 John P. Cooke
John P. Cooke Li Lai
Li Lai
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 02 May 2023
Sec. Atherosclerosis and Vascular Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1155835
This article is part of the Research Topic Emerging Therapeutics for Peripheral Artery Disease View all 9 articles
Tissue repair requires the orchestration of multiple processes involving a multiplicity of cellular effectors, signaling pathways, and cell-cell communication. The regeneration of the vasculature is a critical process for tissue repair and involves angiogenesis, adult vasculogenesis, and often arteriogenesis, which processes enable recovery of perfusion to deliver oxygen and nutrients to the repair or rebuild of the tissue. Endothelial cells play a major role in angiogenesis, whereas circulating angiogenic cells (primarily of hematopoietic origin) participate in adult vasculogenesis, and monocytes/macrophages have a defining role in the vascular remodeling that is necessary for arteriogenesis. Tissue fibroblasts participate in tissue repair by proliferating and generating the extracellular matrix as the structural scaffold for tissue regeneration. Heretofore, fibroblasts were not generally believed to be involved in vascular regeneration. However, we provide new data indicating that fibroblasts may undergo angiogenic transdifferentiation, to directly expand the microvasculature. Transdifferentiation of fibroblasts to endothelial cells is initiated by inflammatory signaling which increases DNA accessibility and cellular plasticity. In the environment of under-perfused tissue, the activated fibroblasts with increased DNA accessibility can now respond to angiogenic cytokines, which provide the transcriptional direction to induce fibroblasts to become endothelial cells. Periphery artery disease (PAD) involves the dysregulation of vascular repair and inflammation. Understanding the relationship between inflammation, transdifferentiation, and vascular regeneration may lead to a new therapeutic approach to PAD.
It is generally recognized that inflammation and inflammatory signaling plays a critical role during tissue repair. In addition, tissue repair requires restoration of tissue perfusion. As described below, these elements of tissue repair, i.e., inflammatory signaling and vascular recovery, are inextricably linked. However, before delving into the new insights linking inflammatory signaling, vascular recovery, and tissue repair, it is useful to review the current paradigm.
It is well-established that in the acute response to tissue injury, immune cells are recruited to combat bacterial infection and to clear necrotic debris. During this stage, M1-like macrophages generate inflammatory cytokines and chemokines, as well as bactericidal products such as peroxynitrite anion. This pro-inflammatory stage gives way to a regenerative phase typified by the polarization of macrophages to the M2 phenotype, which macrophage subset generates anti-inflammatory factors such as arginase, IL-10 and TGFb. At this point, the inflammation begins to subside, while tissue fibroblasts that have migrated into the injury site begin to proliferate and generate extracellular matrix proteins. Concurrently, tissue perfusion is restored, through the processes of angiogenesis and adult vasculogenesis. In addition, if the tissue injury is due to ischemia secondary to occlusive disease of larger conduit arteries, then arteriogenesis is activated to restore perfusion through the growth of collateral channels. The processes of angiogenesis, adult vasculogenesis and arteriogenesis are described elsewhere in this special issue, so we will focus on the mechanisms of angiogenic transdifferentiation, a process we first described several years ago. This process involves the transdifferentiation of a subset of tissue fibroblasts into endothelial cells, triggered by inflammatory signaling and angiogenic factors, which contributes to angiogenesis and restoration of tissue perfusion. Before launching into this discussion, lets review prior knowledge regarding the role of fibroblasts and endothelial cells in tissue repair and response to ischemia.
Tissue repair and response to ischemia involves well-orchestrated teamwork among different cellular effectors and encompasses precisely controlled sequential events. With respect to vascular regeneration in the injured or ischemic tissue, although we appreciate that immune cells, pericytes, and smooth muscle cells are also major contributors to vasculature homeostasis and repair, we will focus on the role of fibroblasts and endothelial cells in this review, because we have discovered an intriguing link between these cell subsets in tissue repair and response to ischemia.
There is increasing evidence of heterogeneity of tissue fibroblasts. Furthermore, the response of specific subsets of fibroblasts to injury or ischemia can determine whether the tissue repair occurs by scarring or regeneration (1–7). With genetic lineage tracing studies and single-cell sequencing, functionally distinct subsets of fibroblasts have been identified. For example, a subset of Engrailed-1(En1) lineage-positive fibroblasts are reported to contribute to scar formation in the skin after injury (8), whereas En-1 negative fibroblasts proote skin regeneration without scarring (9). Also, subpopulations of cardiac fibroblasts that express high levels of collagen triple helix repeat containing 1 (Cthrc1) (10) or Thbs4 (11) emerge after myocardial infarction to promote a profibrotic response. The presence of pro-fibrotic or regenerative fibroblasts is also dependent upon the tissue site and context as well as the developmental stage. Identifying bona fide markers for regenerative or fibrotic fibroblasts, and the molecular pathways that promote these different phenotypes is of great interest in regenerative medicine.
Inflammatory mediators modulate the fibroblast response to injury in the skin, heart, brain, and other organs (12–15). A recent study of fibroblasts from reindeer skin shed light on different fibroblast subsets and their interplay with immune cells. Fibroblasts derived from the skin (velvet) overlying the antlers suppress immune cell infiltration and support cutaneous regeneration. By contrast, fibroblasts derived from the skin on the dorsal surface of the animal are pro-inflammatory, intensify myeloid infiltration, and promote fibrosis (16). Similarly, in response to myocardial infarction, a subset of cardiac fibroblasts remain pathologically activated, generating pro-inflammatory signals and excessive extracellular matrix deposition, causing cardiac fibrosis and impaired ventricular function (14, 17). The activated fibroblasts express FAP (fibroblast activation protein) on their surface. Selective removal of this fibroblast subset can reduce cardiac fibrosis. Specifically, a chimeric antigen receptor (CAR) T cell therapy has recently been leveraged as a novel approach to mitigate cardiac fibrosis (18). In this work, the authors developed an immunotherapy strategy to reprogram CD5+ T cells in vivo with modified messenger RNA encoding a chimeric antigen receptor against FAP. The CAR T cell therapy reduced the number of FAP-expressing fibroblasts, attenuated cardiac fibrosis, and improved cardiac function in a murine model of heart failure. To conclude, different subsets of fibroblasts promote true tissue regeneration whereas others promote fibrosis.
