 Pavel Zhabyeyev
Pavel Zhabyeyev Chandu Sadasivan1,2
Chandu Sadasivan1,2 Saumya Shah
Saumya Shah Gavin Y. Oudit
Gavin Y. Oudit
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med., 21 April 2023
Sec. General Cardiovascular Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1129349
This article is part of the Research TopicMolecular Pathogenesis and Novel Treatments for Inherited CardiomyopathiesView all 11 articles
Background: Iron overload cardiomyopathy (IOC) is a major co-morbidity of genetic hemochromatosis and secondary iron overload with limited therapeutic options. We aim to investigate mechanisms of rescue action of amlodipine in the murine model of iron overload, characterize changes in human cardiac tissue due to IOC, and compare them to the changes in the animal model of IOC.
Methods and results: As an animal model, we used male hemojuvelin knockout (HJVKO) mice, which lacked hemojuvelin (a co-receptor protein for hepcidin expression). The mice were fed a high-iron diet from 4 weeks to 1 year of age. As a rescue, iron-fed mice received the Ca2+ channel blocker, amlodipine, from 9 to 12 months. Iron overload resulted in systolic and diastolic dysfunctions and changes in the cardiac tissue similar to the changes in the explanted human heart with IOC. An IOC patient (β-thalassemia) with left-ventricular ejection fraction (LVEF) 25% underwent heart transplantation. The murine model and the explanted heart showed intra-myocyte iron deposition, fibrosis, hypertrophy, oxidative stress, remodeling of Ca2+ cycling proteins, and metabolic kinases typical of heart failure. Single-myocyte contractility and Ca2+ release were diminished in the murine model. The amlodipine-treated group exhibited normalization of cellular function and reversed fibrosis, hypertrophy, oxidative stress, and metabolic remodeling. We also report a clinical case of primary hemochromatosis successfully treated with amlodipine.
Conclusions: The aged HJVKO murine model on the iron-rich diet reproduced many features of the human case of IOC. The use of amlodipine in the murine model and clinical case reversed IOC remodeling, demonstrating that amlodipine is effective adjuvant therapy for IOC.
Iron is an essential trace element that plays a crucial role in oxygen transport, oxidative phosphorylation, and the production of reactive oxygen species (ROS) (1, 2). Iron homeostasis is achieved by a concerted action of multiple proteins, whose actions converge on the SMAD signaling pathway controlling the expression of the Hamp gene encoding a suppressor of iron uptake, hepcidin (2–5). The two major regulatory proteins involved in the control of hepcidin expression are human hemochromatosis protein (HFE protein) and hemojuvelin (HJV) (4, 5). Mutations in these genes lead to primary hemochromatosis (iron overload) (6). Mutations in the Hfe gene are the most prevalent, constituting approximately 90% of adult hemochromatosis phenotypes in white populations of European descent (4–8). Mutations in Hjv (Hfe2) gene are considerably less frequent but lead to juvenile hemochromatosis with a high degree of iron overload and overt cardiac phenotype (7, 9). Secondary hemochromatosis primarily arises as a side effect of frequent blood transfusions used to treat congenital anemias (2, 10) and intravenous iron supplementation in hemodialysis (11). In the case of primary hemochromatosis, the current approach to treatment is an early screening of susceptible populations, so early interventions (dietary management and phlebotomy) can be used to control iron levels (4, 6). Secondary hemochromatosis is managed by chelation therapy to reduce iron cardiotoxicity (6, 12). Combining chelation therapy with antioxidants was also suggested [for review, see Wongjaikam, Kumfu (13)].
Another route for improving chelation therapy is to combine iron chelators with L-type Ca2+ channel blockers. Ca2+ channels aid iron cardiotoxicity by providing a major entry route for Fe2+ ions into cardiomyocytes (14–16). Clinical trials to investigate the effectiveness of Ca2+ channel blockers as an adjunct therapy to iron chelators have been started (17, 18), and one has been completed (19). Based on subgroup analyses, the AmloThal clinical trial (NCT01395199) concluded that amlodipine combined with chelation therapy reduced cardiac iron more effectively than chelation therapy alone (19). In light of these findings, mechanistic studies on the animal models are important to elucidate the mechanism of action of amlodipine in the settings of iron overload. Amlodipine is a dihydropyridine Ca2+ channel blocker mainly used to treat hypertension (20). Besides channel-blocking, the compound has antioxidant (21) and inhibitory activity on several enzymes at concentrations above 5 µmol/L (22, 23). The typical plasma concentration of amlodipine in patients is 10–20 nmol/L (24, 25), and in the mouse model of iron overload is about 650 nmol/L (15).
In this study, we report two cases of hemochromatosis (primary and secondary) managed by the L-type Ca2+ channel blocker (amlodipine) and compare them to the animal model of iron overload treated with amlodipine. In the case of secondary hemochromatosis, we obtained explanted tissue samples of the heart with iron overload cardiomyopathy (IOC). We compared these samples with non-failing controls to identify IOC-related changes. Then, we compared these changes to the analogous changes in the animal model of iron overload. As the animal model of iron overload, we used a 12-month hemojuvelin knockout (Hjv−/−) on a high-iron diet. That model has a more severe and pronounced phenotype resembling human IOC more closely than wild-type mice on a high-iron diet (26, 27). We also investigated the ability of amlodipine (i) to lower cardiac iron and (ii) to reverse maladaptive cardiac remodeling by introducing amlodipine at the age of 9 months (after 8 months of an iron diet).
Male Hjv−/− (HJVKO) mice (kindly provided by Dr. Nancy C. Andrews, Duke University) were bred in-house at the University of Alberta Health Sciences Laboratory Animal Services facility. All experiments were performed in accordance with the University of Alberta institutional guidelines, which conformed to guidelines published by the Canadian Council on Animal Care and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (revised 2011). We performed advanced iron overload protocol by feeding 4 weeks old HJVKO mice with the high iron diet (28) (Prolab®RHM 3000 with iron 380 ppm) until they were 1 year old. At 9 months, the amlodipine group received amlodipine in drinking water (95 mg/L) for 3 months. The vehicle group comprised male HJVKO mice receiving a regular diet without treatment.
This patient was consented with written informed consent as part of the Heart Function and Cardiomyopathy Clinic registry, which is approved by the Ethics Committee at the University of Alberta (Pro00077124). Data for this patient's case was obtained through an electronic chart review.
The study was approved by the Ethics Committee at the University of Alberta, and all patients provided written informed consents in accordance with the Declaration of Helsinki (2008) of the World Medical Association. Left ventricular tissue was harvested from explanted human failing hearts and donor non-failing human hearts via HELP (Human Explanted Heart Program) at the Mazankowski Alberta Heart Institute and the HOPE (Human Organ Procurement and Exchange) program at the University of Alberta Hospital. The harvested hearts were preserved in cold cardioplegia solution, and collected samples were snap-frozen in liquid nitrogen within 15 min of explantation and stored at −80°C.
