry protein translation information but are involved in gene and protein regulation (40). Based on their length, ncRNAs are divided into long non-coding RNAs (lncRNAs), which are longer than 200 nts, and small ncRNAs (sncRNAs), which are shorter than 200 nts and include miRNA, cirRNA, and piRNA (40). Several roles have been established for lncRNAs: regulation of chromatin structure by interacting with histones, repression or activation of gene expression through hybridization with genomic DNA, regulation of splicing, stabilization of messenger RNAs (mRNAs), and sponging of miRNAs (41). On the other hand, miRNAs are known to interfere with the translation mechanism due to their complementarity with the 3′-untranslated region (3′UTR) of mRNAs (42).
Interestingly, metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was found in the mitochondrial matrix where it interacts with mtDNA in several regions (43). Specifically, this interaction represses the transcription of cytochrome C oxidase II (COX2), NADH: Ubiquinone oxidoreductase core subunit 3 (ND3) and cytochrome B (CYTB) genes encoded at the mitochondrial level (43). In addition, downregulation of MALAT1 in mitochondria leads to altered mtDNA copy number, mitophagy, apoptosis, and abnormalities in ATP production (43).
The RNA component of the RNase MRP ribonucleoprotein (RNP) complex (RMRP) lncRNA has also been found in the mitochondrial matrix where it acts as a primer for mtDNA replication (44). Furthermore, RMRP associates with nucleoporins involved in the export of small nuclear RNAs, highlighting its interaction between nuclear and mitochondrial epigenetic regulation (44).
Recent work reports the presence of mitochondria-specific miRNAs, called MitomiRs, which are subsequently transported into the cytosol to inhibit their target mRNAs or interact with mitochondrial proteins (45). One example is MitomiR-2392, which reduces the expression of mt-ND2, mt-ND4, mt-ND5, mt-CYTB, and mt-COX1 genes through interaction with Argonaute RISC Catalytic Component 2 (AGO2), which is present in the mitochondrial matrix (46). Dysregulation of mitomiR-2392 leads to altered cellular metabolism with decreased oxidative phosphorylation and increased glycolysis (46). Due to AGO2′s dual localization (cytosol and mitochondrial matrix) and ability to bind RNA, it has been proposed as an importer of miRNAs (47). Indeed, many miRNAs synthesized from genomic DNA have been found in mitochondria where they inhibit their targets. One example is miR-181c, which regulates levels of the mitochondrial protein mt-COX1 (48). Interestingly, overexpression of miR-181c did not change mt-COX1 mRNA but significantly decreased mt-COX1 protein, suggesting that miR-181c is primarily a translational regulator of mt-COX1 (48). In addition, overexpression of miR-181c results in increased mt-COX2 mRNA and protein content, with an increase in both mitochondrial respiration and reactive oxygen species generation, causing mitochondrial dysfunction (48). Similarly, human miR-198 and miR-765 have also been found in mitochondria, but their targets have not yet been established (47).
A brief overview about the differences between epigenetics and mitoepigenetics is summarized in Figure 1.
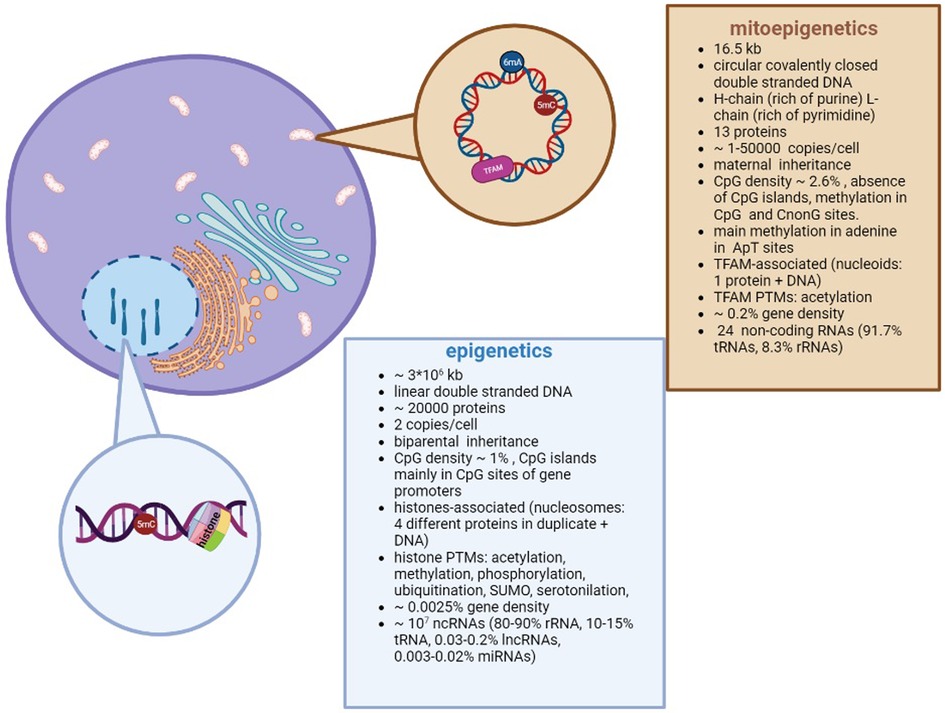
Figure 1. Differences between nuclear and mitochondrial Epigenetics. Despite different DNA structures (circular vs linear) and genome organizations (i.e. nucleoids vs nucleosomes; methylation in adenosine vs cytosine), both types of epigenetics are aimed to regulate the gene expression.
The aging process leads to nuclear epigenetic alterations such as remodelling of chromatin structure caused by changes in DNA methylation and histone PTMs, telomere shortening, and modulation of ncRNAs espression (49, 50, 51).
TFAM plays an essential role in maintenance, expression, and organization of mitochondrial DNA. Indeed, it is required for efficient promoter recognition by mitochondrial RNA polymerase and binds and induces significant conformational changes in mtDNA. In muscles of aged flies, TFAM and mtDNA have been found to form a more condensed structure than in juveniles (52) impeding access to the mitochondrial transcriptomic machinery with a subsequent reduction of gene expression. In detail, from 1 day of age to 12 weeks of age, the size of nucleoids decreases while their number increases (52). Under normal conditions, overexpression of TFAM leads to the reduction of Drosophila lifespan, whereas in the presence of 1% H2O2, flies that up-regulate TFAM resist the environment more, revealing that TFAM is a central factor under severe oxidative stress and mitochondria maintenance (53). Despite the lack of research on TFAM and aging in mammals, the repression of mitochondrial gene transcription in the presence of a high level of TFAM protein is well known (54) suggesting a role of TFAM in aging.
Recently, it has been observed that DNA methyltransferase 3 alpha (DNMT3A) and ten-even-transposon 2 (TET2) regulates the TFAM expression in macrophages (55). Specifically, decreased DNMT3A and/or TET2 results in downregulation of TFAM with subsequent cytosolic mtDNA release (55). The presence of mtDNA in the cytosol is responsible for the activation of cyclic GMP–AMP synthase (cGAS) signalling that triggers interferon alpha (IFNα) production with subsequent increase in inflammation (55).
Notably, in aging is well known the increasing of inflammation state (named inflammaging) which involved several cytokines included IFN (56). This finding suggests that TFAM migh contribute to the burst of inflammation observed in aging. in aging is well known the increasing of inflammation state (named inflammaging) which involved several cytokines included IFN (56). In young mice, lymphocyte-specific Tfam knockout recapitulated the characteristics of mitochondrial dysfunction that occurs in aged (22-month-old) wild-type mice, and this mitochondrial decline is characterized by increased secretion of inflammatory cytokines, such as interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) (57).
