 Laura Rolland
Laura Rolland Jourdano Mancilla Abaroa
Jourdano Mancilla Abaroa Adèle Faucherre
Adèle Faucherre Chris Jopling
Chris Jopling
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 08 January 2024
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1186086
This article is part of the Research Topic Novel Strategies to Repair the Infarcted Heart, Volume II View all 6 articles
Myocardial damage caused, for example, by cardiac ischemia leads to ventricular volume overload resulting in increased stretch of the remaining myocardium. In adult mammals, these changes trigger an adaptive cardiomyocyte hypertrophic response which, if the damage is extensive, will ultimately lead to pathological hypertrophy and heart failure. Conversely, in response to extensive myocardial damage, cardiomyocytes in the adult zebrafish heart and neonatal mice proliferate and completely regenerate the damaged myocardium. We therefore hypothesized that in adult zebrafish, changes in mechanical loading due to myocardial damage may act as a trigger to induce cardiac regeneration. Based on this notion we sought to identify mechanosensors which could be involved in detecting changes in mechanical loading and triggering regeneration. Here we show using a combination of knockout animals, RNAseq and in vitro assays that the mechanosensitive ion channel Trpc6a is required by cardiomyocytes for successful cardiac regeneration in adult zebrafish. Furthermore, using a cyclic cell stretch assay, we have determined that Trpc6a induces the expression of components of the AP1 transcription complex in response to mechanical stretch. Our data highlights how changes in mechanical forces due to myocardial damage can be detected by mechanosensors which in turn can trigger cardiac regeneration.
Following a myocardial infarction, the loss of cardiac tissue results in significant changes in the mechanical loads exerted on the heart. As dynamic/elastic myocardium is replaced by relatively stiff non-contractile scar tissue, the resulting elevated preload and changes in tissue composition increases the amount of stretch exerted on the remaining myocardium, triggering an increased force of contraction and concomitant cardiomyocyte hypertrophy (Frank-Starling law) (1). While this adaptive mechanism initially compensates for the increased mechanical load, prolonged stress will become maladaptive and, without medical intervention, will ultimately result in pathological hypertrophy and heart failure.
In contrast to adult mammals, neonatal mice and adult zebrafish can fully regenerate their hearts after cardiac injury (2, 3). Rather than undergoing compensatory hypertrophy, neonatal mouse and adult zebrafish cardiomyocytes proliferate in response to the loss of myocardium. This suggests that the changes in mechanical loading which occur after cardiac injury in neonates/zebrafish may act as a trigger to induce cardiomyocyte proliferation and ultimately regeneration.
Although decades of research have been devoted to understanding the effects of mechanical stretch on non-proliferative adult cardiomyocytes there is very little information regarding the effects on cardiomyocytes which are capable of proliferation. However, in vitro evidence studying the effects of mechanical loading on embryonic mouse cardiomyocytes indicates an increase in the proliferation index following 24 h of cyclic stretch (4). Likewise, transcriptomic analysis of neonatal rat ventricular cardiomyocytes subjected to cyclic stretch in vitro showed a significant upregulation of genes associated with cell proliferation (5).
To understand whether increased mechanical stretch can induce cardiomyocyte proliferation in vivo, in models which are capable of cardiac regeneration, it is important to assess whether changes in mechanical force occur following cardiac injury. Analysis of adult zebrafish indicates that following cardiac injury, the initial response is similar to that observed in adult humans resulting in a stiffer myocardium due to the extensive fibrosis at the site of injury (6). Furthermore, the resulting volume overload initially induces an elongation of cardiomyocyte sarcomere length which reverts following cardiac regeneration. This indicates that following cardiac injury in adult zebrafish, the heart is subjected to significant changes in mechanical loading. Whether these forces can induce a proliferative response is at present unclear (6). However, other situations that can also increase cardiomyocyte stretch do support this hypothesis. In humans vigorous exercise results in an elevated preload resulting in increased cardiomyocyte stretch. These conditions trigger an adaptive hypertrophic response resulting in an essentially larger more powerful heart (1). On the other hand, in adult zebrafish, exercise can induce cardiomyocytes to proliferate rather than undergo hypertrophy (7). This indicates that increased mechanical load can act as a stimulus to induce cardiomyocyte proliferation. But care must be taken when extrapolating exercise induced cardiomyocyte hypertrophy vs. damage induced hypertrophy as it appears that different mechanisms are at play depending on the conditions (8). Understanding how increased stretch can influence cardiomyocyte proliferation, particularly in vivo, may provide invaluable information on how this phenomenon could be harnessed to induce a regenerative response.
Increased myocardial stretch can be sensed by a wide variety of mechanosensory mechanisms present in cardiomyocytes such as cell surface receptors, sarcomeric components, intercalated discs and stretch activated ion channels (1). The transient receptor potential (Trp) channels are a family of non-selective cation channels which can be regulated by a variety of stimuli including mechanical stretch. Of these, TRPC3 and TRPC6 are highly expressed in the heart and have been directly linked to pathological cardiac hypertrophy in response to chronic overload. In particular, stretch elevates intracellular Ca2+ which activates the CALCINEURIN/NUCLEAR FACTOR OF ACTIVATED T CELLS (NFAT) pathway triggering pathological hypertrophy and remodelling (9, 10). Both TRPC3 and TRPC6 have been shown to be responsible for the stretch induced increase in Ca2+ (9). Indeed, targeting both of these ion channels can inhibit pathological cardiomyocyte hypertrophy (11).
Because of the role TRPC6 plays in regulating the cardiomyocyte hypertrophic response to increased stretch in mammals we surmised that Trpc6 may also regulate cardiomyocyte proliferation in adult zebrafish regenerating hearts in response to the chronic mechanical overload associated with cardiac injury.
In this study we examined how the loss of trpc6a could affect cardiac regeneration in zebrafish. Similar to reports in mammals, we found that trpc6a knockout (KO) did not result in any observable cardiac developmental defects. However, adult trpc6a KO zebrafish failed to regenerate their hearts after 30 days following apical resection due to a reduction in cardiomyocyte proliferation. Transcriptomic analysis indicated that following cardiac injury, trpc6a KO zebrafish did not upregulate the expression of genes required for this process. In particular, this included orthologous components of the ACTIVATOR PROTEIN 1 (AP1) transcription factor complex which is critical for cardiac regeneration in adult zebrafish (12). Furthermore, we demonstrate that the stretch induced expression of AP1 components is markedly reduced in trpc6a KO zebrafish. Taken together these findings indicate that Trpc6a positively regulates the expression of pro-regenerative genes in response to mechanical stretch and that this process is required for successful cardiac regeneration.
Zebrafish possess 2 paralogs of TRPC6, trpc6a and trpc6b. Previous research indicates that trpc6a is expressed in the developing heart of 5 days post fertilisation (dpf) larvae whereas trpc6b expression is restricted to motor neurons in the brain (13). Based on this data we focused on trpc6a as this paralog was likely to play a role similar to mammalian TRPC6 in the heart. To understand the role Trpc6a plays during cardiac development and regeneration, we utilised a KO zebrafish line which harbours a single base pair substitution G637T resulting in a premature stop codon in exon2 of trpc6a (Figure 1A). To confirm that this mutation results in a loss of Trpc6a protein, we performed immunohistochemistry (IHC) using a Trpc6 antibody. In this manner we could detect Trpc6 in the myocardium of adult trpc6a+/+ zebrafish hearts (Figure 1B). In comparison, Trpc6 was absent in adult trpc6a−/− zebrafish hearts indicating that no functional Trpc6a protein was present in the KO line (Figure 1C). In parallel, we also assessed sarcomere structure in trpc6a−/− hearts using Tropomyosin (trpm) and α Sarcomeric actin (αsa) antibodies. We could not detect any observable differences in tropomyosin and sarcomeric actin labelling between trpc6a−/− and their trpc6a+/+ siblings (Figures 1D–G). We next determined whether loss of Trpc6a affects cardiac development in zebrafish larvae as this could potentially impact processes occurring in adulthood, such as cardiac regeneration. Because of the role TRPC6 plays in hypertrophy in mammals we first measured the ventricular wall of 5dpf larvae (n = 10/group) (Figures 1H,I). In this manner we could not detect any significant differences between trpc6a−/− and trpc6a+/+ larvae (Figure 1J). Furthermore, chamber morphology and looping appeared unaffected by the loss of trpc6a (data not shown). Mutations in TRPC6 have also been associated with cardiac arrhythmias which could, if present, disrupt heart regeneration at later stages. Therefore, we analysed a variety of cardiac physiological parameters in both trpc6a−/− and trpc6a+/+ larvae. Measurements of ventricular and atrial heart rates indicated there was no significant difference between trpc6a−/− and trpc6a+/+ larvae (n = 10/group) (Figure 1K). Furthermore, we could not detect any blood regurgitation between the ventricle and atrium indicating that valve development occurred normally in trpc6a−/− larvae (data not shown). Lastly, we measured the blood flow rate and calculated the cardiac output in both trpc6a−/− and trpc6a+/+ larvae. Our data indicates that there are no significant differences in these parameters between trpc6a−/− and trpc6a+/+ larvae indicating that loss of Trpc6a does not appear to affect overall cardiac performance (n = 10/group) (Figures 1L,M). These data indicate that heart development and cardiac performance are not significantly affected in trpc6a−/− zebrafish larvae.
