
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 10 March 2023
Sec. Cardiovascular Genetics and Systems Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1135723
This article is part of the Research Topic Cytokines, Novel Cell Death Models and Pathways in Cardiovascular Diseases View all 11 articles
 Dan Ke1,†
Dan Ke1,† Zhen Zhang2,3,†
Zhen Zhang2,3,† Jieting Liu1,2,†
Jieting Liu1,2,† Peijian Chen1,2
Peijian Chen1,2 Jialing Li1
Jialing Li1 Xinhai Sun4
Xinhai Sun4 Yanhui Chu1,2*
Yanhui Chu1,2* Luxin Li1,2*
Luxin Li1,2*
Diabetes is a common chronic metabolic disease, and its incidence continues to increase year after year. Diabetic patients mainly die from various complications, with the most common being diabetic cardiomyopathy. However, the detection rate of diabetic cardiomyopathy is low in clinical practice, and targeted treatment is lacking. Recently, a large number of studies have confirmed that myocardial cell death in diabetic cardiomyopathy involves pyroptosis, apoptosis, necrosis, ferroptosis, necroptosis, cuproptosis, cellular burial, and other processes. Most importantly, numerous animal studies have shown that the onset and progression of diabetic cardiomyopathy can be mitigated by inhibiting these regulatory cell death processes, such as by utilizing inhibitors, chelators, or genetic manipulation. Therefore, we review the role of ferroptosis, necroptosis, and cuproptosis, three novel forms of cell death in diabetic cardiomyopathy, searching for possible targets, and analyzing the corresponding therapeutic approaches to these targets.
In 2021, worldwide, the number of adults with diabetes reached 537 million (10.5%), with more than one-in-ten adults affected by the disease. The number of people in 2021 is 7.4 million more than that in 2019, an increase of 16%, which highlights the prevalence of diabetes worldwide. The international diabetes federation (IDF) estimates that this number will reach 783 million by 2045, a 46% increase that is more than double the projected population growth of 20% over the same period, with one-in-eight adults likely to have the disease (1).
Diabetic cardiomyopathy (DCM), a disease common to most diabetic patients, is based on changes in the cardiac structure and systolic and diastolic functions. When patients have cardiac insufficiency, other clear causes, such as hypertension and structural or ischemic heart disease can be diagnosed (2). However, due to limits in awareness and a lack of diagnostic means, the current clinical diagnosis rate of diabetic cardiomyopathy is low, and the corresponding targeted clinical intervention is insufficient.
In general, the intervention involves conventional sugar control measures such as diet and hypoglycemic drugs, in the early stage. When the disease progresses to the stage of heart failure, the corresponding treatment is given according to the current guidelines for heart failure (3). However, the pathogenesis of diabetic cardiomyopathy is different from that of ordinary cardiomyopathy, which involves various factors, such as abnormal glucose and lipid metabolism, calcium balance disorder, etc. Therefore, treatment by ordinary means may lead to a poor prognosis (4).
In fact, the major risk factors for the development of diabetic cardiomyopathy are the progressive death of cardiomyocytes, changes in endothelial cells (5), and cardiomyocytes (6) in the high glucose environment. As early as 15 years ago, it was confirmed that myocardial apoptosis in patients with diabetic cardiomyopathy was 85 times more than that in the control group based on clinical biopsies (7). As a recognized form of cell death in diabetic cardiomyopathy, apoptosis has been studied extensively and intensively. However, the pathophysiological and clinical significance of this mode of cell death is inconclusive. Therefore, expanding the study of death modalities beyond apoptosis and pyroptosis will help pinpoint the target of cell death to alleviate or even cure the onset of diabetic cardiomyopathy. Recently, there have been numerous studies on regulatory cell death. More forms of death have been discovered, such as pyroptosis, immunogenic cell death, ferroptosis, necroptosis, cuproptosis, exocytosis, etc. These various forms of cell death have been proven to play a role in many diseases. From these studies, we will mainly discuss ferroptosis, necroptosis, and cuproptosis, three relatively newly identified forms of cell death. We will search for possible targets of these three forms in diabetic cardiomyopathy and analyze the corresponding therapeutic approaches according to these targets.
Much attention has been paid to the mechanisms related to diabetes. In recent years, oxidative stress, inflammation, and Ca2+-related dysfunction have been identified, as well as changes in substrate metabolism, insulin signal transduction, gene regulation, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, neurohumoral activation, and cardiac cell death (8, 9). Oxidative stress is the production of excessive reactive oxygen species (ROS). In addition to the increased ROS production in diabetic patients, the endogenous antioxidant mechanism of diabetic patients is often impaired (10, 11). In diabetic animal models, intervention with appropriate antioxidants, such as superoxide dismutase (SOD) mimics or coenzyme Q10, has determined the relationship between oxidative stress and diabetic cardiomyopathy, whether as an early preventive measure to limit the progression of diabetic cardiomyopathy or after the heart has been damaged (12, 13). In addition to the direct oxidative damage caused by inappropriate cardiac ROS levels, ROS is also a key trigger for the activation of inflammatory bodies (10, 14). ROS activates a variety of other pathological signaling cascades, such as the protein kinase C (PKC), apoptosis signal-regulated kinase-1 (ASK-1), p38 mitogen-activated protein kinase (p38-MAPK), NH2 terminal JUN kinase (JUNK), and JAK-STAT pathways (8, 15–18). These signals themselves are also related to diabetes-induced cardiac complications (19, 20). Some of these signals, such as PKC, p38-MAPK, and JAK-STAT, can also induce the generation of ROS, thus forming a harmful feedforward loop. Therefore, we believe that diabetic cardiomyocytes are in a high glucose microenvironment for a long time, resulting in the activation of a variety of signaling pathways, that causes cell death. Cell death is a normal cellular phenomenon. Since the ultrastructural characteristics of programmed cell suicide were determined in 1972 (21), programmed cell death with cell apoptosis as the “representative name” has gradually come into the view of scientists, and related research has been a hot spot in the field of life science. A large number of new cells are generated every day along with the death of old cells. The normal development and maintenance of the homeostasis of living organisms as well as the elimination of damaged, senescent, and infected cells all depend on a closely regulated programmed cell death. With the deepening of research, it was found that apoptosis is not the only form of cell death. According to the different mechanisms of cell death, regulated cell death can be divided into apoptosis, pyroptosis, ferroptosis, necroptosis, cuproptosis, etc. The purpose of this review is to describe the frontier research of ferroptosis, necroptosis, and cuproptosis in DCM, to sort out the research results and current situation, to point out areas for further exploration and possible research directions, and to provide some reference for future research. Ferroptosis, necroptosis, and cuproptosis differ from other forms of cell death based on morphology, biochemistry and immune status (Table 1).
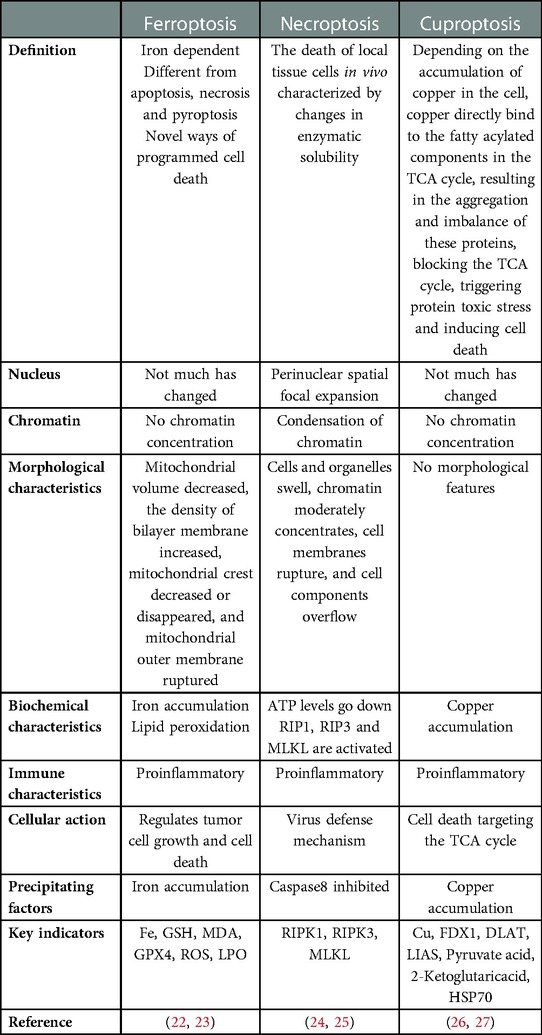
Table 1. Comparison of ferroptosis, necroptosis and cuproptosis.
Ferroptosis, named in 2012, is a new cell death mode and has become a focus of recent research. It is iron-dependent regulatory cell death that is different from other programmed cell death and is characterized by lipid peroxidation and iron overload (28). Since the concept was first proposed a decade ago, we have seen an explosion of research in this field, with more than 5,000 papers on ferroptosis. As a form of regulatory death, ferroptosis is significantly different from classical regulatory cell death, such as pyroptosis, especially in metabolic heart disease (29–31). Therefore, studying the mechanism of ferroptosis may reveal targets to control the progression of DCM and reduce mortality.
Currently, it is believed that the accumulation of phospholipid high oxides (PLOOHs) directly leads to ferroptosis (22). Spontaneous lipid peroxidation is initiated by ROS, and intracellular soluble ROS, including superoxide, hydrogen peroxide, and hydroxyl radicals. Among these, hydroxyl radicals are the most active and toxic ROS (32), which can rob the hydrogen atoms of phosphorylated PUFAs and form peroxides (PUFA-PL-OOH), directly leading to the death of iron. We will describe this process by splitting and tracing.
