
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 02 May 2023
Sec. Atherosclerosis and Vascular Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1116861
This article is part of the Research TopicInsights in Atherosclerosis and Vascular Medicine: 2022View all 23 articles
 Alexandra C. Finney1
Alexandra C. Finney1 Sandeep Das1
Sandeep Das1 Dhananjay Kumar2M. Peyton McKinney1
Dhananjay Kumar2M. Peyton McKinney1 Bishuang Cai3
Bishuang Cai3 Arif Yurdagul Jr1,2*†
Arif Yurdagul Jr1,2*† Oren Rom1,2*†
Oren Rom1,2*†
Therapeutic approaches that lower circulating low-density lipoprotein (LDL)-cholesterol significantly reduced the burden of cardiovascular disease over the last decades. However, the persistent rise in the obesity epidemic is beginning to reverse this decline. Alongside obesity, the incidence of nonalcoholic fatty liver disease (NAFLD) has substantially increased in the last three decades. Currently, approximately one third of world population is affected by NAFLD. Notably, the presence of NAFLD and particularly its more severe form, nonalcoholic steatohepatitis (NASH), serves as an independent risk factor for atherosclerotic cardiovascular disease (ASCVD), thus, raising interest in the relationship between these two diseases. Importantly, ASCVD is the major cause of death in patients with NASH independent of traditional risk factors. Nevertheless, the pathophysiology linking NAFLD/NASH with ASCVD remains poorly understood. While dyslipidemia is a common risk factor underlying both diseases, therapies that lower circulating LDL-cholesterol are largely ineffective against NASH. While there are no approved pharmacological therapies for NASH, some of the most advanced drug candidates exacerbate atherogenic dyslipidemia, raising concerns regarding their adverse cardiovascular consequences. In this review, we address current gaps in our understanding of the mechanisms linking NAFLD/NASH and ASCVD, explore strategies to simultaneously model these diseases, evaluate emerging biomarkers that may be useful to diagnose the presence of both diseases, and discuss investigational approaches and ongoing clinical trials that potentially target both diseases.
Despite the remarkable advances in interventional therapeutics, decades of basic science and clinical research, atherosclerotic cardiovascular disease (ASCVD) remains the leading cause of death worldwide (1). While the overarching pathoetiology largely arises from dyslipidemia, the imbalance of cholesterol and triglyceride lipids, numerous comorbidities complicate and exacerbate ASCVD (1). Of particular significance are metabolic- and obesity-related diseases, which have globally increased prevalence since the 1970s (2). Nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) are also strongly associated with the metabolic syndrome (3), which currently afflicts approximately 90% of obese patients (4) and approximately 55% of individuals with type 2 diabetes (T2D) (5). Globally, the incidence of NAFLD has increased from 25% in 2005 to 32% today (6), highlighting an alarming trend in rising NAFLD burden. Despite this, no FDA-approved drug exists in the treatment of NAFLD/NASH. While NAFLD is associated with increased risk of liver-related mortality, the most common cause of death in patients with NAFLD, particularly those with the more severe NASH, is cardiovascular disease (CVD) (7–12). This, combined with the rising prevalence of both ASCVD and NAFLD has led to extensive discussion of the relationship between these two diseases. In 2022 alone, the increasingly transparent relationship between NAFLD/NASH and ASCVD has piqued interest between multiple scientific fields of expertise (13–17), culminating in a scientific statement from the American Heart Association (8).
Despite this acknowledgement, the specific mechanisms regulating the onset, crosstalk, and exacerbation of NAFLD and ASCVD remain unclear. The reasons for this are multifactorial: (1) there is no single established model to study NAFLD/NASH and ASCVD simultaneously, (2) since most patients with NAFLD/NASH and ASCVD are asymptomatic, diagnosis is often incidental and limited to routine blood screening (e.g., plasma lipids, liver transaminases) (18), calcium imaging (19), or less routinely, biopsy (20), and (3) clinical trials have remained limited in targeting either NASH or atherosclerosis, thus, it is unknown whether current clinical trials for NASH affect cardiovascular outcome or vice versa. For example, obeticholic acid, the most advanced drug candidate for NASH, causes hyperlipidemia, raising concerns about the possible adverse consequences on ASCVD (21). Furthermore, the effect of traditional therapies for ASCVD, e.g., statins, on NASH has shown inconsistent results in improving histological features of NASH (22, 23). Thus, strategies that simultaneously interrogate therapies for both NASH and ASCVD are necessary. This review will provide insight into each of these limitations, offering a comprehensive and current summary of our understanding regarding the relationship between NAFLD/NASH and ASCVD (Figure 1). Below, we (1) summarize the molecular drivers that regulate ASCVD and NAFLD/NASH, (2) discuss which animal models should be considered for evaluating translational interpretation of preclinical findings, (3) review emerging biomarkers for both NASH and atherosclerosis that may also serve as therapeutic strategies, and (4) examine potential limitations and caveats for the concurrent treatment of both NASH and ASCVD.
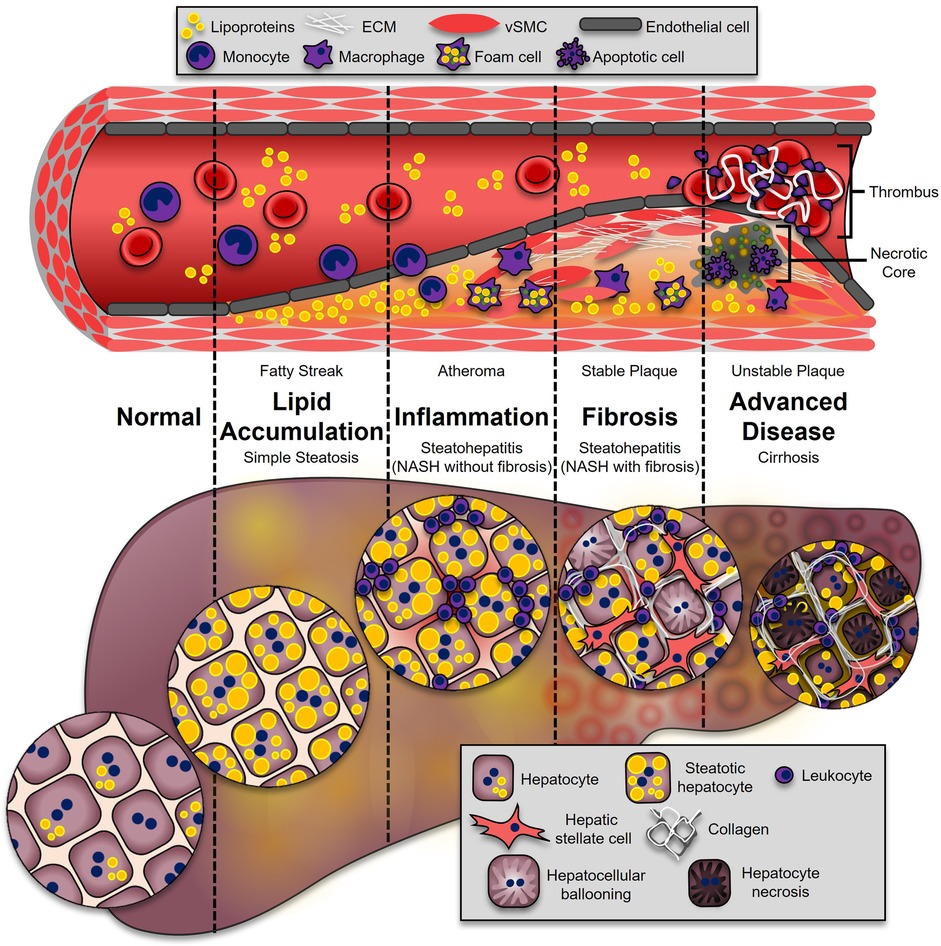
Figure 1. Progression of ASCVD and NASH. The onset of both ASCVD and NASH begins with dysregulated lipid metabolism, leading to their accumulation in the neointimal region of the artery (fatty streak), or the hepatocytes (simple hepatic steatosis). This process enhances inflammatory pathways in both diseases. During atherosclerosis, leukocytes adhere and transmigrate into the developing plaque, where they secrete additional cytokines and chemokines (atheroma). In the liver, leukocytes from the circulation accumulate, leading to NASH (NASH without fibrosis). These immune cells secrete soluble factors to activate collagen-producing cells: synthetic vascular smooth muscle cells (vSMCs) in atherosclerosis (stable plaque), and hepatic stellate cells in the liver (NASH with fibrosis). The most advanced stages of disease are associated with higher mortality. In atherosclerosis, advanced plaques with a large necrotic core and thin fibrous caps are prone to rupture (unstable plaque), which is highly thrombogenic. In the liver, excessive fibrosis and cell death leads to irreversible damage and loss of liver function (cirrhosis).
Most cases of myocardial infarction and stroke are caused by atherosclerosis, the fibrofatty plaques in the arterial branch of the vascular tree (24). The formation of atherosclerotic plaques is driven primarily by the deposition of apolipoprotein (Apo)-B-containing lipoproteins in the subendothelial spaces of the intima that subsequently drive maladaptive, non-resolving inflammation (25). Thus, individuals with familial hypercholesterolemia, particularly in the low-density lipoprotein (LDL) fraction, are disposed to developing atherosclerotic plaques at an early age (26). Other risk factors include insulin resistance and metabolic syndrome (27). Advanced atherosclerotic plaques contain vast amounts of extracellular matrix (ECM) proteins, calcium minerals, and a large necrotic core formed from the death of lipoprotein-rich monocyte-derived macrophages. These advanced atherosclerotic plaques can impede blood flow to downstream tissues through occlusion of the vessel lumen, causing symptomatic ischemia (24). More frequently however, atherosclerotic plaques rupture and leak the highly thrombogenic contents from the necrotic core into the lumen, resulting in an occlusive thrombus. Deaths from ASCVD were declining over the last two decades as treating more individuals for high LDL (∼28% in 1999–2002 to ∼48% in 2005–2008) resulted in twice as many individuals successfully lowering their circulating LDL-cholesterol from ∼15% to ∼33% (28). Despite the advent of potent cholesterol-lowering medicines, such as statins and anti-proprotein convertase subtilisin/kexin type 9 (PCSK9)-blocking antibodies, ASCVD remains the leading cause of death worldwide. More troubling is the recent trend that life expectancy growth has begun to decline, with a substantial rise in CVD deaths having the most impact (29). Thus, a deeper understanding of the cellular and molecular mechanisms driving atherosclerosis is necessary to conceive novel therapeutic strategies.
LDL particles accumulate in the subendothelial intima due to increased endothelial cell permeability caused by disturbed blood flow (30). Apart from the antioxidant environment normally provided by the blood, LDL particles become oxidized (ox-LDL) by unmitigated reactive oxygen species (ROS) production, leading to its uptake by scavenger receptors (31, 32). Unlike the LDL receptor (LDLR), scavenger receptors undergo positive feedback that maintains persistent cellular uptake of ox-LDL (33). Endothelial cells that take up ox-LDL activate the proinflammatory transcription factor nuclear factor-κB (NF-κB) that drive the expression of adhesion molecules, such as intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1) (34). These adhesion molecules presented on the apical surface of endothelial cells bind circulating leukocytes and promote their entry into the vessel wall. The role of endothelial cell activation in promoting atherosclerosis is crucial, as atherosclerosis formation tends to only occur at sites of disturbed blood flow, such as curvatures, branch points, and bifurcations, and experimental strategies that prevent endothelial cell activation prevent atherosclerosis formation (30).
Vascular smooth muscle cells (vSMCs) regulate blood pressure and vessel integrity under normal conditions (35). However, during early atherosclerosis, vSMCs undergo dedifferentiation whereby they lose canonical vSMC markers, such as α smooth muscle actin (αSMA) and transgelin (SM22), and reignite signaling pathways associated with development (36). Furthermore, dedifferentiated vSMCs begin to migrate, proliferate, and synthesize ECM proteins, thereby expanding the growing lesion towards the lumen of the vessel. Interestingly, vSMCs produce a panoply of ECM proteins that can retain growth factors, cytokines, and chemokines (35). Whereas soluble growth factors and cytokines transmit potent signals rapidly, matrix-bound and immobilized factors resist internalization and degradation, sustaining their signaling capabilities and promoting fibroproliferative remodeling (37).
After endothelial cells are activated in regions of disturbed flow, monocytes infiltrate the subendothelial intima, where they differentiate into macrophages. These monocyte-derived macrophages ingest rampant amounts of ox-LDL, transforming them into cholesterol-rich “foam cells” and compromising their beneficial immune cell functions (25). Macrophages are also highly susceptible to cell death owing to the intrinsic lipotoxic properties of ox-LDL that drive endoplasmic reticulum (ER) stress, resulting in their eventual death and release of damage-associated molecular patterns (DAMPs) in the surrounding microenvironment (38). Through various mechanisms, surrounding macrophages lose their ability to clear dying cells (termed “efferocytosis”), substantially expanding necrotic core areas and impairing the production of pro-resolving mediators, such as interleukin (IL)-10 and transforming growth factor beta (TGFβ) (39, 40). Importantly, experimental strategies to restore efferocytosis in settings where it fails, mitigate atherosclerosis and even promote its regression (41–43).
Most acute cardiovascular events leading to myocardial infarction and stroke are caused by plaque rupture. During this process, highly thrombogenic material from the necrotic core, which is particularly rich in tissue factor, are released into the vessel lumen (24). Atheromas with relatively large necrotic cores and thin fibrous caps have often been considered “vulnerable” plaques, whereas “stable” plaques have much thicker fibrous caps (44). Macrophages and vSMCs are particularly sensitive to ox-LDL and undergo cell death, forming necrotic cores. An imbalance in fibrogenesis vs. fibrolysis impedes vSMC-dependent ECM synthesis and assembly and drives thinning of the protective fibrous cap. Inflammatory cells activated in the atherosclerotic milieu also possess robust levels of collagenases that degrade the collagen-rich fibrous cap. Structural weakening of the fibrous cap results in interfacial debonding, characterized as the physical separation of fibrillar matrix (45, 46). Notably, this phenomenon is frequently observed in ruptured atheromas (45, 46).
NAFLD represents a range of liver pathologies beginning with excessive accumulation of lipids, particularly triglycerides, in hepatocytes (47). Additional findings of enhanced cytokine and chemokine production, inflammatory cell recruitment, and hepatocyte death characterize NASH, which may progress into fibrosis, cirrhosis, and liver failure. Importantly, NAFLD is emerging as a leading cause of liver disease, with 20%–30% of the individuals progressing to cirrhosis due to NASH (48, 49). Cardiometabolic disorders, such as insulin resistance and the metabolic syndrome, are risk factors contributing to the progression of NASH (50).
Increased caloric consumption is one of the leading causes of NAFLD, as excessive substrate overload or dysfunction in the ability of adipose tissue to store fats results in lipolysis (51). Consequently, circulating free fatty acids increase and are then taken up by secondary storage organs, particularly the liver, through fatty acid transport protein 5 (FATP5) and the scavenger receptor CD36 (52, 53). This stimulates signaling pathways that ultimately drive intrahepatic triglyceride accumulation. In addition, de novo lipogenesis (DNL) promotes hepatic steatosis by converting carbohydrates into lipids. Thus, the three main pathways, (1) enhanced lipolysis from adipose tissue, (2) triglyceride synthesis from the dietary nutrients, and (3) the conversion of dietary sugars into fatty acids by DNL, drive hepatic steatosis. In this manner, the liver's capacity to adequately process carbohydrates and fatty acids become impaired, and the formation of toxic lipid species, such as lysophosphatidylcholines, diacylglycerols, and ceramides, takes place (51). Consequently, these lipotoxic lipid species elicit a robust unfolded protein response (UPR) and ER stress that promote inflammasome activation and cell death.
Excess accumulation of intrahepatic fatty acids drives ER stress, uncouples mitochondria, and elevates ROS production by the mitochondria (54). Consequently, Jun N-terminal kinase (JNK) becomes activated and promotes intrinsic apoptosis through a caspase-2-BID signaling pathway (55). Also, fatty acid conversion to triglycerides increases the expression of death receptors and their cognate ligands, tumor necrosis factor alpha (TNFα) and Fas, to stimulate extrinsic cell death. Intrinsic or extrinsic apoptosis leads to the release of DAMPs that crosstalk with either resident or recruited macrophages to stimulate toll-like receptor (TLR)-dependent expression of multiple proinflammatory cytokines and chemokines.
A critical feature that distinguishes hepatic steatosis from NASH is the presence of hepatic inflammation, particularly of resident Kupffer cells and recruited monocyte-derived macrophages (56). Meta-analysis of RNA sequencing and single-cell RNA sequencing have revealed critical alterations in the myeloid compartment recruited to livers during NASH. Firstly, turnover and maintenance of embryonically-derived Kupffer cells are diminished during the progression of steatosis to NASH, likely due to lipotoxicity (57). Second, monocyte-derived macrophages recruited to the liver, which highly expresses Trem2, Gpnmb, Cd9, Spp1, and Itgax, genes associated with macrophages in NASH, accumulate in areas near desminhigh hepatic stellate cells, revealing their capability to crosstalk with hepatic stellate cells to drive hepatic fibrosis (58). Importantly, macrophages have been definitively proven to contribute to NASH, as depleting Kupffer cells from mice using liposomal clodronate, deleting the chemokine receptor C-C chemokine receptor type 2 (CCR2), or ablating bone marrow cells from mice using irradiation, mitigates the progression of steatosis to NASH (59–62).
Through a variety of mechanisms, macrophages in the liver exhibit a heightened state of inflammation and produce vast amounts of IL-1β (63). Consequently, peroxisome proliferator-activated receptor alpha (PPARα) becomes suppressed, and oxidation of fatty acids is impaired, ultimately leading to an accumulation of fatty acids (63). Fatty acids not only stimulate triglyceride production in hepatocytes, but they also stimulate inflammatory responses in liver immune cells (56). The saturated fatty acids, palmitate and laurate, drive IL-1β secretion by mediating NLRP3 inflammasome activation during NASH in a TLR2-dependent mechanism (56, 64, 65). Furthermore, palmitate activates NADPH oxidase 2 (NOX2) in hepatic macrophages and induces ROS production (66). Importantly, elevated levels of ROS directly stimulate TNFα expression. Also, macrophages from steatotic livers show enhanced production of toxic lipid mediators, particularly diacylglycerols and ceramides (56).
Persistent deposition of ECM proteins, such as collagens, in the liver drive cirrhosis and liver failure. Excluding CVD, liver fibrosis is the major cause of liver-related mortality in patients with NASH (47, 50). Therefore, hepatic fibrosis is among the most important endpoints in clinical trials. Hepatic fibrosis is largely mediated by the activation of non-parenchymal hepatic stellate cells that leads to their dedifferentiation towards a myofibroblast phenotype, enabling them to robustly synthesize and deposit ECM proteins (67). Evolutionarily conserved developmental programs, including Notch, hedgehog, and Hippo-YAP-TAZ, are “reawakened” in acute liver injury to stimulate hepatocyte regeneration (67). However, growing research in these pathways has revealed that they also critically drive hepatic fibrosis during NASH. For example, transgenic overexpression of Notch in hepatocytes leads to enhanced osteopontin secretion, enhancing fibrosis through hepatic stellate cell activation (68, 69). Consistently, hepatocyte-specific inactivation of Notch signaling protects mice from developing NASH-induced hepatic fibrosis (69). Whereas Hedgehog signaling is inactive in normal livers, it becomes reactivated during NASH and enhanced Hedgehog activity correlates with disease severity and fibrosis staging (67, 70). In addition, Sonic Hedgehog and Indian Hedgehog activates hepatic stellate cells and drive ECM protein synthesis (71). Moreover, hepatocyte YAP and TAZ expression are intimately linked to liver fibrosis and positively correlate with NASH and deleting or silencing TAZ in hepatocytes lowers inflammation and fibrosis in a mouse model of NASH (72–74).
To investigate the pathophysiology of NAFLD/NASH or ASCVD, multiple well-established animal models have been accepted by the scientific community and are commonly utilized for evaluating translational interpretation of preclinical findings. Below, we will discuss dietary models predominantly administered to mice, highlighting potential limitations of current application when investigating both NAFLD/NASH and ASCVD, as well as non-murine models that may have stronger translational potential but comprise their own set of limitations.
Given both NAFLD/NASH and ASCVD arise from dysregulated lipid metabolism and excessive lipid accumulation, the most appropriate models capitalize on genetic and/or dietary lipid loading with additional modifications to exacerbate disease, such as simple carbohydrates or cholesterol. Lipid profiling of mice demonstrates that the majority of their cholesterol is carried in high-density lipoprotein (HDL) particles, contrasting human lipid profile in which HDLs comprise only one-third of total cholesterol (75). Since elevated LDLs and very low-density lipoproteins (VLDLs) are direct contributors to atherogenesis (76), mice will therefore not spontaneously develop atherosclerotic lesions comparable to humans beyond the initial fatty streak (77). Thus genetic (Ldlr−/− (78), and apolipoprotein E deficient [Apoe−/−] mice 79, 80) or viral (PCSK9-AAV 81, 82) manipulation is required for mice to develop atherosclerosis. Implementing a combination of genetic dyslipidemia with dietary models to induce NASH permits simultaneous investigation of both NASH and atherosclerosis.
While administration of a high-fat diet in atherosclerotic models is well-established to induce hyperlipidemia and steatohepatitis (83–85), whether high-fat or Western diets are sufficient to elicit all components of NASH (hepatic steatosis, inflammation, and hepatocellular ballooning) and fibrosis remains unclear. Multiple studies report conflicting phenotypes in Apoe−/− mice following a high-fat diet regimen. For example, Karavia and colleagues demonstrated that despite administration of a high-fat diet (21.2% fat) for 24 weeks, Apoe−/− mice will accumulate less hepatic triglycerides compared with C57BL/6 mice fed the same diet (86). In contrast, others showed that only 8 weeks of high-fat diet in Apoe−/− mice was sufficient to induce hepatocellular ballooning and hepatic fibrosis (87). Additional studies by Matsuzawa et al. found that 12–24 weeks of an “atherogenic diet” in C57BL/6J mice is sufficient to induce hepatocellular ballooning and hepatic fibrosis (88), while Zhang et al., induced steatohepatitis with fibrosis and hepatocellular carcinoma following 14 months of high-fat, high-cholesterol feeding in C57BL/6 mice (89). Furthermore, a study by Schierwagen et al. compared Western diet and methionine-choline deficient (MCD)-diet in Apoe−/− mice, demonstrating significant fibrosis and hepatocellular ballooning in Western diet-fed mice after just 7 weeks (90). Comparisons between diet formulations used in the above studies show that while Karavia and colleagues utilized a Western-type diet, which contains 0.2% cholesterol (86, 91), Schierwagen et al. and Matsuzawa et al. utilized diets containing 1.25% cholesterol (88, 90). Furthermore, Trevaskis and colleagues first described the Amylin liver NASH (AMLN) diet, which contained 2% cholesterol and 40% fat from either partially hydrogenated vegetable oil or lard and induced murine steatohepatitis and fibrosis following 12 weeks feeding (92). Together, these studies highlight the fact that additional components of a high-fat or Western diet, mainly cholesterol, contribute to the NASH phenotype beyond excessive calories from fat (Table 1).