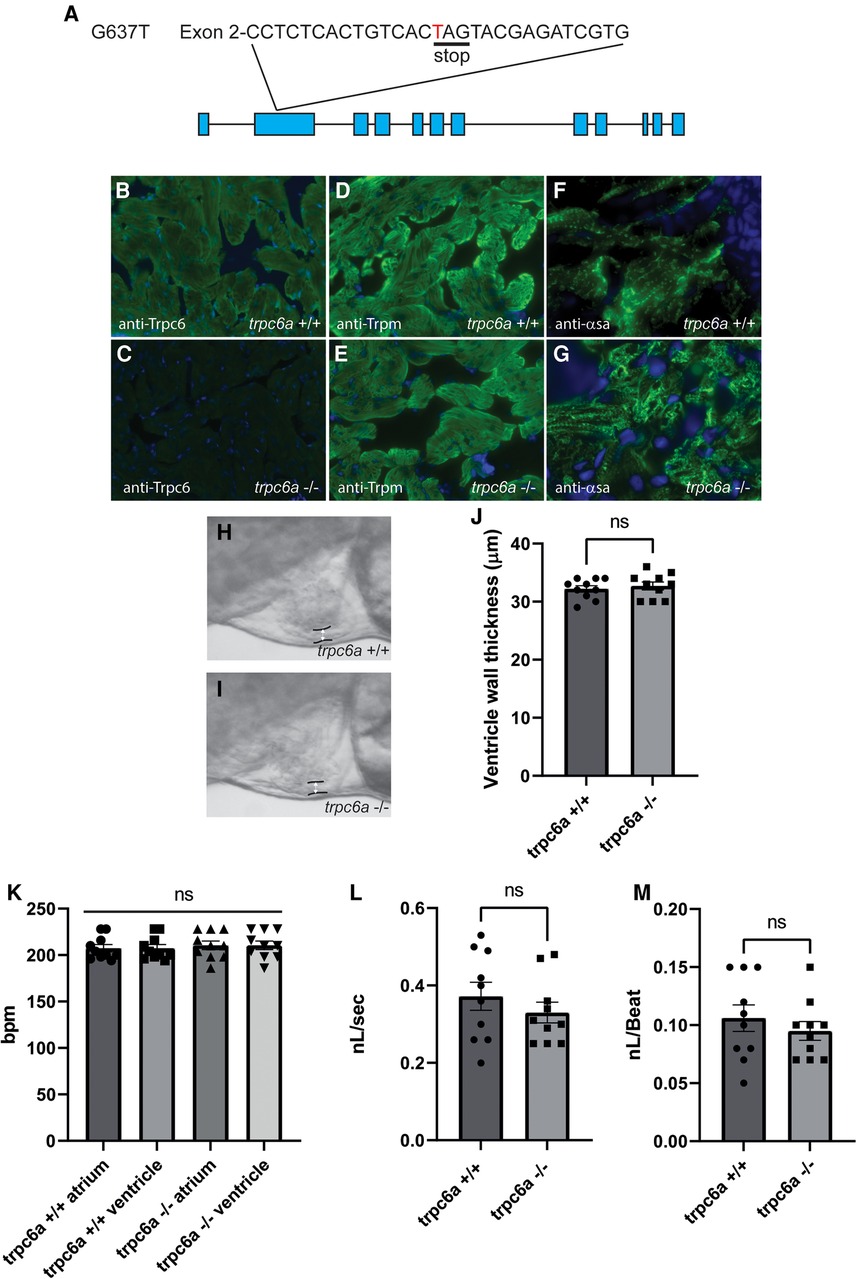
Figure 1. Loss of Trpc6a does not affect cardiac development. (A) Diagram of the point mutation carried by the sa23930 zebrafish transgenic line. The G637T mutation causes a premature stop codon in the Exon 2 of the trpc6a. (B,C) IHC images from adult heart sections showing the presence of Trpc6 in the myocardium of trpc6a+/+ but absent from the myocardium of trpc6a−/− zebrafish. Trpc6: green, DAPI: blue. (D,E) IHC images from adult heart sections showing the organization of tropomyosin (Trpm) in myocardium of trpc6a+/+ and in trpc6a−/− zebrafish. Trpm: green, DAPI: blue. (F,G). IHC images from adult heart sections showing the organization of α-sarcomeric actin (α-sa) in the myocardium of trpc6a+/+ and in trpc6a−/− zebrafish. α-sa: green, DAPI: blue. (H,I) Representative morphology of the ventricular wall of 5dpf larvae from control (trpc6a+/+, H) and trpc6a KO (trpc6a−/−, I) groups. (J) Ventricular wall thickness measurements of 5dpf larvae during diastole. T-test was used for statistical analysis. (K) Atrial and ventricular contraction rates (in bpm) of trpc6a+/+ and trpc6a−/− 5dpf larvae. 1-way ANOVA was used for statistical analysis. (L) Blood flow velocity (in nL/s) measured in the caudal vein at 5dpf. T-test was used for statistical analysis. (M) Cardiac output (in nL/beat) of trpc6a+/+ and trpc6a−/− 3dpf larvae. t-test was used for statistical analysis. (H–M) Data obtained on n = 10 larvae per group.
Because of the role TRPC6 plays in sensing changes in mechanical load after cardiac injury in mammals, we assessed whether the loss of Trpc6 affected cardiac regeneration in adult zebrafish. To achieve this, we performed apical resection of adult trpc6a−/− and trpc6a+/+ zebrafish. At 30 days post amputation (dpa), histological staining using acid fuchsin orange G (AFOG) indicated that the loss of Trpc6a inhibited cardiac regeneration resulting in the presence of a significant fibrin/collagen scar (n = 5/group) (Figures 2A–C). Previous research indicates that TRPC6 can also play a role in angiogenesis (14). During cardiac regeneration in adult zebrafish, revascularization of the wound region is a critical early event which could be affected by the loss of Trpc6a. To address this possibility, we analysed wound revascularization at 7dpa. In this manner we could not detect any obvious differences in vascular plexus formation within the wound region of both trpc6a−/− and trpc6a+/+ zebrafish hearts indicating that this process appears largely unaffected and is unlikely the cause of the defective regeneration we observed in trpc6a−/− hearts (n = 5/group) (Figures 2D–G). We next sought to determine whether cardiomyocyte proliferation had been affected in trpc6a−/− zebrafish. To meet this end, we preformed EdU labelling of resected trpc6a−/− and trpc6a+/+ zebrafish hearts at 14dpa. Our analysis indicates that there is a significant reduction in the number of EdU labelled cardiomyocytes in the trpc6a−/− hearts compared to their trpc6a+/+ siblings (n = 3/group) (Figures 2H–J). Taken together this data indicates that the loss of Trpc6a disrupts cardiac regeneration due to a significant reduction in cardiomyocyte proliferation.