It is well known that nuclear translocation of p53 protein is associated with increased aging due to its inhibition of PPARγ co-activator 1α (PGC1α) and PGC1β (58). It is interesting to see that in skeletal cells after endurance exercise, the presence of nuclear p53 is reduced while its presence is increased in mitochondria where it serves to positively modulate the activity of the mitochondrial transcription factor TFAM (59). Based on this evidence, physical activity in the elderly could also improve mitochondrial function through the p53-TFAM interaction.
Another epigenetic modification that occurs in aging is the global decrease in the level of methylation in genomic DNA, which cooperates with the modification of histones (60). Despite the global decrease in 5mC, it has been observed that some specific CpGs are particularly hypo- or hyper-methylated. This signature has been used to develop so-called “epigenetic clocks” that are able to specifically detect the biological age of individuals (61–65).
Although the most representative methylated base in mtDNA is adenine, recent work shows a correlation between aging and 5 mC methylation of mtDNA in the brain, where some 5 mCs (in this case both CpG and non-CpG) were found to be modulated (66). Unlike genomic DNA, aged mtDNA reveals a pattern of global hypermethylation (66). This feature is likely due to the loss of the ability of ten-eleven transposons (TETs) to eliminate methylation on cytosines, caused by altered metabolism in which the reduction of α-ketoglutarate (the cofactor of TETs) was observed (67). In recent work, a significant increase in DNA methylation levels was found according to age and the administration of the low-calorie diet. Particularly, the increase in methylation level represses genes involved in mitochondrial biogenesis, suggesting that mitochondrial dynamics is driven by age and diet (68). During aging, mitochondrial increase in reactive-oxygenated species (ROS) has been reported to modulate mtDNA (69, 70). Specifically, ROS-induced damage to mtDNA reveals impaired production or mutation of enzymes involved in oxidative phosphorylation, exacerbating mitochondrial dysfunction and enhanced cellular senescence (69).
The correlation between miRNA and histone deacetylase has been demonstrated during aging. Specifically, miR-9 and miR-34a have been found to be up-regulated in aging and both target Sirt1 mRNA, which is found to be down-regulated contributing to enhanced gene expression (71). In addition, Sirt1 is also regulated by miR-181a, which is down-regulated during aging instead (72). Repression of miR-181a in the elderly has been associated with defects in T-cell activation (72). The lncRNAs are also modulated in aging. For example, H19 is down-regulated in senescent endothelial cells (73). Specifically, repression of H19 reflects increased phosphorylation of STAT3, which becomes active and transcribes p16 and p21 reflecting cell cycle inhibition (73). In addition, in cardiomyocytes (CM), lncRNA ENSMUST00000134285 has been observed to control apoptosis. In this case, in aged tissues, the expression of ENSMUST00000134285 is increased and inhibits MAPK11 activity, initiating the apoptotic process (74).
Modulation of ncRNAs has also been observed in aging, and the ncRNAome is tissue-specific. An example is miR-183-5p, miR-199b-5p, miR-205-5p, and miR-200b-3p, which are up-regulated in 3-month-old mice and are involved in the thymus regression process (75). Similarly, modulation of the lncRNAome during aging was found in the liver, where Meg3, Rian and Mirg were found to be up-regulated, revealing increased inflammation, repression of cell proliferation and metabolic changes (76). The lncRNA nuclear-enriched abundant transcript 1 (NEAT1) was found to be modulated in bone marrow mesenchymal stem cell (BMSC) senescence (77). Up-regulation of NEAT1 drives the transition from bone cells to fat cells by hindering mitochondrial function, while repression of NEAT1 reflects an increase in Sirt3 (77), which, as has been discussed previously, is responsible for deacetylating TFAM (28).
In conclusion, the ncRNAs that are transcribed by nuclear DNA and shifted to mitochondria have been found to modulate oxidative stress response, Sirt1-dependent deacetylation, inflammation, cell cycle and apoptosis confirming their involvement in mitochondrial homeostasis leading to aging process (Table 1).
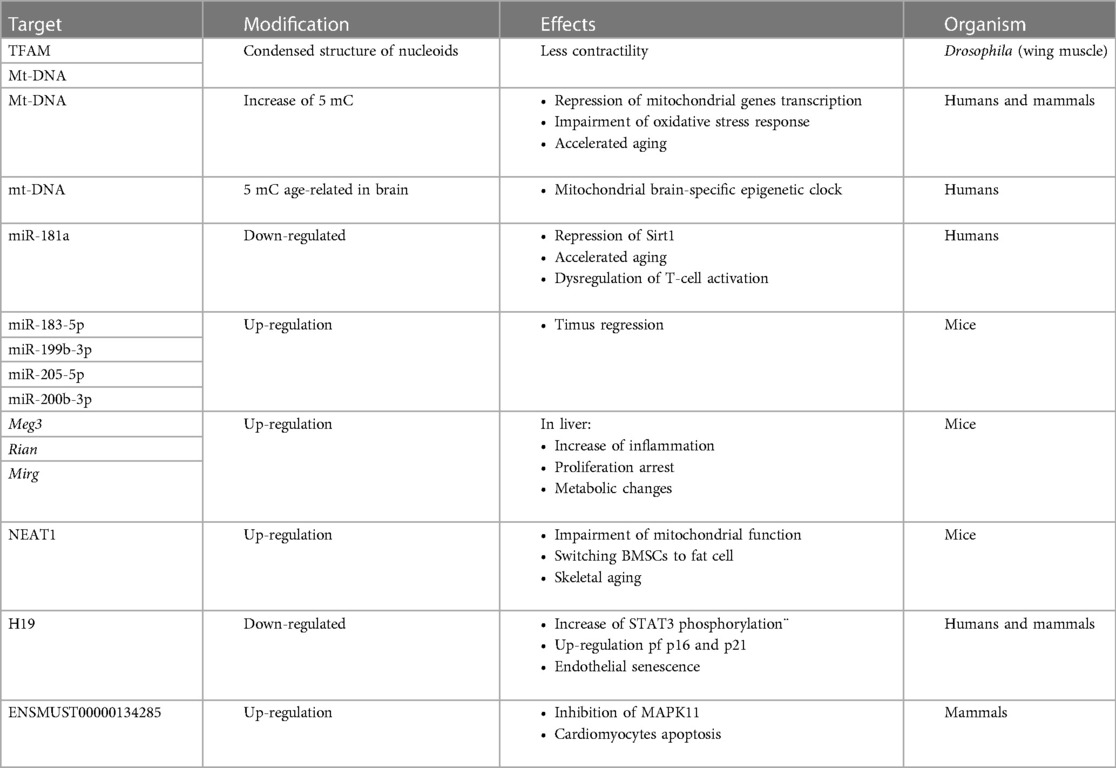
Table 1. Overview of mitoepigenetic modifications in aging.
During aging, the risk of developing cardiovascular disease (CVD) increases (78). On the other hand, various risk factors such as unhealthy dietary regimens, physical inactivity, smoking, and alcohol abuse play a major role in the development and acceleration of cardiovascular diseases (2, 79, 80). Heart failure (HF), coronary heart disease, rheumatic heart disease, and stroke are among the leading causes of death globally (17.9 million per year), and the involvement of epigenetics in the development of these diseases has been extensively studied (81, 82, 83, 84, 85, 86). However, growing evidence suggests the potential contribution of mitoepigenetics in the pathophysiology of CVD.