Endothelial cells (ECs) form the luminal lining of all blood vessels, participating in nutrient exchange (19, 20), immune cell trafficking (21, 22), and mechanotransduction (23, 24). They are also the major contributor to vascular recovery with ischemia or injury (25). Endothelial cells participate in three major processes involved in vascular regeneration: adult vasculogenesis, angiogenesis, and arteriogenesis. These processes are discussed elsewhere in this collection, so they will be only briefly mentioned here.
Vasculogenesis refers to the de novo generation of endothelial cells differentiated from progenitor cells in the mesoderm, which cells participate in the formation of the vasculature during early embryonic development (26). By contrast, adult vasculogenesis refers to a post-natal process by which angiogenic cells are mobilized from the bone marrow, in response to HIF-1α regulated factors generated by ischemic tissue. The bone-marrow-derived angiogenic cells circulate in the blood, homing to the ischemic or injured tissue, and participate in the restoration of the microvasculature. These circulating cells include myeloid angiogenic cells (which can home to the ischemic tissue, and generate angiogenic cytokines and chemokines), as well as true endothelial colony-forming cells, which can incorporate into the newly forming vasculature (27). In a mouse wound healing model bone marrow (BM)–derived endothelial progenitor cells responded to the gradient of hypoxia and were preferentially recruited to the most ischemic region to contribute to the micro-vasculature (28). Similar findings were found in muscle, heart, and other tissues (29–31). Some circulating cells that contribute to adult vasculogenesis may be derived from sites other than the bone marrow, likely the endothelium of other organs (31). In this case, it is likely that endothelial cells are mobilized from the vascular lumen into the circulation by systemic angiogenic factors, home to the ischemic site, and incorporate into the microcirculation.
Angiogenesis is the process of expansion of the microvasculature, where new microvessels form from pre-existing ones (32). Angiogenesis begins with sprouting of capillary endothelial cells into the tissue matrix, proliferation, migration, and luminogenesis to form functional microvessels. Angiogenesis is a pre-requisite for tissue regeneration and remodeling, but may also contribute to pathology. Tumor angiogenesis supports the growth and metastasis of tumors, and pathological neovascularization participates in arteriosclerosis, diabetic retinopathy, and arthritis (33). By contrast, deficient angiogenesis contributes to impaired tissue healing, as seen with chronic ischemia of peripheral arterial disease.
Whereas vasculogenesis and angiogenesis contribute to the restoration of the microvasculature, arteriogenesis is in response to large vessel disease. For example, in patients with an occluded superficial femoral artery, angiography frequently reveals collateral channels that connect branches of the deep femoral artery (which normally supplies the thigh) to branches of the popliteal artery, thus restoring flow to the lower leg. These collateral channels have a characteristic corkscrew appearance, and likely form from small pre-existing high resistance vessels. When the conduit vessels are not diseased, blood flows preferentially through them and not the collateral channels. As the conduit vessels becomes progressively narrowed with the progression of PAD, resistance to flow increases, and some flow now occurs through the collateral channels. The endothelium of the collateral channels release vasodilator factors to functionally increase vessel diameter, and generate adhesion molecules and chemokines that attract macrophages (34). Macrophage infiltration and generation of matrix metalloproteinases cause structural remodeling and enlargement of the collateral channels to complete the process of arteriogenesis (34).
Regeneration of the vasculature requires transcriptional signaling that directs the specialization of arterial, venous, lymphatic and capillary endothelial cells (35). VEGF (vascular endothelial growth factor) and Notch signaling are critical signals for arterial phenotype (36–39), whereas COUP-TFII (COUP transcription factor 2) and BMP (bone morphogenetic protein)-SMAD signaling are required for venous specification (40–43). Altered expression of these factors during development and in the post-natal state can lead to arteriovenous malformations(44, 45). Single cell RNAseq has revealed many subsets of endothelial cells, confirming the heterogeneity of this cell type (46–54). For example, in the cerebrovascular endothelium, ECs are specialized to form tight junctions that provide a relatively impermeable lining. Experimental modification of the brain-barrier, such as that mediated by ANGPT2 (angiopoietin 2) deficiency, causes a disruption of the tight junctions of brain endothelial cells, with vascular leakage (55, 56). Increased permeability of the brain endothelium may contribute to impaired cognition, as in SARS CoV 2 infection. By contrast the fenestrated hepatic endothelium provides for high permeability to support the filtration and metabolic functions of the liver. The transcriptional factor c-Maf plays a key role in the development of increased EC permeability (57). The heterogeneity of ECs provides for a microvasculature that supports specialized tissue functions. Furthermore, in response to ischemia or injury, the endothelium exhibits great plasticity, and assumes new functions to support tissue and vascular regeneration (58–61).
We have discovered a novel form of vascular regeneration in our murine model of peripheral artery disease. Angiogenic transdifferentiation is a form of mesenchyme-to-endothelial transition that occurs in the setting of limb ischemia. In the setting of limb ischemia, we have observed that a subset of fibroblasts transdifferentiates into endothelial cells that incorporate into the microvasculature. Another set of fibroblasts generates angiogenic cytokines to support the process. Furthermore, this process requires both inflammatory signaling and angiogenic cytokines. The inflammatory signaling within a somatic cell triggers epigenetic changes that favor DNA accessibility, in a process that we have termed “transflammation”. In this state, the chromatin is in a more open configuration, and permissive to changes in cell fate. Cell fate transitions are then determined by the cellular milieu, e.g., angiogenic cytokines in the setting of hypoxia, which can promote mesenchyme-to-endothelial transition.
