
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 02 June 2023
Sec. Thrombosis and Haemostasis
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1113827
This article is part of the Research Topic Potential Mechanism of Thrombogenesis and Promising Therapeutic Strategy View all 7 articles
 Zhongyu Han1,†
Zhongyu Han1,† Qiong Liu1,†
Qiong Liu1,† Hongpeng Li1,†Meiqi Zhang1Luling You1Yumeng Lin2Ke Wang2Qiaoyin Gou2Zhanzhan Wang3Shuwei Zhou4YiJin Cai1Lan Yuan1*Haoran Chen5*
Hongpeng Li1,†Meiqi Zhang1Luling You1Yumeng Lin2Ke Wang2Qiaoyin Gou2Zhanzhan Wang3Shuwei Zhou4YiJin Cai1Lan Yuan1*Haoran Chen5*
Cardiovascular and cerebrovascular diseases are the number one killer threatening people's life and health, among which cardiovascular thrombotic events are the most common. As the cause of particularly serious cardiovascular events, thrombosis can trigger fatal crises such as acute coronary syndrome (myocardial infarction and unstable angina), cerebral infarction and so on. Circulating monocytes are an important part of innate immunity. Their main physiological functions are phagocytosis, removal of injured and senescent cells and their debris, and development into macrophages and dendritic cells. At the same time, they also participate in the pathophysiological processes of pro-coagulation and anticoagulation. According to recent studies, monocytes have been found to play a significant role in thrombosis and thrombotic diseases of the immune system. In this manuscript, we review the relationship between monocyte subsets and cardiovascular thrombotic events and analyze the role of monocytes in arterial thrombosis and their involvement in intravenous thrombolysis. Finally, we summarize the mechanism and therapeutic regimen of monocyte and thrombosis in hypertension, antiphospholipid syndrome, atherosclerosis, rheumatic heart disease, lower extremity deep venous thrombosis, and diabetic nephropathy.
Thrombotic diseases, which are mostly caused by thrombosis and thromboembolic obstruction, are the main challenges in clinical practice (1, 2). At present, thrombotic diseases have become a kind of circulatory system disease that seriously endangers human health. Based on research data from the World Health Organization, CVDs are responsible for the highest number of deaths globally, with 17.9 million people dying from these diseases each year. A significant proportion of these deaths result from cardio-cerebrovascular embolism (3). According to the location of the disease, thrombotic diseases can be classified into arterial thrombosis, venous thrombosis and microcirculatory disorders.
For decades, it has been recognized that there is a complex interaction between coagulation and inflammation, and the coagulation cascade and platelet activation can trigger the immune system, leading to leukocyte recruitment, adhesion, extravasation and activation, and the immune system can in turn influence haemostatic system, a process referred to as immunothrombosis (4). Monocytes, which are myeloid cells throughout an individual's lifetime, contribute to regulating immune responses and serve multiple functions in the body, such as tissue development and maintaining vascular homeostasis, host defense, and playing a role in initiating and resolving inflammation and tissue remodeling (5). Monocytes in an immature state are released from the bone marrow into the bloodstream, after which some relocate to various tissues to differentiate into resident macrophages (MΦ) or dendritic cells (DC) (6, 7). Under the action of different factors and environments, macrophages can be polarized into different subtypes, and the common ones are classically activated MΦ1 (M1) or alternately activated MΦ2 (M2). M1 macrophages polarized into a pro-inflammatory state, and exhibited high expression of proinflammatory miRNA, promoting Th1 response (8). On the other hand, M2 macrophages have been found to promote tissue repair and cell proliferation, display anti-inflammatory characteristics, and encourage a Th2 response (8). The influence of the two macrophage subsets on angiogenesis is divergent, with M1 macrophages generally suppressing cell proliferation and reducing the ability of endothelial cells (ECs) to undergo angiogenesis, while M2 macrophages often oppose this effect (8, 9). Studies have demonstrated that M2 macrophages can facilitate angiogenesis by upregulating basic fibroblast growth factor (bFGF), insulin-like growth factor-1 (IGF1), and placental growth factor signaling pathways (10).
Different monocytes subset play diverse roles in cardiovascular physiology and pathophysiology. Though now, there are many subgroups (Table 1) to be researched through various classification methods and the mechanisms are still not enough to understand, several experiments have shown changes in the absolute and relative concentrations of these subsets can serve as exquisite markers of different inflammatory states in CVDs (15–18). In cases of thrombosis, the formation of circulating monocyte-platelet aggregates (MPA) can indicate platelet activation and the presence of an inflammatory response. MPA act as a mediator between inflammation and thrombosis, highlighting their significant role (19). Multiple receptor ligands bind between monocytes and platelets, leading to activation of MPA. P-selectin/PSGL-1 mediated aggregation of platelet and monocytes leads to NF-κB activation/translocation, superoxide anion production and promotion of monocyte chemotactic protein-1 (MCP-1), interleukin-8 (IL-8), IL-1β and tissue factor (TF) release, which trigger coagulation and accelerate thrombosis (20, 21). Moreover, multiple studies have found that heightened levels of MPA in individuals with coronary heart disease, unstable angina pectoris, and acute myocardial infarction contribute significantly to the acceleration of thromboembolic events (22–24). This highlights the importance of MPA in the progression of CVD.
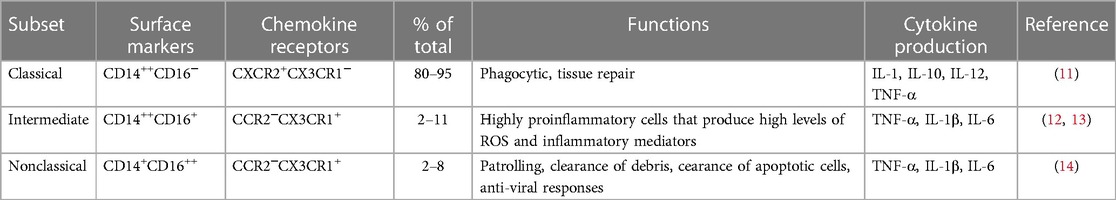
Table 1. Phenotype and function of circulating monocyte subsets in human.
Neutrophil extracellular traps (NET) are the network ultrastructure released into the extracellular after polymorphonuclear neutrophils (PMN) activation, which serves as the first line of defense against microbial infection in the early stage. Studies have shown that NET is associated with thrombus-inflammation-related diseases including Sepsis, systemic lupus erythematosus, and coronary heart disease, which is a hot topic in current research (25, 26). At present, Monocyte extracellular trap (ET) shows similar morphology to NET, exhibited procoagulant activity, and was associated with myeloperoxidase (MPO), lactoferrin (LF), citrullinated histones, and elastase (27, 28). In an inflammatory setting, extracellular trap cell death (ETosis) occurs when peripheral blood mononuclear cells encounter elastase and citrullinated portions of NET, and monocytes preferentially take up apoptotic bodies from PMN, thereby removing apoptotic bodies and NET-DNA (28, 29). We increasingly recognize the importance of monocytes in participating in thrombotic diseases such as CVDs, and increasing research condenses into monocyte-specific molecular mechanisms, signaling pathways, and gene expression, providing direct evidence to elucidate some diseases (21, 30–33).
Indeed, the role of monocytes in the link between inflammation and the pro-thrombotic state is a topic of interest and relevance in the field of cardiovascular disease. Monocytes are important immune cells that can differentiate into different subsets of macrophages that play distinct roles in the body's immune response. During inflammation, monocytes can become activated and release pro-inflammatory cytokines, which can contribute to the development of a pro-thrombotic state. Additionally, monocytes can interact with platelets and endothelial cells, further contributing to the development of thrombosis. There has been some research on the specific mechanisms by which monocytes contribute to the link between inflammation and the pro-thrombotic state, and further studies in this area could potentially lead to the development of new therapeutic approaches for the prevention and treatment of cardiovascular disease.
It should be mentioned that roles of moncytes in arterial thrombosis “itself” has been rarely investigated. Pathophysiology of thrombotic diseases are complex, because arterial atherosclerosis (or venous blood stasis), plaque (and rupture), tissue injury (such as cerebral infarction and myocardial infarction), and systemic stress (related to cardiovascular events in humans and animals or modeling-related surgical stress in animals) are interwined, affecting each other. Therefore, under the subtitle of monocytes and arterial thrombosis, we do not have much data; monocyte-related features in the literature for “thrombotic diseases” are not necessarily about “thrombosis” itself.
Monocytes are the largest blood cells in the blood, accounting for 8%–10% of the total number of leukocytes in the body (34). Monocytes are the main component of the autoimmune response and are closely related to the endogenous inflammatory process, its surface sensing changes of the receptor (35). Monocytes can respond quickly when body tissues are damaged or infected, transforming into macrophages or DC to regulate the inflammatory response and safeguard the body from infection and injury (36). Several studies have shown that monocytes are heterogeneous and plastic, and a variety of molecular markers are present on the cell membrane, such as adhesion molecules, complement receptors, and cytokines (31, 37). These molecules jointly participate in the physiopathological processes such as cell migration, thrombosis, and phagocytosis (31, 38, 39). According to the International Federation of Immunology's 2010 classification, human monocytes were divided into three subsets based on the expression of CD14 (an LPS-related receptor) and CD16 (an FcγⅢ receptor). These subsets include Classical Monocytes (CMs, CD14++CD16−), Intermediate Monocytes (IMs, CD14++CD16+), and Non-classical Monocytes (NCMs, CD14+CD16++) (40) (Table 1). Classical monocytes overexpressed genes related to phagocytosis, such as CD93, CD64, CD11B, CD36, CD32, CD14, ficolin-1 (FCN1), and signal regulatory protein alpha (SIRPA) (41, 42). CMs are involved in various immune responses such as inflammation and tissue repair (43). IMs exhibit the highest expression of TLR2, TLR4, CD40 and MHC-class II molecules (HLA-DR), and also possess the greatest antigen presentation ability (42). NCMs perform endothelial cell patrolling, which aids in maintaining cellular integrity, removing dead ECs, repairing the vasculature in atherosclerotic diseases, and removing lipids from the blood (44). In vitro studies have revealed that NCMs demonstrate the most robust response to LPS, and are capable of secreting pro-inflammatory cytokines like tumor necrosis factor-alpha (TNF-α) and IL-1β. The heightened expression of miR-146a observed in NCMs is indicative of the senescence-associated secretory phenotype (SASP) that results from increased basal levels of phosphorylated NF-κB (p65) and IL-1α (45).