In human cells, Acetyl-CoA is a global currency that mediates carbon trading among the metabolic pathways, including glycolysis, the tricarboxylic acid cycle, amino acid metabolism, gluconeogenesis, and fatty acid synthesis (33). Acetyl-CoA in the mitochondria is catalyzed by ACC to produce unsaturated fatty acid PUFA. In addition, dietary habits also affect the content of free unsaturated fatty acids (34). However, PUFA cannot directly cause ferroptosis (35), which needs to be further catalyzed by ACSL family members into phosphorylated PUFA (PUFA-PL), or lipid metabolism that also produces a small amount of PUFA-PL (36). PUFA-PL then enters the cytoplasm as a prelude to ferroptosis. Starting from the initial Acetyl-CoA, energy stress activates AMPK, directly inhibits ACC, and restricts the synthesis of PUFA, thus inhibiting ferroptosis (37). Correspondingly, proteins that inhibit ACSLs can also inhibit ferroptosis, such as E-cadherin, which plays a role through the Hippo-Yap pathway (38).
ROS include a superoxide anion (O2·−), H2O2, and hydroxyl radical (HO·), which are derived from the four-electron reduction reaction of oxygen in the mitochondrial electron transport chain (39). If oxygen is reduced by only one electron in this reduction process, superoxide will be produced (40). Then, superoxide is reduced to a low-reactive substance hydrogen peroxide under the action of SOD (39). Iron bivalent in cells is an active REDOX metal, which is involved in the formation of free atomic groups and the expansion of lipid peroxidation, which is one of the most destructive effects of iron-catalyzing Fenton reaction: H2O2 + Fe2+→ +•OH + OH− + Fe3+ producing highly toxic hydroxyl radicals (41–43). Abnormal uptake, excretion, and storage of iron can lead to increased intracellular free iron and Fenton reactions (44).
Ferric ions transported by the Tf/TfR1 protein and required by the iron uptake pathway cannot directly participate in the Fenton reaction. Ferric ions are released from Tf and become ferric bivalent after acidification from endocytosis (45). Then, ferric bivalent enters the cytoplasm by endocytosis (46), a process mainly due to ESCRT1 on the membrane (47). Therefore, both Tf/TfR1 and ESCRT are ferroptosis-promoting proteins. The role of iron is not only to promote a Fenton reaction but also to use iron as a cofactor for some key enzymes. These include arachidonic lipoxygenase (ALOX), which triggers the formation of lipid hydroperoxides that are substrates for Fenton reactions. ALOX is also involved in the peroxidation of PUFA-PL (48), and 12-lipoxygenase is required for p53-dependent ferroptosis (49). Cytochrome P450 REDOX reductase (POR) also contributes to lipid peroxidation during ferroptosis (50), suggesting that several enzymes using iron as cofactors can promote lipid peroxidation leading to ferroptosis. Of course, iron is an essential trace element in the human body (51), and the emergence of iron does not mean the death of iron. A part of the iron bivalent will be transformed into iron trivalent and stored in ferritin, and another part of it can become a polyvesicular body containing iron (MVB) mediated through a prominin-2 (prom2) solution (52). The balance of iron homeostasis depends on the expression levels and activities of ferri-carriers, ferri-transporters, and ferri-regulatory and storage proteins (53). The sensitivity of cells to ferroptosis can be regulated by controlling the level of ferritin through iron pyroptosis and further regulating the abundance of free iron because the abundance of ferritin determines the size of the labile iron pool (54).
Similar to the regulation of iron, the presence of PUFA-PL-OOH in cells does not directly lead to ferroptosis. In fact, PUFA-PL-OOH can accumulate under the action of iron bivalent, resulting in cell death through damage to cell membranes or the production of lipid-derived electrophilic small molecules (55). This accumulation process can be inhibited by CoQ10, NADPH, and BH4 (56). In addition to the inhibitory process, there is a major antioxidant defense mechanism: GPX4. GPX4, glutathione peroxidase, is a GSH-dependent enzyme that converts reduced GSH to oxidized GSH and reduces PUFA-PL-OOH to non-toxic PUFA-PL-OH (57). Based on the dependence on GPX4, GSH depletion leads to the inactivation of GPX4, causing lipid peroxide accumulation to trigger ferroptosis. GSH is an important antioxidant in cells, so it is critical to find out where GSH comes from. System Xc- on the cell membrane is an amino acid transporter that can transport extracellular cystine into the cell and intracellular glutamate into the cell, and then the intracellular cystine is reduced to cysteine, which is the rate-limiting precursor of GSH synthesis (58). GSH then serves as a cofactor for GPX4, which acts as a lipid hydroperoxidase to reduce lipid peroxides (PUFA-OOH) to the non-toxic PUFA-OH (28, 59). Therefore, GPX4 is one of the important defense mechanisms for the cellular detoxification of lipid peroxides. Normal GPX4 activity is essential for maintaining membrane lipid homeostasis, preventing the excessive accumulation of toxic lipid peroxides and the formation of free radicals (L-OOH and L-O•), thereby reducing ferroptosis (60, 61) Acyl-CoA synthetase long-chain family member 4 (ACSL4) family is an important gene promoting ferroptosis, but the sensitivity of cancer cells to GPX4 inhibitors varies depending on the cancer type, which indicates that there may be another resistance mechanism similar to GPX4-FSP1-CoQ10-NAD(P)H pathway (62). Ferroptosis suppressor protein 1 (FSP1), located on human chromosome 10q22.1, mediates p53-independent apoptosis (63). FSP1 was originally called apoptosis-inducing factor mitochondrial-related gene 2 (AIFM2). It specifically mediates cell death because it is caspase-independent (64). Recently, it was revealed that Ferroptosis is mediated by ubiquinone (CoQ10) (63, 65). FSP1 is enriched in the cell membrane by myristoylation. As an NAD(P)H-dependent CoQ10 oxidoreductase, NAD(P)H can be used to reduce the oxidized ubiquinone. It is known that the reduced form of ubiquinone can capture the free radicals that mediate lipid peroxidation and limit the occurrence of lipid peroxidation. Thus, FSP1 can inhibit the occurrence of ferroptosis (62). All in all, the FSP1-CoQ10-NAD(P) H axis may play an important role in the occurrence of ferroptosis caused by lipid peroxidation.
In a study conducted by Mao C, the effect of a GPX4 inhibitor on ferroptosis in tumor cells was reversed by dihydroorotate dehydrogenase (DHODH), and this reversal was more significant in tumor cells with a low expression of GPX4, suggesting that DHODH may be the third mechanism of inhibiting ferroptosis (66). The anti-ferroptosis mechanism of DHODH is different from that of FSP1. According to research, the function of FSP1 is mainly limited to the cell membrane, and it is not clear whether it affects the mitochondrial membrane. DHODH mainly plays a role in the mitochondria. In the process of mitochondrial pyrimidine synthesis, DHODH oxidizes FMNH2 to FMN and reduces CoQ to CoQ10, thus inhibiting ferroptosis through CoQ10 capture and the scavenging of lipid peroxides (66). In addition, it has been reported that benzene-induced inflammatory anemia is associated with ferroptosis caused by the IRP1-DHODH-ALOX12 axis (67). Sorafenib is a commonly used drug in liver cancer, and its anti-tumor effect is closely related to ferroptosis. However, in recent years, the enrichment of DHODH has been found in some cells and tissues resistant to sorafenib and is related to the poor prognosis of HCC patients (68). The role of DHODH was also found in the occurrence of cervical cancer. In summary, DHODH-mediated ferroptosis in cancer is expected to become a new target for tumor therapy (66), and a nano-platform based on DHODH inhibitors has been reported to block the redox system (69) (Figure 1).
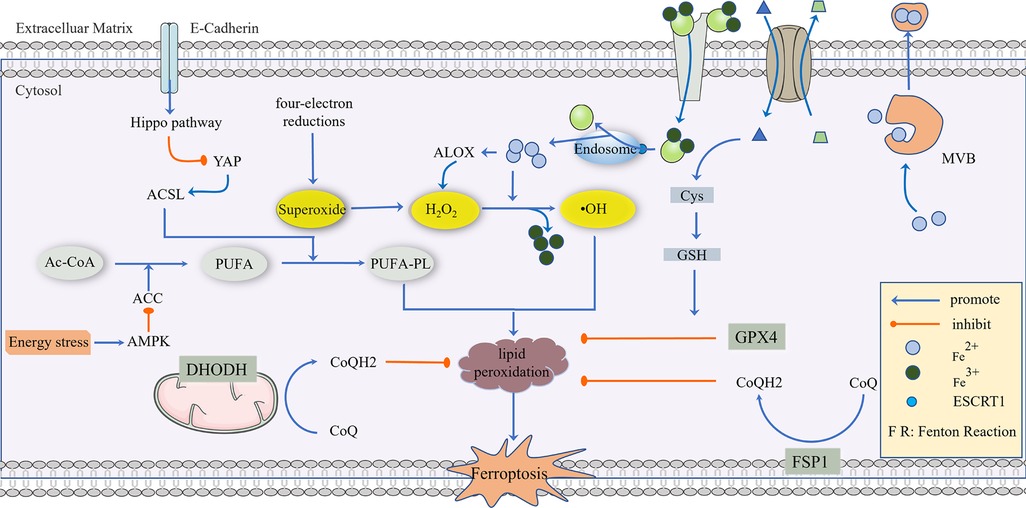
Figure 1. The regulatory mechanism of ferroptosis. AC-CoA in the mitochondria catalyzes the formation of PUFA under the action of ACC, which is then phosphorylated to generate PUFA-PL by the ACSL family and transferred into the cytoplasm to mediate the next stage of ferroptosis, which can be inhibited by energy stress and E-Cadherin, respectively. Superoxide produced in a mitochondrial four-electron reaction is reduced to H2O2 under the action of SOD, and a Fenton Reaction with the participation of Fe2+ produces highly toxic •OH, which mediates PUFA-PL peroxidation. This process can be inhibited by GPX4, CoQ10, NADPH, and BH4. GPX4 is the main antioxidant defense substance, and its inhibition of protein lipidation requires the participation of GSH. Cells transport Fe3+ via the Tf/TfR1 protein, which enters into the body through ESCRT1 on the endosome and is reduced to Fe2+. As a reducing agent, Fe2+ participates in a Fenton Reaction or as a cofactor of some key enzymes (such as ALOX), and part of Fe2+ can be excreted to the extracellular space via MVB. System Xc- can reverse the transport of Glu out of the cell membrane and Cys-Cys into the cell membrane, and Cys-Cys is further converted into Cys as a rate-limiting precursor of GSH synthesis. When excess lipid peroxides accumulate in cells, this damages to the cell membrane and eventually leads to ferroptosis.
