 Fenglan Liu
Fenglan Liu Yanfei Liu
Yanfei Liu Shihan Xu
Shihan Xu Qing Wang
Qing Wang Fengqin Xu1,2*
Fengqin Xu1,2* Yue Liu
Yue Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med., 13 June 2023
Sec. Cardiovascular Genetics and Systems Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1152201
This article is part of the Research TopicCytokines, Novel Cell Death Models and Pathways in Cardiovascular DiseasesView all 11 articles
Background: Growing observational studies have shown that abnormal systemic iron status is associated with Coronary heart disease (CHD). However, these results from observational studies was not entirely consistent.It remains unclear whether this relationship represents causality.It is necessary to explore the causal relationship between iron status and CHD and related cardiovascular diseases (CVD).
Objective: We aimed to investigate the potential casual relationship between serum iron status and CHD and related CVD using a two-sample Mendelian randomization (MR) approach.
Methods: Genetic statistics for single nucleotide polymorphisms (SNPs) between four iron status parameters were identified in a large-scale genome-wide association study (GWAS) conducted by the Iron Status Genetics organization. Three independent single nucleotide polymorphisms (SNPs) (rs1800562, rs1799945, and rs855791) aligned with four iron status biomarkers were used as instrumental variables. CHD and related CVD genetic statistics We used publicly available summary-level GWAS data. Five different MR methods random effects inverse variance weighting (IVW), MR Egger, weighted median, weighted mode, and Wald ratio were used to explore the causal relationship between serum iron status and CHD and related CVD.
Results: In the MR analysis, we found that the causal effect of serum iron (OR = 0.995, 95% CI = 0.992–0.998, p = 0.002) was negatively associated with the odds of coronary atherosclerosis (AS). Transferrin saturation (TS) (OR = 0.885, 95% CI = 0.797–0.982, p = 0.02) was negatively associated with the odds of Myocardial infarction (MI).
Conclusion: This MR analysis provides evidence for a causal relationship between whole-body iron status and CHD development. Our study suggests that a high iron status may be associated with a reduced risk of developing CHD.
Due to an aging population and declining fertility rates, cardiovascular disease mortality continues to rise and imposes a considerable economic and health burden on society (1). As research progresses, more and more studies show the correlation between systemic iron status and heart disease and related CVD (2).
CHD remains one of the major diseases threatening the health of the entire human population (3). The development of CHD involves many associated CVD. Among CVD, MI, a type of coronary heart disease, is a serious consequence of coronary heart disease. Hypertension (HP) is not only a CVD but also one of the risk factors for CHD, while heart failure (HF) is the end-stage disease of most CVD including CHD.
Iron, an essential mineral for maintaining homeostasis in the body, plays a key role in oxygen transport and utilization as well as in mitochondrial function (4). Iron deficiency (ID) is associated with morbidity and mortality in CHD, M I and HF (5–8). Studies have shown that ID is one of the most common complications of HF. Iron supplementation via intravenous can reduce the number of hospitalizations for HF (9). It has been shown that ID impairs the contractility of human cardiomyocytes by reducing mitochondrial function and decreasing energy production, which leads to impaired cardiac function (10). When uncontrolled elevation of iron concentration leads to iron overload, the basic cellular mechanisms and functional composition are disrupted and changed (11). The redox properties of iron enable the generation of reactive oxygen species (ROS), and iron (Fe2+) and iron (Fe3+) mediate lipid peroxidation, leading to the formation of alkoxyl (RO) and peroxyl (RO2) radicals (12, 13). Studies have displayed that in animal models of ischemia/reperfusion (I/R) cardiac tissue samples it can be observed that increased mitochondrial iron-related reactive oxygen species (ROS) production leads to myocardial injury (14). However, the physiopathological mechanisms of ID and iron overload participating in CHD and associated CVD remain unclear. In conclusion both opposite factors, ID and iron overload, can have an impact on CHD and related CVD. However, even in observational studies it is difficult to distinguish which specific association exists between iron status and CHD and associated CVD, as selective bias or other biases inherent in observational studies can still influence the results. Therefore, further studies are necessary to elucidate whether there is a causal or other relationship between iron status and CHD and associated CVD.
MR analysis is a novel method of epidemiological analysis that strengthens causal inferences by using genetic variation as an instrumental variable (IV) such as SNPs for exposure. This method minimizes the effects of residual confounding and strengthens causal inferences about the effects of specific exposure factors on outcomes while overcoming the limitations of traditional epidemiological studies (15). Here, we performed a 2-sample MR study to examine the association of iron status with HP, AS, CHD, MI, and HF based on the effect of systemic iron status and CHD and associated CVD using GWAS data, aiming to provide new evidence on the relationship between iron status and disease progression of CHD.
A genetic tool of four iron status biomarkers: ferritin, iron, transferrin, and transferrin saturation (TS), was selected for a two-sample MR analysis as a way to investigate the association of iron status biomarkers with the chain of cardiovascular disease events including HP, AS, CHD, MI and HF. The screening flowsheet is shown in Figure 1.