The developmental relationship of monocytes goes through three stages, which are typical phenotype, intermediate phenotype and non-classical phenotype. The results of several current studies have shown that researchers have a large controversy about proinflammatory cytokines secreted by monocytes of different phenotypes (42, 46). In a comparison regarding how much reactive oxygen species (ROS) are produced, Cros et al. suggested that IMs do not produce ROS and CMs produce large amounts of ROS, while Zawada et al. suggested that CMs produce the lowest ROS and IMs produce the most ROS (41, 46). In terms of how much TNF-α is secreted, Cros et al. suggested that IMs produce the most TNF-α, whereas Wong et al. claimed that NCMs produce the highest levels of TNF-α (42, 46). In addition, at the level of gene expression subset function, Cormican and Griffin screened genes targeting monocyte subsets while giving a critical evaluation (33). Anika Witten et al. conducted flow cytometric analysis and genome-wide transcriptional profiling to investigate the correlations between miRNA and mRNA species in three distinct monocyte subsets. These subsets were obtained from patients with first acute myocardial infarction (MI), stable coronary artery disease (CAD), and individuals without any previous history of CVD. They found that most MI-specific miRNAs were involved in NCMs, which was consistent with the involvement of NCMs in tissue repair after myocardial infarction. In the inflammatory state of active CAD, the CMs are more inflammation and patrolling. The findings suggest that miRNAs from monocyte subsets might play a significant role in CVD both during and after the disease progression. Due to their stability and abundance in the circulating blood, the miRNAs linked to monocytes could be potentially useful as biomarkers for diagnosis, prevention, and treatment of CVD (47).
In the past few decades, there has been growing evidence pointing to the existence of distinct subpopulations of monocytes that play different roles in the development of CVDs. Changes in the function, number, and proportion of these monocyte subsets have been closely linked to the progress and prognosis of CVD (15, 48–50). A single published literature by Helen Williams very well has reviewed the relationship between the nature and number of monocyte subpopulations developing in CVDs (51). The ratio of IMs and/or NCMs are frequently reported to be elevated in CVD (51). In general, CD16+ monocytes may have more promoting inflammatory and pro-coagulant functions in vascular inflammation. In a clinical trial, IL-6 and IL-8 mRNA levels were elevated in both IMs and NCMs and not detected in CMs after LPS bolus injection in 12 healthy volunteers (52). In addition, CD16 on CMs can be induced to rise by low serum HDL-C in patients with coronary atherosclerosis, resulting in a concomitant increase in IMs (53, 54). Interestingly, R. Cappellari et al. made an intriguing discovery-they found that cardiovascular outcomes could be predicted by the shift of monocyte subsets along their continuum, as opposed to the proportions of these subsets (55). The shift of monocyte subsets along their continuum in patients with major adverse cardiovascular events (MACE) was presented and described, mainly manifested as the increase of classical CD16 fluorescence intensity, the increase of intermediate type proportion, and the increase of non-typical CD14 fluorescence intensity. Such drift changes are particularly helpful in determining cardiovascular events. The findings indicate that the predictive power of monocyte subsets for adverse cardiovascular outcomes lies in the shift along the CD14/CD16 continuum, rather than their frequency. Ramona Vinci et al. investigated whether variations in plaque erosion and rupture, as identified by optical coherence tomography (OCT) studies, were significantly different in non-ST-segment elevation acute coronary syndromes (ACS) patients based on the analysis of monocyte subsets present in circulating blood (56). The study introduced a pre-classical monocyte (PCM) population near the coronary microvasculature with CD14+CD16− surface staining. The distribution of monocyte subsets was analyzed in the study population, and the frequency of the newly identified PCM subsets was found to be higher in patients with non-ST-segment elevation ACS (NSTE-ACS) compared to those with chronic coronary syndrome (CCS). When the plaque phenotype was analyzed by OCT, the PCM subsets were higher in NSTE-ACS patients, especially those with ruptured fibrous cap (RFC) plaques and concomitant local macrophage infiltration (MΦI). Overall, higher rates of circulating PCM may represent a unique marker of specific plaque rupture with local MΦI.
Furthermore, the Monocyte-to-HDL Ratio (MHR), which is a newly emerged biomarker indicating inflammation and oxidative stress, can be conveniently obtained in clinical settings. Currently, it has been considered a marker of drug-resistant hypertension in chronic kidney disease (CKD), coronary heart disease, diabetic retinopathy, pulmonary embolism (PE) and other diseases (57–61).
Arterial thrombosis is mainly due to the blood vessels in unstable atherosclerotic plaque rupture, plaques inside the lipids are released into the bloodstream, causing blood platelet aggregation and adhesion, and attracting white blood cells (WBC) and red blood cell (RBC) aggregation, leading to thrombosis, vascular occlusion and possible acute myocardial infarction, ischemic stroke, lower limb arteriosclerosis occlusion (62). Arterial thrombosis is characterized by high blood flow velocity and high wall shear rate. When ECs are destroyed or atherosclerotic plaque is ruptured, a series of events lead to the formation of platelet-rich thrombosis (63).
At sites of vascular injury, inflammation, or thrombosis, platelet activation plays a crucial role in orchestrating the recruitment of WBC. This intricate process is largely mediated via the release of soluble mediators or through direct cellular interactions. Notably, platelets exhibit the highest binding affinity with monocytes/macrophages, and their potency descends progressively with respect to neutrophils and lymphocytes. This mechanism of cellular recruitment represents a profound component of the immune response and serves to fortify the host's defenses against pathogenic insults (64).
P-selectin (CD62P), a transmembrane glycoprotein of platelets, constitutes a fundamental component of thromboinflammatory cascades. Its exposure on the surface of platelets initiates a series of intricate interactions with leukocytes by binding to O-glycosylated carbohydrate ligands on PSGL1 on the outer surface of myeloid cell membranes. Such binding, synergistic with the CD40-CD40l pathway, elicits intracellular signal transduction, culminating in the upregulation of TF expression in leukocytes (20, 30, 65–67). In turn, PSGL-1 induce up-regulation and activation of integrin β2 [macrophage-1 antigen (Mac-1) and lymphocyte function-associated antigen-1 (LFA-1)] in monocytes, further supporting their interaction with platelets (68). Glycoprotein (GP) VI, a prominent receptor located on the surface of platelets, drives the interaction between platelets and the extracellular matrix metalloproteinase inducer (EMMPRIN, CD147/basigin) expressed on the surface of monocytes, promoting their recruitment to the site of inflammation in arterial walls (69, 70). In addition to the direct interaction, platelet-derived substance influence monocyte recruitment and endothelial cell adhesion and behavior. Intriguingly, platelets activated under conditions of vascular insult then extrude into their surroundings, an array of extracellular vesicles (EVs) derived from the membrane-bound structures of the activated platelets. These EVs exhibit a preference towards binding to blood monocytes in comparison to other leukocyte subsets, a mechanism chiefly contributing to the selective recruitment of monocytes towards sites of inflammation or tissue injury. This intricate process represents an indispensable component of the host's immune response, highlighting the functional importance of these EVs as modulators of both inflammation and thrombosis (Figure 1). Platelet-derived EVs transfer the platelet adhesion receptor GPIbα to the surface of monocytes, which can be recruited in various blood vessels (71). As a perfectly tuned transcellular conveyance mechanism for RANTES, circulating platelet microparticles (PMPs) augment the adhesion of CXCL4 (PF4) to the exterior of monocytes, inducing a robust anti-inflammatory effect. The exquisite intercellular orchestration orchestrated by PMPs not only furthers the targeted transport of RANTES, but also orchestrates an intricate cellular dialogue that culminates in the suppression of the inflammatory signals generated by activated monocytes (72, 73). CXCL4 induces monocyte respiratory burst through rapid activation of phosphoinositide 3-kinase (PI3K), spleen tyrosine kinase (SYK), and p38 mitogen-activated protein kinase (MAPK), mediates ROS formation, cytokine/chemokine expression, and apoptosis rescue depends on continuous activation of SPHK and extracellular regulated protein kinases (ERK) to inhibit caspase activation (74, 75). CXCL4 also promotes monocyte differentiation into macrophages and inhibits CD163 in macrophages to promote atherogenesis (76, 77). Through a precision-driven process, activated platelets demonstrate their nuanced ability to manipulate the MCP-1 and intercellular adhesion molecule-1 (ICAM-1) properties of ECs using a mechanism that is dependent upon the regulation of NF-κB (78). With intricate precision, downstream upregulation of adhesion molecule target genes, namely vascular cell adhesion molecule-1 (VCAM-1), ICAM-1, E-selectin, P-selectin and MCP-1, confers an indirect yet potent effect on monocyte recruitment (79). Overall, the formation of MPA is sophisticated, but it is crucial for thrombosis. With remarkable dexterity, previous studies have postulated that P-selectin may be a key determinant in the initiation of the intricate cellular conjugation between platelets and monocytes, subsequently fueling a deleterious cycle of platelet activation that manifests as a cascade of platelet activator synthesis. By offering an incisive glimpse into this complex interplay, these seminal findings expand our understanding of the intricate molecular pathways that underlie the profound intercellular cooperation observed in platelet-monocyte conjugation. Such knowledge opens up a rich avenue of exploration for the development of novel therapies designed to target the various components involved in this process for better clinical outcomes.