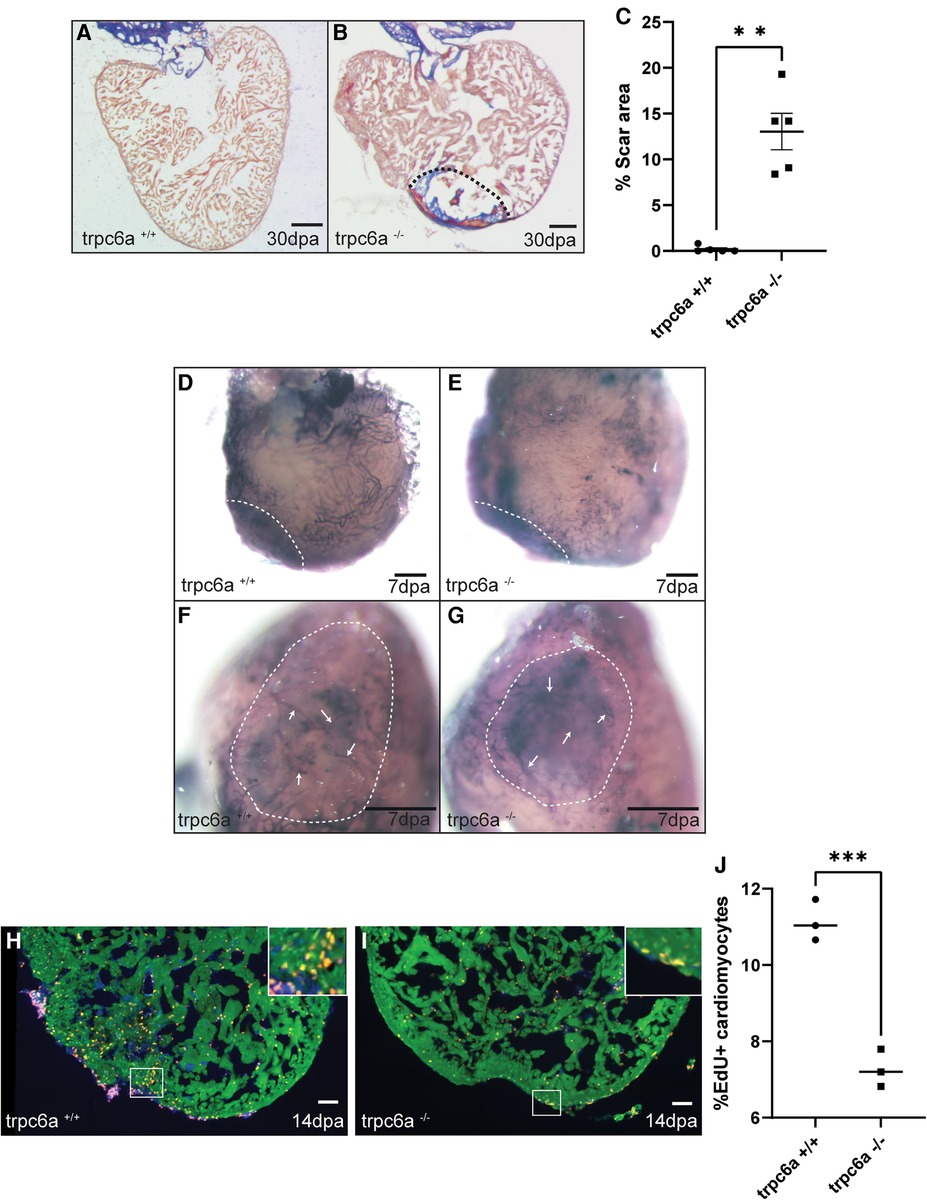
Figure 2. Trpc6a is required for cardiac regeneration. (A-C) AFOG staining images and quantification of the scar area at 30dpa. Representative image of AFOG staining obtained for trpc6a+/+ (A) and trpc6a−/− (B) Scale bars: 200 μm. The dashed line outlines the scar region. (C) Histogram depicting the quantification of the scar area (n=5/group). Students t-test was used for statistical analysis. **: p value < 0.01. (D–G) Representative images of alkaline phosphatase staining showing the vasculature of 7dpa whole mount hearts. Low (D,E) and high (F,G) magnification of the vascular plexus present in the wound region of trpc6a+/+ (D,F) and trpc6a−/− (E,G) zebrafish hearts. Scale bars: 200 μm. (H–J) Cardiomyocyte proliferation measured at 14dpa. Representative IHC images showing Mef2c (green), EdU (red) and DAPI (blue) for trpc6a+/+ (H) and trpc6a−/− (I). The white box depicts a higher magnification image in the upper right corner. Scale bars: 100 μm. (J). Quantification of EdU+ cardiomyocytes (n=3/group). Students t-test was used for statistical analysis. ***: p value < 0.001.
To determine what effect the loss of Trpc6a had on the cardiac transcriptome during heart regeneration, we performed bulk RNA sequencing of sham operated and 7dpa resected trpc6a−/− and trpc6a+/+ hearts (n = 5/group). Bi-clustering heatmaps were generated to visualize the expression profile of the top 30 differentially expressed genes sorted by their adjusted p-value by plotting their log2 transformed expression values (Figures 3A,B). Analysis of sham vs. 7dpa samples indicates that there are 515 differentially expressed genes (DEGS) specific to 7dpa trpc6a+/+ regenerating hearts compared with 451 DEGS specific to 7dpa trpc6a−/− hearts. We next performed GO analysis of the DEGS specific to either trpc6a+/+ 7dpa hearts or trpc6a−/− 7dpa hearts (Tables 1, 2). From this data we were able to determine that there are numerous pathways which are associated with regenerating trpc6a+/+ 7dpa hearts (30 in total) including a number of signalling pathways (Adipocytokine, PPAR, Toll-like receptor, NOD-like receptor, C-type lectin, Insulin, MTOR, TGF-beta and VEGF), metabolic processes and the cell cycle (Table 1). In contrast there are 15 pathways associated with trpc6a−/− 7dpa hearts, these include a different set of signalling pathways (Hedgehog, FoxO, Apelin and Adrenergic) and, notably, a lack of cell cycle pathways (Table 2). We next focused on the DEGS which were specific for regenerating trpc6a+/+ hearts as these likely include genes which may be induced by Trpc6a activation. In this manner we identified a number of transcription factors which were significantly upregulated in the trpc6a+/+ hearts but not in trpc6a−/− hearts (Figure 3C). Of particular interest were components of the AP1 transcription factor complex (june and fosl1a), a critical regulator of cardiac regeneration in adult zebrafish and a known downstream target of Trpc6 (Figure 3C) (15). We next questioned whether the upregulation of june and fosl1a was a common mechanism associated with cardiac injury which also occurred in adult mammals. To address this, we reanalysed single nuclei RNAseq (snRNAseq) data performed on infarcted adult mouse hearts from Yamada et al. (16). Although adult mice do not appear to possess a june orthologue we were able to determine that the expression of Fosl1 did not change significantly in border zone cardiomyocytes following myocardial infarction in adult mice (Supplementary Figures S1B,C). This indicates that the upregulation of fosl1a we observed is likely specific to regenerating zebrafish hearts. Trpc6 also classically induces calcineurin/NFAT signalling (10) and in this respect we observed a significant increase in expression of nfatc2b specifically in regenerating trpc6a+/+ hearts (Figure 3C and Supplementary Figure S2A) in conjunction with a significant increase in expression of the downstream target nppb (p-adj 1.87E-05) (Supplementary Figure S3A). In comparison, in adult mice Nfatc2 does not appear to be significantly upregulated by cardiomyocytes following myocardial infarction (Supplementary Figures S2B,C), however there is a significant increase in the cardiomyocyte expression of Nppb in infarcted mouse hearts (Supplementary Figures S3B,C). Taken together, this indicates that the calcineurin/NFAT signalling pathway appears to be activated by Trpc6a in regenerating zebrafish hearts. Lastly, we also assessed whether trpc6a expression is upregulated during regeneration. Analysis of trpc6a+/+ sham vs. 7dpa samples indicates there is a significant upregulation of trpc6a expression during regeneration which is not the case in the trpc6a−/− samples (Supplementary Figure S4A). In contrast adult mice do not appear to upregulate the expression of Trpc6 following myocardial infarction (Supplementary Figures S4D,E). We subsequently performed IHC for Trpc6a on regenerating trpc6a+/+ hearts in an attempt to localise the upregulation in expression (Supplementary Figures S4B,C). Although there appears to be a moderate elevation of Trpc6a in the cardiomyocytes adjacent to the wound region compared to distal regions (Supplementary Figures S4B,C) we were unable to quantify this satisfactorily and this will require higher resolution analysis such as single nuclei RNA sequencing.