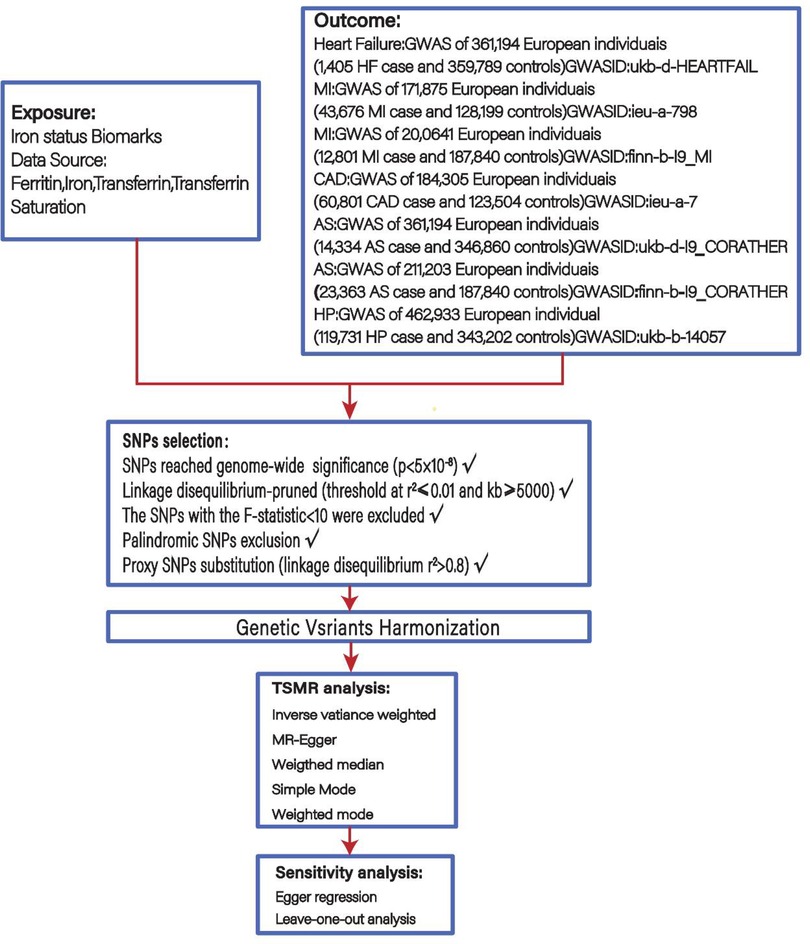
Figure 1. Flowsheet of Mendelian randomisation in this study.
Genetic variants associated with serum iron status were identified through a meta-analysis of 19 GWAS, which included 48,972 Europeans (16). A higher level of systemic iron was associated with higher iron levels, higher transferrin saturation, and higher ferritin levels, but decreased transferrin levels (4, 16, 17). Thus, genetic tools for iron status should be consistently related to each of these four markers, and thus three loci (rs1800562 and rs1799945 in the HFE gene, and rs855791 in TMPRSS6) could be identified in the meta-analysis performed by the GIS Consortium as being significantly associated with all four iron status markers genome-wide (p < 5 × 10−8) in a pattern consistent with effects on systemic iron status (i.e., increased serum iron, transferrin saturation and ferritin levels and decreased transferrin levels) (16), and these three were suggested as tools for systemic iron status in our MR analysis. Three SNPs had p < 5 × 10−8 and r2 ≤ 0.01, and their F-statistics were calculated to quantify the intensity of the selected instrument (F-value > 10) (18).
Characteristics and summary data of the SNPs and iron status parameters shown in Table 1 and Supplementary Table S1, respectively.
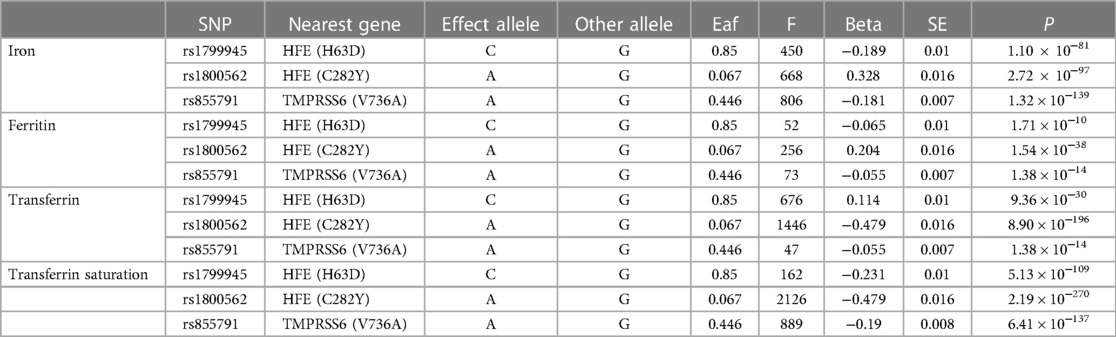
Table 1. The characteristics and summary data of the exposed SNPs and iron status parameters.
GWAS statistics related CHD and related CVD can be extracted from the corresponding authoritative consortium or cohort studies.
Data for both HP and HF were obtained from UKBiobank, with a final sample of 462,933 people of European descent for HP (119,731 HP cases and 343,202 non-cases), and a final sample of 361,194 people of European descent for HF (1,405 HF cases and 359,789 non-cases) (19). GWAS resources for coronary atherosclerosis are based on data from the UKBiobank consortium for a total of 361,194 individuals of European descent including 14,334 AS patients and 346,860 healthy controls (20), and FinnGen for a total of 211,203 individuals of European descent including 23,363 AS patients and 187,840 healthy controls. The second is based on FinnGen data for 211,203 individuals of European descent, including 23,363 AS patients and 187,840 healthy controls, available on the FinnGen study website (https://finngen.gitbook.io/documentation/) (20, 21). CHD Statistical data were obtained from the coronary artery genome-wide replication and meta-analysis (CARDIoGRAM) and coronary artery disease genetics (CARDIoGRAMplusC4D) GWASmeta-analysis, which included 60,801 cases and 123,504 controls (22). Statistics for MI were also obtained from the Coronary Artery Whole Genome Replication and Meta-Analysis (CARDIoGRAM) and Coronary Artery Disease Genetics (CARDIoGRAMplusC4D) GWASmeta-analysis, which included 43,678 cases and 128,199 controls (22), and FinnGen which included 12,801 cases and 187,840 controls (21). We used aggregated data from published GWAS that referenced the original definitions of the diseases in their GWAS without any modifications. The specific data sources used are in Supplementary Table S4.
MR analysis was performed using five methods, inverse variance weighting (IVW) under a multiplicative random effects model, MR-Egger, Weighted median, Simple mode, and Weighted mode. IVW was performed by combining the Wald ratio estimates for each individual SNP will be one causal estimate for each risk factor (23). A sensitivity analysis is required to test the validity and robustness of the IVW estimates due to invalid instrumental bias and polymorphism.