Samples (20 mg) of frozen tissue from LV were subjected to inductively coupled plasma resonance mass spectrometry to quantify iron levels at the Trace Metals Laboratory, London, Western Ontario. The samples were analyzed in triplicates, and the average values were used (15, 26).
Images were obtained on Hitachi S5500 SEM. Energy-dispersive X-ray spectra were acquired using Oxford INCA Energy in STEM mode with a 1.5 nm probe.
Transthoracic echocardiography was performed at 1 year of iron overload phenotype mice with the Vevo 2100 high-resolution imaging system equipped with a 30-MHz transducer using 1.5%–2% isoflurane (26, 29).
Mice were anesthetized with 1.5% isoflurane, and the right common carotid artery was cannulated. 1.4F Scisense catheter (Scisense Inc.) was advanced through the aortic valve and placed into the LV chamber. Pressure-volume loops were recorded via TCP-500 amplifier (Scisense Inc.) and analyzed offline using LabScribe 2 software (IWorks Inc.) as described previously (26, 29).
Myocytes were enzymatically isolated as described previously (30) without blebbistatin to preserve contractility. After isolation, myocytes were kept in the perfusion buffer solution (pH 7.4). An aliquot of isolated cardiomyocytes was transferred to a bath atop of an inverted microscope to measure excitability, contractility, or Ca2+ transients (loaded with FURA-2AM). The measurements were done in myocytes superfused with Tyrode's solution (1.2 mM Ca2+) at 35–36°C and paced with field stimulation at 1 Hz as previously described (26, 30, 31). Action potentials were recorded in the whole-cell ruptured-patch configuration with K+ pipette solution (31).
Mice were placed under isoflurane anesthesia (1.5%–2%) on a heated pad (body temperature maintained at 37°C, measured by the rectal probe). ECG leads were placed in Lead I configuration. The signal was digitized using acquisition interface ACQ-7700 (Data Science International, USA) with P3 Plus software (ver. 5.0, Data Science International, USA).
The excised hearts from anesthetized mice were arrested in diastole using saline with 15 mM KCl, fixed in 10% buffered formalin, and embedded in paraffin. Thin sections (5 µm) were stained with Prussian blue, picro-sirius red (PSR), Masson trichrome, or wheat germ agglutinin (WGA) stain as described previously (26, 32). Iron depositions were visualized as blue depositions using a bright field view. Myocardial fibrosis was evaluated by quantifying PSR stained sections, and myocyte cross-sectional area (MCSA) was assessed from WGA staining using an Olympus IX81 microscope. Image analysis was done using MetaMorph software (26, 32).
Immunofluorescence was performed on formalin-fixed paraffin-embedded heart sections (5 µm). Briefly, the sections were deparaffinized, followed by antigen retrieval and blocked with blocking buffer (1% BSA in 1X PBS) for 1 h. Similarly, OCT-embedded sections were fixed with 4% paraformaldehyde for 20 min and rehydrated in PBS for 30 min. Then, sections were incubated overnight in a humidified chamber at 4°C with primary antibody against rat anti-mouse neutrophil (Serotec), rat anti-mouse F4/80 (Serotec), mouse anti-nitrotyrosine (Santa Cruz), mouse anti-4-HNE (Abcam). Finally, sections were incubated with different fluorophore-conjugated secondary antibodies (Invitrogen USA), as described previously (26, 32).
The levels of malondialdehyde (MDA), an indicator of lipid peroxidation, were measured in myocardial tissue (100–150 mg) by using a commercially available kit (Bioxytech, MDA-586TM assay, Oxis International Inc., Foster City, CA). Myocardial reduced (GSH) and oxidized glutathione (GSSG) levels were measured as described previously (26, 33).
mRNA expression levels were evaluated using TaqMan real-time PCR. Total RNA was extracted from flash-frozen LV tissue using the TRIzol RNA extraction method. RNA (1 µg) was subjected to reverse transcription to synthesize cDNA. Samples were loaded in triplicate, and the data were analyzed by Roche's Light cycler® 480 system.
Transcriptome sequencing, including sample preparation, library construction, and Illumina sequencing, were carried out by Novogene Corporation Inc. (California, USA). The reported methods below were modified based on the standard procedures provided by Novogene. Total RNAs from left ventricles (4 hearts/group) were extracted. The pathway analysis was performed using iDEP.94 http://bioinformatics.sdstate.edu/idep94/ (34).
Western blot analysis was performed on flash-frozen LV tissue samples as previously described (26, 32). Briefly, we extracted protein from LV tissues and performed immunoblotting for various proteins using the following primary antibodies: SERCA2a, NCX1 (Thermo Fisher Scientific), PLN-PSer16, total PLN (Badrilla Ltd), Akt-PSer473, Akt-PThr308, total Akt (Cell Signaling), AMPK-PThr172 and total AMPK (Cell Signaling) and subsequently incubated with HRP conjugated secondary antibodies respectively.
All data are presented as mean ± SEM. The explanted heart was assessed using the one-sample t-test. In the case of multiple group comparisons, differences were evaluated by one-way ANOVA followed by Tukey's post-test. Statistical analysis was performed using Origin 2020 software (OriginLab).
HJVKO mice were fed a high-iron diet to achieve iron overload as mice aged to 12 months. In the rescue group, amlodipine was introduced at 9 months (Figure 1A). Prussian blue histological staining showed a marked increase in iron deposition in groups receiving the high-iron diet (Figure 1B). Direct measurement of iron levels with inductively-coupled plasma mass spectrometry showed that the iron-fed group had markedly elevated iron levels and the amlodipine group had significantly reduced iron levels (Figure 1C). At the cellular level, iron deposition occurred as sub-micrometer particles (Figure 1D). The composition of the deposits was confirmed using energy-dispersive X-ray spectra, which showed a characteristic Fe peak at 6.4 keV (Figure 1D). In contrast to the heart, liver iron levels were increased at 12 months in both iron-fed groups (placebo and amlodipine; see Figures 1E, F). These results demonstrate that amlodipine reduces the iron load in the heart but not in the liver.
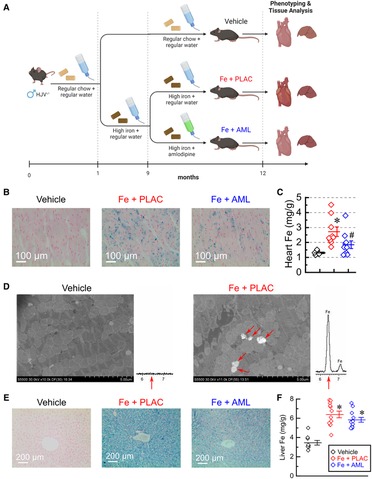
Figure 1. Amlodipine reduces cardiac iron levels. (A) The schematic of the study design (created with BioRender.com). (B) Representative images of Prussian blue staining. (C) Total myocardial iron levels (per dry weight; n = 7–10 per group) measured by inductively-coupled plasma mass spectrometry. (D) Representative scanning electron microscopy images (iron deposits marked by red arrows) and energy-dispersive X-ray spectra for vehicle and iron groups. Spectrum from the iron group, but not from the vehicle group, has a characteristic Fe peak at 6.4 keV (red arrow). (E) Representative images of Prussian Blue staining of liver sections. (F) Quantification of iron levels in the liver by inductive plasma mass-spectrometry (n = 10–14 per group). ppm = parts per million; *p < 0.05 compared with the vehicle group. #p < 0.05 compared with the Iron (Fe) group.