As mentioned earlier, the system Xc-/GSH/GPX4 axis is an important pathway for ferroptosis. Many drugs function in the system Xc-/GSH/GPX4 axis, such as the classic Erastin, when ferroptosis was originally associated with this compound. Erastin inactivates GPX4 by inhibiting the system Xc- and reducing the production of GSH, promoting ferroptosis (70–72). Skipping the system Xc-, reagents that promote ferroptosis through the depletion of GSH, including acetaminophen, etc., are commonly used in clinics (73). In addition, ML162 (74) and DPI compounds (75) can also directly inhibit GPX4 and promote ferroptosis. As one of the forms of cell death, ferroptosis is involved in diabetic cardiomyopathy.
The accumulation of advanced glycation end products (AGEs) caused by hyperglycemia increases lipid peroxidation and the ferroptosis of cardiomyocytes (44), which then leads to cardiac inflammation and remodeling (cardiomyocyte hypertrophy, pro-fibrotic response and fibrosis), and ultimately leads to cardiac dysfunction, namely DCM (76). DCM inhibits the expression of SLC7A11 (system Xc- gene) and ferritin, thereby reducing GSH levels and increasing active iron levels, leading to mitochondrial oxidative damage (77). SFN plays a shielding role by activating AMPK to stimulate the downstream expression of SLC7A11 and ferritin, and at the same time, SFN enhances the AMPK dependence of the heart in age-induced ECT dysfunction and diabetic heart remodeling and dysfunction (78). These results suggest that the selective removal of excess iron inhibits ferroptosis, has a protective effect against chronic oxidative stress and that a modest reduction in plasma iron levels may be a viable treatment for the prevention of DCM in diabetic patients. From a clinical point of view, studies can test the combination of SFN with low-dose iron chelating agents, and further explore more targets of iron in the mechanism of ferroptosis depending on the effect and dose of the drug.
Increasing evidence shows that ferroptosis is involved in the onset and progression of DCM, and ferroptosis also affects the pathological consequences of cardiac function (79–82). These findings identify ferroptosis as a promising cellular target for correcting heart function in patients with DCM and possibly also for delaying the degenerative progression of DCM. However, the pathophysiological role of ferroptosis is not fully understood. Because the concept of ferroptosis is only ten years old and the data are limited, the current review does not provide any further insight into the mechanisms associated with ferroptosis and DCM, leaving many questions unanswered. For example, what role does ferroptosis play in the development of DCM, and does it play the same role at different stages in the development of DCM? In addition, all current treatments for diabetic patients are based on the premise of controlling blood sugar levels. However, the effect of controlling blood sugar levels on ferroptosis has not been determined. Whether ferroptosis affects other tissue cells in the presence of DCM has not been studied, and clearly these questions need to be further explored. Elucidating the mechanism of ferroptosis in DCM will help us develop better strategies to precisely target ferroptosis in order to effectively alleviate DCM and prevent its progression to more severe conditions. However, no such studies have been done in the literature to date, and this is a very valuable conclusion and recommendation. But up to now, no such studies have been conducted. Thus, this is a rather valuable field waiting to be explored.
Initially, necroptosis was not considered a new form of cell death. It was morphologically similar to necrosis and shared an upstream pathway with apoptosis. Later, researchers gradually distinguished the differences between them, which led to the emergence of necroptosis (83). As a well-known mode of cell death, necrosis has long been found to be an unregulated form of cell death, caused by external physical and chemical stress, and a subsequent reaction of injury to the body (84). In contrast, necroptosis is highly regulated and serves as a defense mechanism or escape route for cells facing viral infection (85). When a viral caspase inhibitor is present, the cell can only choose to commit suicide in a way that is not caspase-dependent. The two cell death pathways, necroptosis and apoptosis, share some upstream signaling elements and ultimately lead to plasma membrane rupture. Thus, at first there was no distinction between the two forms of regulatory death, but as research progressed, it was revealed that there was a distinct difference in the cellular morphology of each process. Necroptosis is characterized by increased cell size, organelle swelling, and membrane perforation, followed by cell disintegration, the release of contents, initiation of innate and adaptive immune responses, and clearance of necrotic cells through giant cytostome (86). Meanwhile, apoptosis is characterized by cell shrinkage, cell membrane blisters, chromatin concentration, apoptotic body formation, and immunogenic protein phagocytosis through phagocytes and macrophages (87). Next, we will introduce the mechanism of necroptosis to draw out the role of necroptosis in DCM, present possible targets, and provide some suggestions for future research.
Necroptosis is a unique cell death mode in vertebrate cells, which is the second line of defense established by cells to resist pathogens. When apoptosis fails, necroptosis becomes a fail-safe mechanism. Abnormal regulation of necroptosis is associated with these diseases. The expression and activity of necroptosis signaling proteins are low in cancers, such as breast cancer (88) and ovarian cancer (89), whereas the occurrence of certain inflammatory diseases, such as myocardial ischemia-reperfusion injury (90) and DCM (91), is associated with an increased expression of the signaling proteins.
Recently, necroptosis signaling pathways have been extensively studied. These pathways share some upstream signaling elements with apoptosis (92). Here we highlight well-studied tumor necrosis factor receptor 1 (TNFR1). The powerful TNF family has a homeostasis function (93) to defend against pathogens, in which TNF-α binds to the transmembrane protein TNFR1, allowing the TNF receptor-associated death domain (TRADD) to signal RIPK1 and recruit RIPK3 to form bad dead bodies (94). Either in terms of apoptosis or necrosis, caspase is an indissoluble protein. Thus, here, we discuss necroptosis from the caspase protein. In 2019, the journal Nature reported that caspase-8 induces and inhibits cell death at the same time. It induces apoptosis through death receptors such as TNFR1, and inhibits necroptosis through lysis and the inactivation of RIPK1 and RIPK3 (95). If caspase-8 is active in cells, the formation of a complex between activated caspase-8 and RIPK1 and FADD could induce apoptosis (96). If caspase-8 is inhibited, RIPK1 and RIPK3 are phosphorylated and bind to the phosphorylated MLKL, eventually leading to necroptosis (97). Caspase-8 acts like a shunt, with different gates opening and different flow directions. As a type of regulatory cell death independent of caspase, necroptosis is due to a series of automatic and cross-phosphorylation interactions between RIPK1 and RIPK3. Upon phosphorylation of RIPK3, MLKL aggregates and subsequently phosphorylates, penetrates the plasma membrane and organelles, and the membrane breakdown results in the cell contents spilling into organs, further leading to inflammatory phenotypes and damage-related DAMP releases, such as IL-1α, IL-β, and IL-33, which induce immune responses (98). DAMP sends signals to the circulatory system to recruit immune cells to damaged tissues, the component in macrophages, macro pinocytosis, and clears necrotic apoptotic cells by pinocytosis (99) (Figure 2).
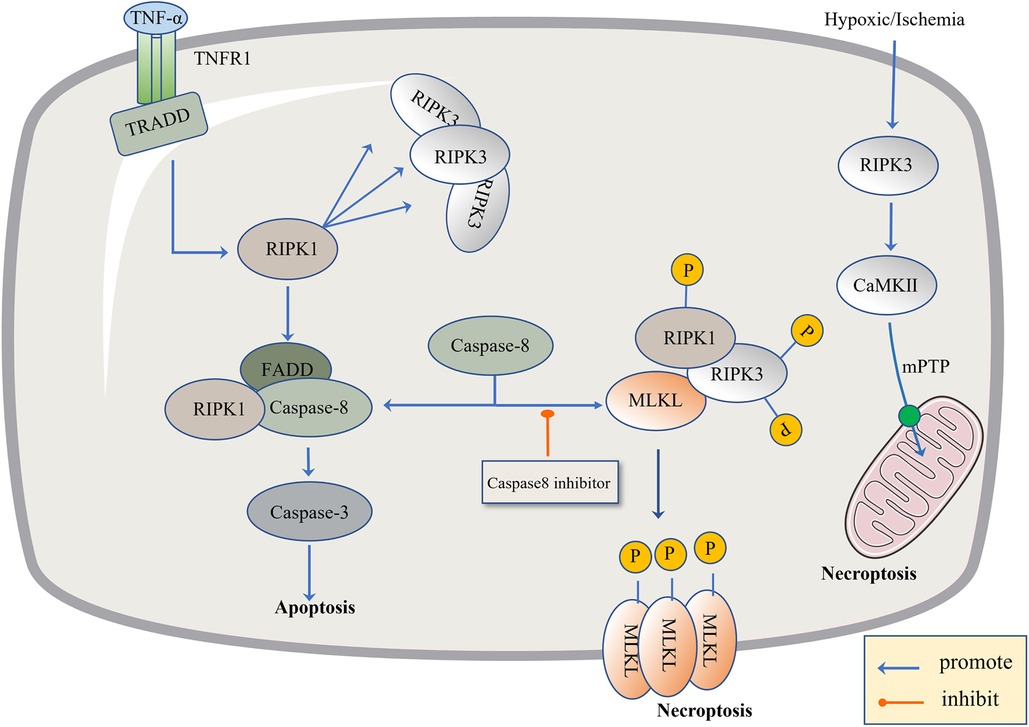
Figure 2. The regulatory mechanism of necroptosis. The pathogen inhibits Caspase-8, leading to apoptosis failure and the initiation of necroptosis. The level of TNF-α is up-regulated, TNF-α binds to TNFR1, activates TRADD and RIPK1, and there is the recruitment of RIPK3. When Caspase-8 is active, it promotes the combination of FADD, RIPK1 and Caspase-8, and then activates Caspase-3 to induce apoptosis. When Caspase-8 is inhibited, RIPK1 and RIPK3 are phosphorylated and bind to the phosphorylated MLKL. Phosphorylated MLKL is inserted into organelles and cell membranes, eventually leading to necroptosis. In addition, necroptosis can be activated through RIPK1-independent pathways, where RIPK3 activates CAMK II and the mitochondrial permeability transition pore (MPTP), leading to the loss of membrane potential and integrity, resulting in necroptosis.