Our discovery of transflammation owes much to the work of Shinyi Yamanaka. In 2006, he electrified the field of stem cell biology with his generation of induced pluripotent stem cells (iPSCs) (62), which work garnered the Nobel Prize in 2012. The iPSCs facilitated studies of the determinants of pluripotency; elucidation of the development and differentiation of different progenitor and somatic cell types; a greater understanding of the mechanisms of disease; and a potentially unlimited resource for cellular therapies (63, 64). The forced expression of the 4 transcriptional factors Oct3/4, Sox2, Klf4, and c-Myc (OSKM) using a retroviral vector was highly effective in generating iPSCs and widely used (65). However, to avoid the off-target effects of a strategy that integrated these exogenous genes into the human genome, we attempted to generate iPSCs using OSKM in the form of cell-permeant peptides (CPPs). This approach failed after many attempts to refine the dosing and timing, with poor induction of genes downstream of OSKM and little or no generation of iPSCs. Why were OSKM ineffective as cell-permeant proteins when a viral vector encoding OSKM was effective at generating iPSCs? Previous work by our group had shown that viral vectors could alter endothelial cell phenotype (66). Was it possible that the viral vector itself could be playing a role in a more dramatic change in cell phenotype, i.e., induction of pluripotency? To answer this question, we added a retroviral vector encoding the green fluorescent protein (GFP) together with the transcriptional factors. With some amazement, we noted that when the CPPs were combined with a retroviral vector encoding GFP, they were as effective at inducing the downstream transcriptional program as retroviral vectors encoding the pluripotency factors (67). This observation indicated that the retroviral vector itself, and not the Yamanaka factors alone, contributed to reprogramming. We subsequently showed that inflammatory activation of a somatic cell (in this case by the retroviral vector) shifted the balance of epigenetic factors to promote an open chromatin configuration. In an open chromatin configuration, OSKM could gain greater access to the chromatin to activate downstream genes. For example, histone acetyltransferase family members were upregulated, and histone deacetylase family members were reduced in their expression. These changes were associated with a gain of activating histone marks, and a loss of repressive marks, on downstream pluripotency genes. In gain- and loss-of-function studies, we found that the toll-like receptor 3 (TLR3) pathway which senses double-stranded viral RNA, was required for efficient induction of pluripotency by retroviral vectors encoding OSKM (67). Other pattern recognition receptors (e.g., RIG-I) which are stimulated by viral RNA, also involved in the induction of pluripotency by retroviral vectors encoding OSKM (68). In fact, it seems likely that any activation of inflammatory signaling within a somatic cell is likely to promote DNA accessibility and phenotypic fluidity because the knockdown of NFkB or IRF3 each reduced the generation of iPSCs (67).
Because inflammatory signaling is associated with the generation of reactive oxygen species (ROS) signaling, we were interested to understand if there was a role of ROS signaling in iPSC generation. We examined ROS production during the reprogramming of mouse embryonic fibroblasts (MEFs) employing a doxycycline-inducible cassette encoding OSKM (69). With induction of reprogramming, ROS generation increased. Scavenging the ROS with antioxidants reduced the generation of iPSCs. Pharmacological antagonists of NADPH oxidase (the major generator of ROS in fibroblasts), or genetic knockdown of the enzyme substantially decreased reprogramming efficiency. On the other hand, when we increased ROS generation substantially using genetic and pharmacological approaches the generation of iPSCs was also impaired (69). These studies indicate that there is a Goldilock's zone for inflammatory signaling and generation of ROS, where cell fate transitions are facilitated.
The activation of TLR3 also increases the expression of inducible nitric oxide synthase (iNOS), which produces nitrogen oxide radicals that can S-nitrosylate proteins to modify their activity. During transdifferentiation of fibroblasts to endothelial cells, we have observed that iNOS expression is increased in both the cytoplasm and nucleus, indicating that the enzyme translocates to the nucleus. There it binds RING1a of the polycomb repressive complex 1,and S-nitrosylates RING1a. RING1a subsequently dissociates from the chromatin, with a concomitant reduction in its suppressive histone marks (70). In addition, iNOS binds to, and S-nitrosylates, a component of the NURD complex (a deacetylase), thereby reducing histone deacetylation (71). These effects of inflammatory signaling, e.g., iNOS translocation to the nucleus and S-nitrosylation (and inhibition) of repressive epigenetic modifiers, promotes an open chromatin configuration that facilitates cell fate transitions and tissue regeneration (70, 71). Transflammation is equally critical for the induction of pluripotency, as well as the transdifferentiation of a somatic cell into another cell type (67–74).
With respect to transdifferentiation, we have been able to generate induced endothelial cells (iECs) from human fibroblasts simply by activating inflammatory signaling (with the TLR3 agonist poly inosinic:cytidylic acid, i.e., Poly I:C), and adding to the medium a cocktail of endothelial differentiating factors (i.e., VEGF, BMP4, FGF, and 8Br-cAMP, together with a TGF β inhibitor) (73). In the absence of Poly I:C, the endothelial differentiating factors do not induce fibroblasts to become iECs. However, with activation of inflammatory signaling, the increase in DNA accessibility facilitates the transcriptional direction provided by the endothelial differentiation factors, and iECs are generated.
It seems likely that in the setting of tissue injury or ischemia, the subsequent inflammatory signaling increases DNA accessibility in the cells of the damaged tissue. In our conceptual model the increased DNA accessibility would promote cellular plasticity, and adaptive changes in cell phenotype. Indeed, we have tested this hypothesis in the murine model of limb ischemia, using single cell RNA sequencing and cell lineage tracing (74). In this model, we were able to define 8 subsets of fibroblasts in the mouse hindlimb, each of which subsets were detected by lineage tracing and expressed a set of typical fibroblast genes such as Col1a. Two of these fibroblast subsets increased in the ischemic limb. One subset (cluster 5) expressed some typical endothelial identity genes, whereas another (cluster 8) expressed some angiogenic cytokines. Using FACS sorting and antibodies to surface markers for these clusters, we pulled out cells that resembled fibroblasts in 2D culture. However, when we added the cells to Matrigel, cluster 5 cells formed a network very similar to that formed by genuine endothelial cells and expressed endothelial surface markers, whereas cluster 8 continued to morphologically resemble fibroblasts (which form nodules in Matrigel) but made angiogenic cytokines. The importance of inflammation in the generation of these clusters, and their contribution to perfusion, was demonstrated by pharmacological (dexamethasone) or genetic knockdown (p65 kd) of inflammatory activation. Pharmacological or genetic suppression of inflammatory activation markedly reduced the numbers of iECs detected (by lineage tracing), limited recovery of perfusion (by laser doppler spectroscopy), and impaired healing of the hindlimb (74). To conclude, a subset of fibroblasts in the hindlimb appears to be poised to become endothelial cells under the influence of inflammatory activation and angiogenic factors generated during hypoxia. Furthermore, these endogenous iECs appear to contribute significantly to the restoration of perfusion and tissue healing. We call this process Angiogenic Transdifferentiation (74) (Figure 1).