Sensitivity analyses include heterogeneity tests, genetic pleiotropy tests, and the “leave-one-out” method (19). In sensitivity analysis, we can use the weighted median method to check for invalid instrumental bias to estimate the multiplicity of potential causal effects or the inclusion of invalid instruments (24), while the use of MR-Egger regression can explain both the dilution bias of the skewed regression, with the mean level of multiplicity consisting of the intercept term (24, 25). Subsequently, symmetries can be visualized using funnel plots, and if they are skewed in one direction, they indicate a potential multiplicative effect (26). Cochran's Q test was also used to estimate the heterogeneity between the Wald ratio estimated for the different SNPs (27). Finally, to identify all genetic variants potentially affecting SNPs, we performed a “leave-one-out” analysis, whose fluctuations in results before and after removal of SNPs may reflect unstable associations.
To further investigate the relationship between iron status biomarkers and CHD and related CVD, we separately selected the iron status biomarkers with positive MR analysis results as described above and each SNP of the corresponding disease for two-sample MR analysis again, respectively, to obtain a more accurate estimate of the causal effect of each iron status biomarker and disease. The characteristics and summary data of the separately selected SNPs are shown in Supplementary Table S2.
“Two SampleMR” (version 0.5.6) of R software (version 4.2.1) was used for all analyses. P values less than 0.05 were considered statistically significant.
Genetically determined higher serum iron was negatively associated with higher odds of AS (OR = 0.995, 95% CI = 0.992–0.998, p = 0.002). The same results were obtained again using each SNP (OR = 0.996,95% CI = 0.992–0.998, p = 0.0009). Higher TS was negatively associated with higher odds of MI(Finn) (OR = 0.885, 95% CI = 0.797–0.982, p = 0.02). A repeat MR analysis using each SNP yielded no causal relationship between TS and MI (Finn) (p = 0.657) while higher TS was negatively associated with higher odds of MI (OR = 0.939, 95% CI = 0.886–0,996, p = 0.037), using the inverse variance-weighted approach. Moreover, the sensitivity analysis revealed that the selected instruments did not differ horizontally (p-values >0.05 for MR-Egger intercepts) or heterogeneously (p-values >0.05 for Cochran's Q statistic). In addition, MR-Egger regression and funnel plot appearance analyses showed a poor possibility of horizontal polymorphism (all p-values for MR-Egger intercept > 0.05) and visually, the leave-one-out analysis plot proved that the results were not altered by the removal of any SNPs and the results remained quite robust. The remaining MR analyses were negative that iron status markers were not causally associated with HP, CHD or HF.
Complete results are presented in Supplementary Figure S2. And the positive results were presented in Figure 2. The IVW results are shown in Table 2. Results of MR Analysis are expressed as the ORs for a positive result per one standard deviation (SD) increase for each iron biomarker, as shown in Figure 3 and Supplementary Figure S4. The leave-one-out analysis plot proves that removing any SNP does not change the results and is quite robust. The leave-one-out analysis graph of positive results are presented in Figure 4, and all results are presented in Supplementary Figure S3. The results of MR Analysis of AS and MI with the separately selected SNPs associated with Iron and TS are presented in Supplementary Table S3 and Supplementary Figure S3. The results of the positive results reanalysis are expressed as ORs per one standard deviation (SD) increase in positive results for each iron biomarker, as shown Supplementary Figure S4.
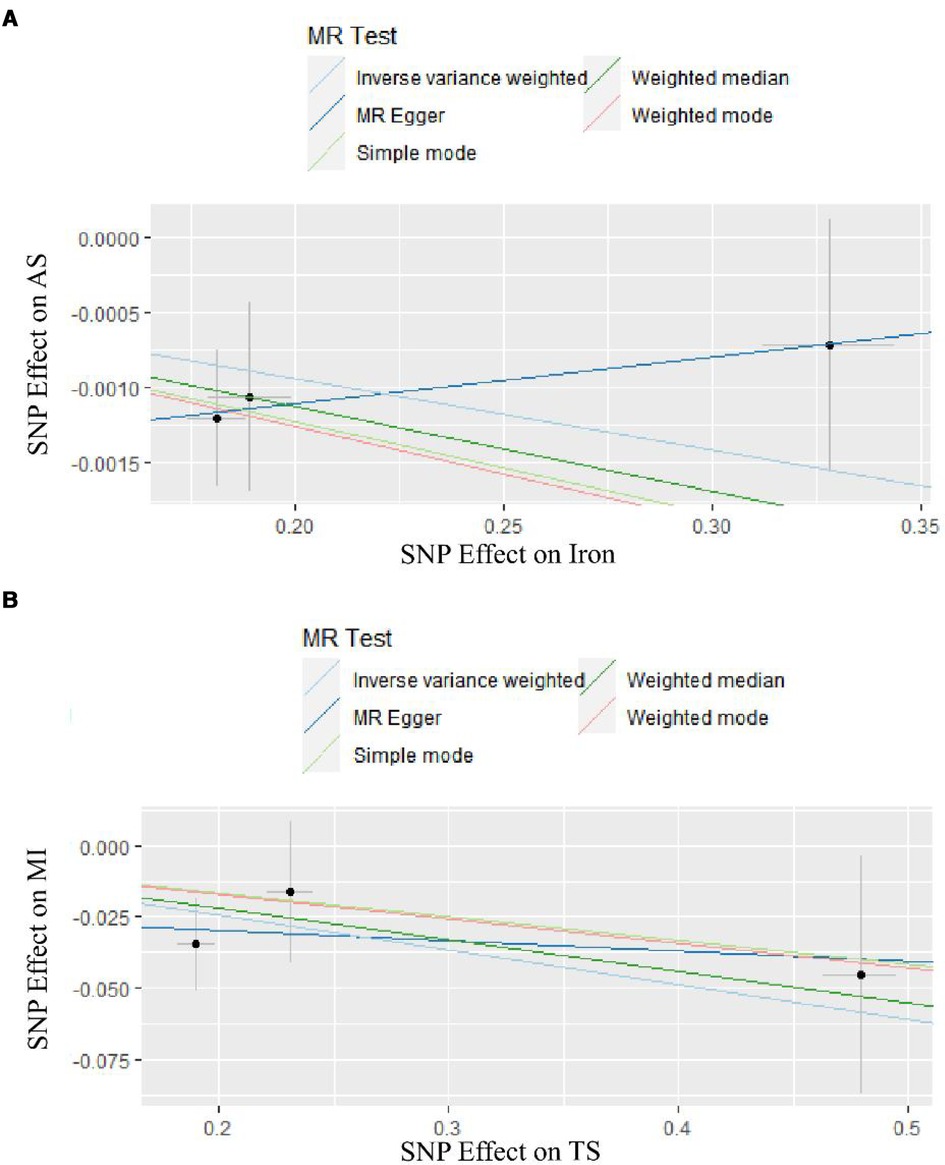
Figure 2. The result of two-sample MR analyses. (A) is the result of two samples MR analyses of Iron. (B) is the result of two samples MR analyses of TS.
