
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 30 March 2023
Sec. Cardiovascular Genetics and Systems Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1148041
This article is part of the Research TopicTranscriptional Regulation in Cardiovascular DiseasesView all 5 articles
 Keyan Wang1
Keyan Wang1 Xiaoqian Sun1
Xiaoqian Sun1 Ying Sun1
Ying Sun1 Boyang Jiao1Junkai Yao1
Boyang Jiao1Junkai Yao1 Yueyao Hu1
Yueyao Hu1 Qiong Deng1Jianteng Dong1Wei Wang1,2,3*
Qiong Deng1Jianteng Dong1Wei Wang1,2,3* Yong Wang1,2,4*
Yong Wang1,2,4* Chun Li2,5*
Chun Li2,5*
Adverse cardiac remodeling after acute myocardial infarction is the most important pathological mechanism of heart failure and remains a major problem in clinical practice. Cardiac macrophages, derived from tissue resident macrophages and circulating monocyte, undergo significant phenotypic and functional changes following cardiac injury and play crucial roles in inflammatory response and tissue repair response. Currently, numerous studies indicate that epigenetic regulatory factors and transcription factors can regulate the transcription of inflammatory and reparative genes and timely conversion of inflammatory macrophages into reparative macrophages and then alleviate cardiac remodeling. Accordingly, targeting transcriptional regulation of macrophages may be a promising option for heart failure treatment. In this review, we not only summarize the origin and function of cardiac macrophages, but more importantly, describe the transcriptional regulation of macrophages in heart failure, aiming to provide a potential therapeutic target for heart failure.
Acute myocardial infarction (AMI) has always been a disease with high morbidity and mortality worldwide. Although reperfusion therapy is currently the main therapeutic strategy, the risk of progression to adverse cardiac remodeling and subsequently heart failure (HF) remains (1, 2). Therefore, it is necessary to explore novel therapeutic targets to effectively alleviate cardiac remodeling in order to protect the heart from failure.
Studies revealed convincing results to the effect that immunotherapy represented by macrophages has significant implications for the treatment of ischemic heart disease (3). Following AMI, dead cardiomyocytes release endogenous damage-associated molecular patterns (DAMPs), which interact with toll-like receptors to activate inflammatory signaling pathways, produce large amounts of proinflammatory factors and chemokines, recruit and activate immune cells including monocyte macrophages to infiltrate the infarct site, and exacerbate the inflammatory response. In the advanced stages of cardiac injury, macrophages undergo phenotypic changes and mediated tissue repair response (4, 5). Notably, an imbalance between inflammatory and reparative responses contributes to pathological cardiac remodeling (6–8).
A large number of studies have recently demonstrated that the expression of genes related to cardiac hypertrophy, fibrosis, and inflammation can be regulated by transcription, and inhibited harmful transcription and promoted beneficial transcription can effectively improve cardiac function after cardiac injury (9–11). Therefore, it is meaningful to explore the transcriptional regulatory mechanisms of macrophages in adverse cardiac remodeling after AMI.
In recent years, scientists have widely recognized that tissues establish a certain number of macrophages during embryonic development, called resident macrophages, which has revolutionized previous clinical beliefs of circulating monocytes entering the tissue and differentiating into macrophages (12–15). Therefore, the heart contains subpopulations of macrophages from different origins.
It has been reported that cardiac resident macrophages in mice account for approximately 7%–8% of non-cardiomyocytes in the stable state. Cardiac resident macrophages can be divided into C-C motif receptor-2 (CCR2)− and CCR2+ macrophage subset according to CCR2. CCR2− resident macrophages derived from embryonic yolk sac and fetal liver hematopoietic stem cells maintain their numbers by local proliferation. A subset of CCR2+ resident macrophages is supplemented by monocyte recruitment and proliferation (13, 16). In addition, based on the expression of major histocompatibility complex II (MHCII) and CCR2, there are three classifications of cardiac resident macrophages, namely, MHCIIlow CCR2−, MHCIIhigh CCR2−, and MHCIIhigh CCR2+ (17).
Monocytes play an important role in cardiac damage. Monocytes can be divided into classical monocyte lymphocyte antigen 6C (Ly6C)high and non-classical monocyte Ly6Clow in mice according to the Ly6C expression. Furthermore, mice monocytes have also been classified into classical monocytes Ly6Chigh CCR2high CX3C chemokine receptor 1 (CX3CR1)low and non-classical monocytes Ly6Clow CCR2highCX3CR1high (18). In vivo microscopy, Ly6Chigh monocytes have been observed to circulate rapidly and recruit to the site of injury, while Ly6Clow monocytes circulate more slowly and crawl along the endothelium to maintain homeostasis (19).
The number of macrophages at the infarct site increases significantly after AMI, and their infiltration into the infarct site occurs in two consecutive phases. In the early stage of AMI, Ly6Chigh monocytes are recruited to the infarct site under the action of chemokine CCL2 and differentiated into recruited CCR2+ macrophages, resulting in inflammatory response. Subsequently, Ly6Chigh macrophages differentiate into Ly6Clow macrophages with the assistance of the orphan nuclear receptor 4a (Nr4a) and coordinated tissue repair response (20, 21).
Resident macrophages play a significant role in cardiac electrical conduction. Hulsmans et al. proved that there are abundant resident macrophages in the atrioventricular (AV) node that are connected to cardiomyocytes via Cx43-containing gap junctions, and they promote cardiac electrical conduction (22). Another study reported that Amphiregulin (AREG) produced by cardiac macrophages leads to a phosphorylation of Cx43 via the EGFR/MEK/ERK pathway, mediating the normal formation of gap junctions, thus promoting cardiac electrical conduction (23).
Cardiomyocytes contain a large number of mitochondria to provide energy for the heart. However, the continuous supply of energy induces the accumulation of defective mitochondria. Timely removal of detrimental mitochondria is highly important to maintain cardiac homeostasis. Melentijevic et al. discovered a novel garbage removal mechanism. Caenorhabditis elegans neurons secrete a specialized membrane vesicle, called exophers, which can package and transport dysfunctional proteins and organelles (24). Research has found that cardiomyocytes release dysfunctional mitochondria via the exophers, and cardiac resident macrophages recognize and phagocytic these exophers with the assistance of the receptor Mertk, thus preventing inflammasome activation and autophagy block, eventually maintaining cardiac homeostasis (25).
Research revealed that the population of macrophages expanded significantly in response to cardiac injury and neonatal mice increased the number of CCR2− resident macrophages. However, in adult mice, there was a dramatic loss of CCR2− resident macrophages and a large number of infiltrated circulating monocyte-derived inflammatory CCR2+ macrophages. Compared with adult mice, neonatal mice induced a weaker inflammatory response, accompanied by angiogenesis and cardiomyocyte proliferation. In addition, the inhibition of cardiac monocyte recruitment in adult mice reduced inflammation response and improved cardiac function (26). Another research reported that cardiac resident macrophages repressed fibrosis and promoted angiogenesis under the condition of cardiac pressure overload (27). These results suggest that resident macrophages derived from embryonic development are key mediators in promoting cardiac repair.
