 Lance N. Benson
Lance N. Benson Yunping Guo
Yunping Guo Katherine Deck
Katherine Deck Christoph Mora
Christoph Mora Yunmeng Liu
Yunmeng Liu Shengyu Mu
Shengyu Mu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 09 March 2023
Sec. Hypertension
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1129384
This article is part of the Research Topic Novel Mechanisms and Therapies of Immunity and Inflammation in Hypertension and Target Organ Damage View all 5 articles
Hypertension is the primary cause of cardiovascular disease, which is a leading killer worldwide. Despite the prevalence of this non-communicable disease, still between 90% and 95% of cases are of unknown or multivariate cause (“essential hypertension”). Current therapeutic options focus primarily on lowering blood pressure through decreasing peripheral resistance or reducing fluid volume, but fewer than half of hypertensive patients can reach blood pressure control. Hence, identifying unknown mechanisms causing essential hypertension and designing new treatment accordingly are critically needed for improving public health. In recent years, the immune system has been increasingly implicated in contributing to a plethora of cardiovascular diseases. Many studies have demonstrated the critical role of the immune system in the pathogenesis of hypertension, particularly through pro-inflammatory mechanisms within the kidney and heart, which, eventually, drive a myriad of renal and cardiovascular diseases. However, the precise mechanisms and potential therapeutic targets remain largely unknown. Therefore, identifying which immune players are contributing to local inflammation and characterizing pro-inflammatory molecules and mechanisms involved will provide promising new therapeutic targets that could lower blood pressure and prevent progression from hypertension into renal or cardiac dysfunction.
Classically understood to activate as the “first line” in host defense without developing memory, innate immunity is comprised of dendritic cells, natural killer cells, neutrophils, macrophages, and other granulocytes (1). Although these cells typically function in infection clearance and homeostasis, a myriad of inflammatory responses have been identified when any of these immune cells become dysregulated such as in respiratory diseases (2) and aging (3). In the context of hypertension, monocytes, neutrophils, natural killer cells, and dendritic cells have been identified as present within the kidney and contributing to blood pressure elevation through several inflammatory mechanisms (4, 5). In contrast to innate immunity, adaptive immune cells (T and B lymphocytes) are classically understood to take longer for recruitment and activation through antigen-specific interactions, resulting in formation of memory and enabling more rapid and robust response following re-challenge with the infectious agent. Further sub-categories of T cells have been identified—each fulfilling a unique and critical biological niche, including CD4+, CD8+, and CD4−/CD8− γδ T cells, within which further sub-classes have been identified (6). T cells, in particular, have been demonstrated to play a critical role in the development of hypertension (7), but B cell involvement cannot be ruled out (8). Certain cytokines, proteins that regulate the immune cellular function, have been directly linked to pro-inflammatory immune responses; however, the specific response can vary between target cell, surrounding tissue, timing, and cytokine concentration (9). In hypertension models, many cytokines characterized classically as “pro-inflammatory” have been found to arbitrate at least some of the animal model's blood pressure elevation or organ damage, including IFNγ (10–12), TNFα (13), RANTES (14), IL-1 (15, 16), IL-6 (17), IL-17 (12), IL-18 (18), and, potentially, IL-4 (18–20). IFNγ and IL-17, especially, have been confirmed to be elevated in clinical hypertension (21). We further demonstrated that IFNγ is critical for mediating CD8+ T cell infiltration and interaction within the kidney, driving increased sodium retention (10). Adoptive transfer of primary CD8+ T cells from hypertensive mice is able to induce salt sensitivity hypertension(SSH) in WT mice, however, adoptive transfer of primary CD8+ T cells from IFNγ KO hypertensive mice failed to induce SSHTN in WT mice (22). Collectively these data evidence the critical role of IFNγ in mediating immune-driven hypertension. For a thorough discussion of other cytokines within the kidney contributing to hypertension, we refer the reader to this review by Drs. Wen and Crowley (23).
Cardiovascular disease is the leading global killer, and hypertension is the primary contributing cause to this premature mortality (24). Of note, untreated or unsuccessfully managed hypertension has been demonstrated to result in organ dysfunction and damage—particularly within the heart and kidney (25). Either successful reduction of blood pressure or attenuation of hypertension induced organ dysfunction are key targets to prevent this premature mortality. Reducing blood pressure globally has proven challenging: not only due to resource scarcity -be that diet or medical- in certain areas (24) but also due to the clinical findings that the origin of elevated blood pressure is unknown in 90%–95% of cases (“essential hypertension”) (26) and up to 20%–30% of patient cases are resistant to current treatments (“resistant hypertension”) (27). Although patient compliance to drug regimens complicates treatment of this disease, true diagnosis of “resistant hypertension” requires assurance of adherence to administered therapeutics; as such, other confounding factors are clearly involved (28). Furthermore, the gut microbiota may further contribute to treatment resistance through direct enhancement of drug metabolism thereby reducing therapeutic effect (29, 30).
In light of recent studies, hypertension is presently understood as an inflammatory disease, involving immune cell invasion into organ tissue resulting in a myriad of cytokine expression changes, eventually driving endothelial dysfunction and organ damage (31–34). Through a combination of depletion and adoptive transfer studies, researchers were able to identify cells from both the innate and adaptive immunity that contributed to the development of hypertension (35) and subsequent tissue inflammation, especially the contributive roles of macrophages (36) and T cells (7). Other pre-clinical studies contributed to our current understanding that dendritic cells (37–40) and neutrophils (41, 42), are also participating in the pathogenesis of hypertension. Such pre-clinical studies have been corroborated clinically through the association of pro-inflammatory biomarkers with increased risk of treatment resistant hypertension in patients with chronic kidney disease (43), an independent association between several pro-inflammatory markers and higher blood pressure in healthy patients (44), and more than 50 years of observations of a myriad of immune cells accumulating in the blood vessels and kidneys of hypertensive patients (45). The key regulatory role of the kidney in the cardiovascular system has been well established (46); as such, identifying which immune cells are infiltrating the tissue and elucidating the signaling mechanisms involved are critical in improving treatment of this non-communicable disease. Indeed, recent advances have highlighted several new molecular targets involving the immune system that may serve as new targets for the treatment of resistant hypertension (10, 28, 47), but many pathways have not yet been elucidated. Additionally, unsuccessful management of hypertension has long been associated with progression to left ventricular hypertrophy and diastolic dysfunction within the heart (48–51), and the immune system has been implicated in this progression—particularly cardiac macrophages (52). Based on the critical functions of the kidney and heart in the cardiovascular system, this review will focus on the currently understood link between specific immune cells and hypertension through their interactions with -and within- the kidneys and heart, highlighting “key players” mediating these interactions.
Kidneys and blood pressure control have been intricately linked since Guyton's studies highlighted the importance of kidneys in pressure regulation through natriuresis (53). Defects in kidney function directly correlate to blood pressure dysregulation wherein transplantation of hypertensive kidneys into normotensive animals leads to the development of hypertension within that animal (54–59). Inversely, transplantation of a normotensive kidney into a previously hypertensive animal also can attenuate previously high blood pressure (57, 58). Mechanistically, several factors within the kidneys lead to high blood pressure through inappropriate sodium handling driving greater water retention. The generation of this pathology remains incompletely understood, but several mechanisms leading to this inappropriate handling have been hypothesized ranging from microvascular modeling to the interaction of various immune cells (Figure 1) (35, 59).
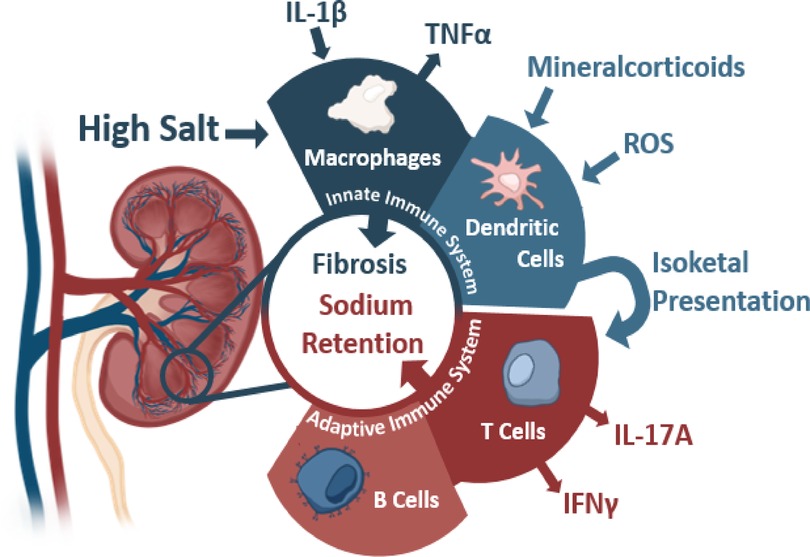
Figure 1. The contributions of the innate and adaptive immunity to kidney damage during hypertension with various activating stimuli and subsequent cytokine production. Sources below: Macrophages: IL-1β (Veiras et al., 2022, Circulation Research, secondary hypertension), TNFα (Singh et al., 2013 American Journal of Physiology: Renal Physiology, essential hypertension). Dendritic Cells: Mineralcorticoids (Araos et al., 2019 Journal of Hypertension, secondary hypertension), ROS (Lu et al., 2020 Hypertension, essential hypertension), and Isoketals (Kirabo et al., 2014, Journal of Hypertension, essential hypertension). T Cells: IL-17 (Kamat et al., 2015, Hypertension, essential hypertension), IFNγ (Benson et al., 2022 Circulation Research and Kamat et al., 2015, Hypertension, essential hypertension). B Cells: (Chan et al., 2015 Hypertension.)
During the onset of hypertension, initial high blood pressure can cause permanent damage to the nephrons through arterial injury and glomerular ischemia, which progresses to nephrosclerosis. Within spontaneously hypertensive rats (SHR), renal vascular resistance increases within the afferent renal arterioles with arterial hypertrophy resulting in glomerular ischemia and reduced glomerular filtration (60, 61). Clinical evidence correlates these findings in several patient studies linking increased arteriole pressure with elevated hyaline deposits and arteriole wall thickness, indicative of renal damage (62, 63). Lower glomerular pressure, sensed via juxtaglomerular feedback, and lower luminal chloride, sensed via macula densa (64), promote increased renin production and activation of the renin-angiotensin-aldosterone system (RAAS). Increased secretion of renin leads through a series of hormones to increased angiotensin II, which acts as a vasoconstrictor and stimulates aldosterone secretion. Aldosterone will act on the distal nephron to increase sodium and water retention thereby increasing blood pressure (65). Damage from renal ischemia due to systemic hypertension can lead to secretion of various inflammatory cytokines, which, in turn, drives infiltration of immune cells and tubular atrophy.
Concurrently, the sympathetic nervous system (SNS) also plays a role in immune cell infiltration. Sensory neurons sense changes in blood pressure leading to the activation of the SNS, and chronic activation of SNS may lead to the development of hypertension as described in these reviews referenced here (65–67). One such experiment by Xiao et al., found that renal denervation blunted immune activation and blood pressure elevation in mice receiving angiotensin II (68), indicating some neural regulation of inflammation in hypertension. SNS activation also leads to increased release of renin, which elevates the blood pressure sensed by the kidney and is hypothesized to stimulate prostaglandin production (69). Catecholamines signal through the β2-adrenergic receptors on innate and adaptive immune cells which are involved in the development of cancer and autoimmune disorder. However, the clear pathway of how Catecholamines regulates immune cells is still unknown (65). Overall, overactivation of the SNS is often seen with hypertension, and its activity can lead to cytokine production and release (e.g., IFNγ and/or TNFα), which is associated with various tissue and organ damage in hypertensive patients resulting in subsequent damage as both the innate and adaptive immune systems accumulate and are key players in kidney damage (7, 35, 68, 70–75). An inverse relationship wherein inflammation influences nervous system function and signaling has been observed in viral infection (76), endometriosis (77), and arthritis (78). Considering the interplay between SNS activity and hypertension, it is possible that renal inflammation may, in turn, dysregulate SNS function in a feedback loop; however, further research is necessary to confirm if this relationship exists in hypertension.
Prostaglandins(PGs), which increase in production with salt loading, indirectly reduce sodium reabsorption and contribute to vasodilation beneficial for hypertensive individuals (65). In addition to their beneficial effects, PGs also increase vascular permeability and their chemotactic activity may attract leukocytes to the locally inflamed zone (79). Moreover, PGE2 has been found to stimulate IFNγ receptor expression on human lymphocytes (80), further implicating IFNγ signaling mechanisms in inflammation and hypertension. PGE2 has been shown to increase blood pressure and disrupt endothelial signaling in EP2/4 (E-prostanoid receptor) deficient mouse models (81–83) or in the presence of enhanced EP3 expression (84, 85), complicating the role of prostaglandins in hypertension.