Modulation of the mtDNA methylation of the mt-COX2 gene has been observed in human cardiac mesenchymal stem cells (HMSCs) (87). In particular, senescent HMSCs reveal hypermethylation of the COX2 gene, which appears to be downregulated (87). On the other hand, overexpression of mt-COX2 and inhibition of DNMT1 by 5-aza-2′-deoxycytidine delay the senescence process (87). It has also been shown that, in the rat model, depletion of mt-COX2 reduces ATP and acetyl-CoA production. Consistently, the expression of genes related to mitochondrial oxidation, were downregulated, while glycolytic hexokinase 1 (HK1) was upregulated. These observations indicate that COX2-deficient rats develop hypertension, heart failure, and increased thrombotic events, probably as a result of dysregulation of cardiac energy metabolism (88).
Analysis of mtDNA methylation was also performed in platelets from CVD patients, where an increase in 5mC methylation was found in comparison with healthy controls (89). In particular, an increase in the methylation level of the mt-COX1/2/3 gene and mitochondrial leucine 1 (mt-TL1) tRNA was found (89). In line with these findings, in vascular smooth muscle cells (VSMCs) platelet-derived growth factor-BB (PDGF-BB) triggers the translocation of DNMT1 from the nucleus to the mitochondria, where mtDNA methylation increases resulting in suppression of gene expression (90). These increased levels of methylation are also associated with mitochondrial dysfunction, altered contractility of VSMCs, and aberrant cell growth (90).
Mitoepigenetics also seems to play an important role in CAD. Indeed, comparing mtDNA methylation levels of peripheral blood leukocytes from patients with stable coronary artery disease (SCAD) and acute coronary syndrome (ACS), a lower level of global 5 mC mtDNA was found in patients with ACS. In line with these findings, hypermethylation of the D-loop region was also detected in these patients, which is associated with reduced mtDNA synthesis (91).
In myocardial infarction, alteration of physiological functions of mitochondria leads to modification of proteins and lipids and inhibition of energy production, contractile capacity, cell necrosis, or apoptosis. In the myocardium, oxidative stress caused by ischemia/reperfusion (I/R) injury triggers nuclear translocation of nuclear respiratory factor 1 (NRF1) and upregulation of PPARG coactivator 1 alpha (PGC-1α), which increase TFAM transcription and contribute to mitochondrial biogenesis and repair, acting as a compensatory mechanism(92). Interestingly, TFAM depletion reflects the loss of cellular ability to respond to I/R damage. In CMs after the decrease in oxygen level, the amount of TFAM increases as a compensatory mechanism. Thereafter, its level progressively decreases revealing the increase in ROS production and calcium mismanagement. This phenomenon has been particularly observed in the later stages of HF. Therefore, restoring TFAM levels by increasing mitochondrial biogenesis and reducing ROS production could protect cardiomyocytes from the oxidative damage of mtDNA induced by I/R-injury (93).
Mitochondrial dysfunction is also a feature of hypertension that leads to increase vascular oxidative stress. In particular, Sirt3 has been found to play a key role in maintaining endothelium function (94). In the aortas of hypertensive mice, Sirt3 down-regulation was found to result in worsened blood pressure, vascular relaxation, superoxide and nitric oxide production, as well as increased hypoxia-induced factor 1 alpha (HIF1α), pro-inflammatory gene expression as well as vascular permeability (94). In addition, reduction of Sirt3 leads to increased acetylation of superoxide dismutase 2 (SOD2), resulting in loss of its antioxidant activity (94).
Consistently, beneficial effects of Sirt3 have also been observed (94). In a rat model, activation of the AMP-activated protein kinase (AMPK)-PGC1α-Sirt3 signaling pathway was observed (94). Specifically, PGC1α increases the expression of Sirt3, which in turn deacetylates NRF1, the transcription factor responsible for TFAM transcription (94). On the other hand, Sirt3 also acts directly on TFAM allowing its translocation into mitochondria and subsequently transcription of mitochondrial genes.
Activation of this pathway, thus on the one hand enhances mitochondrial biogenesis and function while on the other hand reduces ROS-dependent cellular stress (94). Interestingly, this pathway can be stimulated by melatonin administration through AMPK activation (94).
Consistently, beneficial effects of Sirt3 have also been observed in myocardial I/R injury (95). In a rat model of I/R, activation of the AMP-activated protein kinase (AMPK)-PGC1α-Sirt3 signalling pathway has been observed (95).. Specifically, PGC1α increases the expression of Sirt3 which in turn deacetylates NRF1 the transcription factor responsible for TFAM transcription (95). Moreover, Sirt3 also acts directly to TFAM allowing its translocation into mitochondria and subsequently transcription of mitochondrial genes. As a result,the biogenesis and function are gained and ROS-dependent cellular stress is reduced (95). Interestingly, this pathway can be stimulated by melatonin administration, which acts by activating AMPK (95). Similarly, the PGC1α-Sirt3-TFAM/NRF1 pathway was also found to be repressed in a rat model of isoprotenerol-induced HF (96). Of note, administration of perindopril improves cardiac function through SIRT3 and PGC1α signalling pathway activation (96).
In CVDs, several mitomiRs have been found to be involved in the pathogenesis. An example is miR-181c, a nuclear-encoded miRNA, which is translocated from nucleus to mitochondria and targets mt-COX1 mRNA (97). To mimic the HF condition, miR-181c has been up-regulated targeting nanoparticles to heart (97). The miR-181c- treated rats revealed a significant decrease in left ventricular fractional shortening and markedly lower ejection fraction (97). MiR-181c has also been found to regulate calcium uptake in cardiomyocytes (98). The miR-181c loss can protect the heart from I/R injury by modulating calcium transport through the upregulation of mitochondrial calcium uptake 1 (MICU1) (98). In fact, in miR-181c−/− mouse model, the molecular mechanism found is the involvement of mt-COX1 which up-regulates the transcription factor specificity protein 1 (Sp1) that in turn triggers the expression of MICU1 (98).
Several miRNAs are involved in the pathogenesis of CVD. One example is miR-181c, a nuclear-encoded miRNA that is translocated from the nucleus to the mitochondria and targets mt-COX1 mRNA (97). To recapitulate an HF phenotype, miR-181c was up-regulated by targeting nanoparticles to the heart (97). Rats treated with miR-181c revealed a significant decrease in fractional shortening and ejection fraction (97). It was also found that miR-181c regulates calcium uptake in cardiomyocytes (98). Thus, suppression of miR-181c may protect the heart from I/R injury by modulating calcium transport through upregulation of mitochondrial calcium uptake 1 (MICU1) (98). Indeed, in the miR-181c−/− mouse model, the molecular mechanism found is the involvement of mt-COX1 regulating the transcription factor specificity protein 1 which in turn triggers the expression of MICU1 preserving Ca2+ uptake in CMs after I/R injury (98). Interestingly, in the human heart, up-regulation of miR-181c leads to ventricular septal defects (99). In this condition, the molecular mechanism of miR181c involves inhibition of bone morphogenetic protein type 2 receptor (BMPR2) (99), which is critical for septal formation and valvulogenesis (100).
Small RNA-seq performed in mitochondria isolated from mouse failing hearts, showed that mitochondria-enriched microRNAs in HF were associated with energy metabolism and oxidative stress pathway (101). Little is known about these mitomiRs in CVDs. However, miR-696 has been observed to down-regulate PGC1α with subsequent impairment of mitochondrial function in skeletal muscle (102). An analogous mechanism might occur also during I/R injury in CMs, however no information is currently available.
On the other hand, lncRNA growth arrest specific 5 (GAS5) is known to sponge miR-532-5p and is up-regulated during I/R (103). Specifically, this lncRNA is increased in I/R injury and is responsible for phosphoinositide-3-kinase (PI3K)/protein kinase B (AKT) downregulation, thus resulting in apoptosis (103). Overexpression of GAS5 lncRNA during ischemic injury prevents the development of an adverse cardiac phenotype (103). Similarly, during stroke, miR-532-5p expression is strongly reduced and is associated with increased infarct area, neuronal apoptosis, and worsening neurological score (104). The up-regulation of miR-532-5p enhances the PI3K/AKT pathway that increases cell viability by attenuating neurological damage (104).
