
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 04 August 2023
Sec. Cardiovascular Genetics and Systems Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1179018
This article is part of the Research Topic Electrical Management of Heart Failure: Shaping the future of cardiac pacing and electrophysiology View all 8 articles
 Kirstine Calloe1*
Kirstine Calloe1* Helena B. D. Magnusson1
Helena B. D. Magnusson1 Dorte Launholt Lildballe2
Dorte Launholt Lildballe2 Morten Krogh Christiansen3
Morten Krogh Christiansen3 Henrik Kjærulf Jensen3,4
Henrik Kjærulf Jensen3,4
In the past 20 years, genetic variants in SCN5A encoding the cardiac voltage-gated sodium channel Nav1.5 have been linked to a range of inherited cardiac arrhythmias: variants resulting in loss-of-function of Nav1.5 have been linked to sick sinus syndrome, atrial stand still, atrial fibrillation (AF) impaired pulse generation, progressive and non-progressive conduction defects, the Brugada Syndrome (BrS), and sudden cardiac death. SCN5A variants causing increased sodium current during the plateau phase of the cardiac action potential is associated with Long QT Syndrome type 3 (LQTS3), Torsade de Pointes ventricular tachycardia and SCD. Recently, gain-of-function variants have been linked to complex electrical phenotypes, such as the Multifocal Ectopic Purkinje-related Premature Contractions (MEPPC) syndrome. MEPPC is a rare condition characterized by a high burden of premature atrial contractions (PACs) and/or premature ventricular contractions (PVCs) often accompanied by dilated cardiomyopathy (DCM). MEPPC is inherited in an autosomal dominant fashion with an almost complete penetrance. The onset is often in childhood. The link between SCN5A variants, MEPPC and DCM is currently not well understood, but amino acid substitutions resulting in gain-of-function of Nav1.5 or introduction of gating pore currents potentially play an important role. DCM patients with a MEPPC phenotype respond relatively poorly to standard heart failure medical therapy and catheter ablation as the PVCs originate from all parts of the fascicular Purkinje fiber network. Class 1c sodium channel inhibitors, notably flecainide, have a remarkable positive effect on the ectopic burden and the associated cardiomyopathy. This highlights the importance of genetic screening of DCM patients to identify patients with SCN5A variants associated with MEPPC. Here we review the MEPPC phenotype, MEPPC-SCN5A associated variants, and pathogenesis as well as treatment options.
The depolarization phase of the cardiac action potential is due to the activation of the voltage-gated sodium channel Nav1.5 encoded by the gene SCN5A.
Genetic variants in SCN5A causing a loss-of-function of Nav1.5 have been linked to sick sinus syndrome, atrial stand still, atrial fibrillation (AF), progressive and non-progressive cardiac conduction disease, the Brugada syndrome (BrS) and sudden cardiac death (SCD). SCN5A variants causing increased sodium current during the plateau phase of the cardiac action potential is associated with Long QT Syndrome type 3 (LQTS3), Torsade de pointes ventricular tachycardia and SCD. The different disease mechanisms are excellently reviewed by Wilde and Amin (1). These pathologies are often inherited in an autosomal dominant manner with incomplete penetrance. Furthermore, the manifestations of SCN5A variants are highly variably and the same genotype can be associated with multiple and overlapping phenotypes (2).
Recently, the Multifocal Ectopic Purkinje-related Premature Contractions (MEPPC) syndrome has been added to the list of SCN5A associated phenotypes. MEPPC is a rare, but likely underdiagnosed, syndrome in which the presence of multiple ectopic polymorphic ventricular complexes (PVCs) with relatively narrow QRS complexes is a central feature. MEPPC was first described by Laurent and coworkers in 2012 based on three families with the c.665G > A SCN5A variant resulting in the R222Q amino acid substitution in Nav1.5. At almost the same time, other reports on R222Q in families with complex arrhythmias and dilated cardiomyopathy (DCM) were published (3, 4). After these initial reports, additional SCN5A variants have been suggested to cause an MEPPC phenotype or an MEPPC-like phenotype.
Here, we review the MEPPC phenotype, MEPPC-SCN5A associated variants, and pathogenesis as well as management options.
MEPPC patients typically present with palpitations, dyspnea, syncope, or SCD (5). The age of onset varies, but the clinical symptoms are often found at a young age (6) or during childhood (5). The electrical phenotype is characterized by multiple premature ventricular complexes (PVCs) originating from the fascicular Purkinje system, including narrow complexes (high septal) and wide complexes (left or right bundle branch block patterns) morphologies. It is not uncommon for the patients to have more than 50.000 PVCs/24 h (5, 6). Often premature atrial complexes (PACs), AF and varying conduction abnormalities are concomitant (Figure 1). Occasionally, ventricular tachycardia (predominantly non-sustained) and associated syncope or SCD may be observed (5).