Depletion of mononuclear phagocytes (MPs, includes macrophages and dendritic cells)) with liposome-encapsulated clodronate reduces fibrosis and glomerular sclerosis typical of salt-sensitive hypertension models (86). Activation of these cells can stem from a variety of effectors from the high salt environment to the activity of the SNS (68, 75, 87). High salt, specifically high sodium rather than high chloride or high osmolarity, triggers a pro-inflammatory state similar to the macrophage M1 state through p38 and ERK (87). Interestingly, this specific activation does not completely match the pro-inflammatory macrophage response seen with LPS stimulation. Macrophages activated by LPS eventually resolve towards an anti-inflammatory state whereas high salt-activated macrophages experienced suppression of this resolution (87). Activated MPS within hypertension leads to ROS production and generation of the cytokines IL-1β and TNFα (88), which is covered in greater detail in this review referenced here (89). Tubular IL-1β, in particular, has been demonstrated to recruit macrophages to the kidney leading to upregulation of ENaC and salt-sensitive hypertension in diabetic mice which are characterized by renal inflammation (16). By knocking out the IL-1R receptor in immune cells or knocking down IL-1β, the authors were able to prevent the switch to pro-inflammatory macrophages thereby preventing macrophage induced salt-sensitivity (16). Additionally, mineralocorticoid receptor may also participate in activated MPs-induced fibrosis during acute kidney injury (AKI), because mineralocorticoid receptor antagonist Finerenone providing protection against this development of renal fibrosis (90); SGK1, a commonly known downstream signaling molecule of ROS and mineralocorticoids, has also been linked to antigen presentation as a salt sensor in CD11c+ cells (expressed on dendritic cells or macrophages) (91) which leads to an increase in Epithelial sodium channel (ENaC) in these cells (92). Upregulation of ENaC in CD11c+ cells, in turn, correlated with increased activation, NADPH oxidase expression, and isolevuglandin-protein adducts (92).
Bridging the gap between innate and adaptive immunity, high salt not only promotes activation of MPs but also leads to the formation of isolevuglandins, also referred to as isoketals (37, 92). Isolevuglandins are a product of arachidonic acid generated as the result of oxidative stress, which is both a product of -and contributor to- hypertension and renal dysfunction (93). Harrison and colleagues identified the accumulation of isolevuglandins during hypertension and the development of these into neoantigens by dendritic cells. Hence, these dendritic cells have the capability of presenting isolevuglandins to T cells leading to their activation, which plays an important role in the changes within the kidney during hypertension (37).
In his landmark study, Guzik et al., demonstrated adoptive transfer of T cells restored the hypertensive response to angiotensin II in RAG1−/– (immunodeficient) mice (7). Many other studies confirmed the critical role of the T cell in the development of hypertension, including alternative animal models (94). De Miguel et al., for example, found that T cells invaded into the kidney of Dahl salt-sensitive rats on high salt diet, and administration of the immunosuppressant tacrolimus blunted T cell invasion within the kidney, reduced renal damage, and lowered blood pressure (95). Subsequent studies were conducted to identify the specific subtypes of contributing T cells, resulting in the identification of a variety of T cells that become active within hypertension including γδ, CD4+, and CD8+ T cells (5). γδ T cells hold the most similarities with the innate immune system and are capable of antigen presentation. Within hypertension, these γδ T cells as well as CD4+ T cells function by aiding in the activation of CD8+ T cells or through cytokine production, including IFNγ and IL-17A (5). In particular, γδ T cell deficient mice showed blunted hypertensive response and endothelial dysfunction to angiotensin II, and increased numbers of these T cells have been found in the spleens of hypertensive mice (96).
CD8+ T cells not only contribute to cytokine production in general inflammatory responses but we also found they interact directly with the nephron to increase sodium retention by stimulating an increase in the sodium chloride cotransporter (NCC) in the DOCA + salt model of hypertension (97). Additionally, splenic CD8Ts from hypertensive mice can generate salt sensitive hypertension in recipients (97). We further found the cytokine IFNγ was critical for this infiltration to take place in the DOCA + Salt model (10) and adoptive transfer models of hypertension (22). Of note, IFNγ from CD8+Ts stimulates the IFNγ receptor expressed within the distal convoluted tubule to promote interaction between the CD8+T and tubule (10). Additionally, Carnevale et al., found CD8Ts not only increased resistance in arteries in a three-dimensional culture model of hypertension but also exhibit enhanced gene pathway upregulation to chemotaxis and response to IFNγ in pre-hypertensive mice (98). The crucial role of IFNγ—along with IL-17—in immune-cell mediated hypertension was previously characterized by Kamat et al., in the angiotensin II model of hypertension (12). Clinically, hypertensive human CD8+T IFNγ production and CD4+T IL-17 has been found to be significantly elevated (21). Youn et al., also found pro-inflammatory immunosenescent CD8+Ts (expressing higher perforin, granzyme B, IFNγ, and TNFα) are increased in peripheral blood of hypertensive patients (99).
Within CD8+ T cells a variety of memory populations are generated upon activation including an effector memory T cell population that can enter and exit from lymphoid tissue to non-lymphoid tissue. Once activated, these T cells remain primed to the stimulus and become easier to activate. In a variety of models, several studies have found a dendritic cell-mediated increase in this effector memory population during hypertension (100–102). Trott et al., confirmed that an oligoclonal population of CD8+Ts can be found in the kidney of angiotensin II-induced hypertensive mice (103); however, the subtypes of CD8+Ts present have not yet been characterized. The development of these populations lead to the potential for continual T cell activation and the exertion of their effects on the kidney. Accordingly, outside of the physical damage due to the high pressure within the kidney, CD8+ T cells may have a long-lasting ability to become activated and lead directly to the inappropriate sodium handling within the nephron that drives water retention.
Regulatory T cells (Tregs)—CD4+ T cells that express CD4, CD25, and FOXP3—are classically understood to reduce inflammation and play an anti-proliferative role in immune responses (104); as such, these cells may play a protective role in the kidney during the development of hypertension. Expansion of T regulatory cells through low-dose IL-2 administration has been shown to lower blood pressure induced via systemic lupus erythematosus in mice; as such, these cells may play a protective role in hypertension (105). Indeed, increased numbers of Tregs in female rats may explain the reduced DOCA + Salt induced hypertension in female rats compared to males, and depletion of Tregs increased female but not male rat blood pressure elevation and renal fibrosis to DOCA (106). Such a protective role may involve an immunosuppression independent mechanism; as such, further research is needed to clarify the protective, antihypertensive role of regulatory T cells in the kidney (107).
In addition to T cells, B cells have been observed to increase within- and invade- the hypertensive kidneys. B cells alone may not lead to the development of hypertension, as adoptive transfer of T cells and not B cells from hypertensive mice into normotensive Rag1−/– mice (which lack B and T cells) resulted in hypertension (7). However, in mice that lack mature B cells through B-cell activating factor receptor deficiency, hypertension was attenuated in the Ang II model (8). B cells play a role in the accumulation of MPS and T cells, which may lead to the kidney pathologies observed during hypertension.
Chronic hypertension has been identified as the leading cause of congestive heart failure (108). The progression of hypertension into cardiac dysfunction, and that of cardiac dysfunction into heart failure, is a multi-faceted progression which often involves morphological and molecular biological changes in the cardiomyocytes, alterations within the surrounding microenvironment, and physical changes to the heart. In addition to playing a role in the perturbation of blood pressure, often through salt-sensitive inappropriate sodium handling (109), decades of research have shown that immune cells are partially culprit in the onset of hypertension, the progression of cardiac dysfunction to heart failure and, possibly, even the progression of hypertension to cardiac dysfunction (41).
Either hypervolemia or increased peripheral resistance increase the pumping force requirements of the heart, especially within the left ventricle (110), to overcome the pressure in the aorta to successfully deliver blood to the rest of the body. This increase in pressure within the ventricle causes excessive stretching, resulting in cardiomyocyte injury and subsequent cardiac hypertrophy—a process mediated by the immune system (111). Furthermore, immune regulation of T cells, macrophages, fibroblasts, and dendritic cells all play a role in the worsening of cardiac function in hypertension and heart failure; as such, immunological targets appear to be promising targets to increase heart health and function in treatment of heart disease (112).
Collagen deposition in cardiac fibrosis plays an important role in the maintenance of the heart. Fibrosis functions in both maintaining homeostasis—especially within the interstitium—and in repairing physical injury that occurs. Fibrotic remodeling is a complex function that involves the secretion and deposition of connective molecules, most importantly Collagens I and III, into the interstitial and perivascular spaces. However, excessive deposition of these connective molecules can impair cardiac function. The increased deposition of molecules within the extracellular matrix (ECM) stiffens the heart chambers, thereby inhibiting the heart from expanding and contracting efficiently, and, ultimately, resulting in increased cardiac stress to maintain homeostatic output. As this fibrotic deposition progresses, cardiac output becomes sufficiently reduced to cause damage to other organs and damage to cardiac cells due to impaired circulation and increased pressure, a pathology so common that 50% of heart failure patients present with this increased cardiac load due to fibrosis (52, 113). Therefore, preventing fibrosis in heart failure remains an attractive target to help preserve cardiac function.
Matrix Metalloproteinases (MMPs) are a family of proteins that help regulate ECM deposition and degradation, many of which are produced by macrophages and other immune cells (114). In particular, MMP-9 has been implicated in a wide range of functions, including the degradation of ECM (115). During cardiac dysfunction, when the fibrotic response is excessive, MMP9 is produced to chaperone the correct degradation and disposition of molecules for proper ECM development and scar formation. However, it has been noted that the expression of MMP9 is reduced in patients with congestive heart failure (116), potentially indicating an impaired ability to degrade excess collagen and further highlighting the importance of extracellular matrix homeostasis.
Macrophages, originating from monocytes found in the circulation, express a variety of markers and serve a great deal of roles in innate immunity as well as mediate adaptive immune response by cross-talking with B and T cells (117). These cells are recruited to injured or infected cells via chemokine signaling (e.g., CCL2,CXCL1, CX3CL1) from a number of other cells including fibroblasts, endothelial cells, and epithelial cells (118). Of note, cardiac-resident macrophages appear to play a more protective role against fibrosis than monocyte-derived macrophages that infiltrate the heart and promote hypertrophy, but further research is needed to clarify the distinction in responses between monocyte derived macrophages and cardiac resident (119). There are traditionally understood to be two main macrophage phenotypes: M1 and M2 (120).
M1 macrophages are considered “traditionally activated” and become active upon exposure to IFN-γ and TLR stimulation (120). These macrophages are considered pro-inflammatory and secrete inflammatory cytokines such as IL-1β, TNF, and IL-12 (121). In a neonatal injured heart model, inhibition of cyclooxygenase-2 resulted in increased M1 macrophage recruitment at the wound site which may contribute to the treatment induced suppressed cardiac hypertrophy and fibrosis (122); however, resolution-phase macrophages can exhibit M1 markers yet express a unique inflammatory phenotype (123), making the role of M1 macrophages in cardiac fibrosis unclear. Depletion of macrophages (both phenotypes) has been shown to reduce cardiac remodeling in the Dahl salt-sensitive rat (124), but did not identify the key contributing phenotype.
In contrast to M1 macrophages, M2 macrophages are anti-inflammatory and pro-fibrotic in function; as such, the M2 phenotype may especially contribute to the fibrogenesis in hypertensive hearts.
M2 macrophages are “alternatively activated”(activated by IL-13, CSF-1, IL-4, IL-10, and TGF-β) (121, 125, 126). These cytokines, commonly produced by Th2 T cells, have been shown to be directly linked to the production of collagens and the onset of fibrosis (126), potentially through their stimulation of M2 macrophages. Furthermore Angiotensin II and cations have been implicated in the transition of fibroblasts to myofibroblasts capable of collagen production and deposition (127). The M2 macrophage, alone, is not capable of depositing the collagen seen in fibrosis; rather, these cells are able to promote the transition of the homeostatic fibroblasts into fibrosis-inducing myofibroblasts (128). Macrophages, once activated at the site of injury, can recruit more macrophages and circulating fibroblasts, leading to increased inflammation and fibrosis (127).