In hypoxia, the potentiation of miR-210 and its involvement in CVD is well known (105). Iron-sulfur cluster assembly proteins 1 and 2 (ISCU1/2) have been found to be a target of miR-210 in mitochondria (106). Increased miR-210, under oxygen-deficient conditions, reduces the level of ISCU1/2, which is involved in iron-sulphur clusters for electron transport and mitochondrial redox reactions (106). Consequently, downregulation of ISCU1/2 impairs metabolism and cell survival.
MitomiRs are also involved in cardiac hypertrophy. MiR-485-5p has been found repressed in mouse hypertrophic heart where it interacts with the mitochondrial anchored protein ligase (MAPL). In turn, the downregulation of MAPL reveals the over-expression of the mitochondrial fusion protein 2 (MFN2) which induces the mitochondrial fission (107). In hypertrophic hearts, miR-485-5p up-regulation was shown to reduce LV wall thickness (107). miR-485-5p is also regulated by metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) (108) that is over-expressed in hypoxic condition (109). In CVD patients, high level of circulating MALAT1 has been observed in association with inflammation and hypoxia (110). Notably, in hepatocellular carcinoma it has been found that MALAT1 binds the mtDNA modifying the mitochondrial structure, reducing oxidative phosphorylation and ATP production, decreasing mtDNA copy number, and activating the apoptosis (43). Despite the increase of MALAT1 in CVDs, no work reports the modulation of mitochondrial gene expression upon MALAT1-mtDNA interaction.
Figure 2 reports a brief overview of mitochondrial epigenetics in cardiovascular diseases.
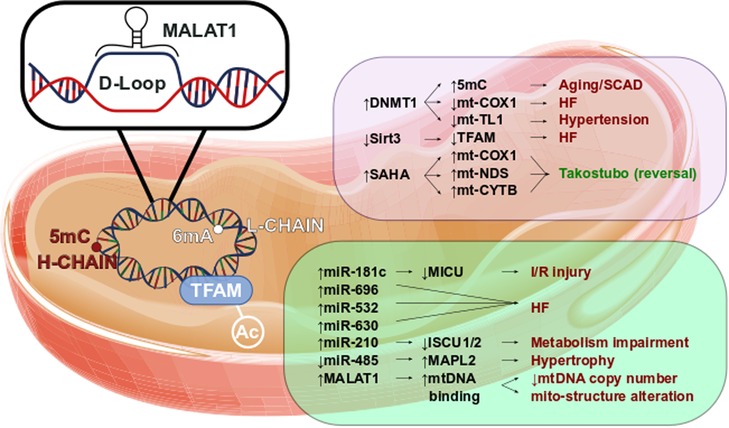
Figure 2. Mitoepigenetics of cardiovascular diseases. Several epigenetic modifications might occur in mitochondrial matrix. The mt-DNA methylation occurs mainly in Adenine of L-chain. However, the 5mC in H-chain has been found to have a regulatory function. In fact, the increase of 5mC has been observed in aged mitochondria as well in mitochondria leucocyte of SCAD patients. In parallel, high level of 5mC in mt-COXQ and in mt-TL1 promoters triggers the HF and hypertension. Additionally, the retains of TFAM acetylation, caused by the decrease of Sirt3 activity, has been observed in HF. Little is known of SAHA effects in mitochondrial regulation, however we might suppose that SAHA modulates the expression of mt-COX1, mt-NDS, and mt-CYTB contributing to Takotsubo disease reversion. Pivotal roles have also been observed in mitochondrial ncRNA regulation. Higher level of miR-181c is leaded to the worsening of I/R injury as well the up-regulation of miR-696, miR-532 and miR-630 triggers the HF. The impairment of metabolism has been associated to the increase of miR-210 through the repression of ISCU1/2. The down-regulation of mitochondrial miR-485 increases MAPL2 expression which stimulates the hypertrophic growth. lncRNAs are involved in CVD too. In fact, MALAT1 has been found to bind the D-Loop of mt-DNA inhibiting the synthesis of new mt-DNA. The reduction of mt-DNA copy number reflects the alteration of mitochondrial structure and the impairment of the oxidative phosphorylation.
Mitoepigenetics is arising as a new marker of aging (51) and CVDs (13). However, therapeutic interventions, that specifically target a mitoepigenetic factor, are difficult to develop due to the strict correlation to the genomic regulation. An example is represented by DNMT1 which methylates nuclear DNA and it translocates into mitochondria where acts on the mtDNA (36, 66, 90, 111). The inhibition of DNMT1 with 5-aza-2′-deoxycytidine has been observed to decrease the amount of 5 mC in mitochondria associated to the improvement of mt-COX2 expression (87). However, the hypomethylation has been observed in genomic DNA in response to 5-aza-2′-deoxycytidine administration which induces genomic instability and tumor growth (112). Similarly, the inhibition of HDACs by SAHA reverts the heart remodelling through increased H3 acetylation (113). However, no data are available on SAHA effects on mitochondrial deacetylases that regulate TFAM. Analogously, several miRNAs have shown an active interplay between cytosol and mitochondrial matrix (45). Hence, modulation of their activity within a specific cellular compartment (without altering the other) remains challenging.
Physical activity is known to influence nuclear epigenetics by increasing lifespan through activation of sirtuins, including the mitochondrial protein SIRT3 (114). During the exercise, there is upregulation of mitochondrial TFAM as well as of protein involved in beta-oxidation, Krebs cycle and electron transport chain. This results in improvement of ROS clearance, enhancing the fusion process between functional organelles and the removal of dysfunctional mitochondria reduce thus reducing the onset of senescence (115). Interestingly, in the elderly population (65 ± 7 years), exercise alters the mtDNA methylome in skeletal muscle making it similar to that of younger men (116).
In conclusion, mitoepigenetics and epigentetics seem to cooperate for the cellular homeostasis. In fact, both systems share proteins (i.e., DNMT1) and ncRNAs (i.e., MALAT1, miR485 which are synthetized in the nucleus and shuttled in mitochondia and an alteration of one mechanism might modulate the other (43, 87, 107). As an example, Dunham-Snary (112, 116) demonstrated that mtDNA can modulate nuclear gene expression in adipose tissue (117).
Application of machine learning to identify novel cardiac biomarkers reveals that total nuclear methylation and methylation in a specific CpG island of TFAM were the best diagnostic measures related to diabetes progression (118). Also this study support the theory that epigenetics and mitoepigenetics are two processes interconnected.
AM: Conceptualization; AM, FP and SC: writing and original draft preparation; AM and MG: Figures and tables preparation; SM, EG, FR, FP, and SC: review and editing. All authors have read and agreed to the published version of the manuscript.
AM is the recipient of an International Grant from the Italian Society of Arterial Hypertension. SAM is the recipients of a Forschungskredit Candoc grant from the University of Zürich. FP is the recipient of a H.H. Sheikh Khalifa bin Hamad Al Thani Foundation Assistant Professorship at the Faculty of Medicine, University of Zürich. This work was supported by the Swiss National Science Foundation (n. 310030_197557), the Swiss Heart Foundation (n. FF19045), the Stiftung für wissenschaftliche Forschung, the Olga Mayenfisch Foundation, the Swiss Life Foundation, the Kurt und Senta-Hermann Stiftung, the EMDO Stiftung and the Schweizerische Diabetes-Stiftung (to FP); and the Holcim Foundation, the Swiss Heart Foundation, the Swiss Life Foundation and the Gebauer Stiftung (to SC).