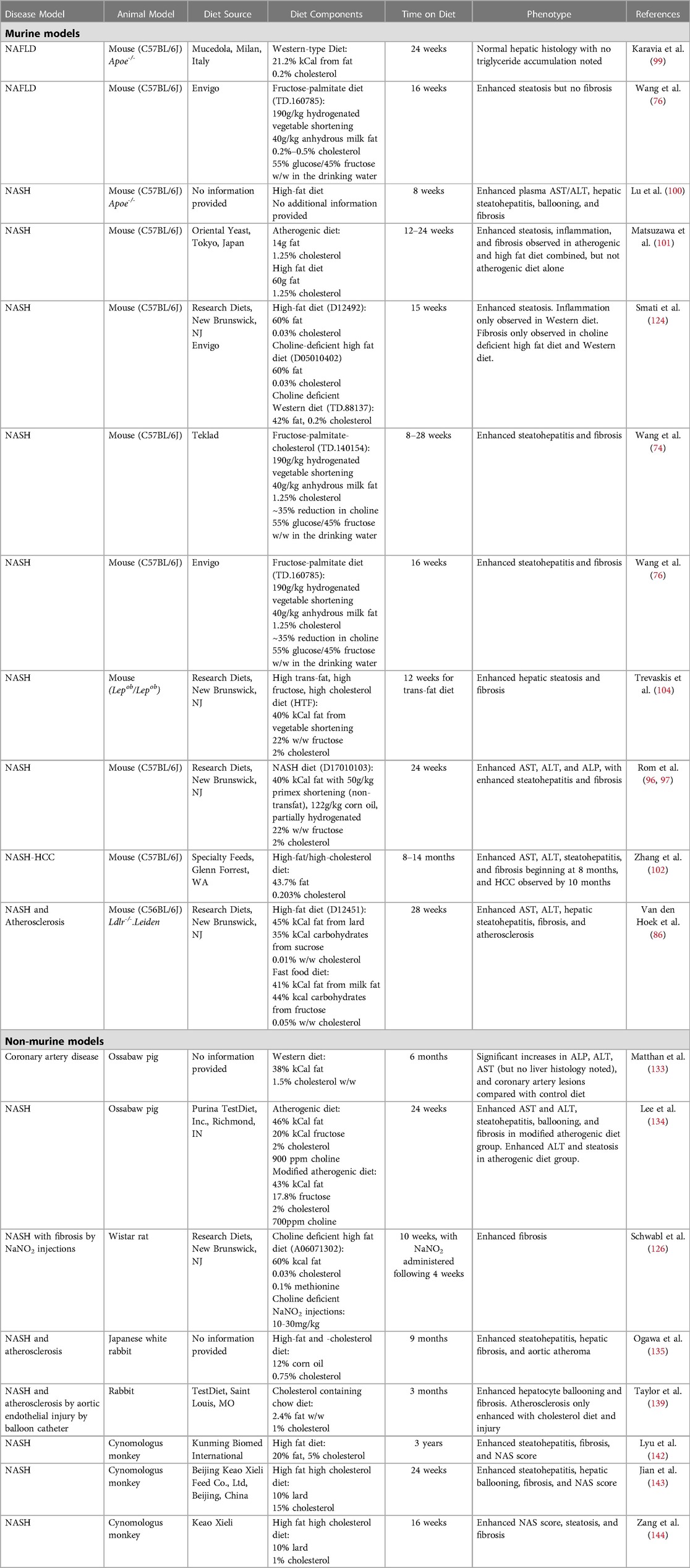
Table 1.. Murine and non-murine models of NAFLD/NASH with or without atherosclerosis
Supplementation of a high-fat diet with cholesterol appears to be a major contributor to the pathogenesis of NASH. Analysis of liver biopsies from patients with NASH demonstrated that free cholesterol accumulation associates with hepatic steatosis and continues to increase with the progression of NASH (93). In addition, unlike triglycerides or free fatty acids, cholesterol loading is sufficient to deplete mitochondrial glutathione in hepatocytes resulting in sensitivity to inflammatory cytokines (94). Following extended high-fat, high-cholesterol feeding for 14 months, cholesterol induces gut microbiota dysbiosis, enhanced gut leakiness, endotoxemia, and bile acid biosynthesis in C57BL/6 mice, which result in NASH with fibrosis and HCC (89). However, the effects of dietary cholesterol and the risk of CVD remains unclear (95). Since conventional atherogenic diets parallel human consumption of cholesterol (96, 97), but typically contain approximately one-tenth that of murine NASH diets (0.2% and 2%, respectively (83, 92)), excessive cholesterol supplementation may be inappropriate for the comparative studies of CVD and NASH together. Thus, other components such as dietary sugars may be considered when addressing models for concurrent NASH and ASCVD.
Since fructose largely replaced sucrose as a source of sweeteners in soft drinks in the 1970's, an association between high-fructose corn syrup consumption and obesity became increasingly observed (98). In addition, beyond increasing hepatic steatosis, fructose enhances aortic wall thickness and foam cell count in Sprague-Dawley rats fed a high-fat diet (99). Van den Hoek and colleagues fed Ldlr−/−.Leiden mice an obesogenic diet for 28 weeks containing 41% calories from fat, 0.05% cholesterol, and 44% calories from fructose (100), which recapitulated multiple aspects of NASH like inflammation (100), fibrosis (101), and circulating AST and ALT (102), as well as established atherosclerotic lesions (100). Since Ldlr−/−.Leiden mice are susceptible to diet-induced obesity and metabolic syndrome compared with conventional Ldlr−/− mice (103), this model proved effective in examining both fibrotic NASH and atherosclerosis (100). While normal consumption of fructose feeds into glycogen biosynthesis (104), excessive fructose consumption suppresses fatty acid β-oxidation (FAO) in the liver (105) and induces DNL by the induction of sterol regulatory element-binding protein-1 (SREBP1), acetyl-CoA carboxylase-1 (ACC1), and fatty acid synthase (FAS) (105, 106). By comparing the supplementation of fructose to glucose in humans and mice, Stanhope et al. and Softic et al. demonstrated that inhibition of FAO and induction of DNL are caused specifically by high intake of fructose, and not glucose (107–109). In the gastrointestinal tract, fructose deteriorates the gut barrier and promotes chronic inflammation by endotoxemia (110). Since endotoxemia is associated with liver disease and atherosclerosis (111, 112), the effects of fructose on the development of NAFLD/NASH and ASCVD may be due to chronic inflammation secondary to enhanced gastrointestinal permeability. Thus, the contribution of high-fructose intake for the concurrent development on NASH and atherosclerosis warrants further research.
Although diets with excess nutrients elicit NASH or ASCVD pathology, diets lacking key nutrients are an additional avenue for inducing disease. Choline and methionine deficiency diminishes VLDL assembly and reduces triglyceride clearance but results in weight loss (113), contrasting with typical weight gain associated with most human NASH. The MCD diet was previously viewed as a conventional NASH model; however, multiple groups demonstrated that MCD does not cause insulin resistance (114) and enhances weight loss despite hepatic steatosis (115), highlighting the disconnect between human disease characteristics and disease in MCD diet-fed mice. Since the MCD model clearly has its deficiencies in application with NASH pathology, researchers have developed modifications of this model to align the diet-induced phenotype more closely with human NASH. For example, the high-fat, choline-deficient diet induces steatosis, inflammation, and fibrosis over a 15-week period; however, it does not induce ballooning (116). Choline deficiency reduces pro-atherogenic VLDL assembly (113) but choline supplementation has no effect on atherosclerotic plaque area (117). The choline-deficient high-fat diet with no added choline but 0.1% methionine has approximately 0.03% cholesterol and induces steatohepatitis (116); however, to develop fibrosis the addition of 25 mg/kg NaNO2 (118) is required to induce hypoxemia (119). Enhancing methionine to 0.2% does prevent weight loss while enhancing NASH and hepatic fibrosis (113). While enhancing liver fat accumulation, the choline-deficient high-fat diet actually attenuates fasting plasma insulin and improves glucose tolerance (120). Patients with NAFLD develop hyperinsulinemia as a result of impaired whole-body insulin clearance, which may further drive hepatic steatosis (121). The positive correlation between hyperinsulinemia and atherosclerosis has been long-established (122). Therefore, models mimicking hyperinsulinemia, should be considered in appropriate models of both NASH and ASCVD.
The utilization of mice for pathological modelling has its benefits. For example, mice gestate and grow rapidly, require small spaces for housing, are relatively inexpensive to care for, and are easily genetically manipulated. While numerous mouse models have been implemented to study NAFLD/NASH or ASCVD, each provides a unique set of limitations. For example, atheroprone mice must first be “humanized” with genetic manipulation to shift their endogenous plasma cholesterol composition. Furthermore, as outlined in section 2.1, many mouse models of NASH do not completely mimic all aspects of the human disease, particularly hepatocellular ballooning and fibrosis (123). Additionally, dietary models alone are insufficient to induce atherosclerosis in mice due to their plasma lipid composition (77). Therefore, the use of non-murine or large animal models that spontaneously develop atherosclerosis may provide a more accurate representation of both human NASH and ASCVD.
Porcine models of atherosclerosis are closely related to the human disease due to similar lipoprotein composition; thus, pigs do not require genetic modification to induce ASCVD (124). In addition to their use as an atherogenic model (125), miniature Ossabaw pigs develop metabolic syndrome with abnormal liver pathology indicative of NASH when fed a modified high-fat, low-choline diet for 24 weeks (126). However, pigs require larger housing facilities, utilize more resources, and are therefore not as cost-effective. Rabbits may be a useful alternative to pigs or mice because they require less resources than pigs and are able to develop NASH with fibrosis following 9 months of a modified diet containing 0.75% cholesterol and 12% corn oil (127). Rabbits were pivotal in the initial discovery of atherosclerosis in which the Russian physician Ignatowski observed aortic plaques in rabbits fed an enriched animal fat and protein diet (128). Since then, rabbits are widely used for atherosclerosis studies due to their similarities to human lipoproteins, and both diet-and genetically-induced atherosclerotic models have been implemented (129). Furthermore, 1% cholesterol-fed rabbits develop both atherosclerosis (130) and fibrotic NASH, representing a simple model to investigate both diseases simultaneously (131). However, rabbits show wide genetic variability compared with mice (129) and therefore require larger cohorts to observe meaningful differences between treatment groups. Perhaps the most translatable model of either NASH or atherosclerosis is the use of nonhuman primates. For example, cholesterol metabolism between humans and Baboons is remarkably similar (132), and baboons given a high-sugar, high-fat diet leads to weight gain and hyperlipidemia similar to humans (133). Cynomolgus monkeys given a diet containing 20% fat with 5% cholesterol developed NASH with fibrosis (134). In addition, a high-fat, high-cholesterol (1%) diet can accelerate NASH in Cynomolgus monkeys with spontaneously-developed NASH symptoms (135, 136). However, the ethical considerations of these animals should be heavily weighed when deciding which models are the most appropriate. Despite their obvious similarities with humans, the advanced cognition of nonhuman primates sheds light on the moral obligations of scientific researchers (137).
The circulating levels of liver enzymes (aspartate transaminase [AST], and alanine transaminases [ALT]), other nonenzymatic proteins (albumin) and metabolites of heme (bilirubin) are routinely used to diagnose and monitor liver diseases, including NAFLD/NASH (138). While liver function tests are routinely preformed, their interpretation is often challenging and their relevance to CVD, the main cause of death in patients with NASH (7–12, 139–141) is limited. Furthermore, predictive biomarkers of NASH are lacking, resulting in invasive biopsy as the only method for diagnosis (51). Established biomarkers for CVD including C-reactive protein (CRP), cardiac troponins I and T, B-type natriuretic peptides, and D-dimer, are widely used for diagnosis and management of various CVDs including atherosclerosis, myocardial infarction, acute coronary syndrome, cardiac arrest, thrombosis, and ischemic cardiac diseases (142–144). Despite the wide use of these biomarkers for diagnosis and monitoring, there remains a need to identify new pathological pathways and pertinent biomarkers that can be useful for concurrent diagnosis and monitoring of NAFLD and CVD. Herein, we explore established and newly identified biomarkers that are closely related to NAFLD/NASH and ASCVD (Figure 2).
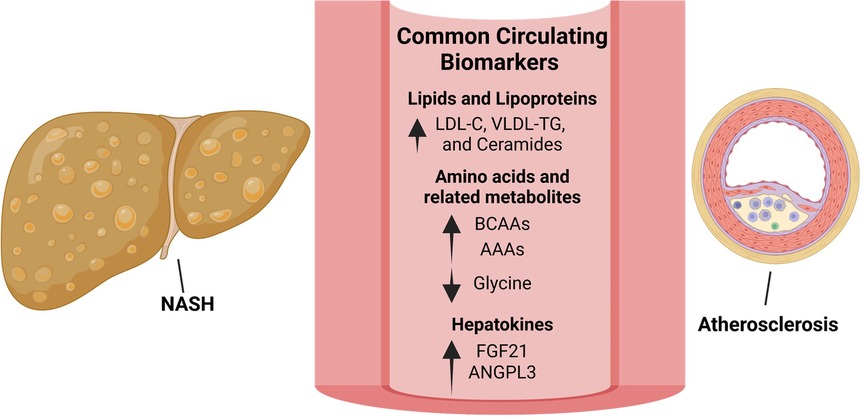
Figure 2. Biomarkers linking NAFLD/NASH and ASCVD offer potential therapeutic strategies. Lipid species that are increased in both NAFLD/NASH and ASCVD include low-density lipoprotein-cholesterol (LDL-C), very low-density lipoprotein (VLDL) and triglycerides (TG) as well as ceramides. Although significantly lowering ASCVD, LDL-C reduction using statins has shown inconsistent results with regards to NASH treatment. Targeting ceramide synthesis have shown promising results in rodent models of NAFLD/NASH and ASCVD and warrants clinical evaluation. Amino acid metabolism is commonly dysregulated in NAFLD/NASH and ASCVD with circulating branched-chain amino acids (BCAAs) and aromatic amino acids (AAAs) increased and glycine decreased in both diseases. In rodent models and small-scale clinical studies, glycine-based treatments reduced steatohepatitis and atherosclerosis warranting clinical evaluation in larger cohorts. The hepatokines, fibroblast growth factor-21 (FGF-21) and angiopoietin like 3 (ANGPTL3), are increased in both NAFLD/NASH and ASCVD. Interestingly, approaches to inhibit hepatic ANGPTL3 have shown promise in treating dyslipidemia but were associated with increased hepatic steatosis and markers of liver injury. Despite being increased in both diseases, FGF21 analogues are protective in rodent models of NASH and atherosclerosis as well as in patients with NASH.
The liver is the major site of lipid and lipoprotein metabolism and regulates the production and clearance of all classes of lipoprotein particles (145). In addition, the liver regulates the metabolism of the major lipoprotein components including triglycerides and cholesterol (146, 147). Dysregulation of hepatic lipid metabolism leading to excess lipid accumulation is a hallmark feature of NAFLD which further promotes atherogenic dyslipidemia and the risk of ASCVD (147, 148). Thus, alteration in circulating lipoproteins in patients with NAFLD is considered an early biomarker to predict the risk of ASCVD. Preclinical and clinical reports showed that improvement in NAFLD improves dyslipidemia (149–151); however, statins or other lipid-lowering agents did not reduce the risk of cardiovascular mortality in patients with NAFLD (152). In contrast, pemafibrate, a PPARα modulator that lowers triglycerides, VLDL, and cholesterol, did not reduce the incidence of cardiovascular events but lowered the incidence of NAFLD (153). These studies highlight the need to improve our understanding of the role of other lipid and non-lipid metabolites, not only as biomarkers linking these two diseases, but also as potential targets for concurrent therapy.
Enhanced influx of free fatty acids to the liver, oxidative stress and inflammatory stimuli promotes the synthesis of hepatic ceramides in NAFLD (154, 155). Ceramides are active lipid intermediates of the sphingolipid family that are produced mainly in the liver (156). Beyond their increased levels in the liver, circulating ceramides are elevated in animal models and patients with NAFLD (157, 158), particularly in those with NASH (159), where they are found mainly in VLDL and LDL particles (159, 160). Moreover, various ceramide species (mainly Cer16:0, Cer18:0 and Cer24:1) are consistently associated with adverse cardiovascular outcomes and mortality (161–163), and have been suggested as biomarkers for ASCVD beyond the currently exciting risk factors (164). Ceramides are not only associated with ASCVD but can also increase atherosclerosis by promoting endothelial dysfunction (154, 165). Pharmacological (myriocin) and genetic (hepatic deletion of dihydroceramide desaturase-1) approaches targeting ceramide synthesis not only lowered hepatic steatosis and fibrosis (166, 167), but also reversed endothelial dysfunction and atherosclerosis in rodent models (168, 169).
In NAFLD, and particularly in NASH, hepatic mitochondrial dysfunction augments ROS production promoting lipid peroxidation, the oxidation of polyunsaturated fatty acids via lipid-peroxyl radical reaction (54, 170–172). Beyond enhanced hepatic lipid peroxidation, an increase in systemic markers of lipid peroxidation (e.g., malonaldehyde, MDA) is well-documented in both experimental models and in patients with NAFLD (173–175). Furthermore, higher circulating MDA in patients with NAFLD is associated with lower antioxidant capacity of HDL and subclinical atherosclerosis (176). Peroxidation of lipoproteins (mainly ox-LDL) plays critical roles in various steps of atherosclerosis development (177), including endothelial activation and dysfunction (178, 179), monocyte adhesion (180, 181), macrophage-foam cell formation (182–185), and proliferation and migration of vSMCs (186, 187). Indeed, circulating ox-LDL is a useful marker in predicting the risk of coronary artery diseases (CAD) (188) as well as NAFLD severity (189). In addition, circulating ox-LDL in the form of MDA-LDL is not only increased in individuals with NAFLD, but is also associated with high-risk atherosclerotic plaques in the same patients (190).
While dysregulated lipid metabolism in NAFLD/NASH and ASCVD has been extensively studied, recent evidence strongly suggests altered amino acid metabolism as a common factor in both diseases (83, 191–203). Gaggini et al. (192) found that most circulating amino acids were elevated among obese subjects with NAFLD and further increased in the presence of insulin resistance (IR) and obesity. Patients with more advanced liver damage and fibrosis had higher levels of the branched-chain amino acids (BCAAs, leucine, isoleucine and valine) (204) and aromatic amino acids (AAAs tryptophan, phenylalanine, and tyrosine) (192, 205). Furthermore, BCAAs and AAAs are consistently reported to be positively associated with increased risk for ASCVD independent of hypertension and metabolic disease (206–209).
Despite the associations between elevated circulating amino acids and NAFLD or ASCVD, a causative role of BCAAs and AAAs remains unclear. In mice with NAFLD, BCAAs promote liver injury and apoptosis by downregulating lipid-induced autophagy (210). In contrast, BCAA supplementation to mice fed high-fat or choline-deficient, high-fat diets lowered hepatic steatosis and injury through suppression of hepatic lipogenic genes and modulation of intestinal microbiota-mediated production of acetic acid (211, 212). These contrasting effects may be due to specific BCAAs, since the adverse metabolic effects in obese mice appear to be mediated by isoleucine and valine but not by leucine, whose restriction aggravated hepatic steatosis (213). In addition, leucine protects against macrophage foam cell formation by inhibiting lipid biosynthesis, promoting cholesterol efflux and enhancing mitochondrial respiration (191, 197, 214, 215). Furthermore, Apoe−/− mice supplemented with leucine showed enhanced hepatic cholesterol efflux, which effectively reduced circulating LDL and atherosclerosis (216). The effects of BCAAs on different cell types may differentially regulate the pathogenesis of atherosclerosis. For example, supraphysiological levels of BCAAs (6 mmol/L) enhanced ROS and activated endothelial cells (217). In contrast, physiological levels of leucine (0.2 mmol/L) protect against macrophage foam cell formation by inhibiting lipid biosynthesis, promoting cholesterol efflux and enhancing mitochondrial respiration (191, 197, 214, 215). Thus, future studies are warranted to clarify the causative role of exogenous BCAAs and determine the effects of individual BCAAs in NAFLD/NASH and ASCVD.
In individuals with histologically confirmed NAFLD, plasma phenylalanine was increased only in those with NASH, while tyrosine was increased in both patients with simple steatosis and NASH (218). Tyrosine and total AAAs were associated with NAFLD severity assessed by hepatocellular ballooning, inflammation and fibrosis in patients with NASH (192, 205). Also, serum AAAs were reported to be higher in patients with NASH, but when compared to patients with simple steatosis, only tryptophan was higher in those with NASH. In addition, serum tryptophan and tyrosine were positively correlated with total and LDL-cholesterol (219), suggesting that alterations in circulating AAAs are associated with the risk of NAFLD-associated CVD. Indeed, in a large cohort of adults Finns, circulating tyrosine was positively associated with subclinical atherosclerosis assessed by carotid intima-media thickness (IMT) (200). In addition, phenylalanine and tyrosine were associated with CAD, ischemic stroke, and cardiovascular events (220). While the above studies demonstrate increased circulating AAAs in NAFLD/NASH and ASCVD, studies addressing the causative role of altered AAA metabolism and the effects of individual AAAs in the development of these diseases are lacking.
Whereas circulating BCAAs and AAAs are increased, glycine, the simplest amino acid, is consistently reported to be lower in association with suppressed hepatic glycine biosynthetic genes (e.g., alanine-glyoxylate aminotransferase [AGXT] and serine hydroxymethyltransferase [SHMT]) and inversely associated with the risk or severity of NAFLD/NASH, CVD and related cardiometabolic diseases in both mouse models and patients (83, 192, 195, 196, 199, 201, 221–225). While these reports highlight lower circulating glycine as an emerging biomarker for both NAFLD/NASH and ASCVD, studies in humans and mice support a causative role of reduced glycine availability and the potential of glycine-based treatment in both diseases (83, 199, 201). Glycine is a nonessential amino acid mainly synthesized in the liver (226). In patients and mice with NAFLD, glycine is a limiting substrate for de novo synthesis of glutathione (GSH), the most abundant endogenous antioxidant (83, 199). Therefore, the decrease in circulating glycine in NAFLD may be explained by insufficient hepatic production coupled with enhanced demand for GSH biosynthesis. Furthermore, glycine restriction aggravates atherosclerosis in Apoe−/− mice (83, 195). Glycine or glycine-based treatments [e.g., serine, trimethylglycine (betaine) and a glycine-based tripeptide, DT-109] lowered hepatic steatosis, inflammation and fibrosis as well as atherosclerosis in various rodent models (83, 195, 227) and humans (199) through mechanisms involving hepatic GSH biosynthesis, enhanced fatty acid utilization, suppression of proinflammatory/fibrotic responses and modulation of the gut microbiome. In addition to glycine, glutamate, another component of GSH, is increased in NAFLD/NASH, which has been attributed to gamma-glutamyltransferase-mediated glutamate release during GSH transamination and upregulation of hepatic glutaminase-1 (192, 203). This, together with alternations is serine metabolism in NAFLD (192, 198), serve as a basis for the glutamate-serine-glycine (GSG) index, which recently emerged as a potential biomarker for the severity of NAFLD and fibrosis (192, 228).
Polyamines including putrescine, spermidine, and spermine are present in all living organisms. These aliphatic polycation compounds play a role in various biological events including maintenance of chromatin structure, gene transcription and translation, cell growth, and proliferation. The biological effects of polyamines are believed to be mediated by modulation of protein-protein and protein-DNA interactions (229–231). Emerging evidence suggests that polyamines modulate the risk of CVD, metabolic diseases, neurological disorders, and cancer (232–235). Nevertheless, the role of polyamine metabolism as a potential link between NAFLD/NASH and CVD remains to be explored.
Dysregulated metabolism of polyamines in NASH has been identified in human and rodent studies. A metabolomics-based study demonstrated that circulating spermidine was more than 2-fold lower in individuals with advanced NASH and fibrosis compared to those with the early disease (236). Alternations in polyamine metabolism during NASH could be attributed to the availability of S-adenosylmethionine (SAMe), a universal methyl donor and a polyamine precursor. In NASH, glycine-N-methyl transferase (GNMT), which catalyzes the transfer of a methyl group from SAMe to glycine, is reduced, promoting an increase in SAMe and subsequent accumulation of putrescine associated with enhanced lipid peroxidation (237). While changes in circulating putrescine in NAFLD/NASH have not been reported yet and the evidence for decreased spermidine is limited (236), a number of studies reported a protective role of spermidine in mouse models of NAFLD. In diet-induced obese mice, supplementation with spermidine lowered hepatic steatosis associated with downregulation of lipogenic genes and upregulation of genes driving FAO, including Ppara (233, 238). Also, spermidine ameliorated obesity-associated NAFLD in mice by increasing the phosphorylation of hepatic AMP-activated protein kinase (AMPK), which in turn inhibited the expression of the lipogenic genes Srebf1c and Fas (239). In addition, spermidine treatment restored the hypusination of translation factor EIF5A, which was decreased in NASH, leading to enhanced mitochondrial FAO and protection against diet-induced NASH in mice (240). While the studies above suggest dysregulated polyamine metabolism in NASH and indicate a protective role of spermidine, further research is needed to establish the use of polyamines as biomarkers for NAFLD/NASH.