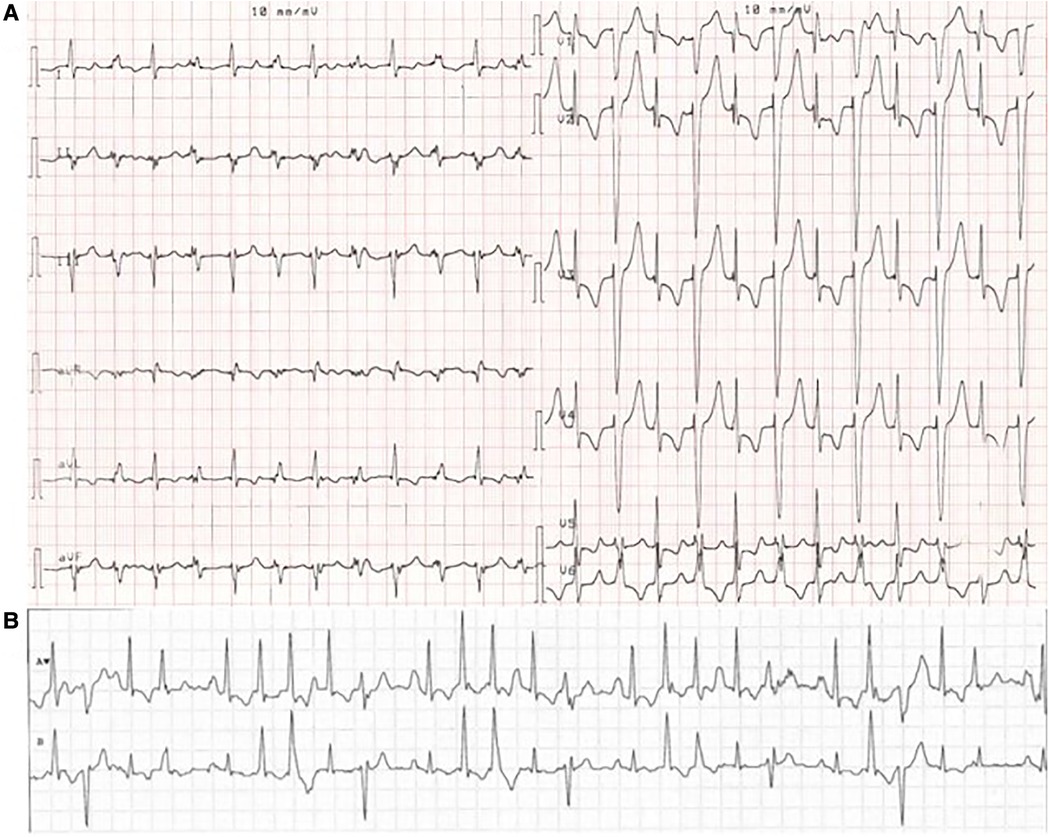
Figure 1. ECG from a MEPPC patient with the G213D variant. (A) ECG with PVCs as bigemini. (B) Telemetry of the patient showing a chaotic ECG with rare sinus beats interpolated between frequent PVCs, premature atrial complexes and premature junctional complexes. The figure is reprinted from International Journal of Cardiology, Calloe et al. (6) with permission from Elsevier.
The diagnostic criteria for MEPPC have not been defined and often MEPPC-like syndromes are reported as complex arrhythmia and DCM. A central feature of MEPPC is the presence of multiple PVCs with relatively narrow QRS complexes or typical left/right branch block patterns indicating an origin from the fascicular Purkinje system, in which case a diagnosis of MEPPC should be suspected. The diagnosis may further be supported by the presence of a positive family history of PVCs and/or sudden cardiac death and a SCN5A gain-of-function variant. In contrast to other SCN5A-associated arrhythmia phenotypes, ST-segment morphology and QT interval during sinus beats appear to be normal.
For patients with the R222Q (5) variant or one of the two later reported variants G213D (6) and Q419P (7), the number of PVCs decline during exercise, likely due to overdrive suppression. Structural changes with dilation of the atria and ventricles as well as impairment of left ventricular systolic function is frequently found (3–5, 8) and left ventricular fibrosis has been observed in MEPPC patients with left ventricular dysfunction (9, 10).
MEPPC exhibits an autosomal dominant inheritance pattern with almost complete penetrance (3, 5, 6). There is an equal distribution of male and female MEPPC patients, but DCM and reduced left ventricular ejection fraction (LVEF) appear to be more frequently in males than in females. For patients with the R222Q variant almost twice as many males as females (9 vs. 5) presented with DCM (3–5). Of the 39 genotypic positive family members, all except one had cardiac arrhythmic ectopies (3–5). Calloe and coworkers identified 15 cases with the amino acid substitution G213D. DCM was found in three genotype positive male patients and in two male family members, where MEPPC was suspected but genetic testing not possible. DCM was not found in female carriers of this variant, with exception of a 14 year-old female with atrial tachycardia and an enlarged left atrium (6).
Considering the central role of Nav1.5 in cardiac action potential generation and conduction, the association between pathogenic SCN5A variants and cardiac arrhythmia is not surprising. Variants in SCN5A is found in 1.7% of familiar cases of DCM (11) but the mechanistic link between Nav1.5 variants and DCM is still a subject of debate.
A recent systematic review by Peters and coworkers identified 18 DCM-associated Nav1.5 variants (12) and they concluded that the majority of DCM-associated variants were also associated with PVCs. Out of these 18 DCM-associated Nav1.5 variants, A204E (9), G213D (6, 13), R222Q (3–5, 14), R225P (15), Q419P (7), and L828F (16) have been reported as causing MEPPC or MEPPC-like syndromes (Tables 1, 2). M1851V (27) and I141P (26) are causing an atypical MEPPC-like phenotype where PVCs are induced by excise rather than being suppressed (Table 2). Other variants, like R814W (22–24) appear to evoke an MEPPC-like phenotype (Table 1), but are reported as causing “multifocal PVCs and DCM” in the literature.