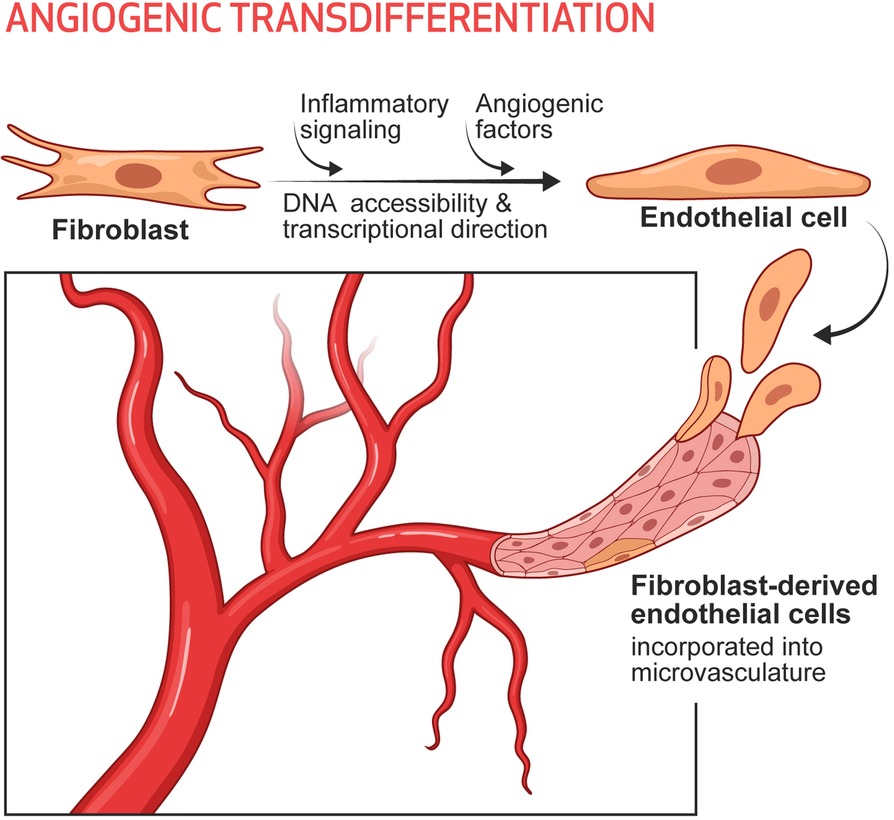
Figure 1. Angiogenic transdifferentiation contributes to the expansion of the microvasculature, particularly in the setting of ischemia. Under the influence of inflammatory signaling (which increases DNA accessibility) and environmental cues (such as angiogenic cytokines, which provide transcriptional direction) some subset(s) of fibroblasts can transdifferentiate into endothelial cells. These induced endothelial cells contribute to the expansion of the microvasculature as a response to ischemia and augment tissue perfusion.
Metabolic regulation of DNA accessibility has emerged as a key regulatory process controlling cell fate (75). It has been intensively investigated regarding the bidirectional regulation between immune cells and metabolism (76–79). However, less is known about the role of cell-autonomous immune signaling on non-immune cells responding to tissue damage (79). We have recently shown that metabolic modulation of epigenetic regulation plays an important role in cell fate decisions in non-immune cells (80). In our study, we found that innate immune activation triggers a glycolytic shift that is required for transdifferentiation. In parallel, the mitochondrial citrate transporter (Slc25a1) is upregulated to facilitate the export of citrate out of the mitochondria. Concurrently, there is an increase in the expression of nuclear ATP citrate lyase (ACL), which converts citrate to acetyl-CoA to support the increase in histone acetyltransferase activity. The subsequent increase in histone acetylation increases DNA accessibility and facilitates the transdifferentiation of fibroblasts to endothelial cells. This work indicates an important role for metabolism during transdifferentiation and vascular regeneration.
Ischemic cardiovascular disease is the leading cause of disability, morbidity, and mortality globally. Of concern, ischemic vascular disorders, such as myocardial infarction, cerebrovascular disease, peripheral vascular disease, and microcirculatory disorder, are increasing worldwide (81). Peripheral artery disease(PAD) occurs in about 12% of the U.S. adult population over the age of 60 (82), and in the most severe form of critical limb ischemia (CLI), is associated with a mortality rate of about 20% within 1 year of diagnosis. Endovascular procedures and surgical bypass for CLI are useful in salvaging limbs but amputation-free survival in the absence of efficacious medical treatment for limb ischemia remains poor (83). Although angiogenesis is critical for perfusion recovery (84), angiogenic therapies have largely failed in improving function (e.g., walking distance) and in alleviating complications (e.g., ischemic ulcers) of CLI (85, 86). We hypothesize that a reduction in DNA accessibility and cellular plasticity may be playing a significant role in the impairment of angiogenesis and arteriogenesis that is observed in CLI patients. Because these patients have evidence of significant inflammation, particularly in the region of ischemia and non-healing ulcers, it is likely that in these regions, inflammatory signaling is excessively activated. As we have shown, a modest amount of inflammatory signaling is necessary for DNA accessibility and cell fate transitions. However, in the face of excessive inflammatory activation, cellular plasticity is impaired (69, 71). Recent clinical trials using anti-inflammation strategies showed promising results in lowering the risk of major adverse cardiovascular events (87–89). Excessive inflammatory signaling is clearly involved in the progression of cardiovascular disease (90–94), and it may also be involved in the impairment of tissue healing that is frequently associated with cardiovascular disease. However, because an optimal amount of inflammatory signaling is also necessary for angiogenic transdifferentiation and wound healing, we feel that caution should be exercised in the use of these agents in patients with healing wounds or tissue damage (95). It would be of great clinical merit to define the optimal range of inflammatory signaling during the healing stages; develop diagnostics that could quantify the degree of inflammatory signaling in the target tissue; and therapeutics that could modulate the level of inflammatory signaling.