Research has revealed that cardiac resident CCR2− macrophages mediate adaptive cardiac remodeling. To be specific, exhaustion of CCR2− macrophages mice impaired adaptive changes to maintain cardiac output after dilated cardiomyopathy surgery, such as depressed coronary angiogenesis and aggravated ventricular remodeling, suggesting that CCR2− resident macrophages are a key protective factor. Further studies have found that CCR2− macrophages interact with adjacent cardiomyocytes through the adhesion complex and sense mechanical stretch, contributing to the expression of the pro-angiogenic growth factor and promoting coronary angiogenesis depending on TRPV4. Therefore, CCR2− resident macrophages may be a key mediator in regulating adaptive cardiac remodeling (28).
Cardiac resident CCR2+ macrophages promote the recruitment of circulating monocytes through the pathway of myeloid differentiation primary response 88 (MyD88). Resident CCR2+ macrophages are inflammatory macrophages but that express lower levels of inflammatory cytokines and chemokines than recruited CCR2+ macrophages. In addition, resident CCR2+ macrophages express different type I IFN-stimulated genes in response to myocardial injury compared with recruited CCR2+ macrophages, suggesting that CCR2+ macrophages respond to type I interferon during myocardial infarction. Notably, the research also found that the inhibition of resident CCR2+ macrophage activity can limit inflammatory response and pathological cardiac remodeling, which may be a therapeutic target to improve outcomes following AMI (29). However, little is known about the function of the resident CCR2+ macrophage subset, and further studies are needed to elucidate the role of cardiac resident CCR2+ macrophages.
Recruited macrophages show functional heterogeneity during AMI. In the early stages after AMI, Ly6Chigh macrophages produce and release a large number of inflammatory cytokines and chemokines such as interleukin IL-1β (IL-1β), IL-6, IL-12, tumor necrosis factor-α (TNF-α), and CXC motif chemokine ligand 9 (CXCL9). All of these factors work together to promote inflammation response. In the advanced stages after AMI, Ly6Chigh macrophages differentiate into Ly6Clow macrophages, mainly secrete anti-inflammatory cytokines and chemokines such as IL-10 and C-C motif chemokine ligand 17 (CCL17), and also produce vascular endothelial growth factor (VEGF) and tumor growth factor β (TGFβ) (30, 31). These mediators jointly promote the production of the extracellular matrix, cell proliferation, and angiogenesis, thereby promoting tissue repair (Figure 1).
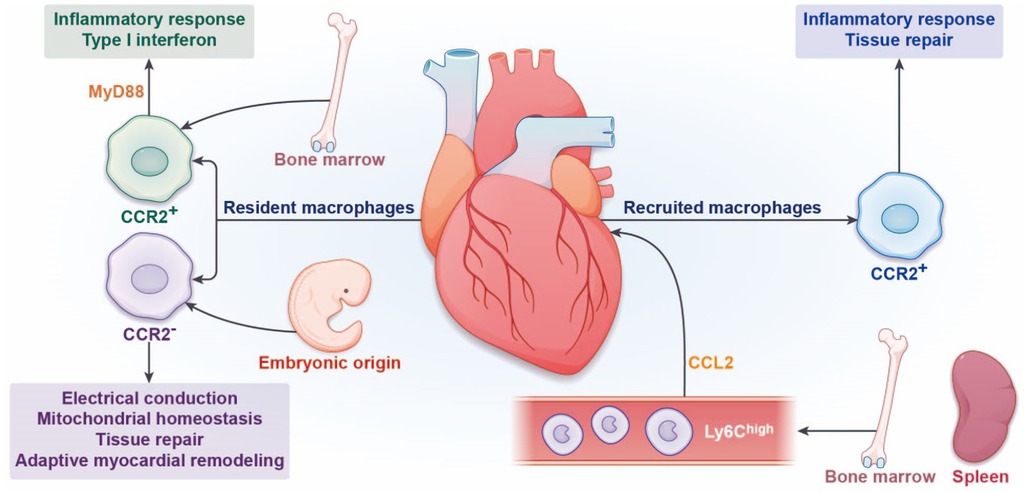
Figure 1. Origin and function of cardiac macrophages. In the steady state, the heart contains a certain number of resident macrophages. According to the expression of C-C motif receptor-2 (CCR2), cardiac resident macrophages can be divided into CCR2− and CCR2+ macrophages. CCR2− resident macrophages are derived from embryonic yolk sac and fetal liver hematopoietic stem cells, which maintain their numbers by local proliferation. CCR2+ resident macrophages promote the recruitment of circulating monocytes mainly through the pathway of MyD88. The functions of CCR2− resident macrophages are facilitated cardiac electrical conduction, mitochondrial homeostasis, tissue repair, and adaptive myocardial remodeling. The functions of resident CCR2+ macrophages are proinflammatory response and type I interferon. During myocardial infarction, Ly6Chigh monocytes derived from the bone marrow and spleen are recruited to the infarct site under the action of chemokine CCL2, differentiate into recruited CCR2+ macrophages, and mediate inflammatory response. Subsequently, repairing Ly6Clow macrophages mediate repair response.
It has also been described that recruited macrophages in the inflammatory phase are M1 macrophages, and macrophages in the repair phase are described as M2 macrophages. However, some researchers believe that the transcriptional heterogeneity of recruited macrophages does not easily conform to the M1/M2 paradigm, and the M1/M2 classification is more suitable for studies of bone marrow-derived macrophages in vitro (6).
Transcription refers to a process in which RNA polymerase (Pol II) and promoters or enhancers on DNA sequences are combined with the assistance of transcription factors (TFs), subsequently regulating gene expression (32). Research studies have demonstrated that the degree of chromatin opening is an essential factor in the regulation of transcription. Nucleosome, the basic structure of chromatin, is composed of the 147 bp segment of DNA and histone octamer formed by two molecules each of H2A, H2B, H3, and H4. Such a compacted nucleosome structure leads to regulatory elements such as transcription factors that cannot bind to DNA-induced repression of gene transcription (33, 34). Thus, the first step in gene transcription is to transform dense heterochromatin into open euchromatin. Histone modification and DNA methylation in epigenetics regulate the structure and function of chromatin and recruit transcriptional activators or inhibitors to regulate transcription.
