 Hiroyuki Tokiwa
Hiroyuki Tokiwa Kazutaka Ueda
Kazutaka Ueda Eiki Takimoto
Eiki Takimoto
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 12 April 2023
Sec. Sex and Gender in Cardiovascular Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1127340
This article is part of the Research Topic Sex Differences and Cardiovascular Therapeutics View all 13 articles
Sexual dimorphism exists in the epidemiology of cardiovascular disease (CVD), which indicates the involvement of sexual hormones in the pathophysiology of CVD. In particular, ample evidence has demonstrated estrogen's protective effect on the cardiovascular system. While estrogen receptors, bound to estrogen, act as a transcription factor which regulates gene expressions by binding to the specific DNA sequence, a subpopulation of estrogen receptors localized at the plasma membrane induces activation of intracellular signaling, called “non-nuclear signaling” or “membrane-initiated steroid signaling of estrogen”. Although the precise molecular mechanism of non-nuclear signaling as well as its physiological impact was unclear for a long time, recent development of genetically modified animal models and pathway-selective estrogen receptor stimulant bring new insights into this pathway. We review the published experimental studies on non-nuclear signaling of estrogen, and summarize its role in cardiovascular system, especially focusing on: (1) the molecular mechanism of non-nuclear signaling; (2) the design of genetically modified animals and pathway-selective stimulant of estrogen receptor.
Cardiovascular disease (CVD) is the leading cause of death in many countries, and its total burden is increasing dramatically (1–3). Sexual dimorphism has been observed in various CVDs. Women are less susceptible to coronary artery disease than men; however, their morbidity increases after menopause, reaching male levels (4, 5). Furthermore, menopause is associated with an increased prevalence of metabolic syndrome, a CVD risk factor (6, 7). These findings suggest a cardiovascular protective role of female sex hormones, especially estrogen, which have been consistently reported in basic research. Although hormone replacement therapy (HRT) was expected to decrease CVDs in postmenopausal women, a randomized controlled trial (RCT) by the Women's Health Initiative failed to demonstrate an improvement in CVD morbidity and was terminated early owing to adverse events including breast cancer (8). However, subanalysis of the RCT revealed conjugated equine estrogens had a tendency to lower CVD morbidity in relatively early postmenopausal women (9). Additionally, oral estradiol administration suppressed carotid artery atherosclerosis when treatment was initiated within six years, but not ten or more years after menopause (10). These reports indicate that HRT can induce cardiovascular benefits with careful application and encourage further research on the molecular mechanisms of estrogen signaling.
Estrogen receptors (ERs) regulate gene expression as transcription factors in the nucleus, known as nuclear signaling. However, a subpopulation of ERs is present at the plasma membrane and initiates intracellular signaling, referred to as “non-nuclear signaling” or “membrane-initiated steroid signaling”. Despite their relatively small numbers compared to nuclear ERs (11, 12), an increasing body of evidence suggests the essential role of non-nuclear signaling in various physiological functions, including cardiovascular effects (13, 14). Additionally, G-protein-coupled estrogen receptor (GPER), a distinct subtype of ER, has been identified as another mediator of non-nuclear signaling.
In this article, we first describe the characteristics of ERs and the molecular mechanism of non-nuclear signaling. Next, the role of non-nuclear signaling in cardiovascular systems is discussed through studies using genetically modified animals and pathway-selective stimulators. A concise review of GPER is also provided.
Endogenous estrogens exert physiological effects by binding to their receptors (ERs). Two subtypes of ERs, ERα and ERβ, belong to the nuclear hormone receptor superfamily and share common structural characteristics (15–17). ER consists of six distinct domains (A to F domains) (18). The N-terminal A/B domains contain a transcriptional activation domain (AF1), which facilitates the transcriptional function of ER. The C domain is a DNA-binding domain (DBD) that interacts with a specific DNA sequence called estrogen response elements (EREs) located in the transcriptional regulatory region of estrogen-responsive genes. The D domain is a flexible hinge region between domains C and E, and contains a nuclear localization signal (NLS) and a nuclear export signal (NES). The E domain corresponds to the ligand-binding domain (LBD), which harbors another transcriptional activation domain (AF2). The C-terminal of ER is the F domain. Although ERα and ERβ are encoded by two genes, their C and E domains are highly homologous (19), while the other domains are relatively divergent (20). In addition, splicing variants of ERα and ERβ have distinct physiological functions (18).
Upon binding to their ligands, ERs undergo a conformational change and form a stable dimer (21–23), which then enter the nucleus guided by NLS (24–26). ERs regulate gene expression with associated coregulators (27–30), and phosphorylation of ER also enhances their transcriptional activity in a ligand-independent manner (31–33).
Estradiol (E2) is the most potent endogenous estrogen in pre-menopause women, whereas estrone (E1) plays a larger role after menopause, and estriol (E3) shows a greater importance during pregnancy (34). Estetrol (E4) is synthesized during pregnancy by fetal liver enzymes (35). Additionally, various natural and synthetic exogenous compounds act as ER ligands (36). A group of synthesized estrogenic compounds, known as selective estrogen receptor modulators (SERMs), exhibit dual functionality as both agonist and antagonist of ER in different organs due to tissue- or cell-specific difference in the recruitment of cofactors (36, 37). It has been reported that E4 exhibits the activity of an natural SERM (38).
Some oxysterols, which are oxygenated derivatives of cholesterol, function as ER ligands. 27-hydroxycholesterol (27HC) inhibits E2-induced nitric oxide synthase expression and re-endothelialization of murine carotid artery (39). In contrast, 27HC promotes breast cancer progression in an ER-dependent manner (40, 41), suggesting its characteristic as an endogenous SERM (42). 27HC also regulates bone homeostasis, partially mediated by ERs (43, 44). Similarly, 25-hydroxycholesterol exhibits ERα-mediated breast and ovarian cancer cell proliferation and prevents hypoxia-induced cardiomyocyte apoptosis (45).
In addition to their role in nuclear signaling, ERs also mediate rapid intracellular signaling. In 1967, Szego and Davis showed that estrogen increased cyclic adenosine monophosphate (cAMP) concentration in the rat uterus within minutes (46). Following studies documented rapid calcium uptake of endometrial cells after E2 administration and E2 binding to the cell membrane (47, 48). Further research has identified the existence of membrane-bound ERα and ERβ (11, 49, 50), which are responsible for rapid signaling, such as the activation of extracellular signal-regulated kinase (ERK), protein kinase B (PKB, also known as Akt), and endothelial nitric oxide synthase (eNOS) (51–54). This signaling is referred to as “non-nuclear signaling” or “membrane-initiated steroid signaling”. However, it should be noted that this signaling can also induces the transcriptional response subsequently (55, 56).
Palmitoylation, a posttranslational modification of ER plays an essential role in trafficking to the plasma membrane (57–60). A conserved amino acid motif in the E domain of ERα and ERβ is responsible for palmitoylation (61) by DHHC-7 and −21 (62). Caveolin-1, the main component of caveolae (63), is colocalized with ERα (64) and the amino acid substitution of S522A in ERα impairs the interaction with caveolin-1 and plasma membrane localization (65). Striatin, a scaffold protein, is another component of the signaling complex of membrane-bound ERα (66).
On the plasma membrane, ERs form functional modules with associated proteins (Figure 1), including G-protein. In human umbilical vein endothelial cells, membrane ERα interacts with Gα13, which activates the RhoA/Rho Kinase/Moesin pathway and induces cell migration (67). Furthermore, E2-bound membrane ERα links to Gαi-2/3 and stimulate particulate guanylate cyclase-A, which causes the generation of cyclic guanosine monophosphate (cGMP). In consequence, activated cGMP-dependent protein kinase (PKG)-I stimulates cystathionine γ-lyase in endothelial cells, resulting the rapid release of hydrogen sulfide, which acts as a vasodilator (68). Human ERα binds to Gαi and Gβγ at amino acids of 251–260 and 271–595 of ERα respectively. Disruption of the interaction of ERα with Gαi or Gβγ inhibits E2-induced Src and ERK phosphorylation (69). Point mutations in the Gαi-binding domain of ERα diminish E2-stimulated activation of ERK and eNOS (70).