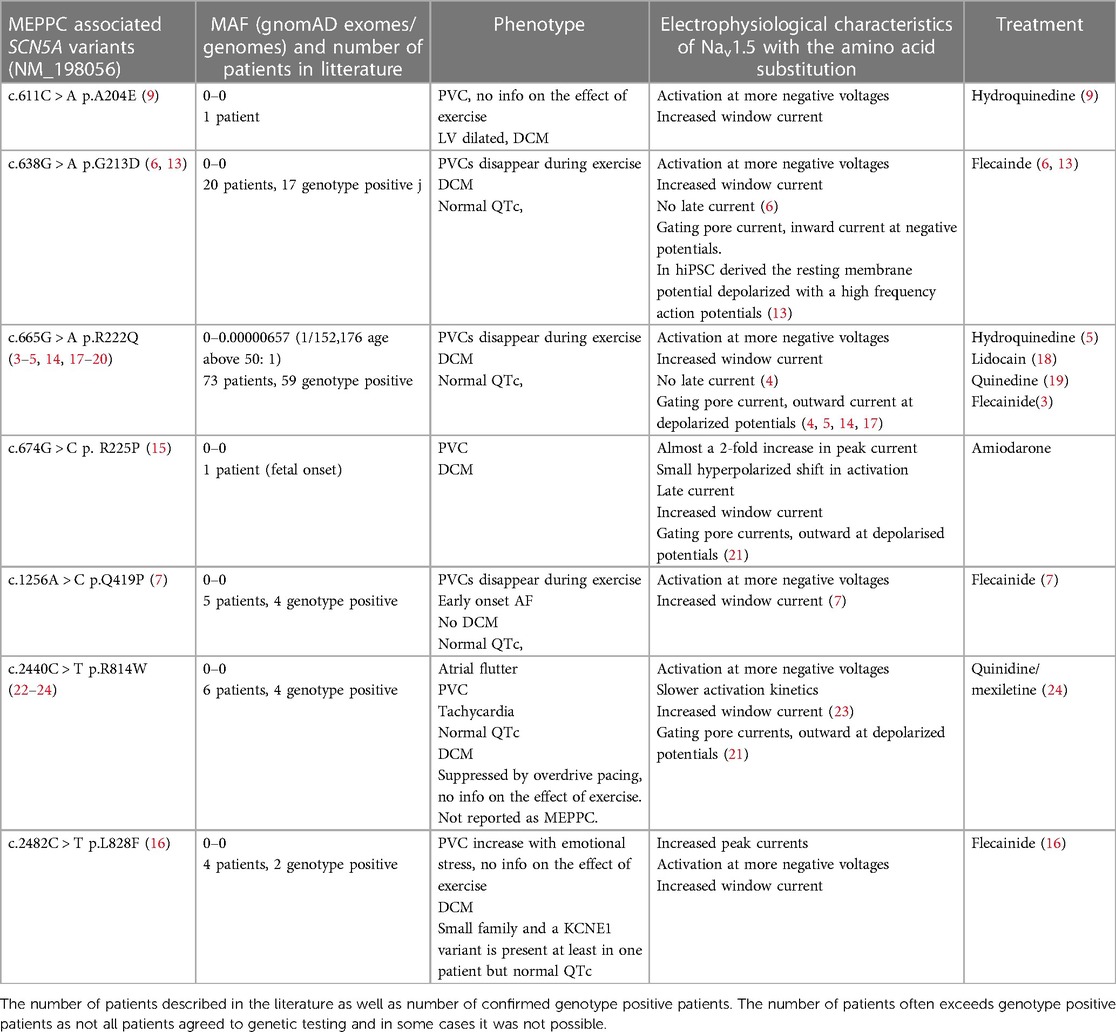
Table 1. SCN5A variants associated with MEPPC. Minor allele frequency (MAF) based on (https://gnomad.broadinstitute.org/gene/ENSG00000183873?dataset = gnomad_r2_1).
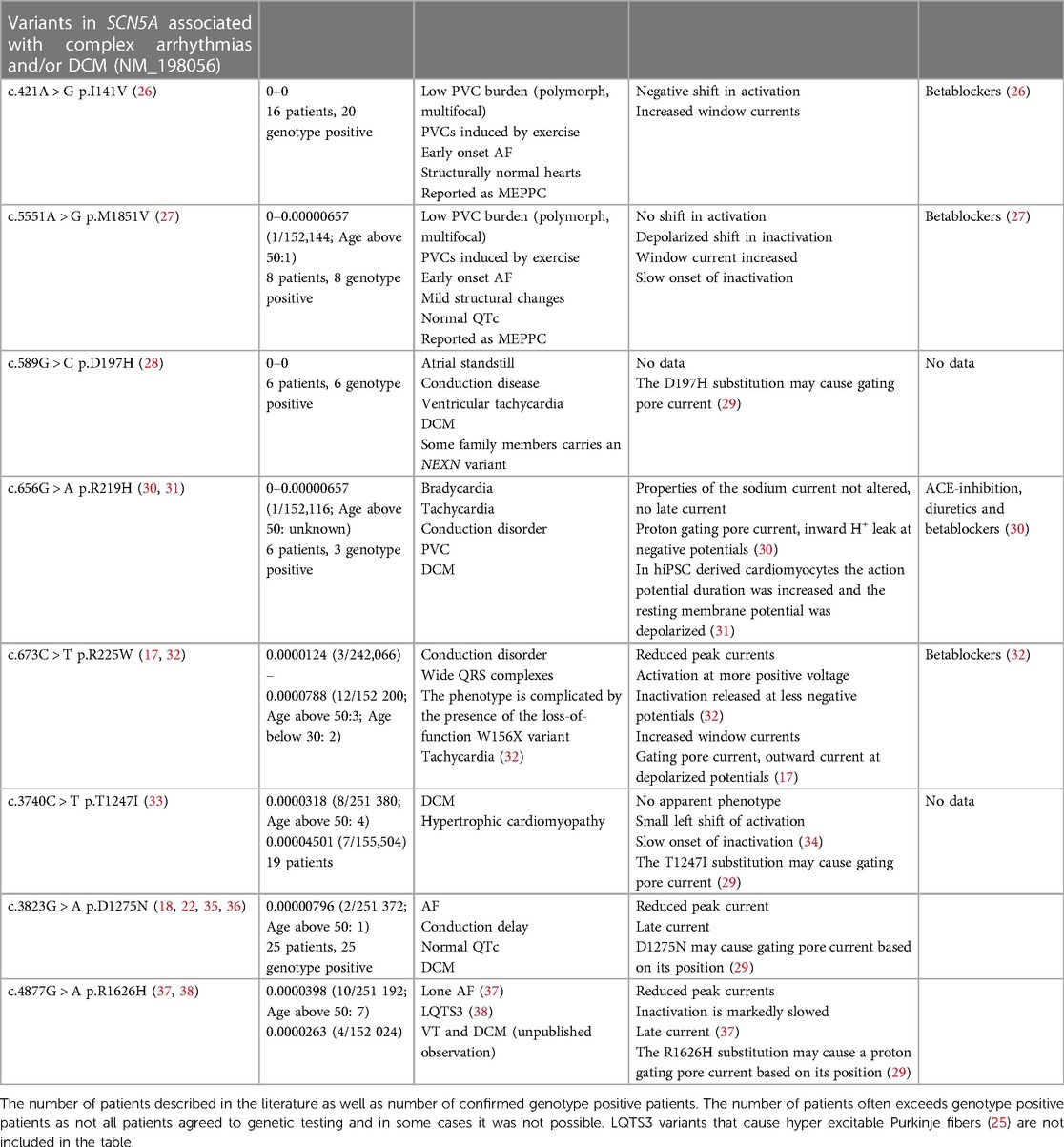
Table 2. SCN5A variants associated with complex arrhythmias and DCM. Minor allele frequency (MAF) based on (https://gnomad.broadinstitute.org/gene/ENSG00000183873?dataset = gnomad_r2_1).