At present, the most studied histone modifications are histone methylation and acetylation. Histone methylation means that histone methyltransferase adds one, two, or three methyl groups to histone lysine or arginine residues, and demethylase regulates the process of histone demethylation (35, 36). Histone methylation is related to transcriptional inhibition as well as transcriptional activation. For example, H3K4me2/3 (37), H3K36me1/3 (38), and H3K79me1/2 (39) are associated with transcriptional activation, and H3K27me2/3 (40) and H3K9me2/3 (41) are associated with transcriptional inhibition (42). Histone acetylation is regulated by histone acetyltransferase (HAT) and histone deacetylase (HDAC) mediates the process of deacetylation. It is well known that histone acetylation is associated with transcription activation, and histone deacetylation leads to transcriptional inhibition (43, 44). DNA methylation refers to binding a methyl group on the C5 position of CpG dinucleotide cytosine under the action of DNA methyltransferase (DNMT) and is modified into 5-methyl cytosine (5-mc). DNA demethylation is catalyzed by the enzyme Ten-Eleven translocation protein 1 (Tet1) to convert 5-mc into 5-hydroxymethylcytosine (5hmc). Most gene promoters are located on the CpG island and when the promoter at the CpG island is at a low DNA methylation level, resulting in gene transcription, Conversely, at high levels of DNA methylation, which inhibits promoter combined with TFs-induced gene expression silenced (45, 46) (Table 1).
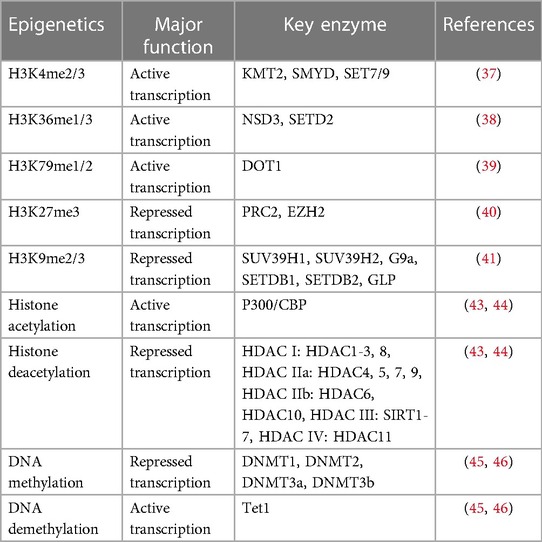
Table 1. Histone modification and DNA methylation in epigenetics associated with transcription.
Numerous studies have shown that epigenetic regulation affects the gene expression related to myocardial fibrosis, hypertrophy, and so on through transcription. G9a, H3K9 histone methyltransferase, combined with enhancer of zeste homolog 2 (EZH2) and transcription factor myocyte-specific enhancer factor 2C (MEF2C), inhibits the transcription of antihypertrophic genes, thereby promoting cardiac hypertrophy (47). Endothelial Kruppel-Like Factor 4 (KLF4) is an important protective regulatory factor against atherosclerosis. There is increased DNA methylation of the KLF4 promoter in the CpG island in atherosclerotic disease, inhibiting KLF4 transcription. In addition, DNMT inhibitors ameliorate atherosclerosis (48). Liu et al. showed that the forkhead box transcription factor P1 (Foxp1) regulated the transcription of transforming growth factor-β1 (TGF-β1)-mediated pathological cardiac remodeling (49). It has been reported that Runt-related transcription factor-1 (RUNX1), which is a key regulatory factor involved in pathologic cardiac remodeling, may be a potential target for the treatment of HF (50).
In summary, it is known that epigenetic and transcription factors regulate the expression of myocardial fibrosis and hypertrophy genes, thus participating in the process of pathological cardiac remodeling. Macrophages play an important role in cardiac remodeling. Therefore, targeting the transcriptional regulation of cardiac macrophages contributes to finding novel targets for HF treatment.
After AMI, there is a decrease in the abundance of CCR2− resident macrophages at the infarct site and subsequently a slow increase by self-proliferation. The deletion of CCR2− resident macrophages using the CX3CR1-based system significantly deteriorates cardiac function and exacerbates adverse cardiac remodeling (28). There are also studies that have found that CCR2− resident macrophages mediate the cardiac repair response by promoting cardiomyocyte proliferation and angiogenesis (27). In conclusion, CCR2− resident macrophages play a protective role in cardiac injury. CCR2+ resident macrophages mainly promote the recruitment of monocytes after cardiac injury, but its role in HF is poorly studied.
There are few studies on the transcriptional regulation of cardiac resident macrophages in HF. The crucial role of resident macrophages in cardiac injury may become a hotspot in future research. It has been reported that the reduction of Legumain (Lgmn) in CCR2− macrophages leads to significantly aggravated adverse cardiac remodeling post-AMI. Mechanistically, the decrease in Lgmn leads to a downregulation of the degradation and clearance of apoptotic cardiomyocytes. In addition, Lgmn deficiency also upregulates the infiltration of inflammatory macrophages, resulting in excessive inflammatory response. Conversely, Lgmn activation significantly improves cardiac function (17), suggesting that Lgmn in CCR2− macrophages is a cardioprotective factor. Several studies have revealed that transcription factors such as CCAAT-enhancer-binding protein (C/EBpβ), p53, and ELK1 bind to the promoter of Lgmn and regulate the transcription of Lgmn (51, 52). Accordingly, more in-depth studies are needed to explore the transcription regulation of Lgmn in cardiac resident macrophages, so as to identify novel targets for improving cardiac function. CCR2− resident macrophages mediate cardiac repair by promoting cardiomyocyte proliferation and angiogenesis. A study has shown that NLR family CARD domain containing 5 (NLRC5) and activator of transcription 3 (STAT3) promote the transcription of vascular enhancing angiopoietin-2 (Ang2) and cyclin D1 (CCND1), thereby promoting angiogenesis (53). Transcription factor GATA1 and histone methyltransferase SET7 promote the transcription of vascular endothelial growth factor–induced angiogenesis (54). In the future, targeting CCR2− resident macrophages to promote cardiomyocyte proliferation and angiogenesis through transcriptional regulation may become a research hotspot.
Persistent inflammatory response interferes with the timely occurrence of the repair response, resulting in pathological cardiac remodeling. Therefore, achieving a balance between the two phases may provide a breakthrough in the treatment of HF.