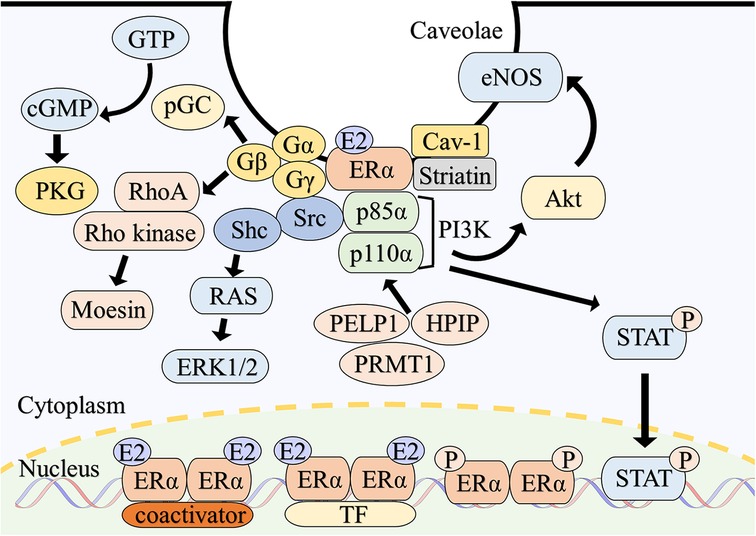
Figure 1. Overview of intracellular signaling of estrogen receptor α. Classically, E2-bound ERα dimerizes and translocate to the nucleus. ERα directly binds to estrogen response elements of the target genes with coactivators and modulates gene expressions. ERα also binds to DNA indirectly in association with other transcription factors. Phosphorylation of ERα also enhances transcriptional activity. A subpopulation of ERα is localized to the caveolae of the plasma membrane through the interaction with caveolin-1 and striatin. ERα on the plasma membrane assembles a functional complex with associated proteins such as G proteins, Src and PI3K, resulting in the rapid activation of multiple intracellular signaling. Abbreviations; Akt, protein kinase B; Cav-1, caveolin-1; cGMP, cyclic guanosine monophosphate; eNOS, endothelial nitric oxide synthase; E2, estradiol; ERK, extracellular signal-regulated kinase; ERα, estrogen receptor α; GTP, guanosine triphosphate; HPIP, hematopoietic PBX-interacting protein; PELP1, proline-, glutamic acid- and leucine-rich protein 1; pGC, particulate guanylate cyclase; PI3K, phosphatidylinositol-3 kinase; PKG, cGMP-dependent protein kinase; PRMT1, protein arginine methyltransferase 1; P, phosphorylation; Shc, src homology and collagen; STAT, signal transducer and activator of transcription; TF, transcription factor.
Src, a proto-oncogene, plays a critical role in Ras/ERK activation by E2-bound ERα (71). Src phosphorylates human ERα at Tyrosine 537 (72), and the SH2 domain of Src subsequently binds to ERα, modulating Src activity (73). A similar mechanism is observed with ERβ (73). Disruption of the ERα/Src association inhibits E2-induced proliferation of MCF-7 cell, which is an estrogen-responsive tumor cell (74). Src homology and collagen (Shc) is also contribute to ERK1/2 activation, with Src acting as an upstream regulator of Shc (75). Additionally, Proline-, glutamic acid- and leucine-rich protein 1 and hematopoietic PBX-interacting protein assist in the complex formation of ERα, Src and p85α subunit of phosphatidylinositol-3-OH kinase (PI3K) (76, 77). Protein arginine methyltransferase 1 is also involved in this process, mediating the methylation of ERα arginine 260 (78).
Nitric oxide production by eNOS is a vital function of endothelial cells (79). E2-bound ERα on the plasma membrane binds to the p85α subunit of PI3K and rapidly stimulates eNOS via the PI3K-Akt pathway (52, 80), primarily in caveolae (81). Gαi and heat shock protein 90 are also involved in E2-induced eNOS activation (82, 83).
While studies on non-nuclear ERβ signaling are limited, it has been reported that ERβ activates eNOS in endothelial cell caveolae (84). Additionally, both ERα and ERβ activate ERK1/2 and Akt in a subtype-specific manner (85).
Several genetically modified mouse models have been generated to investigate the role of non-nuclear signaling of ERα function (Table 1). Although the specific method of inhibiting non-nuclear signaling differ in each mouse model, these mice consistently exhibit a lack of rapid activation of eNOS by E2, which is known to play a pleiotropic role in maintaining cardiovascular homeostasis (107).
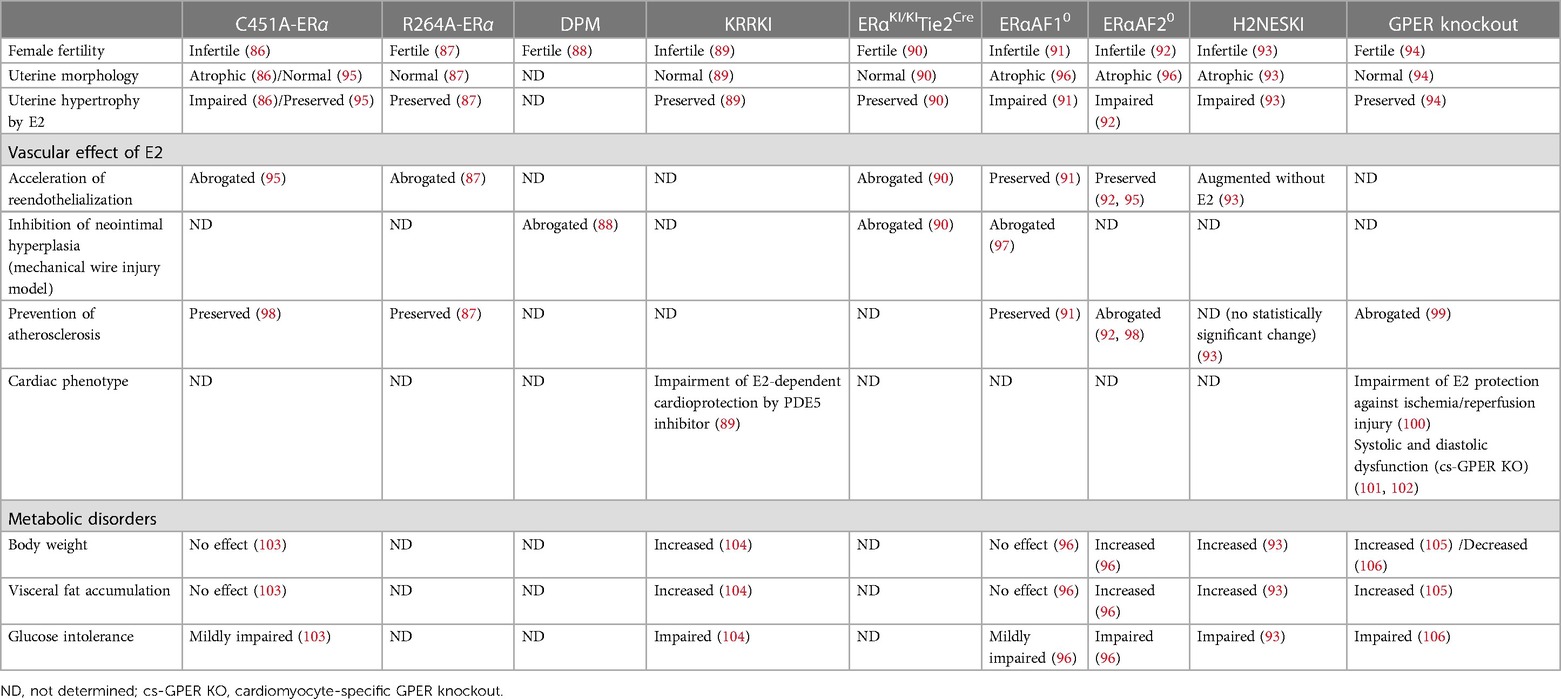
Table 1. Comparison of the phenotypes of genetically modified mouse models.
The C451A-ERα mouse model, which is characterized by the inhibition of palmitoylation and translocation of ERα to the plasma membrane by the substitution of cysteine 451 with alanine, exhibits a complete absence of membrane-localized ERα (86, 95), leading to the abrogation of non-nuclear signaling.
Based on the importance of human ERα amino acids 251–260 in non-nuclear signaling (69, 70), the R264A-ERα mouse model was generated by replacing arginine 264 of murine ERα with alanine, which corresponds to arginine 260 of human ERα (87). While the C451A-ERα female mouse exhibits infertility and impaired reproductive organ development (86), the R264A-ERα female mice remain fertile with intact reproductive organs, which suggests a difference in the degree of non-nuclear signaling inhibition between the two models.
Another mouse model of non-nuclear signaling inhibition was produced by disrupting the interaction between ERα and striatin, which is facilitated by amino acids 183–253 of human ERα and is crucial for the membrane localization of ERα (66). The disrupting peptide mouse (DPM) was generated through transgenic overexpression of a peptide containing the amino acid sequence of ERα 176–253, which disrupts the interaction between ERα and striatin. DPM mice exhibit a lack of rapid E2-induced phosphorylation of Akt or ERK in endothelial cells, resulting in the failure to activate eNOS (88).
By developing the concept of DPM mice, it was discovered the substitution of lysine 231, arginine 233 and 234 into alanine (KRR to AAA) of human ERα inhibits its interaction with striatin (108). In a human endothelial cell line with modified ERα, the rapid activation of ERK, Akt, and eNOS by E2 administration is abrogated, while the direct genomic reaction is preserved. To investigate the effects of this modification in vivo, a mouse model was established in which the endogenous ERα was replaced with the modified ERα (KRR knock-in: KRRKI mice) (104).
Recently, a novel mouse model called ERαKI/KITie2Cre mice was established. In this mouse model, ERα non-nuclear signaling is inactivated by disrupting its binding to the p85α subunit of PI3K through an arginine 263 to alanine mutation in a tissue-specific manner under the Cre-loxP system (90).
In this section, we summarize the findings obtained from these models and compare them with the mouse model with inactivated nuclear signaling, specifically with regards to the cardiovascular and metabolic systems.