Nav1.5 consists of a single protein with four homologue domains DI to DIV (Figure 2). Each of the domains contain six transmembrane segments, S1 to S6 (39, 40). S1 to S4 form the voltage sensing domain (VSD) of the protein. S5 and S6 from the four homologue domains form the central pore and the selectivity filter of the channel selectively allows passage of Na+ (39). The intracellular loop connecting DIII and DIV functions as an inactivation particle. Not surprisingly, LQTS3 variants are often found in this loop or its interaction sites on the cytoplasmic face of the channel. The S4 contains four to seven positive charges due to arginine or lysine residues. At resting membrane potentials, the positive residues are attracted to the negative cytosol. During depolarization of the membrane, the S4 segments are repelled causing a conformational change of the Nav1.5 protein resulting in opening of the activation gate of the central pore. The S1-S3 forms a canal or a “gating pore” that allows S4 to move in response to changes in membrane potential (41, 42). During gating, the S4 moves outward and rotates, while the positive charges on the S4 form ion pairs sequentially with highly conserved negative charged amino acids (aspartate and glutamate) and aromatic amino acids (tryptophan, phenylalanine and tyrosine) in S1, S2 and S3 (29, 41). This ion pairing neutralizes the positive charges on S4 during its transition through the gating pore. Remarkably, genetic variants that result in a substitution of either the positive charges in the S4 or the negative charges in S1–S3, can result in a leak current through the gating pore of the VSD, resulting in a “gating pore current” (29, 41) as illustrated in Figure 2C. Notice that the direction of the gating pore current depends on the position of the substitution (Figure 2C) and that gating pore currents can occur in either the closed or the open state of the channel (29). The gating pores are typically permeable for cations and the charge carrier as well as the direction of the current depends on the electrochemical driving force (29). Variants located in the inner region of S4 induce gating pore current in the open state (42) and the gating pore current is typically carried by Na+ and K+, which may shorten action potential duration. Variants in the outer region of S4 may cause a gating pore current in the closed state and the current is typically carried by Na+ due to electrochemical driving forces (17).
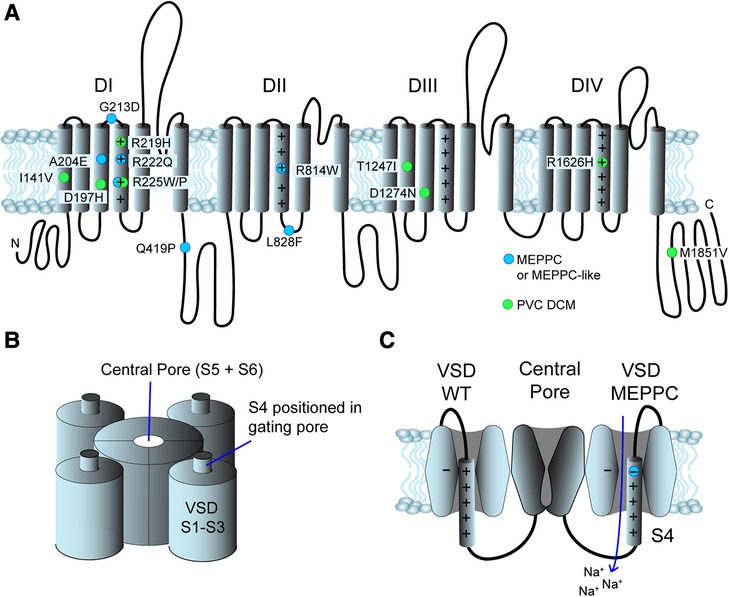
Figure 2. Structure and position of variants in the Nav1.5 channel. (A) Primary structure of the Nav1.5 channel. It consists of 4 domains (DI to DIV), each with 6 transmembrane segments (S1–S6). The positive residues in S4 are indicated by “+”. The N- and C- termini are found in the cytoplasm. Blue dots indicate the position of MEPPC variants and green dots indicate variants with complex arrhythmias and DCM. (B) The S5 and S6 from homologue domains form the central pore and the selectivity filter. S1–S4 form the voltage sensitive domains (VSD), S1–S3 form a gating pore allowing S4 to slide up and down in response to voltage changes. (C) The central pore of Nav1.5 channel in a closed state surrounded by two of the VSDs. On the left, a wild-type VSD is shown. When the S4 moves through the gating pore formed by S1 to S3, the positive charges on S4 interact with negative and aromatic amino acids (indicated by “−“). The VSD on the right has a substitution of the outermost arginine in S4 resulting in disruption of the interaction of the S4 and the gating pore. This results in an inward gating pore current, where mainly Na+ leaks in during closed state and depolarize the membrane potential.