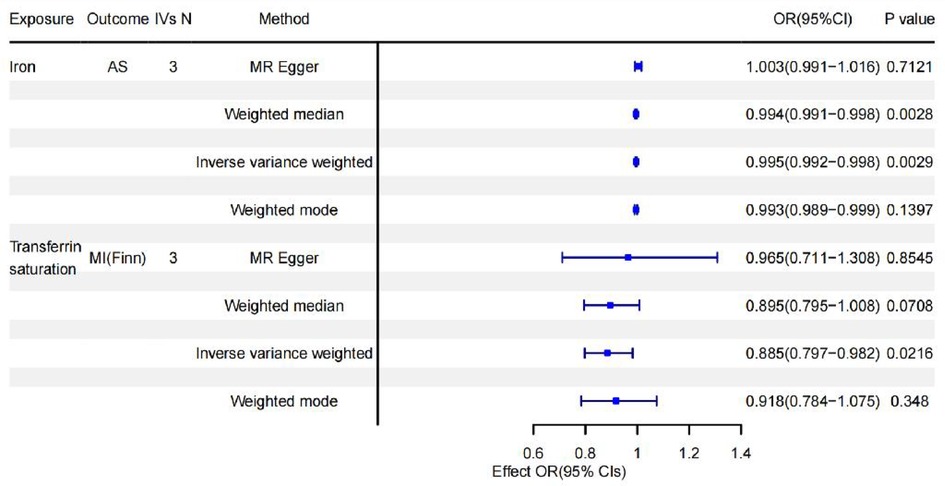
Figure 3. Forest plot summarizing causality of positive results.
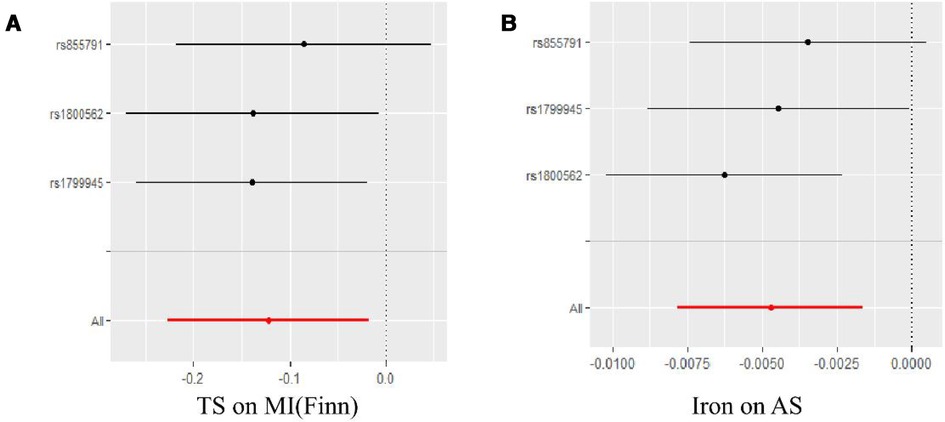
Figure 4. Leave-one-outanalyses of positive results in the causal relationship between iron status and CHD and related CVD. (A) is the result of TS and MI(Finn), and (B) is the result of Iron and AS.
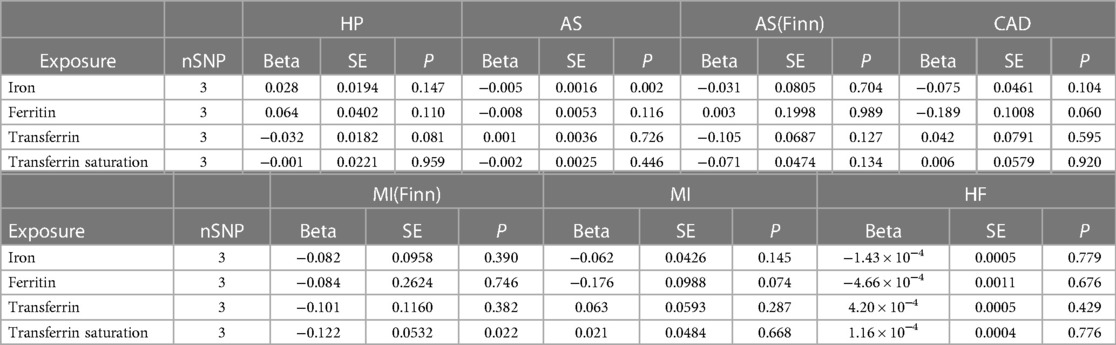
Table 2. The results of IVW.
We applied MR to analyze the causal relationship between four biomarkers of iron status and CHD and related CVD and concluded that there is a partial causal relationship between systemic iron status and CHD and related CVD. In this MR analysis, there was a negative correlation between serum iron levels and coronary AS and between TS and MI. The same conclusion was reached when the MR analysis was repeated for AS and MI using the individual SNPs for serum iron and TS, respectively. It implies that a genetically determined increase in serum iron decreases the risk of AS and a genetically determined increase in TS decreases the risk of MI. Based on the fact that TS is one of the biomarkers reflecting systemic iron status, we can speculate that genetically determined ID may lead to an increased risk of CHD.