Advanced iron overload led to a significant reduction of systolic function, and this reduction was rescued by treating iron-fed mice with amlodipine (Figure 2A). In response to iron overload, the systolic function was substantially reduced (reductions in fractional shortening, FS, and ejection fraction, EF; Figure 2B), and this reduction was accompanied by LV hypertrophy (increase in LV posterior wall thickness, LV PWT; Figure 2B) and LV dilation (increase in LV end-diastolic dimensions, LV EDD; Figure 3B). Amlodipine treated group showed normalization of systolic function and LV dilation (normalization of FS, EF, and LV EDD; Figure 2B). Noticeable change toward normalization of hypertrophic status in the amlodipine group did not achieve significance (p = 0.096; LV PWT, Figure 3B). Mitral and tissue Doppler measurements showed that diastolic function markedly deteriorated in response to iron overload (Figure 2C). Iron overload resulted in impaired cardiac relaxation as evident from (i) an increase in isovolumic relaxation time (IVRT), (ii) an increase in deceleration time (DT), and (iii) a decrease in the ratio of annular velocities (E′/A′) (Figure 2D). These functional changes were also accompanied by left-atrial (LA) dilation (Figure 2D). The amlodipine-treated group exhibited normalization of all diastolic parameters but one (IVRT) (Figure 2D). In short, iron overload diminished both systolic and diastolic function, whereas amlodipine was able to rescue most of these developments.
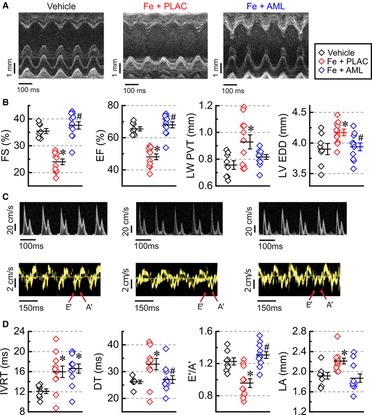
Figure 2. Amlodipine improves systolic and diastolic function. (A) Representative M-mode images. (B) Quantification of systolic function: fractional shortening (FS), ejection fraction (EF), left ventricular posterior wall thickness (LV PWT), and left ventricular end-diastolic dimensions (LV EDD). (C) Representative Doppler (top row) and Tissue Doppler (bottom row) images. (D) Quantification of diastolic function: isovolumic relaxation time (IVRT), deceleration time (DT), E prime (E′) to A prime (A′) ratio (E′/A′) of Tissue Doppler velocities, and left atrial dimensions (LA). n = 8–11 per group. *p < 0.05 compared with the control group; #p < 0.05 compared with the iron group.
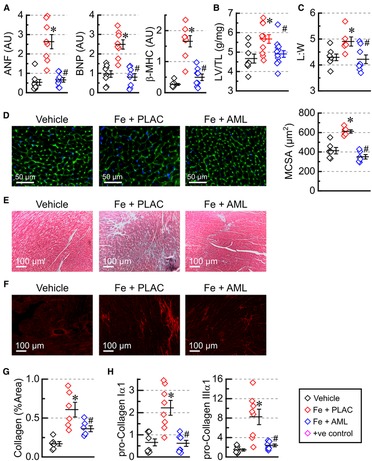
Figure 3. Amlodipine reverses hypertrophic cardiac remodeling. (A) Expression levels of heart failure markers: atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), and myosin heavy chain β isoform (β-MHC) (n = 8 per group). (B) Morphometric index of cardiac hypertrophy (left ventricular weight (LV) adjusted by tibia length (TL); n = 8–13). (C) Morphometric index of cellular hypertrophy: length to width ratio (L:W) for isolated myocytes (6–8 hearts per group 50–150 myocytes per heart). (D) Representative images of wheat germ agglutinin (WGA) staining and myocytes cross-sectional area (MCSA) measured from WGA staining images (6 hearts per group, 4 sections per heart). (E) Representative images of Mason's trichrome staining. (F) Representative images of picrosirius-red (PSR) staining and quantification of PSR stained sections (G); 6 hearts per group; 4 sections per heart. (H) Expression levels of pro-collagens (n = 8 hearts per group). *p < 0.05 compared with the control group; #p < 0.05 compared with the iron group.
Assessment of cardiac function using pressure-volume loop technique showed that (i) iron overload resulted in dilation (increase in LV end-diastolic volume, LV EDV; Table 1), (ii) systolic dysfunction (reductions in ejection fraction, EF; pre-load adjusted contractility, dP/dtmax/EDV; and end-systolic pressure-volume relationship, ESPVR; Table 1), and (iii) diastolic dysfunction [elevated LV end-diastolic pressure, LVEDP; and larger isovolumic relaxation time constant, τ (Glantz); but increase in end-diastolic pressure-volume relation, EDPVR, was not statistically significant; Table 1]. The amlodipine-treated group had (i) normalized cardiac volume (LVEDV; Table 1), (ii) normalized systolic function (normal and nearly-normal values for EF, dP/dtmax/EDV, and ESPVR), and (iii) partial normalization of diastolic function (some reduction in LVEDP (not significant), normalization of isovolumic relaxation, τ (Glantz), and statistically insignificant reduction in EDPVR). In the case of pressure-volume loops, the results were similar to echocardiography: dilation, systolic and diastolic dysfunctions in the iron group, and quasi-normal cardiac function in the amlodipine-treated group).
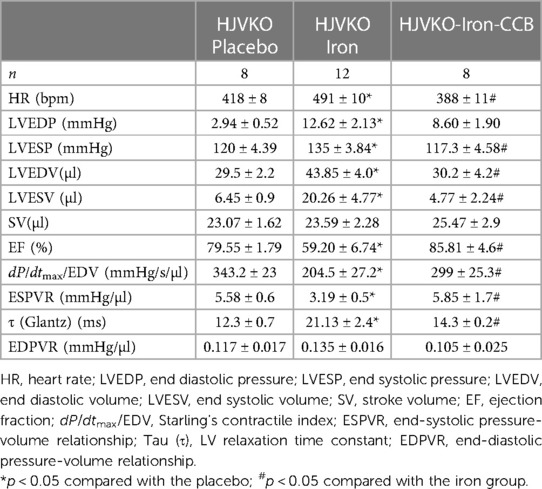
Table 1. Echocardiographic and hemodynamic (pressure-volume loop) assessment.