There is a clear predilection for Nav1.5 variants associated with MEPPC and DCM to localize to the VSD and in particular to the S4 as illustrated in Figure 1 (8, 11, 12, 29). The majority of the variants result in gain-of-function of Nav1.5, due to a shift in the voltage dependence of activation causing the channel to open at more negative voltages and/or increased “window current”, which represents a steady state current due to overlap of activation and inactivation properties of Nav1.5 (1, 12). However, other variants such as R225W (17, 32) and R1626H (37, 38) that cause a loss-of-function of Nav1.5 have also been associated with complex arrhythmias and DCM. This led Moreau and coworkers to suggest that the common modality for the SCN5A variants associated with DCM is the introduction of a gating pore current in Nav1.5 (29, 43). As described above, gating pores are caused by disruption of interaction of the positive residues in the S4 with negative residues in S1–3 in the VSD, allowing ions to leak through the voltage sensitive domain. In support of this hypothesis, gating pore currents have been demonstrated for several S4 substitutions: R222Q (17), R225W (17), R225P (21), and R814W (21) have outward gating pore currents carried by Na+ and K+ at depolarized potentials where the channel is in an open state. Intriguingly, most of these variants also have increased window currents, which is a trademark of LQTS3 (1). Yet, the patients with MEPPC variants tend to have corrected QT (QTc) durations within the normal range (Table 1), suggesting that the increased window current is off-set by outward gating pore currents that will tend to shorten the QTc. For R222Q and R225W it has been demonstrated that prolonged depolarization causes the S4 to freeze in the open configuration resulting in inward gating pore currents carried by Na+ at negative potentials (17). This may explain the unstable resting membrane potential and hyper excitability that have been found in R222Q murine Purkinje fibers (14). For the G213D variant, Calloe and coworkers have recently reported inward gating pore current at negative potentials causing an unstable and depolarized resting membrane potential in cardiomyocytes derived from human induced pluripotent stem cells (13).
Most gating pore allows passage of different cations (29). Interestingly, the R219H variant that has been found in patients with PVCs and DCM (30, 31) has a H+ selective inward gating pore current at negative potentials (30). This proton gating pore may arise when an arginine is replaced by a histidine in the S4 thereby creating a proton wire (30, 31, 44). The R1626H variant has been found in patients with LQTS3 (38), lone AF (37), as well as ventricular tachycardia and DCM (own observation). The position of R1626H in the outer part of the S4 segment in DIV suggests it could induce a proton gating pore current at negative potentials similarly to R219H. Electrophysiological studies revealed a reduction in peak currents, a marked slowing of the onset of inaction and the presence of a late current (37), but the presence of gating pore currents remains to be tested. It should also be noted that the R1626H variant has been seen in the gnomAD allele frequency database at a significant number of individuals, see Table 1.
Even though the majority of DCM and MEPPC variants are found within the VSD, there are several variants found outside the VSD (Figure 2). It is conceivable that some of these regions interact with the VSD and may affect conformation changes or the electrical neutralization of the S4 during transition through the gating pore. Interestingly Barake and coworkers found that a third of LQTS3 patients also exhibited fascicular PVCs and Purkinje fiber hyper-excitability, which led them to suggest that the present of a persistent Na+ current due to a large window current can result in an MEPPC-like phenotype overlapping with LQTS3 (25). This suggest that increased persistent Na+ current, either through a gating pore or due to increased window currents could be the common modality in MEPPC.
It should be noted that gating pore currents are often undetected in routine patch clamp experiments as the gating pore currents are very small and resembles leak currents. It is critical that protocols optimized to detect gating pore currents are applied in functional characterization of SCN5A variants associated with complex arrhythmias and DCM.
Whereas most genetic variants associated with hereditary DCM cause a change in structural genes (45), it is a subject of debate whether SCN5A is truly a DCM gene or the DCM is secondary to the arrhythmic burden. Peters and coworkers concluded that in all patients genotype-positive for an SCN5A variant, the DCM was secondary to the arrhythmic burden and no cases of DCM in the absence of arrhythmias were found, supporting that the arrhythmias are the defining feature of SCN5A variants associated with DCM (12).
Baman and coworkers found that a 24% PVC burden best separated the patient population with impaired left ventricular ejection fraction from patients with a preserved ejection fraction but a PVC burden as low as 10% may cause a reversible cardiomyopathy (46–48). This implicates that the PVCs are responsible for the MEPPC-associated DCM, even though the correlation between PVC burden and reduced LVEF is not linear and males appear to be at higher risk than females (3, 5, 6, 14).
The MEPPC-associated SCN5A variants typically result in a gain-of-function of Nav1.5 and a shift of the voltage dependence of activation making the cells more excitable (5, 13, 15). Increased sodium current either through the central pore or through a gating pore may directly depolarize and destabilize the resting membrane potential as well as lead to a cellular Na+ overload, which can activate the sodium calcium exchanger in reverse-mode, cause early after depolarization (14) and cellular Ca2+ overload (4, 8, 29). Ca2+ overload decreases Ca2+ sensitivity of the sarcomeres (49, 50) which may contribute to the development of cardiomyopathy. The Na+ overload may also predisposes to osmotic swelling and mitochondrial dysfunction (51) as well as activation of the sodium proton exchanger leading to acidification and uncoupling of gap junctions (52). Further, the premature contractions may lead to activation of baroreceptors and thereby increase sympathetic activity and cause myocardial stress (53).
Current guidelines recommend catheter ablation in patients with DCM suspected to be caused by frequent and predominately monomorphic PVCs (54). However, ablation therapy has limited efficacy in eradiating MEPPC-associated PVCs (5, 9) as the whole Purkinje system is involved in triggering the ectopic contractions (5). Treatment with standard heart failure medical therapy including betablockers, ACE inhibitors and diuretics also has modest or no effect on MEPPC-associated DCM (3).
Several studies have reported that class 1c antiarrhythmic drugs, most notably flecainide (7, 13, 55) and quinidine are highly efficacious (5, 24). Flecainide and quinidine suppress PVCs and in longitudinal studies of the effect of flecainide treatment in MEPPC patients with the G213D (13) or R222Q (55) variant it was found that the reduction of the ectopic burden was associated with a reversibility of DCM. The reversible nature of DCM does not indicate that it is a benign cardiomyopathy (12). If not properly managed, the patients risk severe heart failure and sudden cardiac death (5, 12, 24). The presence of DCM in MEPPC patients is also a risk factor for malignant ventricular arrhythmias in itself (12), which underscores the importance of proper treatment and genetic testing including the SCN5A gene should be considered in young patients with a high PVC burden of multifocal origin and/or left bundle branch block morphologies.