Estrogen accelerates re-endothelialization and suppresses neointimal hyperplasia after vascular injury, with ERα playing an essential role (109, 110). Neither the C451A-ERα, R264A-ERα nor ERαKI/KITie2Cre mice exhibits E2-induced acceleration of re-endothelialization after electric perivascular injury (87, 90, 95). Similarly, E2 administration does not improve neointimal hyperplasia after mechanical wire injury in DPM or ERαKI/KITie2Cre mice (88, 90). In contrast, the mouse models with inactivated nuclear signaling by ERα AF1 or AF2 domain deletion (ERαAF10, ERαAF20) have demonstrated preserved E2 acceleration of carotid artery re-endothelialization after electric injury (91, 92, 95).
These findings suggest that ERα non-nuclear signaling plays the predominant role in the vascular protection by estrogen. This idea is supported by previous research demonstrating that E2 suppresses the proliferation of vascular smooth muscle cells (VSMCs), an underlying mechanism of neointimal hyperplasia (111). This effect is mediated through the formation of a complex of membrane ERα and striatin and protein phosphatase 2A (PP2A), leading to subsequent kinase inactivation (112). In VSMCs derived from DPM mice, the estrogen-induced complex formation and anti-proliferative effect is abrogated.
However, it is worth noting that E2-induced suppression of neointimal hyperplasia after mechanical wire injury is abolished in ERαAF10 mice (97). This result suggests that ERα nuclear signaling may also contribute to vascular protection or that there may be differences in the underlying biological mechanism of each vascular injury model.
The mouse models with genetic modification within D-domain provide valuable insight into the function of non-nuclear signaling in vascular protection. Amino acid substitution in the hinge region and NES of ERα D-domain alter the pattern of intracellular distribution of ERα. While wildtype ERα predominantly is localized in the nucleus, ERα with modifications in the putative NLS of the hinge region and NES (H2 + NES ERα) is exclusively localized in the cytoplasm. This altered localization is assumed to be caused by enhanced NES function, which is partially restored by leptomycin B, a nuclear export inhibitor. H2 + NES ERα exhibits impaired transcriptional activity but maintains a non-nuclear response of ERK1/2 phosphorylation after E2 administration (113, 114). The mouse model with mutated ERα (H2NESKI) exhibits an interesting cardiovascular phenotype where the degree of carotid artery re-endothelialization after electric injury is similar to that of E2-treated wild-type female mice, even in the absence of estrogen by ovariectomy. This observation suggests an intrinsically enhanced non-nuclear ERα signaling in H2NESKI mice (93).
Estrogen reduces the development of atherosclerotic lesion through ERα in mouse models of atherosclerosis with apolipoprotein E-deficient (Apoe−/−) or low-density lipoprotein receptor-deficient (Ldlr−/−) (115–118). While ERα non-nuclear signaling appears to play a critical role in protecting against vascular injury, ERα nuclear signaling is essential for preventing atherosclerosis by estrogen. This effect is preserved in C451A-ERα (98) and R264A-ERα mice (87), but abolished in ERαAF20 mice (92) that are crossed with Ldlr−/− mice. Notably, the AF1 domain of ERα appears to be dispensable for atherosclerosis prevention (91), suggesting the individual function of each domain.
Estrogen has been shown to suppress metabolic disorders, which are a common risk factor of CVDs. Female mice with whole-body ERα knockout exhibit several metabolic disorders, including glucose intolerance, body weight gain and visceral fat accumulation (96). Both nuclear and non-nuclear signaling of ERα appear to play a substantial role in these metabolic effects.
KRRKI mice also exhibits these dysfunctions, which are due to the disruption of the signal complex of membrane ERα, striatin, and PP2A in the hypothalamus, resulting in lower levels of physical activity and energy expenditure (104). In contrast, C451A-ERα mice only exhibit partial abnormalities (103), indicating that the mechanism of non-nuclear signaling inhibition may be relevant to the metabolic phenotype.
Regarding nuclear signaling, deletion of ERαAF2 induces similar abnormalities, with the disappearance of estrogen-regulated metabolic gene expression response to estrogen in the liver and adipose tissue. In contrast, deletion of ERαAF1 causes only mild hyperglycemia in the glucose tolerance test, suggesting a minor contribution of the AF1 domain (96).
E2 administration attenuates pressure overload-induced cardiac hypertrophy in female mice (119), as well as inhibits angiotensin II or endothelin-1-induced hypertrophy of neonatal rat cardiomyocytes (120). While ERβ seems to play the predominant role in mediating estrogen's protective effect against hypertrophy (121–124), ERα also contributes to this effect (125).
The KRRKI mouse model has shed light on the role of ERα non-nuclear signaling in pressure overload-induced heart failure (89, 126). Phosphodiesterase 5 (PDE5) inhibitors prevent cardiac remodeling in mice by myocardial PKG activation (127). Interestingly, the efficacy of PDE5 inhibitors in female is dependent on estrogen, which stimulates cGMP synthesis via the eNOS/soluble guanylate cyclase pathway (128). Notably, in female KRRKI mice, PDE5 inhibitors failed to activate PKG and provide cardiac protection against pressure overload-induced heart failure, even in the presence of estrogen (89).
Other mouse models with genetic modification within the DBD (129, 130), LBD (131), and a transgenic mouse expressing only a functional E domain of ERα at the plasma membrane (132) also provide meaningful insights into intracellular estrogen signaling. However, the effect of these genetic modifications on the cardiovascular system have not been determined.
Estrogenic compounds and estrogen derivatives that selectively stimulate a subpopulation of ERs also offer important insights into non-nuclear signaling. Estradiol-bovine serum albumin conjugate (E2-BSA), estrogen-dendrimer conjugate (EDC), and pathway-preferential estrogens (PaPEs) have been developed as selective stimulator of non-nuclear signaling and widely used in various studies, including cardiovascular research.
17β-estradiol conjugated to bovine serum albumin (E2-BSA) is membrane impermeable and therefore selectively stimulates cell-surface ERs. The exposure to E2-BSA leads to an increase in intracellular calcium and NO release in human artery endothelial cells, which is presumed to be mediated by non-nuclear signaling (133).
However, it should be noted that some criticisms have been raised regarding the suitability of E2-BSA as a tool to evaluate membrane-bound ER function: (1) E2-BSA solution contains some free E2 by cleaving from BSA, which may activate nuclear signaling; (2) the pattern of E2-BSA binding to ERs is influenced by the BSA linking site, which leads to different biological responses (134, 135).
A novel conjugate of estrogen and a polyamidoamine dendrimer was developed, in which the estrogens were linked to the dendrimer through a hydrolytically stable bond (136). This conjugate, known as estrogen-dendrimer conjugate (EDC), selectively stimulates ERs localized to the plasma membrane and cytoplasm since its positive charge and large size prevent it from entering the nucleus. EDC rapidly induces phosphorylation of ERK, Shc, and Src in MCF-7 cells, with limited impact on the expression of estrogen-responsive genes.
In bovine artery endothelial cells, EDC activates eNOS and promotes cell proliferation and migration. Moreover, EDC accelerates re-endothelialization of carotid artery after electric injury (137), which is consistent with the results of studies conducted on genetically modified mice.
In an ischemia/reperfusion model, E2 decreases cardiomyocyte apoptosis and infarct size (138, 139). Pretreatment with EDC similarly reduces infarct size and mitigates the decline in left ventricular function (140). This effect is accompanied by S-nitrosylation,of myocardial proteins, which plays an important role in cardioprotection (141).
It is noteworthy that continuous administration of EDC did not activate ER-mediated gene transcription in vivo, as evidenced by the bioluminescence assay using ERE-luciferase reporter mouse and real-time PCR (137). This finding not only confirms the high selectivity of EDC in stimulating non-nuclear signaling but also demonstrates the chemical stability of E2-dendrimer bound in EDC, which prevents the release of free E2.
A novel estrogen compound that preferentially activates a subset of ERs has been developed through a distinct mechanism from EDC (142). Typically, an initial signal triggered by transient ER-ligand binding is sufficient to activate non-nuclear signaling, in which subsequent kinase cascades play a predominant role. In contrast, activation of nuclear signaling often requires sustained ER-ligand binding to induce a series of subcellular processes. Therefore, it is anticipated that the modified estrogen, which possesses appropriately reduced affinity to ERs, will activate non-nuclear signaling effectively while avoiding the stimulation of nuclear signaling.
Pathway-preferential estrogens (PaPEs) are synthesized from estradiol by altering its steroid structure and adding modifications. PaPE-1 binds to ERα and ERβ 50,000 times less effectively than E2, while still retaining essential chemical features. PaPE-1 rapidly stimulates kinase phosphorylation in MCF-7 cells without directly activating genomic target genes. Additionally, PaPE-1 accelerated re-endothelialization of murine carotid arteries after electric injury (142).
In contrast to E2, neither PaPE-1 nor EDC prevents plaque formation in Ldlr−/− female mice fed a hypercholesterolemic diet (98). This result confirms the pivotal role of ER nuclear signaling in the atheroprotective effect of E2, as demonstrated by ERαAF20 mice (98).