Iron accumulation led to elevated expression levels of heart failure markers (natriuretic peptides, ANF and BNP, and myosin heavy chain β, β-MHC) in the iron group. In contrast, the amlodipine-treated group exhibited overall normalization of expression of heart failure markers (Figure 3A). In addition, the morphometric index of hypertrophy, LV mass adjusted by tibia length (LV/TL), was increased in the iron group but normalized in the amlodipine-treated group treated with amlodipine (Figure 3B). At the cellular level, measurement of length (L) and width (W) of isolated cardiomyocytes demonstrated that myocytes were elongated (increased L:W ratio) in the iron group and had an L:W ratio close to normal in the amlodipine-treated group (Figure 3C). Assessment of hypertrophy at the tissue level using WGA staining (Figure 3D) revealed increased MCSA in the iron group and substantially reduced myocyte area in the amlodipine-treated group. The data suggest that iron overload leads to eccentric-hypertrophy remodeling, and amlodipine treatment reversed a significant part of hypertrophic remodeling.
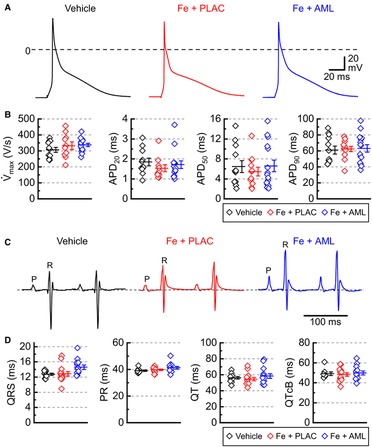
Figure 4. Cardiac electrical activity in murine iron overload. (A) Representative action potentials for the vehicle, iron with placebo (Fe + PLAC), and iron with amlodipine (Fe + AML) groups (left to right). (B) Maximal rate of rise of action potential (V̇max) and action potential durations for 20, 50, and 90% of repolarization (APD20, APD50, and APD90, respectively); 3 hearts /group; 4–6 cells /heart. (C) Representative electrocardiograms for the vehicle, iron with placebo (Fe + PLAC), and iron with amlodipine (Fe + AML) groups. (D) Durations of QRS, PR, QT, and QT Bazett corrected (QTcB) intervals (n = 11–14 per group).
Cardiac fibrosis was assessed using Mason's trichrome (Figure 3E) and PSR (Figure 3F) staining. The quantification of PSR staining (Figure 3G) showed a marked increase in interstitial fibrosis in iron-overloaded hearts and a considerable reduction of fibrosis in the hearts from the amlodipine-treated group. Expression levels of pro-fibrotic markers (pro-collagen Iα1 and IIIα1) followed a similar pattern: upregulation in the iron overload group and normalization in the amlodipine-treated group (Figure 3H).
Measurements of electrical activity (action potentials) in the isolated cardiac myocytes showed no significant change in the major parameters of action potential: the maximal slope of depolarization (V̇max) and action potential duration (APDs corresponding to repolarization levels of 20%, 50%, and 90%; Figures 4A, B) suggesting that iron and amlodipine effects on contractility were driven primarily via changes in Ca2+ cycling. The lack of the effect of iron and amlodipine on cardiac electrical activity was corroborated by electrocardiographic measurements. The Lead I electrocardiogram in anesthetized mice revealed no substantial differences in ECG parameters between the groups (Figures 4C, D).
Single-myocyte contractility was inhibited in the myocytes isolated from iron-overloaded hearts (Figure 5A). Both contractility (FS and maximal velocity of contraction, −dL/dt) and relaxation (maximal velocity of relaxation, +dL/dt) were diminished in the iron group (Figure 5A). Conversely, the amlodipine-treated group showed normalization of both contractility and relaxation parameters (Figure 5A).
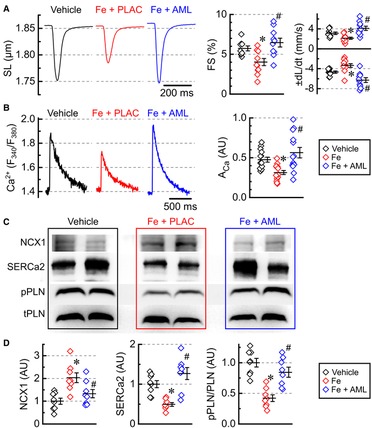
Figure 5. Amlodipine normalizes single-myocyte contractility and Ca2+ cycling. (A) Averaged recording of sarcomeric length (SL), fractional shortening (FS), contractility (−dL/dt), and relaxation (+dL/dt) (2 hearts per group 4–6 cells per heart). (B) Averaged recording of Ca2+ release transients plotted as the ratio of FURA-2 fluorescence (F380/F340) and Ca2+ release amplitudes (ACa). (C) Representative western blots for Na+–Ca2+ exchanger (NCX1), sarcoplasmic reticulum Ca2+ ATPase (SERCa2), Ser16-phosphorylated phospholamban (pPLN), and total phospholamban (tPLN). (D) Protein levels for NCX1 and SERCa2 and phosphorylation ratio for phospholamban (pPLN/tPLN), quantified from western blots (n = 8 hearts per group). *p < 0.05 compared with the control group; #p < 0.05 compared with the iron group.
The amplitude of Ca2+ transients (ACa) was inhibited in the iron-overload group and normalized in the amlodipine-treated group (Figure 5B). To investigate what underlines changes in Ca2+ transients, we assessed levels and phosphorylation state of three major proteins that control the time constant and amplitude of Ca2+ transient: Na+–Ca2+ exchanger (NCX1), sarco-endoplasmic reticulum Ca2+-ATPase (SERCa2), and phospholamban (PLN) (Figure 5C). NCX1 levels were markedly elevated in the iron-overload group and mostly normalized in the amlodipine-treated group (Figure 5D). SERCa2 levels were lowered in the iron-overload group, but roughly normalized in the amlodipine-treated group (Figure 5D), which is consistent with the normalization of Ca2+ transient amplitude (ACa) (Figure 5B). PLN phosphorylation was decreased in the iron group and was normalized in the amlodipine-treated groups (Figure 5D).
The state of the intrinsic antioxidant system was assessed by measuring the concentration of GSH and GSSG. Consistent with a heightened degree of oxidative stress, the iron group had low GSH and high GSSG concentrations (Figure 6A); on the other hand, the amlodipine-treated group had a close-to-normal pattern of GSH-GSSG distribution (high GSH with low GSSG) (Figure 6A). MDA was elevated in the iron group but largely normalized levels in the amlodipine-treated group, corroborating GSH-GSSG results that oxidative stress was elevated in the iron group and was at near-normal levels in the amlodipine-treated hearts (Figure 6A). Assessment of metabolic signaling showed that iron overload was associated with (i) decreased Akt phosphorylation at both Thr308 (Akt-T308) and Ser473 (Akt-S473) and (ii) increased AMPK phosphorylation (Thr172) (Figure 6B). In contrast, the amlodipine-treated group had near-normal phosphorylation levels at all three sites (Figure 6B), suggesting that the amlodipine treatment normalizes pathological metabolic signaling in cardiac tissue.
