 Nazia Hilal
Nazia Hilal Zi Chen
Zi Chen Ming Hui Chen2,5
Ming Hui Chen2,5 Sangita Choudhury
Sangita Choudhury
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 17 July 2023
Sec. Cardiovascular Genetics and Systems Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1176828
This article is part of the Research TopicMolecular Pathogenesis and Novel Treatments for Inherited CardiomyopathiesView all 11 articles
As binary switches, RAS proteins switch to an ON/OFF state during signaling and are on a leash under normal conditions. However, in RAS-related diseases such as cancer and RASopathies, mutations in the genes that regulate RAS signaling or the RAS itself permanently activate the RAS protein. The structural basis of this switch is well understood; however, the exact mechanisms by which RAS proteins are regulated are less clear. RAS/MAPK syndromes are multisystem developmental disorders caused by germline mutations in genes associated with the RAS/mitogen-activated protein kinase pathway, impacting 1 in 1,000–2,500 children. These include a variety of disorders such as Noonan syndrome (NS) and NS-related disorders (NSRD), such as cardio facio cutaneous (CFC) syndrome, Costello syndrome (CS), and NS with multiple lentigines (NSML, also known as LEOPARD syndrome). A frequent manifestation of cardiomyopathy (CM) and hypertrophic cardiomyopathy associated with RASopathies suggest that RASopathies could be a potential causative factor for CM. However, the current supporting evidence is sporadic and unclear. RASopathy-patients also display a broad spectrum of congenital heart disease (CHD). More than 15 genes encode components of the RAS/MAPK signaling pathway that are essential for the cell cycle and play regulatory roles in proliferation, differentiation, growth, and metabolism. These genes are linked to the molecular genetic pathogenesis of these syndromes. However, genetic heterogeneity for a given syndrome on the one hand and alleles for multiple syndromes on the other make classification difficult in diagnosing RAS/MAPK-related diseases. Although there is some genetic homogeneity in most RASopathies, several RASopathies are allelic diseases. This allelism points to the role of critical signaling nodes and sheds light on the overlap between these related syndromes. Even though considerable progress has been made in understanding the pathophysiology of RASopathy with the identification of causal mutations and the functional analysis of their pathophysiological consequences, there are still unidentified causal genes for many patients diagnosed with RASopathies.
Rat Sarcoma Virus, a highly conserved protein, belongs to a class of proteins called small GTPase. The three most widely studied RAS genes in humans are HRAS, KRAS, and NRAS, named after the Harvey Rat sarcoma virus, Kirsten Rat sarcoma virus, and NRAS, for its initial identification in neuroblastoma cells. Since the identification of the RAS protein in 1982, extensive studies have been conducted to identify the RAS-associated pathway and its involvement in human disease. RASopathies refer to multisystem disorders caused by gene mutations that belong to the RAS/MAPK (Mitogen-activated protein kinase) signaling pathway. RAS can be either “switched on” or activated by incoming signals through growth factors binding to receptor tyrosine kinases (RTKs), G-protein-coupled receptors, cytokine receptors, and extracellular matrix receptors, or activated by mutations in RAS genes, which can lead to the production of permanently activated RAS proteins and can cause unintended and overactive signaling inside the cell, even in the absence of incoming signals. The mitogen-activated protein kinase (MAPK) pathway is one of RAS's critical downstream signaling cascades. Activated RAS leads to the phosphorylation of Raf, leading to the activation of the MAPK kinases MEK1 and/or MEK2; these, in turn, phosphorylate and activate ERK1 and/or ERK2. ERK1 and ERK2 are the ultimate effectors which exert their function on many downstream molecules in the cytoplasm and nucleus (Figure 1). RASopathy disorders include wide range of disorders such as neurofibromatosis type 1, Noonan syndrome, Noonan syndrome with multiple lentigines, Costello syndrome, cardio-facio-cutaneous syndrome, and Legius syndrome (1, 2), exhibiting multi-organ dysfunction, including craniofacial dysmorphology, cardiac malformation, cutaneous, musculoskeletal, and ocular abnormalities, neurocognitive impairment, hypotonia and increased cancer risk (1–3). In Table 1, we summarize the critical and cardiac-specific features as well as all other RASopathy-associated malformations. This review will discuss only the cardiac manifestation in RASopathies associated with Noonan syndrome and Neurofibromatosis type 1 (NF1). RASopathy-related heart defects include congenital heart disease (CHD), hypertrophic cardiomyopathy (HCM) as well as dilated cardiomyopathy (DCM).
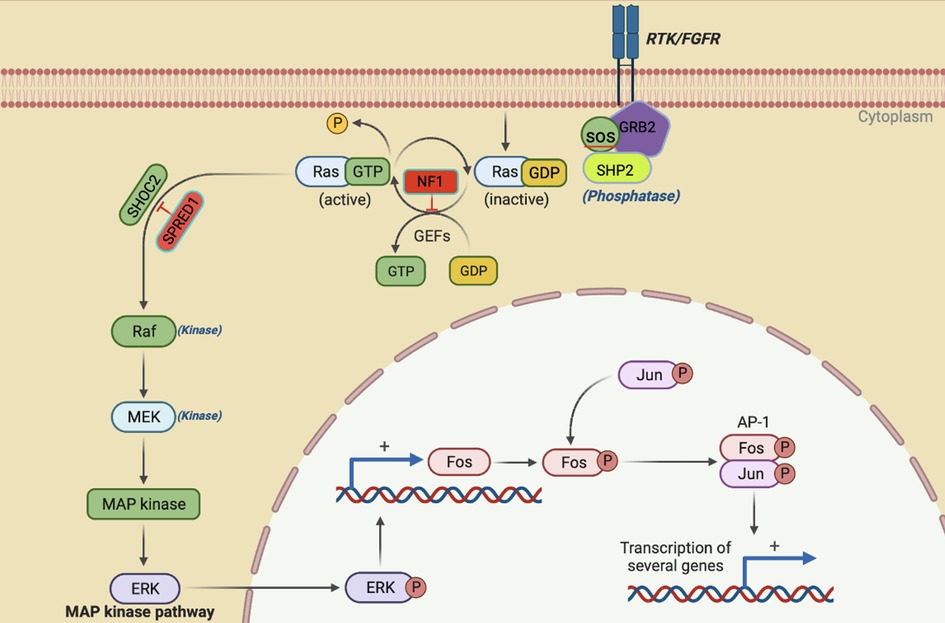
Figure 1. The activation of RAS/ERK occurs when cell survival signals bind to receptor tyrosine kinases (RTK). Once the RTK intracellular domain is phosphorylated following this binding, it triggers a sequence of events that ultimately results in the activation of RAS. NF1 and SPRED 1 act as negative effectors of the pathway. The activation of RAS recruits and activates RAF, which is the first MAPK in this pathway. Then, RAF phosphorylates and activates MEK1/2, which finally activates ERK1/2 through dual phosphorylation on tyrosine and threonine. ERK1/2 then goes on to activate various substrates downstream like FOS and JUN that ultimately leads to transcriptional activation of genes involved in cell proliferation and survival.