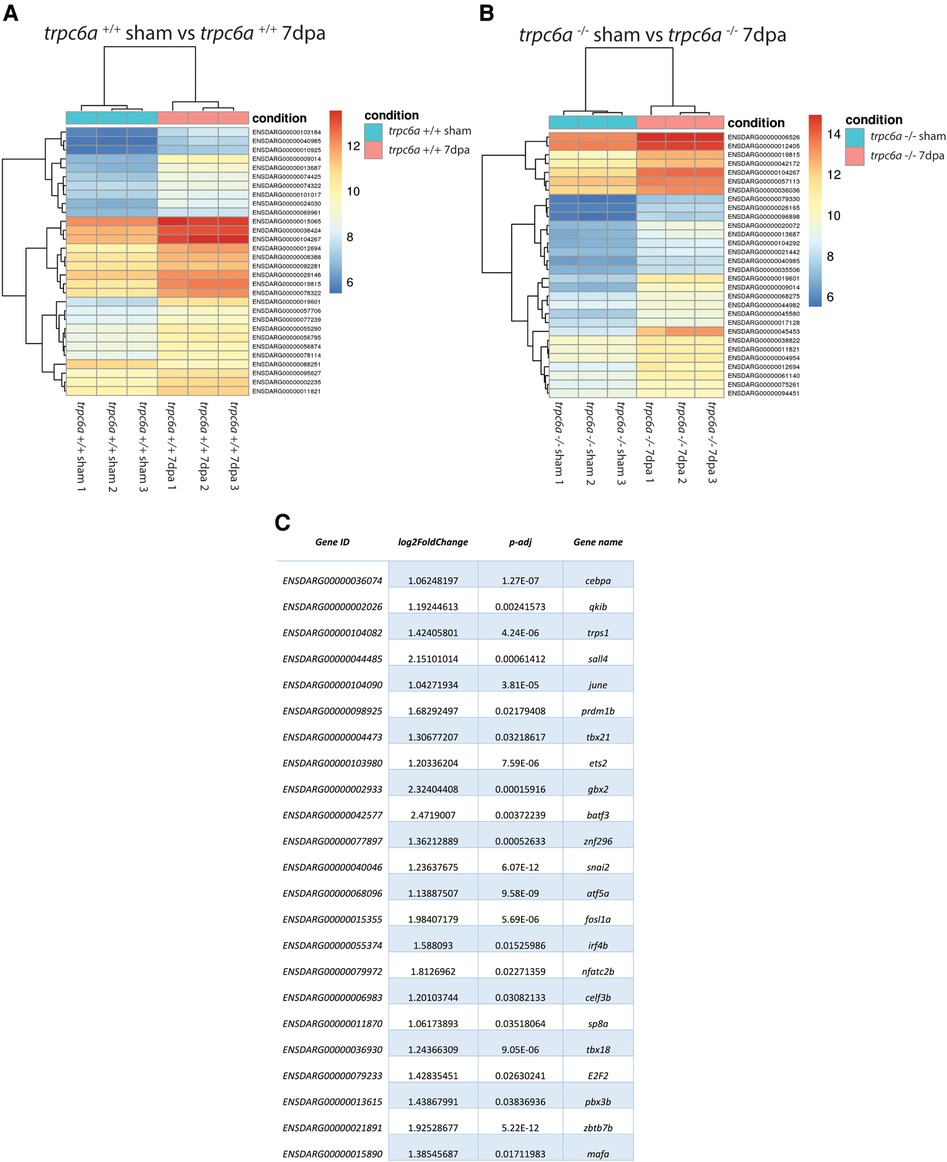
Figure 3. Loss of Trpc6a results in misregulated gene expression during regeneration. (A,B) Heatmaps showing the 30 top differentially regulated genes between sham and 7dpa hearts of trpc6a+/+ (A) and trpc6a−/− (B) zebrafish (C) Table of transcription factors which are significantly upregulated in trpc6a+/+ hearts but not in trpc6a−/− hearts following injury (compared to their respective sham controls).
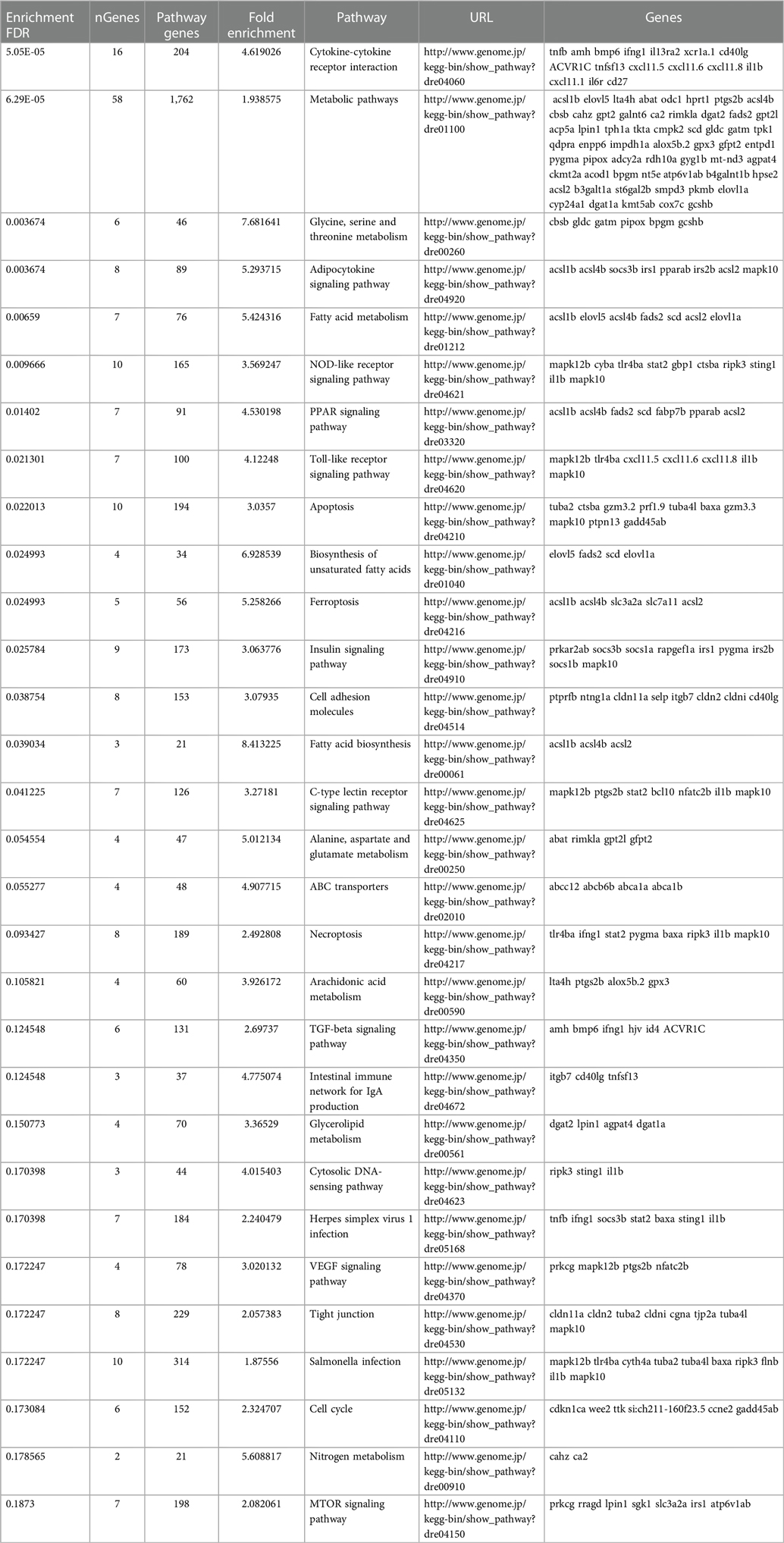
Table 1. GO analysis of DEGS specific to 7dpa trpc6a+/+ hearts.