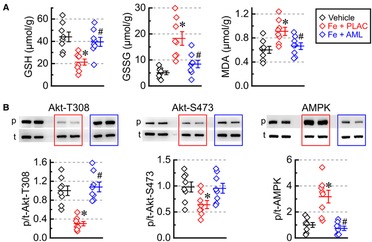
Figure 6. Amlodipine normalizes oxidative stresses and metabolic signaling. (A) Concentrations of reduced glutathione (GSH), oxidized glutathione (GSSG), and malondialdehyde (MDA) (n = 8 per group). (B) Phosphorylation levels (p/t) for Akt [Thr308 phosphorylation site (Akt-T308, p), Ser473 phosphorylation site (Akt-S473, p), and total Akt (t)] as well as AMP-activated protein kinase [AMPK; Thr172 phosphorylation site (p) and total (t)]. *p < 0.05 compared with the control group; #p < 0.05 compared with the iron group.
Iron overload alters the expression of multiple genes involved in cardiac muscle contraction and the citrate cycle. We used bulk RNA sequencing to assess broad changes in expression levels (Data Supplement and Supplementary Figure S1) and found two important pathways being affected: cardiac muscle contraction and citrate cycle. In both pathways, iron overload led to an overall reduction in the expression levels (cardiac muscle contraction pathway Figure 7A and citrate cycle Figure 7B). Amlodipine therapy partially normalized the changes in myocardial gene expression in response to iron overload (Figures 7A, B).
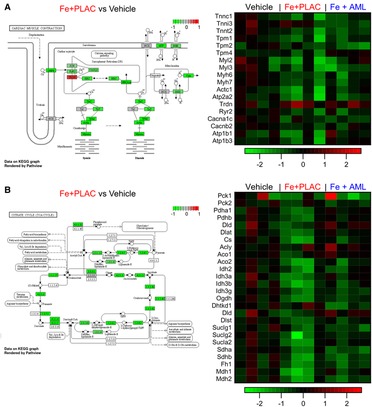
Figure 7. Iron overload reduces RNA expression of proteins in the cardiac muscle contraction pathway and citrate cycle. (A) Relative expression changes in cardiac muscle pathway and heat map of relative expression changes of selected RNA from cardiac muscle pathway. (B) Relative expression changes in citrate cycle and heat map of relative expression changes of selected RNA from citrate cycle.
A 16-year-old girl with β-thalassemia major was admitted with refractory heart failure (HF). Echocardiogram showed dilation with bi-ventricular systolic and diastolic dysfunction with LVEF of 25%. The pre-transplant iron panel showed elevated serum iron (31 µmol/L; normal: 9.0–30 µmol/L), severely high transferrin iron saturation (94%; normal: 25%–35%) and low hemoglobin (94 g/L; normal: 120–160 g/L). Pre-transplant cardiac MRI indicated severe iron overload. With proper consent, samples were collected from the explanted heart for further comparison to non-failing control heart samples obtained from HOPE.
Prussian blue staining showed widespread myocardial iron deposition in the IOC heart but no detectable staining in the non-failing control hearts (Figure 8A). The IOC heart had marked fibrosis, as evident from trichrome staining (Figure 8B) and PSR staining (Figure 8C), in association with marked hypertrophy judging by the increase in MCSA (Figure 8D). Scanning electron microscopy revealed micron-sized iron deposits in the IOC heart but not in non-failing control (Figure 8E), with spectral analysis showing a marked increase of characteristic Fe peak at 6.4 keV (Figure 8F). Assessment of key proteins involved in Ca2+ cycling showed a reduction in the levels of SERCA2 and a compensatory increase in the levels of NCX1 (Figure 8G). Iron overload was also associated with metabolic remodeling, as evidenced by increased phosphorylation ratios of AktThr308 and AMPK (Figure 8H). Assessment of oxidative stress status showed a marked increase in MDA (Figure 8I), reduction in GSH, increase in GSSG, and lower GSH/GSSG ratio (Figure 8J). In this clinical case, IOC exhibited iron deposition (including intracellular micrometer-size particles), fibrosis, hypertrophy, remodeling of Ca2+ cycling, metabolic remodeling, and a heightened degree of oxidative stress compared to non-failing control hearts. One-year post-cardiac transplant, this patient is on chelators (deferiprone and deferoxamine) and L-type Ca2+ channel blocker (amlodipine), has no signs of heart failure, and is followed up regularly. Cardiac MRI showed no evidence of myocardial (T2* = 45.5 ms) or hepatic (T2* = 17.5 ms) iron overload and normal bi-ventricular size and function, with LVEF of 66% and normal right ventricular ejection fraction of 63%.
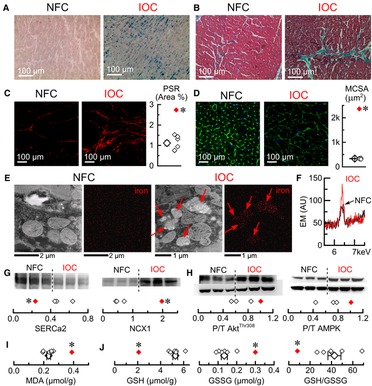
Figure 8. Characterization of the explanted human heart with IOC. (A) Representative images of Prussian blue staining. (B) Representative images of Mason's trichrome staining. (C) Representative images of picrosirius-red (PSR) staining and quantification of PSR stained sections. (D) Representative images of wheat germ agglutinin (WGA) staining and myocytes cross-sectional area (MCSA) measured from WGA staining images. (E) Representative scanning-electron microscopy images for control and iron (Fe) groups (iron deposits marked by red arrows). (F) Superimposed energy-dispersive X-ray spectra for non-failing controls (Ctl, black) and iron overload cardiomyopathy (IOC, red), showing the characteristic Fe peak at 6.4 keV. (G) Western blots and their quantifications (3 non-failing controls vs. an average of IOC) for sarcoplasmic reticulum Ca2+ ATPase (SERCa2) and Na+–Ca2+ exchanger (NCX1). (H) Western blots and their quantifications (3 non-failing controls vs. an average of IOC) for phosphorylation levels of Akt (Thr308 phosphorylation site, AktThr308) and AMP-activated protein kinase (Thr172 phosphorylation site, AMPK). (I) Malondialdehyde (MDA) concentration. (J) Reduced glutathione (GSH), oxidized glutathione (GSSG) concentrations, and GSH/GSSG ratio; open symbols—non-failing controls, filled symbol—explanted heart with IOC; *p < 0.05 compared with the non-failing controls.