In this regard, there is a deficiency of multi-omics information for the affected tissues in PAD. It will be useful to characterize the cell composition, and the genetic, epigenetic, and proteomic features of each cell subtype in patient samples vs. those from healthy individuals; from patients with PAD vs. those with CLI; and from those individuals that do not have PAD but have the pre-requisite risk factors for PAD. Such studies might identify deficiencies or aberrations in cell subtypes that play a role in the disease and might identify pathways that are contributory. Such information could provide a greater understanding of the heterogeneity in the presentation of PAD; could lead to mechanistic insights into etiology; and could provide avenues toward novel therapies.
Vascular regeneration in the adult includes the processes of angiogenesis, arteriogenesis, and adult vasculogenesis. To these, we add the new process of angiogenic transdifferentiation. Angiogenic transdifferentiation involves a cell fate switch that requires an optimal amount of inflammatory signaling (which increases DNA accessibility) and angiogenic factors (that provide transcriptional direction). This newly discovered response to ischemia appears to play an important role in the expansion of the microvasculature and the restoration of perfusion.
Both authors contributed substantially to all aspects of the article. All authors contributed to the article and approved the submitted version.
This work is supported in part by grants to JPC from the National Institutes of Health (NIH) R01s HL133254, HL157790, and HL148338; as well as the Cancer Prevention and Research Institute of Texas CPRIT RP150611). Our manuscript was graced by the artistry of Rachael Whitehead, who created the figure.
JC and LL are inventors on a patent filing assigned to Houston Methodist Hospital dealing with metabolic modulation of cell fate transitions and angiogenesis.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
En1, Engrailed-1; Cthrc1, collagen triple helix repeat containing 1; Thbs4, thrombospondin 4; FAP, fibroblast activation protein; CAR, chimeric antigen receptor; ECs, endothelial cells; HIF1α, hypoxia-inducible factor 1-alpha; BM, bone marrow; PAD, peripheral arterial disease; VEGF, vascular endothelial growth factor; COUP-TFII, COUP transcription factor 2; BMP, bone morphogenetic protein; ANGPT2, angiopoietin 2; iPSCs, induced pluripotent stem cells; OSKM, Oct3/4, Sox2, Klf4, and c-Myc; CPPs, cell-permeant peptides; GFP, green fluorescent protein; TLR3, toll-like receptor 3; ROS, reactive oxygen species; MEFs, mouse embryonic fibroblasts; iNOS, inducible nitric oxide synthase; iECs, induced endothelial cells; Poly I:C, poly inosinic:cytidylic acid; FGF, fibroblast growth factor; 8-Br-cAMP, 8-Bromoadenosine 3′,5′-cyclic adenosine monophosphate; TGF β, transforming growth factor beta; Col1a, collagen, type I, alpha; FACS, fluorescence-activated cell sorting; ACL, ATP citrate lyase; CLI, critical limb ischemia.
1. Muhl L, Genové G, Leptidis S, Liu J, He L, Mocci G. Publisher correction: single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat Commun. (2020) 11:4493. doi: 10.1038/s41467-020-18511-8
2. Wang L, Yang Y, Ma H, Xie Y, Xu J, Near D. Single-cell dual-omics reveals the transcriptomic and epigenomic diversity of cardiac non-myocytes. Cardiovasc Res. (2022) 118:1548–63. doi: 10.1093/cvr/cvab134
3. Hesse J, Owenier C, Lautwein T, Zalfen R, Weber JF, Ding Z. Single-cell transcriptomics defines heterogeneity of epicardial cells and fibroblasts within the infarcted murine heart. Elife. (2021) 10:e65921. doi: 10.7554/eLife.65921
4. Harvey RP, Patrick R, Janbandhu V, Contreras O. Cardiac fibroblast heterogeneity and dynamics through the lens of single-cell dual ‘omics. Cardiovasc Res. (2022) 118:1380–2. doi: 10.1093/cvr/cvac037
5. Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. (2012) 18:1262–70. doi: 10.1038/nm.2848
6. Currie JD, Lidia G, Murawala P, Schuez M, Michel M, Tanaka EM. The Prrx1 limb enhancer marks an adult subpopulation of injury-responsive dermal fibroblasts. Biol Open. (2019) 8(7):bio043711. doi: 10.1242/bio.043711
7. Jiang D, Rinkevich Y. Scars or regeneration?-dermal fibroblasts as drivers of diverse skin wound responses. Int J Mol Sci. (2020) 21:617. doi: 10.3390/ijms21020617
8. Jiang D, Correa-Gallegos D, Christ S, Stefanska A, Liu J, Ramesh P. Two succeeding fibroblastic lineages drive dermal development and the transition from regeneration to scarring. Nat Cell Biol. (2018) 20:422–31. doi: 10.1038/s41556-018-0073-8
9. Mascharak S, desJardins-Park HE, Davitt MF, Griffin M, Borrelli MR, Moore AL. Preventing engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science. (2021) 372:362. doi: 10.1126/science.aba2374
10. Ruiz-Villalba A, Romero JP, Hernández SC, Vilas-Zornoza A, Fortelny N, Castro-Labrador L. Single-cell RNA sequencing analysis reveals a crucial role for CTHRC1 (collagen triple Helix repeat containing 1) cardiac fibroblasts after myocardial infarction. Circulation. (2020) 142:1831–47. doi: 10.1161/CIRCULATIONAHA.119.044557
11. Peisker F, Halder M, Nagai J, Ziegler S, Kaesler N, Hoeft K. Mapping the cardiac vascular niche in heart failure. Nat Commun. (2022) 13:3027. doi: 10.1038/s41467-022-30682-0