GPER, called as GPR30 previously, constitutes a significant part of non-nuclear signaling of estrogen. GPER is a seven transmembrane G protein-coupled receptor (GPCR) of estrogen, identified in 1997 (143). GPER induces rapid activation of protein kinase A (PKA) as well as multiple signaling pathways that are also downstream of ERα (144–150).
Interestingly, GPER is not localized only at plasma membrane but also in intracellular compartments including endoplasmic reticulum and Golgi apparatus (151–99). The cellular distribution of GPER varies depending on the types of tissue and cells (151–154), and dynamically changes through intracellular trafficking (155, 156).
GPER mediates multiple physiological effects of estrogen on various organs, including the cardiovascular system. In contrast, it has been reported that GPER is dispensable for estrogenic effects in the reproductive system (94). Here, we outline the role of GPER in the cardiovascular system. For the detailed function and regulation of GPER, please refer to excellent comprehensive reviews elsewhere (157–159).
G-1, a selective agonist of GPER (160), induces acute eNOS activation in rat aorta and cultured aortic endothelial cells (161). G-1 also activates protein kinase A in vascular smooth muscle cells, phosphorylating myosin light chain kinase (MLCK), while increasing intracellular calcium concentration, resulting in the relaxation of smooth muscle cells and thus vasodilatation (162–164). GPER-deletion in mice abrogates vasodilatory response to G-1 (105) and results in greater arterial constriction after vasoconstrictor exposure (165, 166) and development of high blood pressure with age (106).
GPER also mediates vasculo-protective effects of estrogen against atherosclerosis. In ovary-intact female mice fed on atherogenic diet, GPER deletion reduces vascular NO bioavailability and aggravates aortic inflammation and atherosclerosis (99), which contrasts with the dispensable role of ERα non-nuclear signaling in atherosclerosis (87, 98). G-1 induces differentiation of smooth muscle cell and suppresses proliferation (167), which could lead to the amelioration of atherosclerosis given the major pathogenic role of dedifferentiated smooth muscle cells (168).
GPER stimulation also confers cardiac protection in a PI3K-dependent mechanism. The pretreatment with G-1 attenuates contractile dysfunction and reduces infract size following ischemia/reperfusion injury (169), which is also accompanied by the suppression of proinflammatory cytokines in myocardium (170) and the inhibition of calcium-induced mitochondria permeability transition pore opening (171). Consistently, GPER-deficient male mice lose the protective effect of E2 against ischemia/reperfusion injury (100).
G-1 inhibits angiotensin II-induced cardiomyocyte hypertrophy (172). Studies using genetic models support the role of GPER in the heart. Over-expression of GPER using adeno-associated virus with G-1 stimulation ameliorates cardiac remodeling from chronic pressure-overload (173). Consistently, cardiomyocyte-specific GPER knockout induces cardiac dysfunction with increased cardiac oxidative stress and collagen deposition in female mice (101, 102).
GPER-deficient female mice lack estrogenic response to insulin, exhibiting hyperglycemia and glucose intolerance (106). Another line of GPER-null animals shows obese phenotype with visceral fat accumulation (105).
The series of studies focusing on non-nuclear signaling brought a new perspective on intracellular signaling and expanded our understanding of estrogen function. Further research advance would lead to a therapeutic approach that effectively distinguish cardiovascular protective effects from unfavorable ones such as cancer-progression and thrombosis.
HT wrote the manuscript. KU and ET critically revised the manuscript and contributed to the conception and design of this review paper. All authors contributed to the article and approved the submitted version.
This work was supported by JSPS KAKENHI Grant Number JP22K16064 (HT) and JP21K08048 (ET).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J Am Coll Cardiol. (2020) 76(25):2982–3021. doi: 10.1016/j.jacc.2020.11.010
2. Vos T, Lim SS, Abbafati C, Abbas KM, Abbasi M, Abbasifard M, et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the global burden of disease study 2019. Lancet. (2020) 396(10258):1204–22. doi: 10.1016/s0140-6736(20)30925-9
3. Murray CJL, Aravkin AY, Zheng P, Abbafati C, Abbas KM, Abbasi-Kangevari M, et al. Global burden of 87 risk factors in 204 countries and territories, 1990-2019: a systematic analysis for the global burden of disease study 2019. Lancet. (2020) 396(10258):1223–49. doi: 10.1016/s0140-6736(20)30752-2
4. Kannel WB, Hjortland MC, McNamara PM, Gordon T. Menopause and risk of cardiovascular disease: the Framingham study. Ann Intern Med. (1976) 85(4):447–52. doi: 10.7326/0003-4819-85-4-447
5. Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. (2004) 364(9438):937–52. doi: 10.1016/s0140-6736(04)17018-9
6. Janssen I, Powell LH, Crawford S, Lasley B, Sutton-Tyrrell K. Menopause and the metabolic syndrome: the study of women's health across the nation. Arch Intern Med. (2008) 168(14):1568–75. doi: 10.1001/archinte.168.14.1568
7. Eshtiaghi R, Esteghamati A, Nakhjavani M. Menopause is an independent predictor of metabolic syndrome in Iranian women. Maturitas. (2010) 65(3):262–6. doi: 10.1016/j.maturitas.2009.11.004
8. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women's health initiative randomized controlled trial. Jama. (2002) 288(3):321–33. doi: 10.1001/jama.288.3.321
9. Hsia J, Langer RD, Manson JE, Kuller L, Johnson KC, Hendrix SL, et al. Conjugated equine estrogens and coronary heart disease: the women's health initiative. Arch Intern Med. (2006) 166(3):357–65. doi: 10.1001/archinte.166.3.357
10. Hodis HN, Mack WJ, Henderson VW, Shoupe D, Budoff MJ, Hwang-Levine J, et al. Vascular effects of early versus late postmenopausal treatment with estradiol. N Engl J Med. (2016) 374(13):1221–31. doi: 10.1056/NEJMoa1505241
11. Razandi M, Pedram A, Greene GL, Levin ER. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERalpha and ERbeta expressed in Chinese hamster ovary cells. Mol Endocrinol. (1999) 13(2):307–19. doi: 10.1210/mend.13.2.0239
12. Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. (2006) 20(9):1996–2009. doi: 10.1210/me.2005-0525
13. Puglisi R, Mattia G, Carè A, Marano G, Malorni W, Matarrese P. Non-genomic effects of estrogen on cell homeostasis and remodeling with special focus on cardiac ischemia/reperfusion injury. Front Endocrinol (Lausanne. (2019) 10:733. doi: 10.3389/fendo.2019.00733
14. Ueda K, Adachi Y, Liu P, Fukuma N, Takimoto E. Regulatory actions of estrogen receptor signaling in the cardiovascular system. Front Endocrinol (Lausanne). (2019) 10:909. doi: 10.3389/fendo.2019.00909
15. Evans RM. The steroid and thyroid hormone receptor superfamily. Science. (1988) 240(4854):889–95. doi: 10.1126/science.3283939
16. Giguère V, Yang N, Segui P, Evans RM. Identification of a new class of steroid hormone receptors. Nature. (1988) 331(6151):91–4. doi: 10.1038/331091a0
17. Arnal JF, Lenfant F, Metivier R, Flouriot G, Henrion D, Adlanmerini M, et al. Membrane and nuclear estrogen receptor alpha actions: from tissue specificity to medical implications. Physiol Rev. (2017) 97(3):1045–87. doi: 10.1152/physrev.00024.2016
18. Yaşar P, Ayaz G, User SD, Güpür G, Muyan M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod Med Biol. (2017) 16(1):4–20. doi: 10.1002/rmb2.12006
19. Mosselman S, Polman J, Dijkema R. ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett. (1996) 392(1):49–53. doi: 10.1016/0014-5793(96)00782-x
20. Cowley SM, Parker MG. A comparison of transcriptional activation by ER alpha and ER beta. J Steroid Biochem Mol Biol. (1999) 69(1-6):165–75. doi: 10.1016/s0960-0760(99)00055-2
21. Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engström O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. (1997) 389(6652):753–8. doi: 10.1038/39645
