 Minglu Ma
Minglu Ma Chang Hou
Chang Hou Jian Liu
Jian Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 10 March 2023
Sec. General Cardiovascular Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1148486
This article is part of the Research TopicNovel Strategies for the Prevention and Treatment of Foam Cell Formation and AtherosclerosisView all 7 articles
Atherosclerosis is a basic pathological characteristic of many cardiovascular diseases, and if not effectively treated, patients with such disease may progress to atherosclerotic cardiovascular diseases (ASCVDs) and even heart failure. The level of plasma proprotein convertase subtilisin/kexin type 9 (PCSK9) is significantly higher in patients with ASCVDs than in the healthy population, suggesting that it may be a promising new target for the treatment of ASCVDs. PCSK9 produced by the liver and released into circulation inhibits the clearance of plasma low-density lipoprotein-cholesterol (LDL-C), mainly by downregulating the level of LDL-C receptor (LDLR) on the surface of hepatocytes, leading to upregulated LDL-C in plasma. Numerous studies have revealed that PCSK9 may cause poor prognosis of ASCVDs by activating the inflammatory response and promoting the process of thrombosis and cell death independent of its lipid-regulatory function, yet the underlying mechanisms still need to be further clarified. In patients with ASCVDs who are intolerant to statins or whose plasma LDL-C levels fail to descend to the target value after treatment with high-dose statins, PCSK9 inhibitors often improve their clinical outcomes. Here, we summarize the biological characteristics and functional mechanisms of PCSK9, highlighting its immunoregulatory function. We also discuss the effects of PCSK9 on common ASCVDs.
Atherosclerosis is an early pathological change in many cardiovascular diseases (1). The occurrence and progression of atherosclerosis and atherosclerotic cardiovascular diseases (ASCVDs) are associated with hyperlipidemia but also involves an imbalance between proinflammatory and anti-inflammatory response (2, 3). During this process, the injured arterial endothelium, multiple immune cells (such as macrophages, dendritic cells, B cells, T cells, and mast cells), macrophage-like vascular smooth muscle cells (VSMCs), adhesion molecules, cytokines, and oxidized low-density lipoprotein (ox-LDL) deposited beneath the vascular intima work together to activate the local immune response, while foam cells always have moderate proinflammatory functions and mainly participate in the formation of the lipid core (3–6). Through earlier detection, earlier diagnosis, and more effective treatment, the occurrence and progression of atherosclerosis and ASCVDs can be better controlled.
Statins, as a first-line drug recommended by clinical guidelines for the treatment of atherosclerosis and ASCVDs, inhibits cholesterol synthesis by inhibiting the activation of the rate-limiting enzyme 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, thereby reducing cholesterol concentrations in the endoplasmic reticulum (ER) (7, 8). Statin resistance occurs mainly because the low cholesterol concentration in the ER promotes the activation of sterol regulatory element-binding protein 2 (SREBP2) and the subsequent production of PCSK9 (9). PCSK9 inhibitors have been shown to be beneficial in the treatment of patients with atherosclerosis and ASCVDs (10–15).
PCSK9, also known as neural apoptosis-regulated convertase-1 (NARC-1), was first identified in 2003 (16). As the ninth and last member of the proteinase K subfamily of subtilases, PCSK9 undergoes self-cleavage and multiple rounds of post-translational modifications before becoming the mature form (17–19). PCSK9 functions in a nonenzymatic fashion, which differs from other family members (20, 21). The carboxy-terminal domain (residues 452 to 692) of full-length PCSK9 protein shares structural homology with the resistin, which is essential for the formation of PCSK9-low-density lipoprotein receptor (LDLR) dimer (22). The critical role of PCSK9 in cholesterol regulation was discovered, as its gain-of-function variants could lead to human familial hypercholesterolemia, while its nonsense mutations (Y142X and C679X) in African Americans were associated with 40% lower plasma level of low-density lipoprotein-cholesterol (LDL-C) and lower risk of coronary heart disease (CHD) (23, 24). Then, accumulating evidence has suggested that the effects of PCSK9 on ASCVDs may be associated with immunoregulation, platelet activation, thrombosis, and multiple forms of cell death, which may be independent of its lipid-lowering capacity (25–27). In some clinical trials, the correlation between PCSK9 and immune response has been demonstrated in patients with atherosclerotic disease, coronary artery disease, systemic lupus erythematosus, and human immunodeficiency virus (HIV)-infected (28–31). Experiments in vitro and in mice further support the immunoregulatory function of PCSK9 (32–35).
Here, we used the following keywords to filter corresponding papers in PubMed: “PCSK9 and atherosclerosis”, “PCSK9 and myocardial infarction”, “PCSK9 and inflammation”, “PCSK9 and platelets”, “PCSK9 and autophagy”, “PCSK9 and apoptosis”, as well as “PCSK9 and pyroptosis”. We first introduce the production of PCSK9 and the molecular mechanisms that regulate its expression. We then summarize the functional mechanisms of PCSK9 and its inhibitors in the cardiovascular system, such as the regulatory function in lipid metabolism, immune response, thrombosis, and multiple modes of cell death, with a particular focus on its immunoregulation. Subsequently, we show the effect of PCSK9 and its inhibitors on the prognosis of atherosclerotic and myocardial infarction patients. Finally, we propose several issues that need to be addressed in the future regarding PCSK9 and cardiovascular diseases.
The liver is the major organ that produces PCSK9, and the kidney, the small intestine, the pancreas, the lung, and the central nervous system also produce small amounts of PCSK9 (16, 36). Under physiological conditions, the expression of PCSK9 was detected in cultured human smooth muscle cells (SMCs), while was undetectable in human umbilical vein endothelial cells (HUVECs), monocytes and macrophages (37). Under an inflammatory state [lipopolysaccharide (LPS) treatment], HUVECs can produce PCSK9 (38). When atherosclerosis occurs, various types of cells, especially SMCs, endothelial cells and macrophages in the injured vessels produce large amounts of PCSK9 at the transcriptional and translational levels in response to the stimulation of low shear stress, lipopolysaccharide (LPS), tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β), ox-LDL, reactive oxygen species (ROS), and mitochondrial DNA (mtDNA) and mitochondria-derived reactive oxygen species (mtROS) released from damaged mitochondria in ruptured cells (37, 39–42). When myocardial infarction occurs, the expression level of PCSK9 is also upregulated in the ischemic heart tissue, mostly in the border zone, and hypoxia as well as proinflammatory cytokines may be the key factors that upregulate its expression (43, 44).
The expression of PCSK9 is mainly regulated by SREBP2, hepatocyte nuclear factor-1α (HNF-1α), and forkhead box O3 (FoxO3) at the transcriptional level (45–48). Sequence analysis of the 5′ flanking region from −2,112 to −94 of the PCSK9 gene revealed the presence of the sterol regulatory element (SRE) site (5′-GTGGCGTGAT-3′) in the proximal region of the PCSK9 promoter (49, 50). In the process of sterol-dependent transcriptional regulation of PCSK9, the SRE site and the adjacent upstream nucleotides are critically required (45). The SRE site is the target site for SREBPs (45). After treatment with statins, decreased cholesterol concentrations in the ER promote the production of PCSK9 by activating SREBP2, leading to a weak effect of statin treatment in some ASCVD patients (49). The transcriptional activation of PCSK9 induced by insulin is dependent on SREBP1c (50). Caffeine inhibits the expression of SREBP2 at the transcriptional level by increasing the Ca2+ concentration in the hepatic ER, thereby reducing the expression of PCSK9 and the risk of cardiovascular disease (51). Besides, both HNF1α and HNF1β are positive regulators of PCSK9 transcription, although there is little literature mentioning the role of HNF1β in this process (52). HNF1α regulates the transcription of PCSK9 through hepatocyte nuclear factor 1 (HNF1) site located 28 bp upstream from SRE (53). Mutation of the HNF1 site significantly inhibits the activity of PCSK9 promoter, which is dependent on both its direct effect and its indirect effect of inhibiting the activity of SRE site (46). In mice, the activated mechanistic target of rapamycin complex 1 (mTOR1) pathway suppresses the transcription of PCSK9 by silencing HNF-1α (54). FoxO3 is a negative regulator of PCSK9 transcription (47). Sirtuin-6 (Sirt6) is an NAD + -dependent histone deacetylase (55). After the interaction of FoxO3 with insulin-response element (IRE), Sirt6 binds to the PCSK9 promoter to deacetylate histone H3 at lysines 9 and 56, resulting in the attenuated activity of PCSK9 promoter (47, 56). FoxO3 and Sirt6 also suppress the transcriptional activity of SRE and HNF1 (47). Understanding the molecular mechanisms that regulate the production of PCSK9 is of great value to effectively inhibit the overexpression of PCSK9 and alleviate the risk of ASCVDs (Figure 1).