A 69-year-old man with a history of primary hemochromatosis (PH) (C282Y/H63D compound heterozygote) and myelodysplastic syndrome (MDS) presented with IOC leading to HF and conduction abnormalities. He reported fatigue, shortness of breath, paroxysmal nocturnal dyspnea, and bilateral leg swelling [New York Heart Association (NYHA) Class III]. Blood work demonstrated anemia (hemoglobin = 87 g/L), elevated serum ferritin (1,230 µg/L), and elevated B-type natriuretic peptide (1,224 ng/L). Electrocardiogram showed abnormal conduction delays with underlying atrial flutter (Figure 9A). Cardiac magnetic resonance imaging (CMR) demonstrated dilated left ventricle with moderately reduced systolic function (LVEF = 40%). Cardiac T2 mapping showed evidence of moderate myocardial iron loading (T2* 12–13 ms) and hepatic iron overload (T2* 2 ms) (Figure 9B). Medical therapies were initiated to manage this patient's IOC and subsequent HF. Given this patient's concurrent MDS and PH, therapeutic phlebotomy was not feasible. Oral iron chelation therapy was undertaken with increasing doses of deferasirox; however, iron saturation remained elevated at 70%. Despite improvements, this patient still expressed marked limitations in physical activities (NYHA class III). He was started on amlodipine 2.5 mg orally daily in addition to iron chelation and was titrated to the maximum dose of 10 mg daily. During follow-up 2 years after adding amlodipine, the patient reported marked improvement in HF symptoms (NYHA class I). Comparing initial CMR imaging to 24 months demonstrated improvement in LVEF and RVEF from 40% to 66% and 41% to 64%, respectively, with normalized ventricular volumes and mass. Myocardial T2* improved from 12.5 ms to 30 ms, indicating a marked reduction in myocardial iron loading (Figure 9C).
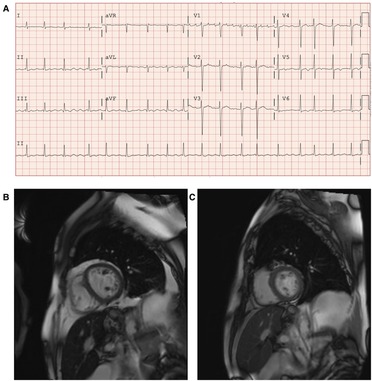
Figure 9. Patient with primary hemochromatosis and IOC treated with amlodipine. (A) Twelve-lead electrocardiogram at initial clinic visit demonstrating irregular conduction with an atrial flutter at 105 beats per minute and non-specific lateral t-wave inversion. (B) Cardiac magnetic resonance imaging (CMR) with T2 mapping at baseline (October 2019), prior to initiation of amlodipine to iron chelation therapy. T2 mapping showed moderate iron loading of the myocardium (T2* 12–13 ms), and hepatic iron overload was also noted (T2* 2 ms). (C) CMR with T2 mapping after iron chelation therapy with adjunctive amlodipine (12 months after baseline CMR, October 2020). Improvement of myocardial T2* to 26 ms, indicating the absence of myocardial iron loading and hepatic T2* improved to 5 ms, indicating mild hepatic iron loading.
Iron homeostasis is essential to maintain cardiac function, and dysfunction of iron metabolism has been implicated in various cardiovascular diseases (35–38). IOC is a major cause of heart failure in patients with hemochromatosis (iron overload) (12, 39–41). Phlebotomy and chelation therapy are the two main approaches to treating and managing iron overload (4, 6, 12, 39–41). However, phlebotomy cannot be used in patients with significant anemia, malignancy, or hemodynamic instability, and chelation therapy has an inherent risk of toxicity (4, 40, 41). Chelation therapies lower overall systemic iron, and amlodipine complements that by hindering the entry of Fe2+ via L-type Ca2+ channels, which are abundant in the heart. Therefore, using amlodipine as an adjunct therapy can improve the effectiveness of chelation therapy (19), creating an opportunity to lower cardiac iron levels faster and with a lower dosage of chelating agents, potentially improving the safety of the therapy.
Hemojuvelin-deficient mice on an iron-rich diet are an established IOC model that exhibits both systolic and diastolic dysfunction (26, 27). Iron levels obtained in the hearts of the animal model (2.7 mg/g dry weight) were approaching myocardial iron levels (3.5–9.2 mg/g dry weight) reported in patients with IOC and associated heart failure. The presented animal model replicated many crucial changes that develop during IOC in patients. We observed deterioration of cardiac function and micron-sized iron particles in cardiac tissue both in the explanted human heart with IOC and in the mouse model. In the case of the explanted heart, the heart failure was very severe (LVEF = 25%), whereas mice on the iron-rich diet had moderate cardiomyopathy (LVEF = 48%). Despite these few differences, both the explanted heart and the mouse model had fibrosis, hypertrophy, remodeling of Ca2+ cycling (decrease in SERCa2 levels accompanied by a compensatory increase in NCX levels), metabolic remodeling (inhibition of Akt activity coupled with upregulation of AMPK activity), and oxidative stress (increased MDA levels and low GSH/GSSG ratio). Accumulation of Fe2+ inside the cardiac cells is known to lead to the excessive production of ROS and trigger cell death due to ferroptosis (42), resulting in heart failure (35). In this animal model of IOC, we observed no changes in cardiac excitability but impaired cardiac cellular contractility and relaxation. The diminished contractility was associated with the reduction in Ca2+ release suggesting that changes in Ca2+ cycling underly reduced cardiac contractility.
As an L-type Ca2+ channel blocker, amlodipine can reduce the entry of non-transferrin-bound iron (Fe2+) via L-type Ca2+ channels, resulting in lower levels of cardiac iron and reduced oxidative stress in cardiac tissue. Mitigated damage from oxidative stress will contribute to the normalization of expression of Ca2+ cycling proteins and collagens, leading to the reversion of fibrosis and normalization of Ca2+ cycling, which translates to the normalization of systolic and diastolic dysfunctions at the whole-heart level. Besides being a channel blocker, amlodipine has been shown to have highly efficient antioxidant properties due to high lipophilicity and the ability to quench free radical formation (21). Antioxidants have been shown to mitigate damage associated with iron toxicity. For example, resveratrol (26, 27), thrombopoetin (43), and ebselen (44) have been shown to protect the heart in the settings of iron overload in mouse models. In our study, amlodipine demonstrated properties that can also be attributed to antioxidant activity (normalization of GSH, GSSG, and MDA levels).
In the reported case of primary hemochromatosis and IOC, the myelodysplastic syndrome prevented therapeutic phlebotomy. Iron chelation alone (deferasirox) was insufficient to reduce the burden of cardiac iron overload. However, adjunctive amlodipine therapy provided objective improvement in cardiac iron overload on imaging and symptomatic improvement in functional status for this patient. Despite randomized clinical trials demonstrating that amlodipine did not exhibit favorable clinical effects for the treatment of chronic heart failure (45, 46), amlodipine therapy could be used as adjunctive therapy in cases of iron overload, as seen in our patient's case and recent randomized clinical trials of amlodipine in patients with iron overload (17, 19). Therefore, any beneficial action from reducing iron deposition in the heart may also be reinforced by the antioxidant properties of amlodipine, suggesting that amlodipine is a good option for an adjuvant agent in chelation therapy.