Lgr4 is a member of the leucine-rich repeat-containing G protein-coupled receptor (LGR) family that is involved in the AMP/protein kinase A (PKA)/cAMP response element binding protein (CREB) signal pathway (55, 56). Activator protein-1 (AP-1) is a proinflammatory transcription factor complex composed of Fos and Jun family members, regulating proinflammatory factors and chemokine expression to modulate macrophage polarization (57–59). Huang et al. found that macrophages highly expressed Lgr4 after AMI. Lgr4 promoted the transcription of Fos and Jun families through the cAMP/PKA/CREB pathway, synergistically increased the activity of AP-1, and promoted the downstream proinflammatory gene transcription, thus aggravating cardiac inflammatory response and poor cardiac remodeling. However, a low expression of Lgr4 plays a cardioprotective role through augmenting reparative macrophage subsets (60). Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) are key regulators of hippo signaling pathways, involved in cardiac regeneration and repair (61, 62). Mia et al. reported that YAP/TAZ bound to the promoter of proinflammatory factors IL-6 and facilitated IL-6 transcription, whereas YAP/TAZ inhibited reparative gene Arginase-1 (Arg1) transcription. This occurs because YAP/TAZ recruits histone deacetylase 3 (HDAC3) and nuclear receptor corepressor 1 (NCoR1) to form the transcriptional repressor complex and then binds to the Arg1 promoter. More importantly, the reduction of YAP/TAZ leads to a polarization of macrophages toward the reparative phenotype, which ameliorates cardiac fibrosis and hypertrophy and enhances angiogenesis after AMI (63). Histone lactylation, a newly discovered epigenetic regulation, is a process by which lactyl groups are added to histone lysine residues to modulate the chromatin structure and facilitate gene transcription (64). Wang et al. reported that an increase in histone H3K18 lactylation (H3K18la) levels in infiltrating macrophages post-AMI and the repair genes Lrg1, Vegf-a, and IL-10 are the downstream targets of H3K18la. Treatment with sodium lactate upregulates H3K18la levels, enhances Lrg1, Vegf-a, and IL-10 expression, inhibits inflammation, improves angiogenesis, and restrains cardiac fibrosis and pathological remodeling after AMI. This research also found that lactate transporter monocarboxylate transporter 1 (MCT1) provides abundant substrates for histone lactylation by transporting extracellular lactates into the cell. In order to further elucidate the upstream regulatory mechanism of H3K181a, it was found that histone acetyltransferase GCN5 could catalyze the process of H3K18la and promote repair gene Lrg1, Vegf-a, and IL-10 transcription, thereby improving cardiac function via anti-inflammatory response and promoting angiogenesis (65). The above findings suggest that H3K181a has cardioprotective effects by promoting the transcription of reparative genes. The timely resolution of inflammation and initiation of cardiac repair after AMI are crucial for heart repair. Cardioprotective transcription factors and epigenetic regulators can promote repair gene transcription, subsequently affecting macrophage phenotype and function. These transcription factors and epigenetic regulators may be potential therapeutic targets for treating HF.
Macrophages interact with adhesion molecules on the surface of endothelial cells through their own specific receptors (e.g., integrins), thereby gaining access to the vasculature and initiating the proinflammatory response (66, 67). Therefore, blocking the adhesion of macrophages to endothelial cells may attenuate the inflammatory response. Liu et al. found that MRTF-A in macrophages collaborates with transcription factor Sp1 and histone H3K9 demethylase KDM3A to promote integrin β2 (ITGB2) transcription, leading to an increase in the adhesion of macrophages to endothelial cells, eventually enhancing inflammatory response and cardiac hypertrophy following cardiac injury. However, MRTF-A deficiency in macrophages reverses the above transcription process, resulting in alleviating inflammatory response and cardiac hypertrophy (68), thereby indicating that suppression of the interaction between macrophages and endothelial cells to attenuate the inflammatory response is a novel therapeutic idea for HF (Figure 2). In addition to the above transcription processes, several widely studied transcription factors have been shown to participate in the process of macrophage phenotype transition, such as nuclear factor-κB (NF-κB), signal transducer and activator of transcription 6 (STAT6), and so on. In the case of pathological stress, the IKK complex is activated, further phosphorylating the IκB protein, ultimately leading to NF-κB entering the nucleus and binding to specific DNA sequence-induced proinflammatory macrophage activation (69, 70). IL-4-activated STAT6 acts as a transcriptional repressor in macrophages, limiting IL-1β transcription and inflammasome activation, resulting in a weaker inflammatory response (71). Therefore, the research and development of novel drugs targeting these transcription factors may be promising for the treatment of HF.
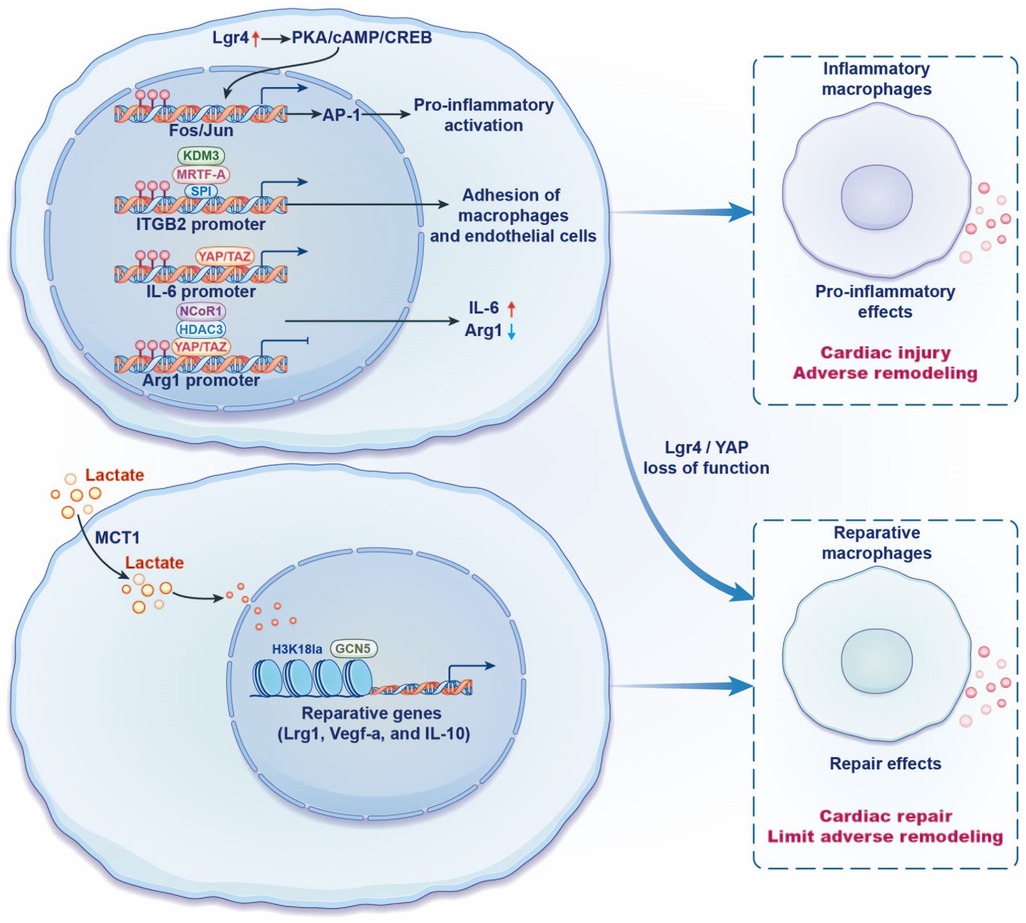
Figure 2. Regulation of recruited macrophage phenotypes and function in heart failure by transcription. Lgr4 promotes the transcription of Fos and Jun families through the cAMP/PKA/CREB pathway, synergistically increases the activity of AP-1, and promotes the downstream proinflammatory gene expression, thus aggravating cardiac inflammatory response and poor cardiac remodeling. YAP/TAZ binds to the proinflammatory cytokine IL-6 promoter, whereas YAP/TAZ inhibits Arg1 expression by binding to the Arg1 promoter and recruiting the histone deacetylase 3 (HDAC3)-nuclear receptor corepressor 1 (NCoR1) inhibition complex. Lgr4/YAP loss of function has cardioprotective effects. MRTF-A in macrophages collaborates with transcription factor Sp1 and histone H3K9 demethylase KDM3A to promote integrin β2 (ITGB2) transcription, leading to an increase in the adhesion of macrophages to endothelial cells, eventually resulting in inflammatory response. Lactate transporter monocarboxylate transporter 1 (MCT1) provides abundant substrates for histone H3K18 lactylation (H3K18la) and promotes the repair genes Lrg1, Vegf-a, and IL-10 transcriptional. It is further found that histone acetyltransferase GCN5 is able to catalyze the process of histone lactylation, thereby improving cardiac function via an anti-inflammatory response.