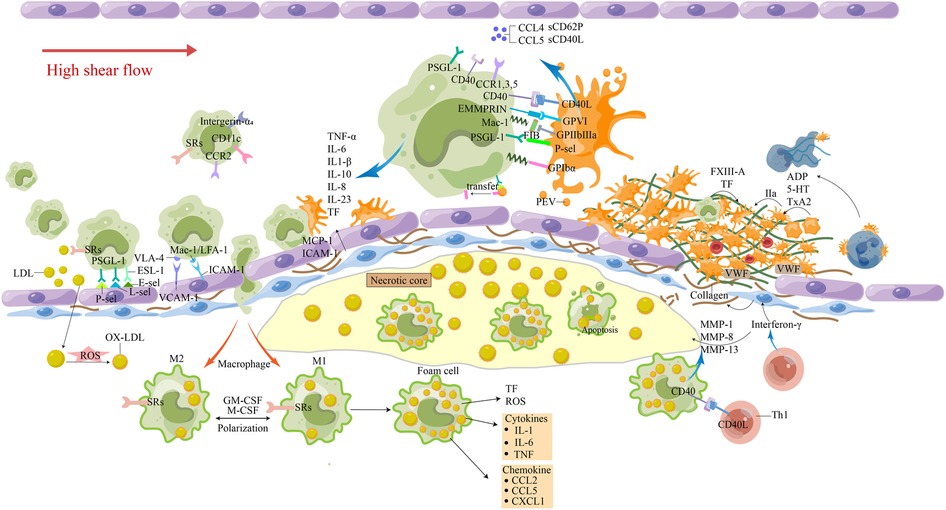
Figure 1. The involvement of monocytes in atherosclerosis-thrombosis. The release of pro-inflammatory factors from cells in the plaque and the damage of ECs caused circulating monocytes to roll, roll slowly, adhere firmly, and travel through the tissue to differentiate. Macrophages phagocytose excessive Ox-LDL and other immune substances, causing apoptosis and necrosis of cells, lipid deposition, the generation of a large number of inflammatory factors and tissue factors, and promoting the transport of more circulating monocytes. Activated Th1 cells produce interferon γ, which inhibits smooth muscle cell synthesis of interstitial collagen. The interaction between Th1 cells and M1 macrophages leads to the overproduction of MMP-1, MMP-8 and MMP-13, and the interstitial collagenase can promote the breakdown of interstitial collagen. At the same time, it can induce M1 macrophages to overexpress tissue factors. These processes make the fibrous cap more vulnerable to rupture, and inflammatory activation leads to the increased potential for thrombosis. Platelet-rich thrombosis forms around atherosclerotic plaque rupture and endothelial damage when endosubcutaneous collagen is exposed to circulation under high shear flow. Platelets, through their glycoproteins, interact with collagen and collagen-deposited vWF to change their shape and adhere to the injured site. Attachment results in the secretion of ADP, serotonin, and thromboxane (TxA2), which recruit and activate more platelets. The platelet-monocyte complex (PMC) is activated by the interaction of various receptor ligands between platelets and monocytes. Platelets and monocytes also shed extracellular vesicles, release cytokines and chemokines, and tissue factors are increased to promote thrombosis. 5-HT, 5-hydroxytryptamine; E-sel, E-selection; FXIII, Factor XIII; FIB, fibrinogen; GM-CSF, granulocyte-macrophage colony stimulating factor; IL-8, interleukin-8; LDL, low-density lipoproteins; L-sel, L-selection; MCP-1, monocyte chemotactic protein-1; oxidized LDL; M-CSF, macrophage colony-stimulating factor; MMP, matrix metalloproteinases; Ox-LDL, oxidized LDL; PEV, platelet-derived extracellular vesicles; PMC, platelet-monocyte complex; P-sel, P-selection; ROS, reactive oxygen species; SRs, scavenger receptors; TF, tissue factor; TNF-α, tumor necrosis factor-α; TxA2, thromboxane; VCAM-1, vascular cell adhesion molecule-1; vWF, von Willebrand factor.
With intricate precision, it has been postulated that leukocytes navigate across the luminal surface of inflammatory ECs via a refined rolling mechanism, only to then stabilize their interaction with the endothelium and progress into the vascular wall. This decisive cellular behavior is regulated by a fine-tuned process of cellular crawling, through which the leukocytes reach the appropriate extravasation site with remarkable specificity. Crucial to this intricate interplay is the dynamic regulation of adhesion molecules, which confer nuanced yet prodigious control over the molecular pathways underlying leukocyte motility and extravasation (31).
With unparalleled finesse, it has been theorized that the exquisite interplay between inflammation and ECs lays the foundations for a highly orchestrated cascade of molecular events. Of primordial import is the rapid induction of the expression of key endothelial adhesion molecules, which are fine-tuned with remarkable precision by the pivotal inflammatory factors, TNF-α and IL-1β. In a study, the authors have expounded on an intricate regulatory scheme governing the rolling rate of inflammatory monocytes. Specifically, their results demonstrate that the resynthesis of E-selectin by ECs plays an indispensable role in activating said regulatory mechanism. Additionally, they have delineated a multifaceted process by which P-selectin, L-selectin, PSGL1, and CD44, intricately intertwine and combine forces to regulate the flux of rolling neutrophils and inflammatory monocytes (80, 81). Very late antigen-4 (LA-4 α4β1 integrin) expressed by monocytes can bind to VCAM-1 on the endothelium, mediating its slow roll to firm adhesion on ECs activated by cytokines such as CXCL1 (82, 83). Monocytes are able to roll over the endothelium and appropriately adapt to the high shear stress that is imposed by blood flow, due to their interactions with C-C and C-X-C chemokines such as C-C chemokine ligand 2 (CCL2) (MCP-1) and CXCL8 (IL-8). These chemokines facilitate the tight adhesion of monocytes to ECs (84, 85). ICAM-1 also promotes leukocyte adhesion and migration through its ligands (LFA-1, MAC-1) and participates in the interaction between ECs and monocytes (86, 87). The risk factor C-reactive protein (CRP) leads to a reduction in endothelial nitric oxide production and drives plaque instability through several mechanisms. These mechanisms include increasing the expression of endothelial cell adhesion molecules, inducing monocyte TF through CRP, promoting the recruitment of monocytes to atherosclerotic plaques, and enzymatically binding modified low-density lipoprotein (88).
TF-positive monocyte-derived microvesicles (MVs) are small, membrane-bound fragments that are released from monocytes into the bloodstream. Monocytes are a type of white blood cell that can differentiate into macrophages, which play a key role in the immune response to infection and inflammation. When monocytes encounter oxidized LDL cholesterol particles in the bloodstream, they can become activated and release TF-positive MVs. These MVs can circulate widely in the bloodstream and contribute to coagulation-related concerns. TF-positive MVs are one of the sources of TF in the bloodstream, along with monocytes and other cells. The upregulation of tissue factor expression in circulating monocytes and macrophages through the TLR4/TLR6/CD36 receptor complex that occurs in the presence of OxLDL is thought to play a key role in the development of atherosclerosis and other vascular inflammatory diseases. As a result, TF-positive MVs are an important biomarker of vascular inflammation and thrombotic risk (89). We summarize the latest factors that stimulate monocytes to produce TF coagulation (90) (Table 2). Plaque rupture exposes the blood to high levels of TF-induced thrombin, initiating a coagulation cascade that subsequently forms a fibrin monolayer covering the exposed damaged surface of the vessel (130) (Figure 1).
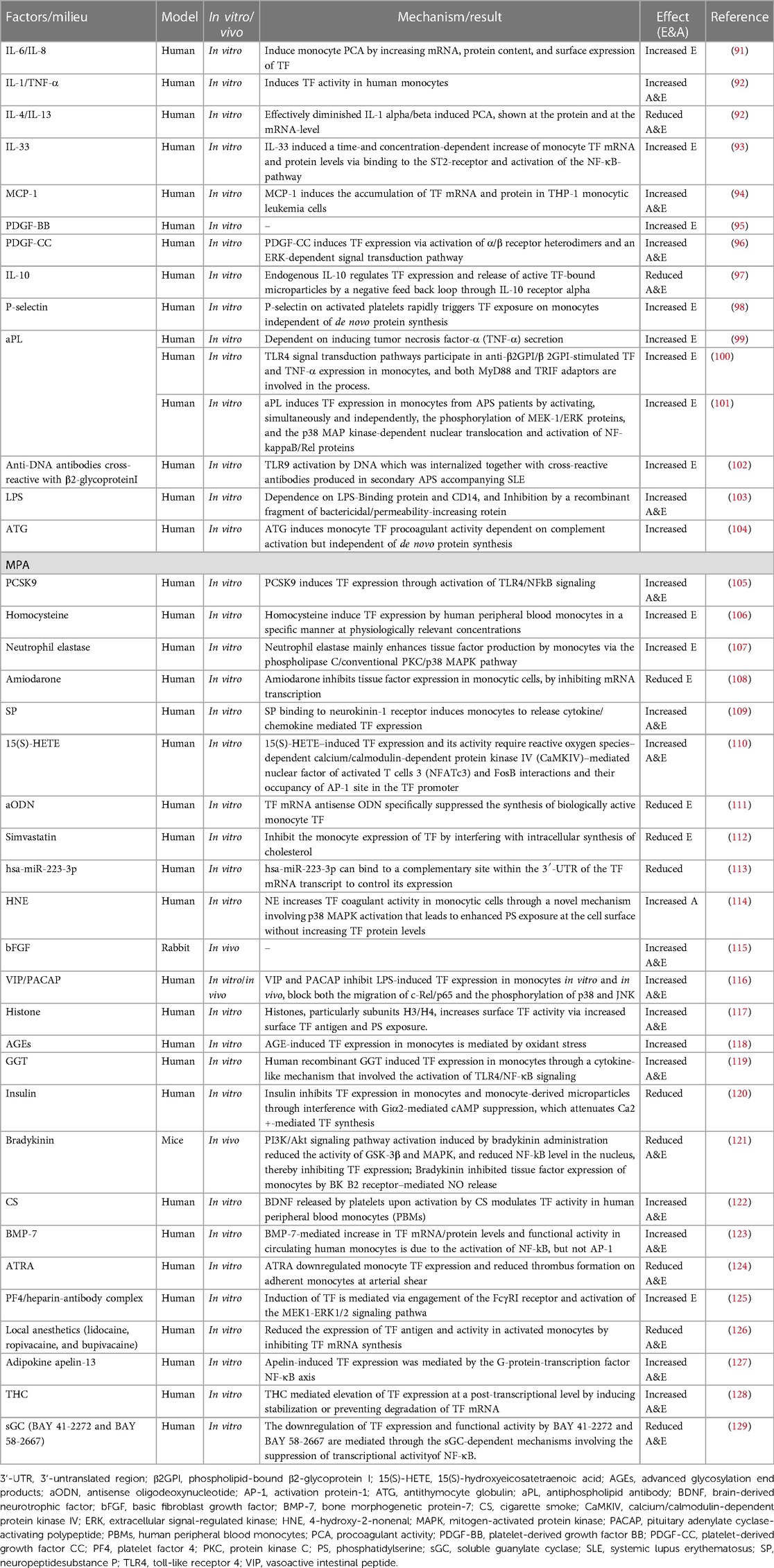
Table 2. Effect of different factors and milieu on monocyte tissue factor expression and procoagulant activity.
In the early stages of atherosclerosis, monocytes are recruited to the site of arterial injury, where they differentiate into macrophages, which engulf LDL, the “bad” cholesterol that contributes to the development of plaques. As the plaques grow larger, they eventually rupture or become unstable, leading to the formation of arterial thrombosis.
Recent research has suggested that monocyte-derived platelets, which are produced by the transformation of monocytes into megakaryocytes, may also play a key role in the development and progression of arterial thrombosis (131). These platelets are smaller than normal platelets and are more reactive, making them more likely to form blood clots.
The relationship between monocytes and arterial thrombosis has been studied extensively, with several mechanisms that underlie the association between these two phenomena being proposed. One of the mechanisms involves the production of various interleukins, such as interleukin 1β (IL-1β), by monocytes, which can induce endothelial cell damage and contribute to the development of arterial thrombosis. Another mechanism is the involvement of monocytes in the activation of the coagulation cascade, leading to the formation of blood clots.
Monocyte-targeted therapies have been proposed as potential treatments for arterial thrombosis, including the use of inhibitors that target interleukins or suppress the activation of the coagulation cascade. In addition, lifestyle modifications such as regular exercise, a healthy diet, and smoking cessation are recommended to reduce the risk of developing arterial thrombosis.