The study of the role of necroptosis in DCM is relatively insufficient but still presents a high correlation. According to an animal model established by Song, using STZ-induced type 1 diabetic mice and HG-cultured rat cardiomyocytes, it was confirmed that Sirtuin 3 (SIRT3) deficiency aggravated hyperglycemia-induced mitochondrial damage, increased ROS accumulation, promoted necrosis, and possibly activated NLRP3 inflammatory. Ultimately, these processes aggravated DCM in mice (91). Recently, the correlation of the RIPK3/MLKL signaling pathway, a key pathway of necroptosis, in DCM has been confirmed. Cao et al. revealed that necroptosis of cardiomyocytes plays an essential role in mediating cardiac pathology in type 1 DCM (100). In addition, Chen et al. reduced myocardial damage, improved cardiac function, and inhibited Ca2+-calmodulin-dependent kinase II (CAMK II) activation through RIPK3 defects. Moreover, alleviating necroptosis in DCM mice demonstrates the existence of severe necroptosis in DCM mice (101).
It is certainly not enough to simply state a correlation. Irbesartan is a common clinical ARB drug. Existing studies have explored how irbesartan acts. Irbesartan has a cardioprotective effect on diabetic rats, and its mechanism may be related to the inhibition of the RIP1-RIP3-MLKL pathway, which alleviates necroptosis in cells. When it comes to the RIP1-RIP3 complex, AMPK inhibits the formation of this complex (102). Based on this, Cao et al. demonstrated that the anti-diabetic drug Engliazine and metformin prevented hyperglycemic-induced cardiomyopathy, which may be related to the activation of AMPK. Metformin reduces the inhibition of AMPK activation by cardiomyocytes in hyperglycemic environments (103). Therefore, the inhibition of necroptosis is a means to control DCM. More studies are needed to explore specific targets. We can consider further exploring the pathway proteins related to necroptosis, such as whether the upstream kinase mediating the regulation of caspase phosphorylation can be blocked by corresponding drugs. Do the blocking drugs have dual regulatory effects and can they regulate apoptosis at the same time? Is double regulation synergistic or antagonistic? What effect does this have on the treatment and prognosis of DCM?.
Similar to iron, copper is an indispensable trace element in all organisms, but it can exhibit cytotoxicity if the concentration of copper ions in cells exceeds the threshold for maintaining homeostasis (104, 105). In fact, as early as 2001, researchers found that compared with non-diabetic patients, plasma copper ion levels and urine copper ionized water are increased to varying degrees on average, while the content of copper in cardiomyocytes is decreased (106, 107). In March 2022, Peter Tsvetkov's team named a controlled cell death mode whose mechanism is clearly different from the known apoptosis by pyroptosis, necroptosis, and ferroptosis as “cuproptosis” (108).
To confirm that this form of death is distinct from other regulatory cell death processes, researchers knocked out the key apoptosis factors BAX and BAK1 and used inhibitors of known types of cell death, including caspase inhibitors for apoptosis, and ferrostatin 1 for ferroptosis. When the cells were treated with necrostatin 1 in response to necroptosis and N-pancreatic cysteine in response to oxidative stress, the cell death induced by a copper ion carrier could not be eliminated. The data suggest that cupric ion-carrier-induced cell death is a mechanism quite different from other known modes of cell death. Interestingly, cells that rely more on mitochondrial respiration are about 1,000 times more sensitive to copper inducers than cells that rely on glycolysis. Copper deficiency damages the function of mitochondria and leads to energy reduction, thus leading to myocardial hypertrophy. A clinical investigation by Oster et al. showed that the copper ion level in cardiomyocytes was positively correlated with ejection fraction in 27 patients after coronary artery bypass grafting (109). Treatment with mitochondrial antioxidants (110), fatty acids (111) and mitochondrial function inhibitors (112) significantly changed the sensitivity of the cells to copper ions. FDX1 is the upstream regulator of protein-lipid acylation, and the loss of FDX1 leads to the complete loss of protein-lipid acylation function, as well as the accumulation of pyruvate and α-ketoglutaric acid and the consumption of succinic acid in cells, indicating that the loss of protein lipid acylation function blocks the progression of the tricarboxylic acid cycle (TCA) (113). FDX1 and protein-lipid acylation are key factors in the induction of cell death by copper ion carriers. Excessive copper promotes the aggregation and function loss of fatty acylation proteins, triggering the instability of Fe-S cluster proteins, leading to toxic protein stress and ultimately cell death (108) (Figure 3).
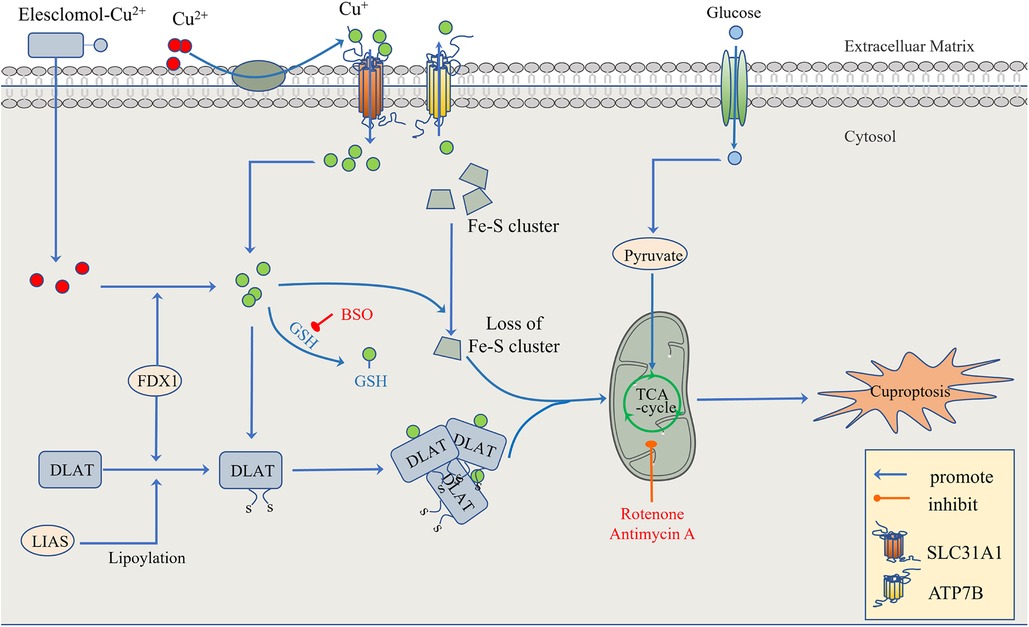
Figure 3. The regulatory mechanism of cuproptosis. The mitochondria are the main organelles involved in Cu-induced cell death. When the mitochondrial membrane is damaged by oxidation, the enzyme function in the TCA cycle is impaired, leading to cuproptosis. On the one hand, FDX1 and protein-lipid acylation are the key factors involved in cupric ion carrier-induced cell death. Excessive copper promotes the aggregation and functional loss of fatty acylation proteins, leading to the instability of Fe-S cluster proteins. On the other hand, FDX1 reduces Cu2+ to Cu + . GSH, a thiol-containing copper chelator, blocks cuproptosis, whereas buthionine sulfoximine (BSO) promotes cuproptosis by depleting GSH. With the participation of Cu+, the lipid acylation and aggregation of enzymes involved in the regulation of the mitochondrial TCA cycle, such as DLAT, are promoted. Together with the instability of Fe-S cluster proteins, the mitochondrial membrane and its TCA cycle are destroyed, leading to the occurrence of cuproptosis. This process can be inhibited by electron transport chain (ETC) complex I/III inhibitors such as rotenone and antimycin A. In addition to Elesclomol-Cu2+, which transports Cu2+ into and out of cells, copper ion channels SLC31A1 and ATP7B regulate the accumulation of copper ions by mediating the entry and exocytosis of copper ions, respectively.
DCM is characterized by myocardial remodeling, including myocardial fibrosis and hypertrophy. As mentioned above, cuproptosis mainly occurs in the mitochondria, which means that cuproptosis affects the course of DCM by destroying mitochondrial structure and function (78). Abnormal copper ion metabolism in cardiomyocytes is a key pathogenic process of diabetes-induced heart failure (114). Before the concept of cuproptosis was proposed, experiments were conducted to detect metabolite levels in cardiomyocytes treated with copper, and the results showed that the overall downregulation of metabolites mainly affected glycerol phospholipid metabolism, fatty acid degradation and other processes, indicating that copper can induce metabolic pathway disorder and lead to myocardial damage (115). More seriously, excessive oxidative stress is activated after copper accumulation, which disrupts the self-regulation and metabolic dynamics of the mitochondria, and the REDOX reaction cannot maintain balance, forming a vicious cycle (116).
Currently, many studies have shown that an increasing copper level can reduce myocardial hypertrophy (117–119), but for patients with DCM, the problem is not copper intake, but copper transport (120). Thus, we have taken into consideration two aspects: First, how can copper transport be regulated in patients with DCM? Is there a target for that? Second, if excessive copper is toxic after all, what is the relationship between copper toxicity and the occurrence and development of DCM? Or does copper toxicity affect the prognosis of heart failure in patients with DCM? Can copper damage be reduced by regulating plasma copper ion levels? TETA, a drug that promotes copper excretion, is currently used as an experimental treatment for diabetes to improve DCM (121) and is undergoing phase II clinical trials (122). Its principle is to promote copper excretion, prevent the excessive deposition of cardiac collagen, and improve cardiac structure and function (123). TETA has been previously approved for the treatment of Wilson disease (124), which to some extent indicates the safety of TETA.