Macrophage metabolism is another important pathway that regulates macrophage phenotype and function through transcriptional pathways. Metabolic reprogramming was first proposed by Otto Warburg, who discovered that tumor cells rapidly produce ATP through the glycolytic pathway even under aerobic conditions, accompanied by a decrease in oxidative phosphorylation (72). This similar process was also found in macrophages. When macrophages are stimulated by lipopolysaccharide (LPS), metabolic pathways are characterized by enhanced glycolysis and a broken tricarboxylic acid (TCA) cycle, and macrophages present a proinflammatory phenotype. When macrophages are stimulated by IL-4, metabolic pathways are characterized by enhanced fatty acid oxidation and oxidative phosphorylation, and macrophages exhibit an anti-inflammatory phenotype (73). Research has demonstrated that α-ketoglutarate (αKG) generated from glutamine is an anti-inflammatory mediator. Specifically, αKG promotes the expression of repair genes Arg1, Yml, Retnla, and Mrcl through JMJD3-dependent H3K27 demethylation epigenetic pathways, resulting in promoting repair macrophage activation. Second, glutamine metabolism regulates IKKβ activity through αKG-PHD, in turn, suppressing the NF-κB pathway, thereby inhibiting inflammatory macrophage activation. In summary, αKG is an important metabolite that regulates macrophage polarization through metabolic and epigenetic mechanisms (74). BMDM stimulated by LPS leads to the accumulation of succinate, an intermediate metabolite in the TCA cycle, which stabilizes hypoxia-inducible factor-1α (HIF-1α) and subsequently promotes the transcription of IL-1β, inducing inflammatory response (75). DeBerge et al. found that HIF-1α mediates poor cardiac remodeling after AMI. In terms of mechanism, HIF-1α promotes myeloid-epithelial-reproductive receptor tyrosine kinase (MerTK) cleavage through macrophage glycolytic reprogramming. MerTK is a cardioprotective receptor that mediates the clearance of apoptotic cardiomyocytes to promote macrophage reparative phenotype activation. Conversely, a knockdown of HIF-1α in bone marrow cells antagonizes the deterioration of cardiac function after AMI (76). The compound dimethyl fumarate (DMF) significantly improves cardiac function post-AMI, which leads to a decrease in the expression of HIF-1α and IL-1β, but is also accompanied by an increase in macrophage oxidative phosphorylation levels (77). The above studies suggest that macrophage metabolic reprogramming is one of the mechanisms regulating macrophage phenotype and function. More importantly, the transcription factors HIF-1α and NF-κB and the epigenetic regulators H3K27 are involved in the process of metabolic reprogramming.
Traditional Chinese medicine (TCM) has the advantages of multiple targets, few side effects, and remarkable therapeutic effects. More importantly, it has been used in clinical practice for thousands of years. The molecular nature of TCM has not yet been fully understood, but it is usually a mixture of multiple compounds that work together to exert therapeutic effects. The sources of TCM are mainly plants, animals, and minerals, which undergo specific processing and preparation processes to enhance therapeutic properties and reduce toxicity. They are usually given to patients in the form of decoction, powder, capsule, or tablet. Although clinical studies have proven the effectiveness of TCM in treating diseases, the specific pathways and modes of action of these drugs are still being studied.
Several studies have found that the treatment of HF with TCM is done through transcriptional regulation. Nuanxinkang (NXK) is composed of Red Ginseng (dry root and rhizome of Panax ginseng C.A. Mey) and Holly (root of Ilex pubescens Hook. et Arn). Dong et al. found that NXK improves cardiac function by inhibiting the NF-κB signaling pathway to suppress the proinflammatory macrophage phenotype and promote anti-inflammatory macrophage phenotype (78). Ankyrin repeat domain 1 (ANKRD1), a nuclear transcription co-factor, induces the transcription of hypertrophic genes and is involved in pathological cardiac remodeling (79). Baoyuan decoction (BYD) consists of Astragalus membranaceus (Fisch.) Bunge, Panax ginseng C. A. Mey., Glycyrrhiza uralensis Fisch., and Cinnamomum cassia Presl. Baoyuan decoction (BYD) inhibits the transcription of myocardial fibrosis and hypertrophy genes through ANKRD1 (80). Curcumin, a polyphenol, originates from the plant curcuma longa, which ameliorates heart hypertrophy and inhibits the development of heart failure by inhibiting p300-HAT activity (81). Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key transcription factor in antioxidant stress response. Tanshinone I (Tan I) is an active ingredient extracted from the Chinese herb Danshen, and it has been shown to upregulate the phosphorylation of AKT and promote the Nrf2 shuttle into the nucleus, activate the expression of its downstream antioxidant enzymes, and then relieve oxidative stress and mitochondrial damage to improve cardiac function (82). On the one hand, the above studies indicate that TCM has a remarkable and beneficial effect in AMI treatment, and on the other hand, transcription factors and epigenetic regulatory factors can be used as key targets to treat diseases. In addition to TCM treatment, biologics and nanoparticles that deliver therapeutic substances and gene editing to macrophages are rapidly evolving, which will contribute to the treatment of heart failure more effectively in the future (3).
Although TCM is not a traditional treatment for cardiovascular disease in Western medicine, a growing number of studies suggest that TCM has remarkable therapeutic effects in lowering blood pressure, improving cholesterol levels, and reducing inflammation (83–85). Therefore, more research is needed to fully understand the mechanisms and potential role of TCM in the treatment of cardiovascular disease, which will contribute to the widespread use of TCM.