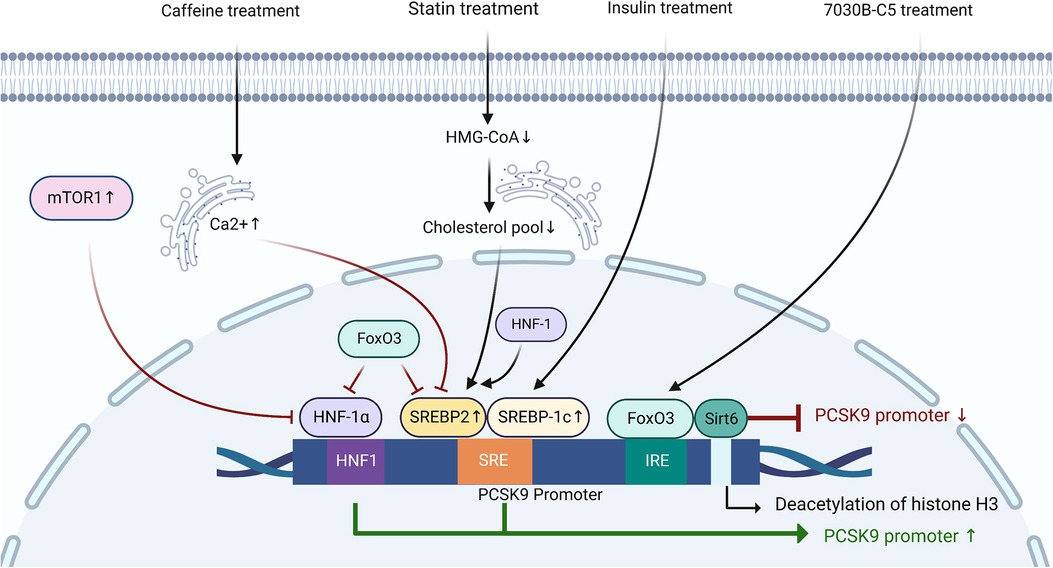
Figure 1. The regulation of PCSK9 expression at the transcriptional level. HNF1, hepatocyte nuclear factor-1; SRE, sterol-regulatory element; SREBP, sterol regulatory element-binding protein; IRE, insulin-response element; FoxO3, forkhead box O3; Sirt6, sirtuin-6; mTOR1, mechanistic target of rapamycin complex 1; HMG-CoA, 3-hydroxy-3-methyl-glutaryl-coenzyme A.
The occurrence and development of atherosclerosis is associated with abnormal lipid metabolism and excessive proinflammatory response (57, 58). Platelet activation and thrombosis secondary to coronary atherosclerotic plaque rupture can lead to myocardial infarction, a common disorder in ASCVDs (59). Subsequently, cardiomyocytes die due to ischemia and hypoxia (59). Here, we describe the molecular functions of PCSK9 and its mechanisms around the above points.
The plasma levels of LDL-C are closely associated with an increased risk of ASCVDs (60). Downregulating LDL-C concentrations can help to reduce the incidence of adverse cardiovascular events (61, 62). Approximately 60%–70% of plasma LDL-C is cleared in the liver after binding to LDLR on the surface of hepatocytes (63). LDL-C-lowering LDLR variants are associated with lower risk of CHD (64). PCSK9 affects the plasma lipid and lipoprotein levels mainly by downregulating LDLR in the liver (65). It has been confirmed that gain-of-function mutations of PCSK9 lead to increased risks of hyperlipidemia and ASCVDs, whereas loss-of-function mutations of PCSK9 reduced plasma levels of LDL-C and ASCVDs risk (Figure 2) (66–68).
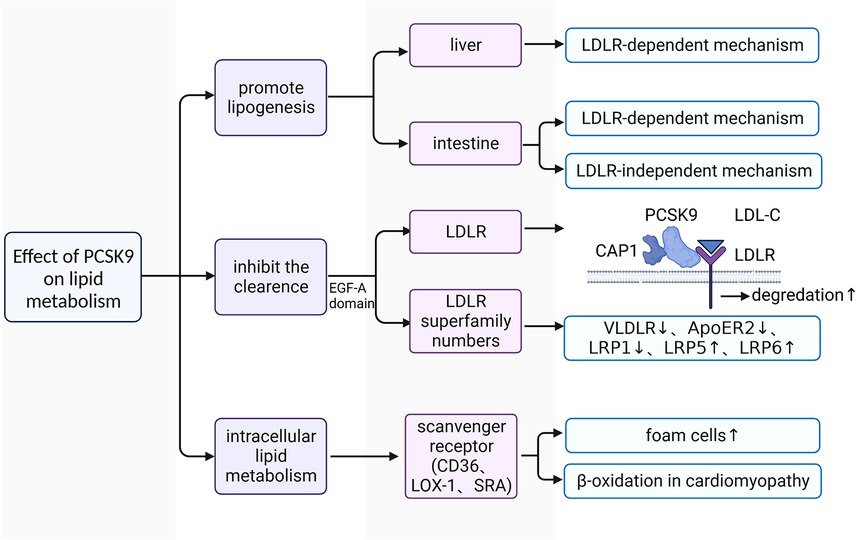
Figure 2. The functional mechanism of PCSK9 in regulating lipid metabolism. LDL-C, low-density lipoprotein-cholesterol; LDLR, low-density lipoprotein receptor; VLDLR, very low-density lipoprotein receptor; ApoER2, apolipoprotein E receptor 2; PCSK9, proprotein convertase subtilisin/kexin type 9; CAP1, cyclase associated protein 1; LOX1, lectin-like oxidized low-density lipoprotein receptor 1; SRA, scavenger receptor type A; EGF-A, epidermal growth factor precursor homology domain A; LRP1, LDLR-related protein 1; LRP5, LDLR-related protein 5; LRP6, LDLR-related protein 6.
PCSK9 promotes the synthesis of lipoprotein (69–72). In C57BL/6 wild-type (WT) and LDLR-/- mice with high fat diet (HFD), researchers found that human (h) PCSK9 promotes the synthesis and secretion of cholesterol and triglycerides in the intestine in both mice, but only significantly increased the expression of key genes involved in lipogenesis at the transcriptional and translational levels in the liver in WT mice (70, 72). These findings indicate that PCSK9 promotes lipoprotein production in the liver only through LDLR-dependent mechanisms, whereas its facilitation of lipid production is achieved through both LDLR-dependent and LDLR-independent mechanisms in the intestine (70, 72).
PCSK9 promotes the upregulation of plasma lipoprotein via reducing their removal. LDLR is a key receptor for PCSK9 to regulate lipid metabolism (73). Under physiological conditions, plasma LDL-C binds to LDLR on the surface of hepatocytes to form the LDL-C-LDLR dimer, which is then transferred into the intracellular space (74). LDL-C is then separated from LDLR in endosomes and degraded in lysosomes (74). Subsequently, the intracellular free LDLR is recycled to the cell surface to participate in a new round of LDL-C transport and degradation (74). Under pathological conditions, the liver produces large amounts of PCSK9 and releases it into the plasma. The increased PCSK9 conjunct with cyclase associated protein 1 (CAP-1) binds to the LDL-C-LDLR dimer and promotes its degradation in lysosomes through a caveolin-dependent mechanism, leading to the downregulation of LDLR on the surface of hepatocytes (75). Some LDL-C-LDLR dimers that are bound to free PCSK9 without CAP-1 are endocytosed and transferred to endosomes through a clathrin-dependent mechanism, and LDLR is then released from the dimer and recycled to the cell surface without the diminution of total LDLR (75). CAP-1 is essential for PCSK9 to regulate lipid metabolism (75). There are also other LDLR family members involved in the lipid regulatory process of PCSK9, such as very low-density lipoprotein receptor (VLDLR), apolipoprotein E receptor 2 (ApoER2), LDLR-related protein 1 (LRP1), LDLR-related protein 5 (LRP5), and LDLR-related protein 6 (LRP6) (76–78). Because VLDLR and ApoER2 share a common epidermal growth factor precursor homology domain A (EGF-A) with LDLR, PCSK9 acts on the EGF-A domain of both receptors and promotes their degradation in the same manner as LDLR (79). PCSK9 is able to mediate the degradation of LRP1 (77). LRP5 and LRP6 act as coreceptors of Wnt ligands and activate Wnt-related signaling (80). Recently, much attention has been paid to the interaction between PCSK9 and LRP5/6 in the development and progression of atherosclerosis (78, 81). LRP5 is capable for promoting the accumulation of cholesterol in macrophages and the formation of foam cells (78). There is a positive feedback effect between LRP5 expression and the level of plasma PCSK9 (78, 82). Despite the lack of direct evidence, LRPs interact with PCSK9 may also rely on the epidermal growth factor-like (EGF-like) domain as it is shared in these LDLR superfamily members (83).
In addition, PCSK9 is also involved in intracellular lipid metabolism. After stimulation with recombinant PCSK9, the expression levels of lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1), class A scavenger receptor (SRA), and CD36 on the surface of macrophages are 2- to 5-fold higher than those of unstimulated macrophages, and the uptake of ox-LDL is also increased by approximately 5-fold (84). LOX-1 is a key receptor for macrophages to engulf ox-LDL (38). There is a positive feedback between LOX-1 and PCSK9, promoting the transformation of macrophages and VSMCs into foam cells (38). PCSK9 also inhibits the expression of ATP-binding cassette transporters [such as (ATP-binding cassette transporter A1) ABCA1] on macrophages, which are key transporters for cholesterol efflux (85). Thus, PCSK9 promotes the formation of foam cells from two different aspects. PCSK9 also affects lipid metabolism in cardiomyocytes. Cardiomyocytes meet their own energy requirement mainly through β-oxidation of fatty acids (FAs), and reactive oxygen species (ROS) is usually produced in this process (86). When oxidative stress occurs, excess cytotoxic ROS results in nonspecific oxidation of proteins, lipids, and DNA (86). The major route that FAs enter cardiomyocytes is active transport, which is mainly mediated by CD36, with fatty acid transporter (FATP) and fatty acid binding protein (FABP) involved (87).