As a newly proposed form of cell death, the current research on cuproptosis still focuses on its intervention in cancer therapy, which destroys the energy system of cancer cells by providing excessive copper to cancer tissues through targeting, leading to the death of cancer cells (108, 125), and its mechanism is still in the stage of exploration. Therefore, regarding the metabolic pathway related to cuproptosis and the influencing mechanism of its related diseases our understanding needs to be improved to find more corresponding targets. In addition, most of the current research is limited to cellular and molecular experiments, and clinical studies are rare. In the future, more studies should be carried out by combining clinical and molecular experiments.
Based on the above review, novel regulatory cell death mechanisms are involved in the cell death and progression of DCM, and it is worth noting that inhibition of any form of cell death can significantly restore damaged myocardial cell function or structure in DCM (80, 126–131), suggesting that different cell death types may be potential therapeutic targets for DCM (Figure 4). It is exciting to note that there are already regulatory cell death-related drug applications, such as Venetoclax (132), a selective Bcl-2 inhibitor in acute myelogenous leukemia, chronic lymphocytic leukemia, and small lymphocytic lymphoma. In addition, inhibiting the necroptosis of hepatocytes is an effective way to improve drug-induced liver injury. The administration of acetaminophen (300 mg/kg) and followed (1 h later) by hydroxyethidine (100 mg/kg) inhibits apoptosis and necroptosis in mice (133). Some anti-cancer drugs appear to target and enhance the ferroptosis process to kill glioma cells, including dihydroartemisinin (134) and ibuprofen (135). Inducing ferroptosis is thought to enhance the effect of traditional anti-cancer treatments that trigger other cell death pathways, such as apoptosis. However, until now, we have not thoroughly studied the mechanism of clearing these regulatory cell deaths, nor understood their pathophysiological effects. The number of studies is small, models are not perfect, and there is no reliable biomarker together with unsolved problems and limited conclusions obtained. Therefore, more studies are needed to answer the relevant questions. For example, we know that the clinical diagnosis and treatment of DM are premised on the control of blood glucose level, and of course, there is a definite relationship between hyperglycemia and DCM (136), but how hyperglycemia affects the various forms of cell death remains unclear. Do these different forms of cell death work independently? If so, what role does each play? If not, what is the connection between these different mechanisms? For example, does oxidation play the same role in each form of death at different stages of the development of DCM, and what effect does it have on the progression of the disease? Unfortunately, we have not found any studies that focus on the proportion of the various forms of death in DCM and their impact on prognosis, which may be one of the directions for further research aiming to improve the effectiveness of the treatments and survival of patients.
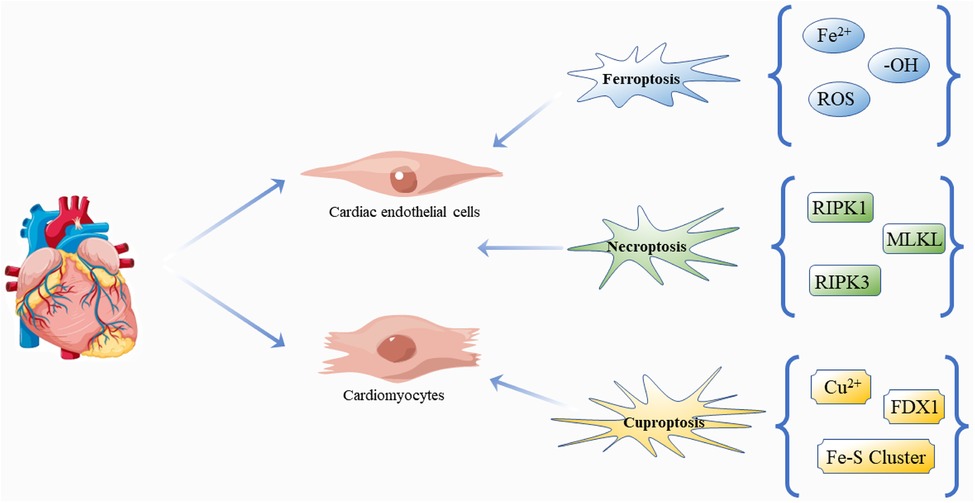
Figure 4. Regulatory cell death: ferroptosis, necroptosis and cuproptosis in DCM. The pathogenesis and progression of DCM are closely related to the effects of three kinds of death on cardiomyocytes and cardiac endothelial cells. Free icons were used from https://www.vecteezy.com (leaders: Shawn Rubel, Adam Gamble, Richard Fontenot).
As mentioned above, the AMPK pathway is also an important pyroptosis pathway (137). Pyroptosis-related proteins promote mitochondrial apoptosis through ROS (138). Therefore, can one mechanism promote or inhibit other mechanisms? In addition, the associations implied by these similarities may also be targets for genetic intervention. We also noted that ferroptosis has many similarities with cuproptosis, such as the oxidative stress response caused by ROS accumulation and GSH depletion, which are related to the synergistic or antagonistic effects of drugs and may be the focus of drug development. In addition, necroptosis is more closely related to apoptosis and necrosis. The determination of the caspase protein may be a potential drug target, and the phosphorylation and subsequent modification of RIPK family may also be one direction for future drug research. In addition, there are also some similarities between death forms, such as pyroptosis, that are worth exploring.
Of particular interest to us is what these similarities represent. Is there an overlap between some forms of death? If there is overlap, are these overlapping processes an opportunity for the onset of DCM? Shedding light on these issues can help to better determine targets and target diseases. Metabolic disorders caused by diabetes are triggers for many forms of cell death. However, as mentioned above, there is currently no specific clinical treatment plan for DCM, and even treatment after heart failure only follows the guidelines for heart failure. If we can further study these forms of death, we may be able to solve this problem. After all, the incidence of DM is high and rising year by year, and cardiovascular diseases are also the main cause of death and disability at present.
Conceptualization, LL; original draft preparation, DK; collected the literatures, PC, JLi and XS; review and editing, LL, ZZ and JLiu; supervision, YC. All authors contributed to the article and approved the submitted version.
This work was supported by the Local Colleges and Universities Talent Development Funding from Heilongjiang Provincial Department of Finance (No. 2020GSP09), the Natural Science Foundation of Heilongjiang Province (No. LH2022H099), the Basic Scientific Research Project which belongs to the University of Heilongjiang (No. 2021-KYYWF-0519; No. 2021-KYYWFMY-0520), the Science and Technology Plan Project of Mudanjiang (No. HT2020JG070), the Education and Teaching Reform Project of Mudanjiang Medical University (No. MYZD20210002), the Torch Program of Mudanjiang Medical University (No. 2022-MYHJ-003) and the Project of Young Innovative Talents Training Program of Regular Undergraduate Colleges and Universities in Heilongjiang Province (No. UNPYSCT-2020064).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
2. Peterson LR, Gropler RJ. Metabolic and molecular imaging of the diabetic cardiomyopathy. Circ Res. (2020) 126(11):1628–45. doi: 10.1161/CIRCRESAHA.120.315899
3. Butler J, Packer M, Greene SJ, Fiuzat M, Anker SD, Anstrom KJ, et al. Heart failure End points in cardiovascular outcome trials of sodium glucose cotransporter 2 inhibitors in patients with type 2 diabetes Mellitus: a critical evaluation of clinical and regulatory issues. Circulation. (2019) 140(25):2108–18. doi: 10.1161/CIRCULATIONAHA.119.042155
4. Seferović PM, Petrie MC, Filippatos GS, Anker SD, Rosano G, Bauersachs J, et al. Type 2 diabetes mellitus and heart failure: a position statement from the heart failure association of the European society of cardiology. Eur J Heart Fail. (2018) 20(5):853–72. doi: 10.1002/ejhf.1170
5. Knapp M, Tu X, Wu R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol Sin. (2019) 40(1):1–8. doi: 10.1038/s41401-018-0042-6
6. Filardi T, Ghinassi B, Baldassarre AD, Tanzilli G, Morano S, Lenzi A, et al. Cardiomyopathy associated with diabetes: the central role of the cardiomyocyte. Int J Mol Sci. (2019) 20(13):3299. doi: 10.3390/ijms20133299
7. Filardi T, Ghinassi B, Baldassarre AD, Tanzilli G, Morano S, Lenzi A, et al. Apoptosis in patients with dilated cardiomyopathy and diabetes: a feature of diabetic cardiomyopathy? Horm Metab Res. (2007) 39(9):672–6. doi: 10.1055/s-2007-985823
8. Huynh K, Bernardo BC, McMullen JR, Ritchie RH. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther. (2014) 142(3):375–415. doi: 10.1016/j.pharmthera.2014.01.003
9. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. (2014) 57(4):660–71. doi: 10.1007/s00125-014-3171-6
10. Wilson AJ, Gill EK, Abudalo RA, Edgar KS, Watson CJ, Grieve DJ, et al. Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart. (2018) 104(4):293–9. doi: 10.1136/heartjnl-2017-311448
11. Hansen SS, Aasum E, Hafstad AD. The role of NADPH oxidases in diabetic cardiomyopathy. Biochim Biophys Acta Mol Basis Dis. (2018) 1864(5 Pt B):1908–13. doi: 10.1016/j.bbadis.2017.07.025