M2 macrophages induce myofibroblast differentiation through a number of mechanisms such as the production of TGF-1β. A recent study by Murray et al., implicated IL-13, TGF-β1, and CCL2 axis in the hyper-stimulation of myofibroblasts in a model of idiopathic pulmonary fibrosis with a usual interstitial pneumonia pathology (IPF/UIP) (129). IL-13, TGF-β1, and CCl2 together appeared to result in a synergistic effect in the activation of myofibroblasts. Though these results have not been explored in the heart specifically, they indicate the M2 macrophage can exacerbate organ fibrosis, which may be relevant in hypertension and heart failure through the same myofibroblastic axis.
TGF-β plays a critical role in the onset and progression of fibrosis through myofibroblast differentiation (130, 131). TGF-β signals through the SMAD pathway inside of the homeostatic fibroblasts and induces the assembly of SMADs 2 and 3 into SMAD 4. SMAD 4 and its R-Smad counterparts then form a complex in the nucleus and modify the gene expression of the fibroblast into a myofibroblast (132). As such, TGF-β and its downstream effects are potential targets for the ablation of fibrosis in hypertension and heart failure. When translated, TGF-β is associated with its negative regulator, the latency associated protein (LAP) (133). TGF-β function can be arbitrated through multiple mechanisms, including through interaction with αVβ6, an inflammation-associated integrin expressed on epithelial cells (134). Munger et al., demonstrated that, though there are multiple methods for TGF-β to be activated, αVβ6 interaction is sufficient to initiate TGF-β signaling (135–138).
IL-10 is an anti-inflammatory cytokine produce by a myriad of cells including macrophages, CD4+ and CD8+ T cells, B cells, other monocytic cells (139). IL-10 targets M2 macrophages and activates them to a collagen-producing, pro-fibrotic phenotype (140). Pro-fibrotic macrophages play a critical role in the pathological progression of hypertension to diastolic dysfunction and heart failure (52). Furthermore, it has been shown that IL-10 deletion in a murine heart failure model attenuates erroneous fibrotic response and reduces mortality (52), suggesting IL-10 signaling cascades may be relevant therapeutic targets to prevent the progression of hypertension into heart failure.
Macrophages, their differentiation, and their signaling molecules present potentially valuable targets for preventing the onset of fibrosis during hypertension and heart failure (outlined in Figure 2). However, the progression of fibrosis in hypertension and heart failure is not yet fully understood; as such, other pro-inflammatory immune cells may be contributing to the pathology. In a pressure overload mouse model of heart failure through transverse aortic constriction, Wang et al., found that depletion of CD11C+ cells blunted left ventricular fibrosis and hypertrophy, implicating dendritic cells in contributing to this pro-inflammatory state (141), but CD11C+ macrophages may be relevant to this model (142). In the DOCA + Salt model of hypertension, depletion of CD11C+ cells attenuated progression into cardiac hypertrophy and fibrosis, further highlighting the role of dendritic cells (143). Natural killer (NK) cells, on the other hand, may play a protective role via attenuating cardiac fibrosis (144). Additional research is needed to clarify the role of other innate immune cells in hypertension induced cardiac fibrosis.
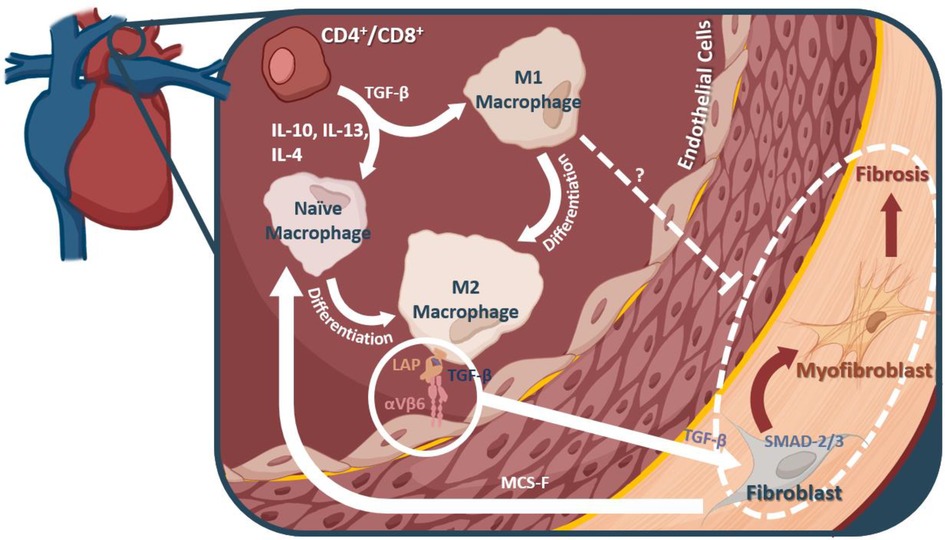
Figure 2. Macrophages and their mechanistic role in fibrosis. M2 macrophages can interact with αVβ6, thereby releasing a TGF-β inhibitor protein, and allowing them to interact with fibroblasts in the heart. TGF-β leads to myofibroblast differentiation from fibroblasts via the SMAD2/3 pathway. It is these myofibroblasts that are thought to account for much of the fibrotic remodeling in many diseases, including diastolic dysfunction and heart failure.
T-helper cells—a subset of CD4+ T cells—are further subdivided into Th1, Th17, Th2, Th3, and Tr1. These cells serve a variety of functions in response to both foreign antigens and injury. Decades of literature, especially recent studies, have identified CD4+T cells in the development and progression of cardiomyopathy (145). Laroumanie et al., recently implicated CD4+T cells in the progression of hypotrophy to heart failure in a myocardial infarction murine model (109). Utilizing multiple mice strains including, RAG2-KO, CD8+-KO, and MHCII-KO, the authors found that CD4+T cells appear to be the primary drivers of cardiac fibrosis after TAC-induced heart failure. Furthermore, adoptive transfer of T cells from CD8+-KO mice (only CD4+T cells transferred) to RAG2-KO (immune incompetent) mice resulted in an increase of fibrosis in the recipients (109).
In the same study, to determine a mechanism by which CD4+T cells increase fibrosis, Lysyl oxidase (LOX), an amine oxidase that influences cross-linking between elastin and collagen, expression was investigated LOX is known to play a role in ECM development and fibrotic disease (146, 147). The researchers found no significant difference between pro-LOX production between WT and RAGII-KO mice; however, they showed that the ratio of mature LOX to pro-LOX increased in WT mice—hinting at some interaction from the CD4+T cells that might induce the maturation of LOX in heart failure thereby driving elastin and collagen cross-linking. While preventing the aberrant increase in LOX maturation may prevent the progression of hypotrophy to heart failure, the production of LOX from pro-LOX is not fully understood and may be dependent on a number of factors, complicating the development of therapies (109).
If pro-inflammatory T cells are contributing to the development of hypertension and subsequent cardiac fibrosis, it would likely follow that regulatory T cells may play a protective effect. Indeed, Wang et al., found that increased Treg presence (induced by IL2/JES6-1 treatment) blunted left ventricular hypertrophy in the transverse aortic constriction model of heart failure (148); as such, Tregs may play a protective role in hypertension induced cardiac fibrosis, but further research is needed to outline the protective mechanisms involved.
The extent of the role of either CD4+ or CD8+ T cells in the progression of hypertension and cardiovascular disease is not yet clear. As previously mentioned, Laroumanie et al., provided evidence pointing toward the role of CD4+ T cells in the progression of heart failure (109); however, in tandem with their CD4+ counterparts, other evidence supports that CD8+ T cells may also play a critical role in cardiovascular disease. In their 2019 study on immune cell activity post myocardial infarction, IIatovskaya et al., examined CD8+ T cell-specific effects by comparing the progression of scar formation after TAC in WT or CD8+-deficient tm1Mak mice. When CD8+ T cells were ablated, TAC mice exhibited abnormal scar formation (149). Additionally, they found a significant increase of mature LOX when CD8+ T cells were present, in contrast to the results found in the study by Laroumanie et al. (109) Regardless of immune cell source, it is evident that LOX production plays some causal role in the development of cardiac-straining scar tissue (109, 149).
Reactive oxygen species (ROS) are a hallmark of many diseases, including cancer, heart disease, and other organ diseases (150). Several potential mechanisms are available for a T cell to induce ROS release or production, such as the rupture of phagocytes that can sequester ROS (151). Excessive ROS production due to T cell activity can negatively affect contractility within the heart, among other dysfunctions. One such mechanism, as depicted in Figure 3, has been identified to involve Myosin II, which relies on myosin phosphatase to be dephosphorylated, leading to the relaxation of the muscle fibers. Myosin phosphatase is activated by protein kinase G, which, previously, had been activated by secondary messenger cGMP. This cGMP is produced by soluble Guanylate Cyclase (sGC) in the heart. sGC is activated by endogenous NO, which is produced by eNOS. eNOS is known to be inhibited by increased levels of intracellular ROS; as such, downstream contractility regulated by eNOS will be negatively impacted by increased ROS (152). By this mechanism, T cell activity in the failing heart might indirectly lead to decreased cardiomyocyte relaxation, which could further exacerbate cardiac stress and the progression to heart failure. Therapies for eNOS inactivity exist in which sGC is stimulated by small molecules to be more reactive to decreased levels of NO (153). Further research is needed to determine whether the ablation of T cells might have a protective role on this function of the cardiomyocytes through reducing inflammatory ROS production (153, 154).
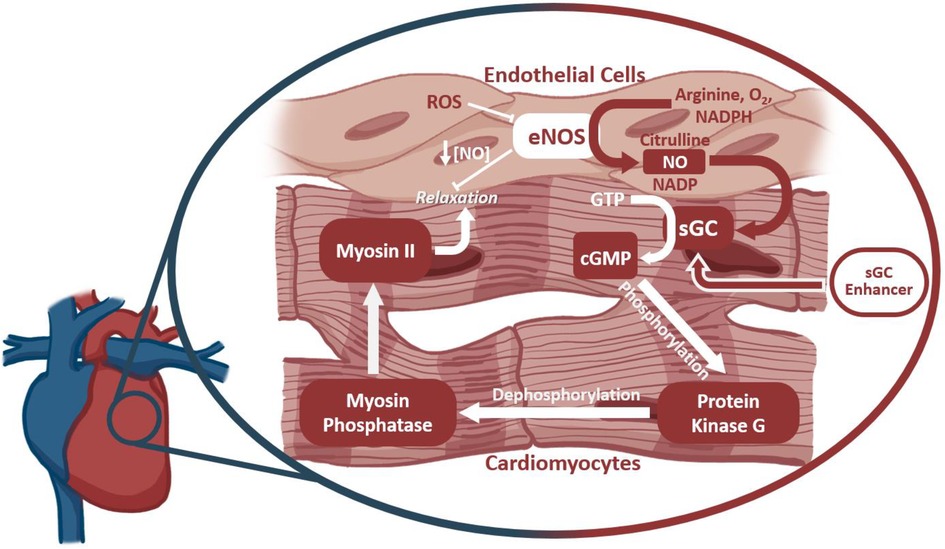
Figure 3. The outcome of ROS production on cardiomyocyte function. Soluble Guanylate Cyclase (sGC) drives Myosin II dephosphorylation and smooth muscle relaxation via production of the secondary messenger cGMP. sGC is activated by intercellular NO produced by endothelial nitric oxide synthase (eNOS). Reactive oxygen species inhibit eNOS activity, thereby affecting cardiomyocyte function further downstream. sGC enhancers target sGC at an allosteric site to increase its sensitivity to NO, thereby rescuing function despite lower NO availability.
Systemic administration of any drug will be expected to result in eventual off-target effects (155); as such, it should be unsurprising to consider that systemic administration of antihypertensives may result in alterations of off-target organ -or cellular- functions. Although such unintended molecule-receptor interactions are typically referred to as toxicities, it is also possible that the desired phenotype (i.e., lowering blood pressure) may be achieved through some of these very same “unintended” effects. To this end, it can become relevant to consider if receptors targeted by antihypertensives are also expressed and functional on other cells—such as immune cells that play a critical role in the development of hypertension (31, 35, 156). An excellent discussion by Felkle et al., regarding the role of ACEi, ARBs, CCBs, β-blockers, and thiazides in immune function modulation can be found referenced here (157). For the purpose of this review, we will provide a brief outline of several receptors and current understanding of expressing immune cell types and potential contributing mechanisms (Figure 4).