The Department of Cardiology (University Hospital of Zurich/University of Zurich) reports research-, educational- and/or travel grants from Abbott, Amgen, Astra Zeneca, Bayer, Berlin Heart, B Braun, Biosense Webster, Biosensors Europe AG, Biotronik, BMS, Boehringer Ingelheim, Boston Scientific, Bracco, Cardinal Health Switzerland, Corteria, Daiichi, Diatools AG, Edwards Lifesciences, Guidant Europe NV (BS), Hamilton Health Sciences, Kaneka Corporation, Kantar, Labormedizinisches Zentrum, Medtronic, MSD, Mundipharma Medical Company, Novartis, Novo Nordisk, Orion, Pfizer, Quintiles Switzerland Sarl, Sahajanand IN, Sanofi, Sarstedt AG, Servier, SIS Medical, SSS International Clinical Research, Terumo Deutschland, Trama Solutions, V-Wave, Vascular Medical, Vifor, Wissens Plus, ZOLL These research and educational grants do not impact FR.'s personal remuneration.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Memme JM, Erlich AT, Phukan G, Hood DA. Exercise and mitochondrial health. J Physiol. (2021) 599(3):803–17. doi: 10.1113/JP278853
2. Costantino S, Libby P, Kishore R, Tardif JC, El-Osta A, Paneni F. Epigenetics and precision medicine in cardiovascular patients: from basic concepts to the clinical arena. Eur Heart J. (2018) 39(47):4150–8. doi: 10.1093/eurheartj/ehx568
3. Carter HN, Hood DA. Contractile activity-induced mitochondrial biogenesis and mTORC1. Am J Physiol Cell Physiol. (2012) 303(5):C540–7. doi: 10.1152/ajpcell.00156.2012
4. Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H, et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature. (2021) 593(7859):435–9. doi: 10.1038/s41586-021-03510-6
5. Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med. (2018) 50(2):121–7. doi: 10.1080/07853890.2017.1417631
6. Jackson DN, Theiss AL. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes. (2020) 11(3):285–304. doi: 10.1080/19490976.2019.1592421
7. Johnson J, Mercado-Ayon E, Mercado-Ayon Y, Dong YN, Halawani S, Ngaba L, et al. Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch Biochem Biophys. (2021) 702:108698. doi: 10.1016/j.abb.2020.108698
8. Costantino S, Mohammed SA, Ambrosini S, Paneni F. Epigenetic processing in cardiometabolic disease. Atherosclerosis. (2019) 281:150–8. doi: 10.1016/j.atherosclerosis.2018.09.029
9. Chinnery PF, Hudson G. Mitochondrial genetics. Br Med Bull. (2013) 106(1):135–59. doi: 10.1093/bmb/ldt017
10. Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. (2005) 6(5):389–402. doi: 10.1038/nrg1606
11. Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. Sequence and gene organization of mouse mitochondrial DNA. Cell. (1981) 26(2 Pt 2):167–80. doi: 10.1016/0092-8674(81)90300-7
12. Pereira F, Soares P, Carneiro J, Pereira L, Richards MB, Samuels DC, et al. Evidence for variable selective pressures at a large secondary structure of the human mitochondrial DNA control region. Mol Biol Evol. (2008) 25(12):2759–70. doi: 10.1093/molbev/msn225
13. Cao K, Feng Z, Gao F, Zang W, Liu J. Mitoepigenetics: an intriguing regulatory layer in aging and metabolic-related diseases. Free Radic Biol Med. (2021) 177:337–46. doi: 10.1016/j.freeradbiomed.2021.10.031
14. Coppede F, Stoccoro A. Mitoepigenetics and neurodegenerative diseases. Front Endocrinol (Lausanne). (2019) 10:86. doi: 10.3389/fendo.2019.00086
15. Dong Z, Pu L, Cui H. Mitoepigenetics and its emerging roles in cancer. Front Cell Dev Biol. (2020) 8:4. doi: 10.3389/fcell.2020.00004
16. Kukat C, Davies KM, Wurm CA, Spahr H, Bonekamp NA, Kuhl I, et al. Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc Natl Acad Sci U S A. (2015) 112(36):11288–93. doi: 10.1073/pnas.1512131112
17. Wang Y, Bogenhagen DF. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J Biol Chem. (2006) 281(35):25791–802. doi: 10.1074/jbc.M604501200
18. Shokolenko IN, Alexeyev MF. Mitochondrial transcription in mammalian cells. Front Biosci (Landmark Ed). (2017) 22(5):835–53. doi: 10.2741/4520
19. King GA, Hashemi Shabestari M, Taris KH, Pandey AK, Venkatesh S, Thilagavathi J, et al. Acetylation and phosphorylation of human TFAM regulate TFAM-DNA interactions via contrasting mechanisms. Nucleic Acids Res. (2018) 46(7):3633–42. doi: 10.1093/nar/gky204
20. Lv T, Zhang Y, Ji X, Sun S, Xu L, Ma W, et al. GCN5L1-mediated TFAM acetylation at K76 participates in mitochondrial biogenesis in acute kidney injury. J Transl Med. (2022) 20(1):571. doi: 10.1186/s12967-022-03782-0
21. Fang Y, Akimoto M, Mayanagi K, Hatano A, Matsumoto M, Matsuda S, et al. Chemical acetylation of mitochondrial transcription factor A occurs on specific lysine residues and affects its ability to change global DNA topology. Mitochondrion. (2020) 53:99–108. doi: 10.1016/j.mito.2020.05.003
22. Hillen HS, Morozov YI, Sarfallah A, Temiakov D, Cramer P. Structural basis of mitochondrial transcription initiation. Cell. (2017) 171(5):1072–81 e10. doi: 10.1016/j.cell.2017.10.036
23. Costantino S, Paneni F, Cosentino F. Targeting chromatin remodeling to prevent cardiovascular disease in diabetes. Curr Pharm Biotechnol. (2015) 16(6):531–43. doi: 10.2174/138920101606150407113644
24. Koentges C, Pfeil K, Schnick T, Wiese S, Dahlbock R, Cimolai MC, et al. SIRT3 Deficiency impairs mitochondrial and contractile function in the heart. Basic Res Cardiol. (2015) 110(4):36. doi: 10.1007/s00395-015-0493-6
25. Laurent G, German NJ, Saha AK, de Boer VC, Davies M, Koves TR, et al. SIRT4 Coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell. (2013) 50(5):686–98. doi: 10.1016/j.molcel.2013.05.012
26. Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, et al. SIRT5 Regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. (2013) 18(6):920–33. doi: 10.1016/j.cmet.2013.11.013
27. Osborne B, Bentley NL, Montgomery MK, Turner N. The role of mitochondrial sirtuins in health and disease. Free Radic Biol Med. (2016) 100:164–74. doi: 10.1016/j.freeradbiomed.2016.04.197
28. Bagul PK, Katare PB, Bugga P, Dinda AK, Banerjee SK. SIRT-3 modulation by resveratrol improves mitochondrial oxidative phosphorylation in diabetic heart through deacetylation of TFAM. Cells. (2018) 7(12):235. doi: 10.3390/cells7120235