12. Blasio MJD, Huynh K, Qin CX, Rosli S, Kiriazis H, Ayer A, et al. Therapeutic targeting of oxidative stress with coenzyme Q10 counteracts exaggerated diabetic cardiomyopathy in a mouse model of diabetes with diminished PI3K(p110alpha) signaling. Free Radic Biol Med. (2015) 87:137–47. doi: 10.1016/j.freeradbiomed.2015.04.028
13. Shen X, Zheng SR, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. (2006) 55(3):798–805. doi: 10.2337/diabetes.55.03.06.db05-1039
14. Sharma A, Tate M, Mathew G, Vince JE, Ritchie RH, Haan JBD, et al. Oxidative stress and NLRP3-inflammasome activity as significant drivers of diabetic cardiovascular complications: therapeutic implications. Front Physiol. (2018) 9:114. doi: 10.3389/fphys.2018.00114
15. Eid RA, Alkhateeb MA, El-Kott AF, Eleawa SM, Zaki MSA, Alaboodi SA, et al. A high-fat diet rich in corn oil induces cardiac fibrosis in rats by activating JAK2/STAT3 and subsequent activation of ANG II/TGF-1beta/Smad3 pathway: the role of ROS and IL-6 trans-signaling. J Food Biochem. (2019) 43(8):e12952. doi: 10.1111/jfbc.12952
16. Yang YC, Tsai CY, Chen CL, Kuo CH, Hou CW, Cheng SY, et al. Pkcdelta activation is involved in ROS-mediated mitochondrial dysfunction and apoptosis in cardiomyocytes exposed to advanced glycation End products (ages). Aging Dis. (2018) 9(4):647–63. doi: 10.14336/AD.2017.0924
17. Ritchie RH, Love JE, Huynh K, Bernardo BC, Henstridge DC, Kiriazis H, et al. Enhanced phosphoinositide 3-kinase(p110alpha) activity prevents diabetes-induced cardiomyopathy and superoxide generation in a mouse model of diabetes. Diabetologia. (2012) 55(12):3369–81. doi: 10.1007/s00125-012-2720-0
18. Luo W, Jin YY, Wu GJ, Zhu WW, Qian YY, Zhang YL, et al. Blockage of ROS and MAPKs-mediated inflammation via restoring SIRT1 by a new compound LF10 prevents type 1 diabetic cardiomyopathy. Toxicol Appl Pharmacol. (2019) 370:24–35. doi: 10.1016/j.taap.2019.03.005
19. Ruiz M, Coderre L, Lachance D, Houde V, Martel C, Legault JT, et al. MK2 Deletion in mice prevents diabetes-induced perturbations in lipid metabolism and cardiac dysfunction. Diabetes. (2016) 65(2):381–92. doi: 10.2337/db15-0238
20. Westermann D, Rutschow S, Linthout SV, Linderer A, Bücker-Gärtner C, Sobirey M, et al. Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia. (2006) 49(10):2507–13. doi: 10.1007/s00125-006-0385-2
21. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. (1972) 26(4):239–57. doi: 10.1038/bjc.1972.33
22. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
23. Ma TL, Chen JX, Zhu P, Zhang CB, Zhou Y, Duan JX, et al. Focus on ferroptosis regulation: exploring novel mechanisms and applications of ferroptosis regulator. Life Sci. (2022) 307:120868. doi: 10.1016/j.lfs.2022.120868
24. Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol. (2020) 13(1):110. doi: 10.1186/s13045-020-00946-7
25. Yan J, Wan P, Choksi S, Liu ZG. Necroptosis and tumor progression. Trends Cancer. (2022) 8(1):21–7. doi: 10.1016/j.trecan.2021.09.003
26. Wang Y, Zhang L, Zhou F. Cuproptosis: a new form of programmed cell death. Cell Mol Immunol. (2022) 19(8):867–8. doi: 10.1038/s41423-022-00866-1
27. Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. (2022) 32(5):417–8. doi: 10.1038/s41422-022-00653-7
28. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
29. Basit F, Oppen LMV, Schöckel L, Bossenbroek HM, Vries SEvE, Hermeling JC, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. (2017) 8(3):e2716. doi: 10.1038/cddis.2017.133
30. Liu M, Fan Y, Li D, Han B, Meng Y, Chen F, et al. Ferroptosis inducer erastin sensitizes NSCLC cells to celastrol through activation of the ROS-mitochondrial fission-mitophagy axis. Mol Oncol. (2021) 15(8):2084–105. doi: 10.1002/1878-0261.12936
31. Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol. (2023) 20(1):7–23. doi: 10.1038/s41569-022-00735-4
32. Igarashi K, Shoji Y, Sekine-Suzuki E, Ueno M, Matsumoto K, Nakanishi I, et al. Importance of locations of iron ions to elicit cytotoxicity induced by a fenton-type reaction. Cancers (Basel). (2022) 14(15):3642. doi: 10.3390/cancers14153642
33. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. (2007) 7(10):763–77. doi: 10.1038/nrc2222
34. Anderwald C, Brunmair B, Stadlbauer K, Krebs M, Fürnsinn C, Roden M, et al. Effects of free fatty acids on carbohydrate metabolism and insulin signalling in perfused rat liver. Eur J Clin Invest. (2007) 37(10):774–82. doi: 10.1111/j.1365-2362.2007.01858.x
35. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417(1):1–13. doi: 10.1042/BJ20081386
36. Zhou Q, Li T, Qin Q, Huang X, Wang Y. Ferroptosis in lymphoma: emerging mechanisms and a novel therapeutic approach. Front Genet. (2022) 13:1039951. doi: 10.3389/fgene.2022.1039951
37. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19(2):121–35. doi: 10.1038/nrm.2017.95
38. Xiong YJ, Dong L, Bai Y, Tang H, Li S, Luo D, et al. Piezo1 activation facilitates ovarian cancer metastasis via hippo/YAP signaling axis. Channels (Austin). (2022) 16(1):159–66. doi: 10.1080/19336950.2022.2099381
39. Fridovich I. Mitochondria: are they the seat of senescence? Aging Cell. (2004) 3(1):13–6. doi: 10.1046/j.1474-9728.2003.00075.x
40. Fridovich I. Superoxide anion radical (O2-.), superoxide dismutases, and related matters. J Biol Chem. (1997) 272(30):18515–7. doi: 10.1074/jbc.272.30.18515
41. Qian ZM, Tang PL. Mechanisms of iron uptake by mammalian cells. Biochim Biophys Acta. (1995) 1269(3):205–14. doi: 10.1016/0167-4889(95)00098-X
42. Thrainsdottir IS, Aspelund T, Thorgeirsson G, Gudnason V, Hardarson T, Malmberg K, et al. The association between glucose abnormalities and heart failure in the population-based Reykjavik study. Diabetes Care. (2005) 28(3):612–6. doi: 10.2337/diacare.28.3.612
43. Drakesmith H, Nemeth E, Ganz T. Ironing out ferroportin. Cell Metab. (2015) 22(5):777–87. doi: 10.1016/j.cmet.2015.09.006
44. Feng B, Cao Y, Chen S, Chu X, Chu Y, Chakrabarti S, et al. miR-200b mediates endothelial-to-mesenchymal transition in diabetic cardiomyopathy. Diabetes. (2016) 65(3):768–79. doi: 10.2337/db15-1033
45. Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC, et al. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci U S A. (1998) 95(3):1148–53. doi: 10.1073/pnas.95.3.1148
46. Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell. (2017) 168(3):344–61. doi: 10.1016/j.cell.2016.12.034
47. Anderson GJ, Wang F. Essential but toxic: controlling the flux of iron in the body. Clin Exp Pharmacol Physiol. (2012) 39(8):719–24. doi: 10.1111/j.1440-1681.2011.05661.x
48. Tomita K, Takashi Y, Ouchi Y, Kuwahara Y, Igarashi K, Nagasawa T, et al. Lipid peroxidation increases hydrogen peroxide permeability leading to cell death in cancer cell lines that lack mtDNA. Cancer Sci. (2019) 110(9):2856–66. doi: 10.1111/cas.14132
49. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 Is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. (2019) 21(5):579–91. doi: 10.1038/s41556-019-0305-6
50. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. (2020) 16(3):302–9. doi: 10.1038/s41589-020-0472-6
51. Margis R, Dunand C, Teixeira FK, Margis-Pinheiro M. Glutathione peroxidase family—an evolutionary overview. FEBS J. (2008) 275(15):3959–70. doi: 10.1111/j.1742-4658.2008.06542.x
52. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. (2019) 51(5):575–586.e4. doi: 10.1016/j.devcel.2019.10.007
53. Forcina GC, Dixon SJ. GPX4 At the crossroads of lipid homeostasis and ferroptosis. Proteomics. (2019) 19(18):e1800311. doi: 10.1002/pmic.201800311
54. Kakhlon O, Gruenbaum Y, Cabantchik ZI. Repression of ferritin expression increases the labile iron pool, oxidative stress, and short-term growth of human erythroleukemia cells. Blood. (2001) 97(9):2863–71. doi: 10.1182/blood.V97.9.2863
55. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. (2016) 113(34):E4966–75. doi: 10.1073/pnas.1603244113
56. Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. (2020) 8:586578. doi: 10.3389/fcell.2020.586578
57. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. (2019) 575(7784):688–92. doi: 10.1038/s41586-019-1705-2
58. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 Promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ. (2020) 27(2):662–75. doi: 10.1038/s41418-019-0380-z
59. Stockwell BR, Angeli JPF, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
60. Baumgardt SL, Paterson M, Leucker TM, Fang J, Zhang DX, Bosnjak ZJ, et al. Chronic co-administration of sepiapterin and L-citrulline ameliorates diabetic cardiomyopathy and myocardial ischemia/reperfusion injury in obese type 2 diabetic mice. Circ Heart Fail. (2016) 9(1):e002424. doi: 10.1161/CIRCHEARTFAILURE.115.002424