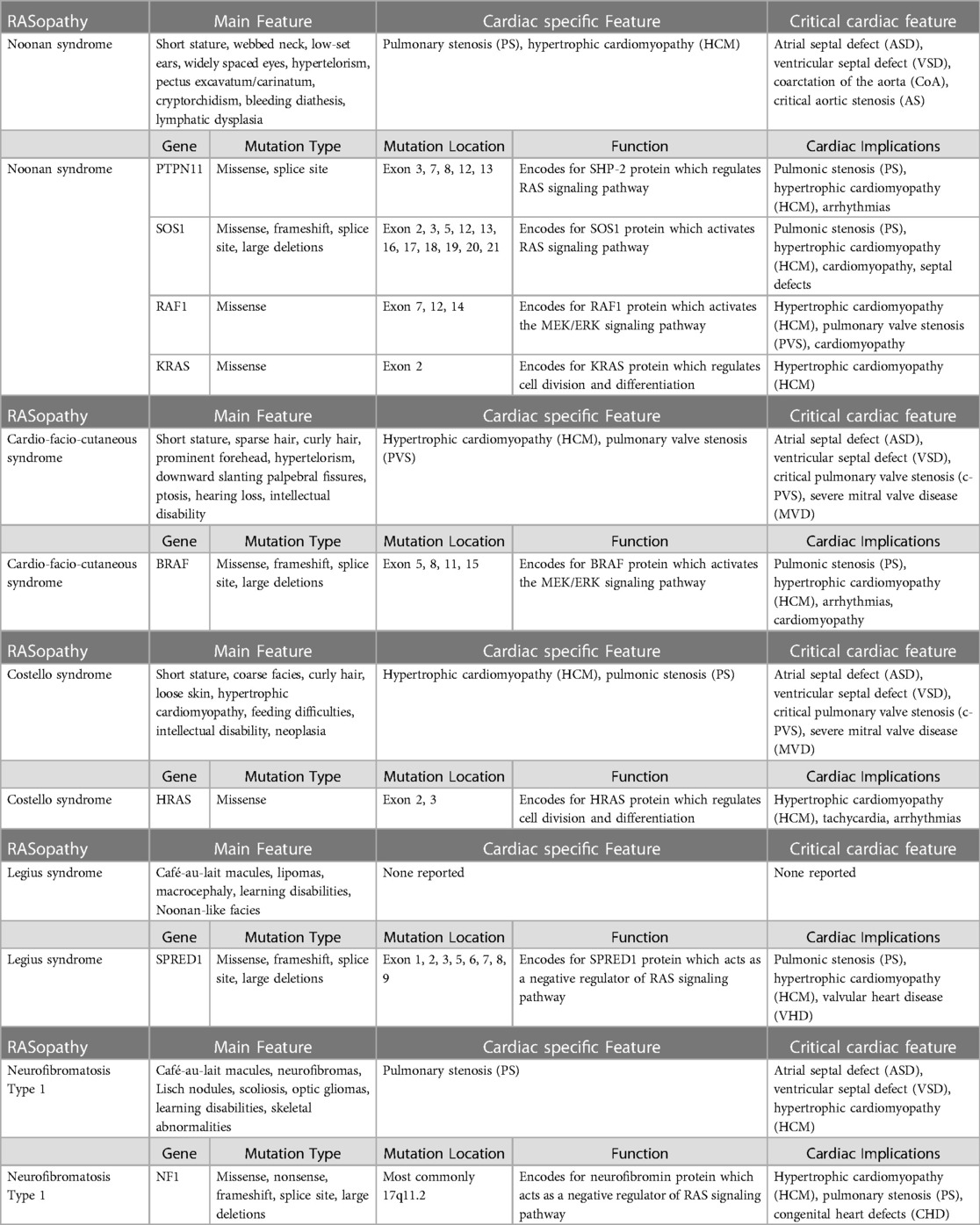
Table 1. Different features of RASopathies, including main features, cardiac-specific features, and critical cardiac features.
Noonan syndrome (NS1, OMIM 163950), caused by mutation and activation of the genes involved in the RAS-MAPK pathway, including PTPN11, SOS1, KRAS, NRAS, RAF1, BRAF, RIT1, and LZTR1, is a common developmental disorder with an autosomal dominant inheritance. The incidence is 1:1,000–2,500 live births. Many patients with NS1 indicate cardiovascular abnormalities, most commonly in the form of congenital heart diseases, such as pulmonary valve stenosis, septal defects, left-sided lesions, and complex forms with multiple anomalies. The most common congenital heart disease (CHD) involves pulmonary valve stenosis in 50%–60% of patients, and a small portion (6%–10%) indicates an atrial septal defect. The other CHDs, such as ventricular septal defect, atrioventricular canal defect, and aortic coarctation, are observed less frequently in NS1 patients (4–7). The second most prevalent cardiovascular anomaly associated with NS1 is HCM, present in approximately 20% of patients. Although NS1 is clinically heterogeneous and can manifest at any age, 80% of NS-1 HCM diagnoses are made early in infancy, and compared to non-syndromic types of HCM, NS1-HCM patients have a greater degree of ventricular hypertrophy, a higher prevalence, and a more severe pattern of left ventricular outflow tract obstruction (LVOTO). A literature survey indicates that a patient's likelihood of NS1-HCM varies significantly according to the gene mutated in the RAS-MAPK pathway. A few studies suggest an association between DCM and NS1 to some extent, where histology/echocardiography showed hypertrophy of myocardial fibers with focal interstitial fibrosis with no evidence of myocardial disarray. The features were consistent with DCM (8–11).
Noonan syndrome with multiple lentigines (NSML), which is also known as LEOPARD syndrome, has the cardiac manifestation of pulmonary valve stenosis and hypertrophic cardiomyopathy along with brown spots on the skin called lentigines, caused by the mutation in one of four genes: BRAF, MAP2K1, PTPN11, and RAF1 (12–14).
PTPN11 was found to be the most studied gene in NS populations (29 studies vs. 16 studies or fewer for other genes). Possible reasons include that PTPN11 was the first gene of the RAS/MAPK pathway to be implicated in NS in 2001, while KRAS was discovered only five years later, followed by SHP-2. Although KRAS was already involved in malignancy disorders through various somatic mutations, its interrelation with NS was found via germline mutations in 2006 (15). Subsequently, in 2007, SOS1, RAF1, and MAP2K1 genes were found to be implicated in NS (16), after which BRAF (12, 16), NRAS (17), and RIT1 (18) (RAS/MAPK kinase genes) were shown to be involved. Notably, the chronology of discovering the involved genes does not correlate with their frequency or the intensity of the phenotypic manifestations but is merely incidental. The most common gene implicated in the causation of NS is still PTPN11 (60%), constituting 52.6% of all mutations detected in Noonan patients to date (Figure 2). The second most found mutated gene in NS is SOS1(16.4% of patients). Furthermore, RIT1 and RAF1 have been found to have a prevalence of 8%, making them the third most-involved genes. Therefore, mutations in PTPN11, SOS1, RAF1, and RIT1 alone comprise 93% of the mutations causing NS. Hence, these genes are included in the first line of genetic screening in patients with the NS phenotype. Table 2 summarizes the genes and domains involved in RASopathies. KRAS (2.8%) and NRAS (0.8%) have the lowest incidence among all reported cases of Noonan syndrome caused by the RAS subfamily of genes involved in the RAS/MAPK pathway, in contrast with the RIT1 gene in the same family. Similarly, BRAF constitutes 2.3% among RAF family members compared to the more prevalent RAF1. Table 1 emphasizes the genes involved in different RASopathies and their normal function, mutation type, mutation location, and cardiac implications.