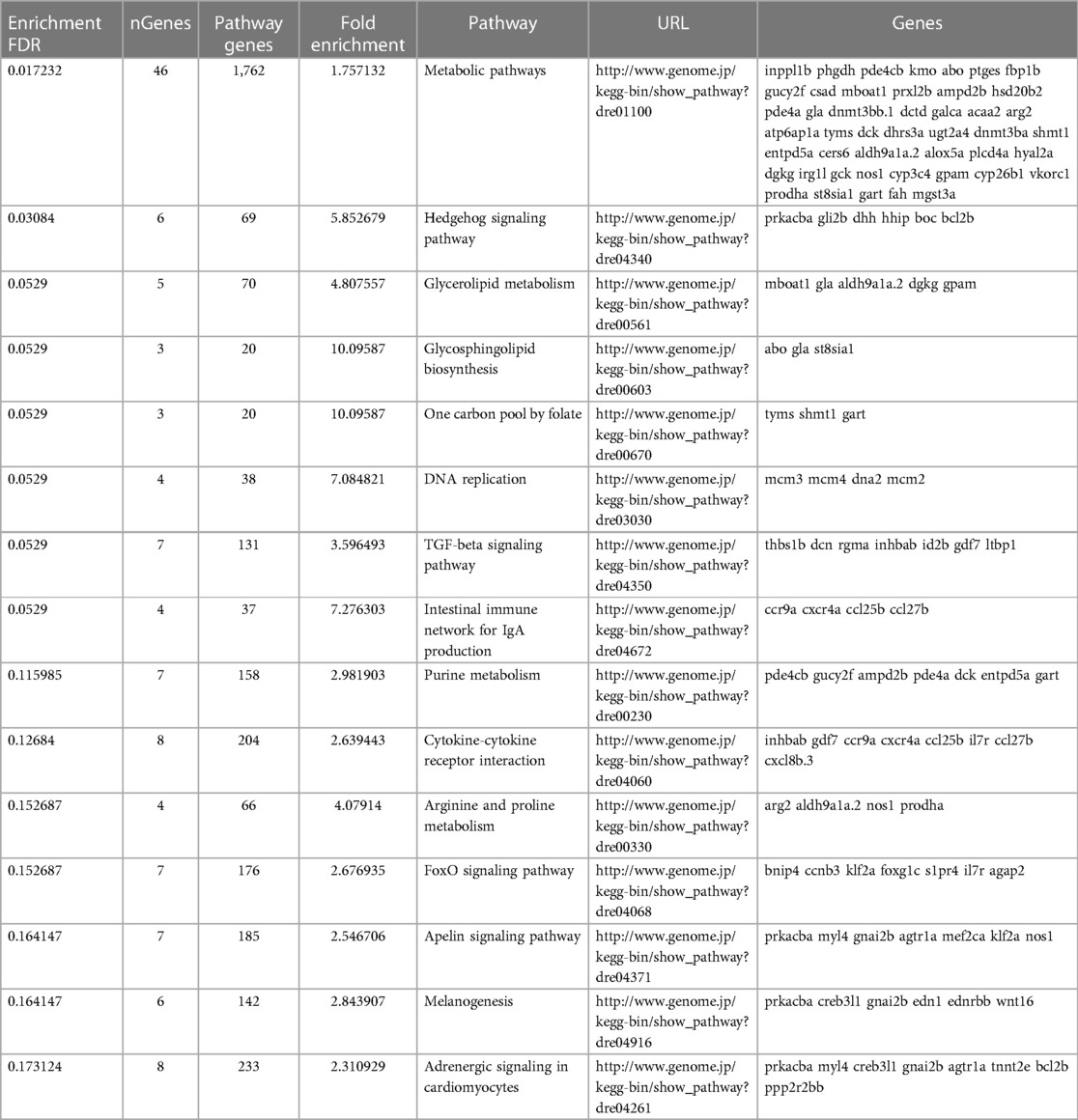
Table 2. GO analysis of DEGS specific to 7dpa trpc6a−/− hearts.
Previous data indicates that pathologically stretching cardiomyocytes in vivo activates TRPC6, which in turn induces downstream gene expression (11). Based on this, we assessed whether Trpc6a regulated the expression of the AP1 transcription factor component fosl1a in response to mechanical stretch. To achieve this, we dissociated and isolated cardiomyocytes from either trpc6−/− or trpc6a+/+ hearts and subjected them to 24 h of cyclic stretch (n = 5 hearts per group, 3 groups per condition) (Figure 4A). Following the completion of this protocol, we harvested the cardiomyocytes and performed RT qPCR for both fosl1a and nppb (Figure 4A). In this manner, we determined that under static conditions there is no significant difference in the relative expression of fosl1a between trpc6a+/+ cardiomyocytes when compared to trpc6−/− cardiomyocytes. However, under stretch conditions the expression of fosl1a was significantly greater in trpc6a+/+ cardiomyocytes when compared to trpc6−/− cardiomyocytes (Figure 4B,C). These data indicate that loss of Trpc6a in cardiomyocytes results in a failure to upregulate the expression of the AP1 transcription factor component fosl1a. We also assessed the expression of the downstream target of calcineurin/NFAT signalling, nppb. Although there appears to be a similar trend in the expression of nppb in response to mechanical stretch this was not significant (Supplementary Figures S5A,B).
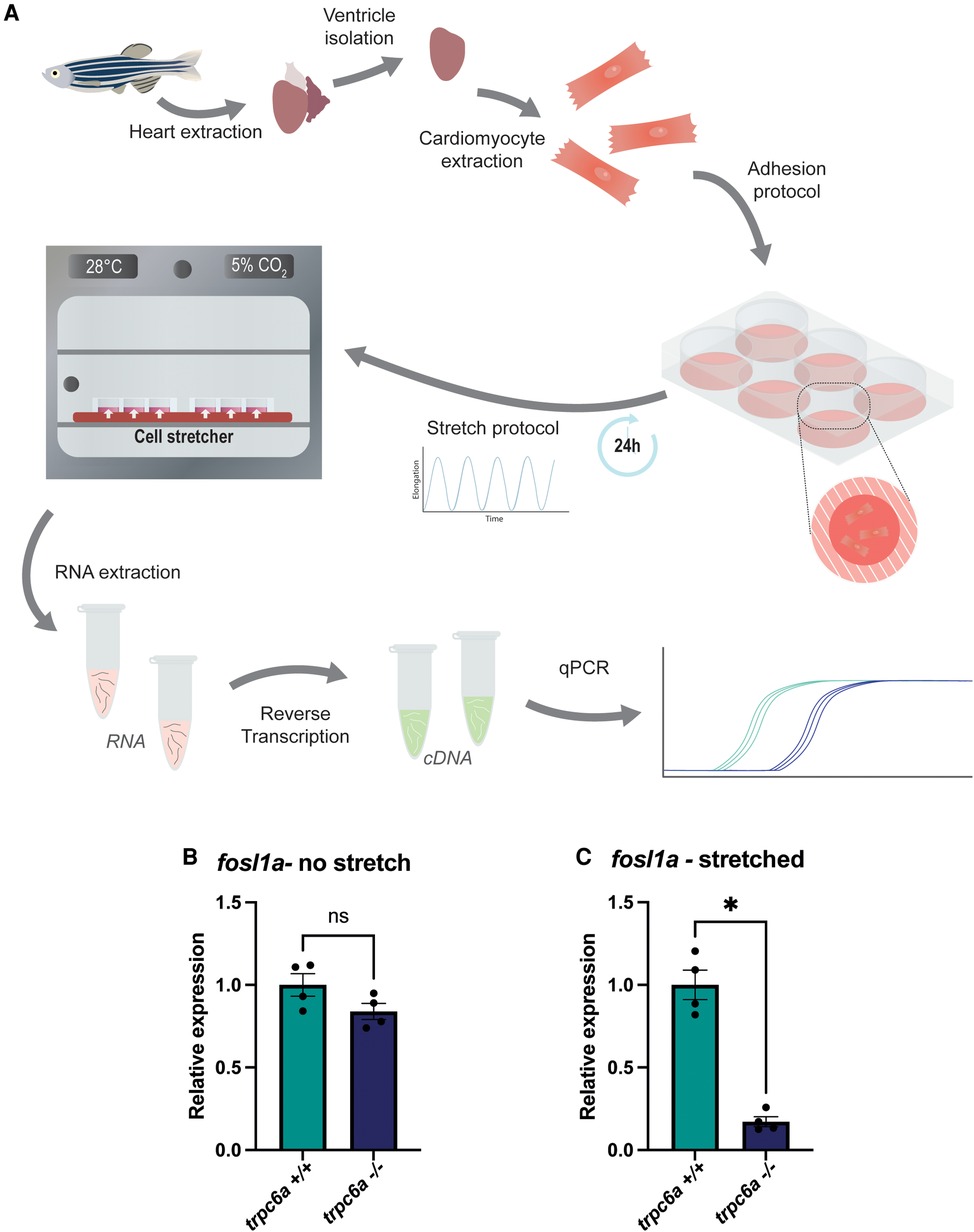
Figure 4. Trpc6a regulates the stretch induced expression of AP1 transcription factor components. (A) Schematic representation of the experimental design. Cardiomyocytes were isolated from extracted hearts and plated onto poly lysine-coated plates. A cyclic stretch protocol was applied for 24 h before RNA extraction and RT-qPCR. (B) Relative expression of fosl1a in unstretched trpc6a+/+ and trpc6a−/− cardiomyocytes. (C) Relative expression of fosl1a in trpc6a+/+ and trpc6a−/− cardiomyocytes subjected to cyclic stretch. Mann–Whitney test was used for statistical analysis. *: p value < 0.05.