In conclusion, the relationship between monocytes and arterial thrombosis is a complex phenomenon that involves multiple aspects, and more research is needed to fully understand the mechanisms and potential therapies for this condition. Nonetheless, the increasing evidence of the significant contribution of monocytes to arterial thrombosis highlights the importance of further investigating their role in this disease. Understanding the relationship between monocytes and arterial thrombosis may ultimately lead to the development of more effective treatments and preventative strategies for this condition, which is a significant cause of morbidity and mortality worldwide.
The development of venous thrombosis is primarily linked to reduced shear flow, typically occurring around intact endothelial walls. Venous thrombi often contain high levels of fibrin, and are surrounded by numerous red blood cells and activated platelets. When blood flow decreases, it can result in hypoxia and lead to an increase in the expression of endothelial adhesion molecules. This increased expression can cause leukocytes to attach to the endothelium (62). The coagulation cascade is initiated by the release of TF from leukocyte microparticles and endothelial-binding monocytes. At the same time, thrombin is activated and fibrinogen is converted to fibrin (132) (Figure 2). Hypoxia can lead to the death of ECs and neutrophils, and the dead cell debris needs to be removed by macrophages (133). Pro-inflammatory Mo/MΦ sampled from blood showed significantly elevated M1-polarization in pathological deep vein thrombosis (DVT) in human patients (134). In both animal and human experimental observations concerning thrombus lysis, the presence of macrophages at the site was detected. Notably, the subsequent inflammatory signals triggered by these macrophages led to a heightened formation of thrombus, thereby instigating the ultimate stage of thrombus resolution (134–138). Faster thrombus resolution is the key to improving the prognosis of the disease. The phenomenon of venous thrombolysis and recanalization parallels the intricate biological process of wound healing, whereby the inflammatory response of the venous wall is provoked, establishing the infiltration of diverse inflammatory cells, fibril growth factors, collagen deposition, and the discernible expression and activation of matrix metalloproteinases (MMP) (139–142). Despite the comparatively diminished platelet count in the manifestly proliferating venous thrombosis than in its arterial counterpart, its activated platelets also duly evoke the expression of P-selection, thereby promoting peripheral leukocyte invasion into the thrombus (143).
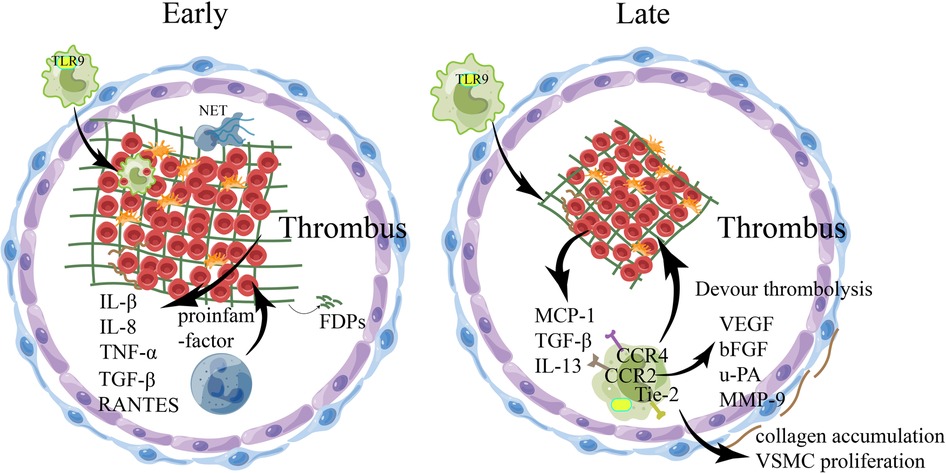
Figure 2. The involvement of relevant immune cells during venous thrombosis. During venous thrombus regression, numerous pro-inflammatory factors are released into the local environment, with an early influx of Neutrophils and macrophages, followed by monocytes, which regulate the production and activity of plasminase and MMPs. In the early stage of thrombolysis, fibrinolysis occurs at a high rate under the action of neutrophils, resulting in the production of fibrin degradation products, the emergence of collagen fibrils within the thrombolus, and the induction of inflammatory cytokines and various proteases by thrombolt-associated immune cells. With the structuring of the thrombus, the dissolution rate of fibrin slows down, collagen proliferation in the thrombus increases, monocytes infiltrate phagocytosis and mobilize EPC, and matrix remodeling of mmp secreted by macrophages occurs, which may eventually recover through the blood flow of the thrombus. bFGF, basic fibroblast growth factor; EPC, endothelial progenitor cells; FDPs, fibrin/fibrinogen degradation products;IL-8, interleukin-8; MCP-1, monocyte chemotactic protein-1; MMP, matrix metalloproteinases; NET, Neutrophil extracellular traps; TNF-α, tumor necrosis factor-α; TGF-β, transforming growth factor-β; TLR-9, toll-like receptor 9;uPA, uridylyl phosphate adenosine; VEGF, vascular endothelial growth factor; VSMC, Vascular smooth muscle cell.
The phenomena of thrombus dissolution release myriad proinflammatory factors into the indigenous milieu, encompassing IL-1β, TNF-α, JE/MCP-1, IL-6, MIP-1α, and ENA-78 (144). Then, leukocytes begin to invade the thrombus, and PMN is the first to reach the thrombus site, promoting fibrinolysis and collagen dissolution, which are critical for early thrombus dissolution (145). In addition, in the context of PMN inflammation in venous thrombosis, NETs can mediate thrombus fibrosis through TGF-β, and then transform into insoluble chronic thrombosis (146). PMN also promotes the entry of monocytes into the thrombu. Wakefield et al. noted that polymorphonuclear leukocytes in the thrombus began to switch from neutrophils to monocytes on day 4 of thrombosis and reached a peak on day 8 (147). As more macrophages accumulate within the thrombus, they become the primary inflammatory cells and start generating a variety of substances such as chemokines, inflammatory cytokines, and matrix-degrading proteases. Examples of these proteases include uridylyl phosphate adenosine (uPA) and MMPs, which promote both fibrinolysis and tissue remodeling. Additionally, the macrophages release multiple vascular-forming factors such as IL-1, TNF, vascular endothelial growth factor (VEGF), FGF-2, and PDGF. These factors can directly or indirectly stimulate endothelial cell proliferation, migration, and promote the formation of tubule-like structures, eventually resulting in the restoration of blood flow within the thrombosis vein (148–151). MMP-9 causes loss of venous wall compliance by increasing collagen elastin fibers and extracellular matrix (ECM), and subsides thrombosis by reducing macrophage and collagen content (152).
During the later stages of venous thromboembolism (VTE), certain cytokines, including IL-10, TNF-α, IL-6, and IL-8, seem to facilitate the resolution of the thrombus (153–156). In an animal experiment, investigators have found that IL-6 may be a therapeutic target to prevent fibrotic complications that arise in post-thrombotic syndrome (PTS) (157). Besides, Mizuho Nosaka's experiment demonstrated that administering an anti-IL-6 antibody slowed down the resolution of the thrombus (156). This evidence suggests that sophisticated cytokine regulation of thrombus also plays a role in post-thrombotic resolution and ensuing complications.
Attracted by high levels of MCP-1 and other factors (such as local inflammatory reactions), monocytes greatly infiltrate the thrombus and promote thrombus recanalization, and venous flow recanalization is mainly dependent on intrathrombotic neovascularization to establish functional flow channels. It is thought that the resolution of inflammation and acceleration of this process restores venous wall patency and reduces the pathology associated with PTS. According to previous research, while exogenous MCP-1 may speed up the resolution of DVT, it has been found to promote fibrosis in organs in vivo (147). In contrast, inhibiting P-selectin and E-selectin has been shown to decrease thrombosis and vein wall fibrosis (158). As such, targeting P-selectin and E-selectin may be promising areas for future research.
Generally speaking, when the normal blood pressure is <120/80 mmHg, the blood passes normally. When the blood pressure increases, it will impact the blood vessels and cause pressure on the blood vessels (159). The long-term effect causes damage to the intima of blood vessels and the formation of red thrombosis. Damaged areas form scarring hyperplasia, which thickens the blood vessel wall and continuously forms proliferation and accumulation, narrowing the lumen and increasing the risk of thrombosis (160).
Increased immune cell infiltration may affect some mechanisms directly associated with the development of hypertension, such as promoting the release of aldosterone, increasing the reabsorption of sodium and water, and increasing circulating blood volume. T cells and macrophages produce a variety of pro-inflammatory factors when stimulated, which can lead to cardiomyocyte fibrosis, vascular dysfunction and end-organ damage. Monocytes and macrophages are particularly involved in antigen presentation to T cells and cytokine production in hypertension. Proteins modified by oxidation of highly reactive γ-ketoaldehydes accumulate in DC in multiple hypertensive mouse models. Monocyte-derived dendritic cells (MDCS) make a difference in promoting hypertension and end-organ damage by producing abundant cytokines, promoting the proliferation of CD8+ T cells releasing IFN-γ and IL-17A (161).
Macrophages are derived from monocytes, and in the context of vascular inflammation, monocytes are recruited to inflamed tissue and differentiate into macrophages. These macrophages then secrete pro-inflammatory mediators (IL-1), promoting vascular inflammation (162). In a review published by Philip Wenzel et al., monocytes as immune targets in the occurrence, development, and manifestation of hypertension may represent a potentially novel therapeutic avenue for treating hypertension, thereby alleviating hypertension-related end-organ damage or preventing the development or deterioration of hypertension (163). Induction of the diphtheria toxin receptor in Lysm-positive macrophages followed by low doses of diphtheria toxin-depleted bone marrow mononuclear cells reduced circulating mononuclear cells and limited ATII-induced macrophage invasion of the vascular wall. Wild-type CD11b+Gr-1+ mononuclear cells transfected into depleted LysMiDTR mice restored Angiotensin II (Ang II)-induced vascular dysfunction and arterial hypertension (164). IL-1 is a typical mononuclear/macrophage cytokine, and activation of IL-1 receptors has been implicated in the promotion of sodium retention and elevation of blood pressure through the release of the NKKC2 sodium cotransporter (165).
Mononuclear cell infiltration, invasion, and differentiation in the heart and blood vessels are associated with the progression and severity of hypertensive diseases. In cell co-culture models, activated platelets from patients with essential hypertension are capable of releasing MCP-1, which is responsible for recruiting monocytes to sites of inflammation in blood vessels, thereby mediating the progression of atherosclerosis (166). Monocyte infiltration into the heart and blood vessels during hypertension is dependent on the CCL2-CCR2 axis, and it has been demonstrated in various studies that manipulating drugs or genes can have positive impacts on the chemokine and chemokine receptor pathway (162, 167, 168). In various rat models of hypertension induced by high-salt diet, deoxycorticosterone acetate, and human renin models, monocyte infiltration and adhesion can lead to increased blood pressure and renal injury (169–171).