Importantly, it is not beneficial to completely remove PCSK9 in vivo (88). Complete knockout of PCSK9 in mice is deleterious, even leading to heart failure with preserved ejection fraction (HFpEF) (88). The pathological deposition of FAs in cardiomyocytes causes lipotoxicity, with a reduced density of mitochondrial cristae (88). Therefore, it is necessary to control the dosage of PCSK9 inhibitors in a reasonable range in the treatment of ASCVDs. Fortunately, in large clinical trials, PCSK9 inhibitors are tolerated and effectively, without the increased risk of heart failure (14, 89).
In recent years, the functional pleiotropy of PCSK9 has been gradually recognized, and its immunoregulatory function has attracted much attention, especially in the fields of autoimmune diseases, cancer, and cardiovascular diseases (25, 29, 90). PCSK9 accelerates the development of ASCVDs through its proinflammatory function independent of its effect on lipid, but the molecular mechanisms by which PCSK9 regulates the immune response are still not well elucidated (Figure 3) (40, 91).
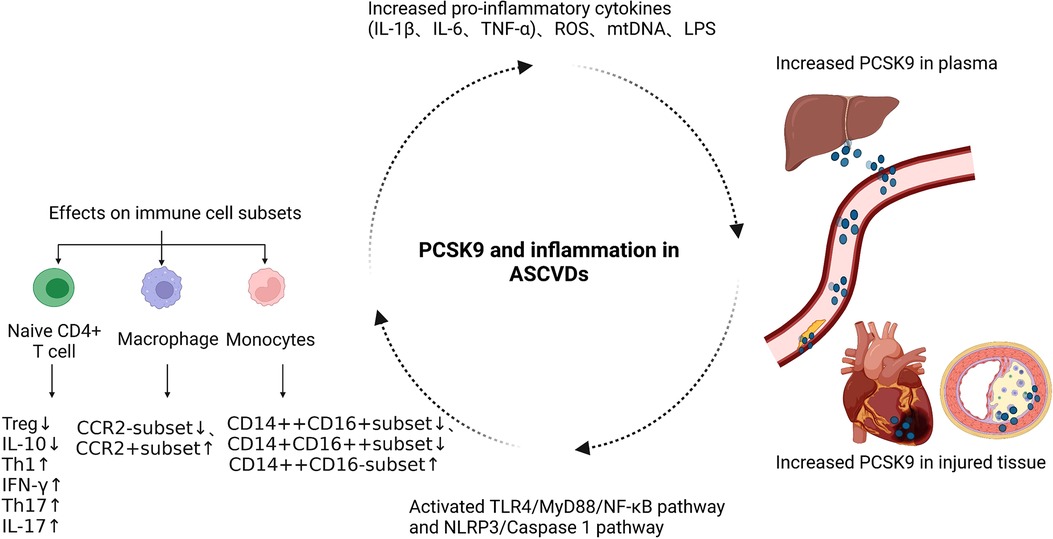
Figure 3. The functional mechanism of PCSK9 in regulating the immune response of ASCVDs. ASCVDs, atherosclerotic cardiovascular diseases; NLRP3, NOD-like receptor family pyrin domain containing 3; TLR4, Toll-Like Receptor 4; ROS, reactive oxygen species; mtDNA, mitochondrial DNA; LPS, lipopolysaccharide.
Several clinical studies have confirmed that PCSK9 promotes the progression of ASCVDs by its proinflammatory function. In the Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk (FOURIER) trial, 27,564 patients with stable ASCVDs and LDL-C ≥ 70 mg/dl were randomly assigned to the evolocumab group and placebo group, both of which were then divided into three subgroups based on the level of high sensitivity C-reactive protein (hs-CRP) (<1, 1–3, and >3 mg/dl) at baseline (10, 92). Patients with higher hs-CRP had a greater risk of the primary and key secondary endpoint events, showing that inflammation may be an independent risk factor in ASCVDs (90). In patients with higher hs-CRP, the reduction in the absolute risk was more significant in the evolocumab group than in the placebo group (93). Thus, the protective effect of evolocumab on ASCVD patients, at least in part, relies on its anti-inflammatory function. In addition, in the ODYSSEY OUTCOMES trial, alirocumab reduced plasma lipoprotein(a) and the risk of major adverse cardiac events (MACEs) in patients with acute coronary syndrome (ACS) (94). Lipoprotein(a) is a biomarker positively correlated with inflammatory response (95). In the PACMAN-AMI trial, the use of alirocumab in addition to high-intensity statin therapy resulted in a significant regression of percent atheroma volume in the two non-infarct-related coronary arteries and increased plaque stability in patients with acute myocardial infarction without a reduction of hs-CRP (14). However, further research revealed that alirocumab caused a greater reduction in the mean angular extension of macrophages, indicating that the local inflammatory response was decreased in this process (14). Therefore, it may not be sufficient to select hs-CRP as the only biomarker reflecting the intensity of the inflammatory response, and PCSK9 inhibitors are useful to inhibit the local inflammatory response in ASCVD patients (14, 96). Moreover, PCSK9 inhibitors suppress the accumulation of the proinflammatory cytokine IL-6 in the plasma of stable coronary artery disease patients with the IL6-74CC genotype and high level of lipoprotein(a) (97). However, in patients with elevated lipoprotein(a) and increased cardiovascular risk, evolocumab only modestly reduces the lipoprotein(a), and residual high level of lipoprotein(a) leads to persistent arterial wall inflammation (98). Thus, whether PCSK9 inhibitors have anti-inflammatory function and the mechanisms involved need to be further explored.
In addition to regulating inflammatory cytokines, numerous studies have linked PCSK9 with immune cell subsets in ASCVDs. Circulating monocytes are usually divided into three categories, including classical monocytes (CD14++CD16−; CMs), intermediate monocytes (CD14++CD16+; IMs) and nonclassical monocytes (CD14+CD16++; NCMs), among which CMs have strong proinflammatory functions (99, 100). When stable coronary artery disease occurs, the proportion of CMs to total monocytes is larger in patients with higher PCSK9 levels. Increased PCSK9 promotes the polarization of monocytes from IM and NCM-like phenotypes to a CM-like phenotype (101). In patients with hyperlipidemia, PCSK9 induces the expression of C-C chemokine receptor type 2 (CCR2) on the surface of monocytes and enhances their migration ability (102).
Previous studies have shown that PCSK9 siRNA can inhibit ox-LDL-induced proinflammatory function of THP-1-derived macrophages via suppressing the activation of the NF-κB pathway (35). Compared with C57BL6/J WT mice, the C57BL6/J PCSK9 knockout (KO) mice have decreased infarct size and improved cardiac function due to inhibition of the polarization of M1-type macrophages, and the suppression of the TLR4/MyD88/NF-κB pathway may be involved in this process, which is consistent with the above results (43). In apolipoprotein E (apoE) KO mice with hyperlipidemia-induced atherosclerosis, PCSK9 silencing inhibits the progression of plaque volume, the accumulation of macrophages in lesion areas, and the secretion of inflammatory cytokines, such as TNF-α and IL-1β, by macrophages, which is accompanied by limited intracellular activation of the TLR4/NF-κB pathway (32). Because the AT04A anti-PCSK9 vaccine reduces the expression of NLR family pyrin domain containing 3 (NLRP3) and proinflammatory biomarkers in macrophages, both the NLRP3 and TLR4/NF-κB pathways may be involved in the PCSK9-mediated regulation of macrophage function (32).
The adaptive immune response is also found to participate in the later stage of atherosclerosis as T cells localize near the ruptured areas of unstable plaques in ischemic heart tissue (103). Increased PCSK9 promotes the maturation of dendritic cells and the differentiation of naive CD4+ T cells toward the Th1 and Th17 subsets, resulting in increased secretion of interferon-γ (IFN-γ) and interleukin 17A (IL-17A) (104). PCSK9 inhibitors promote the differentiation of naive CD4+ T cells into regulatory T cells (Tregs) and the production of anti-inflammatory cytokines, such as interleukin 10 (IL-10) and transforming growth factor beta (TGF-β), which contribute to the resolution of inflammation and good prognosis of ASCVDs (104). Apart from the cardiovascular system, PCSK9 also regulates the adaptive immune response in the tumor microenvironment. The interaction between LDLR and T cell receptor (TCR) regulates TCR recycling and signaling, thus promoting the differentiation of CD8+ T cells into cytotoxic T lymphocytes (90). PCSK9 inhibits the killing function of CD8+ T cells via binding to LDLR and preventing the recycling of LDLR-TCR complex to the plasma membrane (90). Thus, we postulate that PCSK9/LDLR may also be a significant target for regulating the adaptive immune response in ASCVDs, but this specific mechanism has not been confirmed.
Under pathological conditions, such as hyperlipidemia, hyperglycemia, and atherosclerosis, high LDL-C levels are associated with enhanced platelet reactivity and thromboxane production (105, 106). Many factors are involved in the regulation of this process, which is still not completely clear (107). CD36 is associated with platelet reactivity, activation and thrombosis under hyperlipidemic conditions (108). During the processes of plaque formation with inflammation and phospholipid oxidation, LDL is converted to ox-LDL (109). Ox-LDL and hyperlipidemia activate blood platelets via a CD36 mediated pathway (110, 111). In patients with familial hypercholesterolemia, researchers found that ox-LDL induced the activation of platelets via the activation of CD36, LOX-1, and NADPH oxidase 2 (NOX2) (112). PCSK9 is a positive modulator in this process (113). Recently, researchers have found that PCSK9 promotes the aggregation, activation, spreading of platelets and thrombosis by interacting with CD36 on its surface and activating the downstream p38 mitogen-activated protein kinase (MAPK)/cytosolic phospholipase A2/cyclooxygenase-1/thromboxane A2 pathway (26, 113). In C57BL/6J WT mice, PCSK9 injection promotes FeCl3-induced mesenteric artery thrombosis through binding to CD36 receptor in platelets (26). When myocardial infarction occurs, PCSK9 promotes the generation of ROS and activates CD36 in platelets, resulting in microvascular obstruction and enlarged infarct size (26). Therefore, the experimental results mentioned above confirm the positive role of PCSK9 in platelet activation and thrombosis, which may be adverse in ischemic heart disease.