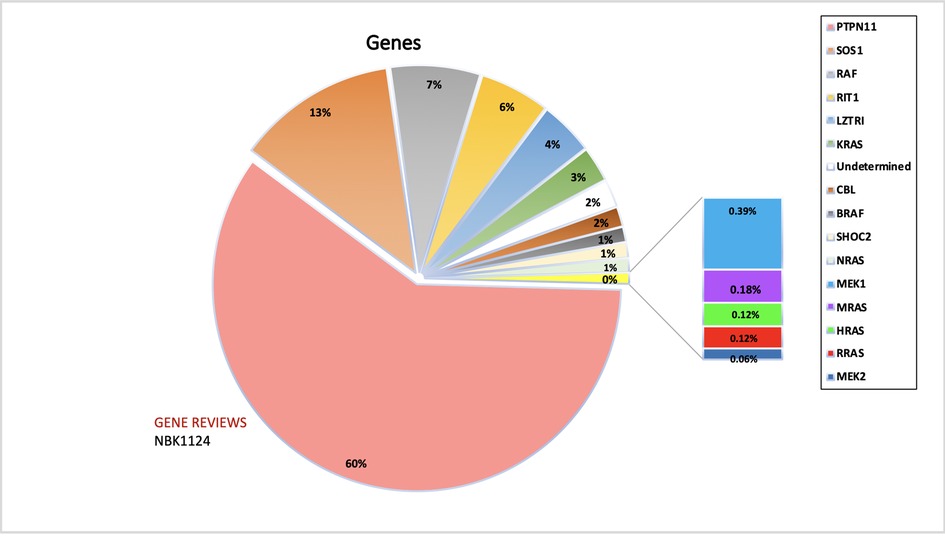
Figure 2. The figure represents the information of the genes implicated in NS and associated RASopathies. PTPN11, SOS1 and RAF alone makes up for more than 90% of the total pathogenic mutations. The data is obtained from the NSEuroNet database.
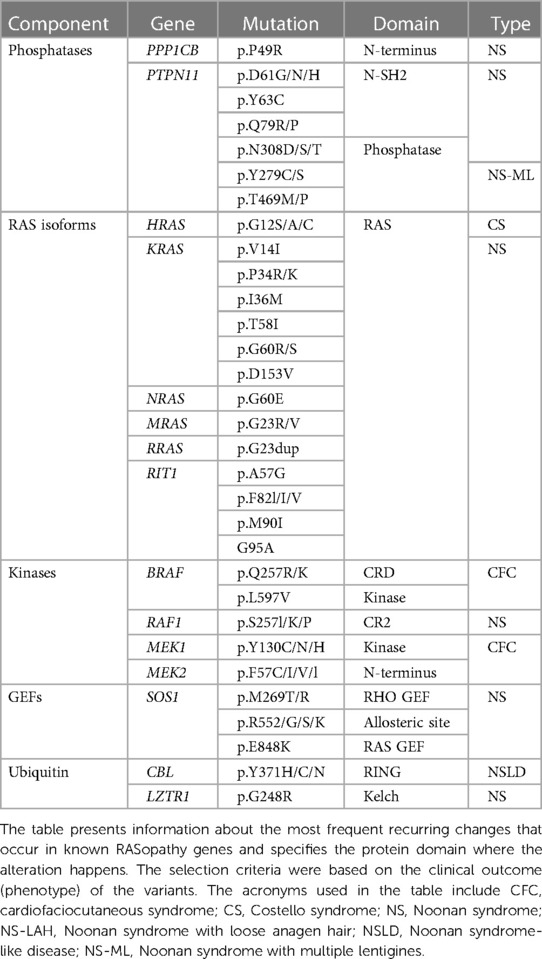
Table 2. Common RASopathy-associated mutations.
PTPN11 is the most common gene associated with NS and accounts for approximately half of all cases. The PTPN11 gene (19) has three domains: the more commonly mutated N-amino terminal src-homology 2 (N-SH2) and phospho-tyrosine phosphatase (PTP) domains, and the C-amino terminal src-homology 2 domain (C-SH2) and carboxy-terminal tail (5, 6). PTPN11 codes for protein SHP-2 which is involved in semilunar valvulogenesis, hemopoietic cell differentiation, and mesodermal patterning (20–23). SHP-2 also regulates the cell proliferation, migration, or differentiation processes during the developmental stage (24) and is widely expressed in several tissues, such as the heart, muscles, and brain. SHP-2 is a pivotal protein in the RAS/MAPK cascade. PTPN11 mutations were mainly seen in cases with pulmonary valve stenosis in NS1 patients (5, 6).
Mutated SOS1 (OMIM 182530) is considered the second-most-common genetic aberration associated with NS, causing NS in up to 20% of patients with absent PTPN11 mutation (25). Its locus is on the 2p22-p21 region, consisting of 23 exons (26, 27) and coding for multiple domains containing: regulatory histone-like folds domain (HF), Dbl homology domains (DH), and Pleckstrin homology domains (PH); catalytic RAS exchanging motif (REM) and Cdc25 domains; helical linker (HL) relating PH and REM, and the Polyproline region (25, 28). SOS1 is a guanine exchange factor (GEF) with a significant role in the RAS/MAPK pathway (26, 27) and mainly implicated in NS patients with ectodermal defects (25, 28–30) and pulmonic stenosis more than that of PTPN1.
The KRAS (OMIM 190070) gene is mapped to the 12p12 region and consists of 6 exons coding for the P loop and switch I and switch II domains (15). Gain of function mutations in KRAS causes approximately 5% of NS cases in the absence of the PTPN11 mutations (16, 31).
The NRAS (OMIM 164790) gene locus on 1p13.2 comprises six coding exons (32). NRAS mutations are involved in less than 1% of NS cases (17).
The RIT1 (OMIM 609591) locus on 1q22, consisting of 6 exons, causes hyperactivation of transcription factor ELK1 in the RAS/MAPK cascade. It is present in 9% of NS cases (18). Prevalence is seen to mainly coexist with cardiac defects such as CHD (94%), HCM (71%), and pulmonic stenosis (65%). This finding was subsequently confirmed by Bertola et al., who found the exact prevalence (9%), and Gos et al., who found a lower mutation rate (3.8%). Those mutation clusters in the G1, Switch I, and more frequently in Switch II domains, were proven to entail a significant activation of the RAS/MAPK pathway by hyper-activating transcription factor ELK1 (33, 34).
RAF1 (OMIM 164760) locus on 3p25, consisting of 17 exons, codes for protein serine-threonine kinase (35–37) and has three conserved regions. Mutations causing failure of autoinhibition of this gene lead to activation of the RAS/MAPK cascade, causing NS (3%–17% of cases). An association of 80% is found with HCM.
Mutated BRAF (OMIM 164757) locus on 7q34 enhances ERK activation (38, 36), causing NS in 1.7%–1.9% of cases.
MAP2K1 (OMIM 176872), with a locus on 15q22, comprises 11 exons encoding the MEK protein, which activates ERK-MAP (39). Among NS cases without PTPN11 and SOS1 mutations, 4.2% are caused by mutated MAP2K1 (40).
Mutation of the homolog of SOS2 (OMIM 601247) locus on 14q21 causes 4% of Noonan cases, closely associated with ectodermal defects like SOS1 (41).
The LZTR1 gene (OMIM 600574) mapped on 22q11.21, consisting of 21 exons, encodes for a protein of the BTB-ketch superfamily, also implicated in neurofibromatosis (42). However, it is not associated with the RAS/MAPK pathway (41).
Mutation of A2ML1 (OMIM 610627), mapped on 12p13 with 35 exons, comprises 1% of Noonan patients negative for other significant genes (43). A2ML1 is a member of the a-macroglobulin superfamily, localized in the 12p13 region with 35 coding exons, and is a protease inhibitor upstream of the MAPK pathway (44). Nevertheless, how its mutation affects the MAPK pathway requires further explication.