29. Wang KZ, Zhu J, Dagda RK, Uechi G, Cherra SJ 3rd, Gusdon AM, et al. ERK-mediated phosphorylation of TFAM downregulates mitochondrial transcription: implications for Parkinson’s disease. Mitochondrion. (2014) 17:132–40. doi: 10.1016/j.mito.2014.04.008
30. Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. (2016) 12(6):999–1014. doi: 10.1080/15548627.2016.1166318
31. Zhao Y, Wang Y, Zhao J, Zhang Z, Jin M, Zhou F, et al. PDE2 inhibits PKA-mediated phosphorylation of TFAM to promote mitochondrial ca(2+)-induced colorectal cancer growth. Front Oncol. (2021) 11:663778. doi: 10.3389/fonc.2021.663778
32. Hong EE, Okitsu CY, Smith AD, Hsieh CL. Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol Cell Biol. (2013) 33(14):2683–90. doi: 10.1128/MCB.00220-13
33. Mechta M, Ingerslev LR, Fabre O, Picard M, Barres R. Evidence suggesting absence of mitochondrial DNA methylation. Front Genet. (2017) 8:166. doi: 10.3389/fgene.2017.00166
34. Bellizzi D, D’Aquila P, Scafone T, Giordano M, Riso V, Riccio A, et al. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. (2013) 20(6):537–47. doi: 10.1093/dnares/dst029
35. Pirola CJ, Gianotti TF, Burgueno AL, Rey-Funes M, Loidl CF, Mallardi P, et al. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut. (2013) 62(9):1356–63. doi: 10.1136/gutjnl-2012-302962
36. Saini SK, Mangalhara KC, Prakasam G, Bamezai RNK. DNA Methyltransferase1 (DNMT1) Isoform3 methylates mitochondrial genome and modulates its biology. Sci Rep. (2017) 7(1):1525. doi: 10.1038/s41598-017-01743-y
37. Koh CWQ, Goh YT, Toh JDW, Neo SP, Ng SB, Gunaratne J, et al. Single-nucleotide-resolution sequencing of human N6-methyldeoxyadenosine reveals strand-asymmetric clusters associated with SSBP1 on the mitochondrial genome. Nucleic Acids Res. (2018) 46(22):11659–70. doi: 10.1093/nar/gky1104
38. Hao Z, Wu T, Cui X, Zhu P, Tan C, Dou X, et al. N(6)-deoxyadenosine methylation in mammalian mitochondrial DNA. Mol Cell. (2020) 78(3):382–95 e8. doi: 10.1016/j.molcel.2020.02.018
39. Xiao CL, Zhu S, He M, Chen D, Zhang Q, Chen Y, et al. N(6)-methyladenine DNA modification in the human genome. Mol Cell. (2018) 71(2):306–18 e7. doi: 10.1016/j.molcel.2018.06.015
40. Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, et al. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput Biol. (2006) 2(4):e33. doi: 10.1371/journal.pcbi.0020033
41. Zhang X, Wang W, Zhu W, Dong J, Cheng Y, Yin Z, et al. Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int J Mol Sci. (2019) 20(22):5573. doi: 10.3390/ijms20225573
42. Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. (2010) 79:351–79. doi: 10.1146/annurev-biochem-060308-103103
43. Zhao Y, Zhou L, Li H, Sun T, Wen X, Li X, et al. Nuclear-encoded lncRNA MALAT1 epigenetically controls metabolic reprogramming in HCC cells through the mitophagy pathway. Mol Ther Nucleic Acids. (2021) 23:264–76. doi: 10.1016/j.omtn.2020.09.040
44. Noh JH, Kim KM, Abdelmohsen K, Yoon JH, Panda AC, Munk R, et al. Hur and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes Dev. (2016) 30(10):1224–39. doi: 10.1101/gad.276022.115
45. Barrey E, Saint-Auret G, Bonnamy B, Damas D, Boyer O, Gidrol X. Pre-microRNA and mature microRNA in human mitochondria. PLoS One. (2011) 6(5):e20220. doi: 10.1371/journal.pone.0020220
46. Fan S, Tian T, Chen W, Lv X, Lei X, Zhang H, et al. Mitochondrial miRNA determines chemoresistance by reprogramming metabolism and regulating mitochondrial transcription. Cancer Res. (2019) 79(6):1069–84. doi: 10.1158/0008-5472.CAN-18-2505
47. Kren BT, Wong PY, Sarver A, Zhang X, Zeng Y, Steer CJ. MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol. (2009) 6(1):65–72. doi: 10.4161/rna.6.1.7534
48. Das S, Ferlito M, Kent OA, Fox-Talbot K, Wang R, Liu D, et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res. (2012) 110(12):1596–603. doi: 10.1161/CIRCRESAHA.112.267732
49. Chen Y, Bravo JI, Son JM, Lee C, Benayoun BA. Remodeling of the H3 nucleosomal landscape during mouse aging. Transl Med Aging. (2020) 4:22–31. doi: 10.1016/j.tma.2019.12.003
50. Guerville F, De Souto Barreto P, Ader I, Andrieu S, Casteilla L, Dray C, et al. Revisiting the hallmarks of aging to identify markers of biological age. J Prev Alzheimers Dis. (2020) 7(1):56–64. doi: 10.14283/jpad.2019.50
51. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. (2013) 153(6):1194–217. doi: 10.1016/j.cell.2013.05.039
52. Wang LJ, Hsu T, Lin HL, Fu CY. Modulation of mitochondrial nucleoid structure during aging and by mtDNA content in Drosophila. Biol Open. (2021) 10(6):bio058553. doi: 10.1242/bio.058553
53. Matsuda T, Kanki T, Tanimura T, Kang D, Matsuura ET. Effects of overexpression of mitochondrial transcription factor A on lifespan and oxidative stress response in Drosophila melanogaster. Biochem Biophys Res Commun. (2013) 430(2):717–21. doi: 10.1016/j.bbrc.2012.11.084
54. Bonekamp NA, Jiang M, Motori E, Garcia Villegas R, Koolmeister C, Atanassov I, et al. High levels of TFAM repress mammalian mitochondrial DNA transcription in vivo. Life Sci Alliance. (2021) 4(11):e202101034. doi: 10.26508/lsa.202101034
55. Cobo I, Tanaka TN, Chandra Mangalhara K, Lana A, Yeang C, Han C, et al. DNA Methyltransferase 3 alpha and TET methylcytosine dioxygenase 2 restrain mitochondrial DNA-mediated interferon signaling in macrophages. Immunity. (2022) 55(8):1386–401 e10. doi: 10.1016/j.immuni.2022.06.022
56. Xia SA-OX, Zhang XA-O, Zheng SA-O, Khanabdali RA-O, Kalionis BA-O, Wu J, et al. An update on inflamm-aging: mechanisms, prevention, and treatment. J Immunol Res. (2016) 2016:8426874. doi: 10.1155/2016/8426874
57. Desdin-Mico G, Soto-Heredero G, Aranda JF, Oller J, Carrasco E, Gabande-Rodriguez E, et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science. (2020) 368(6497):1371–6. doi: 10.1126/science.aax0860
58. Sahin E, DePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol. (2012) 13(6):397–404. doi: 10.1038/nrm3352
59. Saleem A, Hood DA. Acute exercise induces tumour suppressor protein p53 translocation to the mitochondria and promotes a p53-Tfam-mitochondrial DNA complex in skeletal muscle. J Physiol. (2013) 591(14):3625–36. doi: 10.1113/jphysiol.2013.252791
60. Perez RF, Tejedor JR, Bayon GF, Fernandez AF, Fraga MF. Distinct chromatin signatures of DNA hypomethylation in aging and cancer. Aging Cell. (2018) 17(3):e12744. doi: 10.1111/acel.12744