12. Moretti L, Stalfort J, Barker TH, Abebayehu D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J Biol Chem. (2022) 298:101530. doi: 10.1016/j.jbc.2021.101530
13. Dorrier CE, Aran D, Haenelt EA, Sheehy RN, Hoi KK, Pintarić L. CNS Fibroblasts form a fibrotic scar in response to immune cell infiltration. Nat Neurosci. (2021) 24:234–44. doi: 10.1038/s41593-020-00770-9
14. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. (2016) 118:1021–40. doi: 10.1161/CIRCRESAHA.115.306565
15. Shook BA, Wasko RR, Rivera-Gonzalez GC, Salazar-Gatzimas E, López-Giráldez F, Dash BC. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science. (2018) 362:909. doi: 10.1126/science.aar2971
16. Sinha S, Sparks HD, Labit E, Robbins HN, Gowing K, Jaffer A. Fibroblast inflammatory priming determines regenerative versus fibrotic skin repair in reindeer. Cell. (2022) 185:4717–4736.e25. doi: 10.1016/j.cell.2022.11.004
17. Van Linthout S, Miteva K, Tschope C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res. (2014) 102:258–69. doi: 10.1093/cvr/cvu062
18. Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L. CAR T cells produced in vivo to treat cardiac injury. Science. (2022) 375:91–6. doi: 10.1126/science.abm0594
19. Ong YT, Andrade J, Armbruster M, Shi C, Castro M, Costa ASH. A YAP/TAZ-TEAD signalling module links endothelial nutrient acquisition to angiogenic growth. Nat Metab. (2022) 4:672–82. doi: 10.1038/s42255-022-00584-y
20. Abumrad NA, Cabodevilla AG, Samovski D, Pietka T, Basu D, Goldberg IJ. Endothelial cell receptors in tissue lipid uptake and metabolism. Circ Res. (2021) 128:433–50. doi: 10.1161/CIRCRESAHA.120.318003
21. Amersfoort J, Eelen G, Carmeliet P. Immunomodulation by endothelial cells—partnering up with the immune system? Nat Rev Immunol. (2022) 22:576–88. doi: 10.1038/s41577-022-00694-4
22. Shao Y, Saredy J, Yang WY, Sun Y, Lu Y, Saaoud F. Vascular endothelial cells and innate immunity. Arterioscler Thromb Vasc Biol. (2020) 40:e138–52. doi: 10.1161/ATVBAHA.120.314330
23. Cooke JP. Flow, NO, and atherogenesis. Proc Natl Acad Sci U S A. (2003) 100(3):768–70. doi: 10.1073/pnas.0430082100
24. Nakajima H, Yamamoto K, Agarwala S, Terai K, Fukui H, Fukuhara S. Flow-dependent endothelial YAP regulation contributes to vessel maintenance. Dev Cell. (2017) 40:523–536.e6. doi: 10.1016/j.devcel.2017.02.019
25. Luxan G, Dimmeler S. The vasculature: a therapeutic target in heart failure? Cardiovasc Res. (2022) 118:53–64. doi: 10.1093/cvr/cvab047
26. Luttun A, Carmeliet P. De novo vasculogenesis in the heart. Cardiovasc Res. (2003) 58:378–89. doi: 10.1016/s0008-6363(03)00258-x
27. Banno K, Yoder M. Tissue regeneration using endothelial colony-forming cells: promising cells for vascular repair. Pediatr Res. (2018) 83:283–90. doi: 10.1038/pr.2017.231
28. Tepper OM, Capla JM, Galiano RD, Ceradini DJ, Callaghan MJ, Kleinman ME. Adult vasculogenesis occurs through in situ recruitment, proliferation, and tubulization of circulating bone marrow-derived cells. Blood. (2005) 105:1068–77. doi: 10.1182/blood-2004-03-1051
29. Kawada H, Ogawa M. Bone marrow origin of hematopoietic progenitors and stem cells in murine muscle. Blood. (2001) 98:2008–13. doi: 10.1182/blood.v98.7.2008
30. Heeschen C, Chang E, Aicher A, Cooke JP. Endothelial progenitor cells participate in nicotine-mediated angiogenesis. J Am Coll Cardiol. (2006) 48(12):2553–60. doi: 10.1016/j.jacc.2006.07.066.
31. Aicher A, Rentsch M, Sasaki K, Ellwart JW, Fändrich F, Siebert R, et al. Nonbone marrow-derived circulating progenitor cells contribute to postnatal neovascularization following tissue ischemia. Circ Res. (2007) 100(4):581–9. doi: 10.1161/01.RES.0000259562.63718.35
32. Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. (2007) 8:464–78. doi: 10.1038/nrm2183
33. Schwartz BG, Economides C, Mayeda GS, Burstein S, Kloner RA. The endothelial cell in health and disease: its function, dysfunction, measurement and therapy. Int J Impot Res. (2010) 22:77–90. doi: 10.1038/ijir.2009.59
34. Troidl K, Schaper W. Arteriogenesis versus angiogenesis in peripheral artery disease. Diabetes Metab Res Rev. (2012) 28(Suppl 1):27–9. doi: 10.1002/dmrr.2232
35. Trimm E, Red-Horse K. Vascular endothelial cell development and diversity. Nat Rev Cardiol. (2022) 20:1–14. doi: 10.1038/s41569-022-00770-1
36. Wythe JD, Dang LTH, Patrick Devine W, Boudreau E, Artap ST, He D. ETS Factors regulate vegf-dependent arterial specification. Dev Cell. (2013) 26:45–58. doi: 10.1016/j.devcel.2013.06.007
37. Lanner F, Sohl M, Farnebo F. Functional arterial and venous fate is determined by graded VEGF signaling and notch status during embryonic stem cell differentiation. Arterioscler Thromb Vasc Biol. (2007) 27:487–93. doi: 10.1161/01.ATV.0000255990.91805.6d
38. Corada M, Morini MF, Dejana E. Signaling pathways in the specification of arteries and veins. Arterioscler Thromb Vasc Biol. (2014) 34:2372–7. doi: 10.1161/ATVBAHA.114.303218
39. Chen D, Schwartz MA, Simons M. Developmental perspectives on arterial fate specification. Front Cell Dev Biol. (2021) 9:691335. doi: 10.3389/fcell.2021.691335
40. Swift MR, Weinstein BM. Arterial-venous specification during development. Circ Res. (2009) 104:576–88. doi: 10.1161/CIRCRESAHA.108.188805