In a study of patients on hemodialysis, it was found that high doses of intravenous iron reduced the risk of MI compared to lower doses (28). More noteworthy is the finding in another study that patients who were iron deficient at the time of acute coronary syndrome had a significantly higher risk of cardiovascular mortality and nonfatal myocardial infarction over 4 years (6), results that are consistent with those obtained in this paper. However, in patients with st-segment elevation myocardial infarction treated with percutaneous coronary intervention, patients with ID were found to have a better in-hospital prognosis. The investigators speculate that this may be related to the ability of ID to reduce myocardial ischemia-reperfusion injury (29). Based on the above, we hypothesize that the effect of iron on MI is not only that higher iron levels reduce the risk of MI, but also that ID increases the long-term effects of a worse incidence of MI. Considering the small causal effect, these MR-based analyses should be referenced with caution. Previous MR analyses on iron status and carotid plaque have also shown that genetically determined iron levels increase carotid plaque with a protective effect (30). This laterally supports the results of the MR of serum iron with AS in this paper. However, some studies have yielded contradictory results, suggesting that high iron promotes the progression of atherosclerosis and increases its severity (31). This discrepancy may stem from the fact that the occurrence of AS is influenced by genetic and environmental factors, and we analyzed the relationship from a genetic perspective, focusing on lifetime effects rather than short-term effects.
Some of these results contradict the results of the reported papers, which may be explained by the fact that no correction for disease subtyping population stratification and population pedigree was performed when processing GWAS data for CHD, in which cases of acute coronary syndrome, coronary artery bypass grafting, and percutaneous coronary revascularization were included in the CHD cases used (17). As part of the current MR study, three SNPs were selected from a recent large-scale GWAS. Among them, mutations in the gene HFE include rs1800562 (also known as C282Y) and rs1799945 (also known as H63D). The missense mutation in rs1800562 retains the dysfunctional HFE in the endoplasmic reticulum instead of being transported to the plasma membrane, resulting in the parenchymal iron overload disease of hereditary hemochromatosis (HH) (32). A recent study of 2890 European patients with C282Y-pure HH found a significantly lower risk of cardiovascular disease in C282Y-pure HH patients compared to age-matched subjects (33). Moreover, compared to HFE wild-type study participants, C282Y-positive participants had lower total cholesterol and LDL-C levels (34). In addition, a large GWAS study showed that genetic variants on H63D are associated with the prevalence of hypertension (35). It has been hypothesized that H63D causes toxic damage to the vascular endothelium by increasing iron stores and producing oxidative stress (36). However, the exact mechanism by which H63D causes hypertension remains unclear. Hepcidin, a key role of iron homeostasis, is a peptide hormone synthesized in hepatocytes that regulates cellular iron output by interacting with iron transport proteins (37, 38). Mutations in TMPRSS6 result in excessive iron uptake by encoding a type II plasma membrane serine protease, called matriptase-2, which inhibits hepcidin (39). Moreover, rs1800562 and rs1799945 have been reported to affect hepcidin (40). In conclusion, the above indicates that all three SNPs above can be involved in the occurrence and development of CVD by regulating iron metabolism.
Iron, one of the most essential nutrients, has been shown in many previous studies to be associated with abnormal iron status and CVD. In a cohort of 12,164 individuals from three European populations ID was associated with a 24% increased risk of CHD, a 26% increased risk of CVD death and a 12% increased risk of all-cause mortality, with 5.4% of deaths, 11.7% of CVD deaths and 10.7% of CHD events attributable to iron deficiency(ID) (41). ID is also an observable indicator of HF, about half of HF patients having ID according to the definition of ID (42, 43), with a specific prevalence of ID in chronic HF of about 47%–68%, and the more severe the HF, the more likely it is to occur (44). A double-blind randomized trial showed that intravenous iron carboxymaltose supplementation reduced the risk of HF hospitalization in iron-deficient patients with stable left ventricular ejection fraction below 50% after an acute HF episode, but had no significant effect on their risk of cardiovascular death (45). However, although observational studies and MR analyses have indicated that ID increases the risk of CHD, there are no relevant experimental results indicating that improving ID reduces the risk of developing CHD. In an epic-Heidelberg study,serum ferritin concentrations were associated with IM risk and cardiovascular disease mortality, but were not statistically significant after adjustment (46). Thus the effect of iron deficiency on CHD and CVD remains questionable. Therefore, the hypothesis has been put forward that the effect of iron on the heart lies in the increased oxidative stress due to iron overload (47).
Ferroptosis, in which the key factors of iron toxicity are Fe2+ accumulation and lipid peroxidation, is a novel form of cell death with unique genetic, biochemical, morphological and metabolic characteristics in contrast to apoptosis, necroptosis and scorch death (13). As iron plays a key role in catalyzing phospholipid peroxidation in ferroptosis, unrestricted lipid peroxidation is exactly one of the hallmark symptoms of ferroptosis (48) Lipid peroxidation is subject to molecular oxidation reactions that generate peroxyl radicals, and if not eventually reduced to the corresponding alcohols, the propagation of the radical-mediated reactions leads to the formation of numerous secondary products that disrupt cell membrane integrity and eventually lead to cell death (49). On the other hand, excessive intracellular iron accumulation is associated with an overproduction of ROS, which leads to extensive oxidation of polyunsaturated fatty acids and disruption of cell membrane structure, ultimately leading to cell death (50). Iron overload has also been suggested as one of the potential mechanisms of myocardial I/R injury (51). An increase in Fe2+ concentration in cardiomyocytes has been reported to be observed in I/R-treated rats (52, 53). The upregulation of TfR1 in I/R-treated rat hearts was associated with elevated iron content, and the inhibiting of TfR1 expression accompanied a decrease in iron content and reduced I/R injury, so hypoxia may be responsible for causing I/R iron overload (54). Consistently, iron death is observed during atherosclerosis. The likelihood of atherosclerosis can be reduced by inhibiting iron death in aortic endothelial cells to attenuate lipid peroxidation and endothelial dysfunction (55).