The TRP ion channel TRPC6 is responsible for detecting increased mechanical stretch in cardiomyocytes and activating the CALCINEURIN/NFAT pathway (10). Under pathophysiological conditions of chronic elevated cardiomyocyte stretch, for example volume overload caused by a cardiac ischemia, this will lead to pathological hypertrophy and ultimately heart failure (10). Whether TRPC6 could also play a role in detecting increased cardiomyocyte stretch and triggering cardiac regeneration in animal models capable of this feat is currently unknown.
The results of this study indicate that Trpc6a is an essential component of the cardiac regenerative response in adult zebrafish. Our data demonstrates that loss of Trpc6a results in a failure to regenerate the heart after 30 days following cardiac resection. Early regenerative processes such as revascularization appear largely unaffected, however cardiomyocyte proliferation is significantly impeded in the absence of Trpc6a signaling leading to the persistence of extensive scarring. Furthermore, comparative transcriptomic analysis of trpc6a−/− and trpc6a+/+ resected hearts indicates that loss of Trp6a substantially impacts gene expression. In particular components of the AP1 transcription factor complex, which are required for successful cardiac regeneration, are not upregulated in the absence Trpc6a. Lastly, our data indicates that Trpc6a regulates the expression of AP1 transcription factor complex components in response to mechanical stretch. Together these results indicate that, in adult zebrafish, increased/chronic cardiomyocyte stretch associated with cardiac injury is sensed by Trpc6a which subsequently activates downstream signaling pathways resulting in the expression of genes, such as AP1 transcription factor complex components, which are involved in driving cardiac regeneration.
The Frank-Starling law was described over a century ago and explains how elevated ventricular preloading, which stretches cardiomyocytes, results in an increased force of contraction in order to maintain circulatory homeostasis (17). Stretching cardiomyocytes increases the calcium sensitivity of their sarcomeres resulting in enhanced contractility. In situations where ventricular preloading is maintained, there is a further progressive increase in the force of contractility termed the slow force response (SFR) which is driven by elevated, TRPC3 and TRPC6 dependent (9), Ca2+ transients (18).
Chronic ventricular loading, for example after myocardial ischemia, results in cardiac remodeling and pathological cardiomyocyte hypertrophy leading, ultimately, to heart failure. The molecular mechanisms which drive pathological cardiac hypertrophy (as opposed to physiological hypertrophy induced by exercise) are largely driven by CALCINEURIN and its downstream effector NFAT (8). Chronic increases in mechanical load result in elevated intracellular Ca2+ which in turn activates CALCINEURIN. CALCINEURIN subsequently dephosphorylates NFAT which translocates into the nucleus and regulates the expression of genes which drive pathological hypertrophy.
The ion channels TRCP3 and TRPC6 are responsible for detecting increases in cardiomyocyte stretch and generating the sustained Ca2+ transients which drive this pathological process (10). It is apparent then that TRPC6 plays a central role in the cardiac mechanosensitive response to volume overload which results in pathological hypertrophy. Conversely, our data indicates that, in adult zebrafish, it appears that the response to volume overload regulated by Trpc6a results in cardiomyocyte proliferation and ultimately cardiac regeneration. In mice, global KO of Trpc6 results in increased mortality after myocardial infarction, however this is primarily due to the role Trpc6 plays in cardiac fibroblast activation, a process which is essential for early scar formation in order to avoid cardiac rupture (19). Blood pressure in adult zebrafish is around 50 times lower than mice (2.5 mmHg vs. 100 mmHg) (20) and as such the formation of a clot is sufficient to avoid excessive blood loss following damage to the myocardium. Because of the role TRPC6 plays in different cell types in mammals, it will be interesting to determine the effect that conditional, cardiomyocyte specific, deletion of Trpc6 has following myocardial infarction. Furthermore, due to the close relationship between TRPC3 and TRPC6 it will also be interesting to assess whether trpc3 also plays a role in cardiac regeneration in zebrafish.
By taking the opposite approach to genetic KO, constitutive, cardiomyocyte specific, over-expression of Trpc6 in adult mice activates the CALCINEURIN/NFAT pathway resulting in pathological hypertrophy and lethality (10). However, it would also be of interest to assess what effect Trpc6 overexpression has at earlier stages of development when cardiomyocytes are capable of proliferating and regenerating damaged myocardium. Downstream of TRPC6, cardiomyocyte specific overexpression of an active-CALCINEURIN isoform in adult mice is sufficient to trigger pathological hypertrophy and heart failure (21). Furthermore, similar experiments performed in neonatal mice indicates that active-CALCINEURIN induces a switch from proliferation to hypertrophic growth in cardiomyocytes which are normally hyperplastic at this stage of development (22). While these data seem at odds with our finding that Trpc6a plays a beneficial role during cardiac regeneration, our transcriptomic data indicates that other Trpc6a mediated mechanisms are also involved.
Although much focus has been placed on the TRPC6-CALCINEURIN-NFAT axis, TRPC6 also activates other signalling mechanisms such as AP1 mediated gene transcription (15). We found that, during cardiac regeneration, there is a significant increase in the expression the AP1 transcription factor components june and fosl1a which does not occur when Trpc6a is absent. This is in-line with previous in vitro data indicating that activation of Trpc6 results in increased c-fos expression (23). More recently, the AP1 transcription factor complex has been shown to be a critical regulator of cardiac regeneration in adult zebrafish (12). In particular, cardiomyocyte specific expression of a dominant-negative Fos isoform significantly inhibits cardiac regeneration. This loss of AP1 function results in defective cardiomyocyte sarcomere disassembly and proliferation and also affects their ability to extend protrusions into the site of injury (12). Interestingly, although the AP1 components JunB and Fosl1 are upregulated in adult zebrafish hearts after injury, the same is not true for adult mice after myocardial infarction (12, 24). Furthermore, overexpression of JunB and Fosl1 in neonatal rat cardiomyocytes is sufficient to induce proliferation and protrusive behaviour in these cells similar to that observed in zebrafish cardiomyocytes (12). These data indicate a difference in AP1 signalling between adult zebrafish and mammals which may partly explain the differences we observed following KO of trpc6a. Lastly, we have demonstrated that Trpc6a regulates the expression of the AP1 transcription factor component fosl1a in response to mechanical stretch, similar to reports in mammalian cardiomyocytes (5, 25). Although care must be taken when extrapolating in vitro data, it is likely that this is also the situation which occurs during cardiac regeneration in adult zebrafish. The increase in myocardial stretch caused by volume overload following cardiac injury could activate Trpc6a and induce the expression of AP1 components required for cardiac regeneration. In summary we have identified Trpc6a as a critical regulator of cardiac regeneration. Furthermore, we also demonstrate that Trpc6a can induce the expression of AP1 components in response to mechanical stretch in vitro. Future studies will be required to establish exactly why Trpc6a induces a regenerative response in adult zebrafish compared to the pathological response in mammals and also whether this is restricted to cardiomyocytes or also involves other cell types which express TRPC6 such as cardiac fibroblasts.