With regards to CVD, the association with spermidine has been evaluated in a number of recent studies. In individuals with AMI, serum spermidine was associated with improved prognosis and reduced rates of major adverse cardiac events (241). On the other hand, a higher risk of stroke was found with an increasing baseline serum spermidine (242). Moreover, obese and overweight subjects were found to have higher serum spermidine along with increased atherogenic markers including triglycerides, total and LDL-cholesterol (243). While the above association studies appear to be conflicting, intervention studies in mouse models consistently demonstrated athero/cardioprotective properties of spermidine. In Apoe−/− mice, spermidine supplementation lowered plaque lipid accumulation and necrotic cores. Spermidine triggered cholesterol efflux in autophagy-competent but not in autophagy-deficient VSMCs or macrophages lacking autophagy related 7 (Atg7) (244). In addition, spermidine and spermine protected against LDL oxidation resulting in reduced uptake of ox-LDL by macrophages (245). Furthermore, spermidine decreased cardiac hypertrophy and preserved diastolic function in old mice concomitant with enhanced cardiac autophagy, mitophagy and mitochondrial respiration. These cardioprotective effects were abolished in mice lacking Atg5 in cardiomyocytes (232), supporting the notion that induction of autophagy by spermidine may be useful to prevent CVD. Interestingly, in humans, higher consumption of dietary spermidine was associated with lower CVD incidence (232). Together, while spermidine supplementation appears to be protective against NAFLD/NASH and CVD in mouse models, the use of spermidine or other polyamines as biomarkers and the therapeutic potential of spermidine in clinical settings warrant further research.
Oxalate is the ionic form of oxalic acid, and is an end-product of glyoxylate metabolism in the liver, which accounts for 80%–90% of total circulating oxalate (246–248). The vast majority of oxalate (>90%) is eliminated through the kidneys (249, 250). Although humans have no enzymes capable of degrading oxalate (251), specific hepatic enzymes can prevent oxalate overproduction via the detoxification of glyoxylate to glycolate (by glycolate reductase/hydroxypyruvate reductase, GRHPR) or glycine (by AGXT) (252). Genetic defects in these enzymes result in primary hyperoxaluria, in which toxic levels of oxalate are produced by the liver (252). Furthermore, increased systemic oxalate can also be caused by impaired oxalate excretion in chronic kidney disease (250, 253). Beyond this, increased serum or urine oxalate has recently been linked with NAFLD/NASH (83, 248) and CVD (83, 248, 253, 254).
Suppression of glyoxylate detoxifying genes, particularly AGXT, has been consistently reported in both in humans and mice with NAFLD/NASH. Assessment of hepatic gene expression in patients who had undergone bariatric surgery revealed that AGXT is downregulated in those with NASH (255). In support, AGXT and GRHPR were recently reported to be downregulated in steatotic hepatocytes isolated from patients with NAFLD (248). Analysis of liver transcriptomic data from several cohorts of patients with various degrees of liver disease (steatosis, NASH, cirrhosis, and HCC) combined with data from mice with NAFLD or NASH revealed that AGXT was consistently downregulated in all human and mouse cohorts (83, 196, 248, 256). Furthermore, aggravated NASH and fibrosis in Agxt−/− mice fed a NASH-inducing diet suggest a causative role of oxalate in NAFLD (83). Nevertheless, future studies evaluating the liver and circulating levels of oxalate in patients with NAFLD/NASH are warranted.
With regards to CVD, increased circulating oxalate has been associated with increased cardiovascular morbidity and mortality. Among hemodialysis patients, serum oxalate was positively associated with cardiovascular risk factors including elevated pulse wave velocity, central aortic systolic and diastolic blood pressures, and risk for cardiovascular events (253, 257). In patients with end-stage renal disease, increased circulating oxalate was not only associated with CVD events, but also with aggravated dyslipidemia (increased triglycerides and VLDL-cholesterol, and decreased HDL-cholesterol) and proatherogenic cytokines and chemokines (IL-6, TNFα, and monocyte chemoattractant protein-1) (254). In patients with significant CAD and normal kidney function, and in atherosclerotic Apoe−/− mice, we found a significant decrease in the glycine to oxalate ratio aligned with downregulated hepatic AGXT. In mice deficient with both Agxt and Apoe, as well as in Apoe−/− mice challenged with exogenous oxalate, atherosclerosis was increased with enhanced superoxide and CCL5 in atherosclerotic lesions. These effects were reversed by AAV-mediated overexpression of AGXT in livers of Apoe−/− mice, indicating a causative role of oxalate overproduction in atherosclerosis (196). At the cellular level, oxalate was reported to induce mitochondrial dysfunction, oxidative stress and the release of proinflammatory chemokines and cytokines in endothelial cells, monocytes, and macrophages (196, 258–260). Together, the association between circulating oxalate, NAFLD/NASH and ASCVD should be further studied in larger cohorts including patients without kidney disease.
The liver secretes various proteins known as hepatokines that can regulate systemic metabolic homeostasis through a crosstalk with other organs including skeletal muscle, adipose tissue, the central nervous system and blood vessels (261). In addition to their metabolic role, systemic alterations in hepatokines are implicated in several pathological conditions including IR, diabetes and CVD (261, 262); however, evidence regarding the role of hepatokines as modulators of atherosclerosis is limited.
Angiopoietin-like 3 (ANGPTL3) is a glycoprotein that is expressed and secreted primarily by the liver (263). Secreted ANGPTL3 binds lipoprotein lipase and inhibits its activity to hydrolyze lipoprotein triglycerides into fatty acids that are taken up by metabolic tissues. As a result, circulating triglycerides are increased (264, 265). Indeed, individuals with loss-of-function mutations in ANGPTL3 have lower plasma triglycerides (266). In a cross-sectional investigation of obese subjects, both hepatic and plasma ANGPTL3 were higher in individuals with NALFD and positively correlated with hepatic steatosis and histological markers of NASH (267). Among patients with various degrees of NAFLD, serum ANGPTL3 was increased in individuals with NASH, but not in those with simple steatosis (268). With regards to CVD, a study involving 1,493 MI cases and 3,231 controls demonstrated that individuals with lower plasma ANGPTL3 had a reduced risk of MI (269). In line, increased plasma ANGPTL3 was positively associated with the severity of coronary stenosis among patients with angina (270). Beyond its potential as a biomarker, the efficacy of ANGPTL3 inhibition has been studied extensively in preclinical and clinical settings. Both in Ldlr−/− mice treated with antisense oligonucleotides (ASO) targeting Angptl3 and in APOE*3Leiden.CETP mice treated with an antibody against ANGPTL3 (evinacumab), hypercholesterolemia, hypertriglyceridemia and atherosclerosis were significantly decreased (271, 272). Evinacumab also lowered fasting triglycerides and LDL-cholesterol in a phase I trial (271). In a phase IIb trial, administration of vupanorsen, an ASO targeting hepatic ANGPTL3, to patients with hypercholesterolemia and hypertriglyceridemia significantly reduced triglycerides together with a modest decrease in LDL-cholesterol. Unfortunately, at higher doses, vupanorsen administration was associated with increased hepatic fat, and over 3-fold elevations in ALT and AST (273). These studies highlight the potential complications in determining dosage for therapeutics like vupanorsen.
Fibroblast growth factor-21 (FGF-21) is a hormone primarily produced and secreted by the liver (274, 275). The hepatic expression and circulating levels of FGF21 are consistency reported to be higher in NAFLD, and are associated with enhanced hepatic necroinflammation and fibrosis (276–280). Furthermore, FGF21 was positively correlated with total cholesterol and triglycerides, and multivariate regression analysis indicated that FGF21 is an independent risk factor of CAD (281). Moreover, serum FGF21 predicted the incident of ASCVD events independent of NAFLD and other traditional cardiovascular risk factors (282, 283). Despite these findings indicating elevated circulating FGF21 as a common biomarker for NAFLD and ASCVD, FGF21 is known for its protective properties in both diseases. An extensive body of literature have demonstrated the protective effects of recombinant FGF21 or FGF21 analogues in preclinical models of NASH (284, 285) and atherosclerosis (286, 287) as well as in patients with NASH (288, 289), serving as an attractive therapeutic marker for both diseases.
Fetuin-A, also known as α2-Heremans-Schmid glycoprotein (AHSG), is synthesized and secreted predominantly by the liver and is among the first hepatokines identified to regulate metabolism through multiorgan crosstalk (290–292). Elevated fetuin-A levels are positively correlated with liver fat, patients with NAFLD, IR, and hepatic fibrosis (293–295). The link between fetuin-A, NAFLD and other metabolic disorders has sparked interest in its involvement in CVD; however, these studies yielded inconsistent results. In a case-cohort investigation, higher circulating fetuin-A was associated with MI and ischemic stroke after adjustment for confounders (296). In contrast, lower plasma fetuin-A, independent of traditional CVD risk factors, was found to be associated with increased CVD mortality among 1,620 patients with CAD (297). Therefore, while fetuin-A may serve as a potential biomarker in NAFLD, the conflicting findings above indicate that fetuin-A may not be a useful biomarker in ASCVD.
Significant advances in our understanding of the mechanisms that drive NASH have led to the development of numerous of drug candidates that target different pathways in the pathogenesis of NASH. As extensively reviewed (298, 299), these candidates include drugs that target insulin/glucose homeostasis, lipid metabolism, proinflammatory/profibrotic responses, and the gut-liver axis, alongside pharmacological/surgical approaches aimed at lowering body weight. A limited number of drugs that demonstrated efficacy in phase IIb trials were or are currently evaluated in phase III trials. A few drugs approved for other metabolic diseases (e.g., T2D, and obesity) are evaluated as potential treatments for NAFLD/NASH in phase IV trials. While the current therapeutic pipeline in NASH (298, 299) and emerging approaches to treat ASCVD via modifying inflammation (300) have been comprehensively reviewed, in this section we discuss (1) potential cardiovascular consequences of promising drug candidates for NASH, and (2) the effects of commonly used (lipid-lowering) and new (anti-inflammatory) drugs for ASCVD on NASH.
The prevalence of NAFLD and NASH in patients with T2D is higher than the general population and was estimated at 55% and 37%, respectively (5). As T2D is closely associated with NASH, a number of antidiabetic drugs have been considered as potential treatments for NASH. Among these drugs, pioglitazone, a PPARγ agonist and insulin sensitizer, is currently evaluated in a phase IV clinical trial for NASH (NCT00994682). Pioglitazone administered for 18 months to prediabetic or T2D patients with biopsy-proven NASH effectively lowered NAS and fibrosis scores while improving insulin sensitivity (300–302). However, pioglitazone treatment was associated with weight gain compared to placebo (302). Moreover, pioglitazone was associated with other adverse effects including enhanced risk of hospitalization for heart failure due to fluid retention (303–305). Despite this, accumulating evidence suggests a protective effect of pioglitazone on atherosclerosis-driven events including MI and ischemic stroke. In patients with impaired glucose tolerance or T2D, pioglitazone reduced carotid IMT (306, 307) and atherosclerotic plaque inflammation in association with decreased CRP and increased HDL-cholesterol (308, 309). Furthermore, pioglitazone treatment was associated with reduced total and LDL-cholesterol, triglycerides, and lipoprotein (a) (310–312). Therefore, the cardiovascular consequences of pioglitazone in patients with NASH warrant further research in long-term, large clinical trials.
Newer antidiabetic drug classes, including glucagon-like peptide 1 (GLP1) receptor agonists and sodium-glucose cotransporter-2 (SGLT2) inhibitors, have emerged as potential therapies for NASH. GLP1, an incretin secreted from intestinal L-cells, enhances glucose-stimulated insulin secretion and promotes satiety (313–316). Liraglutide is a GLP1 analogue known to lower body weight (317). In a phase II trial including overweight patients with biopsy-confirmed NASH, 48 weeks of liraglutide treatment was associated with higher rates of NASH resolution and attenuation of fibrosis (318). Stable isotope studies in patients treated with liraglutide, supported by lipid flux studies in human primary hepatocytes, demonstrated that liraglutide inhibits hepatic DNL (319), suggesting additional benefits beyond lowering body weight. Semaglutide, another GLP1 receptor agonist, has more pronounced body weight-lowering effects (320). In a phase II trial including patients with biopsy-confirmed NASH and fibrosis, semaglutide administered for 72 weeks led to a 13% reduction in body weight and was associated with higher rates of NASH resolution and improvement of fibrosis (321). With regards to ASCVD, liraglutide administered to patients with T2D has been consistently reported to improve circulating lipid profile (reduce triglycerides, total and LDL-cholesterol, and increase HDL-cholesterol) and reduce carotid IMT (322–324). The effects of semaglutide on atherosclerosis are currently being evaluated in phase IV trials (NCT03985384). Together, the above studies indicate the potential of GLP1 receptor agonists for concurrent treatment of NASH and ASCVD, which should be confirmed in long-term studies assessing cardiovascular outcomes in patients with NASH. Furthermore, considering that the expression of GLP1 receptor is not detected in livers (325, 326) and aortas (327) from mice, monkeys and humans, the mechanisms by which GLP1 receptor agonists protect against NASH and ASCVD, beyond lowering body weight, warrant further investigation.
SGLTs are membrane proteins that regulate nutrient transport across the intestinal epithelium and the proximal renal tubules. While SGLT1 is expressed primarily in enterocytes and absorbs glucose from the gut lumen, SGLT2 is expressed in the proximal tubule and regulates glucose reabsorption from the glomerular filtrate (328). Thus, by decreasing renal glucose reabsorption and increasing urinary glucose excretion, SGLT2 inhibitors, such as empagliflozin, reduce hyperglycemia in patients with T2D (329). Empagliflozin has been evaluated for NAFLD treatment in phase IV trials (NCT02637973, NCT02686476, NCT02964715). In patients with T2D, empagliflozin administrated for 20 weeks reduced circulating ALT and liver fat assessed by MRI-derived proton density fat fraction (MRI-PDFF) (330). Although including a small sample size (n = 9), a study in patients with T2D and biopsy-proven NASH reported that empagliflozin treatment for 24 weeks improved histological components of NASH including steatosis, ballooning and fibrosis while reducing blood glucose, body weight and total cholesterol (331). Dapagliflozin, another SGLT2 inhibitor given to patients with T2D and NAFLD for 12 weeks, lowered circulating ALT and AST together with glucose and body weight. However, compared with placebo, reduction in hepatic fat was found when dapagliflozin was combined with omega-3 carboxylic acids, but not as a monotherapy (332). Also, although lowering body weight, dapagliflozin administered to insulin-resistant overweight/obese individuals for 12 weeks did not improve hepatic steatosis (333). However, when given to patients with T2D and NAFLD for 24 weeks, dapagliflozin lowered circulating ALT, hepatic steatosis and fibrosis assessed by MRI-PDFF and magnetic resonance elastography (MRE) (334). Interestingly, a recent phase II study including patients with NASH reported that 12 weeks of treatment with licogliflozin, a dual SGLT1/2 inhibitor, reduced circulating ALT and hepatic fat assessed by MRI-PDFF (335). Importantly, dramatic beneficial cardiovascular outcomes have been reported in T2D patients treated with SGLT2 inhibitors. In long-term and large phase III trials including patients with T2D with or at risk for ASCVD, treatment with empagliflozin or dapagliflozin was associated with lower rates of cardiovascular death (336, 337). Considering that SGLT2 is primarily expressed in the kidneys, the mechanisms by which SGLT2 inhibitors reduce the cardiovascular risk and directly affect the atherosclerotic plaque, beyond glucose- and body weight-lowering effects, are not completely clear (338, 339). Furthermore, whether long-term treatment with SGLT2 inhibitors concurrently lowers NASH and ASCVD remains unknown.
Lipid overload is central to the pathogenesis of NASH. Fatty acids are supplied in excess to the liver via 1) enhanced flow from lipolysis of triglycerides in adipose tissue, and 2) increased synthesis from carbohydrates, primarily fructose, via DNL (50, 340). In addition to increased lipogenesis, fructose also suppresses hepatic FAO (109). Enhanced DNL coupled with impaired FAO result in the formation of lipotoxic species that induce hepatic oxidative stress, proinflammatory and profibrotic responses to promote NASH (50, 341, 342). Therefore, pharmacological strategies aimed at inhibiting DNL or enhancing FAO can reduce hepatic lipotoxicity and attenuate NASH. Nevertheless, considering the major role of the liver as a regulator of systemic lipids, such approaches may have detrimental or beneficial effects on circulating lipids that may affect ASCVD.
In the initial step of fatty acid biosynthesis, acetyl-CoA is converted to malonyl-CoA by ACC (343). In phase II trials, patients with NASH treated for 12 weeks with the ACC inhibitor, firsocostat (GS-0976), showed reduced circulating ALT, hepatic steatosis and markers of fibrosis (344) mediated by inhibition of hepatic DNL assessed by heavy water labeling (345). However, similar to other ACC inhibitors [MK-4074 (346) or PF-05221304 (347)] treatment with firsocostat increased circulating triglycerides (344), which can be attributed to the upregulation of hepatic SREBP-1, enhanced VLDL production and impaired triglyceride clearance (348). While these findings raise concerns that targeting ACC may aggravate atherogenic dyslipidemia, co-administration of PF-05221304 with a diacylglycerol acyltransferase 2 inhibitor (PF-06865571), reduced liver fat assessed by MRI-PDFF and mitigated the increase in circulating triglycerides in patients with NAFLD (347). Nevertheless, the cardiovascular consequences of ACC inhibition either as a monotherapy or in combination with other drugs warrant further research in long-term clinical trials.
The conversion of acetyl-CoA and malonyl-CoA to palmitate is catalyzed by FAS, which controls the liver capacity to synthesize fatty acids through DNL (349). In a phase IIa trial including individuals with hepatic steatosis and fibrosis, treatment for 12 weeks with a FAS inhibitor, TVB-2640, dose-dependently decreased circulating ALT, AST and liver fat determined by MRI-PDFF. Importantly, TVB-2640 treatment significantly decreased circulating total and LDL-cholesterol. Although HDL-cholesterol was also decreased, lipidomics revealed beneficial effects including reduced triglycerides enriched in palmitate-containing species, diacylglycerols, bile acids and ceramides (350). Therefore, apart from the decrease in HDL-cholesterol, improved circulating lipid profile, reduced markers of hepatic steatosis and injury, indicate TVB-2640 as a promising candidate for dual treatment of NASH and ASCVD. Currently, TVB-2640 is evaluated in a phase IIb trial recruiting patients with NASH that will be treated for 52 weeks (NCT04906421). Longer-term studies are needed to determine the cardiovascular outcomes of TVB-2640 in patients with NASH.
The rate-limiting step in the synthesis of monounsaturated fatty acids is catalyzed by stearoyl-CoA desaturase 1 (SCD1) (351). The partial inhibitor of hepatic SCD1, aramchol, is a conjugate of cholic acid and arachidic acid, and is currently the most advanced drug candidate for NASH among those targeting hepatic DNL. In a 52-weeks, phase IIb trial including 247 patients with NASH, aramchol led to a time- and dose-dependent reduction in circulating ALT and AST. Histological analysis revealed that treatment with aramchol was associated with higher rates of NASH resolution and improvement in fibrosis compared with placebo (352). Of note, no significant differences in circulating lipid profile were found between the groups (352, 353). While the cardiovascular outcomes of SCD1 inhibition have not been addressed in humans, loss of SCD1 in Ldlr−/− mice (354) or its inhibition in Ldlr−/− / Apob 100/100 mice via ASO (355) enhanced atherosclerosis while reducing hepatic steatosis. Plans to test aramchol in the phase III/IV ARMOR trial (NCT04104321) in patients with biopsy-proven NASH and fibrosis for 5 years will shed light on the long-term effects of aramchol treatment on NASH and perhaps its cardiovascular consequences.
In addition to DNL inhibition, drugs that promote FAO can also lower hepatic lipotoxicity and NASH. This approach has been pursued by activation of key regulators of hepatic FAO, mainly PPARɑ and PPARβ/δ. Among the three PPAR isotypes (PPARα, PPARβ/δ and PPARγ), PPARα is the most abundant in hepatocytes where it acts as a master regulator of mitochondrial/peroxisomal FAO (356). In mice, hepatocyte-specific loss of PPARα enhances steatohepatitis, which is aggravated in whole-body Ppara−/− mice, indicating a protective role for both hepatic and extrahepatic PPARα in NASH (357–359). Accordingly, the PPARα agonist, Wy-14,643, lowers MCD diet-induced NASH and fibrosis in mice (360). Few clinical studies evaluated the effects of the PPARα agonists, fibrates, in NASH. In patients with biopsy-confirmed NASH, treatment with fenofibrate for 48 weeks reduced circulating transaminases, triglycerides and glucose while increasing apolipoprotein A1. Histological assessment revealed improved hepatocellular ballooning, but no significant changes in steatosis, inflammation, and fibrosis (361). Interestingly, in patients with NASH and fibrosis, fenofibrate administered 2 weeks before the addition of the ACC inhibitor, firsocostat, not only mitigated hypertriglyceridemia, but also improved liver biochemistry compared to icosapent ethyl (Vascepa) (362). Pemafibrate, a selective PPARα modulator, lowers NASH in mice fed the MCD or AMLN diet (363). In a phase II trial including 117 patients with NAFLD, pemafibrate administered for 48 weeks lowered circulating ALT and LDL-cholesterol. Although liver fat assessed by MRI-PDFF was not altered, MRE-based liver stiffness was significantly reduced (364). The concurrent improvement in plasma lipids and liver biochemistry suggest beneficial effects of PPARα agonism in both NASH and ASCVD. Although this notion was supported by studies in Apoe−/− (365) and ApoE*3Leiden mice (366) in which fenofibrate reduced atherosclerosis, a multinational trial including over 10,000 patients with CVD, demonstrated that pemafibrate was not associated with lower incidence of cardiovascular events although NAFLD incidence was reduced (153).
PPARβ/δ is ubiquitously expressed, including in hepatocytes, Kupffer cells and hepatic stellate cells (367, 368). Studies in mice lacking PPARβ/δ indicated its roles in regulating hepatic FAO and antiinflammatory responses in Kupffer cells (369, 370). The dual PPARα/δ agonist, elafibranor (GFT505), showed promising outcomes in preclinical NASH models (371) and in a phase IIb trial (372) in which 52 weeks of treatment with elafibranor led to higher rates of NASH resolution and reduction in fibrosis. Importantly, elafibranor not only decreased circulating transaminases, but also lowered triglycerides and LDL-cholesterol, increased HDL-cholesterol and improved glycemic control, indicating significant improvement of overall cardiometabolic risk (372). These promising findings led to the evaluation of elafibranor in a phase III trial (RESOLVE IT) including over 2,000 patients with histologically confirmed NASH (NCT02704403). Unfortunately, results of the week 72 interim analysis revealed that elafibranor did not achieve NASH resolution without worsening of fibrosis, and the RESOLVE-IT trial was discontinued.
The beneficial effects of elafibranor and the PPARγ agonist, pioglitazone, have raised interest in pan-PPAR agonism as a potential therapy for NASH. In preclinical studies, selective PPARα (fenofibrate), PPARγ (pioglitazone) and PPARδ (GW501516) were compared to the pan-PPAR agonist, lanifibranor, and indicated that pan-PPAR agonism lowers experimental NASH by combining the beneficial effects of the three PPAR isotypes (373). Indeed, in a phase IIb trial including 247 patients with biopsy-proven NASH, lanifibranor administered for 24 weeks led to higher rates of NASH resolution and improvement in fibrosis compared with placebo. Importantly, in addition to lowering circulating transaminases, lanifibranor had beneficial effects on plasma lipid profile and glycemic control. Nevertheless, a mild increase in body weight (≈3%) was noted (374). Currently, the phase 3 NATiV3 trial (NCT04849728) is recruiting patients with NASH and fibrosis to assess the long-term efficacy of lanifibranor for up to 7 years. Findings from this study will provide important insight of the cardiometabolic consequences of pan-PPAR agonism in patients with NASH.