Recently implicated rare variants in NS include RASA2, MAP3K8, and SPRY (45).
Neurofibromatosis (OMIM 162200) is an autosomal dominant genetic disorder caused by a heterozygous mutation of the NF1 gene located on chromosome 17q11.2. NF1 is a multisystem disease impacting the growth and function of various cell types and organs. Early-onset cerebrovascular disease, pheochromocytomas, and cardiovascular disease frequently cause premature death in individuals with NF1. Neurofibromas, the characteristic tumors of NF1, impact approximately 1/2000 live births (46) and can develop within the heart, obstructing blood flow in the heart or major vessels by compression or invasion, leading to hemorrhage. Fortunately, these are rare complications. NF1 encodes the neurofibromin protein, which belongs to the family of GTPase activating proteins (GAPs) and which negatively regulates RAS signaling. Neurofibromin also positively regulates cyclic adenosine monophosphate (AMP) levels (47, 48). Increased cyclic AMP levels have been associated with reduced cell growth, likely through interference with multiple mitogenic signaling pathways. The most common cardiovascular manifestations of NF1 include vasculopathy (49), hypertension (50), and other congenital heart defects (51). Sørensen found myocardial infarction and cerebrovascular accidents at a younger than-expected age in NF1 patients (52). NF1 vasculopathy includes segmental hypoplasia of the abdominal aorta and fibro cellular intimal proliferation. Both contribute to the luminal stenosis (53), aneurysms, the rupturing of which has been known to cause catastrophic abdominal and retroperitoneal hemorrhage and arteriovenous malformations, and is the second leading cause of death in neurofibromatosis patients (54–56). Coronary heart disease occurs at a higher-than-expected frequency compared with that in the general population, with pulmonary artery stenosis representing 25% of these malformations. Hypertension is common among female NF1 patients during pregnancy (57), and the prevalence increases with age. However, it has not been investigated whether NF1-hypertension is just a coincidental finding often discovered during medical evaluation for other reasons. Based on the previous literature, 10%–15% of NF1 patients have CHD (51). Approximately 50% of NF1 individuals with CHD have PVS. Aortic stenosis, aortic coarctation, atrial septal defects (ASD), and ventricular septal defects (VSD) are detected less frequently in NF-CHDs (58–60).
NF1 is a large and complex gene that carries more than 280 kb of genomic DNA, including 57 constitutive exons and other alternatively spliced exons (61). Now, over 2,800 different NF1 variants have been identified (62). Genetic testing in NF1 is challenging because of the large number of possible mutations in this large gene. Approximately 5% of patients with NF1 have a complete or near-complete deletion of the NF1 gene. These patients display a more severe phenotype, including earlier onset, large load of neurofibromas, greater likelihood of cognitive deficiency, dysmorphic facial features, increased risk of malignancy, and connective tissue involvement, with joint laxity, hyperextensible skin, and mitral valve prolapse. ADAP2 gene, which has been considered as a modifier of NF1, involved in cardiac development, is a reliable candidate gene for the occurrence of congenital valve defects (63). Additionally, CENTA2 and JJAZ1 are two possible candidates for the cardiovascular malformations (64).
The relationship between RASopathies and sex dimorphism is controversial, complex, and likely influenced by many factors. Studies have suggested that males with NS may be more likely to have more severe cardiac manifestations, including a higher incidence of hypertrophic cardiomyopathy and aortic valve stenosis, compared to females with NS (3, 35, 65). Similarly, few studies indicated that males had a higher incidence of pathogenic variants in the RAF1 gene, a less common genetic mutation associated with NS (66–69). However, other studies suggested no significant sex differences in the prevalence or severity of cardiac abnormalities in NS patients (16, 70). A retrospective cohort of 412 children with NS by Romano et al. found that female patients had a higher prevalence of pulmonary valve stenosis and a higher incidence of cardiac surgery compared to male patients. These female patients also indicated a higher incidence of composite cardiovascular events compared to male patients (71).
There is limited evidence regarding sex differences in cardiac manifestations of NF1 (72). But a recent study investigated sex differences in cardiac function in NF1 patients with Left Ventricular (LV) dysfunction and found that males had significantly lower Left ventricular ejection fraction (LV EF) and more severe LV dysfunction than females. In addition, males had a higher incidence of LV remodeling and a higher risk of sudden cardiac death than females (73). Similarly, individuals with NF1 found that males were more likely to have cardiac abnormalities than females and that males had a higher incidence of pulmonary stenosis and atrial septal defects (51). On contrary, an older study found that females with NF1 may be more likely to have cardiac abnormalities than males (58).
Current observations indicate that there may be some sex differences in the prevalence or severity of cardiac manifestations in RASopathies. These differences are not always consistent across studies and may be influenced by other factors such as age, genotype, and environmental factors. Additionally, many individuals with RASopathies have a normal cardiac function. However, the mechanisms underlying these sex differences are not well understood. One possible explanation is the differential expression of RAS-MAPK pathway genes in males and females, which could affect the development and progression of cardiomyopathy in RASopathies. Another possible explanation is the influence of sex hormones on cardiac function and remodeling, which could interact with the RAS-MAPK pathway and contribute to sex differences in RASopathy-induced cardiomyopathy. Despite the growing recognition of sex differences in RASopathy-induced cardiomyopathy, there is a lack of sex-specific guidelines for the diagnosis and management of cardiac complications in these disorders. This highlights the need for further research to understand the mechanisms underlying sex differences in RASopathy-induced cardiomyopathy and develop sex-specific management strategies to improve outcomes for both male and female patients.
Cardiomyopathy, a common cardiovascular complication in patients with NS and NF1, is caused by genetic mutations in the RAS-MAPK pathway. The age of onset and clinical penetrance of cardiomyopathy differ between NS and NF1. NS typically presents in childhood or adolescence, while NF1 may not present until adulthood. The penetrance of cardiomyopathy is also higher in NS than in NF1. Colquitt et al. in 2014 demonstrated that in NS patients severe HCM has an early onset with an increased risk of long-term morbidities (74). Later many studies confirmed the early onset of HCM (75, 76) as well as pulmonary valve stenosis and arterial septal defect in NS patients (77). In contrast, the prevalence of HCM in NF1 patients was only 2%, with a mean age of onset of 26 years. Also, mutations in the NF1 gene have been associated with a decreased risk of cardiomyopathy (59).
Several genetic variants have been associated with an increased risk of cardiomyopathy in NS and NF1. In NS, mutations in the PTPN11 and RAF1 genes have been associated with an increased risk of HCM. Lin et al. in 2000 found that the prevalence of HCM was higher in NS patients with PTPN11 mutations than in those with RAF1 mutations (44% vs. 18%). Overall, 9% of the DCM cohort presenting in childhood or adolescence have RAF1 mutations (59) PTPN11 had common echocardiography features characterized by pulmonary valve stenosis, while HCM is characterized by RAF1. RAF1 genotypes were shown as prognostic factors, eliciting multiple interventions that may be required for NS patients with severe pulmonary stenosis or myectomy for HCM (77). But a recent study indicated that the proportion of RIT1 mutation-positive patients who underwent intervention due to cardiovascular disease was significantly higher than that of patients with PTPN11 mutations (78). A multi-center cohort study to compare the incidence of sudden cardiac death (SCD) and implantable cardioverter-defibrillator (ICD) use between RAS-HCM (n = 188) and P-HCM (n = 567) patients showed a lower median age for RAS-HCM. Nonarrhythmic deaths occurred primarily in infancy, and SCD primarily in adolescence (79). Another study suggested the possibilities of prenatal RASopathy testing by comparing the genotypic variations from 352 chromosomal microarray negative cases for prenatal RASopathy testing with post-natal cohorts (25 patients with available prenatal information and 108 institutional database genotypes). The study supported the view that a subset of RASopathy genes and variants that are more frequently associated with complex prenatal features such as hydrops/effusions or serious cardiopathy should be considered in the prenatal evaluation (80).