Trpc6a KO G637T (sa23930) zebrafish line was purchased from ZIRC and maintained under standardized conditions (26). The sa23930 Trpc6a KO was established and maintained on the ABWT strain. Experiments were conducted in accordance with local approval and the European Communities council directive 2010/63/EU. A mixture of males and females were used in all experiments using adult zebrafish.
5dpf larvae were anaesthetised and mounted in low melt agarose. 30 s videos of either cardiac contractions or blood flow were recorded using a Point Grey GRAS-03K2C-C high speed camera. Heart rate and blood flow were analysed using ViewPoint MicroZebraLab software and ImageJ software. T-test and ANOVA statistical analysis was performed using GraphPad Prism.
Cardiac resection were performed on 6–10-month-old zebrafish as previously described (2), in accordance with local approval (APAFIS#2021021117336492 v5).
Immunohistochemistry and histological staining were performed on 10 μm heart sections as previously described (27). The antibodies used in this manuscript are listed below:
anti-Trpc6 (OST00081W, Osenses)
anti-Trpm (T2780, Sigma)
anti-Mef2c (ab197070 Abcam)
anti-α-Sarcomeric actin (A2172, Sigma)
EdU labelling was performed according to the manufacturer's instructions (Click-iT EdU Kit C10337, Molecular Probes). Acid Fuchsin-Orange G (AFOG) staining was performed as previously described (28) and the size of the scar area was calculated using ImageJ software. T-test statistical analyses was performed using GraphPad Prism. Alkaline phosphate staining was performed on whole-mount heart as previously described (27).
A Zeiss Discovery V20 fluorescence stereomicroscope fitted with a Tucsen FL20 microscope camera was used for histological imaging and either a Zeiss Axio Imager equipped with an Apotome 3 module or a Leica TCS SP-8 confocal microscope were used for imaging immunohistochemistry labelled sections.
To label proliferating cells, amputated fish were anesthetized in Tricaine and injected with 50 μl of 240 μg/mL of EdU solution daily. At 14dpa, fish were euthanized (excess of tricaine), the hearts were collected, and processed for immunohistochemistry. Following imaging, EdU + cardiomyocytes were counted using IMARIS software. T-test statistical analysis was performed using GraphPad Prism.
Adult fish were anesthetized in Tricaine. Each group (trpc6−/− and trpc6a+/+) consists of 3 biological replicates of 5 pooled hearts. For each replicate 5 hearts were pooled and RNA was extracted using Trizol/choloform. Using DESeq2, a comparison of gene expression between the groups of samples was performed. The Wald test was used to generate p-values and log2 fold changes. Genes with an adjusted p-value <0.05 and absolute log2 fold change >1 were called as differentially expressed genes.
For each group (trpc6a+/+ no stretch-3 groups, trpc6a+/+ stretched-3 groups, trpc6a−/− no stretch-3 groups, trpc6a−/− stretched-3 groups), 5 hearts were collected and pooled. Cardiomyocytes were isolated as previously described (29). Cardiomyocytes were plated on BioFlex® culture plates coated with poly lysine and centrifuged briefly (400 G/1 min). Following a period of 2 h to allow the cardiomyocytes to attach, a cyclic stretch protocol was applied for 24 h (Sine, 16% elongation, 0.5 Hz) with the Flexcell Tension System (FX-6000 T, Flexcell®) at 28°C and with 5% CO2.
RNA was extracted from isolated cardiomyocytes using Trizol/chloroform. cDNA was obtained after reverse transcription using a First strand cDNA synthesis RT-PCR kit(Roche) and quantitative PCR was performed using SYBR Green (Roche) and a LightCycler 480 system (Roche). The primer sequences used are as follow:
tubulin alpha Forward: 5’ CGGCCAAGCAACACTACTAGA 3’
tubulin alpha Reverse: 5’ AGTTCCCAGCAGGCATTG 3’
fosl1a Forward: 5’ AAGGGAACGCAACAAAATGG 3’
fosl1a Reverse: 5’ AGCTTCTCCTTTTCCTTCTGG 3’
nppb Forward: 5’ TCCTCAGCGTTCAACACATG 3’
nppb Reverse: 5’ CCGCCTTTACTTCTCTTTCCG 3’
The original contributions presented in the study are publicly available. This data can be found here: ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress#) accession E-MTAB-13603.
The animal study was approved by ministre de l'Enseignement supérieur et de la Recherche et de l'innovation. The study was conducted in accordance with the local legislation and institutional requirements.
LR and CJ were involved in study conception and design. All authors were involved in data collection and analysis. CJ drafted the manuscript. All authors contributed to the article and approved the submitted version.
We would like to acknowledge the imaging facility MRI, member of the France-BioImaging national infrastructure supported by the French National Research Agency (ANR-10-INBS-04, «Investments for the future»)”. We also acknowledge the Zebrafish International Resource Center for providing KO zebrafish lines. The Jopling lab is part of the Laboratory of Excellence Ion Channel Science and Therapeutics supported by a grant from the ANR. Work in the Jopling lab is supported by a grant from the “la Fondation Leducq” and from the ANR (contract ANR-20-CE14-003-02 MetabOx-Heart and ANR-22-CE14-048-02 IONIC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1186086/full#supplementary-material
SUPPLEMENTARY FIGURE S1
Trpc6a regulates the expression of the AP1 transcription complex gene fosl1a during cardiac regeneration. (A) Table showing the difference in expression of fosl1a between trpc6a+/+ sham hearts and trpc6a+/+ 7dpa hearts and trpc6a−/− sham hearts and trpc6a−/− 7dpa hearts, note the significant increase in expression of fosl1a in the trpc6a+/+ hearts. (B) Adult mouse myocardial infarction. Re-analysed UMAP plots depicting the expression Fosl1 in uninjured sham conditions vs. 1, 7 and 14 days post myocardial infarction (MI). (C) Violin plots showing the expression of Fosl1 at different time points after injury (sham, 1,7 and 14 days post myocardial infarction (d pMI) in cardiomyocytes (ns = not significant).
SUPPLEMENTARY FIGURE S2
Trpc6a regulates the expression of nfatc2b during cardiac regeneration. (A) Table showing the difference in expression of nfatc2b between trpc6a+/+ sham hearts and trpc6a+/+ 7dpa hearts and trpc6a−/− sham hearts and trpc6a−/− 7dpa hearts, note the significant increase in expression of nfatc2b in the trpc6a+/+ hearts. (B) Adult mouse myocardial infarction. Re-analysed UMAP plots depicting the expression Nfatc2 in uninjured sham conditions vs. 1, 7 and 14 days post myocardial infarction (MI). (C) Violin plots showing the expression of Nfatc2 at different time points after injury (sham, 1,7 and 14 days post myocardial infarction (d pMI) in cardiomyocytes (ns = not significant).
SUPPLEMENTARY FIGURE S3
Trpc6a regulates the expression of the calcineurin/nfat target gene nppb during cardiac regeneration. (A) Table showing the difference in expression of nppb between trpc6a+/+ sham hearts and trpc6a+/+ 7dpa hearts and trpc6a−/− sham hearts and trpc6a−/− 7dpa hearts, note the significant increase in expression of nppb in the trpc6a+/+ hearts. (B) Adult mouse myocardial infarction. Re-analysed UMAP plots depicting the expression Nppb in uninjured sham conditions vs. 1, 7 and 14 days post myocardial infarction (MI). (C) Violin plots showing the expression of Nppb at different time points after injury (sham, 1,7 and 14 days post myocardial infarction (d pMI) in cardiomyocytes. P values were adjusted using the Benjamini-Hochberg correction **** P < 0.001.
SUPPLEMENTARY FIGURE S4
The expression of trpc6a increases during cardiac regeneration. (A) Table showing the difference in expression of trpc6a between trpc6a+/+ sham hearts and trpc6a+/+ 7dpa hearts and trpc6a−/− sham hearts and trpc6a−/− 7dpa hearts, note the significant increase in expression of trpc6a in the trpc6a+/+ hearts. (B) Immunohistochemistry using an anti-Trpc6 antibody (green) and DAPI (blue) on regenerating 7dpa trpc6a+/+ hearts. White dashed line indicates the border zone between the myocardium and the injury site. (C) Higher magnification of the region denoted by the white square in (B) The yellow dashed circle highlights cardiomyocytes proximal to the border zone. The red dashed circle highlights cardiomyocytes distal to the border zone. (D) Adult mouse myocardial infarction. Re-analysed UMAP plots depicting the expression Trpc6 in uninjured sham conditions vs. 1, 7 and 14 days post myocardial infarction (MI). (C) Violin plots showing the expression of Trpc6 at different time points after injury (sham, 1,7 and 14 days post myocardial infarction (d pMI) in cardiomyocytes (ns = not significant).