In human cohort studies, the expression level of cysteine-rich intestinal protein 1 (CRIP1) in monocytes was associated with an increase in blood pressure, and this association decreased with the expression level of CRIP1 when monocytes differentiated into macrophages (172). This suggests that CRIP1 may affect the interaction between monocytes and the pathogenesis of hypertension through proinflammatory regulation and upregulation. In hypertensive patients, increased vascular stretching promotes endothelial cell activation, which enhances the conversion of monocytes into IMs and CD209-labeled monocytes, it activates STAT3 in monocytes and significantly stimulates the expression of IL-1β, IL-23, CCL4 and TNF-α in monocytes to produce these pro-hypertensive cytokines (173). In patients with poorly controlled hypertension and target organ damage (TOD) characteristics, the surface expression of NCMs costimulatory marker CD86 is higher, and the proportion of VEGF-R2 positive non-classical cells is higher, indicating that phenotypic changes in monocyte subsets are related to the progression and severity of hypertension (168).
Summarizing, in the current clinical trials and animal models related to hypertension that we mentioned, we found that organ damage resulting from hypertensive events is closely related to various immune cells of the immune system. Cells of the innate and adaptive immune systems enter target organs in the body through various routes (such as kidneys, blood vessels, myocardium, etc.) and damage target organs by releasing cytokines, matrix metalloproteinases, and ROS, ultimately leading to increased blood pressure. Several factors can activate immune cells to enter target organs, such as a high sodium diet causing an increase in sodium ions in the immune microenvironment. In addition, ECs can release both ROS and IL-6. These molecules can stimulate monocytes to become APCs, which in turn produce cytokines like IL-1β and IL-23. These cytokines can have a significant impact on T cell function. Therefore, reducing the secretion and production of cytokines by such cells in various ways contributes to improving hypertension and end-organ damage.
APS is a rare and complex systemic disease characterized by the presence of antiphospholipid (aPL) antibodies [including lupus anticoagulant, anticardiolipin antibodies, and anti-β2 glycoprotein i (anti-β2GPI) antibodies], with the main clinical features of thrombosis or pathological pregnancy (174). After antibodies bind to β2GPI protein on monocytes and ECs, the Toll-like receptor 4 (TLR-4) triggers the myeloid differentiation primary response protein 88 (MyD88) signaling pathway which leads to the activation of several protein molecules including p38 MAPK, MEK-1/ERK, and NF-κB (174–178). ECs produce microparticles (MP) and MCP-1, while monocyte adhesion to ECs increases due to the activation of the Toll-like receptor (TLR) expression. Furthermore, the presence of proinflammatory cytokines (IL-1β, IL-6, IL-8, and anti-TNF) and chemokines leads to the release of adhesion molecules including E-selectin, VCAM-1, ICAM-1, and TFs. These factors contribute to the inflammation of the blood vessels and promote the growth of ECs, which can ultimately lead to intimal hyperplasia (176, 179, 180). A study of how WB-6 (a mouse monoclonal anti-β2Gpi antibody) contacts and activates resting-state monocytes showed that monocytes internalizing WB-6 express TF and TNF-α, TNF-α stimulates ECs to express ICAM-1 and VCAM-1 (181). Later, another study identified the signaling pathways within cells involved in this process and suggested that the induction of TF expression was the result of the internalization of DNA-activated TLR9 together with cross-reactive antibodies produced by SLE secondary APS (182). It has proved that the β2GPI dimerization domain V(β2GPI-dV) dimerization of β2GPI-dV is sufficient to induce the up-regulation of procoagulant activity of monocytes (183). Antiphospholipid antibodies can activate monocytes and macrophages through systemic allergic reaction toxins (C3a, C4a) produced during complement activation (184).
Chary Lopez-Pedrera et al. first used proteomic analysis to identify changes in the proteomic pattern of monocytes directly associated with thrombotic events in APS and served as a basis for the mechanism of thrombosis in APS (185). Vera M. Ripoll performed a comprehensive proteomic analysis of human monocytes from APS patients with different manifestations using two-dimensional differential gel electrophoresis (2D DiGE). The regulation of four of these proteins showed significant differences in monocytes treated with thrombotic or obstetric APS IgG compared to those treated with healthy control IgG. The proteome of monocytes treated with thrombotic APS IgG was further described using label-free proteomics. Among the 12 most reliable proteins associated with the cytoskeleton, immune response, and coagulation function, POTEE and b-actin-like protein 3 overlapped with 2D DiGE, Abnormalities in the regulation of these proinflammatory and procoagulant proteins could serve as targets for subsequent treatment and contribute to a deeper understanding of the diversity of APS pathogenesis (186). Urine proteomics has recently been reported as a noninvasive method to distinguish primary thrombotic APS from primary obstetric APS. Urinary CXCL12 and PDGF may be potential noninvasive markers to distinguish between them, but the number of patients is small and may be biased (187).
APL can combine with β2GPI and oxLDL to produce oxLDL/β2GPI/anti-β2GPI complex, which can inhibit autophagy and increase the release of inflammatory factors in ECs through a variety of signaling pathways (such as PI3K/AKT/mTOR and eNOS signaling pathways). When a large number of inflammatory factors accumulate around ECs, monocyte-macrophages activate to release TFs, chemokines, growth factors, and metalloproteinases, and differentiate into foam cells, exacerbating atherothrombosis (177, 188, 189).
At present, it is difficult to diagnose and treat APS patients in clinical practice. Molecular analysis of monocytes in APS patients is helpful to identify unique clinical phenotypes and thus develop treatment plans. Microarray analysis of monocytes with APS revealed 547 differentially expressed genes and identified novel miRNA-mRNA-intracellular signal regulatory networks in monocytes associated with CVD. In patients with APS, the monocytes showed reduced expression of miR-19b-3p and miR-20a-5p, and those with the least miRNA expression had the highest levels of aPL (190, 191). The results of the in vitro experiments revealed that APS patients had significantly higher serum levels of HMGB1 and sRAGE, as compared to healthy individuals. anti-β2-GPI antibody induced RAGE activation and HMGB1 cell relocalization in monocytes and platelets. As an increase in proinflammatory triggers, HMGB1/sRAGE may be involved in monitoring the risk of recurrent miscarriage (192). Monocytes and ECs have a pivotal role in the development of APS, and the interaction between monocytes and ECs has been described in the previous section. Ulštok's study was the first to show a marked increase in VLA4 on monocyte surfaces in patients with APS, and in vitro stimulation of Catastrophic APS (CAPS) showed a more significant increase in VLA4, suggesting that VLA4-enhanced monocyte adhesion is involved in thrombotic pathophysiology in APS (193) (Figure 3).
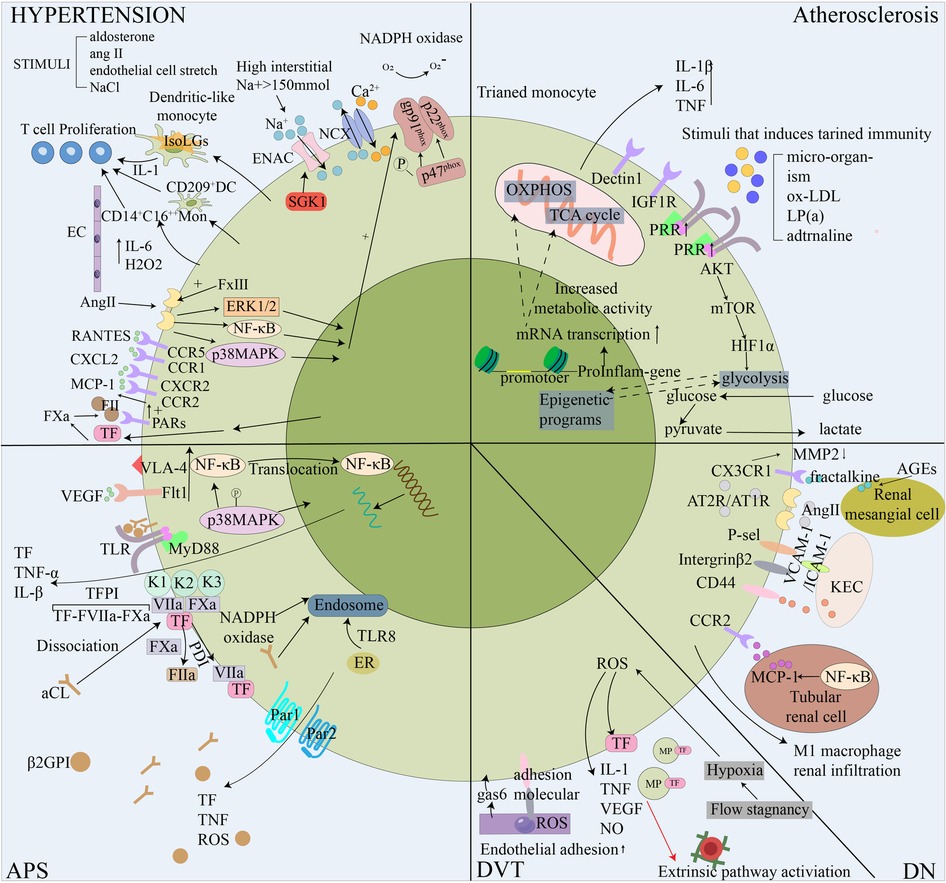
Figure 3. Schematic diagram of the role of monocytes in five thrombotic diseases. In response to the hypertensive stimulant, monocytes transform and activate, release hypertensive cytokines and differentiate into dendritic cells to promote T cell proliferation through the effects of the vascular system, kidney and sympathetic nervous system, elevateing blood pressure, thus increasing the risk of blood vessel rupture. Monocytes are involved in atherosclerosis by training immune processes. This is mediated by epigenetic and metabolic reprogramming. By enhancing cell-related endometabolic pathways, monocytes can develop a long-lasting proinflammatory phenotype. In APS, the anti-β2gpi antibody binds to phospholipid-binding protein β2GPI and acts on monocytes to trigger the Toll-like receptor 4-myeloid differentiation primary response 88 (TLR-4-MyD88) signaling pathway. In addition, aPL triggers coagulation and inflammatory signals by dissociating suppressed TF cell surface complexes. TFPI expressed in myeloid cells specifically supports aPL-induced thrombosis. The hypoxic environment of DVT stimulates the expression of TF and some pro-inflammatory cytokines in monocytes, initiates the exogenous coagulation pathway, activates ECs and promotes the recruitment of monocytes. In the diabetic environment, the accumulation of advanced glycation end products (AGEs) increases the expression of fractalkine in human renal mesangial cells, and the interaction between monocytes and human renal mesangial cells promotes DN inflammation through MMP2 and fractalkine. Ang-II is over-produced and stimulated in the diabetic kidney, the adhesion of monocytes to ECs is enhanced, and M1 macrophages infiltrate the kidney under the regulation of the RAS system. AGEs, Advanced glycation end products; Ang II, Angiotensin II; aPL, antiphospholipid antibody; APS, Antiphospholipid syndrome; AT1R, angiotensin type 1 receptor; β2GPI, beta2glycoprotein I; DC, dendritic cells; DN, Diabetic nephropathy; DVT, Deep vein thrombosis; ENAC, epithelial sodium channel; ER, Endoplasmic reticulum; gas6, growth arrest specific 6; KEC, Kidney endothelial cell; LP(a), Lipoprotein a; IGF1R, insulin like growth factor 1 receptor;MCP-1, monocyte chemotactic protein-1; MMP, matrix metalloproteinases; MP, microparticles; NCX, Sodium-calcium exchangers; NF-κB, nuclear factor κB; NOX-2, NADPH oxidase-2; OXPHOS, oxidative phosphorylation; PARs, protease activated receptors; PRR, pattern recognition receptor; RAS, renin-angiotensin system; ROS, reactive oxygen species; TCA, tricarboxylic acid; TF, tissue factor; TFPI, TF pathway inhibitor; TLR, toll-like receptors; VEGF, vascular endothelial growth facto;VLA-4,very late antigen 4.