In the development of atherosclerosis, ox-LDL is one of the important factors causing the dysfunction of endothelial cells and the primary factor promoting apoptosis in endothelial cells by upregulating the apoptosis-related factors, Bcl2-associated X (Bax), caspase 3, and caspase 9, as well as downregulating the antiapoptotic factor, Bcl2 (114, 115). PCSK9 is a key mediator of ox-LDL-induced apoptosis in HUVECs (116). The proapoptotic function of ox-LDL mainly depends on the upregulation of PCSK9 expression and the activation of its downstream MAPK signaling pathway, especially the phosphorylation of c-Jun N-terminal kinase (JNK) and p38 (116). Upregulated PCSK9 promotes the apoptosis of endothelial cells in atherosclerotic lesions (116). Targeting PCSK9 with short hairpin RNA (shRNA)-PCSK9 inhibits the phosphorylation of p38 and JNK induced by ox-LDL, as well as downregulates the ratio of Bax to Bcl2, thus repressing the apoptosis of endothelial cells (116).
PCSK9 is associated with autophagy (44). Autophagy removes damaged mitochondria, which is of great benefit to maintain cell survival and normal function (117). MtDNA that escapes from autophagy leads to inflammation and heart failure (118). When 3-methyladenine is used to inhibit autophagy, PCSK9 accumulates in the cytoplasm, suggesting that it may be associated with autophagy (41). Under inflammatory conditions, increased PCSK9 destroys mtDNA and promotes the formation of mtROS in SMCs, which further promotes the upregulation of PCSK9 and LOX-1 (41). When myocardial infarction develops in mice with PCSK9 knockdown or with PCSK9 inhibitor Pep2-8 treatment, the infarct size is smaller and the autophagy is reduced compared with WT mice (44).
During the process of chronic myocardial ischemia, upregulated PCSK9 induces mtDNA damage, which activates NLRP3 inflammasome signaling [NLRP3, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), caspase 1, IL-1β, and interleukin-18 (IL-18)] and promotes caspase 1-dependent pyroptosis (27). The pyroptosis marker, N-terminal gasdermin D fragment (GSDMD-NT), is highly expressed in the peripheral area of the infarct zone (27, 119). In PCSK9 knockout mice, the activation of NLRP3 and the upregulation of GSDMD-NT in the ischemic heart are significantly inhibited (27).
In summary, PCSK9 inhibits autophagy but promotes apoptosis and pyroptosis, which is unfavorable for the prognosis of ASCVDs.
There is a significant positive correlation between plasma PCSK9 levels and the risk of atherosclerosis. Feeding a high-fat diet to mice transduced with an adeno-associated virus overexpressing the PCSK9 gene induces hypercholesterolemia and even atherosclerosis (120). Similarly, transgenic pigs with the D374Y gain-of-function mutation in the PCSK9 gene are more prone to develop atherosclerosis than WT pigs when fed a high-fat and high-cholesterol diet (121). In patients with rheumatoid arthritis (RA), the plasma level of PCSK9 and the ratio of PCSK9 to LDLR are positively correlated with the occurrence and development of atherosclerosis (122). LRP5 and LRP6, as coreceptors of PCSK9, promote atherosclerosis by activating the Wnt/β-catenin signaling pathway, resulting in significant proliferation of VSMCs and decreased anti-inflammatory macrophages (78, 81). However, there may still be lack of correlation between the plasma PCSK9 level and the severity of subclinical atherosclerosis in patients without symptoms of cardiovascular diseases (123).
Researchers have found that LincRNA-p21 binding to miR-221 promotes the deacetylation of PCSK9 via negatively regulating the expression of SIRT1, eventually leading to the enhance of the proliferation, migration and angiogenesis of arterial endothelial cells, ultimately diminishing the development of atherosclerosis (124). Treatment of ApoE-/- mice on a high-fat diet with berberine reverses the progression of atherosclerotic plaques by downregulating PCSK9 expression and upregulating LDLR expression through the activation of the ERK1/2 signaling pathway in hepatocytes (125). Similarly, in ApoE-/- mice with a high-cholesterol diet (1.25% w/w), the sirtuin 1 activator [SRT3025 and 20(S)-protopanaxadiol] exerts an anti-atherosclerotic function by reducing plasma PCSK9 and upregulating LDLR (126, 127).
The proatherogenic effect of PCSK9 may be independent of its lipid-regulatory function (32). In ApoE-/- mice fed a high-fat diet, the overexpression of PCSK9 promotes the progression of atherosclerotic plaques without upregulating the plasma cholesterol level (32). The increased PCSK9 could accelerate atherosclerosis through activating the TLR4/NF-κB signaling pathway and promoting inflammation (32). In homocysteine-treated ApoE-/- mice with a methionine diet, upregulated PCSK9 inhibits cholesterol efflux mediated by ABCA1 and ABCG1 in macrophages, thereby accelerating the formation of foam cells (128). Therefore, PCSK9 promotes the development of atherosclerosis dependent on inflammatory regulation in part.
Myocardial infarction is one of the major causes of mortality worldwide (129). Patients with myocardial infarction have a high risk of ischemia reperfusion injury and the “no-reflow” phenomenon after successful percutaneous coronary intervention treatment, and more improvement in therapeutic strategies is still needed (129, 130). Cardiomyocytes from adult mice express PCSK9 at both the transcriptional and translational levels (131). One week after ligation of the left anterior descending coronary artery in C57BL/6J WT mice, the increased expression of PCSK9 in the zone bordering the infarct area leads to the progression of myocardial infarction (44). Non-ST-segment elevation myocardial infarction patients with higher plasma levels of PCSK9 have a higher risk of MACEs than those with moderate or low levels of PCSK9 (132). In young males with myocardial infarction, higher level PCSK9 exacerbates the severity of coronary artery diseases and increases the risk of MACEs (133). Consistently, patients in the Chinese Han population with the PCSK9 R93C variant (PCSK9 loss-of-function mutant) have a lower risk of myocardial infarction (134). Therefore, the level of plasma PCSK9 is positively related to the occurrence risk and the severity of myocardial infarction.
PCSK9 inhibitors can further reduce plasma LDL-C in addition to statins treatment at the maximum dose, which is beneficial to the prognosis of myocardial infarction (135). In the PACMAN-AMI study, 300 patients with acute myocardial infarction treated with rosuvastatin (20 mg/day) were randomly divided into two groups. Patients in the experimental group received alirocumab (150 mg) subcutaneously once every 2 weeks, and those in the control group received an equal dose of placebo. Fifty-two weeks after the initial treatment, intravascular ultrasonography, near-infrared spectroscopy, and optical coherence tomography results all showed that the use of alirocumab significantly reversed the plaque in the two non-infarct-related coronary arteries and improved the stabilization of plaques (14, 136). In the FOURIER study, the reduction of absolute occurrence risk of the endpoint events after using evolocumab in stable atherosclerotic patients with previous myocardial infarction was three times as much as in patients without previous myocardial infarction (137, 138). Similarly, in the ODYSSEY OUTCOMES study, the use of alirocumab in addition to statin treatment significantly reduced the risk of MACEs and death in patients with ACS, which is more significant in patients with previous myocardial infarction (139).
The level of plasma PCSK9 is positively correlated with hs-CRP in myocardial infarction patients (140). Higher plasma levels of PCSK9 and hs-CRP lead to an earlier decline in left ventricular ejection fraction in myocardial infarction patients, further increasing the risk of myocardial infarction-induced heart failure (HF) and even death (140). In C57BL6/J WT mice with myocardial infarction, PCSK9 promotes the polarization of M1-type macrophages and inhibits the polarization of M2-type macrophages by activating the TLR4/MyD88/NF-κB signaling pathway, leading to the significant inflammatory response, increased infarct size, and excessive impairment of cardiac function (43).
In addition, CD36-mediated thrombosis and NLRP3 inflammasome-mediated autophagy are both associated with the effect of PCSK9 on myocardial infarction prognosis (26, 141). Regardless of the specific underlying mechanism, upregulated PCSK9 is unfavorable to the recovery of myocardial infarction.
As the number of patients with ASCVDs increases, many evidences show that PCSK9 has become a new promising therapeutic target for patients who are intolerant of statins or who fail to achieve the plasma lipid goal after treatment with a maximum dose of statins (10). In general, PCSK9 affects homeostasis in vivo through multiple functional mechanisms, including regulating lipid metabolism, promoting the immune response, promoting platelet activation, promoting thrombosis, promoting apoptosis, promoting pyroptosis, and inhibiting autophagy. PCSK9 promotes the progression of ASCVDs via various mechanisms, and it has been confirmed that PCSK9 inhibitors effectively improve the prognosis of patients with ASCVDs (10–15).