Laurent and coworkers reported that cardiac resynchronization therapy (CRT) and amiodarone introduced simultaneously led to a partial normalization of left ventricular function in patients with the R222Q variant (5) but experience with CRT in MEPPC patients with left bundle branch block and heart failure is sparse. One report showed that atrial pacing at 90 beats per minute in combination with mexiletine in a patient carrying the R814W variant was effective in PVC suppression (24). Thus, it is possible that atrial overdrive pacing may also be an effective treatment strategy in patients with a high PVC burden despite medical antiarrhythmic treatment.
Given the rarity of the disease there is a need to establish a registry of SCN5A variants associated with MEPPC and DCM, in order to improve knowledge on clinical symptoms and pharmaceutical management.
SCN5A genetic variants are associated with several cardiac arrhythmia syndromes including MEPPC as well as overlap syndromes. MEPPC is a relatively novel and ultrarare disease entity characterized by multifocal premature complexes originating from the Purkinje system and associated DCM. Increased Na+ current, either through a gating pore or due to increased window currents could be the common modality in MEPPC. MEPPC is important to diagnose as treatment with class 1c agents prevent morbidity and mortality, whereas standard treatment of PVCs and DCM with catheter ablation and heart failure medication is ineffective. Failure to detect MEPPC often results in inappropriate managing with potentially severe consequences for the patient and relatives.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by the Novo Nordisk Foundation (NNF21OC0071188) to KC and Novo Nordisk Foundation (NNF18OC0031258) to HJ.
HJ is member of the European Reference Network for rare, low prevalence, and complex diseases of the heart (Expertise Reference Network GUARD-Heart).
MC received lecture fees from Amgen. HJ received lecture fees from Abbott Denmark, Amgen Denmark and Biosense Webster, Europe.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wilde AAM, Amin AS. Clinical spectrum of SCN5A mutations. JACC Clin Electrophysiol. (2018) 4:569–79. doi: 10.1016/j.jacep.2018.03.006
2. Bezzina C, Veldkamp MW, van den Berg MP, Postma AV, Rook MB, Viersma J-W, et al. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ Res. (1999) 85:1206–13. doi: 10.1161/01.RES.85.12.1206
3. Mann SA, Castro ML, Ohanian M, Guo G, Zodgekar P, Sheu A, et al. R222q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J Am Coll Cardiol. (2012) 60:1566–73. doi: 10.1016/j.jacc.2012.05.050
4. Nair K, Pekhletski R, Harris L, Care M, Morel C, Farid T, et al. Escape capture bigeminy: phenotypic marker of cardiac sodium channel voltage sensor mutation R222Q. Heart Rhythm Off J Heart Rhythm Soc. (2012) 9:1681–1688.e1. doi: 10.1016/j.hrthm.2012.06.029
5. Laurent G, Saal S, Amarouch MY, Béziau DM, Marsman RFJ, Faivre L, et al. Multifocal ectopic Purkinje-related premature contractions: a new SCN5A-related cardiac channelopathy. J Am Coll Cardiol. (2012) 60:144–56. doi: 10.1016/j.jacc.2012.02.052
6. Calloe K, Broendberg AK, Christensen AH, Pedersen LN, Olesen MS, de Los Angeles Tejada M, et al. Multifocal atrial and ventricular premature contractions with an increased risk of dilated cardiomyopathy caused by a Nav1.5 gain-of-function mutation (G213D). Int J Cardiol. (2018) 257:160–7. doi: 10.1016/j.ijcard.2017.11.095
7. Gao X, Ye D, Zhou W, Tester DJ, Ackerman MJ, Giudicessi JR. A novel functional variant residing outside the SCN5A-encoded Nav1.5 voltage-sensing domain causes multifocal ectopic Purkinje-related premature contractions. Hear Case Rep. (2022) 8:54–9. doi: 10.1016/j.hrcr.2021.11.002
8. Gosselin-Badaroudine P, Moreau A, Chahine M. Nav1.5 mutations linked to dilated cardiomyopathy phenotypes. Channels. (2014) 8:90–4. doi: 10.4161/chan.27179
9. Doisne N, Waldmann V, Redheuil A, Waintraub X, Fressart V, Ader F, et al. A novel gain-of-function mutation in SCN5A responsible for multifocal ectopic Purkinje-related premature contractions. Hum Mutat. (2020) 41:850–9. doi: 10.1002/humu.23981
10. Leventopoulos G, Perperis A, Karelas D, Almpanis G. You cannot ablate the lernaean hydra: SCN5A mutation in a patient with multifocal ectopic purkinje-related premature contractions syndrome treated with flecainide and an implant of a subcutaneous defibrillator—a case report. Eur Heart J Case Rep. (2021) 5:ytab158. doi: 10.1093/ehjcr/ytab158
11. McNair WP, Sinagra G, Taylor MRG, Di Lenarda A, Ferguson DA, Salcedo EE, et al. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol. (2011) 57:2160–8. doi: 10.1016/j.jacc.2010.09.084
12. Peters S, Thompson BA, Perrin M, James P, Zentner D, Kalman JM, et al. Arrhythmic phenotypes are a defining feature of dilated cardiomyopathy-associated SCN5A variants: a systematic review. Circ Genomic Precis Med. (2022) 15:e003432. doi: 10.1161/CIRCGEN.121.003432