Co-culture of APS monocytes with LPS increased the expression of IL-1β, IL-6, IL-23, TLR2, CCL2, and CXCL10 genes to improve the sensitivity to LPS and contribute to the formation of thrombosis, but the response was weak in healthy donor cells (194). Reduced levels of ATP result in decreased inflammation among APS monocytes by blocking unidentified signals stimulated by LPS and inhibiting ATP-mediated production of IL-1 through the inflammasome and IL-10 (194, 195). Immunomodulation of plasma exchange reversed the expression of IL-1 β, IL-6, IL-23, CCL2, P2X7, and TNF-α in APS monocytes, with mRNA expression levels returning to the normal range (196). For secondary thromboembolism in APS, it is generally accepted that direct oral anticoagulants can prevent this from occurring. There are currently many anticoagulant drugs of choice in clinical practice, such as DOAC rivaroxaban and warfarin drugs. Compared with warfarin, DOAC rivaroxaban not only provides anticoagulation but also reduces the frequency of bleeding events and does not require patient diet control (197).
Atherosclerosis is a slowly progressing condition of the large and medium-sized arteries, characterized by the gradual build-up of plaques over time. Thrombotic complications of atherosclerotic diseases, such as ACS, occur suddenly and often without warning signs. The long-term process of atherosclerotic plaque initiation and formation is thought to be accelerated by different risk factors, including traditional factors such as smoking, hypertension as mentioned above, hyperglycemia, and severe hyperlipidemia (198). It is increasingly believed that environmental factors, such as air pollution, noise, sleep disorders and stress, contribute to atherosclerotic events in part through the activation of inflammatory pathways (199). The inflammatory mechanisms run through the whole process of atherosclerosis formation, and mononuclear macrophages play a key role. In early atherosclerosis, LDL remains in the intima of blood vessels, mediated by oxidase, lipolysis, proteolytic enzymes, and ROS (200). A variety of danger-associated molecular patterns (DAMP) are modified to obtain immunogenicity. Immunogenic LDL activates vascular ECs and mobilizes immune cells (mainly monocytes and T cells) to inflammatory tissues, mediated by locally produced chemokines, as previously described in the context of arterial thrombosis. Chemokines (CCR2, CCR5, and CX3CR1) bind to receptors on monocytes to promote migration to tissues. When three chemokine receptors, CCR2, CCR5 and CX3CR1 were blocked, atherosclerosis in hypercholesterolemic mice was virtually eliminated by inhibiting the aggregation of monocytes into inflammatory tissue (201). This suggests that all monocyte subsets are involved in the formation of atherosclerosis.
The atherosclerotic process may be a training immune process (Figure 3). Studies have shown that monocytes, as innate immune cells, can form features of immune memory after brief exposure to microorganisms (202). Unlike the specific memory of adaptive immune cells, trained immunity is characterized by its nonspecificity and ability to induce a long-lasting proinflammatory phenotype in monocytes/macrophages through epigenetic reprogramming, particularly at the level of histone methylation (202). In addition, non-microbial endogenous stimuli can also induce trained immunity, and current studies include lipoproteins and adrenal hormones, hyperglycemia, and other factors that contribute to the development of atherosclerotic cardiovascular disease (ASCVD) (203, 204). Monocytes immunized by Ox-LDL training are induced through the Akt-mTOR-HIF-1α pathway, and intracellular metabolic glycolysis and oxidative phosphorylation are up-regulated, which is an important pathway to promote proinflammatory cytokines (205–207). Pharmacological inhibition of the mTOR pathway and related signaling molecules, as well as inhibition of 2-deoxyglucose glycolysis, prevents glycolysis, ROS formation, and proinflammatory initiation in monocytes/macrophages (206, 207). A recent study in 243 healthy volunteers showed that oxLDL-induced training immunity led to changes in the balance of steroid hormones within monocytes. Progesterone inhibits oxLDL-induced training immunity through ribocorticosteroid receptors and minerocorticoid receptors, an effect that may help reduce CVD risk in premenopausal women (208).
In addition to the promotion of cytokine proliferation, macrophages induced by Ox-LDL showed high expression of Ox-LDL recognition receptors CD36 and SR-A and low expression of anticholesterol receptors ABCAI and ABCG1, thus enabling macrophages to have a stronger lipid absorption capacity (209). These processes accelerate the formation of atherosclerosis. Differentiated macrophages, as key cells in atherosclerosis, are transformed into foam cells, and subsequent increase in cholesterol load over time causes the triggering of unfolded protein responses in the ER, from intracellular crystallization to precipitation and activation of inflammasomes, ultimately leading to programmed cell death (210). Foam cells accumulate to form an atherosclerotic core. The properties of lipid material, cholesterol crystallization, accumulation of foam cell fragments to form an atherosclerotic core, and ensuing thinning of the fibrous cap result in a high degree of thrombosis (211, 212). Interestingly, the investigators discovered that neovascularization was prevalent in atherosclerotic plaques, and highly neovascularized plaques were more susceptible to rupturing than their stable counterparts. Matrix metalloproteinases secreted by monocytes/macrophages were responsible for this phenomenon by deteriorating and redesigning the ECM and activating or disintegrating growth factors. As a result, stable atherosclerotic lesions may become unsteady high-risk plaques due to intraplaque hemorrhage.
Macrophages were thought to be major players in the formation of atherosclerosis as well as thrombotic complications. Although the connection between atherosclerosis and immunity in humans is constantly evolving, some of our understanding of this relationship comes from research conducted on animal models of CVD. However, due to inherent biological, genomic, and environmental differences, the results obtained therein cannot be fully transferable to the human environment. Potential therapeutic approaches against atherosclerosis include targeting monocytes/macrophages recruitment, polarization, cytokine profiling, ECM remodeling, cholesterol metabolism, oxidative stress, inflammatory activity, and non-coding RNA. Besides, folic acid can restore hyperlipidemia (HL) and hyperhomocysteinemia (HHcy)-mediated aberrant DNA methylation and decreased ARID5B expression, thereby inhibiting atherosclerotic plaque formation by reducing the proportion of intermediate monocytes (213).
In recent years, there has been significant interest in understanding the role of monocytes and monocyte-derived extracellular vesicles (MDEVs) in the major risk factors for atherosclerosis (214, 215). Several studies have reported a significant association between monocytes and obesity, a major risk factor for atherosclerosis (216, 217). These studies have shown that obesity leads to increased levels of chemokines and cytokines, promoting monocyte recruitment and activation. It also results in increased oxidative stress and inflammation, leading to MDEV release (218). MDEVs have been shown to exert pathogenic effects by promoting endothelial dysfunction and a pro-inflammatory state, exacerbating atherosclerosis (219). In addition, in a clinical trial of obese women without other cardiovascular risk factors, the researchers found that circulating procoagulant microparticles were increased in obese patients compared with healthy controls, and obese patients had an increased risk of thrombosis, suggesting that obesity as a major risk factor for atherosclerosis is associated with the release of MDEVs (220).
Diabetes is another major risk factor for atherosclerosis, with chronic hyperglycemia leading to increased oxidative stress and inflammation (221). High glucose levels induce monocyte activation, leading to MDEV release, and increased endothelial adhesion molecules, resulting in a pro-inflammatory and pro-thrombotic environment (222). The concentration of MDEVs in plasma of patients with type 2 diabetes was much higher than that of healthy individuals, and the amount of MDEVs was positively correlated with platelet activation markers (223, 224). Furthermore, MDEVs have been shown to induce endothelial dysfunction and increase monocyte and macrophage recruitment, aggravating atherosclerosis in diabetes (219).
Hypertension is another major risk factor for atherosclerosis, and monocytes are involved in its pathogenesis. A study showed that shear stress induces monocyte recruitment, activation and migration, promoting inflammation and neointimal formation (225). MDEVs have also been implicated in hypertension. In a clinical trial of 359 hypertensive patients, investigators observed increases in MDEVs and sVCAM-1 regardless of their presence or absence of diabetes (226). In another study, the expression of MDEVs and platelet activation markers (CD62P, CD63, PAC-1, and annexin V) was much higher in hypertensive patients than in healthy individuals, suggesting that hypertensive patients are more prone to atherosclerosis (227).
Hypercholesterolemia is also a cause of atherosclerosis. In response to high levels of cholesterol, monocytes can infiltrate the arterial wall and differentiate into macrophages, which can take up and accumulate lipids, leading to the formation of foam cells and ultimately the development of atherosclerotic plaques (228). Although the exact mechanism is not yet fully understood, recent studies suggest that MDEVs may also be involved in the development of atherosclerosis, and secretion of MDEVs by monocytes can activate endothelial cells and other immune cells and promote inflammation and further atherosclerotic plaque formation (229). Clinical trials have found that elevated oxidized LDL cholesterol in hypercholesterolemia induces increased release of MDEVs (230). MDEVs appear to be markers for the diagnosis of atherosclerotic plaques in patients with hypercholesterolemia, and the number of circulating microparticles before thrombosis can indicate atherosclerotic plaques in the subclinical stage of patients with hypercholesterolemia (231–233).