22. Pike AC, Brzozowski AM, Hubbard RE. A structural biologist's view of the oestrogen receptor. J Steroid Biochem Mol Biol. (2000) 74(5):261–8. doi: 10.1016/s0960-0760(00)00102-3
23. Tamrazi A, Carlson KE, Daniels JR, Hurth KM, Katzenellenbogen JA. Estrogen receptor dimerization: ligand binding regulates dimer affinity and dimer dissociation rate. Mol Endocrinol. (2002) 16(12):2706–19. doi: 10.1210/me.2002-0250
24. Ylikomi T, Bocquel MT, Berry M, Gronemeyer H, Chambon P. Cooperation of proto-signals for nuclear accumulation of estrogen and progesterone receptors. Embo J. (1992) 11(10):3681–94. doi: 10.1002/j.1460-2075.1992.tb05453.x
25. Echeverria PC, Picard D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim Biophys Acta. (2010) 1803(6):641–9. doi: 10.1016/j.bbamcr.2009.11.012
26. Sebastian T, Sreeja S, Thampan RV. Import and export of nuclear proteins: focus on the nucleocytoplasmic movements of two different species of mammalian estrogen receptor. Mol Cell Biochem. (2004) 260(1-2):91–102. doi: 10.1023/b:mcbi.0000026060.81570.35
27. Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. (2001) 29(14):2905–19. doi: 10.1093/nar/29.14.2905
28. Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. (2000) 103(6):843–52. doi: 10.1016/s0092-8674(00)00188-4
29. Safe S, Kim K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol. (2008) 41(5):263–75. doi: 10.1677/jme-08-0103
30. Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, et al. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. (2000) 74(5):311–7. doi: 10.1016/s0960-0760(00)00108-4
31. Ignar-Trowbridge DM, Teng CT, Ross KA, Parker MG, Korach KS, McLachlan JA. Peptide growth factors elicit estrogen receptor-dependent transcriptional activation of an estrogen-responsive element. Mol Endocrinol. (1993) 7(8):992–8. doi: 10.1210/mend.7.8.8232319
32. Márquez DC, Lee J, Lin T, Pietras RJ. Epidermal growth factor receptor and tyrosine phosphorylation of estrogen receptor. Endocrine. (2001) 16(2):73–81. doi: 10.1385/endo:16:2:073
33. Thomas RS, Sarwar N, Phoenix F, Coombes RC, Ali S. Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-alpha activity. J Mol Endocrinol. (2008) 40(4):173–84. doi: 10.1677/jme-07-0165
34. Cui J, Shen Y, Li R. Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends Mol Med. (2013) 19(3):197–209. doi: 10.1016/j.molmed.2012.12.007
35. Fruzzetti F, Fidecicchi T, Montt Guevara MM, Simoncini T. Estetrol: a new choice for contraception. J Clin Med. (2021) 10(23):5625. doi: 10.3390/jcm10235625
36. Farooq A. Structural and functional diversity of estrogen receptor ligands. Curr Top Med Chem. (2015) 15(14):1372–84. doi: 10.2174/1568026615666150413154841
37. Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. (2007) 87(3):905–31. doi: 10.1152/physrev.00026.2006
38. Abot A, Fontaine C, Buscato M, Solinhac R, Flouriot G, Fabre A, et al. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor α modulation, uncoupling nuclear and membrane activation. EMBO Mol Med. (2014) 6(10):1328–46. doi: 10.15252/emmm.201404112
39. Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, et al. 27-Hydroxycholesterol Is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat Med. (2007) 13(10):1185–92. doi: 10.1038/nm1641
40. DuSell CD, Umetani M, Shaul PW, Mangelsdorf DJ, McDonnell DP. 27-hydroxycholesterol Is an endogenous selective estrogen receptor modulator. Mol Endocrinol. (2008) 22(1):65–77. doi: 10.1210/me.2007-0383
41. Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, et al. 27-Hydroxycholesterol Links hypercholesterolemia and breast cancer pathophysiology. Science. (2013) 342(6162):1094–8. doi: 10.1126/science.1241908
42. Umetani M, Shaul PW. 27-Hydroxycholesterol: the first identified endogenous SERM. Trends Endocrinol Metab. (2011) 22(4):130–5. doi: 10.1016/j.tem.2011.01.003
43. DuSell CD, Nelson ER, Wang X, Abdo J, Mödder UI, Umetani M, et al. The endogenous selective estrogen receptor modulator 27-hydroxycholesterol is a negative regulator of bone homeostasis. Endocrinology. (2010) 151(8):3675–85. doi: 10.1210/en.2010-0080
44. Nelson ER, DuSell CD, Wang X, Howe MK, Evans G, Michalek RD, et al. The oxysterol, 27-hydroxycholesterol, links cholesterol metabolism to bone homeostasis through its actions on the estrogen and liver X receptors. Endocrinology. (2011) 152(12):4691–705. doi: 10.1210/en.2011-1298
45. Lappano R, Recchia AG, De Francesco EM, Angelone T, Cerra MC, Picard D, et al. The cholesterol metabolite 25-hydroxycholesterol activates estrogen receptor α-mediated signaling in cancer cells and in cardiomyocytes. PLoS One. (2011) 6(1):e16631. doi: 10.1371/journal.pone.0016631
46. Szego CM, Davis JS. Adenosine 3′,5′-monophosphate in rat uterus: acute elevation by estrogen. Proc Natl Acad Sci U S A. (1967) 58(4):1711–8. doi: 10.1073/pnas.58.4.1711
47. Pietras RJ, Szego CM. Endometrial cell calcium and oestrogen action. Nature. (1975) 253(5490):357–9. doi: 10.1038/253357a0
48. Pietras RJ, Szego CM. Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature. (1977) 265(5589):69–72. doi: 10.1038/265069a0
49. Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. Faseb j. (1995) 9(5):404–10. doi: 10.1096/fasebj.9.5.7896011
50. Norfleet AM, Thomas ML, Gametchu B, Watson CS. Estrogen receptor-alpha detected on the plasma membrane of aldehyde-fixed GH3/B6/F10 rat pituitary tumor cells by enzyme-linked immunocytochemistry. Endocrinology. (1999) 140(8):3805–14. doi: 10.1210/endo.140.8.6936
51. Chen DB, Bird IM, Zheng J, Magness RR. Membrane estrogen receptor-dependent extracellular signal-regulated kinase pathway mediates acute activation of endothelial nitric oxide synthase by estrogen in uterine artery endothelial cells. Endocrinology. (2004) 145(1):113–25. doi: 10.1210/en.2003-0547
52. Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, et al. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res. (2000) 87(8):677–82. doi: 10.1161/01.res.87.8.677
53. Caulin-Glaser T, García-Cardeña G, Sarrel P, Sessa WC, Bender JR. 17 beta-estradiol regulation of human endothelial cell basal nitric oxide release, independent of cytosolic Ca2 + mobilization. Circ Res. (1997) 81(5):885–92. doi: 10.1161/01.res.81.5.885
54. Lantin-Hermoso RL, Rosenfeld CR, Yuhanna IS, German Z, Chen Z, Shaul PW. Estrogen acutely stimulates nitric oxide synthase activity in fetal pulmonary artery endothelium. Am J Physiol. (1997) 273(1 Pt 1):L119–26. doi: 10.1152/ajplung.1997.273.1.L119
55. Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. (2008) 22(9):2116–27. doi: 10.1210/me.2008-0059
56. Björnström L, Sjöberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. (2005) 19(4):833–42. doi: 10.1210/me.2004-0486
57. Li L, Haynes MP, Bender JR. Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proc Natl Acad Sci U S A. (2003) 100(8):4807–12. doi: 10.1073/pnas.0831079100
58. Acconcia F, Ascenzi P, Fabozzi G, Visca P, Marino M. S-palmitoylation modulates human estrogen receptor-alpha functions. Biochem Biophys Res Commun. (2004) 316(3):878–83. doi: 10.1016/j.bbrc.2004.02.129
59. Acconcia F, Ascenzi P, Bocedi A, Spisni E, Tomasi V, Trentalance A, et al. Palmitoylation-dependent estrogen receptor alpha membrane localization: regulation by 17beta-estradiol. Mol Biol Cell. (2005) 16(1):231–7. doi: 10.1091/mbc.e04-07-0547
60. Galluzzo P, Caiazza F, Moreno S, Marino M. Role of ERbeta palmitoylation in the inhibition of human colon cancer cell proliferation. Endocr Relat Cancer. (2007) 14(1):153–67. doi: 10.1677/erc-06-0020
61. Pedram A, Razandi M, Sainson RC, Kim JK, Hughes CC, Levin ER. A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem. (2007) 282(31):22278–88. doi: 10.1074/jbc.M611877200
62. Pedram A, Razandi M, Deschenes RJ, Levin ER. DHHC-7 and -21 are palmitoylacyltransferases for sex steroid receptors. Mol Biol Cell. (2012) 23(1):188–99. doi: 10.1091/mbc.E11-07-0638
63. Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. (2013) 14(2):98–112. doi: 10.1038/nrm3512
64. Razandi M, Oh P, Pedram A, Schnitzer J, Levin ER. ERs associate with and regulate the production of caveolin: implications for signaling and cellular actions. Mol Endocrinol. (2002) 16(1):100–15. doi: 10.1210/mend.16.1.0757