Based on the existing researches on PCSK9 and cardiovascular diseases, we propose several perspectives and directions for future research. First, many studies regarding PCSK9 and immunity in cardiovascular diseases only detected changes in immune cell subsets, and a few studies have demonstrated that the above functions of PCSK9 are mediated by classical pathways, such as TLR4/NF-kB and NLRP3 (27, 43). Thus, it is meaningful to determine whether other pathways associated with inflammation are involved, such as cGAS-STING pathway, Notch pathway et al. (142, 143). Whether PCSK9 regulates the transformation of immune cells subsets by altering their metabolic activity? Which kind of metabolic activity is altered most dramatically? Which kind of immune cells subset is most sensitive to the metabolic effects of PCSK9? These can be further studied. Second, because PCSK9 and CD8+ T cells accumulate in the ischemic heart and the effect of PCSK9 on CD8+ T cells in the tumor microenvironment is already clear, will their cross-talk in the cardiac microenvironment be different from the former (90)? What factors account for this difference? Third, whether PCSK9 inhibitors are useful in improving the prognosis of patient with heart failure (HF) remains controversial. For example, in the multicenter, prospective, observational Biology Study to Tailored Treatment in Chronic Heart Failure (BIOSTAT-CHF) study, the rates of all-cause mortality and the composite of mortality or unscheduled hospitalizations due to HF had a positive association with plasma level of PCSK9 and a negative association with the level of LDLR in HF patients (144). In the ODYSSEY OUTCOMES study, however, it was found that although alirocumab reduced plasma cholesterol levels to the same extent, it did not lower the incidence of MACEs, death, or unscheduled hospitalizations for HF in ACS patients with previous HF (145, 146). The inconsistency among the above findings may be related to the heterogeneity of the enrolled patient population with respect to the cause or severity of the heart injury, or the usage and dosage of PCSK9 inhibitors. Therefore, according to these influencing factors, we can divide the enrolled patients into different subgroups before carrying out our research. Finally, it is valuable to investigate whether PCSK9 inhibitors can improve the prognosis of other common cardiovascular diseases.
JL proposed the topics for the review and revised the manuscript. MM and CH wrote the manuscript. All authors contributed to the article and approved the submitted version.
The review was supported by the National Natural Science Foundation of China (grant no. 11832003 and 81970294) and Beijing Natural Science Foundation (grant no. 7212119). With the support of the funding, we consulted the latest literature, collected data and so on.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer [YW] declared a shared affiliation with the authors to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. (2005) 352:1685–95. doi: 10.1056/NEJMra043430
2. Winkels H, Meiler S, Lievens D, Engel D, Spitz C, Bürger C, et al. CD27 co-stimulation increases the abundance of regulatory T cells and reduces atherosclerosis in hyperlipidaemic mice. Eur Heart J. (2017) 38:3590–9. doi: 10.1093/eurheartj/ehx517
3. Depuydt MAC, Prange KHM, Slenders L, Örd T, Elbersen D, Boltjes A, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. (2020) 127:1437–55. doi: 10.1161/CIRCRESAHA.120.316770
4. Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. (1986) 6:131–8. doi: 10.1161/01.ATV.6.2.131
5. Kaartinen M, Penttilä A, Kovanen PT. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation. (1994) 90:1669–78. doi: 10.1161/01.CIR.90.4.1669
6. Vallejo J, Cochain C, Zernecke A, Ley K. Heterogeneity of immune cells in human atherosclerosis revealed by scRNA-seq. Cardiovasc Res. (2021) 117:2537–43. doi: 10.1093/cvr/cvab260
7. Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American college of cardiology/American heart association task force on practice guidelines. Circulation. (2014) 129:S1–45. doi: 10.1161/01.cir.0000437738.63853.7a
8. Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. (2001) 292:1160–4. doi: 10.1126/science.1059344
9. Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, et al. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res. (2010) 51:1486–95. doi: 10.1194/jlr.M003566
10. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. (2017) 376:1713–22. doi: 10.1056/NEJMoa1615664
11. Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, et al. Effect of evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV randomized clinical trial. JAMA. (2016) 316:2373–84. doi: 10.1001/jama.2016.16951
12. Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. (2015) 372:1489–99. doi: 10.1056/NEJMoa1501031
13. Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. (2018) 379:2097–107. doi: 10.1056/NEJMoa1801174
14. Räber L, Ueki Y, Otsuka T, Losdat S, Häner JD, Lonborg J, et al. Effect of alirocumab added to high-intensity statin therapy on coronary atherosclerosis in patients with acute myocardial infarction: the PACMAN-AMI randomized clinical trial. JAMA. (2022) 327:1771–81. doi: 10.1001/jama.2022.5218
15. O'Donoghue ML, Rosenson RS, Gencer B, López JAG, Lepor NE, Baum SJ, et al. Small interfering RNA to reduce lipoprotein(a) in cardiovascular disease. N Engl J Med. (2022) 387:1855–64. doi: 10.1056/NEJMoa2211023
16. Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. (2003) 100:928–33. doi: 10.1073/pnas.0335507100
17. Siezen RJ, Leunissen JA. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. (1997) 6:501–23. doi: 10.1002/pro.5560060301
18. McNutt MC, Lagace TA, Horton JD. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem. (2007) 282:20799–803. doi: 10.1074/jbc.C700095200
19. Piper DE, Jackson S, Liu Q, Romanow WG, Shetterly S, Thibault ST, et al. The crystal structure of PCSK9: a regulator of plasma LDL-cholesterol. Structure. (2007) 15:545–52. doi: 10.1016/j.str.2007.04.004
20. Seidah NG, Mayer G, Zaid A, Rousselet E, Nassoury N, Poirier S, et al. The activation and physiological functions of the proprotein convertases. Int J Biochem Cell Biol. (2008) 40:1111–25. doi: 10.1016/j.biocel.2008.01.030
21. Seidah NG, Prat A. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov. (2012) 11:367–83. doi: 10.1038/nrd3699
22. Hampton EN, Knuth MW, Li J, Harris JL, Lesley SA, Spraggon G. The self-inhibited structure of full-length PCSK9 at 1.9 A reveals structural homology with resistin within the C-terminal domain. Proc Natl Acad Sci U S A. (2007) 104:14604–9. doi: 10.1073/pnas.0703402104
23. Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. (2003) 34:154–6. doi: 10.1038/ng1161
24. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. (2005) 37:161–5. doi: 10.1038/ng1509
25. Zhang Y, Zhu CG, Xu RX, Li S, Guo YL, Sun J, et al. Relation of circulating PCSK9 concentration to fibrinogen in patients with stable coronary artery disease. J Clin Lipidol. (2014) 8:494–500. doi: 10.1016/j.jacl.2014.07.001
26. Qi Z, Hu L, Zhang J, Yang W, Liu X, Jia D, et al. PCSK9 (proprotein convertase subtilisin/kexin 9) enhances platelet activation, thrombosis, and myocardial infarct expansion by binding to platelet CD36. Circulation. (2021) 143:45–61. doi: 10.1161/CIRCULATIONAHA.120.046290
27. Wang X, Li X, Liu S, Brickell AN, Zhang J, Wu Z, et al. PCSK9 Regulates pyroptosis via mtDNA damage in chronic myocardial ischemia. Basic Res Cardiol. (2020) 115:66. doi: 10.1007/s00395-020-00832-w
28. Cheng JM, Oemrawsingh RM, Garcia-Garcia HM, Boersma E, van Geuns RJ, Serruys PW, et al. PCSK9 In relation to coronary plaque inflammation: results of the ATHEROREMO-IVUS study. Atherosclerosis. (2016) 248:117–22. doi: 10.1016/j.atherosclerosis.2016.03.010
29. Fang C, Luo T, Lin L. Elevation of serum proprotein convertase subtilisin/kexin type 9 (PCSK9) concentrations and its possible atherogenic role in patients with systemic lupus erythematosus. Ann Transl Med. (2018) 6:452. doi: 10.21037/atm.2018.11.04
30. Li S, Zhang Y, Xu RX, Guo YL, Zhu CG, Wu NQ, et al. Proprotein convertase subtilisin-kexin type 9 as a biomarker for the severity of coronary artery disease. Ann Med. (2015) 47:386–93. doi: 10.3109/07853890.2015.1042908