Statins reduce circulating cholesterol through inhibition of HMG-CoA reductase and remain the leading therapeutic in reducing the risk of cardiovascular events (375). Although dyslipidemia is a hallmark of both NAFLD/NASH and atherosclerosis, whether cholesterol-lowering by statin therapy improves NASH outcome remains inconsistent and thus is not a current recommendation for NASH management (376). Despite this, statin therapy may have pleotropic beneficial effects for the treatment of NAFLD/NASH. In MCD diet-fed mice, fluvastatin reduces hepatic steatosis and improves inflammation and fibrosis through activation of PPARɑ and its target genes enhancing FAO (377). Rosuvastatin blunts NASH-induced pro-inflammatory cytokine expression in livers from high-fat diet-fed STAM mice (378), while simvastatin reduces inflammation and fibrosis in Apoe−/− mice fed a high-fat, high-cholesterol diet for 7 weeks with corresponding inhibition of Ras and Rho signaling (379). Treating obese mice with atorvastatin reduces cholesterol accumulation in isolated hepatocytes and reduces cholesterol-induced mitochondrial depletion of GSH (94), and atorvastatin is currently being evaluated in phase II trials for the treatment of NAFLD/NASH (NCT04679376). However, high-intensity atorvastatin therapy appears to enhance insulin secretion in patients with an increased risk of developing T2D (380). Since hyperinsulinemia is an early marker for metabolic disease (381) and is strongly associated with NAFLD (121), chronic use of statins in the treatment of NASH and ASCVD warrants further investigation with potential contraindications. Furthermore, statin users appear to have higher caloric intake, which is associated with weight gain and complicates disease progression (382).
A potential complicating factor is the presence of genetic variants or single-nucleotide polymorphisms (SNPs). In particular, SNPs in patatin-like phospholipase domain-containing protein 3 (PNPLA3), or transmembrane 6 super family 2 (TM6SF2) are known as strong predictors of NAFLD risk independent of associated metabolic confounding factors, despite these variants promoting lipotoxicity (383). However, the presence of the PNPLA and TM6SF2 variants reduces the risk of ASCVD in patients with NAFLD (384). In contrast, mutations in Angiopoietin-like 3 (ANGPTL3) lead to hypolipidemia (385), since circulating ANGPTL3 inhibits lipoprotein lipase and is positively associated with NASH (386). Thus, therapeutics targeting PNPLA3, but not ANGPTL3, may be contraindicated should the outcome yield exacerbated ASCVD. These studies highlight the importance of identifying and considering genetic factors in both NAFLD and ASCVD which has been thoroughly discussed previously (14).
Patients with metabolic disease and obesity who have undergone bariatric surgery have marked improvement in insulin resistance (NCT03853590) and reduced risk of major cardiovascular events (387). Since previous studies demonstrated an association between bariatric surgery-induced weight loss and improved hepatic inflammation and fibrosis (388), a retrospective cohort study of nearly 1,200 patients with NAFLD and obesity was analyzed following bariatric surgery (389). Patients who received gastric bypass or sleeve gastrectomy demonstrated marked improvement in both adverse liver and cardiovascular outcomes (389). Since bariatric surgery effectively achieves weight loss in obese patients (390), the relationship between the effects of bariatric surgery and improved NASH and CVD outcome may be due to the effects of reducing visceral and ectopic adipose tissue. Although the risk of NASH and CVD rise with increasing BMI (391), ectopic fat [the storage of fat in non-adipose tissues (392)] and visceral fat [the storage of fat in the mediastinal and abdominal cavities (393)] appear to be a more reliable correlation between cardiometabolic disease compared with BMI alone (394). Similarly, CAD patients with normal BMI have enhanced visceral fat accumulation (395). Indeed, NAFLD patients with normal BMI have excessive visceral fat compared with non-NAFLD patients (396). The detrimental correlations between visceral fat, NASH and CVD are likely due in part to adipokine secretion, like TNFɑ (397), which mediates inflammatory responses locally and systemically. Independent of BMI, reducing visceral fat improves comorbidities of CVD and NASH (398). Consistent with this, calorie restriction improves NAFLD-related biomarkers such as transaminases, liver steatosis and fibrosis scores (399), as well as reducing the risk for atherosclerosis (400). The benefits for calorie restriction and improvement of NASH and ASCVD are multifactorial. Calorie restriction (1) reduces adipokine release which attenuates systemic inflammatory signaling (401), (2) reduces serum lipids and comorbidities associated with disease exacerbation (e.g., hypertension) (400), (3) activates autophagy, which protects against hepatic steatosis and inflammation (402), and (4) activates various molecular pathways (e.g., AMPK) which are associated with protection against NASH and atherosclerosis (403, 404).
AMPK responds to energy demand by sensing the ratio of ATP to ADP/AMP. Activation of AMPK enhances catabolism and reduces anabolism, but additionally protects against oxidative stress-induced endothelial activation in atherosclerosis (405). AMPK additionally augments reverse cholesterol transport in atherosclerosis and polarizes macrophages to an M2 phenotype (404), which are associated with plaque stability and regression (406). In murine models of NAFLD, AMPK activation is inhibited due to overnutrition (407). Thus, activation of AMPK yields improvement in both CVD and NASH outcome in mouse models. Metformin activates AMPK, which reduces hepatic steatosis (408), and activation of AMPK with PF-06409577 reduces dyslipidemia and liver transaminases in rats and non-human primates (409). Another AMPK activator, PXL770, attenuates DNL, hepatic steatosis, inflammation, and ballooning in mouse NASH models (403). These effects may be due to inhibition of mTORC1, which is inhibited by AMPK through phosphorylation of raptor (410). mTOR activates lipogenesis by inducing SREBP-1c activation (411). Selective inhibition of mTORC1 by folliculin (FCLN) deletion protects against NASH by TFE3 transcription factor-induced inhibition of lipogenesis (412); however, the impact of FCLN deletion has not been investigated in atherosclerotic mice. While clinical trials for NASH are ongoing and activation of AMPK by PXL770 in mice improves atherosclerotic outcome (403, 413, 414), whether these results extend to human atherosclerotic patients has yet to be explored.
Since primary components of the pathophysiology of NASH and atherosclerosis involve the regulation of inflammatory cytokines, leukocyte response, and the crosstalk between these mediators (300, 415), systemic therapy reducing inflammation may yield benefits across both pathologies. Given the potent effects of IL-1β signaling and its central role in inflammation, the monoclonal antibody targeting IL-1β (canakinumab) has been implemented in the CANTOS phase III clinical trials (NCT01327846) for the treatment of CVD (416). It is well-established that the proinflammatory cytokine, IL-1β, activates endothelial cells to express adhesion molecules, secrete chemokines, and vSMC proliferation to augment atherogenesis (417). Furthermore, IL-1β gene expression increases in livers of mice fed a high-fat, high-cholesterol (1.25%) diet for 18 weeks, and deletion of IL-1β reduces steatosis, inflammation, ballooning, and fibrosis in these mice (418). Thus far, the CANTOS trial has proved promising since inhibition of IL-1β reduces the total number of serious cardiovascular events in patients with prior MI history (416); however, it has not examined whether IL-1β inhibition by canakinumab improves characteristics of NASH. Since deletion of IL-1β reduces steatohepatitis, and fibrosis in mice fed a NASH-inducing diet (418), further investigation on the effects of canakinumab in human NASH are warranted. Such studies should also consider the potential risk of infection or sepsis considering that treatment with canakinumab was found to be associated with a higher incidence of fatal infection in the CANTOS trial (419).
While the effects of IL-1β inhibition remain unexplored in NASH clinical trials, several preclinical and clinical studies have analyzed the potential benefits of targeting TNFα for NASH. Antibody therapy against TNFα yielded promising results with diet-induced NASH in mouse and rat models showing improvement in circulating AST, ALT, steatohepatitis, and fibrosis (420–422). However, retrospective studies of patients receiving anti-TNFα for immune-related diseases reported no reduction in the incidence of new onset NAFLD, NASH, or cirrhosis (423). While antagonism of TNFα with monoclonal antibodies yielded effective results in patients with rheumatoid arthritis (424) and inflammatory bowel disease (425), clinical trials for anti-TNFα in patients with chronic heart failure (RECOVER and RENAISSANCE) were terminated prematurely due to no observable benefit (426). Furthermore, the anti-TNFα monoclonal antibody CNTO5048 (CNT) enhanced plasma triglycerides, VLDL, and atherosclerosis in Ldlr−/− mice (427), suggesting the use of anti-TNFα antibodies for atherosclerosis may be contraindicated.
While the effects of targeting cytokines in NASH and ASCVD warrant further investigation, chemokine signaling appears to be a promising direction in targeting inflammation. Since mice fed a choline-deficient diet have enhanced hepatic CCR2 (60) and clinical trials demonstrated efficacy and safety with antagonism of CCR2 and CCR5 in patients with HIV (428), the CENTAUR clinical trial (NCT02217475) proceeded with the CCR2/CCR5 dual antagonist cenicriviroc over the course of 2 years (429). At the completion of this phase IIb trial, patients with NASH and fibrosis who received cenicriviroc demonstrated marked improvement in fibrosis without worsening of NASH (429). Despite the completion of phase II clinical trial, phase III (AURORA, NCT03028740) was terminated early due to lack of efficacy. CCL2 signals through its receptor CCR2, which is required for monocyte emigration from the bone marrow during an inflammatory response (430), and deletion of CCR2 significantly reduces atherosclerosis (431). Inhibition of CCR2 with the MLN1202 monoclonal antibody reduced the levels circulating CRP, a marker of cardiovascular risk (432). Although the effects of cenicreviroc on CVD were studied in an early clinical trial (NCT01474954), the trial was terminated due to low enrollment. Taken together, the effects of CCR2/CCR5 inhibition may improve some aspects of ASCVD; however, since activation of both CCR2 and CCR5 receptors directly activate hepatic stellate cells which promote hepatic fibrosis (433), further investigation on the effects of cenicreviroc on vSMCs are indicated to determine whether treatment affects atherosclerotic plaque stability.
Despite the numerous studies investigating the mechanisms behind NASH or ASCVD, those that utilize genetic manipulation often do so with subsequent onset of disease. These studies, while informative, may not be appropriate for identifying therapeutic targets since intervention of NASH and ASCVD does not occur until they become symptomatic. More advanced disease, both for NASH and ASCVD, involve the accumulation of fibrous tissue in the liver and vessels, respectively. In NASH, advanced fibrosis is correlated with worse prognosis (434). In contrast, fibrous or fibrocellular atherosclerotic plaques confer stability against plaque rupture and catastrophic events (435). Therefore, systemic targeting of fibrosis to improve NASH may be contraindicated for maintaining stable fibroatheromas. Currently, several clinical trials for the treatment of NASH seek to improve clinical outcome which includes lowering fibrosis (374, 436, 437). However, it is unknown whether these will affect cardiovascular morbidity and mortality.
PPARs regulate lipid homeostasis through transcriptional control of FAO and DNL (438). Since the relationship between dysregulated lipid metabolism and NASH or ASCVD is well-established, therapeutics targeting of PPARs may yield successful results. Indeed, the PPARγ agonist pioglitazone and PPARα fenofibrate reduced atherosclerosis and hepatic steatosis in mice lacking both ApoE and Farnesoid x receptor (FXR) (439), which modeled NASH and atherosclerosis simultaneously. FXR, a nuclear receptor responsible for bile acid and cholesterol synthesis, suppresses hepatic lipogenesis and VLDL assembly by attenuating SREBP1-c (440). In addition, FXR activity promotes PPARα transcription through binding directly to the PPARα reporter (441). Currently the non-steroidal FXR agonist Cilofexor is undergoing Phase II clinical trials for NASH treatment and has shown promising results in the reduction of hepatic steatosis, inflammation, and fibrosis (436). While deletion of FXR from Apoe−/− mice enhanced atherosclerosis (442), it remains unknown if the anti-fibrotic effects of FXR in stellate cells is conserved in plaque associated vSMCs. Since Cilofexor primarily acts in the intestine (443), it may reduce the potential side effects of systemic FXR activation; however, further investigation on its effects on atherosclerotic plaques is warranted. FXR additionally induces FGF-19, which enhances cholesterol efflux and HDL assembly through modulation of hepatic ABCA1 and ApoA1 (444). Administration of the FGF-19 analog NGM282 reduces atherosclerosis in Apoe−/− mice, enhances plasma HDL-cholesterol in healthy subjects (444), and improved NASH and fibrosis in phase II clinical trials (445). However, NGM282 increases plasma LDL-cholesterol (437), suggesting a potentially exacerbating factor in atherosclerosis. Furthermore, NGM282 reduces atherosclerotic fibrosis in mice (444), implicating the potential for plaque rupture with sustained therapy. In addition to NGM282, the synthetic bile acid obetacholic acid agonizes FXR and has been implemented in phase III clinical trials for the treatment of NASH (NCT02548351). Since patients receiving obetacholic acid have enhanced circulating VLDL and LDL but reduced circulating HDL (21), its administration in the context of atherosclerosis may be contraindicated. Since these studies terminated at 72 weeks (21), the long-term effects of obetacholic acid on atherosclerosis remain unknown. Overall, the long-term effects of these NASH/fibrosis-targeting drugs must consider the potential effects on atherosclerotic plaque instability.
In this review, we highlighted our current gaps in knowledge with particular emphasis on modelling both diseases, common biomarkers and potential therapeutics, and the potential caveats we currently face by targeting specific aspects of each disease. In the past decade cardiovascular-related mortality rates are steadily increasing concomitant with a rapid rise in obesity and NAFLD/NASH incidences (1, 2), currently afflicting one-third of the population worldwide (6). Despite this prevalence, no FDA-approved drugs exist for the treatment of NASH. Since NASH serves as an independent risk factor for ASCVD, and individuals with NASH are at a greater risk of ASCVD-related mortality compared with liver-related mortality (7–12, 139–141), further understanding of the link between these two diseases is clearly indicated (8). Future studies establishing accepted models of NASH and atherosclerosis will provide a translational understanding of the relationship between NASH and ASCVD. By identifying new biomarkers shared between NASH and ASCVD, early detection and intervention will help to reverse the incline in NASH- and ASCVD-related mortality. Lastly, clinical trials seeking an effective therapeutic for NASH must heavily consider the potential influences on atherosclerotic plaque burden.
AF designed the graphic in Figure 1 and Table 1. AY and OR designed Figure 2 with Biorender. All authors contributed to drafting and editing of the review. All authors contributed to the article and approved the submitted version.
This work was partly supported by the National Institutes of Health grants DK134011 and HL150233 (OR), HL145131 and HL167758 (AY), and DK115778 and GM147269 (BC); Collaborative Intramural Research Program (LSUHS and Ochsner Clinic Foundation, AY and OR); an American Heart Association Postdoctoral Fellowship 23POST1026505 (AF), and the LSUHS Center for Cardiovascular Diseases and Sciences Malcolm Feist Fellowships (AF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, et al. Heart disease and stroke statistics-2023 update: a report from the American heart association. Circulation. (2023) 147(8):e93–621. doi: 10.1161/CIR.0000000000001123
2. Loos RJF, Yeo GSH. The genetics of obesity: from discovery to biology. Nat Rev Genet. (2022) 23(2):120–33. doi: 10.1038/s41576-021-00414-z
3. Marchesini G, Marzocchi R. Metabolic syndrome and NASH. Clin Liver Dis. (2007) 11(1):105–17; ix. doi: 10.1016/j.cld.2007.02.013
4. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. (2016) 64(1):73–84. doi: 10.1002/hep.28431
5. Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. J Hepatol. (2019) 71(4):793–801. doi: 10.1016/j.jhep.2019.06.021
6. Riazi K, Azhari H, Charette JH, Underwood FE, King JA, Afshar EE, et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. (2022) 7(9):851–61. doi: 10.1016/S2468-1253(22)00165-0
7. Targher G, Byrne CD, Lonardo A, Zoppini G, Barbui C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta-analysis. J Hepatol. (2016) 65(3):589–600. doi: 10.1016/j.jhep.2016.05.013
8. Duell PB, Welty FK, Miller M, Chait A, Hammond G, Ahmad Z, et al. Nonalcoholic fatty liver disease and cardiovascular risk: a scientific statement from the American heart association. Arterioscler Thromb Vasc Biol. (2022) 42(6):e168–85. doi: 10.1161/ATV.0000000000000153
9. Stepanova M, Younossi ZM. Independent association between nonalcoholic fatty liver disease and cardiovascular disease in the US population. Clin Gastroenterol Hepatol. (2012) 10(6):646–50. doi: 10.1016/j.cgh.2011.12.039
10. Oni ET, Agatston AS, Blaha MJ, Fialkow J, Cury R, Sposito A, et al. A systematic review: burden and severity of subclinical cardiovascular disease among those with nonalcoholic fatty liver; should we care? Atherosclerosis. (2013) 230(2):258–67. doi: 10.1016/j.atherosclerosis.2013.07.052
11. Pais R, Giral P, Khan JF, Rosenbaum D, Housset C, Poynard T, et al. Fatty liver is an independent predictor of early carotid atherosclerosis. J Hepatol. (2016) 65(1):95–102. doi: 10.1016/j.jhep.2016.02.023
12. Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. (2020) 69(9):1691–705. doi: 10.1136/gutjnl-2020-320622
13. Barbarroja N, Ruiz-Ponce M, Cuesta-Lopez L, Perez-Sanchez C, Lopez-Pedrera C, Arias-de la Rosa I, et al. Nonalcoholic fatty liver disease in inflammatory arthritis: relationship with cardiovascular risk. Front Immunol. (2022) 13:997270. doi: 10.3389/fimmu.2022.997270
14. Baratta F, D'Erasmo L, Bini S, Pastori D, Angelico F, Del Ben M, et al. Heterogeneity of non-alcoholic fatty liver disease (NAFLD): implication for cardiovascular risk stratification. Atherosclerosis. (2022) 357:51–9. doi: 10.1016/j.atherosclerosis.2022.08.011
15. Chew NWS, Chong B, Ng CH, Kong G, Chin YH, Xiao W, et al. The genetic interactions between non-alcoholic fatty liver disease and cardiovascular diseases. Front Genet. (2022) 13:971484. doi: 10.3389/fgene.2022.971484
16. Cazac GD, Lacatusu CM, Mihai C, Grigorescu ED, Onofriescu A, Mihai BM. New insights into non-alcoholic fatty liver disease and coronary artery disease: the liver-heart axis. Life (Basel). (2022) 12(8):1–25. doi: 10.3390/life12081189
17. Wang Z, Ye M, Zhang XJ, Zhang P, Cai J, Li H, et al. Impact of NAFLD and its pharmacotherapy on lipid profile and CVD. Atherosclerosis. (2022) 355:30–44. doi: 10.1016/j.atherosclerosis.2022.07.010
18. Galvin Z, Rajakumar R, Chen E, Adeyi O, Selzner M, Grant D, et al. Predictors of de novo nonalcoholic fatty liver disease after liver transplantation and associated fibrosis. Liver Transpl. (2019) 25(1):56–67. doi: 10.1002/lt.25338
19. Meng H, Ruan J, Yan Z, Chen Y, Liu J, Li X, et al. New progress in early diagnosis of atherosclerosis. Int J Mol Sci. (2022) 23(16):1–14. doi: 10.3390/ijms23168939
20. Spengler EK, Loomba R. Recommendations for diagnosis, referral for liver biopsy, and treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mayo Clin Proc. (2015) 90(9):1233–46. doi: 10.1016/j.mayocp.2015.06.013
21. Siddiqui MS, Van Natta ML, Connelly MA, Vuppalanchi R, Neuschwander-Tetri BA, Tonascia J, et al. Impact of obeticholic acid on the lipoprotein profile in patients with non-alcoholic steatohepatitis. J Hepatol. (2020) 72(1):25–33. doi: 10.1016/j.jhep.2019.10.006
22. Nelson A, Torres DM, Morgan AE, Fincke C, Harrison SA. A pilot study using simvastatin in the treatment of nonalcoholic steatohepatitis: a randomized placebo-controlled trial. J Clin Gastroenterol. (2009) 43(10):990–4. doi: 10.1097/MCG.0b013e31819c392e
23. Bril F, Portillo Sanchez P, Lomonaco R, Orsak B, Hecht J, Tio F, et al. Liver safety of statins in prediabetes or T2DM and nonalcoholic steatohepatitis: post hoc analysis of a randomized trial. J Clin Endocrinol Metab. (2017) 102(8):2950–61. doi: 10.1210/jc.2017-00867
24. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5(1):56. doi: 10.1038/s41572-019-0106-z
25. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. (2013) 13(10):709–21. doi: 10.1038/nri3520
26. Hu P, Dharmayat KI, Stevens CAT, Sharabiani MTA, Jones RS, Watts GF, et al. Prevalence of familial hypercholesterolemia among the general population and patients with atherosclerotic cardiovascular disease: a systematic review and meta-analysis. Circulation. (2020) 141(22):1742–59. doi: 10.1161/CIRCULATIONAHA.119.044795
27. Fruchart JC, Nierman MC, Stroes ES, Kastelein JJ, Duriez P. New risk factors for atherosclerosis and patient risk assessment. Circulation. (2004) 109(23 Suppl 1):III15–9. doi: 10.1161/01.CIR.0000131513.33892.5b
28. Centers for Disease C, Prevention. Vital signs: prevalence, treatment, and control of high levels of low-density lipoprotein cholesterol–United States, 1999–2002 and 2005–2008. MMWR Morb Mortal Wkly Rep. (2011) 60(4):109–14.21293326
29. Mehta NK, Abrams LR, Myrskyla M. US life expectancy stalls due to cardiovascular disease, not drug deaths. Proc Natl Acad Sci U S A. (2020) 117(13):6998–7000. doi: 10.1073/pnas.1920391117
30. Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. (2009) 10(1):53–62. doi: 10.1038/nrm2596
31. Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. (2011) 108(2):235–48. doi: 10.1161/CIRCRESAHA.110.223875
32. Navab M, Ananthramaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, Fonarow GC, et al. The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J Lipid Res. (2004) 45(6):993–1007. doi: 10.1194/jlr.R400001-JLR200
33. Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. (2006) 26(8):1702–11. doi: 10.1161/01.ATV.0000229218.97976.43
34. Yurdagul A Jr, Green J, Albert P, McInnis MC, Mazar AP, Orr AW. Alpha5beta1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol. (2014) 34(7):1362–73. doi: 10.1161/ATVBAHA.114.303863
35. Yurdagul A Jr. Crosstalk between macrophages and vascular smooth muscle cells in atherosclerotic plaque stability. Arterioscler Thromb Vasc Biol. (2022) 42(4):372–80. doi: 10.1161/ATVBAHA.121.316233
36. Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. (2012) 95(2):156–64. doi: 10.1093/cvr/cvs115
37. Davies MJ, Richardson PD, Woolf N, Katz DR, Mann J. Risk of thrombosis in human atherosclerotic plaques: role of extracellular lipid, macrophage, and smooth muscle cell content. Br Heart J. (1993) 69(5):377–81. doi: 10.1136/hrt.69.5.377
38. Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. (2013) 339(6116):166–72. doi: 10.1126/science.1230720
39. Doran AC, Yurdagul A Jr, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. (2020) 20(4):254–67. doi: 10.1038/s41577-019-0240-6
40. Yurdagul A Jr, Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med. (2017) 4:86. doi: 10.3389/fcvm.2017.00086
41. Ampomah PB, Cai B, Sukka SR, Gerlach BD, Yurdagul A Jr, Wang X, et al. Macrophages use apoptotic cell-derived methionine and DNMT3A during efferocytosis to promote tissue resolution. Nat Metab. (2022) 4(4):444–57. doi: 10.1038/s42255-022-00551-7
42. Gerlach BD, Ampomah PB, Yurdagul A Jr, Liu C, Lauring MC, Wang X, et al. Efferocytosis induces macrophage proliferation to help resolve tissue injury. Cell Metab. (2021) 33(12):2445–63.e8. doi: 10.1016/j.cmet.2021.10.015
43. Yurdagul A Jr, Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. (2020) 31(3):518–33.e10. doi: 10.1016/j.cmet.2020.01.001
44. Back M, Yurdagul A Jr, Tabas I, Oorni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. (2019) 16(7):389–406. doi: 10.1038/s41569-019-0169-2
45. Maldonado N, Kelly-Arnold A, Cardoso L, Weinbaum S. The explosive growth of small voids in vulnerable cap rupture; cavitation and interfacial debonding. J Biomech. (2013) 46(2):396–401. doi: 10.1016/j.jbiomech.2012.10.040
46. Vengrenyuk Y, Carlier S, Xanthos S, Cardoso L, Ganatos P, Virmani R, et al. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc Natl Acad Sci U S A. (2006) 103(40):14678–83. doi: 10.1073/pnas.0606310103
47. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. (2021) 184(10):2537–64. doi: 10.1016/j.cell.2021.04.015
48. Loomba R, Adams LA. The 20% rule of NASH progression: the natural history of advanced fibrosis and cirrhosis caused by NASH. Hepatology. (2019) 70(6):1885–8. doi: 10.1002/hep.30946
49. Grgurevic I, Podrug K, Mikolasevic I, Kukla M, Madir A, Tsochatzis EA. Natural history of nonalcoholic fatty liver disease: implications for clinical practice and an individualized approach. Can J Gastroenterol Hepatol. (2020) 2020:9181368. doi: 10.1155/2020/9181368
50. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. (2018) 24(7):908–22. doi: 10.1038/s41591-018-0104-9
51. Brunt EM, Wong VW, Nobili V, Day CP, Sookoian S, Maher JJ, et al. Nonalcoholic fatty liver disease. Nat Rev Dis Primers. (2015) 1:15080. doi: 10.1038/nrdp.2015.80
52. Mitsuyoshi H, Yasui K, Harano Y, Endo M, Tsuji K, Minami M, et al. Analysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver disease. Hepatol Res. (2009) 39(4):366–73. doi: 10.1111/j.1872-034X.2008.00464.x
53. He J, Lee JH, Febbraio M, Xie W. The emerging roles of fatty acid translocase/CD36 and the aryl hydrocarbon receptor in fatty liver disease. Exp Biol Med (Maywood). (2011) 236(10):1116–21. doi: 10.1258/ebm.2011.011128
54. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. (2015) 21(5):739–46. doi: 10.1016/j.cmet.2015.04.004
55. Johnson ES, Lindblom KR, Robeson A, Stevens RD, Ilkayeva OR, Newgard CB, et al. Metabolomic profiling reveals a role for caspase-2 in lipoapoptosis. J Biol Chem. (2013) 288(20):14463–75. doi: 10.1074/jbc.M112.437210
56. Huby T, Gautier EL. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat Rev Immunol. (2022) 22(7):429–43. doi: 10.1038/s41577-021-00639-3
57. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gelineau A, et al. Impaired kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity. (2020) 53(3):627–40.e5. doi: 10.1016/j.immuni.2020.06.003
58. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol Cell. (2019) 75(3):644–60.e5. doi: 10.1016/j.molcel.2019.07.028
59. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of liver kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. (2010) 59(2):347–57. doi: 10.2337/db09-0016
60. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. (2012) 302(11):G1310–21. doi: 10.1152/ajpgi.00365.2011
61. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 Modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. (2006) 116(1):115–24. doi: 10.1172/JCI24335
62. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated kupffer cells. Nat Commun. (2016) 7:10321. doi: 10.1038/ncomms10321
63. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, et al. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology. (2010) 51(2):511–22. doi: 10.1002/hep.23337
64. Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. (2013) 57(2):577–89. doi: 10.1002/hep.26081
65. Snodgrass RG, Huang S, Choi IW, Rutledge JC, Hwang DH. Inflammasome-mediated secretion of IL-1beta in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol. (2013) 191(8):4337–47. doi: 10.4049/jimmunol.1300298
66. Kim SY, Jeong JM, Kim SJ, Seo W, Kim MH, Choi WM, et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun. (2017) 8(1):2247. doi: 10.1038/s41467-017-02325-2
67. Zhu C, Tabas I, Schwabe RF, Pajvani UB. Maladaptive regeneration—the reawakening of developmental pathways in NASH and fibrosis. Nat Rev Gastroenterol Hepatol. (2021) 18(2):131–42. doi: 10.1038/s41575-020-00365-6
68. Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. (2013) 27(7):719–24. doi: 10.1101/gad.207803.112
69. Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, et al. Hepatocyte notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci Transl Med. (2018) 10(468):1–13. doi: 10.1126/scitranslmed.aat0344
70. Guy CD, Suzuki A, Abdelmalek MF, Burchette JL, Diehl AM, Nash CRN. Treatment response in the PIVENS trial is associated with decreased hedgehog pathway activity. Hepatology. (2015) 61(1):98–107. doi: 10.1002/hep.27235
71. Chung SI, Moon H, Ju HL, Cho KJ, Kim DY, Han KH, et al. Hepatic expression of sonic hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. J Hepatol. (2016) 64(3):618–27. doi: 10.1016/j.jhep.2015.10.007
72. Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. (2016) 24(6):848–62. doi: 10.1016/j.cmet.2016.09.016
73. Wang X, Sommerfeld MR, Jahn-Hofmann K, Cai B, Filliol A, Remotti HE, et al. A therapeutic silencing RNA targeting hepatocyte TAZ prevents and reverses fibrosis in nonalcoholic steatohepatitis in mice. Hepatol Commun. (2019) 3(9):1221–34. doi: 10.1002/hep4.1405
74. Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R, et al. Cholesterol stabilizes TAZ in hepatocytes to promote experimental non-alcoholic steatohepatitis. Cell Metab. (2020) 31(5):969–86.e7. doi: 10.1016/j.cmet.2020.03.010
75. Gordon SM, Li H, Zhu X, Shah AS, Lu LJ, Davidson WS. A comparison of the mouse and human lipoproteome: suitability of the mouse model for studies of human lipoproteins. J Proteome Res. (2015) 14(6):2686–95. doi: 10.1021/acs.jproteome.5b00213
76. Linton MRF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, et al. The role of lipids and lipoproteins in atherosclerosis. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, et al., editors. Endotext. South Dartmouth (MA, U.S.A.) (2000). p. 1–144.