65. Razandi M, Alton G, Pedram A, Ghonshani S, Webb P, Levin ER. Identification of a structural determinant necessary for the localization and function of estrogen receptor alpha at the plasma membrane. Mol Cell Biol. (2003) 23(5):1633–46. doi: 10.1128/mcb.23.5.1633-1646.2003
66. Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha. Proc Natl Acad Sci U S A. (2004) 101(49):17126–31. doi: 10.1073/pnas.0407492101
67. Simoncini T, Scorticati C, Mannella P, Fadiel A, Giretti MS, Fu XD, et al. Estrogen receptor alpha interacts with Galpha13 to drive actin remodeling and endothelial cell migration via the RhoA/Rho kinase/moesin pathway. Mol Endocrinol. (2006) 20(8):1756–71. doi: 10.1210/me.2005-0259
68. Xu X, Yan Q, Liu X, Li P, Li X, Chen Y, et al. 17β-Estradiol Nongenomically induces vascular endothelial H(2)S release by promoting phosphorylation of cystathionine γ-lyase. J Biol Chem. (2019) 294(43):15577–92. doi: 10.1074/jbc.RA119.008597
69. Kumar P, Wu Q, Chambliss KL, Yuhanna IS, Mumby SM, Mineo C, et al. Direct interactions with G α i and G βγ mediate nongenomic signaling by estrogen receptor α. Mol Endocrinol. (2007) 21(6):1370–80. doi: 10.1210/me.2006-0360
70. Wu Q, Chambliss K, Lee WR, Yuhanna IS, Mineo C, Shaul PW. Point mutations in the ERα Gαi binding domain segregate nonnuclear from nuclear receptor function. Mol Endocrinol. (2013) 27(1):2–11. doi: 10.1210/me.2011-1378
71. Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, et al. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. Embo j. (1996) 15(6):1292–300. doi: 10.1002/j.1460-2075.1996.tb00471.x
72. Arnold SF, Vorojeikina DP, Notides AC. Phosphorylation of tyrosine 537 on the human estrogen receptor is required for binding to an estrogen response element. J Biol Chem. (1995) 270(50):30205–12. doi: 10.1074/jbc.270.50.30205
73. Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. Embo J. (2000) 19(20):5406–17. doi: 10.1093/emboj/19.20.5406
74. Varricchio L, Migliaccio A, Castoria G, Yamaguchi H, de Falco A, Di Domenico M, et al. Inhibition of estradiol receptor/Src association and cell growth by an estradiol receptor alpha tyrosine-phosphorylated peptide. Mol Cancer Res. (2007) 5(11):1213–21. doi: 10.1158/1541-7786.Mcr-07-0150
75. Song RX, McPherson RA, Adam L, Bao Y, Shupnik M, Kumar R, et al. Linkage of rapid estrogen action to MAPK activation by ERalpha-Shc association and Shc pathway activation. Mol Endocrinol. (2002) 16(1):116–27. doi: 10.1210/mend.16.1.0748
76. Barletta F, Wong CW, McNally C, Komm BS, Katzenellenbogen B, Cheskis BJ. Characterization of the interactions of estrogen receptor and MNAR in the activation of cSrc. Mol Endocrinol. (2004) 18(5):1096–108. doi: 10.1210/me.2003-0335
77. Manavathi B, Acconcia F, Rayala SK, Kumar R. An inherent role of microtubule network in the action of nuclear receptor. Proc Natl Acad Sci U S A. (2006) 103(43):15981–6. doi: 10.1073/pnas.0607445103
78. Le Romancer M, Treilleux I, Leconte N, Robin-Lespinasse Y, Sentis S, Bouchekioua-Bouzaghou K, et al. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell. (2008) 31(2):212–21. doi: 10.1016/j.molcel.2008.05.025
79. Loscalzo J, Welch G. Nitric oxide and its role in the cardiovascular system. Prog Cardiovasc Dis. (1995) 38(2):87–104. doi: 10.1016/s0033-0620(05)80001-5
80. Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. (2000) 407(6803):538–41. doi: 10.1038/35035131
81. Chambliss KL, Yuhanna IS, Mineo C, Liu P, German Z, Sherman TS, et al. Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ Res. (2000) 87(11):E44–52. doi: 10.1161/01.res.87.11.e44
82. Wyckoff MH, Chambliss KL, Mineo C, Yuhanna IS, Mendelsohn ME, Mumby SM, et al. Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Galpha(i). J Biol Chem. (2001) 276(29):27071–6. doi: 10.1074/jbc.M100312200
83. Russell KS, Haynes MP, Caulin-Glaser T, Rosneck J, Sessa WC, Bender JR. Estrogen stimulates heat shock protein 90 binding to endothelial nitric oxide synthase in human vascular endothelial cells. Effects on calcium sensitivity and NO release. J Biol Chem. (2000) 275(7):5026–30. doi: 10.1074/jbc.275.7.5026
84. Chambliss KL, Yuhanna IS, Anderson RG, Mendelsohn ME, Shaul PW. ERbeta has nongenomic action in caveolae. Mol Endocrinol. (2002) 16(5):938–46. doi: 10.1210/mend.16.5.0827
85. Majumdar S, Rinaldi JC, Malhotra NR, Xie L, Hu DP, Gauntner TD, et al. Differential actions of estrogen receptor α and β via nongenomic signaling in human prostate stem and progenitor cells. Endocrinology. (2019) 160(11):2692–708. doi: 10.1210/en.2019-00177
86. Pedram A, Razandi M, Lewis M, Hammes S, Levin ER. Membrane-localized estrogen receptor α is required for normal organ development and function. Dev Cell. (2014) 29(4):482–90. doi: 10.1016/j.devcel.2014.04.016
87. Adlanmerini M, Fébrissy C, Zahreddine R, Vessières E, Buscato M, Solinhac R, et al. Mutation of arginine 264 on ERα (estrogen receptor alpha) selectively abrogates the rapid signaling of estradiol in the endothelium without altering fertility. Arterioscler Thromb Vasc Biol. (2020) 40(9):2143–58. doi: 10.1161/atvbaha.120.314159
88. Bernelot Moens SJ, Schnitzler GR, Nickerson M, Guo H, Ueda K, Lu Q, et al. Rapid estrogen receptor signaling is essential for the protective effects of estrogen against vascular injury. Circulation. (2012) 126(16):1993–2004. doi: 10.1161/circulationaha.112.124529
89. Fukuma N, Takimoto E, Ueda K, Liu P, Tajima M, Otsu Y, et al. Estrogen receptor-α non-nuclear signaling confers cardioprotection and is essential to cGMP-PDE5 inhibition efficacy. JACC Basic Transl Sci. (2020) 5(3):282–95. doi: 10.1016/j.jacbts.2019.12.009
90. Liu P-Y, Fukuma N, Hiroi Y, Kunita A, Tokiwa H, Ueda K, et al. Tie2-Cre–Induced inactivation of non-nuclear estrogen receptor-α signaling abrogates estrogen protection against vascular injury. JACC: Basic to Transl Sci. (2023) 8(1):55–67. doi: 10.1016/j.jacbts.2022.07.001
91. Billon-Galés A, Fontaine C, Filipe C, Douin-Echinard V, Fouque MJ, Flouriot G, et al. The transactivating function 1 of estrogen receptor alpha is dispensable for the vasculoprotective actions of 17beta-estradiol. Proc Natl Acad Sci U S A. (2009) 106(6):2053–8. doi: 10.1073/pnas.0808742106
92. Billon-Galés A, Krust A, Fontaine C, Abot A, Flouriot G, Toutain C, et al. Activation function 2 (AF2) of estrogen receptor-alpha is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc Natl Acad Sci U S A. (2011) 108(32):13311–6. doi: 10.1073/pnas.1105632108
93. Arao Y, Gruzdev A, Scott GJ, Ray MK, Donoghue LJ, Neufeld TI, et al. A novel mouse model to analyze non-genomic ERα physiological actions. J Endocrine Soc. (2022) 6(9):1–11. doi: 10.1210/jendso/bvac109
94. Otto C, Fuchs I, Kauselmann G, Kern H, Zevnik B, Andreasen P, et al. GPR30 Does not mediate estrogenic responses in reproductive organs in mice. Biol Reprod. (2009) 80(1):34–41. doi: 10.1095/biolreprod.108.071175
95. Adlanmerini M, Solinhac R, Abot A, Fabre A, Raymond-Letron I, Guihot AL, et al. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci U S A. (2014) 111(2):E283–90. doi: 10.1073/pnas.1322057111
96. Handgraaf S, Riant E, Fabre A, Waget A, Burcelin R, Lière P, et al. Prevention of obesity and insulin resistance by estrogens requires ERα activation function-2 (ERαAF-2), whereas ERαAF-1 is dispensable. Diabetes. (2013) 62(12):4098–108. doi: 10.2337/db13-0282
97. Smirnova NF, Fontaine C, Buscato M, Lupieri A, Vinel A, Valera MC, et al. The activation function-1 of estrogen receptor alpha prevents arterial neointima development through a direct effect on smooth muscle cells. Circ Res. (2015) 117(9):770–8. doi: 10.1161/circresaha.115.306416
98. Guivarc'h E, Buscato M, Guihot AL, Favre J, Vessières E, Grimaud L, et al. Predominant role of nuclear versus membrane estrogen receptor α in arterial protection: implications for estrogen receptor α modulation in cardiovascular prevention/safety. J Am Heart Assoc. (2018) 7(13):e008950. doi: 10.1161/jaha.118.008950
99. Meyer MR, Fredette NC, Howard TA, Hu C, Ramesh C, Daniel C, et al. G protein-coupled estrogen receptor protects from atherosclerosis. Sci Rep. (2014) 4:7564. doi: 10.1038/srep07564