As mentioned above, the targeted regulation of macrophage phenotype by repressing inflammatory gene transcription or promoting repair gene transcription will lead to an amelioration of adverse cardiac remodeling. Accordingly, research and development novel drugs targeting cardioprotective transcription factors and epigenetic regulators factors may hold promise for HF treatment. At present, there are few studies on cardiac resident macrophages. In particular, CCR2− resident macrophages play protective roles in promoting cardiomyocyte proliferation, angiogenesis, cardiac conduction, and mitochondrial homeostasis. Therefore, efforts should be devoted to the transcription regulation of CCR2− resident macrophages in the future.
KW was involved in writing the article, tabulating, and drawing the figures. XQS, YS, BYJ, JKY, YYH, QD, and JTD revised the article and proofread the manuscript. WW, YW, and CL reviewed the article. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (grant no. 82174215 and 82174364), the Fundamental Research Funds for the Central Universities (Distinguished project), and the National Science Fund for Excellent Young Scholars of China (grant no. 82222075).
The authors thank WW, YW, and CL for reviewing the article and XQS, YS, BYJ, JKY, YYH, QD, and JTD for correcting the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Del Buono MG, Moroni F, Montone RA, Azzalini L, Sanna T, Abbate A. Ischemic cardiomyopathy and heart failure after acute myocardial infarction. Curr Cardiol Rep. (2022) 24(10):1505–15. doi: 10.1007/s11886-022-01766-6
2. Ong SB, Hernández-Reséndiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, et al. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. (2018) 186:73–87. doi: 10.1016/j.pharmthera.2018.01.001
3. Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. (2018) 18(12):733–44. doi: 10.1038/s41577-018-0065-8
4. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. (2012) 110(1):159–73. doi: 10.1161/CIRCRESAHA.111.243162
5. Sun K, Li YY, Jin J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal Transduct Target Ther. (2021) 6(1):79. doi: 10.1038/s41392-020-00455-6
6. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res. (2020) 116(6):1101–12. doi: 10.1093/cvr/cvz336
7. Kologrivova I, Shtatolkina M, Suslova T, Ryabov V. Cells of the immune system in cardiac remodeling: main players in resolution of inflammation and repair after myocardial infarction. Front Immunol. (2021) 12:664457. doi: 10.3389/fimmu.2021.664457
8. Halade GV, Lee DH. Inflammation and resolution signaling in cardiac repair and heart failure. EBioMedicine. (2022) 79:103992. doi: 10.1016/j.ebiom.2022.103992
9. Hong JH, Zhang HG. Transcription factors involved in the development and prognosis of cardiac remodeling. Front Pharmacol. (2022) 13:828549. doi: 10.3389/fphar.2022.828549
10. Nomura S, Satoh M, Fujita T, Higo T, Sumida T, Ko T, et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat Commun. (2018) 9(1):4435. doi: 10.1038/s41467-018-06639-7
11. Xu M, Xue RQ, Lu Y, Yong SY, Wu Q, Cui YL, et al. Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and UPRmt through SIRT3-AMPK pathway. Cardiovasc Res. (2019) 115(3):530–45. doi: 10.1093/cvr/cvy217
12. Zaman R, Epelman S. Resident cardiac macrophages: heterogeneity and function in health and disease. Immunity. (2022) 55(9):1549–63. doi: 10.1016/j.immuni.2022.08.009
13. Moskalik A, Niderla-Bielińska J, Ratajska A. Multiple roles of cardiac macrophages in heart homeostasis and failure. Heart Fail Rev. (2022) 27(4):1413–30. doi: 10.1007/s10741-021-10156-z
14. Gentek R, Molawi K, Sieweke MH. Tissue macrophage identity and self-renewal. Immunol Rev. (2014) 262(1):56–73. doi: 10.1111/imr.12224
15. Wang Z, Lu YL, Zhao WT, Zhong J, Lin X, Sun Z, et al. Distinct origins and functions of cardiac orthotopic macrophages. Basic Res Cardiol. (2020) 115(2):8. doi: 10.1007/s00395-019-0769-3
16. Lafuse WP, Wozniak DJ, Rajaram MVS. Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells. (2020) 10(1):51. doi: 10.3390/cells10010051
17. Jia D, Chen S, Bai P, Luo C, Liu J, Sun A, et al. Cardiac resident macrophage-derived legumain improves cardiac repair by promoting clearance and degradation of apoptotic cardiomyocytes after myocardial infarction. Circulation. (2022) 145(20):1542–56. doi: 10.1161/CIRCULATIONAHA.121.057549
18. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. (2003) 19(1):71–82. doi: 10.1016/S1074-7613(03)00174-2
19. Jung K, Kim P, Leuschner F, Gorbatov R, Kim JK, Ueno T, et al. Endoscopic time-lapse imaging of immune cells in infarcted mouse hearts. Circ Res. (2013) 112(6):891–9. doi: 10.1161/CIRCRESAHA.111.300484
20. Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res. (2013) 113(8):1004–12. doi: 10.1161/CIRCRESAHA.113.301198
21. Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. (2014) 114(10):1611–22. doi: 10.1161/CIRCRESAHA.114.303204
22. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell. (2017) 169(3):510–22.e20. doi: 10.1016/j.cell.2017.03.050
23. Sugita J, Fujiu K, Nakayama Y, Matsubara T, Matsuda J, Oshima T, et al. Cardiac macrophages prevent sudden death during heart stress. Nat Commun. (2021) 12(1):1910. doi: 10.1038/s41467-021-22178-0
24. Melentijevic I, Toth ML, Arnold ML, Guasp RJ, Harinath G, Nguyen KC, et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature. (2017) 542(7641):367–71. doi: 10.1038/nature21362
25. Nicolás-Ávila JA, Lechuga-Vieco AV, Esteban-Martínez L, Sánchez-Díaz M, Díaz-García E, Santiago DJ, et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell. (2020) 183(1):94–109.e23. doi: 10.1016/j.cell.2020.08.031
26. Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A. (2014) 111(45):16029–34. doi: 10.1073/pnas.1406508111
27. Revelo XS, Parthiban P, Chen C, Barrow F, Fredrickson G, Wang H, et al. Cardiac resident macrophages prevent fibrosis and stimulate angiogenesis. Circ Res. (2021) 129(12):1086–101. doi: 10.1161/CIRCRESAHA.121.319737