Trametinib, cobimetinib, and binimetinib are examples of medications that have been approved for use in certain tumors to suppress the RAS/MAPK signaling pathway. These medications may benefit NS patients with mutations resulting in gain-of-function alterations in the RAS/MAPK pathway. This has been investigated in mouse models with the RAF1 mutation, which is present in many NS patients. Mek inhibition during postnatal treatment reversed hypertrophy, restored standard cardiomyocyte size, and lowered fractional shortening toward the target range (81). Since then, there have been several case reports highlighting anecdotal successes with MEK inhibition in NS patients. By now, three groups have described the cases of four patients who, after using trametinib, showed improvement from NS and HCM (82–84). Studies have also shown that arrhythmia and lymphatic abnormalities resolve after starting MEK inhibition treatment (83, 85, 86). While there are some promising early reports of this medical therapy for a patient population for which it is typically believed that the only treatment option is cardiac transplantation, more research is still required in this area (87).
Our understanding of the molecular basis of RASopathies continues to expand, along with our knowledge of the various clinical manifestations of these disorders, including cardiomyopathy. Figure 3 indicates the involvement of RAS/MAPK pathway genes in NS and NF1. The age of onset and clinical penetrance of cardiomyopathy in NS and NF1 are important factors that can influence the diagnosis and management of these conditions. However, much is still unknown about the mechanisms underlying the development of cardiomyopathy in RASopathies, and further research is needed to identify novel therapeutic targets and improve outcomes for affected individuals. One potential explanation for the variability in age of onset and clinical penetrance of cardiomyopathy in NS and NF1 is the wide range of genetic mutations that can occur within these genes. As we have seen, specific mutations can result in more severe forms of cardiomyopathy, while others may have little to no effect on the heart. Other genetic and environmental factors may also play a role in determining the severity and timing of cardiomyopathy in these individuals. Another possible explanation is that comorbidities, such as hypertension, diabetes, or obesity, can further exacerbate the risk of developing cardiomyopathy in individuals with RASopathies. It is crucial for clinicians to carefully monitor and manage these conditions to reduce the risk of cardiovascular complications in this patient population.
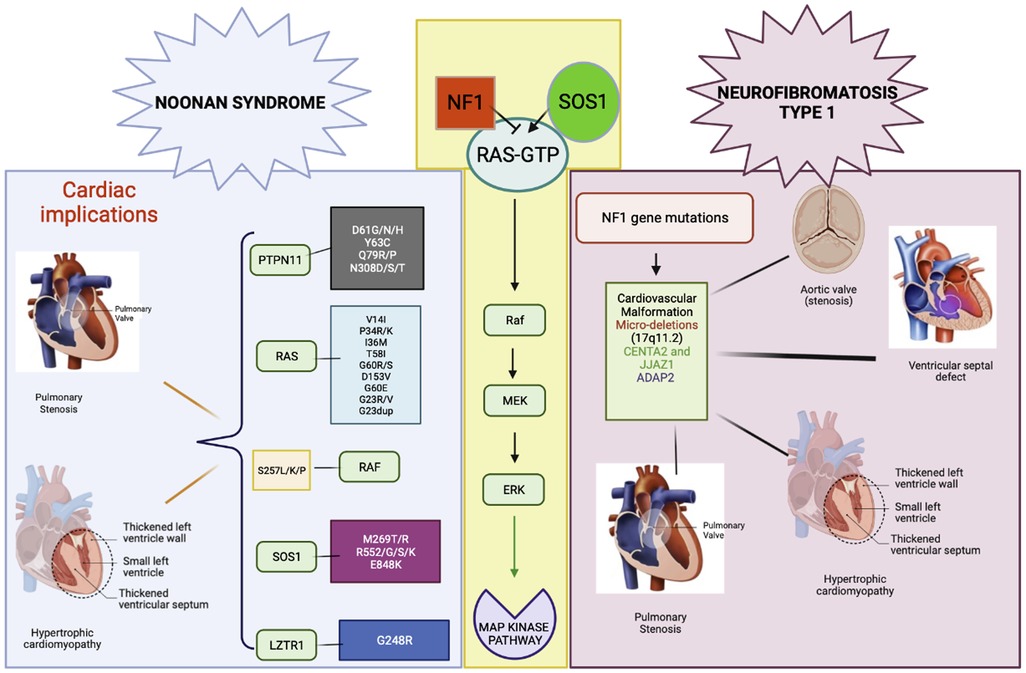
Figure 3. RAS/MAPK pathway gene involvement in NS and NF1. Gene variants and cardiac malformations in NS and NF1.
Despite these challenges, genetic testing and imaging technology advances have greatly improved our ability to diagnose and monitor cardiomyopathy in individuals with RASopathies. Identifying specific genetic mutations associated with cardiomyopathy can help guide treatment decisions and improve outcomes for affected individuals.
Since the RAS/MAPK pathway was discovered in humans, the role of these two molecules has been investigated extensively in a wide range of human diseases, including the role of somatic mutations in RAS/MAPK mediated cancer. RAS/MAPK pathway genes are often activated because of germline mutations, referred to as RASopathies, comprising ectodermal and mesodermal development abnormalities and various neoplasia. Interestingly, mutations in RASopathy genes impact different cellular subsets differently, and the phenotype observed in patients varies widely. This phenotype diversity with the same genotype could be due to secondary events modified by epigenetic, environmental, and yet undetermined factors. Recent sequencing technology advances have enabled us to decipher many genotype-phenotype mysteries. A recent study discovered de novo variants in PTPN11, RAF1, BARF, SHOC2, RASA1, and HRAS in nine sporadic patients, all of whom had cardiovascular abnormalities along with other Noonan syndromic malformations (88). The above study identified six genes harboring eight de novo variants. Two patients with Capillary Malformation-Arteriovenous Malformation (CM-AVM had a novel variant in RASA1. The novel missense variant (NM_002890.2: c.2828T>C, p.Leu943Pro) occurred with an amino acid change from a nonpolar amino acid, leucine (Leu), to another nonpolar amino acid, proline (Pro). This study demonstrates the limitation of phenotype-driven genetics testing and the power of family-based NGS for detecting disorders with a clinically atypical presentation and in severely ill infants with CHDs without known genetic cause. Individuals with RASopathies have been linked to various malformations along with cardiovascular problems. Patients with these illnesses may have improved outcomes when the diagnosis is determined based on phenotype and genotype.
RASopathies are a group of genetic disorders characterized by gene mutations in the RAS/MAPK signaling pathway. These genetic disorders are associated with a broad range of clinical manifestations, including developmental abnormalities, intellectual disabilities, and cardiac defects. Among these disorders' the most common cardiac abnormalities are pulmonary valve stenosis, septal defects, left-sided lesions, and complex forms with multiple anomalies. Early diagnosis and management of these cardiac abnormalities are critical for improving the overall outcome of individuals with RASopathies. With the development of genomic technologies, more details of genetic mutations that result in RASopathies and associated cardiac abnormalities can be identified. The new advancement will provide valuable insights into the pathophysiology of these disorders and may lead to the development of new therapeutics for these debilitating disorders.
NH and SC conceptualized the idea. NH drafted the manuscript. SC, ZC, and MHC edited the drafts. All authors contributed to the article and approved the submitted version.