100. Kabir ME, Singh H, Lu R, Olde B, Leeb-Lundberg LM, Bopassa JC. G protein-coupled estrogen receptor 1 mediates acute estrogen-induced cardioprotection via MEK/ERK/GSK-3β pathway after ischemia/reperfusion. PLoS One. (2015) 10(9):e0135988. doi: 10.1371/journal.pone.0135988
101. Wang H, Sun X, Chou J, Lin M, Ferrario CM, Zapata-Sudo G, et al. Cardiomyocyte-specific deletion of the G protein-coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: a sex-specific gene profiling analysis. Biochim Biophys Acta Mol Basis Dis. (2017) 1863(8):1870–82. doi: 10.1016/j.bbadis.2016.10.003
102. Wang H, Sun X, Lin MS, Ferrario CM, Van Remmen H, Groban L. G protein-coupled estrogen receptor (GPER) deficiency induces cardiac remodeling through oxidative stress. Transl Res. (2018) 199:39–51. doi: 10.1016/j.trsl.2018.04.005
103. Allard C, Morford JJ, Xu B, Salwen B, Xu W, Desmoulins L, et al. Loss of nuclear and membrane estrogen receptor-α differentially impairs insulin secretion and action in male and female mice. Diabetes. (2019) 68(3):490–501. doi: 10.2337/db18-0293
104. Ueda K, Takimoto E, Lu Q, Liu P, Fukuma N, Adachi Y, et al. Membrane-Initiated estrogen receptor signaling mediates metabolic homeostasis via central activation of protein phosphatase 2A. Diabetes. (2018) 67(8):1524–37. doi: 10.2337/db17-1342
105. Haas E, Bhattacharya I, Brailoiu E, Damjanović M, Brailoiu GC, Gao X, et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res. (2009) 104(3):288–91. doi: 10.1161/circresaha.108.190892
106. Mårtensson UE, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology. (2009) 150(2):687–98. doi: 10.1210/en.2008-0623
107. Farah C, Michel LYM, Balligand JL. Nitric oxide signalling in cardiovascular health and disease. Nat Rev Cardiol. (2018) 15(5):292–316. doi: 10.1038/nrcardio.2017.224
108. Lu Q, Schnitzler GR, Ueda K, Iyer LK, Diomede OI, Andrade T, et al. ER alpha rapid signaling is required for estrogen induced proliferation and migration of vascular endothelial cells. PLoS One. (2016) 11(4):e0152807. doi: 10.1371/journal.pone.0152807
109. Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, et al. Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circ Res. (2002) 90(10):1087–92. doi: 10.1161/01.res.0000021114.92282.fa
110. Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-alpha but not estrogen receptor-beta. Circulation. (2001) 103(3):423–8. doi: 10.1161/01.cir.103.3.423
111. Sivritas D, Becher MU, Ebrahimian T, Arfa O, Rapp S, Bohner A, et al. Antiproliferative effect of estrogen in vascular smooth muscle cells is mediated by Kruppel-like factor-4 and manganese superoxide dismutase. Basic Res Cardiol. (2011) 106(4):563–75. doi: 10.1007/s00395-011-0174-z
112. Ueda K, Lu Q, Baur W, Aronovitz MJ, Karas RH. Rapid estrogen receptor signaling mediates estrogen-induced inhibition of vascular smooth muscle cell proliferation. Arterioscler Thromb Vasc Biol. (2013) 33(8):1837–43. doi: 10.1161/atvbaha.112.300752
113. Burns KA, Li Y, Arao Y, Petrovich RM, Korach KS. Selective mutations in estrogen receptor alpha D-domain alters nuclear translocation and non-estrogen response element gene regulatory mechanisms. J Biol Chem. (2011) 286(14):12640–9. doi: 10.1074/jbc.M110.187773
114. Burns KA, Li Y, Liu L, Korach KS. Research resource: comparison of gene profiles from wild-type ERα and ERα hinge region mutants. Mol Endocrinol. (2014) 28(8):1352–61. doi: 10.1210/me.2014-1122
115. Bourassa PA, Milos PM, Gaynor BJ, Breslow JL, Aiello RJ. Estrogen reduces atherosclerotic lesion development in apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A. (1996) 93(19):10022–7. doi: 10.1073/pnas.93.19.10022
116. Elhage R, Arnal JF, Pieraggi MT, Duverger N, Fiévet C, Faye JC, et al. 17 beta-estradiol prevents fatty streak formation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (1997) 17(11):2679–84. doi: 10.1161/01.atv.17.11.2679
117. Hodgin JB, Krege JH, Reddick RL, Korach KS, Smithies O, Maeda N. Estrogen receptor alpha is a major mediator of 17beta-estradiol's atheroprotective effects on lesion size in Apoe-/- mice. J Clin Invest. (2001) 107(3):333–40. doi: 10.1172/jci11320
118. Billon-Galés A, Fontaine C, Douin-Echinard V, Delpy L, Berges H, Calippe B, et al. Endothelial estrogen receptor-alpha plays a crucial role in the atheroprotective action of 17 beta-estradiol in low-density lipoprotein receptor-deficient mice. Circulation. (2009) 120(25):2567–76. doi: 10.1161/circulationaha.109.898445
119. van Eickels M, Grohé C, Cleutjens JP, Janssen BJ, Wellens HJ, Doevendans PA. 17beta-estradiol Attenuates the development of pressure-overload hypertrophy. Circulation. (2001) 104(12):1419–23. doi: 10.1161/hc3601.095577
120. Pedram A, Razandi M, Aitkenhead M, Levin ER. Estrogen inhibits cardiomyocyte hypertrophy in vitro. Antagonism of calcineurin-related hypertrophy through induction of MCIP1. J Biol Chem. (2005) 280(28):26339–48. doi: 10.1074/jbc.M414409200
121. Skavdahl M, Steenbergen C, Clark J, Myers P, Demianenko T, Mao L, et al. Estrogen receptor-beta mediates male-female differences in the development of pressure overload hypertrophy. Am J Physiol Heart Circ Physiol. (2005) 288(2):H469–76. doi: 10.1152/ajpheart.00723.2004
122. Pedram A, Razandi M, Lubahn D, Liu J, Vannan M, Levin ER. Estrogen inhibits cardiac hypertrophy: role of estrogen receptor-beta to inhibit calcineurin. Endocrinology. (2008) 149(7):3361–9. doi: 10.1210/en.2008-0133
123. Pedram A, Razandi M, O'Mahony F, Lubahn D, Levin ER. Estrogen receptor-beta prevents cardiac fibrosis. Mol Endocrinol. (2010) 24(11):2152–65. doi: 10.1210/me.2010-0154
124. Menazza S, Murphy E. The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ Res. (2016) 118(6):994–1007. doi: 10.1161/circresaha.115.305376
125. Pelzer T, Jazbutyte V, Hu K, Segerer S, Nahrendorf M, Nordbeck P, et al. The estrogen receptor-alpha agonist 16alpha-LE2 inhibits cardiac hypertrophy and improves hemodynamic function in estrogen-deficient spontaneously hypertensive rats. Cardiovasc Res. (2005) 67(4):604–12. doi: 10.1016/j.cardiores.2005.04.035
126. Ueda K, Fukuma N, Adachi Y, Numata G, Tokiwa H, Toyoda M, et al. Sex differences and regulatory actions of estrogen in cardiovascular system. Front Physiol. (2021) 12:738218. doi: 10.3389/fphys.2021.738218
127. Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. (2005) 11(2):214–22. doi: 10.1038/nm1175
128. Sasaki H, Nagayama T, Blanton RM, Seo K, Zhang M, Zhu G, et al. PDE5 Inhibitor efficacy is estrogen dependent in female heart disease. J Clin Invest. (2014) 124(6):2464–71. doi: 10.1172/jci70731
129. Jakacka M, Ito M, Martinson F, Ishikawa T, Lee EJ, Jameson JL. An estrogen receptor (ER)alpha deoxyribonucleic acid-binding domain knock-in mutation provides evidence for nonclassical ER pathway signaling in vivo. Mol Endocrinol. (2002) 16(10):2188–201. doi: 10.1210/me.2001-0174
130. Ahlbory-Dieker DL, Stride BD, Leder G, Schkoldow J, Trölenberg S, Seidel H, et al. DNA Binding by estrogen receptor-alpha is essential for the transcriptional response to estrogen in the liver and the uterus. Mol Endocrinol. (2009) 23(10):1544–55. doi: 10.1210/me.2009-0045
131. Sinkevicius KW, Burdette JE, Woloszyn K, Hewitt SC, Hamilton K, Sugg SL, et al. An estrogen receptor-alpha knock-in mutation provides evidence of ligand-independent signaling and allows modulation of ligand-induced pathways in vivo. Endocrinology. (2008) 149(6):2970–9. doi: 10.1210/en.2007-1526
132. Pedram A, Razandi M, Kim JK, O'Mahony F, Lee EY, Luderer U, et al. Developmental phenotype of a membrane only estrogen receptor alpha (MOER) mouse. J Biol Chem. (2009) 284(6):3488–95. doi: 10.1074/jbc.M806249200
133. Stefano GB, Prevot V, Beauvillain JC, Cadet P, Fimiani C, Welters I, et al. Cell-surface estrogen receptors mediate calcium-dependent nitric oxide release in human endothelia. Circulation. (2000) 101(13):1594–7. doi: 10.1161/01.cir.101.13.1594
134. Stevis PE, Deecher DC, Suhadolnik L, Mallis LM, Frail DE. Differential effects of estradiol and estradiol-BSA conjugates. Endocrinology. (1999) 140(11):5455–8. doi: 10.1210/endo.140.11.7247