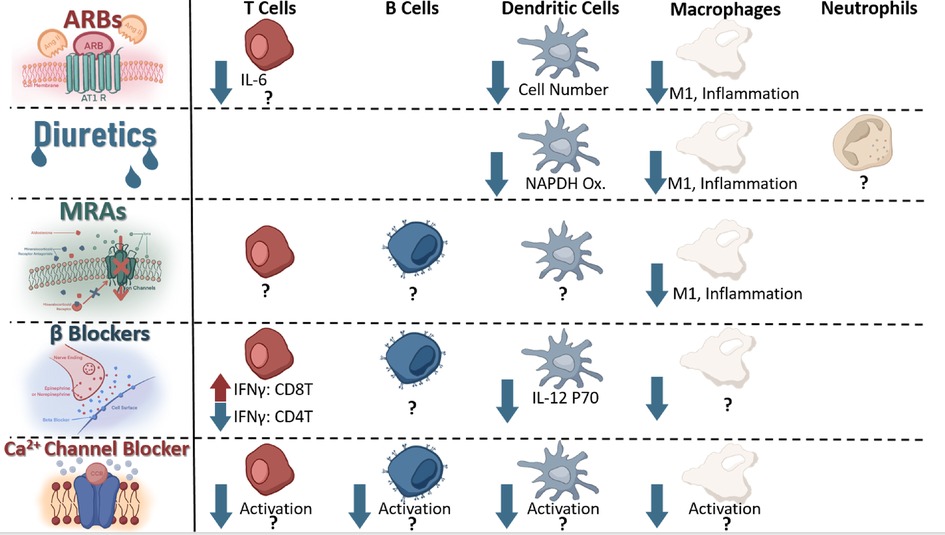
Figure 4. Immune cells known to express receptors antagonized by current antihypertensives and known change in signaling or function as a result of receptor antagonism.
The AT1 receptor, target of ARBs such as Candesartan, was described on splenocytes in 1999 (158), and further experiments have found this receptor on T cells (159), macrophages (160), and dendritic cells (161). Using an [3H] thymidine incorporation assay, Nataraj et al., found that increased Angiotensin II concentration stimulated proliferation of splenocytes, and inhibition of the AT1 receptor using Losartan resulted in reduced AngII binding to splenocytes (158). This link between angiotensin II and splenocyte proliferation indicated a potential for AT1 receptor antagonism to repress immune function. Indeed, Nataraj et al., found that AT1A-receptor deficient mouse graft recipients showed increased allograft survival when coupled with subtherapeutic doses of the calcineurin inhibitor cyclosporine, which showed no significant effect on AT1A receptor proficient mice, indicating a potential AT1-calcinuerin regulation pathway within immune cells (158). Such regulation opens the possibility of ARBs to influence immune function in addition to reducing vasoconstriction. T cells have been further implicated to mediate—at least partially- angiotensin II induced thrombosis through IL-6 by means of an adoptive transfer model into Rag1−/− mice; however, this study did not directly implicate the AT1 receptor on the T cell, indicating a need for further mechanistic confirmation between IL-6 production and AT1 agonism on T cells (17). In the DOCA + salt mouse model of hypertension, the AT1 receptor has been demonstrated to polarize macrophages into the M1 phenotype following agonism with Angiotensin II through the HIF1α/ NF-κβ pathway resulting in vascular dysfunction and injury (162). Likewise, antagonizing AT1R reduced numbers of dendritic cells and reduced chemokine production in an experimental model of autoimmune encephalomyelitis, indicating this receptor can contribute to dendritic cell-mediated inflammation (163). Activated dendritic cell numbers increase in angiotensin-II induced hypertension (164), and Nahmod et al., found that DC differentiation is directly regulated by angiotensin II (165). Taken together, these findings indicate ARBs may elicit some of their antihypertensive effects through direct immune-modulation in addition to blocking the vasoconstrictive effect of angiotensin II. Further studies are needed to clarify some of these alternative mechanisms of action; however, it appears reasonable to anticipate that some of the antihypertensive effects of ARBs may be mediated through non-traditional mechanisms.
The presence of a bumetanide sensitive channel on mouse macrophages (J774.2) was identified as early as 1985 by Bourrit et al. (166) NKCC1, an isotype of NKCC2 (targeted by loop diuretics such as furosemide or bumetanide), has been found on macrophages; indeed, antagonism with bumetanide resulted in blunted macrophage LPS-induced activation in the RAW264.7 mouse macrophage cell line (167). The presence and functionality of a channel on macrophages targetable by bumetanide and resulting in attenuated activation following stimulation suggests a potential for loop diuretics to blunt macrophage involvement in the progression of hypertension.
ENaC, therapeutically targeted by mineralocorticoid receptors (MR) blockers such as spironolactone to reduce expression indirectly or direct blockers such as amiloride, has been suggested to be expressed on dendritic cells (39), macrophages (168), and neutrophils (upregulated in hypertension) (169), playing a crucial role in regulating their involvement in hypertension. On dendritic cells, an amiloride sensitive channel has been demonstrated to regulate NADPH oxidase production, influencing isolevuglandin-protein adduct formation, T cell recruitment, and activation (39). These protein adducts may play a critical role in driving T cell involvement in the pathogenesis of hypertension (37, 170); as such, systemic administration of amiloride may lower blood pressure at least partially by reducing dendritic cell-to-T cell recruitment in hypertension, but further experimentation is necessary for confirmation if such an off-target mechanism is clinically relevant. In macrophages, a similar role for amiloride sensitive channel promotion of the inflammatory response has been characterized as well, indicating that systemic administration of amiloride may reduce macrophage inflammatory responses which have been demonstrated to play a role in the progression of hypertension (36, 86, 89).
The mineralocorticoid receptor has been found to be expressed on cells of T cells, B cells (171), and myeloid lineage (172), contributing to macrophage-induced cardiac hypertrophy in the angiotensin II + L-NAME mouse model of hypertension (172). Deficiency of this receptor on myeloid cells resulted in reduced aortic thickness, vascular damage, and fibrosis despite similar blood pressure elevation to L-NAME + AngII, illustrating the MR receptor on macrophages may be contributing to some of the inflammation observed in the heart during hypertension (172). In like manner, antagonism of the MR receptor with Finerenone or eliminating myeloid mineralocorticoid receptor expression has been shown to reduce the pro-inflammatory macrophage population in mouse and Large White Pig models, protecting against AKI-induced chronic kidney dysfunction (90). This correlation between myeloid MR reduction in function and reduced macrophage inflammation suggests that current mineralocorticoid receptor antagonists may partially reduce blood pressure and tissue inflammation through alteration of macrophage function within the kidneys and heart. If such a non-classically predicted effect of antihypertensives can be verified, it would further highlight the inextricable connection between hypertension and inflammation within the kidneys and heart.
β2-adrenergic receptors, targeted by beta blockers such as propranolol leading to reduced cardiac output and vasodilation (173, 174), have been found on B cells, CD4+ T cells, Th1 cells, CD8+ T cells, and macrophages (1, 175). The role of catecholamines (such as norepinephrine or epinephrine) on these cells due to beta adrenergic stimulation varies based on cell type (1). In CD4+Ts, for example, norepinephrine (NE) may inhibit IFNγ production through the β2 receptor (176); however, human CD8+Ts (particularly memory CD8Ts) responded to NE with increased inflammatory cytokine production (177). T effector function has been modulated successfully through NE-induced reduction in dendritic cell IL12p70 secretion leading to reduced IFNγ and higher IL-17 production in T cells following TCR stimulation by the LPS and NE exposed DCs (178). Although many of the nuances of this regulatory relationship between β2AR signaling and cytokine production are not yet understood, it is clear that such a relationship exists (1) and may influence innate-to-adaptive immunity recruitment in pro-inflammatory situations (178). Further research is needed to identify the effect of β-blockers on influencing the immune interactions contributing to blood pressure elevation.
L-type calcium channels (LTCCs), blocked by calcium channel blockers such as amlodipine resulting in slower depolarization of vascular smooth muscle cells and conduction in the atrioventricular node (173), have been found expressed on macrophages (179), T cells (180), dendritic cells (Cav1.3) (181), and B cells (182). A hallmark signal of activation within immune cells involves an influx of calcium within the cytosol—either from extracellular sources or from a combination of release from intracellular stores and extracellular-to-cytosolic influx (183–187). The presence of these functional calcium channels on immune cells and their potential contribution to calcium influx lends credibility to the possibility that inhibition of LTCCs may influence immune cell proliferation and activation (181, 188). Evidence has been provided clinically by -perhaps somewhat surprisingly- dentistry wherein CCBs result in increased risk of gingivitis in patients, something also associated with immunosuppression (189, 190). This excellent 2013 review by Badou et al., outlines several mechanisms in T cells in particular (180); however, further research is needed to elucidate the precise effects inhibition of LTCCs plays on immune function and if some of the antihypertensive effects of calcium channel blockers are arbitrated through alteration of immune function.
Clinically, elevated immune cell activation/function of CD3+/CD4+ (191), CD3+,CD8+ (99), and monocytes (192) have been described in hypertension. Upregulation of the cytokines IL-17 and IFNγ (21), IL-6 and TNFα (44), and IL-18 (18), has been correlated with clinical manifestation of essential hypertension, among other cytokines (193). To this end, targeting the immune system to reduce cytokine production and immune cell activation and infiltration may reduce blood pressure and subsequent tissue damage. This excellent review by Murray et al., describes the effects of immunosuppressants on blood pressure in animal models (193); however, clinical trials have been complicated through the nephrotoxic (and other tissue) effects of several immunosuppressants -such as calcineurin inhibitors- resulting in blood pressure elevation driven by tissue damage and sodium transporter upregulation (194–196). Our lab has recently identified the critical role tubular PD-L1 plays in mediating CD8+T-distal convoluted tubular interaction and hypertension; by knocking down PD-L1 in mice using renal tubule specific nanoparticles (197, 198) we were able to blunt blood pressure elevation in the DOCA + salt or adoptive transfer models of hypertension, identifying another potential therapeutic target (10). Further studies are needed to address the potential effectiveness of alternative immunosuppressant therapies in reducing blood pressure. Several promising pre-clinical studies have been conducted indicating the feasibility of using FDA-approved nanodrug platforms to deliver tissue specific immunosuppressant therapy (minimizing off-target toxicities), but further validation and studies are required before use in a clinical setting (199). Regardless of current difficulties, targeted immunosuppression to reduce inflammation and immune-mediated organ dysfunction in hypertension continues to be of scientific interest.
The immune system plays a role in the pathogenesis of hypertension through several inflammatory signaling mechanisms involving cells from both the innate and adaptive immune system; however, the signaling molecules and pathways governing these interactions have proven to be complex and not yet fully understood. Within both the heart (109) and kidney (200), current data suggests higher pressure can, alone, drive immune infiltration and subsequent inflammation within the invaded organ leading to dysfunction. Immunodeficient mice (Rag2−/−) exhibited reduced pressure-driven cardiac remodeling (109), and the immunosuppressant tacrolimus reduced T cell infiltration, renal damage, and blood pressure elevation in the Dahl salt-sensitive rat on high salt diet (95). The immune system not only contributes to blood pressure elevation (35) but also mediates organ dysfunction and dysregulation initiated by elevated blood pressure (94). This inextricable connection between immunity, hypertension, inflammation, and organ dysfunction lends high priority to targeting the immune system to lower blood pressure or, at least, reduce inflammation due to hypertension (47). Even current FDA approved anti-hypertensives may mediate some of their beneficial effects through immune modulation, but further studies are needed for confirmation (157). Targeting the immune system to lower blood pressure and reduce organ damage has proven complicated (201, 202), but organ specific immune targeting using nanotechnology appears to be a promising solution to reduce toxicities (155, 199). Recent identification of single nucleotide polymorphisms in SH2B adaptor protein 3 contributing to T cell involvement in hypertension and renal damage (203) further complicates therapeutic targeting as genetic mutations may predispose certain immune cells to promote inflammation, supporting consideration of individual patient genetic predispositions when identifying driving factors of hypertension to design future treatment plans, as is becoming increasingly debated (204). As contributing immune players and inflammation-mediating molecules are characterized, novel pathways can be identified and targeted therapeutically to lower blood pressure and attenuate hypertension-mediated organ dysfunction. Recent studies have highlighted several such immunity-associated relevant receptors or cytokines and confirmed their relevance in several animal models of hypertension.