13. Calloe K, Geryk M, Freude K, Treat JA, Vold VA, Frederiksen HRS, et al. The G213D variant in Nav1.5 alters sodium current and causes an arrhythmogenic phenotype resulting in a multifocal ectopic Purkinje-related premature contraction phenotype in human-induced pluripotent stem cell-derived cardiomyocytes. Eur Eur Pacing Arrhythm Card Electrophysiol J Work Groups Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. (2022) 24:2015–27. doi: 10.1093/europace/euac090
14. Daniel LL. SCN5A variant R222Q generated abnormal changes in cardiac sodium current and action potentials in murine myocytes and Purkinje cells. Heart Rhythm. (2019) 16:1676–85. doi: 10.1016/j.hrthm.2019.05.017
15. Beckermann TM, McLeod K, Murday V, Potet F, George AL. Novel SCN5A mutation in amiodarone-responsive multifocal ventricular ectopy-associated cardiomyopathy. Heart Rhythm. (2014) 11:1446–53. doi: 10.1016/j.hrthm.2014.04.042
16. Bekke Rt, David M, Krapels IPC, Crijns HJGM, Volders PGA. Beauty and the beat: a complicated case of multifocal ectopic Purkinje-related premature contractions. Hear Case Rep. (2018) 4:429–33. doi: 10.1016/j.hrcr.2018.05.010
17. Moreau A, Gosselin-Badaroudine P, Delemotte L, Klein ML, Chahine M. Gating pore currents are defects in common with two Nav1.5 mutations in patients with mixed arrhythmias and dilated cardiomyopathy. J Gen Physiol. (2015) 145:93–106. doi: 10.1085/jgp.201411304
18. McNair WP, Ku L, Taylor MRG, Fain PR, Dao D, Wolfel E, et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. (2004) 110:2163–7. doi: 10.1161/01.CIR.0000144458.58660.BB
19. Zakrzewska-Koperska J, Franaszczyk M, Bilińska Z, Truszkowska G, Karczmarz M, Szumowski Ł, et al. Rapid and effective response of the R222Q SCN5A to quinidine treatment in a patient with Purkinje-related ventricular arrhythmia and familial dilated cardiomyopathy: a case report. BMC Med Genet. (2018) 19:94. doi: 10.1186/s12881-018-0599-4
20. Cheng J, Morales A, Siegfried JD, Li D, Norton N, Song J, et al. SCN5A rare variants in familial dilated cardiomyopathy decrease peak sodium current depending on the common polymorphism H558R and common splice variant Q1077del. Clin Transl Sci. (2010) 3:287–94. doi: 10.1111/j.1752-8062.2010.00249.x
21. Moreau A, Gosselin-Badaroudine P, Boutjdir M, Chahine M. Mutations in the voltage sensors of domains I and II of Nav1.5 that are associated with arrhythmias and dilated cardiomyopathy generate gating pore currents. Front Pharmacol. (2015) 6:301. doi: 10.3389/fphar.2015.00301
22. Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. (2005) 293:447–54. doi: 10.1001/jama.293.4.447
23. Nguyen TP, Wang DW, Rhodes TH, George AL. Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circ Res. (2008) 102:364–71. doi: 10.1161/CIRCRESAHA.107.164673
24. Zakrzewska-Koperska J, Bilińska ZT, Truszkowska GT, Franaszczyk M, Elikowski W, Warmiński G, et al. A combination of quinidine/mexiletine reduces arrhythmia in dilated cardiomyopathy in two patients with R814W SCN5A mutation. ESC Heart Fail. (2020) 7:4326–35. doi: 10.1002/ehf2.12993
25. Barake W, Giudicessi JR, Asirvatham SJ, Ackerman MJ. Purkinje system hyperexcitability and ventricular arrhythmia risk in type 3 long QT syndrome. Heart Rhythm. (2020) 17:1768–76. doi: 10.1016/j.hrthm.2020.05.019
26. Swan H, Amarouch MY, Leinonen J, Marjamaa A, Kucera JP, Laitinen-Forsblom PJ, et al. Gain-of-function mutation of the SCN5A gene causes exercise-induced polymorphic ventricular arrhythmias. Circ Cardiovasc Genet. (2014) 7:771–81. doi: 10.1161/CIRCGENETICS.114.000703
27. Lieve KV, Verkerk AO, Podliesna S, van der Werf C, Tanck MW, Hofman N, et al. Gain-of-function mutation in SCN5A causes ventricular arrhythmias and early onset atrial fibrillation. Int J Cardiol. (2017) 236:187–93. doi: 10.1016/j.ijcard.2017.01.113
28. Kean AC, Helm BM, Vatta M, Ayers MD, Parent JJ, Darragh RK. Clinical characterisation of a novel SCN5A variant associated with progressive malignant arrhythmia and dilated cardiomyopathy. Cardiol Young. (2019) 29:1257–63. doi: 10.1017/S1047951119001860
29. Moreau A, Gosselin-Badaroudine P, Chahine M. Biophysics, pathophysiology, and pharmacology of ion channel gating pores. Front Pharmacol. (2014) 5:53. doi: 10.3389/fphar.2014.00053
30. Gosselin-Badaroudine P, Keller DI, Huang H, Pouliot V, Chatelier A, Osswald S, et al. A proton leak current through the cardiac sodium channel is linked to mixed arrhythmia and the dilated cardiomyopathy phenotype. PLoS One. (2012) 7:e38331. doi: 10.1371/journal.pone.0038331
31. Moreau A, Gosselin-Badaroudine P, Mercier A, Burger B, Keller DI, Chahine M. A leaky voltage sensor domain of cardiac sodium channels causes arrhythmias associated with dilated cardiomyopathy. Sci Rep. (2018) 8:13804. doi: 10.1038/s41598-018-31772-0
32. Bezzina CR, Rook MB, Groenewegen WA, Herfst LJ, van der Wal AC, Lam J, et al. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ Res. (2003) 92:159–68. doi: 10.1161/01.RES.0000052672.97759.36
33. Priganc M, Zigová M, Boroňová I, Bernasovská J, Dojčáková D, Szabadosová V, et al. Analysis of SCN5A gene variants in east Slovak patients with cardiomyopathy. J Clin Lab Anal. (2016) 31:e22037. doi: 10.1002/jcla.22037
34. Ishikawa T, Kimoto H, Mishima H, Yamagata K, Ogata S, Aizawa Y, et al. Functionally validated SCN5A variants allow interpretation of pathogenicity and prediction of lethal events in Brugada syndrome. Eur Heart J. (2021) 42:2854–63. doi: 10.1093/eurheartj/ehab254