Other risk factors for atherosclerosis, such as smoking, aging, and dyslipidemia, have been associated with monocyte activation and MDEV release. Smoking induces oxidative stress and inflammation, leading to increased monocyte activation, and MDEV release (234). In cell experiments simulating smoking environments, tobacco smoke-exposed monocytes/macrophages have been found to induce biologically active procoagulant MDEVs in cells, which may be associated with activation of JNK, p38, ERK, and MAPK (235). Aging is a primary risk factor for atherosclerosis, characterized by declines in tissue and cell functions resulting in increased inflammation and oxidative stress, leading to MDEV release (236). Dyslipidemia, characterized by increased levels of LDL cholesterol, induces monocyte activation and lipid accumulation in macrophages, leading to foam cell formation and atherosclerosis (237). In addition, IL-33 acts as a pro-inflammatory factor, induces TF expression in monocytes, releases procoagulant MDEVs, and ultimately forms a prothrombotic state of atherosclerosis (238).
In summary, monocytes and monocyte-derived extracellular vesicles play a crucial role in the pathogenesis of atherosclerosis, and different major risk factors for the disease have been associated with their activation and release (239). Obesity, diabetes, hypertension, smoking, aging, and dyslipidemia all promote monocyte activation and MDEV release, leading to increased inflammation, oxidative stress, and endothelial dysfunction, culminating in atherosclerosis. A better understanding of the role of monocytes and MDEVs in atherosclerosis could lead to novel therapeutic interventions targeting their recruitment and activation, potentially preventing or slowing the progression of this debilitating disease.
RHD is a condition caused by damage to the heart valves from rheumatic fever (RF), which is an autoimmune response to untreated streptococcal throat infections (240). Left atrial (LA) thrombosis is a common complication with RHD and mitral stenosis (MS) (241). As early as 1991, the luminol-enhanced chemiluminescence technique was used to study the production of oxygen free radicals (OFR) by monocytes and neutrophils in the peripheral blood of patients with RF and RHD (242). It has been observed that in RHD patients, phagocytes are capable of penetrating the myocardium and possibly contributing to the development of cardiac injury by generating OFR (242). In a clinical study of mitral valve resection in patients with rheumatic MS, histological studies have revealed a positive correlation between the degree of infiltration of inflammatory cells in valve tissue and the levels of MMP-1 and IFN-γ. In addition, in an in vitro cell assay, IFN-γ was found to increase MMP-1 expression in monocytes (243). The risk of embolus dislodging and causing thromboembolism may increase due to the easy tearing of valve tissue resulting from the thickening of collagen and structural abnormalities in the valve tissue caused by inflammatory cell infiltration.
The expression of inflammatory cells and inflammatory genes in the aortic valve of female RHD patients was significantly higher than that of male patients. More monocytes and macrophages infiltrate into the aortic valve to produce IFNγ and IL8, which as major Th1 cytokines, participate in the positive feedback between macrophages and Th1 cells to accelerate the secretion of pro-inflammatory cytokines. The NFKB pathway plays a crucial role in enhancing the autoimmune response of the aortic valve in female RHD patients by increasing mononuclear/macrophage infiltration and pro-inflammatory cytokine (especially Th1 cytokine) levels. Upon activation, phosphorylated p65 enters the nucleus and triggers the transcription of inflammatory genes, which further exacerbates the inflammation of the damaged aortic valve (244).
RF is a complex inflammatory autoimmune disease that involves the interaction of multiple pro-inflammatory agents produced by activated neutrophils and macrophages, resulting in a synergistic pathological effect (245). In individuals with rheumatic mitral stenosis and atrial fibrillation, pro-inflammatory M1 macrophages are primarily observed in the atria of those with mitral stenosis, atrial fibrillation, and thrombosis. The inflammatory effect of M1 cells may be related to the up-regulation of eNOS expression. In addition, the polarization of M1 macrophages towards M2 is inhibited due to the down-regulation of M-CSF (246).
DVT is a kind of thrombotic disease. It refers to venous reflux disorders caused by abnormal coagulation of blood in deep veins (247). It often occurs in the lower limbs. It is a common and serious complication of hospitalized patients, especially those after major surgery and long-term bedridden patients, with high incidence and serious consequences. Detachment of the thrombus can cause PE, and both are collectively referred to as VTE.
The mononuclear/macrophage system is essential in both the formation and resolution of DVT. In mice with venous thrombosis, inflammatory bodies are activated, triggering venous thrombosis through pyrodeath and TFs released by inflammatory monocytes and macrophages (248). Lack of caspase-1 but not caspase-11 protects mice from venous thrombosis. The monocyte signaling pathway, key enzymes and thrombolytic regulators regulate the occurrence of DVT, which can be used as a molecular target network to study the treatment of DVT. Protein tyrosine kinases participate in the activation of monocytes and ECs. For example, tyrosine kinase PYK2 regulates platelet activity and TF expression by stimulating monocytes and ECs, and is involved in the regulation of natural immunity and inflammation (249). In a mouse experiment, the number of inflammatory Ly6Chi monocytes controls DVT formation, growth, and regression, and Nur77 agonist may be an ideal candidate for therapeutic intervention with inflammatory monocyte activity in patients with DVT to prevent thrombosis growth and accelerate regression (250). Inhibit the formation of Tbet+ interleukin-12 from bone marrow monocytes and accelerate thrombus regression (251). Modulating inflammatory immune responses may be a way to improve DVT treatment. The effects of MMP-9 on the loss of compliance during thrombolysis and the biomechanics of vein walls contribute to the development of specific molecular therapies for DVT. With the age of thrombosis, IFN-γ is mainly infiltrated by macrophages, and IFN-γ/STAT1 signaling pathway is activated to negatively regulate the expression of Mmp-9 and Vegf genes in PMA-induced macrophages (252). The use of anti-IFN-γ monoclonal antibody can accelerate the resolution of DVT. Recombinant human granulocyte colony-stimulating factor (rhG-CSF) not only mobilized monocyte lineage cells into peripheral blood but also induced higher expression of CCR2 protein, thus enhancing the regression and recalculation of venous thrombosis (253). Circulating monocytes and monocyte-derived macrophages in patients with idiopathic DVT exhibit significantly increased M1 polarization, which can significantly up-regulate the expression of endothelial cell adhesion molecules (134). The thrombus targeting adenovirus uPA (ad-uPA) gene transduction of human blood monocyte-derived macrophages (HBMMs) increases their fibrinolytic activity. In an experimental model of venous thrombosis, systematic administration of uPA up-regulated HBMMs reduced the size of the thrombus (254). Alternatives to delivering fibrinolytic agents are worth exploring. Loss of prostag-landin-endoperoxide synthase (PTGS) alters the natural distribution of ANXA2 in mononuclear/macrophages, increases TF expression and activity, and leads to venous thrombosis. Targeting PTGI2/ANX2/TF pathways, such as treatment with cabpaprost, inhibits nuclear ANXA2 transport, controls monocyte TF activity, and prevents the occurrence of DVT (255).
DN has emerged as a pervasive renal disease, causing end-stage renal failure on a global scale (256). As one of the most serious and harmful chronic complications caused by diabetes, DN is one of the manifestations of diabetic systemic microangiopathy (257). DN has a prethrombotic state and obvious endothelial dysfunction (258). With the increase of urinary protein and the progression of the disease, it is easy to appear hypercoagulation, and then thrombosis.
Inflammatory cell infiltration is a consistent feature of the early stages of DN, particularly the influx of mononuclear cells into the affected tissues and the infiltration of macrophages into the kidney (259, 260). This influx is thought to be due to the activation of innate immunity accompanied by the development of a chronic low-grade inflammatory response, but may also be related to differences in monocyte phenotype and function. Experiments showed that diabetes increased the number of monocytes and had no effect on the total number of white blood cells. At the same time, the number of CD14+CD16++ NCMs was significantly reduced by diabetic status, but only in patients with diabetic complications (261). The circulating monocytes phenotype can be changed by diabetic complication status. Changes in monocyte activation and function have also been reported in inflammatory pathways in diabetes, such as increased monocyte adhesion to ECs and enhanced TLR signaling pathway conduction in monocytes (262). Mouse data showed that monocyte CD163 and sCD163 changes predate DN. CD163+ monocytes with a high circulating proportion have anti-inflammatory effects and may protect against diabetes complications (263).
Macrophage infiltration and activation are evident in the renal biopsies of diabetic animal models and patients with DN (264–266). Therefore, monocytes need to be activated and migrate from the circulation to differentiate and infiltrate the renal mesangium. Under the influence of high glucose, monocyte adhesion, migration, differentiation and MMP expression can be enhanced by increasing the secretion of pro-inflammatory cytokines in mesangial cells, and thus MMPs can mediate the degradation of ECM (260, 267). Mesangial cell-monocyte interactions are important to activate monocyte migration from circulation to the kidney in early DN. The pro-inflammatory cytokines TNF-α and IL-6 can induce the expression and adhesion of MMP-9 in monocytes (260). Moreover, the interaction between monocytes and human renal mesangial cells (HRMCs) is also through MMP-2 and fractalkine. In the Monocyte-HRMC co-culture system, AGEs not only directly down-regulated MMP-2, but also indirectly down-regulated the expression and activity of MMP-2 through up-regulated fractalkine, thus increasing the expression of fractalkine, which is mainly used as chemotactic agent (268) (Figure 3). The presence of such crosstalk between monocytes and HRMCs has been interpreted to be related to abnormal deposition of ECM and migration of monocytes into tissues.
Diabetes can induce renal MCP-1 production, and serum and urinary MCP1 levels are elevated in early DN, which can be used as markers and possible mediators of early DN, as well as to evaluate renal inflammation in diabetic patients (269, 270). Increased expression of MCP-1/CCL2 in the kidney is considered a crucial contributor to the initiation of monocyte recruitment in the tubulointerstitium (271, 272). Exposure to MCP-1 and high-glucose environments induces infiltration and activation of macrophages, resulting in the release of ROS, profibrotic growth factor (TGF-β, PDGF) and pro-inflammatory cytokines (IL-1, TNF-α, MCP-1) (272–274). The amplified inflammatory response mediates parenchymal cell injury and death leading to decreased renal function, and myofibroblast proliferation promotes renal fibrosis. These tandem processes together promote the progression of DN, lead to the development of kidney failure and cause vascular dysfunction. Inhibition of MCP-1 in DN shows strong therapeutic potential by reducing persistent proteinuria. Spiegelmer® emapticap pegol (NOX-E36) continued to restore glomerular endothelial glycocalyx and barrier function, decreased CCR2-expressing Ly6Chi monocytes in peripheral blood, and polarization of tissue macrophages toward anti-inflammatory phenotypes (275). Activation of the renin-angiotensin system (RAS) is a characteristic manifestation of DN (276).