Morever, it has been shown that systemic iron satuts are not equal to cardiac iron levels, so systemic iron disorders do not directly affect cardiac iron satuts (56, 57), and further studies are needed to determine whether systemic iron supplements have a beneficial effect on CVD. Studies have shown that a low-iron diet fed to rats results in reduced levels of iron transport proteins in the rat heart, resulting in reduced iron output from heart cells (58), suggesting that iron levels in the heart are not necessarily affected by a low-iron diet. Consistently, in another study of a mild cardiomyopathy model in FthMCK/MCK mice both showed reduced cardiac iron levels without changes in serum or skeletal muscle iron levels, but after 4 weeks on a high iron diet, cardiac GSH levels in mice were reduced due to increased cardiac iron levels, resulting in cardiomyocyte ferroptosis.
Herein, the decrease in serum iron does not correlate positively with the decrease in cardiac iron levels,which cellular iron levels may be increased and be caused ferroptosis in cardiomyocytes while iron supplementation (59). Combined with the results of the Mendelian study in this paper, we can propose the hypothesis that the effect of decreased systemic iron status on CVD lies in overall cardiomyocyte function. Systemic iron status is negatively correlated with diseasewhen there are more normal cardiomyocytes than damaged cardiomyocytes in a cardiovascular disease event, whereas systemic iron status is not correlated with disease development when the situation is reversed. It is possible that iron supplementation at this point will lead to cellular iron overload, which will promote the development of cellular ferroptosis as a result. Therefore, considering the potentially deleterious effects of ID and iron overload, iron status intervention strategies may not be beneficial for patients in the CHD and related CVD without ID. In conclusion, based on the effect of systemic iron status on cardiac cellular iron levels, future studies should aim to identify CHD and related CVD phenotypes that would benefit from improved ID and to investigate their specific pathophysiological mechanisms.
The study we conducted has several limitations. First, because individual data were not available and we only performed summary statistics, the CHD and related CVD data used in this study were not stratified by disease subtype, such as dividing CHD into stable angina pectoris and acute non-ST-segment elevation myocardial infarction. Indeed, iron status markers may have a stronger association with specific subtypes of CHD or at acute onset. Therefore, further studies are needed to investigate whether similar results exist in patients of different races, different subtypes of CHD and CVD, and different degrees of disease severity. Second, for some exposures, the body may have mechanisms to respond to this exposure level, such as systemic iron status that is not synchronized with cellular iron levels, which hinders our study of iron metabolism and CHD and related CVD from macroscopic regulators to microscopic pathophysiological changes in the present study. Considering the small causal effect of this analysis, the MR Analysis estimates of this study should be interpreted with caution, and the inferences and assumptions in this paper should be referred to with caution. Nevertheless, the present study provides some clues to the pathophysiology and therapeutic exploration of CHD and related CVD. We expect future studies to delve into the relationship between CHD and related CVD and iron status, which may provide new insights into the prevention and treatment of CHD.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
FL and YL: Are joint first co-authors, and oversaw investigation, Resources, Data Curation and Writing—Original Draft; YL and FX: Conceptualization, Methodology and Writing—Review & Editing; QW and SX: Data Curation and Visualization. All authors contributed to the article and approved the submitted version.
This study is supported by The Young Qihuang Scholar of the “Tens of Millions” Talent Project of China, the key project of scientific and technological innovation project sponsored by Chinese Academy of Chinese Medical Sciences (CI2021A01406), and the Special Project for Outstanding Young Talents of China Academy of Chinese Medical Sciences (ZZ15-YQ-017 and ZZ13-YQ-001-A1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1152201/full#supplementary-material
1. Timmis A, Vardas P, Townsend N, Torbica A, Katus H, De Smedt D, et al. European Society of cardiology: cardiovascular disease statistics 2021. Eur Heart J. (2022) 43(8):716–99. doi: 10.1093/eurheartj/ehab892
2. Li S, Zhang X. Iron in cardiovascular disease: challenges and potentials. Front Cardiovasc Med. (2021) 8:707138. doi: 10.3389/fcvm.2021.707138
3. Leong DP, Joseph PG, Mckee M, Anand SS, Teo KK, Schwalm JD, et al. Reducing the global burden of cardiovascular disease, part 2: prevention and treatment of cardiovascular disease. Circ Res. (2017) 121(6):695–710. doi: 10.1161/CIRCRESAHA.117.311849
4. Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of mammalian iron metabolism. Cell. (2010) 142(1):24–38. doi: 10.1016/j.cell.2010.06.028
5. Von Haehling S, Jankowska EA, Van Veldhuisen DJ, Ponikowski P, Anker SD. Iron deficiency and cardiovascular disease. Nat Rev Cardiol. (2015) 12(11):659–69. doi: 10.1038/nrcardio.2015.109
6. Zeller T, Waldeyer C, Ojeda F, Schnabel RB, Schafer S, Altay A, et al. Adverse outcome prediction of iron deficiency in patients with acute coronary syndrome. Biomolecules. (2018) 8(3):60. doi: 10.3390/biom8030060
7. Das De S, Krishna S, Jethwa A. Iron status and its association with coronary heart disease: systematic review and meta-analysis of prospective studies. Atherosclerosis. (2015) 238(2):296–303. doi: 10.1016/j.atherosclerosis.2014.12.018
8. Grammer TB, Scharnagl H, Dressel A, Kleber ME, Silbernagel G, Pilz S, et al. Iron metabolism, hepcidin, and mortality (the ludwigshafen risk and cardiovascular health study). Clin Chem. (2019) 65(7):849–61. doi: 10.1373/clinchem.2018.297242
9. Graham FJ, Pellicori P, Kalra PR, Ford I, Bruzzese D, Cleland JGF. Intravenous iron in patients with heart failure and iron deficiency: an updated meta-analysis. Eur J Heart Fail. (2023) 9(4):528–37.