31. Zanni MV, Stone LA, Toribio M, Rimmelin DE, Robinson J, Burdo TH, et al. Proprotein convertase subtilisin/kexin 9 levels in relation to systemic immune activation and subclinical coronary plaque in HIV. Open Forum Infect Dis. (2017) 4:ofx227. doi: 10.1093/ofid/ofx227
32. Tang ZH, Peng J, Ren Z, Yang J, Li TT, Li TH, et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis. (2017) 262:113–22. doi: 10.1016/j.atherosclerosis.2017.04.023
33. Landlinger C, Pouwer MG, Juno C, van der Hoorn JWA, Pieterman EJ, Jukema JW, et al. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE*3Leiden.CETP mice. Eur Heart J. (2017) 38:2499–507. doi: 10.1093/eurheartj/ehx260
34. Ricci C, Ruscica M, Camera M, Rossetti L, Macchi C, Colciago A, et al. PCSK9 Induces a pro-inflammatory response in macrophages. Sci Rep. (2018) 8:2267. doi: 10.1038/s41598-018-20425-x
35. Tang Z, Jiang L, Peng J, Ren Z, Wei D, Wu C, et al. PCSK9 siRNA suppresses the inflammatory response induced by oxLDL through inhibition of NF-κB activation in THP-1-derived macrophages. Int J Mol Med. (2012) 30:931–8. doi: 10.3892/ijmm.2012.1072
36. Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. (2014) 13:397–406. doi: 10.1074/mcp.M113.035600
37. Ferri N, Tibolla G, Pirillo A, Cipollone F, Mezzetti A, Pacia S, et al. Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis. (2012) 220:381–6. doi: 10.1016/j.atherosclerosis.2011.11.026
38. Ding Z, Liu S, Wang X, Deng X, Fan Y, Shahanawaz J, et al. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc Res. (2015) 107:556–67. doi: 10.1093/cvr/cvv178
39. Ding Z, Liu S, Wang X, Deng X, Fan Y, Sun C, et al. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid Redox Signal. (2015) 22:760–71. doi: 10.1089/ars.2014.6054
40. Tang ZH, Li TH, Peng J, Zheng J, Li TT, Liu LS, et al. PCSK9: a novel inflammation modulator in atherosclerosis? J Cell Physiol. (2019) 234:2345–55. doi: 10.1002/jcp.27254
41. Ding Z, Liu S, Wang X, Mathur P, Dai Y, Theus S, et al. Cross-Talk between PCSK9 and damaged mtDNA in vascular smooth muscle cells: role in apoptosis. Antioxid Redox Signal. (2016) 25:997–1008. doi: 10.1089/ars.2016.6631
42. Giunzioni I, Tavori H, Covarrubias R, Major AS, Ding L, Zhang Y, et al. Local effects of human PCSK9 on the atherosclerotic lesion. J Pathol. (2016) 238:52–62. doi: 10.1002/path.4630
43. Wang F, Li M, Zhang A, Li H, Jiang C, Guo J. PCSK9 Modulates macrophage polarization-mediated ventricular remodeling after myocardial infarction. J Immunol Res. (2022) 2022:7685796. doi: 10.1155/2022/7685796
44. Ding Z, Wang X, Liu S, Shahanawaz J, Theus S, Fan Y, et al. PCSK9 Expression in the ischaemic heart and its relationship to infarct size, cardiac function, and development of autophagy. Cardiovasc Res. (2018) 114:1738–51. doi: 10.1093/cvr/cvy128
45. Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D, Park SW. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res. (2008) 49:399–409. doi: 10.1194/jlr.M700443-JLR200
46. Li H, Dong B, Park SW, Lee HS, Chen W, Liu J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J Biol Chem. (2009) 284:28885–95. doi: 10.1074/jbc.M109.052407
47. Tao R, Xiong X, DePinho RA, Deng CX, Dong XC. Foxo3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J Biol Chem. (2013) 288:29252–9. doi: 10.1074/jbc.M113.481473
48. Lin YK, Yeh CT, Kuo KT, Yadav VK, Fong IH, Kounis NG, et al. Pterostilbene increases LDL metabolism in HL-1 cardiomyocytes by modulating the PCSK9/HNF1α/SREBP2/LDLR signaling cascade, upregulating epigenetic hsa-miR-335 and hsa-miR-6825, and LDL receptor expression. Antioxidants (Basel). (2021) 10:1280. doi: 10.3390/antiox10081280
49. Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. (2004) 24:1454–9. doi: 10.1161/01.ATV.0000134621.14315.43
50. Costet P, Cariou B, Lambert G, Lalanne F, Lardeux B, Jarnoux AL, et al. Hepatic PCSK9 expression is regulated by nutritional status via insulin and sterol regulatory element-binding protein 1c. J Biol Chem. (2006) 281:6211–8. doi: 10.1074/jbc.M508582200
51. Lebeau PF, Byun JH, Platko K, Saliba P, Sguazzin M, MacDonald ME, et al. Caffeine blocks SREBP2-induced hepatic PCSK9 expression to enhance LDLR-mediated cholesterol clearance. Nat Commun. (2022) 13:770. doi: 10.1038/s41467-022-28240-9
52. Dong B, Singh AB, Shende VR, Liu J. Hepatic HNF1 transcription factors control the induction of PCSK9 mediated by rosuvastatin in normolipidemic hamsters. Int J Mol Med. (2017) 39:749–56. doi: 10.3892/ijmm.2017.2879
53. Li H, Liu J. The novel function of HINFP as a co-activator in sterol-regulated transcription of PCSK9 in HepG2 cells. Biochem J. (2012) 443:757–68. doi: 10.1042/BJ20111645
54. Ai D, Chen C, Han S, Ganda A, Murphy AJ, Haeusler R, et al. Regulation of hepatic LDL receptors by mTORC1 and PCSK9 in mice. J Clin Invest. (2012) 122:1262–70. doi: 10.1172/JCI61919
55. Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM. Structure and biochemical functions of SIRT6. J Biol Chem. (2011) 286:14575–87. doi: 10.1074/jbc.M111.218990
56. Wang X, Chen X, Zhang X, Su C, Yang M, He W, et al. A small-molecule inhibitor of PCSK9 transcription ameliorates atherosclerosis through the modulation of FoxO1/3 and HNF1α. EBioMedicine. (2020) 52:102650. doi: 10.1016/j.ebiom.2020.102650
57. Zhang S, Hong F, Ma C, Yang S. Hepatic lipid metabolism disorder and atherosclerosis. Endocr Metab Immune Disord Drug Targets. (2022) 22:590–600. doi: 10.2174/1871530321666211119143653
58. Bäck M, Yurdagul A Jr., Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. (2019) 16:389–406. doi: 10.1038/s41569-019-0169-2
59. Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol. (2015) 5:1841–75. doi: 10.1002/cphy.c150006
60. Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. (2015) 161:161–72. doi: 10.1016/j.cell.2015.01.036
61. Silverman MG, Ference BA, Im K, Wiviott SD, Giugliano RP, Grundy SM, et al. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA. (2016) 316:1289–97. doi: 10.1001/jama.2016.13985
62. Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. (2010) 376:1670–81. doi: 10.1016/S0140-6736(10)61350-5
63. Anderson RG, Brown MS, Goldstein JL. Role of the coated endocytic vesicle in the uptake of receptor-bound low density lipoprotein in human fibroblasts. Cell. (1977) 10:351–64. doi: 10.1016/0092-8674(77)90022-8
64. Ference BA, Kastelein JJP, Ray KK, Ginsberg HN, Chapman MJ, Packard CJ, et al. Association of triglyceride-lowering LPL variants and LDL-C-lowering LDLR variants with risk of coronary heart disease. JAMA. (2019) 321:364–73. doi: 10.1001/jama.2018.20045
65. Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. (2007) 32:71–7. doi: 10.1016/j.tibs.2006.12.008
66. Leren TP. Mutations in the PCSK9 gene in Norwegian subjects with autosomal dominant hypercholesterolemia. Clin Genet. (2004) 65:419–22. doi: 10.1111/j.0009-9163.2004.0238.x
67. Timms KM, Wagner S, Samuels ME, Forbey K, Goldfine H, Jammulapati S, et al. A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum Genet. (2004) 114:349–53. doi: 10.1007/s00439-003-1071-9
68. Kudo T, Sasaki K, Tada H. Familial hypobetalipoproteinemia caused by homozygous loss-of-function mutations in PCSK9: a case report. J Clin Lipidol. (2022) 16:596–600. doi: 10.1016/j.jacl.2022.07.010
69. Sun H, Samarghandi A, Zhang N, Yao Z, Xiong M, Teng BB. Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler Thromb Vasc Biol. (2012) 32:1585–95. doi: 10.1161/ATVBAHA.112.250043
70. Tavori H, Giunzioni I, Predazzi IM, Plubell D, Shivinsky A, Miles J, et al. Human PCSK9 promotes hepatic lipogenesis and atherosclerosis development via apoE- and LDLR-mediated mechanisms. Cardiovasc Res. (2016) 110:268–78. doi: 10.1093/cvr/cvw053
71. Levy E, Ben Djoudi Ouadda A, Spahis S, Sane AT, Garofalo C, Grenier É, et al. PCSK9 Plays a significant role in cholesterol homeostasis and lipid transport in intestinal epithelial cells. Atherosclerosis. (2013) 227:297–306. doi: 10.1016/j.atherosclerosis.2013.01.023
72. Rashid S, Tavori H, Brown PE, Linton MF, He J, Giunzioni I, et al. Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms. Circulation. (2014) 130:431–41. doi: 10.1161/CIRCULATIONAHA.113.006720
73. Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A. (2005) 102:2069–74. doi: 10.1073/pnas.0409736102
74. Luquero A, Badimon L, Borrell-Pages M. PCSK9 Functions in atherosclerosis are not limited to plasmatic LDL-cholesterol regulation. Front Cardiovasc Med. (2021) 8:639727. doi: 10.3389/fcvm.2021.639727