41. Li RF, Wu TY, Mou YZ, Wang YS, Chen CL, Wu CY. Nr2f1b control venous specification and angiogenic patterning during zebrafish vascular development. J Biomed Sci. (2015) 22:104. doi: 10.1186/s12929-015-0209-0
42. Cui X, Lu YW, Lee V, Kim D, Dorsey T, Wang Q. Venous endothelial marker COUP-TFII regulates the distinct pathologic potentials of adult arteries and veins. Sci Rep. (2015) 5:16193. doi: 10.1038/srep16193
43. Chavkin NW, Genet G, Poulet M, Jeffery ED, Marziano C, Genet N. Endothelial cell cycle state determines propensity for arterial-venous fate. Nat Commun. (2022) 13:5891. doi: 10.1038/s41467-022-33324-7
44. Lee HW, Xu Y, He L, Choi W, Gonzalez D, Jin SW. Role of venous endothelial cells in developmental and pathologic angiogenesis. Circulation. (2021) 144:1308–22. doi: 10.1161/CIRCULATIONAHA.121.054071
45. Hwa JJ, Beckouche N, Huang L, Kram Y, Lindskog H, Wang RA. Abnormal arterial-venous fusions and fate specification in mouse embryos lacking blood flow. Sci Rep. (2017) 7:11965. doi: 10.1038/s41598-017-12353-z
46. Su T, Stanley G, Sinha R, D'Amato G, Das S, Rhee S. Single-cell analysis of early progenitor cells that build coronary arteries. Nature. (2018) 559:356–62. doi: 10.1038/s41586-018-0288-7
47. Garcia FJ, Sun N, Lee H, Godlewski B, Mathys H, Galani K. Single-cell dissection of the human brain vasculature. Nature. (2022) 603:893–9. doi: 10.1038/s41586-022-04521-7
48. Tabula Sapiens C, Jones RC, Karkanias J, Krasnow MA, Pisco AO, Quake SR. The tabula sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science. (2022) 376:eabl4896. doi: 10.1126/science.abl4896
49. Winkler EA, Kim CN, Ross JM, Garcia JH, Gil E, Oh I. A single-cell atlas of the normal and malformed human brain vasculature. Science. (2022) 375:eabi7377. doi: 10.1126/science.abi7377
50. Bondareva O, Rodríguez-Aguilera JR, Oliveira F, Liao L, Rose A, Gupta A. Single-cell profiling of vascular endothelial cells reveals progressive organ-specific vulnerabilities during obesity. Nat Metab. (2022) 4:1591–610. doi: 10.1038/s42255-022-00674-x
51. Marcu R, Choi YJ, Xue J, Fortin CL, Wang Y, Nagao RJ. Human organ-specific endothelial cell heterogeneity. iScience. (2018) 4:20–35. doi: 10.1016/j.isci.2018.05.003
52. Urbanczyk M, Zbinden A, Schenke-Layland K. Organ-specific endothelial cell heterogenicity and its impact on regenerative medicine and biomedical engineering applications. Adv Drug Deliv Rev. (2022) 186:114323. doi: 10.1016/j.addr.2022.114323
53. Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature. (2016) 529:316–25. doi: 10.1038/nature17040
54. Gomez-Salinero JM, Rafii S. Endothelial cell adaptation in regeneration. Science. (2018) 362:1116–7. doi: 10.1126/science.aar4800
55. Mäe MA, Liqun L, Nordling S, Vazquez-Liebanas E, Nahar K, Jung B, et al. Correction to: single-cell analysis of blood-brain barrier response to pericyte loss. Circ Res. (2021) 128:e123. doi: 10.1161/RES.0000000000000484
56. Claesson-Welsh L, Dejana E, McDonald DM. Permeability of the endothelial barrier: identifying and reconciling controversies. Trends Mol Med. (2021) 27:314–31. doi: 10.1016/j.molmed.2020.11.006
57. Gómez-Salinero JM, Izzo F, Lin Y, Houghton S, Itkin T, Geng F. Specification of fetal liver endothelial progenitors to functional zonated adult sinusoids requires c-maf induction. Cell Stem Cell. (2022) 29:593–609.e7. doi: 10.1016/j.stem.2022.03.002
58. Pasut A, Becker LM, Cuypers A, Carmeliet P. Endothelial cell plasticity at the single-cell level. Angiogenesis. (2021) 24:311–26. doi: 10.1007/s10456-021-09797-3
59. Greenspan LJ, Weinstein BM. To be or not to be: endothelial cell plasticity in development, repair, and disease. Angiogenesis. (2021) 24:251–69. doi: 10.1007/s10456-020-09761-7
60. Tombor LS, John D, Glaser SF, Luxán G, Forte E, Furtado M. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat Commun. (2021) 12:681. doi: 10.1038/s41467-021-20905-1
61. Rohlenova K, Goveia J, García-Caballero M, Subramanian A, Kalucka J, Treps L. Single-Cell RNA sequencing maps endothelial metabolic plasticity in pathological angiogenesis. Cell Metab. (2020) 31:862–877.e14. doi: 10.1016/j.cmet.2020.03.009
62. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
63. Yamanaka S. Induced pluripotent stem cells: past, present, and future. Cell Stem Cell. (2012) 10:678–84. doi: 10.1016/j.stem.2012.05.005
65. Hu K. All roads lead to induced pluripotent stem cells: the technologies of iPSC generation. Stem Cells Dev. (2014) 23:1285–300. doi: 10.1089/scd.2013.0620
66. Weis M, Kledal TN, Lin KY, Panchal SN, Gao SZ, Valantine HA, et al. Cytomegalovirus infection impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine in transplant arteriosclerosis. Circulation. (2004) 109(4):500–5. doi: 10.1161/01.CIR.0000109692.16004.AF
67. Lee J, Sayed N, Hunter A, Au KF, Wong WH, Mocarski ES. Activation of innate immunity is required for efficient nuclear reprogramming. Cell. (2012) 151:547–58. doi: 10.1016/j.cell.2012.09.034
68. Sayed N, Ospino F, Himmati F, Lee J, Chanda P, Mocarski ES. Retinoic acid inducible gene 1 protein (RIG1)-like receptor pathway is required for efficient nuclear reprogramming. Stem Cells. (2017) 35:1197–207. doi: 10.1002/stem.2607