28. Wong NR, Mohan J, Kopecky BJ, Guo S, Du L, Leid J, et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity. (2021) 54(9):2072–88.e7. doi: 10.1016/j.immuni.2021.07.003
29. Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, et al. Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res. (2019) 124(2):263–78. doi: 10.1161/CIRCRESAHA.118.314028
30. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
31. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. (2014) 5:514. doi: 10.3389/fimmu.2014.00514
32. Bennett H, Troutman TD, Sakai M, Glass CK. Epigenetic regulation of Kupffer cell function in health and disease. Front Immunol. (2020) 11:609618. doi: 10.3389/fimmu.2020.609618
33. Kujirai T, Kurumizaka H. Transcription through the nucleosome. Curr Opin Struct Biol. (2020) 61:42–9. doi: 10.1016/j.sbi.2019.10.007
34. Onufriev AV, Schiessel H. The nucleosome: from structure to function through physics. Curr Opin Struct Biol. (2019) 56:119–30. doi: 10.1016/j.sbi.2018.11.003
35. Jambhekar A, Dhall A, Shi Y. Roles and regulation of histone methylation in animal development. Nat Rev Mol Cell Biol. (2019) 20(10):625–41. doi: 10.1038/s41580-019-0151-1
36. Yang C, Zhang J, Ma Y, Wu C, Cui W, Wang L. Histone methyltransferase and drug resistance in cancers. J Exp Clin Cancer Res. (2020) 39(1):173. doi: 10.1186/s13046-020-01682-z
37. Yang L, Jin M, Jeong KW. Histone H3K4 methyltransferases as targets for drug-resistant cancers. Biology. (2021) 10(7):581. doi: 10.3390/biology10070581
38. Lam UTF, Tan BKY, Poh JJX, Chen ES. Structural and functional specificity of H3K36 methylation. Epigenetics Chromatin. (2022) 15(1):17. doi: 10.1186/s13072-022-00446-7
39. Wood K, Tellier M, Murphy S. DOT1L and H3K79 methylation in transcription and genomic stability. Biomolecules. (2018) 8(1):11. doi: 10.3390/biom8010011
40. Guo Y, Zhao S, Wang GG. Polycomb gene silencing mechanisms: PRC2 chromatin targeting, H3K27me3 “readout”, and phase separation-based compaction. Trends Genet. (2021) 37(6):547–65. doi: 10.1016/j.tig.2020.12.006
41. Maeda R, Tachibana M. HP1 maintains protein stability of H3K9 methyltransferases and demethylases. EMBO Rep. (2022) 23(4):e53581. doi: 10.15252/embr.202153581
42. Mandumpala JJ, Baby S, Tom AA, Godugu C, Shankaraiah N. Role of histone demethylases and histone methyltransferases in triple-negative breast cancer: epigenetic mnemonics. Life Sci. (2022) 292:120321. doi: 10.1016/j.lfs.2022.120321
43. Tang J, Zhuang S. Histone acetylation and DNA methylation in ischemia/reperfusion injury. Clin Sci. (2019) 133(4):597–609. doi: 10.1042/CS20180465
44. Shen Y, Wei W, Zhou DX. Histone acetylation enzymes coordinate metabolism and gene expression. Trends Plant Sci. (2015) 20(10):614–21. doi: 10.1016/j.tplants.2015.07.005
45. Meng H, Cao Y, Qin J, Song X, Zhang Q, Shi Y, et al. DNA Methylation, its mediators and genome integrity. Int J Biol Sci. (2015) 11(5):604–17. doi: 10.7150/ijbs.11218
46. Chen Z, Zhang Y. Role of mammalian DNA methyltransferases in development. Annu Rev Biochem. (2020) 89:135–58. doi: 10.1146/annurev-biochem-103019-102815
47. Papait R, Serio S, Pagiatakis C, Rusconi F, Carullo P, Mazzola M, et al. Histone methyltransferase G9a is required for cardiomyocyte homeostasis and hypertrophy. Circulation. (2017) 136(13):1233–46. doi: 10.1161/CIRCULATIONAHA.117.028561
48. Jiang YZ, Jiménez JM, Ou K, McCormick ME, Zhang LD, Davies PF. Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-like factor 4 promoter in vitro and in vivo. Circ Res. (2014) 115(1):32–43. doi: 10.1161/CIRCRESAHA.115.303883
49. Liu J, Zhuang T, Pi J, Chen X, Zhang Q, Li Y, et al. Endothelial forkhead box transcription factor P1 regulates pathological cardiac remodeling through transforming growth factor-β1-endothelin-1 signal pathway. Circulation. (2019) 140(8):665–80. doi: 10.1161/CIRCULATIONAHA.119.039767
50. Riddell A, McBride M, Braun T, Nicklin SA, Cameron E, Loughrey CM, et al. RUNX1: an emerging therapeutic target for cardiovascular disease. Cardiovasc Res. (2020) 116(8):1410–23. doi: 10.1093/cvr/cvaa034
51. Yamane T, Murao S, Kato-Ose I, Kashima L, Yuguchi M, Kozuka M, et al. Transcriptional regulation of the legumain gene by p53 in HCT116 cells. Biochem Biophys Res Commun. (2013) 438(4):613–8. doi: 10.1016/j.bbrc.2013.08.007
52. Yan Q, Ni C, Lin Y, Sun X, Shen Z, Zhang M, et al. ELK1 enhances pancreatic cancer progression via LGMN and correlates with poor prognosis. Front Mol Biosci. (2021) 8:764900. doi: 10.3389/fmolb.2021.764900
53. Xu X, Shi Y, Luan P, Kou W, Li B, Zhai M, et al. The subcellular redistribution of NLRC5 promotes angiogenesis via interacting with STAT3 in endothelial cells. Theranostics. (2021) 11(9):4483–501. doi: 10.7150/thno.54473
54. Zhang Y, Liu J, Lin J, Zhou L, Song Y, Wei B, et al. The transcription factor GATA1 and the histone methyltransferase SET7 interact to promote VEGF-mediated angiogenesis and tumor growth and predict clinical outcome of breast cancer. Oncotarget. (2016) 7(9):9859–75. doi: 10.18632/oncotarget.7126
55. Wang F, Hong J, Li X, Chen M, Li X, Chen M, et al. Ablation of LGR4 promotes energy expenditure by driving white-to-brown fat switch. Nat Cell Biol. (2013) 15(12):1455–63. doi: 10.1038/ncb2867
56. Wang J, Li X, Ke Y, Lu Y, Wang F, Fan N, et al. GPR48 increases mineralocorticoid receptor gene expression. J Am Soc Nephrol. (2012) 23(2):281–93. doi: 10.1681/ASN.2011040351
57. Ye N, Ding Y, Wild C, Shen Q, Zhou J. Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem. (2014) 57(16):6930–48. doi: 10.1021/jm5004733
58. Tugal D, Liao X, Jain MK. Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol. (2013) 33(6):1135–44. doi: 10.1161/ATVBAHA.113.301453
59. Srivastava M, Saqib U, Naim A, Roy A, Liu D, Bhatnagar D, et al. The TLR4-NOS1-AP1 signaling axis regulates macrophage polarization. Inflamm Res. (2017) 66(4):323–34. doi: 10.1007/s00011-016-1017-z
60. Huang CK, Dai D, Xie H, Zhu Z, Hu J, Su M, et al. Lgr4 governs a pro-inflammatory program in macrophages to antagonize post-infarction cardiac repair. Circ Res. (2020) 127(8):953–73. doi: 10.1161/CIRCRESAHA.119.315807