ZC is funded by the National Science Foundation (2025434). SC is funded by the American Heart Association (SDG) and by NHLBI (R01HL152063). MHC, is funded by NHLBI (R01HL152063).
The figures 1 and 3 were created using biorendor tool. We gratefully acknowledge Sonia Epstein's help in proofreading the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of ras/MAPK pathway dysregulation. Curr Opin Genet Dev. (2009) 19(3):230–6. doi: 10.1016/j.gde.2009.04.001
2. Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. (2013) 14:355–69. doi: 10.1146/annurev-genom-091212-153523
3. Gelb BD, Roberts AE, Tartaglia M. Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog Pediatr Cardiol. (2015) 39(1):13–9. doi: 10.1016/j.ppedcard.2015.01.002
4. Sanchez-Cascos A. The Noonan syndrome. Eur Heart J. (1983) 4(4):223–9. doi: 10.1093/oxfordjournals.eurheartj.a061452
5. Stein-Gerlach M, Wallasch C, Ullrich A. SHP-2, SH2-containing protein tyrosine phosphatase-2. Int J Biochem Cell Biol. (1998) 30(5):559–66. doi: 10.1016/s1357-2725(98)00002-8
6. Feng GS. Shp-2 tyrosine phosphatase: signaling one cell or many. Exp Cell Res. (1999) 253(1):47–54. doi: 10.1006/excr.1999.4668
7. Shaw AC, Kalidas K, Crosby AH, Jeffery S, Patton MA. The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child. (2007) 92(2):128–32. doi: 10.1136/adc.2006.104547
8. Takazawa A, Hashimoto A, Aomi S, Imamaki M, Noji S, Koyanagi H, et al. Successful mitral valve plasty for mitral regurgitation combined with dilated cardiomyopathy in Noonan’s syndrome. Nihon Kyobu Geka Gakkai. (1995) 43(6):894–7.
9. Yu CM, Chow LT, Sanderson JE. Dilated cardiomyopathy in Noonan’s syndrome. Int J Cardiol. (1996) 56(1):83–5. doi: 10.1016/0167-5273(96)02713-1
10. Kurose A, Oyama K, Murakami Y, Ohyama K, Segawa I, Sawai T, et al. Dilated cardiomyopathy in Noonan’s syndrome: a first autopsy case. Hum Pathol. (2000) 31(6):764–7. doi: 10.1053/hupa.2000.7635
11. Aljeaid D, Sanchez AI, Wakefield E, Chadwell SE, Moore N, Prada CE, et al. Prevalence of pathogenic and likely pathogenic variants in the RASopathy genes in patients who have had panel testing for cardiomyopathy. Am J Med Genet A. (2019) 179(4):608–14. doi: 10.1002/ajmg.a.61072
12. Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis. (2008) 3:13. doi: 10.1186/1750-1172-3-13
13. Carcavilla A, Santomé JL, Pinto I, Sánchez-Pozo J, Guillén-Navarro E, Martín-Frías M, et al. LEOPARD Syndrome: a variant of Noonan syndrome strongly associated with hypertrophic cardiomyopathy. Rev Esp Cardiol. (2013) 66(5):350–6. doi: 10.1016/j.rec.2012.09.015
14. Wang J, Chandrasekhar V, Abbadessa G, Yu Y, Schwartz B, Kontaridis MI. In vivo efficacy of the AKT inhibitor ARQ 092 in Noonan syndrome with multiple lentigines-associated hypertrophic cardiomyopathy. PloS One. (2017) 12(6):e0178905. doi: 10.1371/journal.pone.0178905
15. Carta C, Pantaleoni F, Bocchinfuso G, Stella L, Vasta I, Sarkozy A, et al. Germline missense mutations affecting KRAS isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet. (2006) 79(1):129–35. doi: 10.1086/504394
16. Lee BH, Kim JM, Jin HY, Kim GH, Choi JH, Yoo HW. Spectrum of mutations in Noonan syndrome and their correlation with phenotypes. J Pediatr. (2011) 159(6):1029–35. doi: 10.1016/j.jpeds.2011.05.024
17. Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet. (2010) 42(1):27–9. doi: 10.1038/ng.497
18. Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. (2013) 93(1):173–80. doi: 10.1016/j.ajhg.2013.05.021
19. Schade RW, van't Laar A, Majoor CL, Jansen AP. A comparative study of the effects of cholestyramine and neomycin in the treatment of type II hyperlipoproteinaemia. Acta Med Scand. (1976) 199(3):175–80. doi: 10.1111/j.0954-6820.1976.tb06712.x
20. Tang TL, Freeman RM, O'Reilly AM, Neel BG, Sokol SY. The SH2-containing protein-tyrosine phosphatase SH-PTP2 is required upstream of MAP kinase for early Xenopus development. Cell. (1995) 80(3):473–83. doi: 10.1016/0092-8674(95)90498-0
21. Qu CK, Yu WM, Azzarelli B, Cooper S, Broxmeyer HE, Feng GS. Biased suppression of hematopoiesis and multiple developmental defects in chimeric mice containing shp-2 mutant cells. Mol Cell Biol. (1998) 18(10):6075–82. doi: 10.1128/MCB.18.10.6075
22. Chen B, Bronson RT, Klaman LD, Hampton TG, Wang JF, Green PJ. Mice mutant for egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat Genet. (2000) 24(3):296–9. doi: 10.1038/73528
23. Saxton TM, Ciruna BG, Holmyard D, Kulkarni S, Harpal K, Rossant J. The SH2 tyrosine phosphatase shp2 is required for mammalian limb development. Nat Genet. (2000) 24(4):420–3. doi: 10.1038/74279
24. Neel BG, Gu H, Pao L. The ‘Shp’ing news: sH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. (2003) 28(6):284–93. doi: 10.1016/S0968-0004(03)00091-4
25. Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. (2007) 39(1):70–4. doi: 10.1038/ng1926
26. Webb GC, Jenkins NA, Largaespada DA, Copeland NG, Fernandez CS, Bowtell DD. Mammalian homologues of the Drosophila son of sevenless gene map to murine chromosomes 17 and 12 and to human chromosomes 2 and 14, respectively. Genomics. (1993) 18(1):14–9. doi: 10.1006/geno.1993.1421
27. Hart TC, Zhang Y, Gorry MC, Hart PS, Cooper M, Marazita ML, et al. A mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. Am J Hum Genet. (2002) 70(4):943–54. doi: 10.1086/339689
28. Lepri F, De Luca A, Stella L, Rossi C, Baldassarre G, Pantaleoni F, et al. SOS1 Mutations in Noonan syndrome: molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations. Hum Mutat. (2011) 32(7):760–72. doi: 10.1002/humu.21492
29. Nimnual A, Bar-Sagi D. The two hats of SOS. Sci STKE. (2002) 2002(145):pe36. doi: 10.1126/stke.2002.145.pe36