61. Ohno S, Kohjitani A, Miyata M, Tohya A, Yamashita K, Hashiguchi T, et al. Recovery of endothelial function after Minor-to-moderate surgery is impaired by diabetes Mellitus, obesity, hyperuricemia and sevoflurane-based anesthesia. Int Heart J. (2018) 59(3):559–65. doi: 10.1536/ihj.17-143
62. Doll S, Freitas FP, Shah R, Aldrovandi M, Silva MCD, Ingold I, et al. FSP1 Is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
63. Ohiro Y, Garkavtsev I, Kobayashi S, Sreekumar KR, Nantz R, Higashikubo BT, et al. A novel p53-inducible apoptogenic gene, PRG3, encodes a homologue of the apoptosis-inducing factor (AIF). FEBS Lett. (2002) 524(1-3):163–71. doi: 10.1016/S0014-5793(02)03049-1
64. Wu M, Xu LG, Li X, Zhai Z, Shu HB. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J Biol Chem. (2002) 277(28):25617–23. doi: 10.1074/jbc.M202285200
65. Gong M, Hay S, Marshall KR, Munro AW, Scrutton NS. DNA Binding suppresses human AIF-M2 activity and provides a connection between redox chemistry, reactive oxygen species, and apoptosis. J Biol Chem. (2007) 282(41):30331–40. doi: 10.1074/jbc.M703713200
66. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. (2021) 593(7860):586–90. doi: 10.1038/s41586-021-03539-7
67. Zhang W, Wang J, Liu ZY, Zhang L, Jing J, Han L, Gao A, et al. Iron-dependent ferroptosis participated in benzene-induced anemia of inflammation through IRP1-DHODH-ALOX12 axis. Free Radic Biol Med. (2022) 193(Pt 1):122–33. doi: 10.1016/j.freeradbiomed.2022.10.273
68. Li X, Yu Q, Zhao R, Guo X, Liu C, Zhang K, et al. Designer exosomes for targeted delivery of a novel therapeutic cargo to enhance sorafenib-mediated ferroptosis in hepatocellular carcinoma. Front Oncol. (2022) 12:898156. doi: 10.3389/fonc.2022.898156
69. Chen S, Yang J, Liang Z, Li Z, Xiong W, Fan Q, et al. Synergistic functional nanomedicine enhances ferroptosis therapy for breast tumors by a blocking defensive redox system. ACS Appl Mater Interfaces. (2023) 15(2):2705–13. doi: 10.1021/acsami.2c19585
70. Yang Y, Luo M, Zhang K, Zhang J, Gao T, Connell DO, et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat Commun. (2020) 11(1):433. doi: 10.1038/s41467-020-14324-x
71. Li Y, Zeng X, Lu D, Yin M, Shan M, Gao Y, et al. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum Reprod. (2021) 36(4):951–64. doi: 10.1093/humrep/deaa363
72. Zhao Y, Li Y, Zhang R, Wang F, Wang T, Jiao Y, et al. The role of erastin in ferroptosis and its prospects in cancer therapy. Onco Targets Ther. (2020) 13:5429–41. doi: 10.2147/OTT.S254995
73. Gai C, Yu M, Li Z, Wang Y, Ding D, Zheng J, et al. Acetaminophen Sensitizing erastin-induced ferroptosis via modulation of Nrf2/heme oxygenase-1 signaling pathway in non-small-cell lung cancer. J Cell Physiol. (2020) 235(4):3329–39. doi: 10.1002/jcp.29221
74. Shin D, Kim EH, Lee J, Roh JL. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic Biol Med. (2018) 129:454–62. doi: 10.1016/j.freeradbiomed.2018.10.426
75. Sampilvanjil A, Karasawa T, Yamada N, Komada T, Higashi T, Baatarjav C, et al. Cigarette smoke extract induces ferroptosis in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. (2020) 318(3):H508–18. doi: 10.1152/ajpheart.00559.2019
76. Wang X, Chen X, Zhou W, Men H, Bao T, Sun Y, et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. (2022) 12(2):708–22. doi: 10.1016/j.apsb.2021.10.005
77. Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. (2019) 116(7):2672–80. doi: 10.1073/pnas.1821022116
78. Strassburger M, Bloch W, Sulyok S, Schüller J, Keist AF, Schmidt A, et al. Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic Biol Med. (2005) 38(11):1458–70. doi: 10.1016/j.freeradbiomed.2005.02.009
79. Du S, Shi H, Xiong L, Wang P, Shi Y. Canagliflozin mitigates ferroptosis and improves myocardial oxidative stress in mice with diabetic cardiomyopathy. Front Endocrinol (Lausanne). (2022) 13:1011669. doi: 10.3389/fendo.2022.1011669
80. Ni T, Huang X, Pan S, Lu Z. Inhibition of the long non-coding RNA ZFAS1 attenuates ferroptosis by sponging miR-150-5p and activates CCND2 against diabetic cardiomyopathy. J Cell Mol Med. (2021) 25(21):9995–10007. doi: 10.1111/jcmm.16890
81. Wei Z, Shaohuan Q, Pinfang K, Chao S. Curcumin attenuates ferroptosis-induced myocardial injury in diabetic cardiomyopathy through the Nrf2 pathway. Cardiovasc Ther. (2022) 2022:3159717. doi: 10.1155/2022/3159717
82. Miao W, Chen M, Chen M, Cui C, Zhu Y, Luo X, et al. Nr2f2 overexpression aggravates ferroptosis and mitochondrial dysfunction by regulating the PGC-1alpha signaling in diabetes-induced heart failure mice. Mediators Inflamm. (2022) 2022:8373389. doi: 10.1155/2022/8373389
83. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. (2019) 26(1):99–114. doi: 10.1038/s41418-018-0212-6
84. Ruffolo PR. The pathogenesis of necrosis. I. Correlated light and electron microscopic observations of the myocardial necrosis induced by the intravenous injection of papain. Am J Pathol. (1964) 45(5):741–56.14223579
85. Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol. (2017) 12:103–30. doi: 10.1146/annurev-pathol-052016-100247
86. Dhuriya YK, Sharma D. Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflammation. (2018) 15(1):199. doi: 10.1186/s12974-018-1235-0
87. Doonan F, Cotter TG. Morphological assessment of apoptosis. Methods. (2008) 44(3):200–4. doi: 10.1016/j.ymeth.2007.11.006
88. Baik JY, Liu Z, Jiao D, Kwon H, Yan J, Kadigamuwa C, et al. ZBP1 Not RIPK1 mediates tumor necroptosis in breast cancer. Nat Commun. (2021) 12(1):2666. doi: 10.1038/s41467-021-23004-3
89. Chefetz I, Grimley E, Yang K, Hong L, Vinogradova EV, Suciu R, et al. A pan-ALDH1A inhibitor induces necroptosis in ovarian cancer stem-like cells. Cell Rep. (2019) 26(11):3061–3075.e6. doi: 10.1016/j.celrep.2019.02.032
90. Chen AQ, Fang Z, Chen XL, Yang S, Zhou YF, Mao L, et al. Microglia-derived TNF-alpha mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis. (2019) 10(7):487. doi: 10.1038/s41419-019-1716-9
91. Song S, Ding Y, Dai GL, Zhang Y, Xu MT, Shen JR, et al. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol Sin. (2021) 42(2):230–41. doi: 10.1038/s41401-020-0490-7
92. Khoury MK, Gupta K, Franco SR, Liu B. Necroptosis in the pathophysiology of disease. Am J Pathol. (2020) 190(2):272–85. doi: 10.1016/j.ajpath.2019.10.012
93. Dostert C, Grusdat M, Letellier E, Brenner D. The TNF family of ligands and receptors: communication modules in the immune system and beyond. Physiol Rev. (2019) 99(1):115–60. doi: 10.1152/physrev.00045.2017
94. Qi W, Yuan J. RIPK1 And RIPK3 form mosaic necrosomes. Nat Cell Biol. (2022) 24(4):406–7. doi: 10.1038/s41556-022-00879-y
95. Fritsch M, Günther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. (2019) 575(7784):683–7. doi: 10.1038/s41586-019-1770-6
96. O'Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. (2011) 13(12):1437–42. doi: 10.1038/ncb2362
97. Zhuang C, Chen F. Small-Molecule inhibitors of necroptosis: current Status and perspectives. J Med Chem. (2020) 63(4):1490–510. doi: 10.1021/acs.jmedchem.9b01317
98. Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J, et al. A RIPK3-PGE(2) circuit mediates myeloid-derived suppressor cell-potentiated colorectal carcinogenesis. Cancer Res. (2018) 78(19):5586–99. doi: 10.1158/0008-5472.CAN-17-3962
99. Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 Mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. (2016) 353(6299):603–8. doi: 10.1126/science.aaf6803
100. Cao T, Ni R, Ding W, Ji X, Li L, Liao G, et al. MLKL-mediated necroptosis is a target for cardiac protection in mouse models of type-1 diabetes. Cardiovasc Diabetol. (2022) 21(1):165. doi: 10.1186/s12933-022-01602-9
101. Wei J, Zhao Y, Liang H, Du W, Wang L. Preliminary evidence for the presence of multiple forms of cell death in diabetes cardiomyopathy. Acta Pharm Sin B. (2022) 12(1):1–17. doi: 10.1016/j.apsb.2021.08.026
102. Lee SB, Kim JJ, Han SA, Fan Y, Guo LS, Aziz K, et al. The AMPK-parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat Cell Biol. (2019) 21(8):940–51. doi: 10.1038/s41556-019-0356-8
103. Xu Q, Tan X, Xian W, Geng J, Li H, Tang B, et al. Changes of necroptosis in irbesartan medicated cardioprotection in diabetic rats. Diabetes Metab Syndr Obes. (2021) 14:3851–63. doi: 10.2147/DMSO.S300388
104. Vetchy M. Biological role of copper as an essential trace element in the human organism. Ceska Slov Farm. (2018) 67(4):143–53.30646728