LNB wrote the original draft and edited the manuscript; KD and CM contributed subsections to this review; YG generated figures; YL edited the manuscript; SM was responsible for the funding acquisition, provided supervision, and reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by NIH/NHLBI [Grant R01-HL146713] (SM), AHA [Grant 15BGIA25730047] (SM), UAMS Medical Research Endowment Awards (SM), AHA [23PRE1023017] (LNB) as well as the Systems Pharmacology and Toxicology Training Program [Grant T32-GM106999] (LNB and KD). The content of this article reflects the personal experience and views of the author(s) and should not be considered medical advice or recommendation. Responsibility for the information and views expressed herein lies entirely with the author(s).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kin NW, Sanders VM. It takes nerve to tell T and B cells what to do. J Leukoc Biol. (2006) 79(6):1093–104. doi: 10.1189/JLB.1105625
2. Eid R, Yan CH, Stevens W, Doherty TA, Borish L, Charlottesville V, et al. Innate immune cell dysregulation drives inflammation and disease in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. (2021) 148(2):309–18. doi: 10.1016/J.JACI.2021.06.016
3. Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. (2013) 13(12):875. doi: 10.1038/NRI3547
4. Lu X, Crowley SD. Actions of immune cells in the hypertensive kidney. Curr Opin Nephrol Hypertens. (2020) 29(5):515. doi: 10.1097/MNH.0000000000000635
5. Caillon A, Paradis P, Schiffrin EL. Role of immune cells in hypertension. Br J Pharmacol. (2019) 176(12):1818–28. doi: 10.1111/BPH.14427
6. Wu Z, Zheng Y, Sheng J, Han Y, Yang Y, Pan H, et al. CD3+CD4-CD8- (double-negative) T cells in inflammation, immune disorders and cancer. Front Immunol. (2022) 13:388. doi: 10.3389/FIMMU.2022.816005/BIBTEX
7. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II-induced hypertension and vascular dysfunction. J Exp Med. (2007) 204(10):2449–60. doi: 10.1084/jem.20070657
8. Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, et al. Obligatory role for B cells in the development of angiotensin II-dependent hypertension. Hypertens (Dallas, Tex 1979). (2015) 66(5):1023–33. doi: 10.1161/HYPERTENSIONAHA.115.05779
9. Jiang P, Zhang Y, Ru B, Yang Y, Vu T, Paul R, et al. Systematic investigation of cytokine signaling activity at the tissue and single-cell level HHS public access. Nat Methods. (2021) 18(10):1181–91. doi: 10.1038/s41592-021-01274-5
10. Benson LN, Liu Y, Wang X, Xiong Y, Rhee SW, Guo Y, et al. The IFNγ-PDL1 pathway enhances CD8T-DCT interaction to promote hypertension. Circ Res. (2022) 130(10):1550–64. doi: 10.1161/CIRCRESAHA.121.320373
11. Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M, et al. Angiotensin II-induced vascular dysfunction depends on interferon-γ-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb Vasc Biol. (2013) 33(6):1313–9. doi: 10.1161/ATVBAHA.113.301437
12. Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, et al. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-γ-/- and interleukin-17A-/- mice. Hypertension. (2015) 65(3):569–76. doi: 10.1161/HYPERTENSIONAHA.114.04975
13. Zhang J, Patel MB, Griffths R, Mao A, Song YS, Karlovich NS, et al. Tumor necrosis factor-α produced in the kidney contributes to angiotensin II-dependent hypertension. Hypertens (Dallas, Tex 1979). (2014) 64(6):1275–81. doi: 10.1161/HYPERTENSIONAHA.114.03863.
14. Mikolajczyk TP, Nosalski R, Szczepaniak P, Budzyn K, Osmenda G, Skiba D, et al. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J. (2016) 30(5):1987–99. doi: 10.1096/fj.201500088R
15. Reza Mirhafez S, Mohebati M, Disfani F, Karimian MS, Ebrahimi M, Avan A, et al. An imbalance in serum concentrations of inflammatory and anti-inflammatory cytokines in hypertension. J Am Soc Hypertens. (2014) 8(9):614–23. doi: 10.1016/j.jash.2014.05.007
16. Veiras LC, Bernstein EA, Cao D, Okwan-Duodu D, Khan Z, Gibb DR, et al. Tubular IL-1β induces salt sensitivity in diabetes by activating renal macrophages. Circ Res. (2022) 131(1):59–73. doi: 10.1161/CIRCRESAHA.121.320239
17. Senchenkova EY, Russell J, Yildirim A, Granger DN, Gavins FNE. Novel role of T cells and IL-6 (interleukin-6) in angiotensin II-induced microvascular dysfunction. Hypertens (Dallas, Tex 1979). (2019) 73(4):829–38. doi: 10.1161/HYPERTENSIONAHA.118.12286
18. Thomas JM, Ling YH, Huuskes B, Jelinic M, Sharma P, Saini N, et al. IL-18 (Interleukin-18) produced by renal tubular epithelial cells promotes renal inflammation and injury during deoxycorticosterone/salt-induced hypertension in mice. Hypertension. (2021) 78:1296–309. doi: 10.1161/HYPERTENSIONAHA.120.16437
19. Van Heuven-Nolsen D, De Kimpe SJ, Muis T, Van Ark I, Savelkoul H, Beems RB, et al. Opposite role of interferon-gamma and interleukin-4 on the regulation of blood pressure in mice. Biochem Biophys Res Commun. (1999) 254(3):816–20. doi: 10.1006/BBRC.1998.8742
20. Rodriguez-Iturbe B, Pons H, Johnson RJ. Role of the immune system in hypertension. Physiol Rev. (2017) 97(3):1127. doi: 10.1152/PHYSREV.00031.2016
21. Itani HA, McMaster WG, Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM, et al. Activation of human T cells in hypertension: studies of humanized mice and hypertensive humans. Hypertension. (2016) 68(1):123–32. doi: 10.1161/HYPERTENSIONAHA.116.07237
22. Benson LN, Liu Y, Deck KS, Mora C, Mu S. Interferon Gamma contributes to the immune mechanisms of hypertension. Kidney360. Published online October 26, 2022:10.34067/KID.0001292022. doi: 10.34067/KID.0001292022
23. Wen Y, Crowley SD. Renal effects of cytokines in hypertension. Adv Exp Med Biol. (2019) 1165:443–54. doi: 10.1007/978-981-13-8871-2_21
24. Mills KT, Stefanescu A, He J. The global epidemiology of hypertension. Nat Rev Nephrol. (2020) 16(4):223–37. doi: 10.1038/s41581-019-0244-2
25. Nadar S, Tayebjee M, Messerli F, Lip G. Target organ damage in hypertension: pathophysiology and implications for drug therapy. Curr Pharm Des. (2006) 12(13):1581–92. doi: 10.2174/138161206776843368
26. Carretero OA, Oparil S. Essential hypertension. Circulation. (2000) 101(3):329–35. doi: 10.1161/01.CIR.101.3.329
27. Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, et al. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation. (2008) 117(25):1403–19. doi: 10.1161/CIRCULATIONAHA.108.189141
28. Carey RM, Calhoun DA, Bakris GL, Brook RD, Daugherty SL, Dennison-Himmelfarb CR, et al. Resistant hypertension: detection, evaluation, and management: a scientific statement from the American heart association. Hypertens (Dallas, Tex 1979). (2018) 72(5):e53. doi: 10.1161/HYP.0000000000000084
29. Kyoung J, Yang T. Depletion of the gut microbiota enhances the blood pressure-lowering effect of captopril: implication of the gut microbiota in resistant hypertension. Hypertens Res. (2022) 45(9):1505–10. doi: 10.1038/S41440-022-00921-4
30. Yang T, Mei X, Tackie-Yarboi E, Akere MT, Kyoung J, Mell B, et al. Identification of a gut commensal that compromises the blood pressure-lowering effect of ester angiotensin-converting enzyme inhibitors. Hypertens (Dallas, Tex 1979). (2022) 79(8):1591–601. doi: 10.1161/HYPERTENSIONAHA.121.18711
31. McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive End-organ damage. Circ Res. (2015) 116(6):1022–33. doi: 10.1161/CIRCRESAHA.116.303697
32. Lu X, Crowley SD. Inflammation in salt-sensitive hypertension and renal damage. Curr Hypertens Rep. (2018) 20(12):103. doi: 10.1007/s11906-018-0903-x
33. Rodríguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ. Oxidative stress, renal infiltration of immune cells, and salt-sensitive hypertension: all for one and one for all. Am J Physiol Renal Physiol. (2004) 286(4):F606–16. doi: 10.1152/AJPRENAL.00269.2003
34. Rodríguez-Iturbe B, Pons H, Quiroz Y, Johnson RJ. The immunological basis of hypertension. Am J Hypertens. (2014) 27(11):1327–37. doi: 10.1093/AJH/HPU142
35. Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of doca and salt hypertension in mice. Acta Pathol Microbiol Scand Sect A Pathol. (2009) 84A(6):523–8. doi: 10.1111/j.1699-0463.1976.tb00150.x
36. Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. (2011) 124(12):1370–81. doi: 10.1161/CIRCULATIONAHA.111.034470
37. Kirabo A, Fontana V, De Faria APC, Loperena R, Galindo CL, Wu J, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. (2014) 124(10):4642–56. doi: 10.1172/JCI74084
38. Pitzer A, Elijovich F, Laffer CL, Ertuglu LA, Sahinoz M, Saleem M, et al. DC ENaC-dependent inflammasome activation contributes to salt-sensitive hypertension. Circ Res. (2022) 131(4):328–44. doi: 10.1161/CIRCRESAHA.122.320818
39. Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. (2017) 21(4):1009–20. doi: 10.1016/J.CELREP.2017.10.002
40. Hevia D, Araos P, Prado C, Luppichini EF, Rojas M, Alzamora R, et al. Myeloid CD11c+antigen-presenting cells ablation prevents hypertension in response to angiotensin II plus high-salt diet. Hypertens (Dallas, Tex 1979). (2018) 71(4):709–18. doi: 10.1161/HYPERTENSIONAHA.117.10145
41. Rai A, Narisawa M, Li P, Piao L, Li Y, Yang G, et al. Adaptive immune disorders in hypertension and heart failure: focusing on T-cell subset activation and clinical implications. J Hypertens. (2020) 38(10):1878–89. doi: 10.1097/HJH.0000000000002456
42. Araos P, Figueroa S, Amador CA. The role of neutrophils in hypertension. Int J Mol Sci. (2020) 21(22):1–16. doi: 10.3390/IJMS21228536
43. Chen J, Bundy JD, Hamm LL, Hsu CY, Lash J, Miller ER, et al. Inflammation and apparent treatment-resistant hypertension in patients with chronic kidney disease. Hypertens (Dallas, Tex 1979). (2019) 73(4):785–93. doi: 10.1161/HYPERTENSIONAHA.118.12358
44. Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. (2005) 19(2):149–54. doi: 10.1038/SJ.JHH.1001785
45. Harrison DG, Coffman TM, Wilcox CS. Pathophysiology of Hypertension. Circ Res. 2021;128:847–63. doi: 10.1161/CIRCRESAHA.121.318082
46. Crowley SD, Coffman TM. The inextricable role of the kidney in hypertension. J Clin Invest. (2014) 124(6):2341. doi: 10.1172/JCI72274
47. Bomfim GF, Cau SBA, Bruno AS, Fedoce AG, Carneiro FS. Hypertension: a new treatment for an old disease? Targeting the immune system. Br J Pharmacol. (2019) 176(12):2028–48. doi: 10.1111/BPH.14436
48. Oh GC, Cho HJ. Blood pressure and heart failure. Clin Hypertens. (2020) 26(1):1. doi: 10.1186/s40885-019-0132-x
49. Levy D. The progression from hypertension to congestive heart failure. JAMA J Am Med Assoc. (1996) 275(20):1557. doi: 10.1001/jama.1996.03530440037034