30. Zenker M, Horn D, Wieczorek D, Allanson J, Pauli S, van der Burgt I, et al. SOS1 Is the second most common Noonan gene but plays no major role in cardio-facio-cutaneous syndrome. J Med Genet. (2007) 44(10):651–6. doi: 10.1136/jmg.2007.051276
31. Schubbert S, Zenker M, Rowe SL, Böll S, Klein C, Bollag G, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. (2006) 38(3):331–6. doi: 10.1038/ng1748
32. Mitchell EL, Jones D, White GR, Varley JM, Santibanez Koref MF. Determination of the gene order of the three loci CD2, NGFB, and NRAS at human chromosome band 1p13 and refinement of their localisation at the subband level by fluorescence in situ hybridisation. Cytogenet Cell Genet. (1995) 70(3–4):183–5. doi: 10.1159/000134028
33. Bertola DR, Yamamoto GL, Almeida TF, Buscarilli M, Jorge AAL, Malaquias AC, et al. Further evidence of the importance of RIT1 in Noonan syndrome. Am J Med Genet A. (2014) 164A(11):2952–7. doi: 10.1002/ajmg.a.36722
34. Gos M, Fahiminiya S, Poznański J, Klapecki J, Obersztyn E, Piotrowicz M, et al. Contribution of RIT1 mutations to the pathogenesis of Noonan syndrome: four new cases and further evidence of heterogeneity. Am J Med Genet A. (2014) 164A(9):2310–6. doi: 10.1002/ajmg.a.36646
35. Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. (2007) 39(8):1007–12. doi: 10.1038/ng2073
36. Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. (2007) 39(8):1013–7. doi: 10.1038/ng2078
37. Kobayashi T, Aoki Y, Niihori T, Cavé H, Verloes A, Okamoto N, et al. Molecular and clinical analysis of RAF1 in Noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum Mutat. (2010) 31(3):284–94. doi: 10.1002/humu.21187
38. Wan PTC, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. (2004) 116(6):855–67. doi: 10.1016/s0092-8674(04)00215-6
39. Giroux S, Tremblay M, Bernard D, Cardin-Girard JF, Aubry S, Larouche L, et al. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr Biol. (1999) 9(7):369–72. doi: 10.1016/s0960-9822(99)80164-x
40. Nava C, Hanna N, Michot C, Pereira S, Pouvreau N, Niihori T, et al. Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype relationships and overlap with costello syndrome. J Med Genet. (2007) 44(12):763–71. doi: 10.1136/jmg.2007.050450
41. Yamamoto GL, Aguena M, Gos M, Hung C, Pilch J, Fahiminiya S, et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet. (2015) 52(6):413–21. doi: 10.1136/jmedgenet-2015-103018
42. Piotrowski A, Xie J, Liu YF, Poplawski AB, Gomes AR, Madanecki P, et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet. (2014) 46(2):182–7. doi: 10.1038/ng.2855
43. Vissers LELM, Bonetti M, Paardekooper Overman J, Nillesen WM, Frints SGM, de Ligt J, et al. Heterozygous germline mutations in A2ML1 are associated with a disorder clinically related to Noonan syndrome. Eur J Hum Genet. (2015) 23(3):317–24. doi: 10.1038/ejhg.2014.115
44. Galliano MF, Toulza E, Gallinaro H, Jonca N, Ishida-Yamamoto A, Serre G, et al. A novel protease inhibitor of the alpha2-macroglobulin family expressed in the human epidermis. J Biol Chem. (2006) 281(9):5780–9. doi: 10.1074/jbc.M508017200
45. Chen PC, Yin J, Yu HW, Yuan T, Fernandez M, Yung CK, et al. Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc Natl Acad Sci U S A. (2014) 111(31):11473–8. doi: 10.1073/pnas.1324128111
46. Uusitalo E, Leppävirta J, Koffert A, Suominen S, Vahtera J, Vahlberg T, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. (2015) 135(3):904–6. doi: 10.1038/jid.2014.465
47. Dasgupta B, Dugan LL, Gutmann DH. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature. (2000) 403(6772):895–8. doi: 10.1038/35002593
48. Guo HF, Tong J, Hannan F, Luo L, Zhong Y. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci. (2003) 23(26):8949–54. doi: 10.1523/JNEUROSCI.23-26-08949.2003
49. İncecik F, Hergüner ÖM, Alınç Erdem S, Altunbaşak Ş. Neurofibromatosis type 1 and cardiac manifestations. Turk Kardiyol Dern Ars. (2015) 43(8):714–6. doi: 10.5543/tkda.2015.27557
50. Nguyen R, Mir TS, Kluwe L, Jett K, Kentsch M, Mueller G, et al. Cardiac characterization of 16 patients with large NF1 gene deletions. Clin Genet. (2013) 84(4):344–9. doi: 10.1111/cge.12072
51. Pinna V, Daniele P, Calcagni G, Mariniello L, Criscione R, Giardina C, et al. Prevalence, type, and molecular Spectrum of NF1 mutations in patients with neurofibromatosis type 1 and congenital heart disease. Genes (Basel). (2019) 10(9):675. doi: 10.3390/genes10090675
52. Sørensen SA, Mulvihill JJ, Nielsen A. Long-term follow-up of von recklinghausen neurofibromatosis. Survival and malignant neoplasms. N Engl J Med. (1986) 314(16):1010–5. doi: 10.1056/NEJM198604173141603
53. Baradhi KM, Bream P. Fibromuscular dysplasia. In: Statpearls. Treasure Island (FL): StatPearls Publishing (2022). Available at: http://www.ncbi.nlm.nih.gov/books/NBK493204/ (Accessed February 28, 2023).
54. Keenan RA, Robinson DJ, Briggs PC. Fatal spontaneous retroperitoneal haemorrhage caused by von Recklinghausen’s neurofibromatosis. J R Coll Surg Edinb. (1982) 27(5):310.6815324
55. Shelton NP. Fatal spontaneous retroperitoneal hemorrhage in a patient with von Recklinghausen’s disease. A case report. J Indiana State Med Assoc. (1983) 76(12):831.6418827
56. Poston GJ, Grace PA, Venn G, Spencer J. Recurrent near-fatal haemorrhage in von Recklinghausen’s disease. Br J Clin Pract. (1990) 44(12):755–6.2129328
57. Friedman JM. Neurofibromatosis 1. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, et al. editors. Genereviews®. Seattle (WA): University of Washington (1993). Available at: http://www.ncbi.nlm.nih.gov/books/NBK1109/ (Accessed February 27, 2023).