We used a genetic model of primary hemochromatosis, human explanted heart tissue with iron overload and a patient with primary hemochromatosis to reveal convergent pathways of iron-mediated injury and heart disease. IOC had a similar pathogenic profile (iron accumulation pattern, histological changes, regulatory protein alterations and ROS status) in the animal model and the explanted human heart. Amlodipine was shown to rescue IOC in the animal model and the reported clinical case of primary hemochromatosis. Amlodipine's ability to suppress Fe2+ entry and effectively quench the free radical formation and oxidative damage makes it an effective agent to improve the effectiveness of chelation therapy. The clinical significance of our findings supports the use of amlodipine as an adjuvant therapy in patients with IOC.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/sra/PRJNA922223.
The studies involving human participants were reviewed and approved by HREB Biomedical, University of Alberta (Pro00077124). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. The animal study was reviewed and approved by ACUC Health Sciences, University of Alberta, Canada.
PZ performed animal experiments and data collection. CS and SS collected clinical data. SS collected explanted heart samples. FW performed histological studies and molecular analysis. PZ, CS, SS, and GO were responsible for data analysis and manuscript writing. All authors contributed to the article and approved the submitted version.
Heart and Stroke Foundation (Canada), Alberta Innovates-Health Solutions (Alberta, Canada), Canadian Institutes of Health Research (Canada).
We thank the National Institute of Nanotechnology (NINT) (University of Alberta, Canada) for technical help in acquiring electron microscopy images and spectra.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The handling editor [JLJ] declared a past co-authorship with the author [GO].
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1129349/full#supplementary-material.
1. Xu W, Barrientos T, Andrews NC. Iron and copper in mitochondrial diseases. Cell Metab. (2013) 17(3):319–28. doi: 10.1016/j.cmet.2013.02.004
2. Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. (2004) 117(3):285–97. doi: 10.1016/S0092-8674(04)00343-5
3. Zhabyeyev P, Oudit GY. Unravelling the molecular basis for cardiac iron metabolism and deficiency in heart failure. Eur Heart J. (2017) 38(5):373–5. doi: 10.1093/eurheartj/ehw386
4. Zhabyeyev P, Oudit GY. Hemochromatosis protein (HFE) knockout mice as a novel model of hemochromatosis: implications for study and management of iron-overload cardiomyopathy. Can J Cardiol. (2017) 33(7):835–7. doi: 10.1016/j.cjca.2017.04.013
5. Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. (2016) 388(10045):706–16. doi: 10.1016/S0140-6736(15)01315-X
6. Olynyk JK, Ramm GA. Hemochromatosis. N Engl J Med. (2022) 387(23):2159–70. doi: 10.1056/NEJMra2119758
7. Bardou-Jacquet E, Brissot P. Diagnostic evaluation of hereditary hemochromatosis (HFE and non-HFE). Hematol Oncol Clin North Am. (2014) 28(4):625–35; v. doi: 10.1016/j.hoc.2014.04.006
8. Barton JC, Edwards CQ, Acton RT. HFE gene: structure, function, mutations, and associated iron abnormalities. Gene. (2015) 574(2):179–92. doi: 10.1016/j.gene.2015.10.009
9. Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. (2003) 33(1):21–2. doi: 10.1038/ng1053
10. Farmakis D, Triposkiadis F, Lekakis J, Parissis J. Heart failure in haemoglobinopathies: pathophysiology, clinical phenotypes, and management. Eur J Heart Fail. (2017) 19(4):479–89. doi: 10.1002/ejhf.708
11. Rostoker G, Vaziri ND, Fishbane S. Iatrogenic iron overload in dialysis patients at the beginning of the 21st century. Drugs. (2016) 76(7):741–57. doi: 10.1007/s40265-016-0569-0
12. Pennell DJ, Udelson JE, Arai AE, Bozkurt B, Cohen AR, Galanello R, et al. Cardiovascular function and treatment in beta-thalassemia major: a consensus statement from the American heart association. Circulation. (2013) 128(3):281–308. doi: 10.1161/CIR.0b013e31829b2be6
13. Wongjaikam S, Kumfu S, Chattipakorn SC, Fucharoen S, Chattipakorn N. Current and future treatment strategies for iron overload cardiomyopathy. Eur J Pharmacol. (2015) 765:86–93. doi: 10.1016/j.ejphar.2015.08.017
14. Tanaka H, Shigenobu K. Efonidipine hydrochloride: a dual blocker of L- and T-type Ca2+ channels. Cardiovasc Drug Rev. (2002) 20(1):81–92. doi: 10.1111/j.1527-3466.2002.tb00084.x
15. Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, Ackerley C, et al. L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med. (2003) 9(9):1187–94. doi: 10.1038/nm920
16. Kumfu S, Chattipakorn SC, Fucharoen S, Chattipakorn N. Dual T-type and L-type calcium channel blocker exerts beneficial effects in attenuating cardiovascular dysfunction in iron-overloaded thalassaemic mice. Exp Physiol. (2016) 101(4):521–39. doi: 10.1113/EP085517
17. Fernandes JL, Sampaio EF, Fertrin K, Coelho OR, Loggetto S, Piga A, et al. Amlodipine reduces cardiac iron overload in patients with thalassemia major: a pilot trial. Am J Med. (2013) 126(9):834–7. doi: 10.1016/j.amjmed.2013.05.002
18. Shakoor A, Zahoor M, Sadaf A, Alvi N, Fadoo Z, Rizvi A, et al. Effect of L-type calcium channel blocker (amlodipine) on myocardial iron deposition in patients with thalassaemia with moderate-to-severe myocardial iron deposition: protocol for a randomised, controlled trial. BMJ Open. (2014) 4(12):e005360. doi: 10.1136/bmjopen-2014-005360
19. Fernandes JL, Loggetto SR, Verissimo MP, Fertrin KY, Baldanzi GR, Fioravante LA, et al. A randomized trial of amlodipine in addition to standard chelation therapy in patients with thalassemia major. Blood. (2016) 128(12):1555–61. doi: 10.1182/blood-2016-06-721183
20. Officers A, Coordinators for the ACRGTA, Lipid-Lowering Treatment to Prevent Heart Attack T. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). JAMA. (2002) 288(23):2981–97. doi: 10.1001/jama.288.23.2981
21. Mason RP, Walter MF, Trumbore MW, Olmstead EG, Mason PE. Membrane antioxidant effects of the charged dihydropyridine calcium antagonist amlodipine. J Mol Cell Cardiol. (1999) 31(1):275–81. doi: 10.1006/jmcc.1998.0867