105. Kahlson MA, Dixon SJ. Copper-induced cell death. Science. (2022) 375(6586):1231–2. doi: 10.1126/science.abo3959
106. Ito S, Fujita H, Narita T, Yaginuma T, Kawarada Y, Kawagoe M, et al. Urinary copper excretion in type 2 diabetic patients with nephropathy. Nephron. (2001) 88(4):307–12. doi: 10.1159/000046013
107. Cooper GJS, Chan YK, Dissanayake AM, Leahy FE, Keogh GF, Frampton CM, et al. Demonstration of a hyperglycemia-driven pathogenic abnormality of copper homeostasis in diabetes and its reversibility by selective chelation: quantitative comparisons between the biology of copper and eight other nutritionally essential elements in normal and diabetic individuals. Diabetes. (2005) 54(5):1468–76. doi: 10.2337/diabetes.54.5.1468
108. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. (2022) 375(6586):1254–61. doi: 10.1126/science.abf0529
109. Oster O, Dahm M, Oelert H. Element concentrations (selenium, copper, zinc, iron, magnesium, potassium, phosphorous) in heart tissue of patients with coronary heart disease correlated with physiological parameters of the heart. Eur Heart J. (1993) 14(6):770–4. doi: 10.1093/eurheartj/14.6.770
110. Borchard S, Bork F, Rieder T, Eberhagen C, Popper B, Lichtmannegger J, et al. The exceptional sensitivity of brain mitochondria to copper. Toxicol in Vitro. (2018) 51:11–22. doi: 10.1016/j.tiv.2018.04.012
111. Cui L, Gouw AM, LaGory EL, Guo S, Attarwala N, Tang Y, et al. Mitochondrial copper depletion suppresses triple-negative breast cancer in mice. Nat Biotechnol. (2021) 39(3):357–67. doi: 10.1038/s41587-020-0707-9
112. Shi LM, Jiang H, Wang J, Ma ZG, Xie JX. Mitochondria dysfunction was involved in copper-induced toxicity in MES23.5 cells. Neurosci Bull. (2008) 24(2):79–83. doi: 10.1007/s12264-008-0079-5
113. Zhang Z, Ma Y, Guo X, Du Y, Zhu Q, Wang X, et al. FDX1 Can impact the prognosis and mediate the metabolism of lung adenocarcinoma. Front Pharmacol. (2021) 12:749134. doi: 10.3389/fphar.2021.749134
114. Zhang S, Liu H, Amarsingh GV, Cheung CCH, Wu D, Narayanan U, et al. Restoration of myocellular copper-trafficking proteins and mitochondrial copper enzymes repairs cardiac function in rats with diabetes-evoked heart failure. Metallomics. (2020) 12(2):259–72. doi: 10.1039/c9mt00223e
115. Li Q, Liao J, Lei C, Shi J, Zhang H, Han Q, et al. Metabolomics analysis reveals the effect of copper on autophagy in myocardia of pigs. Ecotoxicol Environ Saf. (2021) 213:112040. doi: 10.1016/j.ecoenv.2021.112040
116. Wang Y, et al. Copper or/and arsenic induces autophagy by oxidative stress-related PI3K/AKT/mTOR pathways and cascaded mitochondrial fission in chicken skeletal muscle. J Inorg Biochem. (2018) 188:1–8. doi: 10.1016/j.jinorgbio.2018.08.001
117. Zheng L, Han P, Liu J, Li R, Yin W, Wang T, et al. Role of copper in regression of cardiac hypertrophy. Pharmacol Ther. (2015) 148:66–84. doi: 10.1016/j.pharmthera.2014.11.014
118. Liu J, Chen C, Liu Y, Sun X, Ding X, Qiu L, et al. Trientine selectively delivers copper to the heart and suppresses pressure overload-induced cardiac hypertrophy in rats. Exp Biol Med (Maywood). (2018) 243(14):1141–52. doi: 10.1177/1535370218813988
119. Liu Y, Xiao Y, Liu J, Feng L, Kang YJ. Copper-induced reduction in myocardial fibrosis is associated with increased matrix metalloproteins in a rat model of cardiac hypertrophy. Metallomics. (2018) 10(1):201–8. doi: 10.1039/C7MT00165G
120. Cui X, Wang Y, Liu H, Shi M, Wang J, Wang Y, et al. The molecular mechanisms of defective copper metabolism in diabetic cardiomyopathy. Oxid Med Cell Longev. (2022) 2022:5418376. doi: 10.1155/2022/5418376
121. Zhang L, Ward ML, Phillips ARJ, Zhang S, Kennedy J, Barry B, et al. Protection of the heart by treatment with a divalent-copper-selective chelator reveals a novel mechanism underlying cardiomyopathy in diabetic rats. Cardiovasc Diabetol. (2013) 12:123. doi: 10.1186/1475-2840-12-123
122. Cooper GJS, Phillips ARJ, Choong SY, Leonard BL, Crossman DJ, Brunton DH, et al. Regeneration of the heart in diabetes by selective copper chelation. Diabetes. (2004) 53(9):2501–8. doi: 10.2337/diabetes.53.9.2501
123. Lu J, Gong D, Choong SY, Xu H, Chan YK, Chen X, et al. Copper(II)-selective chelation improves function and antioxidant defences in cardiovascular tissues of rats as a model of diabetes: comparisons between triethylenetetramine and three less copper-selective transition-metal-targeted treatments. Diabetologia. (2010) 53(6):1217–26. doi: 10.1007/s00125-010-1698-8
124. Aggarwal A, Bhatt M. Advances in treatment of wilson disease. Tremor Other Hyperkinet Mov (N Y). (2018) 8:525. doi: 10.5334/tohm.435
125. Yuan M, Gong M, Zhang Z, Meng L, Tse G, Zhao Y, et al. Hyperglycemia induces endoplasmic Reticulum stress in atrial cardiomyocytes, and mitofusin-2 downregulation prevents mitochondrial dysfunction and subsequent cell death. Oxid Med Cell Longev. (2020) 2020:6569728. doi: 10.1155/2020/6569728
126. Ren BC, Zhang YF, Liu SS, Cheng XJ, Yang X, Cui XG, et al. Curcumin alleviates oxidative stress and inhibits apoptosis in diabetic cardiomyopathy via Sirt1-Foxo1 and PI3K-akt signalling pathways. J Cell Mol Med. (2020) 24(21):12355–67. doi: 10.1111/jcmm.15725
127. Yang F, Qin Y, Wang Y, Meng S, Xian H, Che H, et al. Metformin inhibits the NLRP3 inflammasome via AMPK/mTOR-dependent effects in diabetic cardiomyopathy. Int J Biol Sci. (2019) 15(5):1010–9. doi: 10.7150/ijbs.29680
128. Yang F, Qin Y, Lv J, Wang Y, Che H, Chen X, et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis. (2018) 9(10):1000. doi: 10.1038/s41419-018-1029-4
129. Ivanović-Matić S, Bogojević D, Martinović V, Petrović A, Jovanović-Stojanov S, Poznanović G, et al.. Catalase inhibition in diabetic rats potentiates DNA damage and apoptotic cell death setting the stage for cardiomyopathy. J Physiol Biochem. (2014) 70(4):947–59. doi: 10.1007/s13105-014-0363-y
130. Wang X, Pan J, Liu D, Zhang M, Li X, Tian J, et al. Nicorandil alleviates apoptosis in diabetic cardiomyopathy through PI3K/akt pathway. J Cell Mol Med. (2019) 23(8):5349–59. doi: 10.1111/jcmm.14413
131. Gong W, Zhang S, Chen Y, Shen J, Zheng Y, Liu X, et al. Protective role of hydrogen sulfide against diabetic cardiomyopathy via alleviating necroptosis. Free Radic Biol Med. (2022) 181:29–42. doi: 10.1016/j.freeradbiomed.2022.01.028
132. Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discov. (2019) 9(7):890–909. doi: 10.1158/2159-8290.CD-19-0117
133. Liao Y, Yang Y, Wang X, Wei M, Guo Q, Zhao L, et al. Oroxyloside ameliorates Acetaminophen-induced hepatotoxicity by inhibiting JNK related apoptosis and necroptosis. J Ethnopharmacol. (2020) 258:112917. doi: 10.1016/j.jep.2020.112917
134. Yi R, Wang H, Deng C, Wang X, Yao L, Niu W, et al. Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Biosci Rep. (2020) 40(6):BSR20193314. doi: 10.1042/BSR20193314
135. Gao X, Guo N, Xu H, Pan T, Lei H, Yan A, et al. Ibuprofen induces ferroptosis of glioblastoma cells via downregulation of nuclear factor erythroid 2-related factor 2 signaling pathway. Anticancer Drugs. (2020) 31(1):27–34. doi: 10.1097/CAD.0000000000000825
136. Huang Y, Cai X, Mai W, Li M, Hu Y. Association between prediabetes and risk of cardiovascular disease and all cause mortality: systematic review and meta-analysis. Br Med J. (2016) 355:i5953. doi: 10.1136/bmj.i5953
137. Naik PP, Mukhopadhyay S, Praharaj PP, Bhol CS, Panigrahi DP, Mahapatra KK, et al. Secretory clusterin promotes oral cancer cell survival via inhibiting apoptosis by activation of autophagy in AMPK/mTOR/ULK1 dependent pathway. Life Sci. (2021) 264:118722. doi: 10.1016/j.lfs.2020.118722
Keywords: diabetic cardiomyopathy, ferroptosis, necroptosis, cuproptosis, regulatory cell death
Citation: Ke D, Zhang Z, Liu J, Chen P, Li J, Sun X, Chu Y and Li L (2023) Ferroptosis, necroptosis and cuproptosis: Novel forms of regulated cell death in diabetic cardiomyopathy. Front. Cardiovasc. Med. 10:1135723. doi: 10.3389/fcvm.2023.1135723
Received: 1 January 2023; Accepted: 22 February 2023;
Published: 10 March 2023.
Edited by:
Yuli Huang, Southern Medical University, ChinaReviewed by:
Xuexian Fang, Hangzhou Normal University, China© 2023 Ke, Zhang, Liu, Chen, Li, Sun, Chu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanhui Chu eWFuaHVpX2NodUBzaW5hLmNvbQ== Luxin Li bGlsdXhpbkBtZGptdS5lZHUuY24=
†These authors have contributed equally to this work.
Specialty Section: This article was submitted to Cardiovascular Genetics and Systems Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.