69. Zhou G, Meng S, Li Y, Ghebre YT, Cooke JP. Optimal ROS signaling is critical for nuclear reprogramming. Cell Rep. (2016) 15(5):919–25. doi: 10.1016/j.celrep.2016.03.084
70. Meng S, Zhou G, Gu Q, Chanda PK, Ospino F, Cooke JP. Transdifferentiation requires iNOS activation: role of RING1A S-nitrosylation. Circ Res. (2016) 119:e129–38. doi: 10.1161/CIRCRESAHA.116.308263
71. Chanda PK, Meng S, Lee J, Leung HE, Chen K, Cooke JP. Nuclear S-nitrosylation defines an optimal zone for inducing pluripotency. Circulation. (2019) 140:1081–99. doi: 10.1161/CIRCULATIONAHA.119.042371
72. Matrone G, Jung SY, Choi JM, Jain A, Eastwood Leung HC, Rajapakshe K. Nuclear S-nitrosylation impacts tissue regeneration in zebrafish. Nat Commun. (2021) 12:6282. doi: 10.1038/s41467-021-26621-0 Sayed N. et al. Transdifferentiation of human fibroblasts to endothelial cells: role of innate immunity. Circulation. (2015) 131, 300–9. doi: 10.1161/CIRCULATIONAHA.113.007394.34725362
73. Sayed N, Wong WT, Ospino F, Meng S, Lee J, Jha A. Transdifferentiation of human fibroblasts to endothelial cells: role of innate immunity. Circulation. (2015) 131:300–9. doi: 10.1161/CIRCULATIONAHA.113.007394
74. Meng S, Lv J, Chanda PK, Owusu I, Chen K, Cooke JP. Reservoir of fibroblasts promotes recovery from limb ischemia. Circulation. (2020) 142:1647–62. doi: 10.1161/CIRCULATIONAHA.120.046872
75. Li X, Egervari G, Wang Y, Berger SL, Lu Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol. (2018) 19:563–78. doi: 10.1038/s41580-018-0029-7
76. Kedia-Mehta N, Finlay DK. Competition for nutrients and its role in controlling immune responses. Nat Commun. (2019) 10:2123. doi: 10.1038/s41467-019-10015-4
77. Hu C, Pang B, Lin G, Zhen Y, Yi H. Energy metabolism manipulates the fate and function of tumour myeloid-derived suppressor cells. Br J Cancer. (2020) 122:23–9. doi: 10.1038/s41416-019-0644-x
78. Ahl PJ, Hopkins RA, Xiang WW, Au B, Kaliaperumal N, Fairhurst AM. Met-Flow, a strategy for single-cell metabolic analysis highlights dynamic changes in immune subpopulations. Commun Biol. (2020) 3:305. doi: 10.1038/s42003-020-1027-9
79. Adamik J, Munson PV, Hartmann FJ, Combes AJ, Pierre P, Krummel MF. Distinct metabolic states guide maturation of inflammatory and tolerogenic dendritic cells. Nat Commun. (2022) 13:5184. doi: 10.1038/s41467-022-32849-1
80. Lai L, Reineke E, Hamilton DJ, Cooke JP. Glycolytic switch is required for transdifferentiation to endothelial lineage. Circulation. (2019) 139:119–33. doi: 10.1161/CIRCULATIONAHA.118.035741
81. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/reperfusion. Compr Physiol. (2016) 7:113–70. doi: 10.1002/cphy.c160006
82. Conte MS, Bradbury AW, Kolh P, White JV, Dick F, Fitridge R. Global vascular guidelines on the management of chronic limb-threatening ischemia. Eur J Vasc Endovasc Surg. (2019) 58:S1–S109.e33. doi: 10.1016/j.ejvs.2019.05.006
83. Cooke JP, Losordo DW. Modulating the vascular response to limb ischemia: angiogenic and cell therapies. Circ Res. (2015) 116:1561–78. doi: 10.1161/CIRCRESAHA.115.303565
84. Cooke JP, Meng S. Vascular regeneration in peripheral artery disease. Arterioscler Thromb Vasc Biol. (2020) 40:1627–34. doi: 10.1161/ATVBAHA.120.312862
85. Annex BH, Cooke JP. New directions in therapeutic angiogenesis and arteriogenesis in peripheral arterial disease. Circ Res. (2021) 128:1944–57. doi: 10.1161/CIRCRESAHA.121.318266
86. Annex BH. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol. (2013) 10:387–96. doi: 10.1038/nrcardio.2013.70
87. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
88. D'Amario D, Rodolico D, Galli M. Colchicine in patients with chronic coronary disease. N Engl J Med. (2021) 384:778. doi: 10.1056/NEJMc2034992
89. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. (2019) 381:2497–505. doi: 10.1056/NEJMoa1912388
90. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis—from experimental insights to the clinic. Nat Rev Drug Discov. (2021) 20:589–610. doi: 10.1038/s41573-021-00198-1
91. Stark K, Massberg S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol. (2021) 18:666–82. doi: 10.1038/s41569-021-00552-1
92. Engelen SE, Robinson AJB, Zurke YX, Monaco C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat Rev Cardiol. (2022) 19:522–42. doi: 10.1038/s41569-021-00668-4
93. Murphy AJ, Febbraio MA. Immune-based therapies in cardiovascular and metabolic diseases: past, present and future. Nat Rev Immunol. (2021) 21:669–79. doi: 10.1038/s41577-021-00580-5
94. Kong P, Cui ZY, Huang XF, Zhang DD, Guo RJ, Han M. Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal Transduct Target Ther. (2022) 7:131. doi: 10.1038/s41392-022-00955-7
Keywords: endothelial cells, fibroblasts, nuclear reprogramming, transflammation, angiogenesis
Citation: Cooke JP and Lai L (2023) Role of angiogenic transdifferentiation in vascular recovery. Front. Cardiovasc. Med. 10:1155835. doi: 10.3389/fcvm.2023.1155835
Received: 31 January 2023; Accepted: 10 April 2023;
Published: 2 May 2023.
Edited by:
Roberto Vazquez-Padron, University of Miami, United StatesReviewed by:
Brian Annex, Augusta University, United States© 2023 Cooke and Lai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John P. Cooke anBjb29rZUBob3VzdG9ubWV0aG9kaXN0Lm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.