77. Schreyer SA, Wilson DL, LeBoeuf RC. C57BL/6 mice fed high fat diets as models for diabetes-accelerated atherosclerosis. Atherosclerosis. (1998) 136(1):17–24. doi: 10.1016/s0021-9150(97)00165-2
78. Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. (1993) 92(2):883–93. doi: 10.1172/JCI116663
79. Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. (1992) 258(5081):468–71. doi: 10.1126/science.1411543
80. Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. (1992) 71(2):343–53. doi: 10.1016/0092-8674(92)90362-g
81. Goettsch C, Hutcheson JD, Hagita S, Rogers MA, Creager MD, Pham T, et al. A single injection of gain-of-function mutant PCSK9 adeno-associated virus vector induces cardiovascular calcification in mice with no genetic modification. Atherosclerosis. (2016) 251:109–18. doi: 10.1016/j.atherosclerosis.2016.06.011
82. Bjorklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG, et al. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. (2014) 114(11):1684–9. doi: 10.1161/CIRCRESAHA.114.302937
83. Rom O, Liu Y, Liu Z, Zhao Y, Wu J, Ghrayeb A, et al. Glycine-based treatment ameliorates NAFLD by modulating fatty acid oxidation, glutathione synthesis, and the gut microbiome. Sci Transl Med. (2020) 12(572):1–15. doi: 10.1126/scitranslmed.aaz2841
84. Rom O, Xu G, Guo Y, Zhu Y, Wang H, Zhang J, et al. Nitro-fatty acids protect against steatosis and fibrosis during development of nonalcoholic fatty liver disease in mice. EBioMedicine. (2019) 41:62–72. doi: 10.1016/j.ebiom.2019.02.019
85. Rom O, Liu Y, Chang L, Chen YE, Aviram M. Editorial: nitro-fatty acids: novel drug candidates for the co-treatment of atherosclerosis and nonalcoholic fatty liver disease. Curr Opin Lipidol. (2020) 31(2):104–7. doi: 10.1097/MOL.0000000000000666
86. Karavia EA, Papachristou DJ, Kotsikogianni I, Giopanou I, Kypreos KE. Deficiency in apolipoprotein E has a protective effect on diet-induced nonalcoholic fatty liver disease in mice. FEBS J. (2011) 278(17):3119–29. doi: 10.1111/j.1742-4658.2011.08238.x
87. Lu W, Mei J, Yang J, Wu Z, Liu J, Miao P, et al. Apoe deficiency promotes non-alcoholic fatty liver disease in mice via impeding AMPK/mTOR mediated autophagy. Life Sci. (2020) 252:117601. doi: 10.1016/j.lfs.2020.117601
88. Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. (2007) 46(5):1392–403. doi: 10.1002/hep.21874
89. Zhang X, Coker OO, Chu ES, Fu K, Lau HCH, Wang YX, et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. (2021) 70(4):761–74. doi: 10.1136/gutjnl-2019-319664
90. Schierwagen R, Maybuchen L, Zimmer S, Hittatiya K, Back C, Klein S, et al. Seven weeks of western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci Rep. (2015) 5:12931. doi: 10.1038/srep12931
91. Muniz LB, Alves-Santos AM, Camargo F, Martins DB, Celes MRN, Naves MMV. High-lard and high-cholesterol diet, but not high-lard diet, leads to metabolic disorders in a modified dyslipidemia model. Arq Bras Cardiol. (2019) 113(5):896–902. doi: 10.5935/abc.20190149
92. Trevaskis JL, Griffin PS, Wittmer C, Neuschwander-Tetri BA, Brunt EM, Dolman CS, et al. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol. (2012) 302(8):G762–72. doi: 10.1152/ajpgi.00476.2011
93. Caballero F, Fernandez A, De Lacy AM, Fernandez-Checa JC, Caballeria J, Garcia-Ruiz C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol. (2009) 50(4):789–96. doi: 10.1016/j.jhep.2008.12.016
94. Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and fas-mediated steatohepatitis. Cell Metab. (2006) 4(3):185–98. doi: 10.1016/j.cmet.2006.07.006
95. Soliman GA. Dietary cholesterol and the lack of evidence in cardiovascular disease. Nutrients. (2018) 10(6):1–14. doi: 10.3390/nu10060780
96. Shekelle RB, Stamler J. Dietary cholesterol and ischaemic heart disease. Lancet. (1989) 1(8648):1177–9. doi: 10.1016/s0140-6736(89)92759-1
97. McNamara DJ. Dietary cholesterol and atherosclerosis. Biochim Biophys Acta. (2000) 1529(1–3):310–20. doi: 10.1016/s1388-1981(00)00156-6
98. Lancaster KJ. Current intake and demographic disparities in the association of fructose-rich foods and metabolic syndrome. JAMA Netw Open. (2020) 3(7):e2010224. doi: 10.1001/jamanetworkopen.2020.10224
99. Handayani D, Febrianingsih E, Desi Kurniawati A, Kusumastuty I, Nurmalitasari S, Widyanto RM, et al. High-fructose diet initially promotes increased aortic wall thickness, liver steatosis, and cardiac histopathology deterioration, but does not increase body fat index. J Public Health Res. (2021) 10(2):1–7. doi: 10.4081/jphr.2021.2181
100. van den Hoek AM, Verschuren L, Worms N, van Nieuwkoop A, de Ruiter C, Attema J, et al. A translational mouse model for NASH with advanced fibrosis and atherosclerosis expressing key pathways of human pathology. Cells. (2020) 9(9):1–20. doi: 10.3390/cells9092014
101. Morrison MC, Mulder P, Salic K, Verheij J, Liang W, van Duyvenvoorde W, et al. Intervention with a caspase-1 inhibitor reduces obesity-associated hyperinsulinemia, non-alcoholic steatohepatitis and hepatic fibrosis in LDLR-/-.Leiden mice. Int J Obes (Lond). (2016) 40(9):1416–23. doi: 10.1038/ijo.2016.74
102. Morrison MC, Verschuren L, Salic K, Verheij J, Menke A, Wielinga PY, et al. Obeticholic acid modulates serum metabolites and gene signatures characteristic of human NASH and attenuates inflammation and fibrosis progression in Ldlr-/-.Leiden mice. Hepatol Commun. (2018) 2(12):1513–32. doi: 10.1002/hep4.1270
103. Liang W, Menke AL, Driessen A, Koek GH, Lindeman JH, Stoop R, et al. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS One. (2014) 9(12):e115922. doi: 10.1371/journal.pone.0115922
104. Herman MA, Samuel VT. The sweet path to metabolic demise: fructose and lipid synthesis. Trends Endocrinol Metab. (2016) 27(10):719–30. doi: 10.1016/j.tem.2016.06.005
105. Softic S, Gupta MK, Wang GX, Fujisaka S, O'Neill BT, Rao TN, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest. (2018) 128(3):1199. doi: 10.1172/JCI99009
106. Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. (2014) 26(3):331–43. doi: 10.1016/j.ccr.2014.07.001
107. Softic S, Gupta MK, Wang GX, Fujisaka S, O'Neill BT, Rao TN, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest. (2017) 127(11):4059–74. doi: 10.1172/JCI94585
108. Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. (2009) 119(5):1322–34. doi: 10.1172/JCI37385
109. Softic S, Meyer JG, Wang GX, Gupta MK, Batista TM, Lauritzen H, et al. Dietary sugars alter hepatic fatty acid oxidation via transcriptional and post-translational modifications of mitochondrial proteins. Cell Metab. (2019) 30(4):735–53.e4. doi: 10.1016/j.cmet.2019.09.003
110. Kavanagh K, Wylie AT, Tucker KL, Hamp TJ, Gharaibeh RZ, Fodor AA, et al. Dietary fructose induces endotoxemia and hepatic injury in calorically controlled primates. Am J Clin Nutr. (2013) 98(2):349–57. doi: 10.3945/ajcn.112.057331
111. Wiedermann CJ, Kiechl S, Dunzendorfer S, Schratzberger P, Egger G, Oberhollenzer F, et al. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the bruneck study. J Am Coll Cardiol. (1999) 34(7):1975–81. doi: 10.1016/s0735-1097(99)00448-9
112. Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. (2009) 50(2):638–44. doi: 10.1002/hep.23009
113. Chiba T, Suzuki S, Sato Y, Itoh T, Umegaki K. Evaluation of methionine content in a high-fat and choline-deficient diet on body weight gain and the development of non-alcoholic steatohepatitis in mice. PLoS One. (2016) 11(10):e0164191. doi: 10.1371/journal.pone.0164191
114. Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. (2004) 40(1):47–51. doi: 10.1016/j.jhep.2003.09.020
115. Rizki G, Arnaboldi L, Gabrielli B, Yan J, Lee GS, Ng RK, et al. Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J Lipid Res. (2006) 47(10):2280–90. doi: 10.1194/jlr.M600198-JLR200
116. Smati S, Polizzi A, Fougerat A, Ellero-Simatos S, Blum Y, Lippi Y, et al. Integrative study of diet-induced mouse models of NAFLD identifies PPARalpha as a sexually dimorphic drug target. Gut. (2022) 71(4):807–21. doi: 10.1136/gutjnl-2020-323323
117. Collins HL, Adelman SJ, Butteiger DN, Bortz JD. Choline supplementation does not promote atherosclerosis in CETP-expressing male apolipoprotein E knockout mice. Nutrients. (2022) 14(8):1–12. doi: 10.3390/nu14081651
118. Schwabl P, Hambruch E, Budas GR, Supper P, Burnet M, Liles JT, et al. The non-steroidal FXR agonist cilofexor improves portal hypertension and reduces hepatic fibrosis in a rat NASH model. Biomedicines. (2021) 9(1):1–11. doi: 10.3390/biomedicines9010060
119. Takayama F, Egashira T, Kawasaki H, Mankura M, Nakamoto K, Okada S, et al. A novel animal model of nonalcoholic steatohepatitis (NASH): hypoxemia enhances the development of NASH. J Clin Biochem Nutr. (2009) 45(3):335–40. doi: 10.3164/jcbn.09-29
120. Raubenheimer PJ, Nyirenda MJ, Walker BR. A choline-deficient diet exacerbates fatty liver but attenuates insulin resistance and glucose intolerance in mice fed a high-fat diet. Diabetes. (2006) 55(7):2015–20. doi: 10.2337/db06-0097
121. Bril F, Lomonaco R, Orsak B, Ortiz-Lopez C, Webb A, Tio F, et al. Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology. (2014) 59(6):2178–87. doi: 10.1002/hep.26988
123. Santhekadur PK, Kumar DP, Sanyal AJ. Preclinical models of non-alcoholic fatty liver disease. J Hepatol. (2018) 68(2):230–7. doi: 10.1016/j.jhep.2017.10.031
124. Skold BH, Getty R, Ramsey FK. Spontaneous atherosclerosis in the arterial system of aging swine. Am J Vet Res. (1966) 27(116):257–73.4161799
125. Matthan NR, Solano-Aguilar G, Meng H, Lamon-Fava S, Goldbaum A, Walker ME, et al. The ossabaw pig is a suitable translational model to evaluate dietary patterns and coronary artery disease risk. J Nutr. (2018) 148(4):542–51. doi: 10.1093/jn/nxy002
126. Lee L, Alloosh M, Saxena R, Van Alstine W, Watkins BA, Klaunig JE, et al. Nutritional model of steatohepatitis and metabolic syndrome in the ossabaw miniature swine. Hepatology. (2009) 50(1):56–67. doi: 10.1002/hep.22904
127. Ogawa T, Fujii H, Yoshizato K, Kawada N. A human-type nonalcoholic steatohepatitis model with advanced fibrosis in rabbits. Am J Pathol. (2010) 177(1):153–65. doi: 10.2353/ajpath.2010.090895
128. Ignatowski A. Influence of animal food on the organsim of rabbits. Izvest Imper Voennomed Akad St Petersburg. (1908) 16:154–73.
129. Fan J, Kitajima S, Watanabe T, Xu J, Zhang J, Liu E, et al. Rabbit models for the study of human atherosclerosis: from pathophysiological mechanisms to translational medicine. Pharmacol Ther. (2015) 146:104–19. doi: 10.1016/j.pharmthera.2014.09.009
130. Koike T, Koike Y, Yang D, Guo Y, Rom O, Song J, et al. Human apolipoprotein A-II reduces atherosclerosis in knock-in rabbits. Atherosclerosis. (2021) 316:32–40. doi: 10.1016/j.atherosclerosis.2020.11.028
131. Taylor E, Huang N, Bodde J, Ellison A, Killiany R, Bachschmid MM, et al. MRI of atherosclerosis and fatty liver disease in cholesterol fed rabbits. J Transl Med. (2018) 16(1):215. doi: 10.1186/s12967-018-1587-3
132. Eggen DA. Cholesterol metabolism in rhesus monkey, squirrel monkey, and baboon. J Lipid Res. (1974) 15(2):139–45. doi: 10.1016/S0022-2275(20)36816-4
133. Higgins PB, Bastarrachea RA, Lopez-Alvarenga JC, Garcia-Forey M, Proffitt JM, Voruganti VS, et al. Eight week exposure to a high sugar high fat diet results in adiposity gain and alterations in metabolic biomarkers in baboons (papio hamadryas sp.). Cardiovasc Diabetol. (2010) 9:71. doi: 10.1186/1475-2840-9-71
134. Lyu L, Liu XL, Rui MP, Yang LC, Wang GZ, Fan D, et al. Liver extracellular volume fraction values obtained with magnetic resonance imaging can quantitatively stage liver fibrosis: a validation study in monkeys with nonalcoholic steatohepatitis. Eur Radiol. (2020) 30(10):5748–57. doi: 10.1007/s00330-020-06902-w
135. Jian C, Fu J, Cheng X, Shen LJ, Ji YX, Wang X, et al. Low-dose sorafenib acts as a mitochondrial uncoupler and ameliorates nonalcoholic steatohepatitis. Cell Metab. (2020) 31(5):892–908.e11. doi: 10.1016/j.cmet.2020.04.011
136. Zhang XJ, Ji YX, Cheng X, Cheng Y, Yang H, Wang J, et al. A small molecule targeting ALOX12-ACC1 ameliorates nonalcoholic steatohepatitis in mice and macaques. Sci Transl Med. (2021) 13(624):eabg8116. doi: 10.1126/scitranslmed.abg8116
137. Wendler D. Should protections for research with humans who cannot consent apply to research with nonhuman primates? Theor Med Bioeth. (2014) 35(2):157–73. doi: 10.1007/s11017-014-9285-5
138. Targher G, Byrne CD. Circulating markers of liver function and cardiovascular disease risk. Arterioscler Thromb Vasc Biol. (2015) 35(11):2290–6. doi: 10.1161/ATVBAHA.115.305235
139. Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension, diabetes, atherosclerosis and NASH: cause or consequence? J Hepatol. (2018) 68(2):335–52. doi: 10.1016/j.jhep.2017.09.021
140. Adams LA, Anstee QM, Tilg H, Targher G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut. (2017) 66(6):1138–53. doi: 10.1136/gutjnl-2017-313884
141. Allen AM, Therneau TM, Larson JJ, Coward A, Somers VK, Kamath PS. Nonalcoholic fatty liver disease incidence and impact on metabolic burden and death: a 20 year-community study. Hepatology. (2018) 67(5):1726–36. doi: 10.1002/hep.29546
142. Xi L, Kouvelos G, Paolocci N. Circulating biomarkers for cardiovascular diseases: the beats never stop. Acta Pharmacol Sin. (2018) 39(7):1065–7. doi: 10.1038/aps.2018.43
143. Lowe GD, Yarnell JW, Rumley A, Bainton D, Sweetnam PM. C-reactive protein, fibrin D-dimer, and incident ischemic heart disease in the speedwell study: are inflammation and fibrin turnover linked in pathogenesis? Arterioscler Thromb Vasc Biol. (2001) 21(4):603–10. doi: 10.1161/01.atv.21.4.603
144. Pfutzner A, Forst T. High-sensitivity C-reactive protein as cardiovascular risk marker in patients with diabetes mellitus. Diabetes Technol Ther. (2006) 8(1):28–36. doi: 10.1089/dia.2006.8.28
145. Heeren J, Scheja L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol Metab. (2021) 50:101238. doi: 10.1016/j.molmet.2021.101238
146. Pei K, Gui T, Kan D, Feng H, Jin Y, Yang Y, et al. An overview of lipid metabolism and nonalcoholic fatty liver disease. Biomed Res Int. (2020) 2020:4020249. doi: 10.1155/2020/4020249
147. Deprince A, Haas JT, Staels B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol Metab. (2020) 42:101092. doi: 10.1016/j.molmet.2020.101092
148. Manjunath CN, Rawal JR, Irani PM, Madhu K. Atherogenic dyslipidemia. Indian J Endocrinol Metab. (2013) 17(6):969–76. doi: 10.4103/2230-8210.122600
149. Liao X, Ma Q, Wu T, Shao C, Lin Y, Sun Y, et al. Lipid-lowering responses to dyslipidemia determine the efficacy on liver enzymes in metabolic dysfunction-associated fatty liver disease with hepatic injuries: a prospective cohort study. Diabetes Metab Syndr Obes. (2022) 15:1173–84. doi: 10.2147/DMSO.S356371
150. Morrow MR, Batchuluun B, Wu J, Ahmadi E, Leroux JM, Mohammadi-Shemirani P, et al. Inhibition of ATP-citrate lyase improves NASH, liver fibrosis, and dyslipidemia. Cell Metab. (2022) 34(6):919–36.e8. doi: 10.1016/j.cmet.2022.05.004
151. Abdallah M, Brown L, Provenza J, Tariq R, Gowda S, Singal AK. Safety and efficacy of dyslipidemia treatment in NAFLD patients: a meta-analysis of randomized controlled trials. Ann Hepatol. (2022) 27(6):100738. doi: 10.1016/j.aohep.2022.100738
152. Shahab O, Biswas R, Paik J, Bush H, Golabi P, Younossi ZM. Among patients with NAFLD, treatment of dyslipidemia does not reduce cardiovascular mortality. Hepatol Commun. (2018) 2(10):1227–34. doi: 10.1002/hep4.1241
153. Das Pradhan A, Glynn RJ, Fruchart JC, MacFadyen JG, Zaharris ES, Everett BM, et al. Triglyceride lowering with pemafibrate to reduce cardiovascular risk. N Engl J Med. (2022) 387(21):1923–34. doi: 10.1056/NEJMoa2210645
154. Tanase DM, Gosav EM, Petrov D, Jucan AE, Lacatusu CM, Floria M, et al. Involvement of ceramides in non-alcoholic fatty liver disease (NAFLD) atherosclerosis (ATS) development: mechanisms and therapeutic targets. Diagnostics (Basel). (2021) 11(11):1–20. doi: 10.3390/diagnostics11112053
155. Chaurasia B, Summers SA. Ceramides—lipotoxic inducers of metabolic disorders: (trends in endocrinology and metabolism 26, 538-550; 2015). Trends Endocrinol Metab. (2018) 29(1):66–7. doi: 10.1016/j.tem.2017.09.005
156. Sokolowska E, Blachnio-Zabielska A. The role of ceramides in insulin resistance. Front Endocrinol (Lausanne). (2019) 10:577. doi: 10.3389/fendo.2019.00577
157. Wasilewska N, Bobrus-Chociej A, Harasim-Symbor E, Tarasow E, Wojtkowska M, Chabowski A, et al. Increased serum concentration of ceramides in obese children with nonalcoholic fatty liver disease. Lipids Health Dis. (2018) 17(1):216. doi: 10.1186/s12944-018-0855-9
158. Kasumov T, Li L, Li M, Gulshan K, Kirwan JP, Liu X, et al. Ceramide as a mediator of non-alcoholic fatty liver disease and associated atherosclerosis. PLoS One. (2015) 10(5):e0126910. doi: 10.1371/journal.pone.0126910
159. Gorden DL, Myers DS, Ivanova PT, Fahy E, Maurya MR, Gupta S, et al. Biomarkers of NAFLD progression: a lipidomics approach to an epidemic. J Lipid Res. (2015) 56(3):722–36. doi: 10.1194/jlr.P056002
160. Wiesner P, Leidl K, Boettcher A, Schmitz G, Liebisch G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. J Lipid Res. (2009) 50(3):574–85. doi: 10.1194/jlr.D800028-JLR200
161. Anroedh S, Hilvo M, Akkerhuis KM, Kauhanen D, Koistinen K, Oemrawsingh R, et al. Plasma concentrations of molecular lipid species predict long-term clinical outcome in coronary artery disease patients. J Lipid Res. (2018) 59(9):1729–37. doi: 10.1194/jlr.P081281
162. Laaksonen R, Ekroos K, Sysi-Aho M, Hilvo M, Vihervaara T, Kauhanen D, et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur Heart J. (2016) 37(25):1967–76. doi: 10.1093/eurheartj/ehw148
163. Wang DD, Toledo E, Hruby A, Rosner BA, Willett WC, Sun Q, et al. Plasma ceramides, Mediterranean diet, and incident cardiovascular disease in the PREDIMED trial (prevencion con dieta Mediterranea). Circulation. (2017) 135(21):2028–40. doi: 10.1161/CIRCULATIONAHA.116.024261
164. Meeusen JW, Donato LJ, Kopecky SL, Vasile VC, Jaffe AS, Laaksonen R. Ceramides improve atherosclerotic cardiovascular disease risk assessment beyond standard risk factors. Clin Chim Acta. (2020) 511:138–42. doi: 10.1016/j.cca.2020.10.005
165. Akhiyat N, Vasile V, Ahmad A, Sara JD, Nardi V, Lerman LO, et al. Plasma ceramide levels are elevated in patients with early coronary atherosclerosis and endothelial dysfunction. J Am Heart Assoc. (2022) 11(7):e022852. doi: 10.1161/JAHA.121.022852
166. Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, Wang L, et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science. (2019) 365(6451):386–92. doi: 10.1126/science.aav3722
167. Jiang M, Li C, Liu Q, Wang A, Lei M. Inhibiting ceramide synthesis attenuates hepatic steatosis and fibrosis in rats with non-alcoholic fatty liver disease. Front Endocrinol (Lausanne). (2019) 10:665. doi: 10.3389/fendo.2019.00665
168. Chun L, Junlin Z, Aimin W, Niansheng L, Benmei C, Minxiang L. Inhibition of ceramide synthesis reverses endothelial dysfunction and atherosclerosis in streptozotocin-induced diabetic rats. Diabetes Res Clin Pract. (2011) 93(1):77–85. doi: 10.1016/j.diabres.2011.03.017
169. Park TS, Rosebury W, Kindt EK, Kowala MC, Panek RL. Serine palmitoyltransferase inhibitor myriocin induces the regression of atherosclerotic plaques in hyperlipidemic ApoE-deficient mice. Pharmacol Res. (2008) 58(1):45–51. doi: 10.1016/j.phrs.2008.06.005
170. Pessayre D, Mansouri A, Fromenty B. Nonalcoholic steatosis and steatohepatitis. V. Mitochondrial dysfunction in steatohepatitis. Am J Physiol Gastrointest Liver Physiol. (2002) 282(2):G193–9. doi: 10.1152/ajpgi.00426.2001
171. Ore A, Akinloye OA. Oxidative stress and antioxidant biomarkers in clinical and experimental models of non-alcoholic fatty liver disease. Medicina (Kaunas). (2019) 55(2):1–13. doi: 10.3390/medicina55020026
172. Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, Loguercio C, et al. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid Med Cell Longev. (2018) 2018:9547613. doi: 10.1155/2018/9547613