SUPPLEMENTARY FIGURE S5
Trpc6a does not significantly regulate the expression of nppb in response to mechanical stretch. Relative expression of nppb in unstretched trpc6a+/+ and trpc6a−/− cardiomyocytes. (B) Relative expression of nppb in trpc6a+/+ and trpc6a−/− cardiomyocytes subjected to cyclic stretch.
1. Neves JS, Leite-Moreira AM, Neiva-Sousa M, Almeida-Coelho J, Castro-Ferreira R, Leite-Moreira AF. Acute myocardial response to stretch: what we (don’t) know. Front Physiol. (2015) 6:408. doi: 10.3389/fphys.2015.00408
2. Jopling C, Sleep E, Raya M, Martí M, Raya A, Belmonte JCI. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. (2010) 464:606–9. doi: 10.1038/nature08899
3. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. (2011) 331:1078–80. doi: 10.1126/science.1200708
4. Banerjee I, Carrion K, Serrano R, Dyo J, Sasik R, Lund S, et al. Cyclic stretch of embryonic cardiomyocytes increases proliferation, growth, and expression while repressing tgf-beta signaling. J Mol Cell Cardiol. (2015) 79:133–44. doi: 10.1016/j.yjmcc.2014.11.003
5. Rysä J, Tokola H, Ruskoaho H. Mechanical stretch induced transcriptomic profiles in cardiac myocytes. Sci Rep. (2018) 8:4733. doi: 10.1038/s41598-018-23042-w
6. Yu JK, Sarathchandra P, Chester A, Yacoub M, Brand T, Butcher JT. Cardiac regeneration following cryoinjury in the adult zebrafish targets a maturation-specific biomechanical remodeling program. Sci Rep. (2018) 8:15661. doi: 10.1038/s41598-018-33994-8
7. Rovira M, Borràs DM, Marques IJ, Puig C, Planas JV. Physiological responses to swimming-induced exercise in the adult zebrafish regenerating heart. Front Physiol. (2018) 9:1362. doi: 10.3389/fphys.2018.01362
8. Wilkins BJ, Dai Y-S, Bueno OF, Parsons SA, Xu J, Plank DM, et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. (2004) 94:110–8. doi: 10.1161/01.RES.0000109415.17511.18
9. Yamaguchi Y, Iribe G, Nishida M, Naruse K. Role of TRPC3 and TRPC6 channels in the myocardial response to stretch: linking physiology and pathophysiology. Prog Biophys Mol Biol. (2017) 130:264–72. doi: 10.1016/j.pbiomolbio.2017.06.010
10. Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. (2006) 116:3114–26. doi: 10.1172/JCI27702
11. Seo K, Rainer PP, Hahn VS, Lee D-I, Jo S-H, Andersen A, et al. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci U S A. (2014) 111:1551–6. doi: 10.1073/pnas.1308963111
12. Beisaw A, Kuenne C, Guenther S, Dallmann J, Wu C-C, Bentsen M, et al. AP-1 contributes to chromatin accessibility to promote sarcomere disassembly and cardiomyocyte protrusion during zebrafish heart regeneration. Circ Res. (2020) 126:1760–78. doi: 10.1161/CIRCRESAHA.119.316167
13. Von Niederhäusern V, Kastenhuber E, Stäuble A, Gesemann M, Neuhauss SCF. Phylogeny and expression of canonical transient receptor potential (TRPC) genes in developing zebrafish. Dev Dyn. (2013) 242:1427–41. doi: 10.1002/dvdy.24041
14. Ge R, Tai Y, Sun Y, Zhou K, Yang S, Cheng T, et al. Critical role of TRPC6 channels in VEGF-mediated angiogenesis. Cancer Lett. (2009) 283:43–51. doi: 10.1016/j.canlet.2009.03.023
15. Thiel G, Lesch A, Rubil S, Backes TM, Rössler OG. Regulation of gene transcription following stimulation of transient receptor potential (TRP) channels. Int Rev Cell Mol Biol. (2018) 335:167–89. doi: 10.1016/bs.ircmb.2017.07.010
16. Yamada S, Ko T, Hatsuse S, Nomura S, Zhang B, Dai Z, et al. Spatiotemporal transcriptome analysis reveals critical roles for mechano-sensing genes at the border zone in remodeling after myocardial infarction. Nat Cardiovasc Res. (2022) 1:1072–83. doi: 10.1038/s44161-022-00140-7
17. Patterson SW, Piper H, Starling EH. The regulation of the heart beat. J Physiol. (1914) 48:465–513. doi: 10.1113/jphysiol.1914.sp001676
18. Alvarez BV, Pérez NG, Ennis IL, de Hurtado C, & Cingolani MC, E H. Mechanisms underlying the increase in force and ca(2+) transient that follow stretch of cardiac muscle: a possible explanation of the anrep effect. Circ Res. (1999) 85:716–22. doi: 10.1161/01.RES.85.8.716
19. Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. (2012) 23:705–15. doi: 10.1016/j.devcel.2012.08.017
20. Hu N, Yost HJ, Clark EB. Cardiac morphology and blood pressure in the adult zebrafish. Anat Rec. (2001) 264:1–12. doi: 10.1002/ar.1111
21. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. (1998) 93:215–28. doi: 10.1016/S0092-8674(00)81573-1
22. Nguyen NUN, Canseco DC, Xiao F, Nakada Y, Li S, Lam NT, et al. A calcineurin-Hoxb13 axis regulates growth mode of mammalian cardiomyocytes. Nature. (2020) 582:271–6. doi: 10.1038/s41586-020-2228-6
23. Thiel G, Rössler OG. Hyperforin activates gene transcription involving transient receptor potential C6 channels. Biochem Pharmacol. (2017) 129:96–107. doi: 10.1016/j.bcp.2017.01.007
24. van Duijvenboden K, de Bakker DEM, Man JCK, Janssen R, Günthel M, Hill MC, et al. Conserved NPPB+ border zone switches from MEF2- to AP-1-driven gene program. Circulation. (2019) 140:864–79. doi: 10.1161/CIRCULATIONAHA.118.038944
25. Komuro I, Kaida T, Shibazaki Y, Kurabayashi M, Katoh Y, Hoh E, et al. Stretching cardiac myocytes stimulates protooncogene expression. J Biol Chem. (1990) 265:3595–8. doi: 10.1016/S0021-9258(19)39631-0
26. Aleström P, D’Angelo L, Midtlyng PJ, Schorderet DF, Schulte-Merker S, Sohm F, et al. Zebrafish: housing and husbandry recommendations. Lab Anim. (2020) 54:213–24. doi: 10.1177/0023677219869037
27. Lai S-L, Marín-Juez R, Moura PL, Kuenne C, Lai JKH, Tsedeke AT, et al. Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. eLife. (2017) 6:e25605. doi: 10.7554/eLife.25605
28. Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. (2002) 298:2188–90. doi: 10.1126/science.1077857
Keywords: TRPC6 channel, heart regeneration, AP1 complex, mechanosensation, calcineurin/NFAT pathway
Citation: Rolland L, Abaroa JM, Faucherre A, Drouard A and Jopling C (2024) The ion channel Trpc6a regulates the cardiomyocyte regenerative response to mechanical stretch. Front. Cardiovasc. Med. 10:1186086. doi: 10.3389/fcvm.2023.1186086
Received: 14 March 2023; Accepted: 26 October 2023;
Published: 8 January 2024.
Edited by:
Anke Smits, Leiden University Medical Center (LUMC), NetherlandsReviewed by:
Atze Van Der Pol, Eindhoven University of Technology, Netherlands© 2024 Rolland, Abaroa, Faucherre, Drouard and Jopling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chris Jopling Y2hyaXMuam9wbGluZ0BpZ2YuY25ycy5mcg==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.