61. Bekaert B, Kamalandua A, Zapico SC, Van de Voorde W, Decorte R. Improved age determination of blood and teeth samples using a selected set of DNA methylation markers. Epigenetics. (2015) 10(10):922–30. doi: 10.1080/15592294.2015.1080413
62. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. (2013) 49(2):359–67. doi: 10.1016/j.molcel.2012.10.016
63. Horvath S. DNA Methylation age of human tissues and cell types. Genome Biol. (2013) 14(10):R115. doi: 10.1186/gb-2013-14-10-r115
64. Zbiec-Piekarska R, Spolnicka M, Kupiec T, Parys-Proszek A, Makowska Z, Paleczka A, et al. Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci Int Genet. (2015) 17:173–9. doi: 10.1016/j.fsigen.2015.05.001
65. Mongelli A, Panunzi S, Nesta M, Gottardi Zamperla M, Atlante S, Barbi V, et al. Distinguishable DNA methylation defines a cardiac-specific epigenetic clock. Clin Epigenetics. (2023) 15(1):53. doi: 10.1186/s13148-023-01467-z
66. Huang CH, Chang MC, Lai YC, Lin CY, Hsu CH, Tseng BY, et al. Mitochondrial DNA methylation profiling of the human prefrontal cortex and nucleus accumbens: correlations with aging and drug use. Clin Epigenetics. (2022) 14(1):79. doi: 10.1186/s13148-022-01300-z
67. Asadi Shahmirzadi A, Edgar D, Liao CY, Hsu YM, Lucanic M, Asadi Shahmirzadi A, et al. Alpha-ketoglutarate, an endogenous metabolite, extends lifespan and compresses morbidity in aging mice. Cell Metab. (2020) 32(3):447–56 e6. doi: 10.1016/j.cmet.2020.08.004
68. D’Aquila P, De Rango F, Guarasci F, Mandala M, Corsonello A, Bellizzi D, et al. Multi-tissue DNA methylation remodeling at mitochondrial quality control genes according to diet in rat aging models. Nutrients. (2020) 12(2):460. doi: 10.3390/nu12020460
69. Martin-Fernandez B, Gredilla R. Mitochondria and oxidative stress in heart aging. Age (Dordr). (2016) 38(4):225–38. doi: 10.1007/s11357-016-9933-y
70. Stadtman ER. Protein oxidation and aging. Science. (1992) 257(5074):1220–4. doi: 10.1126/science.1355616
71. Owczarz M, Budzinska M, Domaszewska-Szostek A, Borkowska J, Polosak J, Gewartowska M, et al. miR-34a and miR-9 are overexpressed and SIRT genes are downregulated in peripheral blood mononuclear cells of aging humans. Exp Biol Med (Maywood). (2017) 242(14):1453–61. doi: 10.1177/1535370217720884
72. Ye Z, Li G, Kim C, Hu B, Jadhav RR, Weyand CM, et al. Regulation of miR-181a expression in T cell aging. Nat Commun. (2018) 9(1):3060. doi: 10.1038/s41467-018-05552-3
73. Hofmann P, Sommer J, Theodorou K, Kirchhof L, Fischer A, Li Y, et al. Long non-coding RNA H19 regulates endothelial cell aging via inhibition of STAT3 signalling. Cardiovasc Res. (2019) 115(1):230–42. doi: 10.1093/cvr/cvy206
74. Yang XC, Zhao DH, Lau WB, Liu KQ, Tian JY, Cheng ZC, et al. lncRNA ENSMUST00000134285 increases MAPK11 activity, regulating aging-related myocardial apoptosis. J Gerontol A Biol Sci Med Sci. (2018) 73(8):1010–7. doi: 10.1093/gerona/gly020
75. Jia HL, Zeng XQ, Huang F, Liu YM, Gong BS, Zhang KZ, et al. Integrated microRNA and mRNA sequencing analysis of age-related changes to mouse thymic epithelial cells. IUBMB Life. (2018) 70(7):678–90. doi: 10.1002/iub.1864
76. White RR, Milholland B, MacRae SL, Lin M, Zheng D, Vijg J. Comprehensive transcriptional landscape of aging mouse liver. BMC Genomics. (2015) 16:899. doi: 10.1186/s12864-015-2061-8
77. Zhang H, Xu R, Li B, Xin Z, Ling Z, Zhu W, et al. LncRNA NEAT1 controls the lineage fates of BMSCs during skeletal aging by impairing mitochondrial function and pluripotency maintenance. Cell Death Differ. (2022) 29(2):351–65. doi: 10.1038/s41418-021-00858-0
78. Rodgers JL, Jones J, Bolleddu SI, Vanthenapalli S, Rodgers LE, Shah K, et al. Cardiovascular risks associated with gender and aging. J Cardiovasc Dev Dis. (2019) 6(2):19. doi: 10.3390/jcdd6020019
79. Buja LM, Ottaviani G, Mitchell RN. Pathobiology of cardiovascular diseases: an update. Cardiovasc Pathol. (2019) 42:44–53. doi: 10.1016/j.carpath.2019.06.002
80. Hamdani N, Costantino S, Mugge A, Lebeche D, Tschope C, Thum T, et al. Leveraging clinical epigenetics in heart failure with preserved ejection fraction: a call for individualized therapies. Eur Heart J. (2021) 42(20):1940–58. doi: 10.1093/eurheartj/ehab197
81. Barwari T, Joshi A, Mayr M. MicroRNAs in cardiovascular disease. J Am Coll Cardiol. (2016) 68(23):2577–84. doi: 10.1016/j.jacc.2016.09.945
82. Li P, Ge J, Li H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat Rev Cardiol. (2020) 17(2):96–115. doi: 10.1038/s41569-019-0235-9
83. Prasher D, Greenway SC, Singh RB. The impact of epigenetics on cardiovascular disease. Biochem Cell Biol. (2020) 98(1):12–22. doi: 10.1139/bcb-2019-0045
84. Uchida S, Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ Res. (2015) 116(4):737–50. doi: 10.1161/CIRCRESAHA.116.302521
85. Westerman KE, Ordovas JM. DNA Methylation and incident cardiovascular disease. Curr Opin Clin Nutr Metab Care. (2020) 23(4):236–40. doi: 10.1097/MCO.0000000000000659
86. Mohammed SA, Ambrosini S, Luscher T, Paneni F, Costantino S. Epigenetic control of mitochondrial function in the vasculature. Front Cardiovasc Med. (2020) 7:28. doi: 10.3389/fcvm.2020.00028
87. Sun X, Wang Z, Cong X, Lv Y, Li Z, Rong L, et al. Mitochondrial gene COX2 methylation and downregulation is a biomarker of aging in heart mesenchymal stem cells. Int J Mol Med. (2021) 47(1):161–70. doi: 10.3892/ijmm.2020.4799
88. Wan Q, Kong D, Liu Q, Guo S, Wang C, Zhao Y, et al. Congestive heart failure in COX2 deficient rats. Sci China Life Sci. (2021) 64(7):1068–76. doi: 10.1007/s11427-020-1792-5
89. Baccarelli AA, Byun HM. Platelet mitochondrial DNA methylation: a potential new marker of cardiovascular disease. Clin Epigenetics. (2015) 7(1):44. doi: 10.1186/s13148-015-0078-0
90. Liu YF, Zhu JJ, Yu Tian X, Liu H, Zhang T, Zhang YP, et al. Hypermethylation of mitochondrial DNA in vascular smooth muscle cells impairs cell contractility. Cell Death Dis. (2020) 11(1):35. doi: 10.1038/s41419-020-2240-7
91. Park SH, Lee SY, Kim SA. Mitochondrial DNA methylation is higher in acute coronary syndrome than in stable coronary artery disease. In Vivo. (2021) 35(1):181–9. doi: 10.21873/invivo.12247