75. Jang HD, Lee SE, Yang J, Lee HC, Shin D, Lee H, et al. Cyclase-associated protein 1 is a binding partner of proprotein convertase subtilisin/kexin type-9 and is required for the degradation of low-density lipoprotein receptors by proprotein convertase subtilisin/kexin type-9. Eur Heart J. (2020) 41:239–52. doi: 10.1093/eurheartj/ehz566
76. Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, et al. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem. (2008) 283:2363–72. doi: 10.1074/jbc.M708098200
77. Canuel M, Sun X, Asselin MC, Paramithiotis E, Prat A, Seidah NG. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One. (2013) 8:e64145. doi: 10.1371/journal.pone.0064145
78. Badimon L, Luquero A, Crespo J, Peña E, Borrell-Pages M. PCSK9 And LRP5 in macrophage lipid internalization and inflammation. Cardiovasc Res. (2021) 117:2054–68. doi: 10.1093/cvr/cvaa254
79. Shan L, Pang L, Zhang R, Murgolo NJ, Lan H, Hedrick JA. PCSK9 Binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Commun. (2008) 375:69–73. doi: 10.1016/j.bbrc.2008.07.106
80. Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, et al. LDL-receptor-related proteins in wnt signal transduction. Nature. (2000) 407:530–5. doi: 10.1038/35035117
81. Desita SR, Hariftyani AS, Jannah AR, Setyobudi AK, Oktaviono YH. PCSK9 And LRP6: potential combination targets to prevent and reduce atherosclerosis. J Basic Clin Physiol Pharmacol. (2022) 33:529–34. doi: 10.1515/jbcpp-2021-0291
82. Awan Z, Denis M, Bailey D, Giaid A, Prat A, Goltzman D, et al. The LDLR deficient mouse as a model for aortic calcification and quantification by micro-computed tomography. Atherosclerosis. (2011) 219:455–62. doi: 10.1016/j.atherosclerosis.2011.08.035
83. Hey PJ, Twells RC, Phillips MS, Yusuke N, Brown SD, Kawaguchi Y, et al. Cloning of a novel member of the low-density lipoprotein receptor family. Gene. (1998) 216:103–11. doi: 10.1016/S0378-1119(98)00311-4
84. Ding Z, Liu S, Wang X, Theus S, Deng X, Fan Y, et al. PCSK9 Regulates expression of scavenger receptors and ox-LDL uptake in macrophages. Cardiovasc Res. (2018) 114:1145–53. doi: 10.1093/cvr/cvy079
85. Adorni MP, Cipollari E, Favari E, Zanotti I, Zimetti F, Corsini A, et al. Inhibitory effect of PCSK9 on Abca1 protein expression and cholesterol efflux in macrophages. Atherosclerosis. (2017) 256:1–6. doi: 10.1016/j.atherosclerosis.2016.11.019
86. Murray TV, Ahmad A, Brewer AC. Reactive oxygen at the heart of metabolism. Trends Cardiovasc Med. (2014) 24:113–20. doi: 10.1016/j.tcm.2013.09.003
87. van der Vusse GJ, van Bilsen M, Glatz JF. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc Res. (2000) 45:279–93. doi: 10.1016/S0008-6363(99)00263-1
88. Da Dalt L, Castiglioni L, Baragetti A, Audano M, Svecla M, Bonacina F, et al. PCSK9 Deficiency rewires heart metabolism and drives heart failure with preserved ejection fraction. Eur Heart J. (2021) 42:3078–90. doi: 10.1093/eurheartj/ehab431
89. Santos RD, Stein EA, Hovingh GK, Blom DJ, Soran H, Watts GF, et al. Long-Term evolocumab in patients with familial hypercholesterolemia. J Am Coll Cardiol. (2020) 75:565–74. doi: 10.1016/j.jacc.2019.12.020
90. Yuan J, Cai T, Zheng X, Ren Y, Qi J, Lu X, et al. Potentiating CD8(+) T cell antitumor activity by inhibiting PCSK9 to promote LDLR-mediated TCR recycling and signaling. Protein Cell. (2021) 12:240–60. doi: 10.1007/s13238-021-00821-2
91. Liu X, Suo R, Chan CZY, Liu T, Tse G, Li G. The immune functions of PCSK9: local and systemic perspectives. J Cell Physiol. (2019) 234:19180–8. doi: 10.1002/jcp.28612
92. Sabatine MS, Giugliano RP, Keech A, Honarpour N, Wang H, Liu T, et al. Rationale and design of the further cardiovascular OUtcomes research with PCSK9 inhibition in subjects with elevated risk trial. Am Heart J. (2016) 173:94–101. doi: 10.1016/j.ahj.2015.11.015
93. Bohula EA, Giugliano RP, Leiter LA, Verma S, Park JG, Sever PS, et al. Inflammatory and cholesterol risk in the FOURIER trial. Circulation. (2018) 138:131–40. doi: 10.1161/CIRCULATIONAHA.118.034032
94. Szarek M, Bittner VA, Aylward P, Baccara-Dinet M, Bhatt DL, Diaz R, et al. Lipoprotein(a) lowering by alirocumab reduces the total burden of cardiovascular events independent of low-density lipoprotein cholesterol lowering: oDYSSEY OUTCOMES trial. Eur Heart J. (2020) 41:4245–55. doi: 10.1093/eurheartj/ehaa649
95. Dangas G, Mehran R, Harpel PC, Sharma SK, Marcovina SM, Dube G, et al. Lipoprotein(a) and inflammation in human coronary atheroma: association with the severity of clinical presentation. J Am Coll Cardiol. (1998) 32:2035–42. doi: 10.1016/S0735-1097(98)00469-0
96. Vergallo R, Patrono C. The PACMAN-AMI trial: game over for the “vulnerable plaque”? Eur Heart J. (2022) 43:2179–80. doi: 10.1093/eurheartj/ehac222
97. Levstek T, Podkrajšek N, Rehberger Likozar A, Šebeštjen M, Trebušak Podkrajšek K. The influence of treatment with PCSK9 inhibitors and variants in the CRP (rs1800947), TNFA (rs1800629), and IL6 (rs1800795) genes on the corresponding inflammatory markers in patients with very high lipoprotein(a) levels. J Cardiovasc Dev Dis. (2022) 9:127. doi: 10.3390/jcdd9050127.35621838
98. Stiekema LCA, Stroes ESG, Verweij SL, Kassahun H, Chen L, Wasserman SM, et al. Persistent arterial wall inflammation in patients with elevated lipoprotein(a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur Heart J. (2019) 40:2775–81. doi: 10.1093/eurheartj/ehy862
99. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. (2010) 116:e74–80. doi: 10.1182/blood-2010-02-258558
100. Kapellos TS, Bonaguro L, Gemünd I, Reusch N, Saglam A, Hinkley ER, et al. Human monocyte subsets and phenotypes in Major chronic inflammatory diseases. Front Immunol. (2019) 10:2035. doi: 10.3389/fimmu.2019.02035
101. Krychtiuk KA, Lenz M, Hohensinner P, Distelmaier K, Schrutka L, Kastl SP, et al. Circulating levels of proprotein convertase subtilisin/kexin type 9 (PCSK9) are associated with monocyte subsets in patients with stable coronary artery disease. J Clin Lipidol. (2021) 15:512–21. doi: 10.1016/j.jacl.2021.02.005
102. Bernelot Moens SJ, Neele AE, Kroon J, van der Valk FM, Van den Bossche J, Hoeksema MA, et al. PCSK9 Monoclonal antibodies reverse the pro-inflammatory profile of monocytes in familial hypercholesterolaemia. Eur Heart J. (2017) 38:1584–93. doi: 10.1093/eurheartj/ehx002
103. Wolf D, Gerhardt T, Winkels H, Michel NA, Pramod AB, Ghosheh Y, et al. Pathogenic autoimmunity in atherosclerosis evolves from initially protective apolipoprotein B(100)-reactive CD4(+) T-regulatory cells. Circulation. (2020) 142:1279–93. doi: 10.1161/CIRCULATIONAHA.119.042863
104. Liu A, Frostegård J. PCSK9 Plays a novel immunological role in oxidized LDL-induced dendritic cell maturation and activation of T cells from human blood and atherosclerotic plaque. J Intern Med. (2018) 284:193–210. doi: 10.1111/joim.12758
105. Carvalho AC, Colman RW, Lees RS. Platelet function in hyperlipoproteinemia. N Engl J Med. (1974) 290:434–8. doi: 10.1056/NEJM197402212900805
106. Davì G, Averna M, Catalano I, Barbagallo C, Ganci A, Notarbartolo A, et al. Increased thromboxane biosynthesis in type IIa hypercholesterolemia. Circulation. (1992) 85:1792–8. doi: 10.1161/01.CIR.85.5.1792
107. Lacoste L, Lam JY, Hung J, Letchacovski G, Solymoss CB, Waters D. Hyperlipidemia and coronary disease. Correction of the increased thrombogenic potential with cholesterol reduction. Circulation. (1995) 92:3172–7. doi: 10.1161/01.CIR.92.11.3172
108. Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. (2007) 13:1086–95. doi: 10.1038/nm1626
109. Carnevale R, Bartimoccia S, Nocella C, Di Santo S, Loffredo L, Illuminati G, et al. LDL Oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis. (2014) 237:108–16. doi: 10.1016/j.atherosclerosis.2014.08.041
110. Magwenzi S, Woodward C, Wraith KS, Aburima A, Raslan Z, Jones H, et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood. (2015) 125:2693–703. doi: 10.1182/blood-2014-05-574491
111. Yang M, Cooley BC, Li W, Chen Y, Vasquez-Vivar J, Scoggins NO, et al. Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions. Blood. (2017) 129:2917–27. doi: 10.1182/blood-2016-11-750133
112. Cammisotto V, Baratta F, Castellani V, Bartimoccia S, Nocella C, D'Erasmo L, et al. Proprotein convertase subtilisin kexin type 9 inhibitors reduce platelet activation modulating ox-LDL pathways. Int J Mol Sci. (2021) 22:7193. doi: 10.3390/ijms22137193