173. Narasimhan S, Gokulakrishnan K, Sampathkumar R, Farooq S, Ravikumar R, Mohan V, et al. Oxidative stress is independently associated with non-alcoholic fatty liver disease (NAFLD) in subjects with and without type 2 diabetes. Clin Biochem. (2010) 43(10–11):815–21. doi: 10.1016/j.clinbiochem.2010.04.003
174. Zelber-Sagi S, Ivancovsky-Wajcman D, Fliss-Isakov N, Hahn M, Webb M, Shibolet O, et al. Serum malondialdehyde is associated with non-alcoholic fatty liver and related liver damage differentially in men and women. Antioxidants (Basel). (2020) 9(7):1–15. doi: 10.3390/antiox9070578
175. Lasker S, Rahman MM, Parvez F, Zamila M, Miah P, Nahar K, et al. High-fat diet-induced metabolic syndrome and oxidative stress in obese rats are ameliorated by yogurt supplementation. Sci Rep. (2019) 9(1):20026. doi: 10.1038/s41598-019-56538-0
176. Karami S, Poustchi H, Sarmadi N, Radmard AR, Ali Yari F, Pakdel A, et al. Association of anti-oxidative capacity of HDL with subclinical atherosclerosis in subjects with and without non-alcoholic fatty liver disease. Diabetol Metab Syndr. (2021) 13(1):121. doi: 10.1186/s13098-021-00741-5
177. Yang X, Li Y, Li Y, Ren X, Zhang X, Hu D, et al. Oxidative stress-mediated atherosclerosis: mechanisms and therapies. Front Physiol. (2017) 8:600. doi: 10.3389/fphys.2017.00600
178. Dimmeler S, Haendeler J, Galle J, Zeiher AM. Oxidized low-density lipoprotein induces apoptosis of human endothelial cells by activation of CPP32-like proteases. A mechanistic clue to the ‘response to injury’ hypothesis. Circulation. (1997) 95(7):1760–3. doi: 10.1161/01.cir.95.7.1760
179. Cominacini L, Rigoni A, Pasini AF, Garbin U, Davoli A, Campagnola M, et al. The binding of oxidized low density lipoprotein (ox-LDL) to ox-LDL receptor-1 reduces the intracellular concentration of nitric oxide in endothelial cells through an increased production of superoxide. J Biol Chem. (2001) 276(17):13750–5. doi: 10.1074/jbc.M010612200
180. Li D, Mehta JL. Antisense to LOX-1 inhibits oxidized LDL-mediated upregulation of monocyte chemoattractant protein-1 and monocyte adhesion to human coronary artery endothelial cells. Circulation. (2000) 101(25):2889–95. doi: 10.1161/01.cir.101.25.2889
181. Chen H, Li D, Saldeen T, Mehta JL. Transforming growth factor-beta(1) modulates oxidatively modified LDL-induced expression of adhesion molecules: role of LOX-1. Circ Res. (2001) 89(12):1155–60. doi: 10.1161/hh2401.100598
182. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34(8):1731–8. doi: 10.1161/ATVBAHA.114.303887
183. Rom O, Korach-Rechtman H, Hayek T, Danin-Poleg Y, Bar H, Kashi Y, et al. Acrolein increases macrophage atherogenicity in association with gut microbiota remodeling in atherosclerotic mice: protective role for the polyphenol-rich pomegranate juice. Arch Toxicol. (2017) 91(4):1709–25. doi: 10.1007/s00204-016-1859-8
184. Rom O, Jeries H, Hayek T, Aviram M. Supplementation with linoleic acid-rich soybean oil stimulates macrophage foam cell formation via increased oxidative stress and diacylglycerol acyltransferase1-mediated triglyceride biosynthesis. Biofactors. (2017) 43(1):100–16. doi: 10.1002/biof.1319
185. Rom O, Aviram M. Endogenous or exogenous antioxidants vs. pro-oxidants in macrophage atherogenicity. Curr Opin Lipidol. (2016) 27(2):204–6. doi: 10.1097/MOL.0000000000000287
186. Auge N, Garcia V, Maupas-Schwalm F, Levade T, Salvayre R, Negre-Salvayre A. Oxidized LDL-induced smooth muscle cell proliferation involves the EGF receptor/PI-3 kinase/Akt and the sphingolipid signaling pathways. Arterioscler Thromb Vasc Biol. (2002) 22(12):1990–5. doi: 10.1161/01.atv.0000043453.21629.3b
187. Liu J, Ren Y, Kang L, Zhang L. Oxidized low-density lipoprotein increases the proliferation and migration of human coronary artery smooth muscle cells through the upregulation of osteopontin. Int J Mol Med. (2014) 33(5):1341–7. doi: 10.3892/ijmm.2014.1681
188. Holvoet P, Mertens A, Verhamme P, Bogaerts K, Beyens G, Verhaeghe R, et al. Circulating oxidized LDL is a useful marker for identifying patients with coronary artery disease. Arterioscler Thromb Vasc Biol. (2001) 21(5):844–8. doi: 10.1161/01.atv.21.5.844
189. Ho CM, Ho SL, Jeng YM, Lai YS, Chen YH, Lu SC, et al. Accumulation of free cholesterol and oxidized low-density lipoprotein is associated with portal inflammation and fibrosis in nonalcoholic fatty liver disease. J Inflamm (Lond). (2019) 16:7. doi: 10.1186/s12950-019-0211-5
190. Nishihara T, Miyoshi T, Ichikawa K, Osawa K, Nakashima M, Miki T, et al. Association of oxidized low-density lipoprotein in nonalcoholic fatty liver disease with high-risk plaque on coronary computed tomography angiography: a matched case-control study. J Clin Med. (2022) 11(10):1–9. doi: 10.3390/jcm11102838
191. Rom O, Aviram M. It is not just lipids: proatherogenic vs. antiatherogenic roles for amino acids in macrophage foam cell formation. Curr Opin Lipidol. (2017) 28(1):85–7. doi: 10.1097/MOL.0000000000000377
192. Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, et al. Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology. (2018) 67(1):145–58. doi: 10.1002/hep.29465
193. Solon-Biet SM, Cogger VC, Pulpitel T, Wahl D, Clark X, Bagley E, et al. Branched chain amino acids impact health and lifespan indirectly via amino acid balance and appetite control. Nat Metab. (2019) 1(5):532–45. doi: 10.1038/s42255-019-0059-2
194. McGarrah RW, White PJ. Branched-chain amino acids in cardiovascular disease. Nat Rev Cardiol. (2023) 20(2):77–89. doi: 10.1038/s41569-022-00760-3
195. Rom O, Liu Y, Finney AC, Ghrayeb A, Zhao Y, Shukha Y, et al. Induction of glutathione biosynthesis by glycine-based treatment mitigates atherosclerosis. Redox Biol. (2022) 52:102313. doi: 10.1016/j.redox.2022.102313
196. Liu Y, Zhao Y, Shukha Y, Lu H, Wang L, Liu Z, et al. Dysregulated oxalate metabolism is a driver and therapeutic target in atherosclerosis. Cell Rep. (2021) 36(4):109420. doi: 10.1016/j.celrep.2021.109420
197. Grajeda-Iglesias C, Rom O, Aviram M. Branched-chain amino acids and atherosclerosis: friends or foes? Curr Opin Lipidol. (2018) 29(2):166–9. doi: 10.1097/MOL.0000000000000494
198. Mardinoglu A, Agren R, Kampf C, Asplund A, Uhlen M, Nielsen J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat Commun. (2014) 5:3083. doi: 10.1038/ncomms4083
199. Mardinoglu A, Bjornson E, Zhang C, Klevstig M, Soderlund S, Stahlman M, et al. Personal model-assisted identification of NAD(+) and glutathione metabolism as intervention target in NAFLD. Mol Syst Biol. (2017) 13(3):916. doi: 10.15252/msb.20167422
200. Wurtz P, Raiko JR, Magnussen CG, Soininen P, Kangas AJ, Tynkkynen T, et al. High-throughput quantification of circulating metabolites improves prediction of subclinical atherosclerosis. Eur Heart J. (2012) 33(18):2307–16. doi: 10.1093/eurheartj/ehs020
201. Wittemans LBL, Lotta LA, Oliver-Williams C, Stewart ID, Surendran P, Karthikeyan S, et al. Assessing the causal association of glycine with risk of cardio-metabolic diseases. Nat Commun. (2019) 10(1):1060. doi: 10.1038/s41467-019-08936-1
202. Zaric BL, Radovanovic JN, Gluvic Z, Stewart AJ, Essack M, Motwalli O, et al. Atherosclerosis linked to aberrant amino acid metabolism and immunosuppressive amino acid catabolizing enzymes. Front Immunol. (2020) 11:551758. doi: 10.3389/fimmu.2020.551758
203. Simon J, Nunez-Garcia M, Fernandez-Tussy P, Barbier-Torres L, Fernandez-Ramos D, Gomez-Santos B, et al. Targeting hepatic glutaminase 1 ameliorates non-alcoholic steatohepatitis by restoring very-low-density lipoprotein triglyceride assembly. Cell Metab. (2020) 31(3):605–22.e10. doi: 10.1016/j.cmet.2020.01.013
204. Hasegawa T, Iino C, Endo T, Mikami K, Kimura M, Sawada N, et al. Changed amino acids in NAFLD and liver fibrosis: a large cross-sectional study without influence of insulin resistance. Nutrients. (2020) 12(5):1–11. doi: 10.3390/nu12051450
205. Kawanaka M, Nishino K, Oka T, Urata N, Nakamura J, Suehiro M, et al. Tyrosine levels are associated with insulin resistance in patients with nonalcoholic fatty liver disease. Hepat Med. (2015) 7:29–35. doi: 10.2147/HMER.S79100
206. Shah SH, Bain JR, Muehlbauer MJ, Stevens RD, Crosslin DR, Haynes C, et al. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ Cardiovasc Genet. (2010) 3(2):207–14. doi: 10.1161/CIRCGENETICS.109.852814
207. Yang R, Dong J, Zhao H, Li H, Guo H, Wang S, et al. Association of branched-chain amino acids with carotid intima-media thickness and coronary artery disease risk factors. PLoS One. (2014) 9(6):e99598. doi: 10.1371/journal.pone.0099598
208. Ruiz-Canela M, Toledo E, Clish CB, Hruby A, Liang L, Salas-Salvado J, et al. Plasma branched-chain amino acids and incident cardiovascular disease in the PREDIMED trial. Clin Chem. (2016) 62(4):582–92. doi: 10.1373/clinchem.2015.251710
209. Bhattacharya S, Granger CB, Craig D, Haynes C, Bain J, Stevens RD, et al. Validation of the association between a branched chain amino acid metabolite profile and extremes of coronary artery disease in patients referred for cardiac catheterization. Atherosclerosis. (2014) 232(1):191–6. doi: 10.1016/j.atherosclerosis.2013.10.036
210. Zhang F, Zhao S, Yan W, Xia Y, Chen X, Wang W, et al. Branched chain amino acids cause liver injury in obese/diabetic mice by promoting adipocyte lipolysis and inhibiting hepatic autophagy. EBioMedicine. (2016) 13:157–67. doi: 10.1016/j.ebiom.2016.10.013
211. Honda T, Ishigami M, Luo F, Lingyun M, Ishizu Y, Kuzuya T, et al. Branched-chain amino acids alleviate hepatic steatosis and liver injury in choline-deficient high-fat diet induced NASH mice. Metab Clin Exp. (2017) 69:177–87. doi: 10.1016/j.metabol.2016.12.013
212. Iwao M, Gotoh K, Arakawa M, Endo M, Honda K, Seike M, et al. Supplementation of branched-chain amino acids decreases fat accumulation in the liver through intestinal microbiota-mediated production of acetic acid. Sci Rep. (2020) 10(1):18768. doi: 10.1038/s41598-020-75542-3
213. Yu D, Richardson NE, Green CL, Spicer AB, Murphy ME, Flores V, et al. The adverse metabolic effects of branched-chain amino acids are mediated by isoleucine and valine. Cell Metab. (2021) 33(5):905–22.e6. doi: 10.1016/j.cmet.2021.03.025
214. Rom O, Grajeda-Iglesias C, Najjar M, Abu-Saleh N, Volkova N, Dar DE, et al. Atherogenicity of amino acids in the lipid-laden macrophage model system in vitro and in atherosclerotic mice: a key role for triglyceride metabolism. J Nutr Biochem. (2017) 45:24–38. doi: 10.1016/j.jnutbio.2017.02.023
215. Grajeda-Iglesias C, Rom O, Hamoud S, Volkova N, Hayek T, Abu-Saleh N, et al. Leucine supplementation attenuates macrophage foam-cell formation: studies in humans, mice, and cultured macrophages. Biofactors. (2018) 44(3):245–62. doi: 10.1002/biof.1415
216. Zhao Y, Dai XY, Zhou Z, Zhao GX, Wang X, Xu MJ. Leucine supplementation via drinking water reduces atherosclerotic lesions in apoE null mice. Acta Pharmacol Sin. (2016) 37(2):196–203. doi: 10.1038/aps.2015.88
217. Zhenyukh O, Gonzalez-Amor M, Rodrigues-Diez RR, Esteban V, Ruiz-Ortega M, Salaices M, et al. Branched-chain amino acids promote endothelial dysfunction through increased reactive oxygen species generation and inflammation. J Cell Mol Med. (2018) 22(10):4948–62. doi: 10.1111/jcmm.13759
218. Kalhan SC, Guo L, Edmison J, Dasarathy S, McCullough AJ, Hanson RW, et al. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metab Clin Exp. (2011) 60(3):404–13. doi: 10.1016/j.metabol.2010.03.006
219. de Mello VD, Sehgal R, Mannisto V, Klavus A, Nilsson E, Perfilyev A, et al. Serum aromatic and branched-chain amino acids associated with NASH demonstrate divergent associations with serum lipids. Liver Int. (2021) 41(4):754–63. doi: 10.1111/liv.14743
220. Jauhiainen R, Vangipurapu J, Laakso A, Kuulasmaa T, Kuusisto J, Laakso M. The association of 9 amino acids with cardiovascular events in finnish men in a 12-year follow-up study. J Clin Endocrinol Metab. (2021) 106(12):3448–54. doi: 10.1210/clinem/dgab562
221. Rom O, Villacorta L, Zhang J, Chen YE, Aviram M. Emerging therapeutic potential of glycine in cardiometabolic diseases: dual benefits in lipid and glucose metabolism. Curr Opin Lipidol. (2018) 29(5):428–32. doi: 10.1097/MOL.0000000000000543
222. Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. (2009) 9(4):311–26. doi: 10.1016/j.cmet.2009.02.002
223. Guasch-Ferre M, Hruby A, Toledo E, Clish CB, Martinez-Gonzalez MA, Salas-Salvado J, et al. Metabolomics in prediabetes and diabetes: a systematic review and meta-analysis. Diabetes Care. (2016) 39(5):833–46. doi: 10.2337/dc15-2251
224. Li X, Sun L, Zhang W, Li H, Wang S, Mu H, et al. Association of serum glycine levels with metabolic syndrome in an elderly Chinese population. Nutr Metab (Lond). (2018) 15:89. doi: 10.1186/s12986-018-0325-4
225. Ding Y, Svingen GF, Pedersen ER, Gregory JF, Ueland PM, Tell GS, et al. Plasma glycine and risk of acute myocardial infarction in patients with suspected stable angina pectoris. J Am Heart Assoc. (2015) 5(1):1–9. doi: 10.1161/JAHA.115.002621
226. Wang W, Wu Z, Dai Z, Yang Y, Wang J, Wu G. Glycine metabolism in animals and humans: implications for nutrition and health. Amino Acids. (2013) 45(3):463–77. doi: 10.1007/s00726-013-1493-1
227. Kathirvel E, Morgan K, Nandgiri G, Sandoval BC, Caudill MA, Bottiglieri T, et al. Betaine improves nonalcoholic fatty liver and associated hepatic insulin resistance: a potential mechanism for hepatoprotection by betaine. Am J Physiol Gastrointest Liver Physiol. (2010) 299(5):G1068–77. doi: 10.1152/ajpgi.00249.2010
228. Leonetti S, Herzog RI, Caprio S, Santoro N, Trico D. Glutamate-serine-glycine index: a novel potential biomarker in pediatric non-alcoholic fatty liver disease. Children (Basel). (2020) 7(12):1–7. doi: 10.3390/children7120270
229. Zwighaft Z, Aviram R, Shalev M, Rousso-Noori L, Kraut-Cohen J, Golik M, et al. Circadian clock control by polyamine levels through a mechanism that declines with age. Cell Metab. (2015) 22(5):874–85. doi: 10.1016/j.cmet.2015.09.011
230. Wallace HM, Fraser AV, Hughes A. A perspective of polyamine metabolism. Biochem J. (2003) 376(Pt 1):1–14. doi: 10.1042/BJ20031327
231. Madeo F, Eisenberg T, Pietrocola F, Kroemer G. Spermidine in health and disease. Science. (2018) 359(6374):1–11. doi: 10.1126/science.aan2788
232. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. (2016) 22(12):1428–38. doi: 10.1038/nm.4222
233. Wang D, Yin J, Zhou Z, Tao Y, Jia Y, Jie H, et al. Oral spermidine targets brown fat and skeletal muscle to mitigate diet-induced obesity and metabolic disorders. Mol Nutr Food Res. (2021) 65(19):e2100315. doi: 10.1002/mnfr.202100315
234. Fernandes J, Chandler JD, Liu KH, Uppal K, Go YM, Jones DP. Putrescine as indicator of manganese neurotoxicity: dose-response study in human SH-SY5Y cells. Food Chem Toxicol. (2018) 116(Pt B):272–80. doi: 10.1016/j.fct.2018.04.042
235. Holbert CE, Cullen MT, Casero RA Jr, Stewart TM. Polyamines in cancer: integrating organismal metabolism and antitumour immunity. Nat Rev Cancer. (2022) 22(8):467–80. doi: 10.1038/s41568-022-00473-2
236. Ioannou GN, Nagana Gowda GA, Djukovic D, Raftery D. Distinguishing NASH histological severity using a multiplatform metabolomics approach. Metabolites. (2020) 10(4):1–15. doi: 10.3390/metabo10040168
237. Quinn C, Rico MC, Merali C, Merali S. Dysregulation of S-adenosylmethionine metabolism in nonalcoholic steatohepatitis leads to polyamine flux and oxidative stress. Int J Mol Sci. (2022) 23(4):1–14. doi: 10.3390/ijms23041986
238. Ma L, Ni Y, Hu L, Zhao Y, Zheng L, Yang S, et al. Spermidine ameliorates high-fat diet-induced hepatic steatosis and adipose tissue inflammation in preexisting obese mice. Life Sci. (2021) 265:118739. doi: 10.1016/j.lfs.2020.118739
239. Gao M, Zhao W, Li C, Xie X, Li M, Bi Y, et al. Spermidine ameliorates non-alcoholic fatty liver disease through regulating lipid metabolism via AMPK. Biochem Biophys Res Commun. (2018) 505(1):93–8. doi: 10.1016/j.bbrc.2018.09.078
240. Zhou J, Pang J, Tripathi M, Ho JP, Widjaja AA, Shekeran SG, et al. Spermidine-mediated hypusination of translation factor EIF5A improves mitochondrial fatty acid oxidation and prevents non-alcoholic steatohepatitis progression. Nat Commun. (2022) 13(1):5202. doi: 10.1038/s41467-022-32788-x
241. Yu Z, Jiao Y, Zhang J, Xu Q, Xu J, Li R, et al. Effect of serum spermidine on the prognosis in patients with acute myocardial infarction: a cohort study. Nutrients. (2022) 14(7):1–12. doi: 10.3390/nu14071394
242. Zheng L, Xie Y, Sun Z, Zhang R, Ma Y, Xu J, et al. Serum spermidine in relation to risk of stroke: a multilevel study. Front Nutr. (2022) 9:843616. doi: 10.3389/fnut.2022.843616
243. Gao H, Zhang Q, Xu J, Yuan W, Li R, Guo H, et al. Elevation of serum spermidine in obese patients: results from a cross-sectional and follow-up study. Nutrients. (2022) 14(13):1–12. doi: 10.3390/nu14132613
244. Michiels CF, Kurdi A, Timmermans JP, De Meyer GRY, Martinet W. Spermidine reduces lipid accumulation and necrotic core formation in atherosclerotic plaques via induction of autophagy. Atherosclerosis. (2016) 251:319–27. doi: 10.1016/j.atherosclerosis.2016.07.899
245. Balderas FL, Quezada-Larios M, Garcia Latorre EA, Mendez JD. Increased uptake of oxidized LDL by macrophages from type 2 diabetics is inhibited by polyamines. Biomed Pharmacother. (2016) 77:59–64. doi: 10.1016/j.biopha.2015.11.006
246. Holmes RP, Goodman HO, Assimos DG. Contribution of dietary oxalate to urinary oxalate excretion. Kidney Int. (2001) 59(1):270–6. doi: 10.1046/j.1523-1755.2001.00488.x
247. Brzica H, Breljak D, Burckhardt BC, Burckhardt G, Sabolic I. Oxalate: from the environment to kidney stones. Arh Hig Rada Toksikol. (2013) 64(4):609–30. doi: 10.2478/10004-1254-64-2013-2428
248. Gianmoena K, Gasparoni N, Jashari A, Gabrys P, Grgas K, Ghallab A, et al. Epigenomic and transcriptional profiling identifies impaired glyoxylate detoxification in NAFLD as a risk factor for hyperoxaluria. Cell Rep. (2021) 36(8):109526. doi: 10.1016/j.celrep.2021.109526
249. Elder TD, Wyngaarden JB. The biosynthesis and turnover of oxalate in normal and hyperoxaluric subjects. J Clin Invest. (1960) 39(8):1337–44. doi: 10.1172/JCI104151
250. Ermer T, Nazzal L, Tio MC, Waikar S, Aronson PS, Knauf F. Oxalate homeostasis. Nat Rev Nephrol. (2023) 19(2):123–38. doi: 10.1038/s41581-022-00643-3
251. Crivelli JJ, Mitchell T, Knight J, Wood KD, Assimos DG, Holmes RP, et al. Contribution of dietary oxalate and oxalate precursors to urinary oxalate excretion. Nutrients. (2020) 13(1):1–13. doi: 10.3390/nu13010062
252. Salido E, Pey AL, Rodriguez R, Lorenzo V. Primary hyperoxalurias: disorders of glyoxylate detoxification. Biochim Biophys Acta. (2012) 1822(9):1453–64. doi: 10.1016/j.bbadis.2012.03.004
253. Pfau A, Ermer T, Coca SG, Tio MC, Genser B, Reichel M, et al. High oxalate concentrations correlate with increased risk for sudden cardiac death in dialysis patients. J Am Soc Nephrol. (2021) 32(9):2375–85. doi: 10.1681/ASN.2020121793
254. Stepanova N, Driianska V, Korol L, Snisar L, Lebed L. Plasma oxalic acid and cardiovascular risk in end-stage renal disease patients: a prospective, observational cohort pilot study. Korean J Intern Med. (2022) 37(1):167–78. doi: 10.3904/kjim.2020.561
255. Stepanova M, Hossain N, Afendy A, Perry K, Goodman ZD, Baranova A, et al. Hepatic gene expression of caucasian and African-American patients with obesity-related non-alcoholic fatty liver disease. Obes Surg. (2010) 20(5):640–50. doi: 10.1007/s11695-010-0078-2
256. Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol. (2016) 65(3):579–88. doi: 10.1016/j.jhep.2016.05.005
257. Gulhan B, Turkmen K, Aydin M, Gunay M, Cikman A, Kara M. The relationship between serum oxalic acid, central hemodynamic parameters and colonization by Oxalobacter formigenes in hemodialysis patients. Cardiorenal Med. (2015) 5(3):164–74. doi: 10.1159/000381219