10. Hoes MF, Grote Beverborg N, Kijlstra JD, Kuipers J, Swinkels DW, Giepmans BNG, et al. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur J Heart Fail. (2018) 20(5):910–9. doi: 10.1002/ejhf.1154
11. He H, Qiao Y, Zhou Q, Wang Z, Chen X, Liu D, et al. Iron overload damages the endothelial mitochondria via the ROS/ADMA/DDAHII/eNOS/NO pathway. Oxid Med Cell Longev. (2019) 2019:2340392. doi: 10.1155/2019/2340392
12. Li N, Jiang W, Wang W, Xiong R, Wu X, Geng Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol Res. (2021) 166:105466. doi: 10.1016/j.phrs.2021.105466
13. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
14. Jiang L, Yin X, Chen YH, Chen Y, Jiang W, Zheng H, et al. Proteomic analysis reveals ginsenoside Rb1 attenuates myocardial ischemia/reperfusion injury through inhibiting ROS production from mitochondrial complex I. Theranostics. (2021) 11(4):1703–20. doi: 10.7150/thno.43895
15. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. Br Med J. (2018) 362:k601. doi: 10.1136/bmj.k601
16. Benyamin B, Esko T, Ried JS, Radhakrishnan A, Vermeulen SH, Traglia M, et al. Novel loci affecting iron homeostasis and their effects in individuals at risk for hemochromatosis. Nat Commun. (2014) 5:4926. doi: 10.1038/ncomms5926
17. Gill D, Del Greco MF, Walker AP, Srai SKS, Laffan MA, Minelli C. The effect of iron Status on risk of coronary artery disease: a Mendelian randomization study-brief report. Arterioscler Thromb Vasc Biol. (2017) 37(9):1788–92. doi: 10.1161/ATVBAHA.117.309757
18. Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol. (2016) 40(7):597–608. doi: 10.1002/gepi.21998
19. Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-base platform supports systematic causal inference across the human phenome. Elife. (2018) 7:e34408. doi: 10.7554/eLife.34408
20. Van Der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. (2018) 122(3):433–43. doi: 10.1161/CIRCRESAHA.117.312086
21. Rosoff DB, Davey Smith G, Mehta N, Clarke TK, Lohoff FW. Evaluating the relationship between alcohol consumption, tobacco use, and cardiovascular disease: a multivariable Mendelian randomization study. PLoS Med. (2020) 17(12):e1003410. doi: 10.1371/journal.pmed.1003410
22. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. (2015) 47(10):1121–30. doi: 10.1038/ng.3396
23. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. (2013) 37(7):658–65. doi: 10.1002/gepi.21758
24. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. (2016) 40(4):304–14. doi: 10.1002/gepi.21965
25. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-egger method. Eur J Epidemiol. (2017) 32(5):377–89. doi: 10.1007/s10654-017-0255-x
26. Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology. (2017) 28(1):30–42. doi: 10.1097/EDE.0000000000000559
27. Cohen JF, Chalumeau M, Cohen R, Korevaar DA, Khoshnood B, Bossuyt PM. Cochran's Q test was useful to assess heterogeneity in likelihood ratios in studies of diagnostic accuracy. J Clin Epidemiol. (2015) 68(3):299–306. doi: 10.1016/j.jclinepi.2014.09.005
28. Petrie MC, Jhund PS, Connolly E, Mark PB, Macdonald MR, Robertson M, et al. High-dose intravenous iron reduces myocardial infarction in patients on haemodialysis. Cardiovasc Res. (2023) 119(1):213–20. doi: 10.1093/cvr/cvab317
29. Cosentino N, Campodonico J, Pontone G, Guglielmo M, Trinei M, Sandri MT, et al. Iron deficiency in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Int J Cardiol. (2020) 300:14–9. doi: 10.1016/j.ijcard.2019.07.083
30. Gill D, Brewer CF, Monori G, Trégouët DA, Franceschini N, Giambartolomei C, et al. Effects of genetically determined iron Status on risk of venous thromboembolism and carotid atherosclerotic disease: a Mendelian randomization study. J Am Heart Assoc. (2019) 8(15):e012994. doi: 10.1161/JAHA.119.012994
31. Vinchi F, Porto G, Simmelbauer A, Altamura S, Passos ST, Garbowski M, et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur Heart J. (2020) 41(28):2681–95. doi: 10.1093/eurheartj/ehz112
32. Demetz E, Tymoszuk P, Hilbe R, Volani C, Haschka D, Heim C, et al. The haemochromatosis gene hfe and kupffer cells control LDL cholesterol homeostasis and impact on atherosclerosis development. Eur Heart J. (2020) 41(40):3949–59. doi: 10.1093/eurheartj/ehaa140
33. Pilling LC, Tamosauskaite J, Jones G, Wood AR, Jones L, Kuo CL, et al. Common conditions associated with hereditary haemochromatosis genetic variants: cohort study in UK biobank. Br Med J. (2019) 364:k5222. doi: 10.1136/bmj.k5222
34. Adams PC, Pankow JS, Barton JC, Acton RT, Leiendecker-Foster C, Mclaren GD, et al. HFE C282y homozygosity is associated with lower total and low-density lipoprotein cholesterol: the hemochromatosis and iron overload screening study. Circ Cardiovasc Genet. (2009) 2(1):34–7. doi: 10.1161/CIRCGENETICS.108.813089
35. Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. (2011) 478(7367):103–9. doi: 10.1038/nature10405
36. Selvaraj S, Seidelmann S, Silvestre OM, Claggett B, Ndumele CE, Cheng S, et al. HFE H63d polymorphism and the risk for systemic hypertension, myocardial remodeling, and adverse cardiovascular events in the ARIC study. Hypertension. (2019) 73(1):68–74. doi: 10.1161/HYPERTENSIONAHA.118.11730
37. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. (2004) 306(5704):2090–3. doi: 10.1126/science.1104742