113. Camera M, Rossetti L, Barbieri SS, Zanotti I, Canciani B, Trabattoni D, et al. PCSK9 As a positive modulator of platelet activation. J Am Coll Cardiol. (2018) 71:952–4. doi: 10.1016/j.jacc.2017.11.069
114. Napoli C. Oxidation of LDL, atherogenesis, and apoptosis. Ann N Y Acad Sci. (2003) 1010:698–709. doi: 10.1196/annals.1299.127
115. Lu J, Mitra S, Wang X, Khaidakov M, Mehta JL. Oxidative stress and lectin-like ox-LDL-receptor LOX-1 in atherogenesis and tumorigenesis. Antioxid Redox Signal. (2011) 15:2301–33. doi: 10.1089/ars.2010.3792
116. Li J, Liang X, Wang Y, Xu Z, Li G. Investigation of highly expressed PCSK9 in atherosclerotic plaques and ox-LDL-induced endothelial cell apoptosis. Mol Med Rep. (2017) 16:1817–25. doi: 10.3892/mmr.2017.6803
117. Choumar A, Tarhuni A, Lettéron P, Reyl-Desmars F, Dauhoo N, Damasse J, et al. Lipopolysaccharide-induced mitochondrial DNA depletion. Antioxid Redox Signal. (2011) 15:2837–54. doi: 10.1089/ars.2010.3713
118. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. (2012) 485:251–5. doi: 10.1038/nature10992
119. Kong N, Xu Q, Cui W, Feng X, Gao H. PCSK9 Inhibitor inclisiran for treating atherosclerosis via regulation of endothelial cell pyroptosis. Ann Transl Med. (2022) 10:1205. doi: 10.21037/atm-22-4652
120. Bjørklund MM, Bernal JA, Bentzon JF. Atherosclerosis induced by adeno-associated virus encoding gain-of-function PCSK9. Methods Mol Biol. (2022) 2419:461–73. doi: 10.1007/978-1-0716-1924-7_27
121. Al-Mashhadi RH, Sørensen CB, Kragh PM, Christoffersen C, Mortensen MB, Tolbod LP, et al. Familial hypercholesterolemia and atherosclerosis in cloned minipigs created by DNA transposition of a human PCSK9 gain-of-function mutant. Sci Transl Med. (2013) 5:166ra1. doi: 10.1126/scitranslmed.3004853
122. Arida A, Legaki AI, Kravvariti E, Protogerou A, Sfikakis PP, Chatzigeorgiou A. PCSK9/LDLR System and rheumatoid arthritis-related atherosclerosis. Front Cardiovasc Med. (2021) 8:738764. doi: 10.3389/fcvm.2021.738764
123. Coggi D, Frigerio B, Bonomi A, Ruscica M, Ferri N, Sansaro D, et al. Relationship between circulating PCSK9 and markers of subclinical atherosclerosis-the IMPROVE study. Biomedicines. (2021) 9:841. doi: 10.3390/biomedicines9070841
124. Wang H, He F, Liang B, Jing Y, Zhang P, Liu W, et al. LincRNA-p21 alleviates atherosclerosis progression through regulating the miR-221/SIRT1/Pcsk9 axis. J Cell Mol Med. (2021) 25:9141–53. doi: 10.1111/jcmm.16771
125. Ma CY, Shi XY, Wu YR, Zhang Y, Yao YH, Qu HL, et al. Berberine attenuates atherosclerotic lesions and hepatic steatosis in ApoE(-/-) mice by down-regulating PCSK9 via ERK1/2 pathway. Ann Transl Med. (2021) 9:1517. doi: 10.21037/atm-20-8106
126. Miranda MX, van Tits LJ, Lohmann C, Arsiwala T, Winnik S, Tailleux A, et al. The Sirt1 activator SRT3025 provides atheroprotection in apoe-/- mice by reducing hepatic Pcsk9 secretion and enhancing ldlr expression. Eur Heart J. (2015) 36:51–9. doi: 10.1093/eurheartj/ehu095
127. Huang YW, Zhang M, Wang LT, Nie Y, Yang JB, Meng WL, et al. 20(S)-Protopanaxadiol Decreases atherosclerosis in ApoE KO mice by increasing the levels of LDLR and inhibiting its binding with PCSK9. Food Funct. (2022) 13:7020–8. doi: 10.1039/D2FO00392A
128. Jin P, Gao D, Cong G, Yan R, Jia S. Role of PCSK9 in homocysteine-accelerated lipid accumulation in macrophages and atherosclerosis in ApoE(-/-) mice. Front Cardiovasc Med. (2021) 8:746989. doi: 10.3389/fcvm.2021.746989
129. Su Q, Nyi TS, Li L. Adenosine and verapamil for no-reflow during primary percutaneous coronary intervention in people with acute myocardial infarction. Cochrane Database Syst Rev. (2015) 2015:Cd009503. doi: 10.1002/14651858.CD009503.pub3
130. Hausenloy DJ, Chilian W, Crea F, Davidson SM, Ferdinandy P, Garcia-Dorado D, et al. The coronary circulation in acute myocardial ischaemia/reperfusion injury: a target for cardioprotection. Cardiovasc Res. (2019) 115:1143–55. doi: 10.1093/cvr/cvy286
131. Wolf A, Kutsche HS, Schreckenberg R, Weber M, Li L, Rohrbach S, et al. Autocrine effects of PCSK9 on cardiomyocytes. Basic Res Cardiol. (2020) 115:65. doi: 10.1007/s00395-020-00824-w
132. Wang J, Han P, Gao M, Xiao J, Li X, Zhang N, et al. Prognostic value of PCSK9 levels in patients with non-ST elevation myocardial infarction undergoing percutaneous coronary intervention (PCI). Hellenic J Cardiol. (2022) 63:22–31. doi: 10.1016/j.hjc.2021.05.002
133. Gao J, Yang YN, Cui Z, Feng SY, Ma J, Li CP, et al. Pcsk9 is associated with severity of coronary artery lesions in male patients with premature myocardial infarction. Lipids Health Dis. (2021) 20:56. doi: 10.1186/s12944-021-01478-w
134. Yang L, Pu T, Zhang Y, Yan H, Yu H, Gao W. The R93C variant of PCSK9 reduces the risk of premature MI in a Chinese han population. Front Genet. (2022) 13:875269. doi: 10.3389/fgene.2022.875269
135. Andreadou I, Tsoumani M, Vilahur G, Ikonomidis I, Badimon L, Varga ZV, et al. PCSK9 In myocardial infarction and cardioprotection: importance of lipid metabolism and inflammation. Front Physiol. (2020) 11:602497. doi: 10.3389/fphys.2020.602497
136. Zanchin C, Koskinas KC, Ueki Y, Losdat S, Häner JD, Bär S, et al. Effects of the PCSK9 antibody alirocumab on coronary atherosclerosis in patients with acute myocardial infarction: a serial, multivessel, intravascular ultrasound, near-infrared spectroscopy and optical coherence tomography imaging study-rationale and design of the PACMAN-AMI trial. Am Heart J. (2021) 238:33–44. doi: 10.1016/j.ahj.2021.04.006
137. Schwartz GG, Giugliano RP. Proprotein convertase subtilisin/kexin type 9 inhibition after acute coronary syndrome or prior myocardial infarction. Curr Opin Lipidol. (2022) 33:147–59. doi: 10.1097/MOL.0000000000000830
138. Gencer B, Mach F, Murphy SA, De Ferrari GM, Huber K, Lewis BS, et al. Efficacy of evolocumab on cardiovascular outcomes in patients with recent myocardial infarction: a prespecified secondary analysis from the FOURIER trial. JAMA Cardiol. (2020) 5:952–7. doi: 10.1001/jamacardio.2020.0882
139. Chiang CE, Schwartz GG, Elbez Y, Szarek M, Bhatt DL, Bittner VA, et al. Alirocumab and cardiovascular outcomes in patients with previous myocardial infarction: prespecified subanalysis from ODYSSEY OUTCOMES. Can J Cardiol. (2022) 38:1542–9. doi: 10.1016/j.cjca.2022.05.021
140. Silva-Bermúdez LS, Vargas-Villanueva A, Sánchez-Vallejo CA, Palacio AC, Buitrago AF, Mendivil CO. Peri-event plasma PCSK9 and hsCRP after an acute myocardial infarction correlate with early deterioration of left ventricular ejection fraction: a cohort study. Lipids Health Dis. (2022) 21:61. doi: 10.1186/s12944-022-01672-4
141. Huang G, Lu X, Zhou H, Li R, Huang Q, Xiong X, et al. PCSK9 Inhibition protects against myocardial ischemia-reperfusion injury via suppressing autophagy. Microvasc Res. (2022) 142:104371. doi: 10.1016/j.mvr.2022.104371
142. Liubomirski Y, Ben-Baruch A. Notch-Inflammation networks in regulation of breast cancer progression. Cells. (2020) 9:1576. doi: 10.3390/cells9071576
143. Motwani M, Pesiridis S, Fitzgerald KA. DNA Sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. (2019) 20:657–74. doi: 10.1038/s41576-019-0151-1
144. Bayes-Genis A, Núñez J, Zannad F, Ferreira JP, Anker SD, Cleland JG, et al. The PCSK9-LDL receptor axis and outcomes in heart failure: bIOSTAT-CHF subanalysis. J Am Coll Cardiol. (2017) 70:2128–36. doi: 10.1016/j.jacc.2017.08.057
145. White HD, Schwartz GG, Szarek M, Bhatt DL, Bittner VA, Chiang CE, et al. Alirocumab after acute coronary syndrome in patients with a history of heart failure. Eur Heart J. (2022) 43:1554–65. doi: 10.1093/eurheartj/ehab804
Keywords: PCSK9, atherosclerotic cardiovascular diseases, functional mechanism, immune regulation, low-density lipoprotein-cholesterol
Citation: Ma M, Hou C and Liu J (2023) Effect of PCSK9 on atherosclerotic cardiovascular diseases and its mechanisms: Focus on immune regulation. Front. Cardiovasc. Med. 10:1148486. doi: 10.3389/fcvm.2023.1148486
Received: 20 January 2023; Accepted: 24 February 2023;
Published: 10 March 2023.
Edited by:
Shaoyi Zheng, Southern Medical University, ChinaReviewed by:
Maria Borrell-Pages, Sant Pau Institute for Biomedical Research, Spain© 2023 Ma, Hou and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Liu ZHJqaWFubGl1QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Specialty Section: This article was submitted to General Cardiovascular Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.