50. Shehata IE, Eldamanhory AS, Shaker A. Early predictors of left ventricular dysfunction in hypertensive patients: comparative cross-section study. Int J Cardiovasc Imaging. (2020) 36(6):1031–40. doi: 10.1007/s10554-020-01790-z
51. Aziz. Diastolic heart failure: a concise review. J Clin Med Res. (2013) 5(5):327. doi: 10.4021/jocmr1532w
52. Hulsmans M, Sager HB, Roh JD, Valero-Muñoz M, Houstis NE, Iwamoto Y, et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med. (2018) 215(2):423–40. doi: 10.1084/JEM.20171274
53. Guyton AC, Coleman TG, Cowley AW, Scheel KW, Manning RD, Norman RA. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med. (1972) 52(5):584–94. doi: 10.1016/0002-9343(72)90050-2
54. Morgan DA, DiBona GF, Mark AL. Effects of interstrain renal transplantation on NaCl-induced hypertension in Dahl rats. Hypertens (Dallas, Tex 1979). (1990) 15(4):436–42. doi: 10.1161/01.HYP.15.4.436
55. Li XB, Wang Z, Liu BC, Zhu YC, Yao T. Changes in blood pressure in normal rats transplanted with kidney of SHR. Sheng Li Xue Bao. (1999) 51(6):630–6. PMID: 11498932
56. Grisk O, Rettig R. Renal transplantation studies in genetic hypertension. News Physiol Sci. (2001) 16(6):262–5. doi: 10.1152/PHYSIOLOGYONLINE.2001.16.6.262
57. Churchill PC, Churchill MC, Bidani AK. Kidney cross transplants in Dahl salt-sensitive and salt-resistant rats. Am J Physiol. (1992) 262(6 Pt 2):H1809–17. doi: 10.1152/AJPHEART.1992.262.6.H1809
58. Itoh H, Kurihara I, Miyashita K. Organ memory: a key principle for understanding the pathophysiology of hypertension and other non-communicable diseases. Hypertens Res. (2018) 41(10):771–9. doi: 10.1038/S41440-018-0081-X
59. Oguchi H, Sasamura H, Shinoda K, Morita S, Kono H, Nakagawa K, et al. Renal arteriolar injury by salt intake contributes to salt memory for the development of hypertension. Hypertens (Dallas, Tex 1979). (2014) 64(4):784–91. doi: 10.1161/HYPERTENSIONAHA.113.02973
60. Gattone VH, Evan AP, Willis LR, Luft FC. Renal afferent arteriole in the spontaneously hypertensive rat. Hypertens (Dallas, Tex 1979). (1983) 5(1):8–16. doi: 10.1161/01.HYP.5.1.8
61. Skov K, Mulvany MJ. Structure of renal afferent arterioles in the pathogenesis of hypertension. Acta Physiol Scand. (2004) 181(4):397–405. doi: 10.1111/J.1365-201X.2004.01311.X
62. Miyaoka Y, Okada T, Tomiyama H, Morikawa A, Rinno S, Kato M, et al. Structural changes in renal arterioles are closely associated with central hemodynamic parameters in patients with renal disease. Hypertens Res. (2021) 44(9):1113–21. doi: 10.1038/S41440-021-00656-8
63. Hill GS, Heudes D, Jacquot C, Gauthier É, Bariéty J. Morphometric evidence for impairment of renal autoregulation in advanced essential hypertension. Kidney Int. (2006) 69(5):823–31. doi: 10.1038/SJ.KI.5000163
64. Lorenz JN, Weihprecht H, Schnermann J, Skott O, Briggs JP. Renin release from isolated juxtaglomerular apparatus depends on macula densa chloride transport. Am J Physiol. (1991) 260(4 Pt 2):F486–93. doi: 10.1152/AJPRENAL.1991.260.4.F486
65. Wadei HM, Textor SC. The role of the kidney in regulating arterial blood pressure. Nat Rev Nephrol. (2012) 8(10):602–9. doi: 10.1038/NRNEPH.2012.191
66. Ivy JR, Bailey MA. Pressure natriuresis and the renal control of arterial blood pressure. J Physiol. (2014) 592(18):3955–67. doi: 10.1113/JPHYSIOL.2014.271676
67. Campese VM, Mitra N, Sandee D. Hypertension in renal parenchymal disease: why is it so resistant to treatment? Kidney Int. (2006) 69(6):967–73. doi: 10.1038/SJ.KI.5000177
68. Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, et al. Renal denervation prevents immune cell activation and renal inflammation in angiotensin II-induced hypertension. Circ Res. (2015) 117(6):547–57. doi: 10.1161/CIRCRESAHA.115.306010
69. Sata Y, Head GA, Denton K, May CN, Schlaich MP. Role of the sympathetic nervous system and its modulation in renal hypertension. Front Med. (2018) 5:82. doi: 10.3389/FMED.2018.00082
70. Bucsek MJ, Giridharan T, MacDonald CR, Hylander BL, Repasky EA. An overview of the role of sympathetic regulation of immune responses in infectious disease and autoimmunity. Int J Hyperthermia. (2018) 34(2):135–43. doi: 10.1080/02656736.2017.1411621
71. Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. (2010) 298(4):R1089–97. doi: 10.1152/AJPREGU.00373.2009
72. Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol. (2013) 304(6):R407–14. doi: 10.1152/AJPREGU.00304.2012
73. Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL. CD247 Modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertens (Dallas, Tex 1979). (2014) 63(3):559–64. doi: 10.1161/HYPERTENSIONAHA.113.02191
74. Kim J, Padanilam BJ. Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J Am Soc Nephrol. (2013) 24(2):229–42. doi: 10.1681/ASN.2012070678
75. Zaldivia MTK, Rivera J, Hering D, Marusic P, Sata Y, Lim B, et al. Renal denervation reduces monocyte activation and monocyte-platelet Aggregate formation: an anti-inflammatory effect relevant for cardiovascular risk. Hypertens (Dallas, Tex 1979). (2017) 69(2):323–31. doi: 10.1161/HYPERTENSIONAHA.116.08373
76. Garber C, Vasek MJ, Vollmer LL, Sun T, Jiang X, Klein RS. Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via IL-1. Nat Immunol. (2018) 19(2):151–61. doi: 10.1038/S41590-017-0021-Y
77. Arnold J, Barcena de Arellano ML, Rüster C, Vercellino GF, Chiantera V, Schneider A, et al. Imbalance between sympathetic and sensory innervation in peritoneal endometriosis. Brain Behav Immun. (2012) 26(1):132–41. doi: 10.1016/J.BBI.2011.08.004
78. Schrepf A, Kaplan CM, Ichesco E, Larkin T, Harte SE, Harris RE, et al. A multi-modal MRI study of the central response to inflammation in rheumatoid arthritis. Nat Commun. (. 2018) 9(1):1–11. doi: 10.1038/s41467-018-04648-0
79. Johnston MG, Hay JB, Movat HZ. The modulation of enhanced vascular permeability by prostaglandins through alterations in blood flow (hyperemia). Agents Actions. (1976) 6(6):705–11. doi: 10.1007/BF02026092
80. ElMasry MN, Rich RR. Prostaglandin E2 selectively increases interferon gamma receptor expression on human CD8+ lymphocytes. J Clin Invest. (1989) 83(4):1436–40. doi: 10.1172/JCI114035
81. Kennedy CRJ, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, et al. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med. (1999) 5(2):217–20. doi: 10.1038/5583
82. Zhang Y, Guan YF, Schneider A, Brandon S, Breyer RM, Breyer MD. Characterization of murine vasopressor and vasodepressor prostaglandin E(2) receptors. Hypertens (Dallas, Tex 1979). (2000) 35(5):1129–34. doi: 10.1161/01.HYP.35.5.1129
83. Xu H, Fang B, Du S, Wang S, Li Q, Jia X, et al. Endothelial cell prostaglandin E2 receptor EP4 is essential for blood pressure homeostasis. JCI Insight. (2020) 5(13):e138505. doi: 10.1172/JCI.INSIGHT.138505
84. Xiao L, Itani HA, Do Carmo LS, Carver LS, Breyer RM, Harrison DG. Central EP3 (E prostanoid 3) receptors mediate salt-sensitive hypertension and immune activation. Hypertens (Dallas, Tex 1979). (2019) 74(6):1507–15. doi: 10.1161/HYPERTENSIONAHA.119.13850
85. Yang T, Song C, Ralph DL, Andrews P, Sparks MA, Koller BH, et al. Cell-specific actions of the prostaglandin E-prostanoid receptor 4 attenuating hypertension: a dominant role for kidney epithelial cells compared with macrophages. J Am Heart Assoc. (2022) 11(19):e026581. doi: 10.1161/JAHA.122.026581
86. Huang L, Wang A, Hao Y, Li W, Liu C, Yang Z, et al. Macrophage depletion lowered blood pressure and attenuated hypertensive renal injury and fibrosis. Front Physiol. (2018) 9:473. doi: 10.3389/fphys.2018.00473
87. Zhang WC, Zheng XJ, Du LJ, Sun JY, Shen ZX, Shi C, et al. High salt primes a specific activation state of macrophages, M(Na). Cell Res. (2015) 25(8):893–910. doi: 10.1038/cr.2015.87
88. Singh P, Bahrami L, Castillo A, Majid DSA. TNF-α type 2 receptor mediates renal inflammatory response to chronic angiotensin II administration with high salt intake in mice. Am J Physiol Renal Physiol. (2013) 304(7):F991–9. doi: 10.1152/AJPRENAL.00525.2012
89. Justin Rucker A, Crowley SD. The role of macrophages in hypertension and its complications. Pflugers Arch. (2017) 469(3–4):419–30. doi: 10.1007/S00424-017-1950-X
90. Barrera-Chimal J, Estrela GR, Lechner SM, Giraud S, El Moghrabi S, Kaaki S, et al. The myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int. (2018) 93(6):1344–55. doi: 10.1016/J.KINT.2017.12.016
91. Wu H, Perrard XD, Wang Q, Perrard JL, Polsani VR, Jones PH, et al. CD11c expression in adipose tissue and blood and its role in diet-induced obesity. Arterioscler Thromb Vasc Biol. (2010) 30(2):186–92. doi: 10.1161/ATVBAHA.109.198044
92. Van Beusecum JP, Barbaro NR, McDowell Z, Aden LA, Xiao L, Pandey AK, et al. High salt activates CD11c+antigen-presenting cells via SGK (Serum glucocorticoid kinase) 1 to promote renal inflammation and salt-sensitive hypertension. Hypertens (Dallas, Tex 1979). (2019) 74(3):555–63. doi: 10.1161/HYPERTENSIONAHA.119.12761
93. Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension. (2017) 70(4):660–7. doi: 10.1161/HYPERTENSIONAHA.117.07802
94. Madhur MS, Kirabo A, Guzik TJ, Harrison DG. From rags to riches: moving beyond Rag1 in studies of hypertension. Hypertension. (2020) 75(4):930–4. doi: 10.1161/HYPERTENSIONAHA.119.14612
95. de Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol - Ren Physiol. (2011) 300(3):F734–42. doi: 10.1152/ajprenal.00454.2010
96. Caillon A, Mian MOR, Fraulob-Aquino JC, Huo KG, Barhoumi T, Ouerd S, et al. γδ T cells mediate angiotensin II-induced hypertension and vascular injury. Circulation. (2017) 135(22):2155–62. doi: 10.1161/CIRCULATIONAHA.116.027058
97. Liu Y, Rafferty TM, Rhee SW, Webber JS, Song L, Ko B, et al. CD8+ T cells stimulate Na-Cl co-transporter NCC in distal convoluted tubules leading to salt-sensitive hypertension. Nat Commun. (2017) 8:14037. doi: 10.1038/ncomms14037
98. Carnevale D, Carnevale L, Perrotta S, Pallante F, Migliaccio A, Iodice D, et al. Chronic 3D vascular-immune interface established by coculturing pressurized resistance arteries and immune cells. Hypertens (Dallas, Tex 1979). (2021) 78(5):1648. doi: 10.1161/HYPERTENSIONAHA.121.17447
99. Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, et al. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension. (2013) 62(1):126–33. doi: 10.1161/HYPERTENSIONAHA.113.00689
100. Lu X, Rudemiller NP, Privratsky JR, Ren J, Wen Y, Griffiths R, et al. Classical dendritic cells mediate hypertension by promoting renal oxidative stress and fluid retention. Hypertens (Dallas, Tex 1979). (2020) 75(1):131–8. doi: 10.1161/HYPERTENSIONAHA.119.13667
101. Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, et al. CD70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. (2016) 118(8):1233–43. doi: 10.1161/CIRCRESAHA.115.308111
102. Lu X, Rudemiller NP, Wen Y, Ren J, Hammer GE, Griffiths R, et al. A20 in myeloid cells protects against hypertension by inhibiting dendritic cell-mediated T-cell activation. Circ Res. (2019) 125(12):1055–66. doi: 10.1161/CIRCRESAHA.119.315343
103. Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, et al. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension. (2014) 64(5):1108–15. doi: 10.1161/HYPERTENSIONAHA.114.04147
104. Kondělková K, Vokurková D, Krejsek J, Borská L, Fiala Z, Ctirad A. Regulatory T cells (TREG) and their roles in immune system with respect to immunopathological disorders. Acta medica (Hradec Kral. (2010) 53(2):73–7. doi: 10.14712/18059694.2016.63
105. Taylor EB, Sasser JM, Maeda KJ, Ryan MJ. Expansion of regulatory T cells using low-dose interleukin-2 attenuates hypertension in an experimental model of systemic lupus erythematosus. Am J Physiol Renal Physiol. (2019) 317(5):F1274–84. doi: 10.1152/AJPRENAL.00616.2018
106. Belanger KM, Crislip GR, Gillis EE, Abdelbary M, Musall JB, Mohamed R, et al. Greater T regulatory cells in females attenuate DOCA-salt induced increases in blood pressure vs. Males. Hypertens (Dallas, Tex 1979). (2020) 75(6):1615. doi: 10.1161/HYPERTENSIONAHA.119.14089
107. Fabbiano S, Menacho-Márquez M, Robles-Valero J, Pericacho M, Matesanz-Marín A, García-Macías C, et al. Immunosuppression-independent role of regulatory T cells against hypertension-driven renal dysfunctions. Mol Cell Biol. (2015) 35(20):3528–46. doi: 10.1128/MCB.00518-15/ASSET/68F2FCC0-ACCF-4DBE-B738-5ED2E13FC455/ASSETS/GRAPHIC/ZMB9991009790009.JPEG
108. Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics-2022 update: a report from the American heart association. Circulation. (2022) 145(8):E153–639. doi: 10.1161/CIR.0000000000001052
109. Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. (2014) 129(21):2111–24. doi: 10.1161/CIRCULATIONAHA.113.007101
110. Cuspidi C, Sala C, Negri F, Mancia G, Morganti A. Prevalence of left-ventricular hypertrophy in hypertension: an updated review of echocardiographic studies. J Hum Hypertens. (2012) 26(6):343–9. doi: 10.1038/jhh.2011.104
111. Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. (2004) 109(13):1580–9. doi: 10.1161/01.CIR.0000120390.68287.BB
112. Rurik JG, Aghajanian H, Epstein JA. Immune cells and immunotherapy for cardiac injury and repair. Circ Res. (2021) 128(11):1766–79. doi: 10.1161/CIRCRESAHA.121.318005
113. Sharma K, Kass DA. Heart failure with preserved ejection fraction. Circ Res. (2014) 115(1):79–96. doi: 10.1161/CIRCRESAHA.115.302922
114. Lepidi S, Kenagy RD, Raines EW, Chiu ES, Chait A, Ross R, et al. MMP9 Production by human monocyte-derived macrophages is decreased on polymerized type I collagen. J Vasc Surg. (2001) 34(6):1111–8. doi: 10.1067/mva.2001.119401
115. Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase-9: many shades of function in cardiovascular disease. Physiology. (2013) 28(6):391–403. doi: 10.1152/PHYSIOL.00029.2013/ASSET/IMAGES/LARGE/PHY0061301900006.JPEG
116. Batlle M, Pérez-Villa F, García-Pras E, Lázaro A, Orús J, Roqué M, et al. Down-regulation of matrix metalloproteinase-9 (MMP-9) expression in the myocardium of congestive heart failure patients. Transplant Proc. (2007) 39(7):2344–6. doi: 10.1016/J.TRANSPROCEED.2007.06.046
117. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164(12):6166–73. doi: 10.4049/JIMMUNOL.164.12.6166
118. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. (2014) 5:514. doi: 10.3389/FIMMU.2014.00514/BIBTEX
119. Revelo XS, Parthiban P, Chen C, Barrow F, Fredrickson G, Wang H, et al. Cardiac resident macrophages prevent fibrosis and stimulate angiogenesis. Circ Res. (2021) 129(12):1086–101. doi: 10.1161/CIRCRESAHA.121.319737
120. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
121. Chávez-Galán L, Olleros ML, Vesin D, Garcia I. Much more than M1 and M2 macrophages, there are also CD169+ and TCR+ macrophages. Front Immunol. (2015) 6:263. doi: 10.3389/FIMMU.2015.00263/BIBTEX
122. Zhao Y, Zheng Q, Gao H, Cao M, Wang H, Chang R, et al. Celecoxib alleviates pathological cardiac hypertrophy and fibrosis via M1-like macrophage infiltration in neonatal mice. iScience. (2021) 24(3):102233. doi: 10.1016/J.ISCI.2021.102233
123. Bystrom J, Evans I, Newson J, Stables M, Toor I, Van Rooijen N, et al. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood. (2008) 112(10):4117–27. doi: 10.1182/BLOOD-2007-12-129767
124. Liu YY, Luo J, Cai R, Zhang J, Xu Q, Tian Y, Zhou MS. Macrophage depletion improves endothelial insulin resistance and protects against cardiovascular injury in salt-sensitive hypertension. Biomed Res Int. (2020) 2020:5073762. doi: 10.1155/2020/5073762
125. Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med. (2013) 210(11):2477–91. doi: 10.1084/JEM.20121999
126. Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2–dominated inflammatory response. J Clin Invest. (1999) 104(6):777–85. doi: 10.1172/JCI7325
127. Stempien-Otero A, Kim DH, Davis J. Molecular networks underlying myofibroblast fate and fibrosis. J Mol Cell Cardiol. (2016) 97:153–61. doi: 10.1016/J.YJMCC.2016.05.002
128. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. (2010) 30(3):245–57. doi: 10.1055/S-0030-1255354
129. Murray LA, Argentieri RL, Farrell FX, Bracht M, Sheng H, Whitaker B, et al. Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGFβ1, IL-13 and CCL2. Int J Biochem Cell Biol. (2008) 40(10):2174–82. doi: 10.1016/J.BIOCEL.2008.02.016
130. Frangogiannis NG. Transforming growth factor–β in tissue fibrosis. J Exp Med. (2020) 217(3):e20190103. doi: 10.1084/JEM.20190103
131. Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO, et al. Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J Biol Chem. (2013) 288(21):14824. doi: 10.1074/JBC.M113.451336
132. Blank U, Karlsson S. The role of Smad signaling in hematopoiesis and translational hematology. Leukemia. (2011) 25(9):1379–88. doi: 10.1038/leu.2011.95
133. Ali NA, Gaughan AA, Orosz CG, Baran CP, McMaken S, Wang Y, et al. Latency associated peptide has in vitro and in vivo immune effects independent of TGF-beta1. PLoS One. (2008) 3(4):e1914. doi: 10.1371/JOURNAL.PONE.0001914
134. Hahm K, Lukashev ME, Luo Y, Yang WJ, Dolinski BM, Weinreb PH, et al. αvβ6 integrin regulates renal fibrosis and inflammation in alport mouse. Am J Pathol. (2007) 170(1):110. doi: 10.2353/AJPATH.2007.060158
135. Munger JS, Huang X, Kawakatsu H, Griffiths MJD, Dalton SL, Wu J, et al. The integrin αvβ6 binds and activates latent TGFβ1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. (1999) 96(3):319–28. doi: 10.1016/S0092-8674(00)80545-0
136. Horan GS, Wood S, Ona V, Dan JL, Lukashev ME, Weinreb PH, et al. Partial inhibition of integrin αvβ6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. (2012) 177(1):56–65. doi: 10.1164/RCCM.200706-805OC.
137. Chen C, Li R, Ross RS, Manso AM. Integrins and integrin-related proteins in cardiac fibrosis. J Mol Cell Cardiol. (2016) 93:162–74. doi: 10.1016/J.YJMCC.2015.11.010
138. Schinner C, Xu L, Franz H, Zimmermann A, Wanuske MT, Rathod M, et al. Defective desmosomal adhesion causes arrhythmogenic cardiomyopathy by involving an integrin-αVβ6/TGF-β signaling cascade. Circulation. (2022) 146:1610–26. doi: 10.1161/circulationaha.121.057329
139. Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol. (2012) 32(1):23–63. doi: 10.1615/CRITREVIMMUNOL.V32.I1.30
140. Sun ZL, Feng Y, Zou ML, Zhao BH, Liu SY, Du Y, et al. Emerging role of IL-10 in hypertrophic scars. Front Med. (2020) 7:438. doi: 10.3389/FMED.2020.00438/BIBTEX
141. Wang H, Kwak D, Fassett J, Liu X, Yao W, Weng X, et al. Role of bone marrow-derived CD11c+ dendritic cells in systolic overload-induced left ventricular inflammation, fibrosis and hypertrophy. Basic Res Cardiol. (2017) 112(3):25. doi: 10.1007/S00395-017-0615-4
142. Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity. (2015) 42(6):1197–211. doi: 10.1016/J.IMMUNI.2015.05.018
143. Araos P, Prado C, Lozano M, Figueroa S, Espinoza A, Berger T, et al. Dendritic cells are crucial for cardiovascular remodeling and modulate neutrophil gelatinase-associated lipocalin expression upon mineralocorticoid receptor activation. J Hypertens. (2019) 37(7):1482–92. doi: 10.1097/HJH.0000000000002067
144. Ong S, Ligons DL, Barin JG, Wu L, Talor MV, Diny N, et al. Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am J Pathol. (2015) 185(3):847. doi: 10.1016/J.AJPATH.2014.11.023
145. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, et al. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation. (2019) 139(2):206–21. doi: 10.1161/CIRCULATIONAHA.118.036065
146. Smith-Mungo LI, Kagan HM. Lysyl oxidase: properties, regulation and multiple functions in biology. Matrix Biol. (1998) 16(7):387–98. doi: 10.1016/S0945-053X(98)90012-9
147. Nguyen XX, Nishimoto T, Takihara T, Mlakar L, Bradshaw AD, Feghali-Bostwick C. Lysyl oxidase directly contributes to extracellular matrix production and fibrosis in systemic sclerosis. Am J Physiol - Lung Cell Mol Physiol. (2021) 320(1):L29–L40. doi: 10.1152/AJPLUNG.00173.2020/ASSET/IMAGES/LARGE/AJ-ALUN200037F007.JPEG
148. Wang H, Hou L, Kwak D, Fassett J, Xu X, Chen A, et al. Increasing regulatory T cells with interleukin-2 and interleukin-2 antibody complexes attenuates lung inflammation and heart failure progression. Hypertens (Dallas, Tex 1979). (2016) 68(1):114–22. doi: 10.1161/HYPERTENSIONAHA.116.07084
149. Ilatovskaya DV, Pitts C, Clayton J, Domondon M, Troncoso M, Pippin S, et al. CD8+ T-cells negatively regulate inflammation post-myocardial infarction. Am J Physiol - Hear Circ Physiol. (2019) 317(3):H581–96. doi: 10.1152/AJPHEART.00112.2019/ASSET/IMAGES/LARGE/ZH40091928960008.JPEG
150. Yang S, Lian G. ROS And diseases: role in metabolism and energy supply. Mol Cell Biochem. (2020) 467(1–2):1–12. doi: 10.1007/S11010-019-03667-9/TABLES/1
151. Belikov AV, Schraven B, Simeoni L. T cells and reactive oxygen species. J Biomed Sci. (2015) 22(1):1–11. doi: 10.1186/S12929-015-0194-3/FIGURES/3
152. Black SM, Ross PA, Levic S, Hallmark OG. Reactive oxygen Species inhibit endothelial NO synthase by multiple mechanisms. Pediatr Res. (1999) 45(7):296–6. doi: 10.1203/00006450-199904020-01759
153. Kang C, Lamb YN. Vericiguat: a review in chronic heart failure with reduced ejection fraction. Am J Cardiovasc Drugs. (2022) 22(4):451–9. doi: 10.1007/s40256-022-00538-5