258. Schunk SJ, Triem S, Schmit D, Zewinger S, Sarakpi T, Becker E, et al. Interleukin-1alpha is a central regulator of leukocyte-endothelial adhesion in myocardial infarction and in chronic kidney disease. Circulation. (2021) 144(11):893–908. doi: 10.1161/CIRCULATIONAHA.121.053547
259. Patel M, Yarlagadda V, Adedoyin O, Saini V, Assimos DG, Holmes RP, et al. Oxalate induces mitochondrial dysfunction and disrupts redox homeostasis in a human monocyte derived cell line. Redox Biol. (2018) 15:207–15. doi: 10.1016/j.redox.2017.12.003
260. Sun K, Tang X, Song S, Gao Y, Yu H, Sun N, et al. Hyperoxalemia leads to oxidative stress in endothelial cells and mice with chronic kidney disease. Kidney Blood Press Res. (2021) 46(3):377–86. doi: 10.1159/000516013
261. Jensen-Cody SO, Potthoff MJ. Hepatokines and metabolism: deciphering communication from the liver. Mol Metab. (2021) 44:101138. doi: 10.1016/j.molmet.2020.101138
262. Meex RCR, Watt MJ. Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat Rev Endocrinol. (2017) 13(9):509–20. doi: 10.1038/nrendo.2017.56
263. Conklin D, Gilbertson D, Taft DW, Maurer MF, Whitmore TE, Smith DL, et al. Identification of a mammalian angiopoietin-related protein expressed specifically in liver. Genomics. (1999) 62(3):477–82. doi: 10.1006/geno.1999.6041
264. Koishi R, Ando Y, Ono M, Shimamura M, Yasumo H, Fujiwara T, et al. Angptl3 regulates lipid metabolism in mice. Nat Genet. (2002) 30(2):151–7. doi: 10.1038/ng814
265. Shimizugawa T, Ono M, Shimamura M, Yoshida K, Ando Y, Koishi R, et al. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J Biol Chem. (2002) 277(37):33742–8. doi: 10.1074/jbc.M203215200
266. Romeo S, Yin W, Kozlitina J, Pennacchio LA, Boerwinkle E, Hobbs HH, et al. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. (2009) 119(1):70–9. doi: 10.1172/JCI37118
267. Yilmaz Y, Ulukaya E, Atug O, Dolar E. Serum concentrations of human angiopoietin-like protein 3 in patients with nonalcoholic fatty liver disease: association with insulin resistance. Eur J Gastroenterol Hepatol. (2009) 21(11):1247–51. doi: 10.1097/MEG.0b013e32832b77ae
268. Barchetta I, Cimini FA, Chiappetta C, Bertoccini L, Ceccarelli V, Capoccia D, et al. Relationship between hepatic and systemic angiopoietin-like 3, hepatic vitamin D receptor expression and NAFLD in obesity. Liver Int. (2020) 40(9):2139–47. doi: 10.1111/liv.14554
269. Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, et al. ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol. (2017) 69(16):2054–63. doi: 10.1016/j.jacc.2017.02.030
270. Sun T, Zhan W, Wei L, Xu Z, Fan L, Zhuo Y, et al. Circulating ANGPTL3 and ANGPTL4 levels predict coronary artery atherosclerosis severity. Lipids Health Dis. (2021) 20(1):154. doi: 10.1186/s12944-021-01580-z
271. Dewey FE, Gusarova V, Dunbar RL, O'Dushlaine C, Schurmann C, Gottesman O, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med. (2017) 377(3):211–21. doi: 10.1056/NEJMoa1612790
272. Graham MJ, Lee RG, Brandt TA, Tai LJ, Fu W, Peralta R, et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med. (2017) 377(3):222–32. doi: 10.1056/NEJMoa1701329
273. Bergmark BA, Marston NA, Bramson CR, Curto M, Ramos V, Jevne A, et al. Effect of vupanorsen on non-high-density lipoprotein cholesterol levels in statin-treated patients with elevated cholesterol: TRANSLATE-TIMI 70. Circulation. (2022) 145(18):1377–86. doi: 10.1161/CIRCULATIONAHA.122.059266
274. Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta. (2000) 1492(1):203–6. doi: 10.1016/s0167-4781(00)00067-1
275. Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. (2005) 115(6):1627–35. doi: 10.1172/JCI23606
276. Li H, Fang Q, Gao F, Fan J, Zhou J, Wang X, et al. Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J Hepatol. (2010) 53(5):934–40. doi: 10.1016/j.jhep.2010.05.018
277. Wu G, Li H, Fang Q, Zhang J, Zhang M, Zhang L, et al. Complementary role of fibroblast growth factor 21 and cytokeratin 18 in monitoring the different stages of nonalcoholic fatty liver disease. Sci Rep. (2017) 7(1):5095. doi: 10.1038/s41598-017-05257-5
278. Barb D, Bril F, Kalavalapalli S, Cusi K. Plasma fibroblast growth factor 21 is associated with severity of nonalcoholic steatohepatitis in patients with obesity and type 2 diabetes. J Clin Endocrinol Metab. (2019) 104(8):3327–36. doi: 10.1210/jc.2018-02414
279. Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM, et al. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology. (2010) 139(2):456–63. doi: 10.1053/j.gastro.2010.04.054
280. Yilmaz Y, Eren F, Yonal O, Kurt R, Aktas B, Celikel CA, et al. Increased serum FGF21 levels in patients with nonalcoholic fatty liver disease. Eur J Clin Invest. (2010) 40(10):887–92. doi: 10.1111/j.1365-2362.2010.02338.x
281. Shen Y, Ma X, Zhou J, Pan X, Hao Y, Zhou M, et al. Additive relationship between serum fibroblast growth factor 21 level and coronary artery disease. Cardiovasc Diabetol. (2013) 12:124. doi: 10.1186/1475-2840-12-124
282. Chow WS, Xu A, Woo YC, Tso AW, Cheung SC, Fong CH, et al. Serum fibroblast growth factor-21 levels are associated with carotid atherosclerosis independent of established cardiovascular risk factors. Arterioscler Thromb Vasc Biol. (2013) 33(10):2454–9. doi: 10.1161/ATVBAHA.113.301599
283. Wu L, Qian L, Zhang L, Zhang J, Zhou J, Li Y, et al. Fibroblast growth factor 21 is related to atherosclerosis independent of nonalcoholic fatty liver disease and predicts atherosclerotic cardiovascular events. J Am Heart Assoc. (2020) 9(11):e015226. doi: 10.1161/JAHA.119.015226
284. Fisher FM, Chui PC, Nasser IA, Popov Y, Cunniff JC, Lundasen T, et al. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline-deficient diets. Gastroenterology. (2014) 147(5):1073–83.e6. doi: 10.1053/j.gastro.2014.07.044
285. Lee JH, Kang YE, Chang JY, Park KC, Kim HW, Kim JT, et al. An engineered FGF21 variant, LY2405319, can prevent non-alcoholic steatohepatitis by enhancing hepatic mitochondrial function. Am J Transl Res. (2016) 8(11):4750–63.27904677
286. Lin Z, Pan X, Wu F, Ye D, Zhang Y, Wang Y, et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation. (2015) 131(21):1861–71. doi: 10.1161/CIRCULATIONAHA.115.015308
287. Liu C, Schonke M, Zhou E, Li Z, Kooijman S, Boon MR, et al. Pharmacological treatment with FGF21 strongly improves plasma cholesterol metabolism to reduce atherosclerosis. Cardiovasc Res. (2022) 118(2):489–502. doi: 10.1093/cvr/cvab076
288. Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. (2019) 392(10165):2705–17. doi: 10.1016/S0140-6736(18)31785-9
289. Harrison SA, Ruane PJ, Freilich BL, Neff G, Patil R, Behling CA, et al. Efruxifermin in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a trial. Nat Med. (2021) 27(7):1262–71. doi: 10.1038/s41591-021-01425-3
290. Dogru T, Kirik A, Gurel H, Rizvi AA, Rizzo M, Sonmez A. The evolving role of fetuin-A in nonalcoholic fatty liver disease: an overview from liver to the heart. Int J Mol Sci. (2021) 22(12):1–12. doi: 10.3390/ijms22126627
291. Mathews ST, Chellam N, Srinivas PR, Cintron VJ, Leon MA, Goustin AS, et al. Alpha2-HSG, a specific inhibitor of insulin receptor autophosphorylation, interacts with the insulin receptor. Mol Cell Endocrinol. (2000) 164(1–2):87–98. doi: 10.1016/s0303-7207(00)00237-9
292. Denecke B, Graber S, Schafer C, Heiss A, Woltje M, Jahnen-Dechent W. Tissue distribution and activity testing suggest a similar but not identical function of fetuin-B and fetuin-A. Biochem J. (2003) 376(Pt 1):135–45. doi: 10.1042/BJ20030676
293. Stefan N, Hennige AM, Staiger H, Machann J, Schick F, Krober SM, et al. Alpha2-heremans-schmid glycoprotein/fetuin-A is associated with insulin resistance and fat accumulation in the liver in humans. Diabetes Care. (2006) 29(4):853–7. doi: 10.2337/diacare.29.04.06.dc05-1938
294. Reinehr T, Roth CL. Fetuin-A and its relation to metabolic syndrome and fatty liver disease in obese children before and after weight loss. J Clin Endocrinol Metab. (2008) 93(11):4479–85. doi: 10.1210/jc.2008-1505
295. Yilmaz Y, Yonal O, Kurt R, Ari F, Oral AY, Celikel CA, et al. Serum fetuin A/alpha2HS-glycoprotein levels in patients with non-alcoholic fatty liver disease: relation with liver fibrosis. Ann Clin Biochem. (2010) 47(Pt 6):549–53. doi: 10.1258/acb.2010.010169
296. Weikert C, Stefan N, Schulze MB, Pischon T, Berger K, Joost HG, et al. Plasma fetuin-a levels and the risk of myocardial infarction and ischemic stroke. Circulation. (2008) 118(24):2555–62. doi: 10.1161/CIRCULATIONAHA.108.814418
297. Chen X, Zhang Y, Chen Q, Li Q, Li Y, Ling W. Lower plasma fetuin-A levels are associated with a higher mortality risk in patients with coronary artery disease. Arterioscler Thromb Vasc Biol. (2017) 37(11):2213–9. doi: 10.1161/ATVBAHA.117.309700
298. Vuppalanchi R, Noureddin M, Alkhouri N, Sanyal AJ. Therapeutic pipeline in nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. (2021) 18(6):373–92. doi: 10.1038/s41575-020-00408-y
299. Xu X, Poulsen KL, Wu L, Liu S, Miyata T, Song Q, et al. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal Transduct Target Ther. (2022) 7(1):287. doi: 10.1038/s41392-022-01119-3
300. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis—from experimental insights to the clinic. Nat Rev Drug Discov. (2021) 20(8):589–610. doi: 10.1038/s41573-021-00198-1
301. Bril F, Kalavalapalli S, Clark VC, Lomonaco R, Soldevila-Pico C, Liu IC, et al. Response to pioglitazone in patients with nonalcoholic steatohepatitis with vs without type 2 diabetes. Clin Gastroenterol Hepatol. (2018) 16(4):558–66.e2. doi: 10.1016/j.cgh.2017.12.001
302. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med. (2016) 165(5):305–15. doi: 10.7326/M15-1774
303. Erdmann E, Charbonnel B, Wilcox RG, Skene AM, Massi-Benedetti M, Yates J, et al. Pioglitazone use and heart failure in patients with type 2 diabetes and preexisting cardiovascular disease: data from the PROactive study (PROactive 08). Diabetes Care. (2007) 30(11):2773–8. doi: 10.2337/dc07-0717
304. Liao HW, Saver JL, Wu YL, Chen TH, Lee M, Ovbiagele B. Pioglitazone and cardiovascular outcomes in patients with insulin resistance, pre-diabetes and type 2 diabetes: a systematic review and meta-analysis. BMJ Open. (2017) 7(1):e013927. doi: 10.1136/bmjopen-2016-013927
305. Nesti L, Trico D, Mengozzi A, Natali A. Rethinking pioglitazone as a cardioprotective agent: a new perspective on an overlooked drug. Cardiovasc Diabetol. (2021) 20(1):109. doi: 10.1186/s12933-021-01294-7
306. Koshiyama H, Shimono D, Kuwamura N, Minamikawa J, Nakamura Y. Rapid communication: inhibitory effect of pioglitazone on carotid arterial wall thickness in type 2 diabetes. J Clin Endocrinol Metab. (2001) 86(7):3452–6. doi: 10.1210/jcem.86.7.7810
307. Mazzone T, Meyer PM, Feinstein SB, Davidson MH, Kondos GT, D'Agostino RB Sr, et al. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: a randomized trial. JAMA. (2006) 296(21):2572–81. doi: 10.1001/jama.296.21.joc60158
308. Mizoguchi M, Tahara N, Tahara A, Nitta Y, Kodama N, Oba T, et al. Pioglitazone attenuates atherosclerotic plaque inflammation in patients with impaired glucose tolerance or diabetes a prospective, randomized, comparator-controlled study using serial FDG PET/CT imaging study of carotid artery and ascending aorta. JACC Cardiovasc Imaging. (2011) 4(10):1110–8. doi: 10.1016/j.jcmg.2011.08.007
309. Davidson M, Meyer PM, Haffner S, Feinstein S, D'Agostino R Sr, Kondos GT, et al. Increased high-density lipoprotein cholesterol predicts the pioglitazone-mediated reduction of carotid intima-media thickness progression in patients with type 2 diabetes mellitus. Circulation. (2008) 117(16):2123–30. doi: 10.1161/CIRCULATIONAHA.107.746610
310. Nakano K, Hasegawa G, Fukui M, Yamasaki M, Ishihara K, Takashima T, et al. Effect of pioglitazone on various parameters of insulin resistance including lipoprotein subclass according to particle size by a gel-permeation high-performance liquid chromatography in newly diagnosed patients with type 2 diabetes. Endocr J. (2010) 57(5):423–30. doi: 10.1507/endocrj.k10e-006
311. Derosa G, Cicero AF, D'Angelo A, Gaddi A, Ciccarelli L, Piccinni MN, et al. Effects of 1 year of treatment with pioglitazone or rosiglitazone added to glimepiride on lipoprotein (a) and homocysteine concentrations in patients with type 2 diabetes mellitus and metabolic syndrome: a multicenter, randomized, double-blind, controlled clinical trial. Clin Ther. (2006) 28(5):679–88. doi: 10.1016/j.clinthera.2006.05.012
312. Goldberg RB, Kendall DM, Deeg MA, Buse JB, Zagar AJ, Pinaire JA, et al. A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care. (2005) 28(7):1547–54. doi: 10.2337/diacare.28.7.1547
313. Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci U S A. (1987) 84(10):3434–8. doi: 10.1073/pnas.84.10.3434
314. Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest. (1987) 79(2):616–9. doi: 10.1172/JCI112855
315. Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet. (1987) 2(8571):1300–4. doi: 10.1016/s0140-6736(87)91194-9
316. Turton MD, O'Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. (1996) 379(6560):69–72. doi: 10.1038/379069a0
317. Astrup A, Rossner S, Van Gaal L, Rissanen A, Niskanen L, Al Hakim M, et al. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet. (2009) 374(9701):1606–16. doi: 10.1016/S0140-6736(09)61375-1
318. Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. (2016) 387(10019):679–90. doi: 10.1016/S0140-6736(15)00803-X
319. Armstrong MJ, Hull D, Guo K, Barton D, Hazlehurst JM, Gathercole LL, et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J Hepatol. (2016) 64(2):399–408. doi: 10.1016/j.jhep.2015.08.038
320. O'Neil PM, Birkenfeld AL, McGowan B, Mosenzon O, Pedersen SD, Wharton S, et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet. (2018) 392(10148):637–49. doi: 10.1016/S0140-6736(18)31773-2
321. Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. (2021) 384(12):1113–24. doi: 10.1056/NEJMoa2028395
322. Rizzo M, Chandalia M, Patti AM, Di Bartolo V, Rizvi AA, Montalto G, et al. Liraglutide decreases carotid intima-media thickness in patients with type 2 diabetes: 8-month prospective pilot study. Cardiovasc Diabetol. (2014) 13:49. doi: 10.1186/1475-2840-13-49
323. Rizzo M, Rizvi AA, Patti AM, Nikolic D, Giglio RV, Castellino G, et al. Liraglutide improves metabolic parameters and carotid intima-media thickness in diabetic patients with the metabolic syndrome: an 18-month prospective study. Cardiovasc Diabetol. (2016) 15(1):162. doi: 10.1186/s12933-016-0480-8
324. Nikolic D, Giglio RV, Rizvi AA, Patti AM, Montalto G, Maranta F, et al. Liraglutide reduces carotid intima-media thickness by reducing small dense low-density lipoproteins in a real-world setting of patients with type 2 diabetes: a novel anti-atherogenic effect. Diabetes Ther. (2021) 12(1):261–74. doi: 10.1007/s13300-020-00962-3
325. Panjwani N, Mulvihill EE, Longuet C, Yusta B, Campbell JE, Brown TJ, et al. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE(-/-) mice. Endocrinology. (2013) 154(1):127–39. doi: 10.1210/en.2012-1937
326. Pyke C, Heller RS, Kirk RK, Orskov C, Reedtz-Runge S, Kaastrup P, et al. GLP-1 receptor localization in monkey and human tissue: novel distribution revealed with extensively validated monoclonal antibody. Endocrinology. (2014) 155(4):1280–90. doi: 10.1210/en.2013-1934
327. Rakipovski G, Rolin B, Nohr J, Klewe I, Frederiksen KS, Augustin R, et al. The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE(-/-) and LDLr(-/-) mice by a mechanism that includes inflammatory pathways. JACC Basic Transl Sci. (2018) 3(6):844–57. doi: 10.1016/j.jacbts.2018.09.004
328. Wright EM, Turk E. The sodium/glucose cotransport family SLC5. Pflugers Arch. (2004) 447(5):510–8. doi: 10.1007/s00424-003-1063-6
329. Roden M, Weng J, Eilbracht J, Delafont B, Kim G, Woerle HJ, et al. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. (2013) 1(3):208–19. doi: 10.1016/S2213-8587(13)70084-6
330. Kuchay MS, Krishan S, Mishra SK, Farooqui KJ, Singh MK, Wasir JS, et al. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial (E-LIFT trial). Diabetes Care. (2018) 41(8):1801–8. doi: 10.2337/dc18-0165
331. Lai LL, Vethakkan SR, Nik Mustapha NR, Mahadeva S, Chan WK. Empagliflozin for the treatment of nonalcoholic steatohepatitis in patients with type 2 diabetes mellitus. Dig Dis Sci. (2020) 65(2):623–31. doi: 10.1007/s10620-019-5477-1
332. Eriksson JW, Lundkvist P, Jansson PA, Johansson L, Kvarnstrom M, Moris L, et al. Effects of dapagliflozin and n-3 carboxylic acidtys on non-alcoholic fatty liver disease in people with type 2 diabetes: a double-blind randomised placebo-controlled study. Diabetologia. (2018) 61(9):1923–34. doi: 10.1007/s00125-018-4675-2
333. Marjot T, Green CJ, Charlton CA, Cornfield T, Hazlehurst J, Moolla A, et al. Sodium-glucose cotransporter 2 inhibition does not reduce hepatic steatosis in overweight, insulin-resistant patients without type 2 diabetes. JGH Open. (2020) 4(3):433–40. doi: 10.1002/jgh3.12274
334. Shimizu M, Suzuki K, Kato K, Jojima T, Iijima T, Murohisa T, et al. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes Metab. (2019) 21(2):285–92. doi: 10.1111/dom.13520
335. Harrison SA, Manghi FP, Smith WB, Alpenidze D, Aizenberg D, Klarenbeek N, et al. Licogliflozin for nonalcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a study. Nat Med. (2022) 28(7):1432–8. doi: 10.1038/s41591-022-01861-9
336. Sarafidis PA, Tsapas A. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2016) 374(11):1092. doi: 10.1056/NEJMc1600827
337. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2019) 380(4):347–57. doi: 10.1056/NEJMoa1812389
338. Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose co-transporter 2 (SGLT2) inhibitors: a state-of-the-art review. JACC Basic Transl Sci. (2020) 5(6):632–44. doi: 10.1016/j.jacbts.2020.02.004
339. Liu Z, Ma X, Ilyas I, Zheng X, Luo S, Little PJ, et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: from pharmacology to pre-clinical and clinical therapeutics. Theranostics. (2021) 11(9):4502–15. doi: 10.7150/thno.54498
340. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. (2005) 115(5):1343–51. doi: 10.1172/JCI23621
341. Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. (2010) 52(2):774–88. doi: 10.1002/hep.23719
342. Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. (2012) 142(4):711–25.e6. doi: 10.1053/j.gastro.2012.02.003
343. Bianchi A, Evans JL, Iverson AJ, Nordlund AC, Watts TD, Witters LA. Identification of an isozymic form of acetyl-CoA carboxylase. J Biol Chem. (1990) 265(3):1502–9. doi: 10.1016/S0021-9258(19)40045-8
344. Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, et al. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology. (2018) 155(5):1463–73.e6. doi: 10.1053/j.gastro.2018.07.027
345. Lawitz EJ, Coste A, Poordad F, Alkhouri N, Loo N, McColgan BJ, et al. Acetyl-CoA carboxylase inhibitor GS-0976 for 12 weeks reduces hepatic de novo lipogenesis and steatosis in patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. (2018) 16(12):1983–91.e3. doi: 10.1016/j.cgh.2018.04.042
346. Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. (2017) 26(2):394–406.e6. doi: 10.1016/j.cmet.2017.07.009
347. Calle RA, Amin NB, Carvajal-Gonzalez S, Ross TT, Bergman A, Aggarwal S, et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat Med. (2021) 27(10):1836–48. doi: 10.1038/s41591-021-01489-1
348. Goedeke L, Bates J, Vatner DF, Perry RJ, Wang T, Ramirez R, et al. Acetyl-CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology. (2018) 68(6):2197–211. doi: 10.1002/hep.30097
349. Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. (2008) 118(3):829–38. doi: 10.1172/JCI34275
350. Loomba R, Mohseni R, Lucas KJ, Gutierrez JA, Perry RG, Trotter JF, et al. TVB-2640 (FASN inhibitor) for the treatment of nonalcoholic steatohepatitis: FASCINATE-1, a randomized, placebo-controlled phase 2a trial. Gastroenterology. (2021) 161(5):1475–86. doi: 10.1053/j.gastro.2021.07.025
351. Ntambi JM, Miyazaki M. Regulation of stearoyl-CoA desaturases and role in metabolism. Prog Lipid Res. (2004) 43(2):91–104. doi: 10.1016/s0163-7827(03)00039-0