135. Temple JL, Wray S. Bovine serum albumin-estrogen compounds differentially alter gonadotropin-releasing hormone-1 neuronal activity. Endocrinology. (2005) 146(2):558–63. doi: 10.1210/en.2004-1117
136. Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, et al. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol. (2006) 20(3):491–502. doi: 10.1210/me.2005-0186
137. Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. (2010) 120(7):2319–30. doi: 10.1172/jci38291
138. Patten RD, Pourati I, Aronovitz MJ, Baur J, Celestin F, Chen X, et al. 17beta-estradiol Reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ Res. (2004) 95(7):692–9. doi: 10.1161/01.Res.0000144126.57786.89
139. Hale SL, Birnbaum Y, Kloner RA. beta-Estradiol, but not alpha-estradiol, reduced myocardial necrosis in rabbits after ischemia and reperfusion. Am Heart J. (1996) 132(2 Pt 1):258–62. doi: 10.1016/s0002-8703(96)90419-6
140. Menazza S, Sun J, Appachi S, Chambliss KL, Kim SH, Aponte A, et al. Non-nuclear estrogen receptor alpha activation in endothelium reduces cardiac ischemia-reperfusion injury in mice. J Mol Cell Cardiol. (2017) 107:41–51. doi: 10.1016/j.yjmcc.2017.04.004
141. Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. (2007) 101(11):1155–63. doi: 10.1161/circresaha.107.155879
142. Madak-Erdogan Z, Kim SH, Gong P, Zhao YC, Zhang H, Chambliss KL, et al. Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci Signal. (2016) 9(429):ra53. doi: 10.1126/scisignal.aad8170
143. Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. (1997) 45(3):607–17. doi: 10.1006/geno.1997.4972
144. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. (2005) 146(2):624–32. doi: 10.1210/en.2004-1064
145. Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. (2000) 14(10):1649–60. doi: 10.1210/mend.14.10.0532
146. Filardo EJ, Quinn JA, Frackelton AR Jr., Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. (2002) 16(1):70–84. doi: 10.1210/mend.16.1.0758
147. Zhou X, Wang S, Wang Z, Feng X, Liu P, Lv XB, et al. Estrogen regulates Hippo signaling via GPER in breast cancer. J Clin Invest. (2015) 125(5):2123–35. doi: 10.1172/jci79573
148. Petrie WK, Dennis MK, Hu C, Dai D, Arterburn JB, Smith HO, et al. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet Gynecol Int. (2013) 2013:472720. doi: 10.1155/2013/472720
149. Fredette NC, Meyer MR, Prossnitz ER. Role of GPER in estrogen-dependent nitric oxide formation and vasodilation. J Steroid Biochem Mol Biol. (2018) 176:65–72. doi: 10.1016/j.jsbmb.2017.05.006
150. Hsieh YC, Yu HP, Frink M, Suzuki T, Choudhry MA, Schwacha MG, et al. G protein-coupled receptor 30-dependent protein kinase A pathway is critical in nongenomic effects of estrogen in attenuating liver injury after trauma-hemorrhage. Am J Pathol. (2007) 170(4):1210–8. doi: 10.2353/ajpath.2007.060883
151. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. (2005) 307(5715):1625–30. doi: 10.1126/science.1106943
152. Revankar CM, Mitchell HD, Field AS, Burai R, Corona C, Ramesh C, et al. Synthetic estrogen derivatives demonstrate the functionality of intracellular GPR30. ACS Chem Biol. (2007) 2(8):536–44. doi: 10.1021/cb700072n
153. Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. (2006) 346(3):904–10. doi: 10.1016/j.bbrc.2006.05.191
154. Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, et al. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology. (2007) 148(7):3236–45. doi: 10.1210/en.2006-1605
155. Cheng SB, Graeber CT, Quinn JA, Filardo EJ. Retrograde transport of the transmembrane estrogen receptor, G-protein-coupled-receptor-30 (GPR30/GPER) from the plasma membrane towards the nucleus. Steroids. (2011) 76(9):892–6. doi: 10.1016/j.steroids.2011.02.018
156. Cheng SB, Quinn JA, Graeber CT, Filardo EJ. Down-modulation of the G-protein-coupled estrogen receptor, GPER, from the cell surface occurs via a trans-Golgi-proteasome pathway. J Biol Chem. (2011) 286(25):22441–55. doi: 10.1074/jbc.M111.224071
157. Prossnitz ER, Barton M. Estrogen biology: new insights into GPER function and clinical opportunities. Mol Cell Endocrinol. (2014) 389(1-2):71–83. doi: 10.1016/j.mce.2014.02.002
158. Luo J, Liu D. Does GPER really function as a G protein-coupled estrogen receptor in vivo? Front Endocrinol (Lausanne). (2020) 11:148. doi: 10.3389/fendo.2020.00148
159. Rouhimoghadam M, Lu AS, Salem AK, Filardo EJ. Therapeutic perspectives on the modulation of G-protein coupled estrogen receptor, GPER, function. Front Endocrinol (Lausanne). (2020) 11:591217. doi: 10.3389/fendo.2020.591217
160. Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. (2006) 2(4):207–12. doi: 10.1038/nchembio775
161. Li ZL, Liu JC, Liu SB, Li XQ, Yi DH, Zhao MG. Improvement of vascular function by acute and chronic treatment with the GPR30 agonist G1 in experimental diabetes mellitus. PLoS One. (2012) 7(6):e38787. doi: 10.1371/journal.pone.0038787
162. Lindsey SH, Liu L, Chappell MC. Vasodilation by GPER in mesenteric arteries involves both endothelial nitric oxide and smooth muscle cAMP signaling. Steroids. (2014) 81:99–102. doi: 10.1016/j.steroids.2013.10.017
163. Pfitzer G. Invited review: regulation of myosin phosphorylation in smooth muscle. J Appl Physiol (1985). (2001) 91(1):497–503. doi: 10.1152/jappl.2001.91.1.497
164. Meyer MR, Baretella O, Prossnitz ER, Barton M. Dilation of epicardial coronary arteries by the G protein-coupled estrogen receptor agonists G-1 and ICI 182,780. Pharmacology. (2010) 86(1):58–64. doi: 10.1159/000315497
165. Meyer MR, Amann K, Field AS, Hu C, Hathaway HJ, Kanagy NL, et al. Deletion of G protein-coupled estrogen receptor increases endothelial vasoconstriction. Hypertension. (2012) 59(2):507–12. doi: 10.1161/hypertensionaha.111.184606
166. Meyer MR, Field AS, Kanagy NL, Barton M, Prossnitz ER. GPER Regulates endothelin-dependent vascular tone and intracellular calcium. Life Sci. (2012) 91(13-14):623–7. doi: 10.1016/j.lfs.2012.01.007
167. Li F, Yu X, Szynkarski CK, Meng C, Zhou B, Barhoumi R, et al. Activation of GPER induces differentiation and inhibition of coronary artery smooth muscle cell proliferation. PLoS One. (2013) 8(6):e64771. doi: 10.1371/journal.pone.0064771
168. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. (2004) 84(3):767–801. doi: 10.1152/physrev.00041.2003
169. Deschamps AM, Murphy E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Physiol Heart Circ Physiol. (2009) 297(5):H1806–13. doi: 10.1152/ajpheart.00283.2009
170. Weil BR, Manukyan MC, Herrmann JL, Wang Y, Abarbanell AM, Poynter JA, et al. Signaling via GPR30 protects the myocardium from ischemia/reperfusion injury. Surgery. (2010) 148(2):436–43. doi: 10.1016/j.surg.2010.03.011
171. Bopassa JC, Eghbali M, Toro L, Stefani E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. (2010) 298(1):H16–23. doi: 10.1152/ajpheart.00588.2009
172. Pei H, Wang W, Zhao D, Su H, Su G, Zhao Z. G protein-coupled estrogen receptor 1 inhibits angiotensin II-induced cardiomyocyte hypertrophy via the regulation of PI3K-akt-mTOR signalling and autophagy. Int J Biol Sci. (2019) 15(1):81–92. doi: 10.7150/ijbs.28304
173. Wang X, Ma J, Zhang S, Li Z, Hong Z, Jiang L, et al. G protein-coupled estrogen receptor 30 reduces transverse aortic constriction-induced myocardial fibrosis in aged female mice by inhibiting the ERK1/2 -MMP-9 signaling pathway. Front Pharmacol. (2021) 12:731609. doi: 10.3389/fphar.2021.731609
Keywords: estrogen, non-nuclear signaling, cardiovascular disease, genetically modified animal, membrane-initiated steroid signaling
Citation: Tokiwa H, Ueda K and Takimoto E (2023) The emerging role of estrogen's non-nuclear signaling in the cardiovascular disease. Front. Cardiovasc. Med. 10:1127340. doi: 10.3389/fcvm.2023.1127340
Received: 19 December 2022; Accepted: 24 March 2023;
Published: 12 April 2023.
Edited by:
Mohsin Khan, Temple University, United StatesReviewed by:
Michihisa Umetani, University of Houston, United States© 2023 Tokiwa, Ueda and Takimoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eiki Takimoto ZXRha2ltbzFAamhtaS5lZHU=
Specialty Section: This article was submitted to Sex and Gender in Cardiovascular Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.