154. Vericiguat - PubMed. https://pubmed.ncbi.nlm.nih.gov/34662042/ (Accessed December 4, 2022).
155. Zhao Z, Ukidve A, Kim J, Mitragotri S. Targeting strategies for tissue-specific drug delivery. Cell. (2020) 181(1):151–67. doi: 10.1016/J.CELL.2020.02.001
156. Harrison DG, Vinh A, Lob H, Madhur MS. Role of the adaptive immune system in hypertension. Curr Opin Pharmacol. (2010) 10(2):203–7. doi: 10.1016/j.coph.2010.01.006
157. Felkle D, Jarczyński M, Kaleta K, Zięba K, Nazimek K. The immunomodulatory effects of antihypertensive therapy: a review. Biomed Pharmacother. (2022) 153:113287. doi: 10.1016/J.BIOPHA.2022.113287
158. Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS, et al. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest. (1999) 104(12):1693–701. doi: 10.1172/JCI7451
159. Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, et al. A novel role for type 1 angiotensin receptors on T lymphocytes to limit target organ damage in hypertension. Circ Res. (2012) 110(12):1604–17. doi: 10.1161/CIRCRESAHA.111.261768
160. Zhang JD, Patel MB, Griffiths R, Dolber PC, Ruiz P, Sparks MA, et al. Type 1 angiotensin receptors on macrophages ameliorate IL-1 receptor–mediated kidney fibrosis. J Clin Invest. (2014) 124(5):2198. doi: 10.1172/JCI61368
161. Lu X, Zhang J, Wen Y, Ren J, Griffiths R, Rudemiller NP, et al. Type 1 angiotensin receptors on CD11c-expressing cells protect against hypertension by regulating dendritic cell-mediated T cell activation. Hypertens (Dallas, Tex 1979). (2022) 79(6):1227–36. doi: 10.1161/HYPERTENSIONAHA.121.18734
162. Yang XF, Wang H, Huang Y, Huang JH, Ren HL, Xu Q, et al. Myeloid angiotensin II type 1 receptor mediates macrophage polarization and promotes vascular injury in DOCA/salt hypertensive mice. Front Pharmacol. (2022) 13:879693. doi: 10.3389/FPHAR.2022.879693
163. Stegbauer J, Lee DH, Seubert S, Ellrichmann G, Manzel A, Kvakan H, et al. Role of the renin-angiotensin system in autoimmune inflammation of the central nervous system. Proc Natl Acad Sci USA. (2009) 106(35):14942–7. doi: 10.1073/PNAS.0903602106
164. Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, et al. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. (2010) 122(24):2529–37. doi: 10.1161/CIRCULATIONAHA.109.930446
165. Nahmod KA, Vermeulen ME, Raiden S, Salamone G, Gamberale R, Fernández-Calotti P, et al. Control of dendritic cell differentiation by angiotensin II. FASEB J. (2003) 17(3):1–19. doi: 10.1096/FJ.02-0755FJE
166. Bourrit A, Atlan H, Fromer I, Melmed RN, Lichtstein D. Basic characterization of an ouabain-resistant, bumetanide-sensitive K+ carrier-mediated transport system in J774.2 mouse macrophage-like cell line and in variants deficient in adenylate cyclase and cAMP-dependent protein kinase activities. Biochim Biophys Acta - Biomembr. (1985) 817(1):85–94. doi: 10.1016/0005-2736(85)90071-9
167. Hung CM, Peng CK, Wu CP, Huang KL. Bumetanide attenuates acute lung injury by suppressing macrophage activation. Biochem Pharmacol. (2018) 156:60–7. doi: 10.1016/J.BCP.2018.08.013
168. Ni L, Fang P, Hu ZL, Zhou HY, Chen JG, Wang F, et al. Identification and function of acid-sensing Ion channels in RAW 264.7 macrophage cells. Curr Med Sci. (2018) 38(3):436–42. doi: 10.1007/S11596-018-1897-Y
169. Reus-Chavarría E, Martínez-Vieyra I, Salinas-Nolasco C, Chávez-Piña AE, Méndez-Méndez JV, López-Villegas EO, et al. Enhanced expression of the epithelial sodium channel in neutrophils from hypertensive patients. Biochim Biophys Acta Biomembr. (2019) 1861(2):387–402. doi: 10.1016/J.BBAMEM.2018.11.003
170. Aschner M, Nguyen TT, Sinitskii AI, Santamaría A, Bornhorst J, Ajsuvakova OP, et al. Isolevuglandins (isoLGs) as toxic lipid peroxidation byproducts and their pathogenetic role in human diseases. Free Radic Biol Med. (2021) 162:266–73. doi: 10.1016/J.FREERADBIOMED.2020.10.024
171. Armanini D, Endres S, Kuhnle U, Weber PC. Parallel determination of mineralocorticoid and glucocorticoid receptors in T- and B-lymphocytes of human spleen. Acta Endocrinol (Copenh). (1988) 118(4):479–82. doi: 10.1530/ACTA.0.1180479
172. Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schütz G, et al. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. (2010) 120(9):3350–64. doi: 10.1172/JCI41080
173. Laurent S. Antihypertensive drugs. Pharmacol Res. (2017) 124:116–25. doi: 10.1016/J.PHRS.2017.07.026
174. Frishman WH. Beta-adrenergic blockers. Circulation. (2003) 107(18):e117–9. doi: 10.1161/01.CIR.0000070983.15903.A2
175. Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav Immun. (2007) 21(6):736–45. doi: 10.1016/J.BBI.2007.03.008
176. Melnikov M, Rogovskii V, Sviridova A, Lopatina A, Pashenkov M, Boyko A. The dual role of the β2-adrenoreceptor in the modulation of IL-17 and IFN-γ production by T cells in multiple sclerosis. Int J Mol Sci. (2022) 23(2):668. doi: 10.3390/ijms23020668
177. Slota C, Shi A, Chen G, Bevans M, Weng N-p. Norepinephrine preferentially modulates memory CD8 T cell function inducing inflammatory cytokine production and reducing proliferation in response to activation. Brain Behav Immun. (2015) 46:168. doi: 10.1016/J.BBI.2015.01.015
178. Takenaka MC, Araujo LP, Maricato JT, Nascimento VM, Guereschi MG, Rezende RM, et al. Norepinephrine controls effector T cell differentiation through β2-adrenergic receptor-mediated inhibition of NF-κB and AP-1 in dendritic cells. J Immunol. (2016) 196(2):637–44. doi: 10.4049/JIMMUNOL.1501206
179. Das R, Burke T, Van Wagoner DR, Plow EF. L-Type calcium channel blockers exert an anti-inflammatory effect by suppressing expression of plasminogen receptors on macrophages. Circ Res. (2009) 105(2):167. doi: 10.1161/CIRCRESAHA.109.200311
180. Badou A, Jha MK, Matza D, Flavell RA. Emerging roles of L-type voltage-gated and other calcium channels in T lymphocytes. Front Immunol. (2013) 4:243. doi: 10.3389/FIMMU.2013.00243/BIBTEX
181. Kanatani S, Fuks JM, Olafsson EB, Westermark L, Chambers B, Varas-Godoy M, et al. Voltage-dependent calcium channel signaling mediates GABAA receptor-induced migratory activation of dendritic cells infected by Toxoplasma gondii. PLoS Pathog. (2017) 13(12):e1006739. doi: 10.1371/JOURNAL.PPAT.1006739
182. Grafton G, Stokes L, Toellner KM, Gordon J. A non-voltage-gated calcium channel with L-type characteristics activated by B cell receptor ligation. Biochem Pharmacol. (2003) 66(10):2001–9. doi: 10.1016/J.BCP.2003.07.005
183. Michalick L, Kuebler WM. TRPV4-A missing link between mechanosensation and immunity. Front Immunol. (2020) 11:413. doi: 10.3389/FIMMU.2020.00413
184. Quintana A, Griesemer D, Schwarz EC, Hoth M. Calcium-dependent activation of T-lymphocytes. Pflugers Arch. (2005) 450(1):1–12. doi: 10.1007/S00424-004-1364-4
185. Lee MD, Bingham KN, Mitchell TY, Meredith JL, Rawlings JS. Calcium mobilization is both required and sufficient for initiating chromatin decondensation during activation of peripheral T-cells. Mol Immunol. (2015) 63(2):540–9. doi: 10.1016/J.MOLIMM.2014.10.015
186. King LB, Freedman BD. B-lymphocyte calcium influx. Immunol Rev. (2009) 231(1):265–77. doi: 10.1111/J.1600-065X.2009.00822.X
187. Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol. (2015) 33:291–353. doi: 10.1146/ANNUREV-IMMUNOL-032414-112212
188. Davenport B, Li Y, Heizer JW, Schmitz C, Perraud AL. Signature channels of excitability no more: l-type channels in immune cells. Front Immunol. (2015) 6:23. doi: 10.3389/FIMMU.2015.00375
189. Bajkovec L, Mrzljak A, Likic R, Alajbeg I. Drug-induced gingival overgrowth in cardiovascular patients. World J Cardiol. (2021) 13(4):68–75. doi: 10.4330/WJC.V13.I4.68
190. Lo Y, Lin LY, Tsai TF. Use of calcium channel blockers in dermatology: a narrative review. Expert Rev Clin Pharmacol. (2021) 14(4):481–9. doi: 10.1080/17512433.2021.1894128
191. Gackowska L, Michałkiewicz J, Niemirska A, Helmin-Basa A, Kłosowski M, Kubiszewska I, et al. Loss of CD31 receptor in CD4+ and CD8+ T-cell subsets in children with primary hypertension is associated with hypertension severity and hypertensive target organ damage. J Hypertens. (2018) 36(11):2148–56. doi: 10.1097/HJH.0000000000001811
192. Marketou ME, Kontaraki JE, Zacharis EA, Kochiadakis GE, Giaouzaki A, Chlouverakis G, et al. TLR2 and TLR4 gene expression in peripheral monocytes in nondiabetic hypertensive patients: the effect of intensive blood pressure–lowering. J Clin Hypertens. (2012) 14(5):330. doi: 10.1111/J.1751-7176.2012.00620.X
193. Murray EC, Nosalski R, MacRitchie N, Tomaszewski M, Maffia P, Harrison DG, et al. Therapeutic targeting of inflammation in hypertension: from novel mechanisms to translational perspective. Cardiovasc Res. (2021) 117(13):2589–609. doi: 10.1093/CVR/CVAB330
194. Hoorn EJ, Walsh SB, McCormick JA, Fürstenberg A, Yang CL, Roeschel T, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med. (2011) 17(10):1304–9. doi: 10.1038/nm.2497
195. Divac N, Naumović R, Stojanović R, Prostran M. The role of immunosuppressive medications in the pathogenesis of hypertension and efficacy and safety of antihypertensive agents in kidney transplant recipients. Curr Med Chem. (2016) 23(19):1941–52. doi: 10.2174/0929867323666151221150052
196. Rojas-Vega L, Jiménez-Vega AR, Bazúa-Valenti S, Arroyo-Garza I, Jiménez JV, Gómez-Ocádiz R, et al. Increased phosphorylation of the renal Na+-Cl- cotransporter in male kidney transplant recipient patients with hypertension: a prospective cohort. Am J Physiol - Ren Physiol. (2015) 309(10):F836–42. doi: 10.1152/ajprenal.00326.2015
197. Williams RM, Shah J, Tian HS, Chen X, Geissmann F, Jaimes EA, et al. Selective nanoparticle targeting of the renal tubules. Hypertension. (2018) 71(1):87–94. doi: 10.1161/HYPERTENSIONAHA.117.09843
198. Williams RM, Shah J, Ng BD, Minton DR, Gudas LJ, Park CY, et al. Mesoscale nanoparticles selectively target the renal proximal tubule epithelium. Nano Lett. (2015) 15(4):2358. doi: 10.1021/NL504610D
199. Fancher IS, Rubinstein I, Levitan I. Potential strategies to reduce blood pressure in treatment resistant hypertension using FDA-approved nanodrug delivery platforms. Hypertens (Dallas, Tex 1979). (2019) 73(2):250. doi: 10.1161/HYPERTENSIONAHA.118.12005
200. Evans LC, Petrova G, Kurth T, Yang C, Bukowy JD, Mattson DL, et al. Increased perfusion pressure drives renal T-cell infiltration in the dahl salt-sensitive rat. Hypertension. (2017) 70(3):543–51. doi: 10.1161/HYPERTENSIONAHA.117.09208
201. Ferro CJ, Edwards NC, Hutchison C, Cockwell P, Steeds RP, Savage CO, et al. Does immunosuppressant medication lower blood pressure and arterial stiffness in patients with chronic kidney disease? An observational study. Hypertens Res. (2011) 34(1):113–9. doi: 10.1038/hr.2010.193
202. Morales JM. Influence of the new immunosuppressive combinations on arterial hypertension after renal transplantation. Kidney Int. (2002) 62(82):S81–7. doi: 10.1046/J.1523-1755.62.S82.16.X
203. Alexander MR, Hank S, Dale BL, Himmel L, Zhong X, Smart CD, et al. A single nucleotide polymorphism in SH2B3/LNK promotes hypertension development and renal damage. Circ Res. (2022) 131(9):731–47. doi: 10.1161/CIRCRESAHA.121.320625
Keywords: hypertension, immunity, inflammation, kidney, heart
Citation: Benson LN, Guo Y, Deck K, Mora C, Liu Y and Mu S (2023) The link between immunity and hypertension in the kidney and heart. Front. Cardiovasc. Med. 10:1129384. doi: 10.3389/fcvm.2023.1129384
Received: 21 December 2022; Accepted: 20 February 2023;
Published: 9 March 2023.
Edited by:
Xiaohan Lu, Duke University, United StatesReviewed by:
Jing Jason Wu, University of Rochester Medical Center, United States© 2023 Benson, Guo, Deck, Mora, Liu and Mu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lance N. Benson TE5CZW5zb25AdWFtcy5lZHU= Shengyu Mu U011QHVhbXMuZWR1
Specialty Section: This article was submitted to Hypertension, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.