92. Hickson-Bick DL, Jones C, Buja LM. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J Mol Cell Cardiol. (2008) 44(2):411–8. doi: 10.1016/j.yjmcc.2007.10.013
93. Yue R, Xia X, Jiang J, Yang D, Han Y, Chen X, et al. Mitochondrial DNA oxidative damage contributes to cardiomyocyte ischemia/reperfusion-injury in rats: cardioprotective role of lycopene. J Cell Physiol. (2015) 230(9):2128–41. doi: 10.1002/jcp.24941
94. Dikalova AE, Pandey A, Xiao L, Arslanbaeva L, Sidorova T, Lopez MG, et al. Mitochondrial deacetylase Sirt3 reduces vascular dysfunction and hypertension while Sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress. Circ Res. (2020) 126(4):439–52. doi: 10.1161/CIRCRESAHA.119.315767
95. Yu L, Gong B, Duan W, Fan C, Zhang J, Li Z, et al. Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: role of AMPK-PGC-1alpha-SIRT3 signaling. Sci Rep. (2017) 7:41337. doi: 10.1038/srep41337
96. Zhu Z, Li H, Chen W, Cui Y, Huang A, Qi X. Perindopril improves cardiac function by enhancing the expression of SIRT3 and PGC-1alpha in a rat model of isoproterenol-induced cardiomyopathy. Front Pharmacol. (2020) 11:94. doi: 10.3389/fphar.2020.00094
97. Das S, Bedja D, Campbell N, Dunkerly B, Chenna V, Maitra A, et al. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS One. (2014) 9(5):e96820. doi: 10.1371/journal.pone.0096820
98. Banavath HN, Roman B, Mackowski N, Biswas D, Afzal J, Nomura Y, et al. miR-181c activates mitochondrial calcium uptake by regulating MICU1 in the heart. J Am Heart Assoc. (2019) 8(24):e012919. doi: 10.1161/JAHA.119.012919
99. Li J, Cao Y, Ma XJ, Wang HJ, Zhang J, Luo X, et al. Roles of miR-1-1 and miR-181c in ventricular septal defects. Int J Cardiol. (2013) 168(2):1441–6. doi: 10.1016/j.ijcard.2012.12.048
100. Beppu H, Malhotra R, Beppu Y, Lepore JJ, Parmacek MS, Bloch KD. BMP Type II receptor regulates positioning of outflow tract and remodeling of atrioventricular cushion during cardiogenesis. Dev Biol. (2009) 331(2):167–75. doi: 10.1016/j.ydbio.2009.04.032
101. Wang X, Song C, Zhou X, Han X, Li J, Wang Z, et al. Mitochondria associated MicroRNA expression profiling of heart failure. Biomed Res Int. (2017) 2017:4042509. doi: 10.1155/2017/4042509
102. Queiroz AL, Lessard SJ, Ouchida AT, Araujo HN, Goncalves DA, Simoes Froes Guimaraes DSP, et al. The MicroRNA miR-696 is regulated by SNARK and reduces mitochondrial activity in mouse skeletal muscle through Pgc1alpha inhibition. Mol Metab. (2021) 51:101226. doi: 10.1016/j.molmet.2021.101226
103. Han Y, Wu N, Xia F, Liu S, Jia D. Long non-coding RNA GAS5 regulates myocardial ischemia-reperfusion injury through the PI3K/AKT apoptosis pathway by sponging miR-532-5p. Int J Mol Med. (2020) 45(3):858–72. doi: 10.3892/ijmm.2020.4471
104. Mu J, Cheng X, Zhong S, Chen X, Zhao C. Neuroprotective effects of miR-532-5p against ischemic stroke. Metab Brain Dis. (2020) 35(5):753–63. doi: 10.1007/s11011-020-00544-z
105. Devlin C, Greco S, Martelli F, Ivan M. miR-210: more than a silent player in hypoxia. IUBMB Life. (2011) 63(2):94–100. doi: 10.1002/iub.427
106. Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. (2009) 10(4):273–84. doi: 10.1016/j.cmet.2009.08.015
107. Zhao Y, Ponnusamy M, Liu C, Tian J, Dong Y, Gao J, et al. MiR-485-5p modulates mitochondrial fission through targeting mitochondrial anchored protein ligase in cardiac hypertrophy. Biochim Biophys Acta Mol Basis Dis. (2017) 1863(11):2871–81. doi: 10.1016/j.bbadis.2017.07.034
108. Tie W, Ge F. MALAT1 inhibits proliferation of HPV16-positive cervical cancer by sponging miR-485-5p to promote expression of MAT2A. DNA Cell Biol. (2021) 40(11):1407–17. doi: 10.1089/dna.2020.6205
109. Salle-Lefort S, Miard S, Nolin MA, Boivin L, Pare ME, Debigare R, et al. Hypoxia upregulates Malat1 expression through a CaMKK/AMPK/HIF-1alpha axis. Int J Oncol. (2016) 49(4):1731–6. doi: 10.3892/ijo.2016.3630
110. Yan Y, Song D, Song X, Song C. The role of lncRNA MALAT1 in cardiovascular disease. IUBMB Life. (2020) 72(3):334–42. doi: 10.1002/iub.2210
111. Liu B, Du Q, Chen L, Fu G, Li S, Fu L, et al. Cpg methylation patterns of human mitochondrial DNA. Sci Rep. (2016) 6:23421. doi: 10.1038/srep23421
112. Maslov AY, Lee M, Gundry M, Gravina S, Strogonova N, Tazearslan C, et al. 5-aza-2′-deoxycytidine-induced Genome rearrangements are mediated by DNMT1. Oncogene. (2012) 31(50):5172–9. doi: 10.1038/onc.2012.9
113. Khurana I, Maxwell S, Royce S, Mathiyalagan P, Karagiannis T, Mazarakis N, et al. SAHA attenuates takotsubo-like myocardial injury by targeting an epigenetic Ac/Dc axis. Signal Transduct Target Ther. (2021) 6(1):159. doi: 10.1038/s41392-021-00546-y
114. Vargas-Ortiz K, Pérez-Vázquez V, Macías-Cervantes MH. Exercise and sirtuins: a way to mitochondrial health in skeletal muscle. Int J Mol Sci. (2019) 20(11):2717. doi: 10.3390/ijms20112717
115. Memme JA-O, Erlich AT, Phukan G, Hood DA-OX. Exercise and mitochondrial health. J Physiol. (2021) 599(3):803–17. doi: 10.1113/JP278853
116. Ruple BA, Godwin JS, Mesquita PHC, Osburn SC, Vann CG, Lamb DA, et al. Resistance training rejuvenates the mitochondrial methylome in aged human skeletal muscle. FASEB J. (2021) 35(9):e21864. doi: 10.1096/fj.202100873RR
117. Dunham-Snary KJ, Sandel MW, Sammy MJ, Westbrook DG, Xiao R, McMonigle RJ, et al. Mitochondrial—nuclear genetic interaction modulates whole body metabolism, adiposity and gene expression in vivo. EBioMedicine. (2018) 36:316–28. doi: 10.1016/j.ebiom.2018.08.036
Keywords: mitoepigenetics, mtDNA, ncRNAs, mitochondria, aging, cardiovascular diseases, methylation
Citation: Mongelli A, Mengozzi A, Geiger M, Gorica E, Mohammed SA, Paneni F, Ruschitzka F and Costantino S (2023) Mitochondrial epigenetics in aging and cardiovascular diseases. Front. Cardiovasc. Med. 10:1204483. doi: 10.3389/fcvm.2023.1204483
Received: 12 April 2023; Accepted: 29 June 2023;
Published: 13 July 2023.
Edited by:
Hua Zhu, The Ohio State University, United StatesReviewed by:
Lincai Ye, Shanghai Children’s Medical Center, China© 2023 Mongelli, Mengozzi, Geiger, Gorica, Mohammed, Paneni, Ruschitzka and Costantino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah Costantino c2FyYWguY29zdGFudGlub0B1emguY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.