35. Hayano M, Makiyama T, Kamakura T, Watanabe H, Sasaki K, Funakoshi S, et al. Development of a patient-derived induced pluripotent stem cell model for the investigation of SCN5A-D1275N-related cardiac sodium channelopathy. Circ J Off J Jpn Circ Soc. (2017) 81:1783–91. doi: 10.1253/circj.CJ-17-0064
36. Watanabe H, Yang T, Stroud DM, Lowe JS, Harris L, Atack TC, et al. Striking in vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation. (2011) 124:1001–11. doi: 10.1161/CIRCULATIONAHA.110.987248
37. Olesen MS, Yuan L, Liang B, Holst AG, Nielsen N, Nielsen JB, et al. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ Cardiovasc Genet. (2012) 5:450–9. doi: 10.1161/CIRCGENETICS.111.962597
38. Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION® long QT syndrome genetic test. Heart Rhythm. (2009) 6:1297–303. doi: 10.1016/j.hrthm.2009.05.021
39. Jiang D, Shi H, Tonggu L, Gamal El-Din TM, Lenaeus MJ, Zhao Y, et al. Structure of the cardiac sodium channel. Cell. (2020) 180:122–134.e10. doi: 10.1016/j.cell.2019.11.041
40. Wisedchaisri G, Tonggu L, McCord E, Gamal El-Din TM, Wang L, Zheng N, et al. Resting-state structure and gating mechanism of a voltage-gated sodium channel. Cell. (2019) 178:993–1003.e12. doi: 10.1016/j.cell.2019.06.031
41. Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. (2010) 67:915–28. doi: 10.1016/j.neuron.2010.08.021
42. Jiang D, Gamal El-Din TM, Ing C, Lu P, Pomès R, Zheng N, et al. Structural basis for gating pore current in periodic paralysis. Nature. (2018) 557:590–4. doi: 10.1038/s41586-018-0120-4
43. Moreau A, Gosselin-Badaroudine P, Chahine M. Gating pore currents, a new pathological mechanism underlying cardiac arrhythmias associated with dilated cardiomyopathy. Channels. (2015) 9:139–44. doi: 10.1080/19336950.2015.1031937
44. Starace DM, Bezanilla F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature. (2004) 427:548–53. doi: 10.1038/nature02270
45. Rosenbaum AN, Agre KE, Pereira NL. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat Rev Cardiol. (2020) 17:286–97. doi: 10.1038/s41569-019-0284-0
46. Baman TS, Lange DC, Ilg KJ, Gupta SK, Liu T-Y, Alguire C, et al. Relationship between burden of premature ventricular complexes and left ventricular function. Heart Rhythm. (2010) 7:865–9. doi: 10.1016/j.hrthm.2010.03.036
47. Penela D, Acosta J, Aguinaga L, Tercedor L, Ordoñez A, Fernández-Armenta J, et al. Ablation of frequent PVC in patients meeting criteria for primary prevention ICD implant: safety of withholding the implant. Heart Rhythm. (2015) 12:2434–42. doi: 10.1016/j.hrthm.2015.09.011
48. Penela D, Van Huls Van Taxis C, Van Huls Vans Taxis C, Aguinaga L, Fernández-Armenta J, Mont L, et al. Neurohormonal, structural, and functional recovery pattern after premature ventricular complex ablation is independent of structural heart disease status in patients with depressed left ventricular ejection fraction: a prospective multicenter study. J Am Coll Cardiol. (2013) 62:1195–202. doi: 10.1016/j.jacc.2013.06.012
49. Liu B, Tikunova SB, Kline KP, Siddiqui JK, Davis JP. Disease-related cardiac troponins alter thin filament Ca2+ association and dissociation rates. PloS One. (2012) 7:e38259. doi: 10.1371/journal.pone.0038259
50. Clanachan AS. Contribution of protons to post-ischemic Na+ and Ca2+ overload and left ventricular mechanical dysfunction. J Cardiovasc Electrophysiol. (2006) 17:S141–8. doi: 10.1111/j.1540-8167.2006.00395.x
51. Iwai T, Tanonaka K, Inoue R, Kasahara S, Kamo N, Takeo S. Mitochondrial damage during ischemia determines post-ischemic contractile dysfunction in perfused rat heart. J Mol Cell Cardiol. (2002) 34:725–38. doi: 10.1006/jmcc.2002.2002
52. Nielsen MS, Axelsen LN, Sorgen PL, Verma V, Delmar M, Holstein-Rathlou N-H. Gap junctions. Compr Physiol. (2012) 2:1981–2035. doi: 10.1002/cphy.c110051
53. Grassi G, Seravalle G, Bertinieri G, Mancia G. Behaviour of the adrenergic cardiovascular drive in atrial fibrillation and cardiac arrhythmias. Acta Physiol Scand. (2003) 177:399–404. doi: 10.1046/j.1365-201X.2003.01092.x
54. Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel BG, Behr ER, Blom NA, et al. 2022 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. (2022) 43:3997–4126. doi: 10.1093/eurheartj/ehac262
Keywords: dilated cardiomyopathy, premature ventricular contractions, MEPPC, SCN5A, arrhythmia
Citation: Calloe K, Magnusson HBD, Lildballe DL, Christiansen MK and Jensen HK (2023) Multifocal ectopic purkinje-related premature contractions and related cardiomyopathy. Front. Cardiovasc. Med. 10:1179018. doi: 10.3389/fcvm.2023.1179018
Received: 3 March 2023; Accepted: 24 July 2023;
Published: 4 August 2023.
Edited by:
Ahran Arnold, Imperial College London, United KingdomReviewed by:
Elena V. Zaklyazminskaya, Russian National Research Center of Surgery named after B.V. Petrovsky, Russia© 2023 Calloe, Magnusson, Lildballe, Christiansen and Jensen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kirstine Calloe a2lyc3RpbmVjQHN1bmQua3UuZGs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.