61. Liu B, Zheng Y, Yin F, Yu J, Silverman N, Pan D. Toll receptor-mediated hippo signaling controls innate immunity in Drosophila. Cell. (2016) 164(3):406–19. doi: 10.1016/j.cell.2015.12.029
62. Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. (2016) 6(1):80–95. doi: 10.1158/2159-8290.CD-15-0224
63. Mia MM, Cibi DM, Abdul Ghani SAB, Song W, Tee N, Ghosh S, et al. YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction. PLoS Biol. (2020) 18(12):e3000941. doi: 10.1371/journal.pbio.3000941
64. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
65. Wang N, Wang W, Wang X, Mang G, Chen J, Yan X, et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ Res. (2022) 131(11):893–908. doi: 10.1161/CIRCRESAHA.122.320488
66. Barreiro O, Martín P, González-Amaro R, Sánchez-Madrid F. Molecular cues guiding inflammatory responses. Cardiovasc Res. (2010) 86(2):174–82. doi: 10.1093/cvr/cvq001
67. He P. Leucocyte/endothelium interactions and microvessel permeability: coupled or uncoupled? Cardiovasc Res. (2010) 87(2):281–90. doi: 10.1093/cvr/cvq140
68. Liu L, Zhao Q, Kong M, Mao L, Yang Y, Xu Y. Myocardin-related transcription factor a regulates integrin beta 2 transcription to promote macrophage infiltration and cardiac hypertrophy in mice. Cardiovasc Res. (2022) 118(3):844–58. doi: 10.1093/cvr/cvab110
69. Fiordelisi A, Iaccarino G, Morisco C, Coscioni E, Sorriento D. NFkappab is a key player in the crosstalk between inflammation and cardiovascular diseases. Int J Mol Sci. (2019) 20(7):1599. doi: 10.3390/ijms20071599
70. Imam F, Al-Harbi NO, Al-Harbi MM, Ansari MA, Al-Asmari AF, Ansari MN, et al. Apremilast prevent doxorubicin-induced apoptosis and inflammation in heart through inhibition of oxidative stress mediated activation of NF-κB signaling pathways. Pharmacol Rep. (2018) 70(5):993–1000. doi: 10.1016/j.pharep.2018.03.009
71. Czimmerer Z, Daniel B, Horvath A, Rückerl D, Nagy G, Kiss M, et al. The transcription factor STAT6 mediates direct repression of inflammatory enhancers and limits activation of alternatively polarized macrophages. Immunity. (2018) 48(1):75–90.e6. doi: 10.1016/j.immuni.2017.12.010
72. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. (1927) 8(6):519–30. doi: 10.1085/jgp.8.6.519
73. Van den Bossche J, O'Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. (2017) 38(6):395–406. doi: 10.1016/j.it.2017.03.001
74. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. (2017) 18(9):985–94. doi: 10.1038/ni.3796
75. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496(7444):238–42. doi: 10.1038/nature11986
76. DeBerge M, Lantz C, Dehn S, Sullivan DP, van der Laan AM, Niessen HWM, et al. Hypoxia-inducible factors individually facilitate inflammatory myeloid metabolism and inefficient cardiac repair. J Exp Med. (2021) 218(9):e20200667. doi: 10.1084/jem.20200667
77. Mouton AJ, Flynn ER, Moak SP, Aitken NM, Omoto ACM, Li X, et al. Dimethyl fumarate preserves left ventricular infarct integrity following myocardial infarction via modulation of cardiac macrophage and fibroblast oxidative metabolism. J Mol Cell Cardiol. (2021) 158:38–48. doi: 10.1016/j.yjmcc.2021.05.008
78. Dong X, Jiang J, Lin Z, Wen R, Zou L, Luo T, et al. Nuanxinkang protects against ischemia/reperfusion-induced heart failure through regulating IKKβ/IκBα/NF-κB-mediated macrophage polarization. Phytomedicine. (2022) 101:154093. doi: 10.1016/j.phymed.2022.154093
79. Murphy NP, Lubbers ER, Mohler PJ. Advancing our understanding of AnkRD1 in cardiac development and disease. Cardiovasc Res. (2020) 116(8):1402–4. doi: 10.1093/cvr/cvaa063
80. Meng H, Du Z, Lu W, Wang Q, Sun X, Jiang Y, et al. Baoyuan decoction (BYD) attenuates cardiac hypertrophy through ANKRD1-ERK/GATA4 pathway in heart failure after acute myocardial infarction. Phytomedicine. (2021) 89:153617. doi: 10.1016/j.phymed.2021.153617
81. Sunagawa Y, Funamoto M, Sono S, Shimizu K, Shimizu S, Genpei M, et al. Curcumin and its demethoxy derivatives possess p300 HAT inhibitory activity and suppress hypertrophic responses in cardiomyocytes. J Pharmacol Sci. (2018) 136(4):212–7. doi: 10.1016/j.jphs.2017.12.013
82. Jiang Q, Chen X, Tian X, Zhang J, Xue S, Jiang Y, et al. Tanshinone I inhibits doxorubicin-induced cardiotoxicity by regulating Nrf2 signaling pathway. Phytomedicine. (2022) 106:154439. doi: 10.1016/j.phymed.2022.154439
83. Lai X, Dong Z, Wu S, Zhou X, Zhang G, Xiong S, et al. Efficacy and safety of Chinese herbal medicine compared with losartan for mild essential hypertension: a randomized, multicenter, double-blind, noninferiority trial. Circ Cardiovasc Qual Outcomes. (2022) 15(3):e007923. doi: 10.1161/CIRCOUTCOMES
84. Tong X, Xu J, Lian F, Yu X, Zhao Y, Xu L, et al. Structural alteration of gut microbiota during the amelioration of human type 2 diabetes with hyperlipidemia by metformin and a traditional Chinese herbal formula: a multicenter, randomized, open label clinical trial. mBio. (2018) 9(3):e02392-17. doi: 10.1128/mBio.02392-17
Keywords: macrophages, DNA methylation, histone modification, transcription factors, myocardial infarction, adverse cardiac remodeling
Citation: Wang K, Sun X, Sun Y, Jiao B, Yao J, Hu Y, Deng Q, Dong J, Wang W, Wang Y and Li C (2023) Transcriptional regulation of macrophages in heart failure. Front. Cardiovasc. Med. 10:1148041. doi: 10.3389/fcvm.2023.1148041
Received: 19 January 2023; Accepted: 13 March 2023;
Published: 30 March 2023.
Edited by:
Chao Song, Harbin Medical University, ChinaReviewed by:
Zhi Yong Du, Capital Medical University, China© 2023 Wang, Sun, Sun, Jiao, Yao, Hu, Deng, Dong, Wang, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Wang d2FuZ3dlaTI2OTYwQDEyNi5jb20= Yong Wang d2FuZ3lvbmdAYnVjbS5lZHUuY24= Chun Li bGljaHVuMTk4NTAyMDRAMTYzLmNvbQ==
Specialty Section: This article was submitted to Cardiovascular Genetics and Systems Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.