22. Turkes C, Demir Y, Beydemir S. Anti-diabetic properties of calcium channel blockers: inhibition effects on aldose reductase enzyme activity. Appl Biochem Biotechnol. (2019) 189(1):318–29. doi: 10.1007/s12010-019-03009-x
23. Turkes C, Demir Y, Beydemir S. Calcium channel blockers: molecular docking and inhibition studies on carbonic anhydrase I and II isoenzymes. J Biomol Struct Dyn. (2021) 39(5):1672–80. doi: 10.1080/07391102.2020.1736631
24. Setiawati E, Yunaidi DA, Handayani LR, Harinanto G, Santoso ID, Deniati SH. Comparative bioavailability cf two amlodipine formulation in healthy volunteers. Arzneimittelforschung. (2007) 57(7):467–71. PMID 17803060.17803060
25. Wang T, Wang Y, Lin S, Fang L, Lou S, Zhao D, et al. Evaluation of pharmacokinetics and safety with bioequivalence of amlodipine in healthy Chinese volunteers: bioequivalence study findings. J Clin Lab Anal. (2020) 34(6):e23228. doi: 10.1002/jcla.23228
26. Das SK, Wang W, Zhabyeyev P, Basu R, McLean B, Fan D, et al. Iron-overload injury and cardiomyopathy in acquired and genetic models is attenuated by resveratrol therapy. Sci Rep. (2015) 5:18132. doi: 10.1038/srep18132
27. Das SK, Zhabyeyev P, Basu R, Patel VB, Dyck JRB, Kassiri Z, et al. Advanced iron-overload cardiomyopathy in a genetic murine model is rescued by resveratrol therapy. Biosci Rep. (2018) 38(1):BSR20171302. doi: 10.1042/BSR20171302
28. Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest. (2005) 115(8):2187–91. doi: 10.1172/JCI25049
29. Patel VB, Wang Z, Fan D, Zhabyeyev P, Basu R, Das SK, et al. Loss of p47phox subunit enhances susceptibility to biomechanical stress and heart failure because of dysregulation of cortactin and actin filaments. Circ Res. (2013) 112(12):1542–56. doi: 10.1161/CIRCRESAHA.111.300299
30. McLean BA, Zhabyeyev P, Patel VB, Basu R, Parajuli N, DesAulniers J, et al. PI3Kα is essential for the recovery from cre/tamoxifen cardiotoxicity and in myocardial insulin signalling but is not required for normal myocardial contractility in the adult heart. Cardiovasc Res. (2015) 105(3):292–303. doi: 10.1093/cvr/cvv016
31. Zhabyeyev P, McLean B, Chen X, Vanhaesebroeck B, Oudit GY. Inhibition of PI3Kinase-alpha is pro-arrhythmic and associated with enhanced late Na(+) current, contractility, and Ca(2+) release in murine hearts. J Mol Cell Cardiol. (2019) 132:98–109. doi: 10.1016/j.yjmcc.2019.05.008
32. Parajuli N, Patel VB, Wang W, Basu R, Oudit GY. Loss of NOX2 (gp91phox) prevents oxidative stress and progression to advanced heart failure. Clin Sci. (2014) 127(5):331–40. doi: 10.1042/CS20130787
33. Das SK, DesAulniers J, Dyck JR, Kassiri Z, Oudit GY. Resveratrol mediates therapeutic hepatic effects in acquired and genetic murine models of iron-overload. Liver Int. (2016) 36(2):246–57. doi: 10.1111/liv.12893
34. Ge SX, Son EW, Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-seq data. BMC Bioinform. (2018) 19(1):534. doi: 10.1186/s12859-018-2486-6
35. Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol. (2023) 20(1):7–23. doi: 10.1038/s41569-022-00735-4
36. Teng T, Kong CY, Huang R, Ma ZG, Hu C, Zhang X, et al. Mapping current research and identifying hotspots of ferroptosis in cardiovascular diseases. Front Cardiovasc Med. (2022) 9:1046377. doi: 10.3389/fcvm.2022.1046377
37. Zhang H, Jamieson KL, Grenier J, Nikhanj A, Tang Z, Wang F, et al. Myocardial iron deficiency and mitochondrial dysfunction in advanced heart failure in humans. J Am Heart Assoc. (2022) 11(11):e022853. doi: 10.1161/JAHA.121.022853
38. Das SK, Patel VB, Basu R, Wang W, DesAulniers J, Kassiri Z, et al. Females are protected from iron-overload cardiomyopathy independent of iron metabolism: key role of oxidative stress. J Am Heart Assoc. (2017) 6(1):e003456. doi: 10.1161/JAHA.116.003456
39. Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki MB, Nicoll AJ, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. (2008) 358(3):221–30. doi: 10.1056/NEJMoa073286
40. Murphy CJ, Oudit GY. Iron-overload cardiomyopathy: pathophysiology, diagnosis, and treatment. J Card Fail. (2010) 16(11):888–900. doi: 10.1016/j.cardfail.2010.05.009
41. Gujja P, Rosing DR, Tripodi DJ, Shizukuda Y. Iron overload cardiomyopathy: better understanding of an increasing disorder. J Am Coll Cardiol. (2010) 56(13):1001–12. doi: 10.1016/j.jacc.2010.03.083
42. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
43. Chan S, Chan GC, Ye J, Lian Q, Chen J, Yang M. Thrombopoietin protects cardiomyocytes from iron-overload induced oxidative stress and mitochondrial injury. Cell Physiol Biochem. (2015) 36(5):2063–71. doi: 10.1159/000430173
44. Davis MT, Bartfay WJ. Ebselen decreases oxygen free radical production and iron concentrations in the hearts of chronically iron-overloaded mice. Biol Res Nurs. (2004) 6(1):37–45. doi: 10.1177/1099800403261350
45. Packer M, O'Connor CM, Ghali JK, Pressler ML, Carson PE, Belkin RN, et al. Effect of amlodipine on morbidity and mortality in severe chronic heart failure. Prospective randomized amlodipine survival evaluation study group. N Engl J Med. (1996) 335(15):1107–14. doi: 10.1056/NEJM199610103351504
46. Packer M, Carson P, Elkayam U, Konstam MA, Moe G, O'Connor C, et al. Effect of amlodipine on the survival of patients with severe chronic heart failure due to a nonischemic cardiomyopathy: results of the PRAISE-2 study (prospective randomized amlodipine survival evaluation 2). JACC Heart Fail. (2013) 1(4):308–14. doi: 10.1016/j.jchf.2013.04.004
Keywords: iron overload, cardiomyopathy, hemochromatosis, calcium channel blockers, hemojuvelin
Citation: Zhabyeyev P, Sadasivan C, Shah S, Wang F and Oudit GY (2023) Amlodipine rescues advanced iron overload cardiomyopathy in hemojuvelin knockout murine model: Clinical implications. Front. Cardiovasc. Med. 10:1129349. doi: 10.3389/fcvm.2023.1129349
Received: 21 December 2022; Accepted: 28 March 2023;
Published: 21 April 2023.
Edited by:
John Lynn Jefferies, University of Tennessee Health Science Center (UTHSC), United States© 2023 Zhabyeyev, Sadasivan, Shah, Wang and Oudit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gavin Y. Oudit Z2F2aW4ub3VkaXRAdWFsYmVydGEuY2E=
Specialty Section: This article was submitted to General Cardiovascular Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.