352. Ratziu V, de Guevara L, Safadi R, Poordad F, Fuster F, Flores-Figueroa J, et al. Aramchol in patients with nonalcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase 2b trial. Nat Med. (2021) 27(10):1825–35. doi: 10.1038/s41591-021-01495-3
353. Safadi R, Konikoff FM, Mahamid M, Zelber-Sagi S, Halpern M, Gilat T, et al. The fatty acid-bile acid conjugate aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2014) 12(12):2085–91.e1. doi: 10.1016/j.cgh.2014.04.038
354. MacDonald ML, van Eck M, Hildebrand RB, Wong BW, Bissada N, Ruddle P, et al. Despite antiatherogenic metabolic characteristics, SCD1-deficient mice have increased inflammation and atherosclerosis. Arterioscler Thromb Vasc Biol. (2009) 29(3):341–7. doi: 10.1161/ATVBAHA.108.181099
355. Brown JM, Chung S, Sawyer JK, Degirolamo C, Alger HM, Nguyen T, et al. Inhibition of stearoyl-coenzyme A desaturase 1 dissociates insulin resistance and obesity from atherosclerosis. Circulation. (2008) 118(14):1467–75. doi: 10.1161/CIRCULATIONAHA.108.793182
356. Kersten S. Integrated physiology and systems biology of PPARalpha. Mol Metab. (2014) 3(4):354–71. doi: 10.1016/j.molmet.2014.02.002
357. Montagner A, Polizzi A, Fouche E, Ducheix S, Lippi Y, Lasserre F, et al. Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut. (2016) 65(7):1202–14. doi: 10.1136/gutjnl-2015-310798
358. Brocker CN, Patel DP, Velenosi TJ, Kim D, Yan T, Yue J, et al. Extrahepatic PPARalpha modulates fatty acid oxidation and attenuates fasting-induced hepatosteatosis in mice. J Lipid Res. (2018) 59(11):2140–52. doi: 10.1194/jlr.M088419
359. Regnier M, Polizzi A, Smati S, Lukowicz C, Fougerat A, Lippi Y, et al. Hepatocyte-specific deletion of Pparalpha promotes NAFLD in the context of obesity. Sci Rep. (2020) 10(1):6489. doi: 10.1038/s41598-020-63579-3
360. Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. (2004) 39(5):1286–96. doi: 10.1002/hep.20170
361. Fernandez-Miranda C, Perez-Carreras M, Colina F, Lopez-Alonso G, Vargas C, Solis-Herruzo JA. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis. (2008) 40(3):200–5. doi: 10.1016/j.dld.2007.10.002
362. Lawitz EJ, Bhandari BR, Ruane PJ, Kohli A, Harting E, Ding D, et al. Fenofibrate mitigates hypertriglyceridemia in nonalcoholic steatohepatitis patients treated with cilofexor/firsocostat. Clin Gastroenterol Hepatol. (2023) 21(1):143–52.e3. doi: 10.1016/j.cgh.2021.12.044
363. Honda Y, Kessoku T, Ogawa Y, Tomeno W, Imajo K, Fujita K, et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci Rep. (2017) 7:42477. doi: 10.1038/srep42477
364. Nakajima A, Eguchi Y, Yoneda M, Imajo K, Tamaki N, Suganami H, et al. Randomised clinical trial: pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha), versus placebo in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. (2021) 54(10):1263–77. doi: 10.1111/apt.16596
365. Duez H, Chao YS, Hernandez M, Torpier G, Poulain P, Mundt S, et al. Reduction of atherosclerosis by the peroxisome proliferator-activated receptor alpha agonist fenofibrate in mice. J Biol Chem. (2002) 277(50):48051–7. doi: 10.1074/jbc.M206966200
366. Kooistra T, Verschuren L, de Vries-van der Weij J, Koenig W, Toet K, Princen HM, et al. Fenofibrate reduces atherogenesis in ApoE*3Leiden mice: evidence for multiple antiatherogenic effects besides lowering plasma cholesterol. Arterioscler Thromb Vasc Biol. (2006) 26(10):2322–30. doi: 10.1161/01.ATV.0000238348.05028.14
367. Chen J, Montagner A, Tan NS, Wahli W. Insights into the role of PPARbeta/delta in NAFLD. Int J Mol Sci. (2018) 19(7):1–23. doi: 10.3390/ijms19071893
368. Hoekstra M, Kruijt JK, Van Eck M, Van Berkel TJ. Specific gene expression of ATP-binding cassette transporters and nuclear hormone receptors in rat liver parenchymal, endothelial, and kupffer cells. J Biol Chem. (2003) 278(28):25448–53. doi: 10.1074/jbc.M301189200
369. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. (2008) 7(6):496–507. doi: 10.1016/j.cmet.2008.04.003
370. Sanderson LM, Boekschoten MV, Desvergne B, Muller M, Kersten S. Transcriptional profiling reveals divergent roles of PPARalpha and PPARbeta/delta in regulation of gene expression in mouse liver. Physiol Genomics. (2010) 41(1):42–52. doi: 10.1152/physiolgenomics.00127.2009
371. Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. (2013) 58(6):1941–52. doi: 10.1002/hep.26461
372. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. (2016) 150(5):1147–59.e5. doi: 10.1053/j.gastro.2016.01.038
373. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages(⋆). J Hepatol. (2020) 73(4):757–70. doi: 10.1016/j.jhep.2020.04.025
374. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. N Engl J Med. (2021) 385(17):1547–58. doi: 10.1056/NEJMoa2036205
375. Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Circulation. (2019) 140(11):e596–646. doi: 10.1161/CIR.0000000000000678
376. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American association for the study of liver diseases. Hepatology. (2018) 67(1):328–57. doi: 10.1002/hep.29367
377. Park HS, Jang JE, Ko MS, Woo SH, Kim BJ, Kim HS, et al. Statins increase mitochondrial and peroxisomal fatty acid oxidation in the liver and prevent non-alcoholic steatohepatitis in mice. Diabetes Metab J. (2016) 40(5):376–85. doi: 10.4093/dmj.2016.40.5.376
378. Yokohama K, Fukunishi S, Ii M, Nakamura K, Ohama H, Tsuchimoto Y, et al. Rosuvastatin as a potential preventive drug for the development of hepatocellular carcinoma associated with non-alcoholic fatty liver disease in mice. Int J Mol Med. (2016) 38(5):1499–506. doi: 10.3892/ijmm.2016.2766
379. Schierwagen R, Maybuchen L, Hittatiya K, Klein S, Uschner FE, Braga TT, et al. Statins improve NASH via inhibition of RhoA and Ras. Am J Physiol Gastrointest Liver Physiol. (2016) 311(4):G724–33. doi: 10.1152/ajpgi.00063.2016
380. Abbasi F, Lamendola C, Harris CS, Harris V, Tsai MS, Tripathi P, et al. Statins are associated with increased insulin resistance and secretion. Arterioscler Thromb Vasc Biol. (2021) 41(11):2786–97. doi: 10.1161/ATVBAHA.121.316159
381. Thomas DD, Corkey BE, Istfan NW, Apovian CM. Hyperinsulinemia: an early indicator of metabolic dysfunction. J Endocr Soc. (2019) 3(9):1727–47. doi: 10.1210/js.2019-00065
382. Sugiyama T, Tsugawa Y, Tseng CH, Kobayashi Y, Shapiro MF. Different time trends of caloric and fat intake between statin users and nonusers among US adults: gluttony in the time of statins? JAMA Intern Med. (2014) 174(7):1038–45. doi: 10.1001/jamainternmed.2014.1927
383. Romeo S, Sanyal A, Valenti L. Leveraging human genetics to identify potential new treatments for fatty liver disease. Cell Metab. (2020) 31(1):35–45. doi: 10.1016/j.cmet.2019.12.002
384. Wu JT, Liu SS, Xie XJ, Liu Q, Xin YN, Xuan SY. Independent and joint correlation of PNPLA3 I148M and TM6SF2 E167K variants with the risk of coronary heart disease in patients with non-alcoholic fatty liver disease. Lipids Health Dis. (2020) 19(1):29. doi: 10.1186/s12944-020-01207-9
385. Minicocci I, Montali A, Robciuc MR, Quagliarini F, Censi V, Labbadia G, et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J Clin Endocrinol Metab. (2012) 97(7):E1266–75. doi: 10.1210/jc.2012-1298
386. Morinaga J, Zhao J, Endo M, Kadomatsu T, Miyata K, Sugizaki T, et al. Association of circulating ANGPTL 3, 4, and 8 levels with medical status in a population undergoing routine medical checkups: a cross-sectional study. PLoS One. (2018) 13(3):e0193731. doi: 10.1371/journal.pone.0193731
387. Sutanto A, Wungu CDK, Susilo H, Sutanto H. Reduction of major adverse cardiovascular events (MACE) after bariatric surgery in patients with obesity and cardiovascular diseases: a systematic review and meta-analysis. Nutrients. (2021) 13(10):1–19. doi: 10.3390/nu13103568
388. Lassailly G, Caiazzo R, Buob D, Pigeyre M, Verkindt H, Labreuche J, et al. Bariatric surgery reduces features of nonalcoholic steatohepatitis in morbidly obese patients. Gastroenterology. (2015) 149(2):379–88; quiz e15–6. doi: 10.1053/j.gastro.2015.04.014
389. Aminian A, Al-Kurd A, Wilson R, Bena J, Fayazzadeh H, Singh T, et al. Association of bariatric surgery with major adverse liver and cardiovascular outcomes in patients with biopsy-proven nonalcoholic steatohepatitis. JAMA. (2021) 326(20):2031–42. doi: 10.1001/jama.2021.19569
390. Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA. (2004) 292(14):1724–37. doi: 10.1001/jama.292.14.1724
391. Held C, Hadziosmanovic N, Aylward PE, Hagstrom E, Hochman JS, Stewart RAH, et al. Body mass index and association with cardiovascular outcomes in patients with stable coronary heart disease—a STABILITY substudy. J Am Heart Assoc. (2022) 11(3):e023667. doi: 10.1161/JAHA.121.023667
392. Snel M, Jonker JT, Schoones J, Lamb H, de Roos A, Pijl H, et al. Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. Int J Endocrinol. (2012) 2012:983814. doi: 10.1155/2012/983814
393. Nauli AM, Matin S. Why do men accumulate abdominal visceral fat? Front Physiol. (2019) 10:1486. doi: 10.3389/fphys.2019.01486
394. Neeland IJ, Ross R, Despres JP, Matsuzawa Y, Yamashita S, Shai I, et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: a position statement. Lancet Diabetes Endocrinol. (2019) 7(9):715–25. doi: 10.1016/S2213-8587(19)30084-1
395. Nakamura T, Tokunaga K, Shimomura I, Nishida M, Yoshida S, Kotani K, et al. Contribution of visceral fat accumulation to the development of coronary artery disease in non-obese men. Atherosclerosis. (1994) 107(2):239–46. doi: 10.1016/0021-9150(94)90025-6
396. Saponaro C, Sabatini S, Gaggini M, Carli F, Rosso C, Positano V, et al. Adipose tissue dysfunction and visceral fat are associated with hepatic insulin resistance and severity of NASH even in lean individuals. Liver Int. (2022) 42(11):2418–27. doi: 10.1111/liv.15377
397. Sewter CP, Digby JE, Blows F, Prins J, O'Rahilly S. Regulation of tumour necrosis factor-alpha release from human adipose tissue in vitro. J Endocrinol. (1999) 163(1):33–8. doi: 10.1677/joe.0.1630033
398. Guo X, Xu Y, He H, Cai H, Zhang J, Li Y, et al. Visceral fat reduction is positively associated with blood pressure reduction in overweight or obese males but not females: an observational study. Nutr Metab (Lond). (2019) 16:44. doi: 10.1186/s12986-019-0369-0
399. Johari MI, Yusoff K, Haron J, Nadarajan C, Ibrahim KN, Wong MS, et al. A randomised controlled trial on the effectiveness and adherence of modified alternate-day calorie restriction in improving activity of non-alcoholic fatty liver disease. Sci Rep. (2019) 9(1):11232. doi: 10.1038/s41598-019-47763-8
400. Fontana L, Meyer TE, Klein S, Holloszy JO. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc Natl Acad Sci U S A. (2004) 101(17):6659–63. doi: 10.1073/pnas.0308291101
401. Mitchell SE, Delville C, Konstantopedos P, Hurst J, Derous D, Green C, et al. The effects of graded levels of calorie restriction: II. Impact of short term calorie and protein restriction on circulating hormone levels, glucose homeostasis and oxidative stress in male C57BL/6 mice. Oncotarget. (2015) 6(27):23213–37. doi: 10.18632/oncotarget.4003
402. Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. (2014) 5:e1179. doi: 10.1038/cddis.2014.162
403. Gluais-Dagorn P, Foretz M, Steinberg GR, Batchuluun B, Zawistowska-Deniziak A, Lambooij JM, et al. Direct AMPK activation corrects NASH in rodents through metabolic effects and direct action on inflammation and fibrogenesis. Hepatol Commun. (2022) 6(1):101–19. doi: 10.1002/hep4.1799
404. Ma A, Wang J, Yang L, An Y, Zhu H. AMPK activation enhances the anti-atherogenic effects of high density lipoproteins in ApoE(-/-) mice. J Lipid Res. (2017) 58(8):1536–47. doi: 10.1194/jlr.M073270
405. Zhang Y, Qiu J, Wang X, Zhang Y, Xia M. AMP-activated protein kinase suppresses endothelial cell inflammation through phosphorylation of transcriptional coactivator p300. Arterioscler Thromb Vasc Biol. (2011) 31(12):2897–908. doi: 10.1161/ATVBAHA.111.237453
406. Barrett TJ. Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol. (2020) 40(1):20–33. doi: 10.1161/ATVBAHA.119.312802
407. Zhao P, Sun X, Chaggan C, Liao Z, In Wong K, He F, et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science. (2020) 367(6478):652–60. doi: 10.1126/science.aay0542
408. Song YM, Lee YH, Kim JW, Ham DS, Kang ES, Cha BS, et al. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy. (2015) 11(1):46–59. doi: 10.4161/15548627.2014.984271
409. Esquejo RM, Salatto CT, Delmore J, Albuquerque B, Reyes A, Shi Y, et al. Activation of liver AMPK with PF-06409577 corrects NAFLD and lowers cholesterol in rodent and primate preclinical models. EBioMedicine. (2018) 31:122–32. doi: 10.1016/j.ebiom.2018.04.009
410. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. (2008) 30(2):214–26. doi: 10.1016/j.molcel.2008.03.003
411. Eid W, Dauner K, Courtney KC, Gagnon A, Parks RJ, Sorisky A, et al. mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc Natl Acad Sci U S A. (2017) 114(30):7999–8004. doi: 10.1073/pnas.1705304114
412. Gosis BS, Wada S, Thorsheim C, Li K, Jung S, Rhoades JH, et al. Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1. Science. (2022) 376(6590):eabf8271. doi: 10.1126/science.abf8271
413. Lee MKS, Cooney OD, Lin X, Nadarajah S, Dragoljevic D, Huynh K, et al. Defective AMPK regulation of cholesterol metabolism accelerates atherosclerosis by promoting HSPC mobilization and myelopoiesis. Mol Metab. (2022) 61:101514. doi: 10.1016/j.molmet.2022.101514
414. Cusi K, Alkhouri N, Harrison SA, Fouqueray P, Moller DE, Hallakou-Bozec S, et al. Efficacy and safety of PXL770, a direct AMP kinase activator, for the treatment of non-alcoholic fatty liver disease (STAMP-NAFLD): a randomised, double-blind, placebo-controlled, phase 2a study. Lancet Gastroenterol Hepatol. (2021) 6(11):889–902. doi: 10.1016/S2468-1253(21)00300-9
415. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. (2018) 15(6):349–64. doi: 10.1038/s41575-018-0009-6
416. Everett BM, MacFadyen JG, Thuren T, Libby P, Glynn RJ, Ridker PM. Inhibition of interleukin-1beta and reduction in atherothrombotic cardiovascular events in the CANTOS trial. J Am Coll Cardiol. (2020) 76(14):1660–70. doi: 10.1016/j.jacc.2020.08.011
417. Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol. (2017) 70(18):2278–89. doi: 10.1016/j.jacc.2017.09.028
418. Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol. (2011) 55(5):1086–94. doi: 10.1016/j.jhep.2011.01.048
419. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
420. Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. (2003) 37(2):343–50. doi: 10.1053/jhep.2003.50048
421. Barbuio R, Milanski M, Bertolo MB, Saad MJ, Velloso LA. Infliximab reverses steatosis and improves insulin signal transduction in liver of rats fed a high-fat diet. J Endocrinol. (2007) 194(3):539–50. doi: 10.1677/JOE-07-0234
422. Koca SS, Bahcecioglu IH, Poyrazoglu OK, Ozercan IH, Sahin K, Ustundag B. The treatment with antibody of TNF-alpha reduces the inflammation, necrosis and fibrosis in the non-alcoholic steatohepatitis induced by methionine- and choline-deficient diet. Inflammation. (2008) 31(2):91–8. doi: 10.1007/s10753-007-9053-z
423. Tang KT, Dufour JF, Chen PH, Hernaez R, Hutfless S. Antitumour necrosis factor-alpha agents and development of new-onset cirrhosis or non-alcoholic fatty liver disease: a retrospective cohort. BMJ Open Gastroenterol. (2020) 7(1):e000349. doi: 10.1136/bmjgast-2019-000349
424. Radner H, Aletaha D. Anti-TNF in rheumatoid arthritis: an overview. Wien Med Wochenschr. (2015) 165(1–2):3–9. doi: 10.1007/s10354-015-0344-y
425. Cui G, Fan Q, Li Z, Goll R, Florholmen J. Evaluation of anti-TNF therapeutic response in patients with inflammatory bowel disease: current and novel biomarkers. EBioMedicine. (2021) 66:103329. doi: 10.1016/j.ebiom.2021.103329
426. Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the randomized etanercept worldwide evaluation (RENEWAL). Circulation. (2004) 109(13):1594–602. doi: 10.1161/01.CIR.0000124490.27666.B2
427. Oberoi R, Vlacil AK, Schuett J, Schosser F, Schuett H, Tietge UJF, et al. Anti-tumor necrosis factor-alpha therapy increases plaque burden in a mouse model of experimental atherosclerosis. Atherosclerosis. (2018) 277:80–9. doi: 10.1016/j.atherosclerosis.2018.08.030
428. Thompson M, Saag M, DeJesus E, Gathe J, Lalezari J, Landay AL, et al. A 48-week randomized phase 2b study evaluating cenicriviroc versus efavirenz in treatment-naive HIV-infected adults with C–C chemokine receptor type 5-tropic virus. AIDS. (2016) 30(6):869–78. doi: 10.1097/QAD.0000000000000988
429. Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology. (2020) 72(3):892–905. doi: 10.1002/hep.31108
430. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. (2006) 7(3):311–7. doi: 10.1038/ni1309
431. Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. (1998) 394(6696):894–7. doi: 10.1038/29788
432. Gilbert J, Lekstrom-Himes J, Donaldson D, Lee Y, Hu M, Xu J, et al. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am J Cardiol. (2011) 107(6):906–11. doi: 10.1016/j.amjcard.2010.11.005
433. Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F, et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS One. (2016) 11(6):e0158156. doi: 10.1371/journal.pone.0158156
434. Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. (2006) 44(4):865–73. doi: 10.1002/hep.21327
435. van der Wal AC, Becker AE. Atherosclerotic plaque rupture–pathologic basis of plaque stability and instability. Cardiovasc Res. (1999) 41(2):334–44. doi: 10.1016/s0008-6363(98)00276-4
436. Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. (2020) 72(1):58–71. doi: 10.1002/hep.31205
437. Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. (2018) 391(10126):1174–85. doi: 10.1016/S0140-6736(18)30474-4
438. Grimaldi PA. Peroxisome proliferator-activated receptors as sensors of fatty acids and derivatives. Cell Mol Life Sci. (2007) 64(19–20):2459–64. doi: 10.1007/s00018-007-7278-5
439. Lee Y, Kim BR, Kang GH, Lee GJ, Park YJ, Kim H, et al. The effects of PPAR agonists on atherosclerosis and nonalcoholic fatty liver disease in ApoE-/-FXR-/- mice. Endocrinol Metab (Seoul). (2021) 36(6):1243–53. doi: 10.3803/EnM.2021.1100
440. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. (2004) 113(10):1408–18. doi: 10.1172/JCI21025
441. Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. (2003) 17(2):259–72. doi: 10.1210/me.2002-0120
442. Hanniman EA, Lambert G, McCarthy TC, Sinal CJ. Loss of functional farnesoid X receptor increases atherosclerotic lesions in apolipoprotein E-deficient mice. J Lipid Res. (2005) 46(12):2595–604. doi: 10.1194/jlr.M500390-JLR200
443. Yin Y, Wang M, Gu W, Chen L. Intestine-specific FXR agonists as potential therapeutic agents for colorectal cancer. Biochem Pharmacol. (2021) 186:114430. doi: 10.1016/j.bcp.2021.114430
444. Zhou M, Learned RM, Rossi SJ, Tian H, DePaoli AM, Ling L. Therapeutic FGF19 promotes HDL biogenesis and transhepatic cholesterol efflux to prevent atherosclerosis. J Lipid Res. (2019) 60(3):550–65. doi: 10.1194/jlr.M089961
Keywords: atherosclerosis, animal models, biomarkers, nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, pathophysiology, therapeutics
Citation: Finney AC, Das S, Kumar D, McKinney MP, Cai B, Yurdagul A Jr and Rom O (2023) The interplay between nonalcoholic fatty liver disease and atherosclerotic cardiovascular disease. Front. Cardiovasc. Med. 10:1116861. doi: 10.3389/fcvm.2023.1116861
Received: 5 December 2022; Accepted: 23 March 2023;
Published: 2 May 2023.
Edited by:
Masanori Aikawa, Brigham and Women's Hospital, Harvard Medical School, United StatesReviewed by:
Robert N. Helsley, University of Kentucky, United States© 2023 Finney, Das, Kumar, Mckinney, Cai, Yurdagul and Rom. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arif Yurdagul YXJpZi55dXJkYWd1bEBsc3Vocy5lZHU= Oren Rom b3Jlbi5yb21AbHN1aHMuZWR1
†These authors share senior authorship
Specialty Section: This article was submitted to Atherosclerosis and Vascular Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.