58. Friedman JM, Arbiser J, Epstein JA, Gutmann DH, Huot SJ, Lin AE, et al. Cardiovascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am J Med Genet. (2000) 95(2):108–17. doi: 10.1002/1096-8628(20001113)95:2-108::aid-ajmg4%3E3.0.co;2-0
59. Lin AE, Birch PH, Korf BR, Tenconi R, Niimura M, Poyhonen M, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 cardiovascular task force. Genet Med. (2002) 4(3):105–11. doi: 10.1097/00125817-200205000-00002
60. Tedesco MA, Di Salvo G, Natale F, Pergola V, Calabrese E, Grassia C, et al. The heart in neurofibromatosis type 1: an echocardiographic study. Am Heart J. (2002) 143(5):883–8. doi: 10.1067/mhj.2002.122121
61. Gutmann DH, Wood DL, Collins FS. Identification of the neurofibromatosis type 1 gene product. Proc Natl Acad Sci U S A. (1991) 88(21):9658–62. doi: 10.1073/pnas.88.21.9658
62. Prasad BCM, Chandra VVR, Sudarsan A, Kumar PS, Sarma PVGK. Clinical characteristics and NF1 gene mutation analysis of three successive generations in three different Indian families with neurofibromatosis type 1 and peripheral nerve sheath tumours. J Clin Neurosci. (2018) 53:62–8. doi: 10.1016/j.jocn.2018.04.006
63. Venturin M, Carra S, Gaudenzi G, Brunelli S, Gallo GR, Moncini S, et al. ADAP2 In heart development: a candidate gene for the occurrence of cardiovascular malformations in NF1 microdeletion syndrome. J Med Genet. (2014) 51(7):436–43. doi: 10.1136/jmedgenet-2013-102240
64. Venturin M, Guarnieri P, Natacci F, Stabile M, Tenconi R, Clementi M, et al. Mental retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2. J Med Genet. (2004) 41(1):35–41. doi: 10.1136/jmg.2003.014761
65. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. (2001) 29(4):465–8. doi: 10.1038/ng772
66. Ratola A, Silva HM, Guedes A, Mota C, Braga AC, Oliveira D, et al. A novel Noonan syndrome RAF1 mutation: lethal course in a preterm infant. Pediatr Rep. (2015) 7(2):5955. doi: 10.4081/pr.2015.5955
67. Harms FL, Alawi M, Amor DJ, Tan TY, Cuturilo G, Lissewski C, et al. The novel RAF1 mutation p.(Gly361Ala) located outside the kinase domain of the CR3 region in two patients with Noonan syndrome, including one with a rare brain tumor. Am J Med Genet A. (2018) 176(2):470–6. doi: 10.1002/ajmg.a.38569
68. Hagino M, Ota C, Onoki T, Iwasawa S. Male infant with Noonan syndrome with RAF-1 gene mutation who survived hypertrophic cardiomyopathy-induced fatal heart failure and uncontrollable arrhythmias. BMJ Case Rep. (2022) 15(9):e250342. doi: 10.1136/bcr-2022-250342
69. Lan J, Zeng T, Liu S, Lan J, Qian L. Noonan syndrome with RAF1 gene mutations in a newborn with cerebral haemorrhage. Eur J Med Res. (2022) 27(1):146. doi: 10.1186/s40001-022-00772-2
70. Lee BH, Yoo H-W. Noonan syndrome and RASopathies: clinical features, diagnosis and management. J Genet Med,. (2019) 16(1):1–9. doi: 10.5734/JGM.2019.16.1.1
71. Romano A, Kaski JP, Dahlgren J, Kelepouris N, Pietropoli A, Rohrer TR, et al. Cardiovascular safety of growth hormone treatment in Noonan syndrome: real-world evidence. Endocr Connect. (2022) 11(1):e210549. doi: 10.1530/EC-21-0549
72. Hamoy-Jimenez G, Elahmar HA, Mendoza M, Kim RH, Bril V, Barnett C. A cross-sectional study of gender differences in quality of life domains in patients with neurofibromatosis type 1. Orphanet J Rare Dis. (2022) 17(1):40. doi: 10.1186/s13023-022-02195-y
73. Cutruzzolà A, Irace C, Frazzetto M, Sabatino J, Gullace R, De Rosa S, et al. Functional and morphological cardiovascular alterations associated with neurofibromatosis 1. Sci Rep. (2020) 10(1):12070. doi: 10.1038/s41598-020-68908-0
74. Colquitt JL, Noonan JA. Cardiac findings in Noonan syndrome on long-term follow-up. Congenit Heart Dis. (2014) 9(2):144–50. doi: 10.1111/chd.12102
75. Lioncino M, Monda E, Verrillo F, Moscarella E, Calcagni G, Drago F, et al. Hypertrophic cardiomyopathy in RASopathies: diagnosis, clinical characteristics, prognostic implications, and management. Heart Fail Clin. (2022) 18(1):19–29. doi: 10.1016/j.hfc.2021.07.004
76. Chen H, Li X, Liu X, Wang J, Zhang Z, Wu J, et al. Clinical and mutation profile of pediatric patients with RASopathy-associated hypertrophic cardiomyopathy: results from a Chinese cohort. Orphanet J Rare Dis. (2019) 14(1):29. doi: 10.1186/s13023-019-1010-z
77. Sun L, Xie YM, Wang SS, Zhang ZW. Cardiovascular abnormalities and gene mutations in children with Noonan syndrome. Front Genet. (2022) 13:915129. doi: 10.3389/fgene.2022.915129
78. Ichikawa Y, Kuroda H, Ikegawa T, Kawai S, Ono S, Kim KS, et al. Cardiac features of Noonan syndrome in Japanese patients. Cardiol Young. (2023) 33(4):564–9. doi: 10.1017/S104795112200124X
79. Lynch A, Tatangelo M, Ahuja S, Steve Fan CP, Min S, Lafreniere-Roula M, et al. Risk of sudden death in patients with RASopathy hypertrophic cardiomyopathy. J Am Coll Cardiol. (2023) 81(11):1035–45. doi: 10.1016/j.jacc.2023.01.012
80. Scott A, Di Giosaffatte N, Pinna V, Daniele P, Corno S, D'Ambrosio V, et al. When to test fetuses for RASopathies? Proposition from a systematic analysis of 352 multicenter cases and a postnatal cohort. Genet Med. (2021) 23(6):1116–24. doi: 10.1038/s41436-020-01093-7
81. Wu X, Simpson J, Hong JH, Kim KH, Thavarajah NK, Backx PH, et al. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J Clin Invest. (2011) 121(3):1009–25. doi: 10.1172/JCI44929
82. Andelfinger G, Marquis C, Raboisson MJ, Théoret Y, Waldmüller S, Wiegand G, et al. Hypertrophic cardiomyopathy in Noonan syndrome treated by MEK-inhibition. J Am Coll Cardiol. (2019) 73(17):2237–9. doi: 10.1016/j.jacc.2019.01.066
83. Meisner JK, Bradley DJ, Russell MW. Molecular management of multifocal atrial tachycardia in Noonan’s syndrome with MEK1/2 inhibitor trametinib. Circ Genom Precis Med. (2021) 14(5):e003327. doi: 10.1161/CIRCGEN.121.003327
84. Mussa A, Carli D, Giorgio E, Villar AM, Cardaropoli S, Carbonara C, et al. MEK Inhibition in a newborn with RAF1-associated Noonan syndrome ameliorates hypertrophic cardiomyopathy but is insufficient to revert pulmonary vascular disease. Genes (Basel). (2021) 13(1):6. doi: 10.3390/genes13010006
85. Dori Y, Smith C, Pinto E, Snyder K, March ME, Hakonarson H, et al. Severe lymphatic disorder resolved with MEK inhibition in a patient with Noonan syndrome and SOS1 mutation. Pediatrics. (2020) 146(6):e20200167. doi: 10.1542/peds.2020-0167
86. Nakano TA, Rankin AW, Annam A, Kulungowski AM, McCallen LM, Hill LR, et al. Trametinib for refractory chylous effusions and systemic complications in children with Noonan syndrome. J Pediatr. (2022) 248:81–88.e1. doi: 10.1016/j.jpeds.2022.05.030
87. Wilkinson JD, Lowe AM, Salbert BA, Sleeper LA, Colan SD, Cox GF, et al. Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: a study from the pediatric cardiomyopathy registry. Am Heart J. (2012) 164(3):442–8. doi: 10.1016/j.ahj.2012.04.018
Keywords: rasopathy, cardiac abnormalities, congenital heart disease, hypertrophic cardiomyopathy, ras/MAPK
Citation: Hilal N, Chen Z, Chen MH and Choudhury S (2023) RASopathies and cardiac manifestations. Front. Cardiovasc. Med. 10:1176828. doi: 10.3389/fcvm.2023.1176828
Received: 28 February 2023; Accepted: 20 June 2023;
Published: 17 July 2023.
Edited by:
Michael T. Chin, Tufts Medical Center, United StatesReviewed by:
Paolo Versacci, Sapienza University of Rome, Italy© 2023 Hilal, Chen, Chen and Choudhury. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sangita Choudhury c2FuZ2l0YS5jaG91ZGh1cnlAY2hpbGRyZW5zLmhhcnZhcmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.