38. Buerkli S, Pei SN, Hsiao SC, Lee CT, Zeder C, Zimmermann MB, et al. The TMPRSS6 variant (SNP rs855791) affects iron metabolism and oral iron absorption—a stable iron isotope study in Taiwanese women. Haematologica. (2021) 106(11):2897–905. doi: 10.3324/haematol.2020.264556
39. Silvestri L, Pagani A, Nai A, De Domenico I, Kaplan J, Camaschella C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. (2008) 8(6):502–11. doi: 10.1016/j.cmet.2008.09.012
40. Gao J, Chen J, Kramer M, Tsukamoto H, Zhang AS, Enns CA. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. (2009) 9(3):217–27. doi: 10.1016/j.cmet.2009.01.010
41. Schrage B, Rubsamen N, Ojeda FM, Thorand B, Peters A, Koenig W, et al. Association of iron deficiency with incident cardiovascular diseases and mortality in the general population. ESC Heart Fail. (2021) 8(6):4584–92. doi: 10.1002/ehf2.13589
42. Von Haehling S, Gremmler U, Krumm M, Mibach F, Schön N, Taggeselle J, et al. Prevalence and clinical impact of iron deficiency and anaemia among outpatients with chronic heart failure: the PrEP registry. Clin Res Cardiol. (2017) 106(6):436–43. doi: 10.1007/s00392-016-1073-y
43. Bekfani T, Pellicori P, Morris D, Ebner N, Valentova M, Sandek A, et al. Iron deficiency in patients with heart failure with preserved ejection fraction and its association with reduced exercise capacity, muscle strength and quality of life. Clin Res Cardiol. (2019) 108(2):203–11. doi: 10.1007/s00392-018-1344-x
44. Masini G, Graham FJ, Pellicori P, Cleland JGF, Cuthbert JJ, Kazmi S, et al. Criteria for iron deficiency in patients with heart failure. J Am Coll Cardiol. (2022) 79(4):341–51. doi: 10.1016/j.jacc.2021.11.039
45. Ponikowski P, Kirwan BA, Anker SD, Mcdonagh T, Dorobantu M, Drozdz J, et al. Ferric carboxymaltose for iron deficiency at discharge after acute heart failure: a multicentre, double-blind, randomised, controlled trial. Lancet. (2020) 396(10266):1895–904. doi: 10.1016/S0140-6736(20)32339-4
46. Quintana Pacheco DA, Sookthai D, Wittenbecher C, Graf ME, Schübel R, Johnson T, et al. Red meat consumption and risk of cardiovascular diseases-is increased iron load a possible link? Am J Clin Nutr. (2018) 107(1):113–9. doi: 10.1093/ajcn/nqx014
47. Galaris D, Pantopoulos K. Oxidative stress and iron homeostasis: mechanistic and health aspects. Crit Rev Clin Lab Sci. (2008) 45(1):1–23. doi: 10.1080/10408360701713104
48. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
49. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. (2019) 15(12):1137–47. doi: 10.1038/s41589-019-0408-1
50. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
51. Shan X, Lv ZY, Yin MJ, Chen J, Wang J, Wu QN. The protective effect of cyanidin-3-glucoside on myocardial ischemia-reperfusion injury through ferroptosis. Oxid Med Cell Longev. (2021) 2021:8880141. doi: 10.1155/2021/8880141
52. Li T, Tan Y, Ouyang S, He J, Liu L. Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene. (2022) 808:145968. doi: 10.1016/j.gene.2021.145968
53. Li S, Lei Z, Yang X, Zhao M, Hou Y, Wang D, et al. Propofol protects myocardium from ischemia/reperfusion injury by inhibiting ferroptosis through the AKT/p53 signaling pathway. Front Pharmacol. (2022) 13:841410. doi: 10.3389/fphar.2022.841410
54. Tang LJ, Zhou YJ, Xiong XM, Li NS, Zhang JJ, Luo XJ, et al. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TfR1 pathway in the rat hearts after ischemia/reperfusion. Free Radic Biol Med. (2021) 162:339–52. doi: 10.1016/j.freeradbiomed.2020.10.307
55. Guo Y, Lu C, Hu K, Cai C, Wang W. Ferroptosis in cardiovascular diseases: current Status, challenges, and future perspectives. Biomolecules. (2022) 12(3):390. doi: 10.3390/biom12030390
56. Paterek A, Oknińska M, Chajduk E, Polkowska-Motrenko H, Mączewski M, Mackiewicz U. Systemic iron deficiency does not affect the cardiac iron content and progression of heart failure. J Mol Cell Cardiol. (2021) 159:16–27. doi: 10.1016/j.yjmcc.2021.06.005
57. Hirsch VG, Tongers J, Bode J, Berliner D, Widder JD, Escher F, et al. Cardiac iron concentration in relation to systemic iron status and disease severity in non-ischaemic heart failure with reduced ejection fraction. Eur J Heart Fail. (2020) 22(11):2038–46. doi: 10.1002/ejhf.1781
58. Berezovsky B, Frýdlová J, Gurieva I, Rogalsky DW, Vokurka M, Krijt J. Heart ferroportin protein content is regulated by heart iron concentration and systemic hepcidin expression. Int J Mol Sci. (2022) 23(11):5899. doi: 10.3390/ijms23115899
Keywords: iron status, cardiovascular disease, causal effect, genome-wide association studies, Mendelian randomization
Citation: Liu F, Liu Y, Xu S, Wang Q, Xu F and Liu Y (2023) Mendelian randomization study reveals a causal relationship between serum iron status and coronary heart disease and related cardiovascular diseases. Front. Cardiovasc. Med. 10:1152201. doi: 10.3389/fcvm.2023.1152201
Received: 27 January 2023; Accepted: 29 May 2023;
Published: 13 June 2023.
Edited by:
Peter Barany, Karolinska University Hospital, SwedenReviewed by:
Nils Bomer, University of Groningen, University Medical Center Groningen, Netherlands© 2023 Liu, Liu, Xu, Wang, Xu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue Liu bGl1eXVlaGVhcnRAaG90bWFpbC5jb20= Fengqin Xu ZHIueHVmZW5ncWluQCBob3RtYWlsLmNvbQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.