Activation of RAS is pro-inflammatory, and pro-fibrotic and enhances oxidative stress. By increasing the production of MCP-1 and osteopontin in renal tubule cells, it facilitates the adhesion of monocytes to renal ECs and thus their entry into the kidney. Under the activation of various proinflammatory cytokines and RAS, macrophages differentiate into M1 proinflammatory phenotypes, leading to the development of DN. Under the antagonistic drugs of RAS, the phenotypic transformation of macrophages and the number of infiltrates can be improved, thus reducing proteinuria and correcting renal disorders (277, 278). Furthermore, recent studies have shown that signal communication between macrophages and renal tubular epithelial cells (TECs) can be used as targets in the treatment of DN. For example, the exosome/miR19b-3p/SOCS1 axis mediates the communication between injured TECs and macrophages, leading to the activation of M1 macrophages (279). During DN progression, IRG1 enrichment, lipotoxic tec derived EVe activates macrophages through TGFβR1-dependent pathways and subsequently up-regulates the expression of many inflammatory genes, thereby inducing inflammation and damage in DN. Meanwhile, the LRG1/TGFβR1 signaling pathway can also increase the expression of TRAIL in macrophages, and the macrophage-derived EVs enriched with TRAIL can promote the apoptosis of TECs, acting as a feedback loop in DN (280). Proteinuria induces glycolysis in renal macrophages by stabilizing HIF-1α through tubular epithelial-derived EVs (281). These targets may be some important links to subsequent regulation, which needs further in-depth study.
Indicators related to monocytes can be an important predictor of DN, and increased Monocyte-lymphocyte ratio (MLR) is significantly associated with the risk of DN (282). Higher monocyte to high-density lipoprotein cholesterol ratio (MHR) levels for patients with DN compared to others (283).
Monocytes undoubtedly have a significant role in the pathophysiology of various thrombotic diseases. Compared with tissue macrophages, circulating monocytes are easier to detect and characterize. As the medical field advances into the 21st century, with an augmented emphasis on forecasting and averting future acute phenomena, the enumeration and functionality of monocytes and their respective subsets have the potential to serve as an alluring biomarker. Nevertheless, crucial parameters of monocytes that have the capability to act as prognostic indicators and targeted therapies necessitate identification and authentication.
While monocytes have been implicated in the link between inflammation and the pro-thrombotic state, their potential use as disease biomarkers or therapeutic agents in thrombosis is still an area of ongoing research. While there have been some promising findings, there are also challenges associated with the use of monocytes in these contexts. For example, monocyte populations are heterogeneous, and their pro-thrombotic or anti-thrombotic properties may vary depending on their differentiation state and activation status. Additionally, the interaction between monocytes and other immune cells, such as platelets and neutrophils, can further complicate the picture. Therefore, while the role of monocytes in thrombotic disease is an important area of investigation, it may be premature to consider them as biomarkers or therapeutic agents without further research and clinical validation. Further studies on the specific molecular mechanisms underlying the association between monocyte activation and thrombosis could inform the potential development of new targeted interventions for thrombotic disease.
In these thrombotic diseases, we found that each disease can also be closely related to each other, and monocytes act as an indispensable part as a close link in this network. Clarifying the role of monocytes in thrombotic diseases is helpful to prevent and diagnose thrombotic diseases, to reduce the incidence and mortality of thrombotic diseases.
ZH, LY, and HC were involved in the conception of the study. ZH, QL, HL, MZ and LY were involved in writing the article. CH, LY, YL, KW, QG, ZW, SZ and QG critically revised the manuscript. All authors contributed to the article and approved the submitted version.
The work was supported by Chengdu University of Traditional Chinese Medicine undergraduate research practice innovation project (ky-2023015; ky-2023081; ky-2023083).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schafer AI, Levine MN, Konkle BA, Kearon C. Thrombotic disorders: diagnosis and treatment. Hematology Am Soc Hematol Educ Program. (2003) 1:520–39. doi: 10.1182/asheducation-2003.1.520
2. Muñoz Tovar RA, Alvarez Perdomo LC, Rojas Molina SM, Salazar SJ. Submassive pulmonary thromboembolism in a patient with thrombocytopenia: therapeutic challenge. Case Rep Crit Care. (2019) 2019:1919401. doi: 10.1155/2019/1919401
3. GBD 2017 SDG Collaborators: Lozano R, Fullman N, Abate D, Abay SM, Abbafati C, Abbasi N, et al. Measuring progress from 1990 to 2017 and projecting attainment to 2030 of the health-related sustainable development goals for 195 countries and territories: a systematic analysis for the global burden of disease study 2017. Lancet. (2018) 392(10159):2091–138. doi: 10.1016/s0140-6736(18)32281-5
4. Boyd AN. An inflammatory basis for coronary thrombosis. Am J Pathol. (1928) 4(2):159–66.5.19969785
5. Wolf AA, Yáñez A, Barman PK, Goodridge HS. The ontogeny of monocyte subsets. Front Immunol. (2019) 10:1642. doi: 10.3389/fimmu.2019.01642
6. Trzebanski S, Jung S. Plasticity of monocyte development and monocyte fates. Immunol Lett. (2020) 227:66–78. doi: 10.1016/j.imlet.2020.07.007
7. Liu Z, Gu Y, Chakarov S, Bleriot C, Kwok I, Chen X, et al. Fate mapping via ms4a3-expression history traces monocyte-derived cells. Cell. (2019) 178(6):1509–25.e19. doi: 10.1016/j.cell.2019.08.009
8. Mills CD. M1 and m2 macrophages: oracles of health and disease. Crit Rev Immunol. (2012) 32(6):463–88. doi: 10.1615/critrevimmunol.v32.i6.10
9. Liu S, Chen J, Shi J, Zhou W, Wang L, Fang W, et al. M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic Res Cardiol. (2020) 115(2):22. doi: 10.1007/s00395-020-0781-7
10. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory m2, but not pro-inflammatory m1 macrophages promote angiogenesis in vivo. Angiogenesis. (2014) 17(1):109–18. doi: 10.1007/s10456-013-9381-6
11. Swirski FK. The spatial and developmental relationships in the macrophage family. Arterioscler Thromb Vasc Biol. (2011) 31(7):1517–22. doi: 10.1161/atvbaha.110.221150
12. Merino A, Buendia P, Martin-Malo A, Aljama P, Ramirez R, Carracedo J. Senescent CD14+CD16+ monocytes exhibit proinflammatory and proatherosclerotic activity. J Immunol. (2011) 186(3):1809–15. doi: 10.4049/jimmunol.1001866
13. Zawada AM, Rogacev KS, Rotter B, Winter P, Marell RR, Fliser D, et al. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood. (2011) 118(12):e50–61. doi: 10.1182/blood-2011-01-326827
14. Thomas G, Tacke R, Hedrick CC, Hanna RN. Nonclassical patrolling monocyte function in the vasculature. Arterioscler Thromb Vasc Biol. (2015) 35(6):1306–16. doi: 10.1161/atvbaha.114.304650
15. Kologrivova I, Suslova T, Koshelskaya O, Trubacheva O, Haritonova O, Vinnitskaya I. Frequency of monocyte subsets is linked to the severity of atherosclerosis in patients with ischemic heart disease: a case-control study. Biomedicine (Taipei). (2020) 10(2):36–47. doi: 10.37796/2211-8039.1015
16. Shantsila E, Ghattas A, Griffiths HR, Lip GYH. Mon2 predicts poor outcome in st-elevation myocardial infarction. J Intern Med. (2019) 285(3):301–16. doi: 10.1111/joim.12847
17. Gao C, Ge L, Chen D, Zhang M, Zhao L, Liu W, et al. Increased frequency of circulating classical monocytes in patients with rosacea. Clin Cosmet Investig Dermatol. (2021) 14:1629–36. doi: 10.2147/ccid.S336194
18. Krychtiuk KA, Lenz M, Richter B, Hohensinner PJ, Kastl SP, Mangold A, et al. Monocyte subsets predict mortality after cardiac arrest. J Leukoc Biol. (2021) 109(6):1139–46. doi: 10.1002/jlb.5a0420-231rr
19. Allen N, Barrett TJ, Guo Y, Nardi M, Ramkhelawon B, Rockman CB, et al. Circulating monocyte-platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis. (2019) 282:11–8. doi: 10.1016/j.atherosclerosis.2018.12.029
20. Hui H, Fuller KA, Erber WN, Linden MD. Imaging flow cytometry in the assessment of leukocyte-platelet aggregates. Methods. (2017) 112:46–54. doi: 10.1016/j.ymeth.2016.10.002
21. Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, et al. Cell type-specific roles of nf-κb linking inflammation and thrombosis. Front Immunol. (2019) 10:85. doi: 10.3389/fimmu.2019.00085
22. Brown RA, Lip GYH, Varma C, Shantsila E. Impact of mon2 monocyte-platelet aggregates on human coronary artery disease. Eur J Clin Invest. (2018) 48(5):e12911. doi: 10.1111/eci.12911
23. Kamińska J, Lisowska A, Koper-Lenkiewicz OM, Mikłasz P, Grubczak K, Moniuszko M, et al. Differences in monocyte subsets and monocyte-platelet aggregates in acute myocardial infarction-preliminaryresults. Am J Med Sci. (2019) 357(5):421–34. doi: 10.1016/j.amjms.2019.02.010
24. Pfluecke C, Tarnowski D, Plichta L, Berndt K, Schumacher P, Ulbrich S, et al. Monocyte-platelet aggregates and cd11b expression as markers for thrombogenicity in atrial fibrillation. Clin Res Cardiol. (2016) 105(4):314–22. doi: 10.1007/s00392-015-0922-4
25. Klopf J, Brostjan C, Eilenberg W, Neumayer C. Neutrophil extracellular traps and their implications in cardiovascular and inflammatory disease. Int J Mol Sci. (2021) 22(2):559. doi: 10.3390/ijms22020559
26. Chen Z, Zhang H, Qu M, Nan K, Cao H, Cata JP, et al. Review: the emerging role of neutrophil extracellular traps in sepsis and sepsis-associated thrombosis. Front Cell Infect Microbiol. (2021) 11:653228. doi: 10.3389/fcimb.2021.653228
27. Granger V, Faille D, Marani V, Noël B, Gallais Y, Szely N, et al. Human blood monocytes are able to form extracellular traps. J Leukoc Biol. (2017) 102(3):775–81. doi: 10.1189/jlb.3MA0916-411R
28. Han T, Tang H, Lin C, Shen Y, Yan D, Tang X, et al. Extracellular traps and the role in thrombosis. Front Cardiovasc Med. (2022) 9:951670. doi: 10.3389/fcvm.2022.951670
29. Haritha VH, Seena P, Shaji BV, Nithin TU, Hazeena VN, Anie Y. Monocyte clearance of apoptotic neutrophils is unhindered in the presence of netosis, but proteins of net trigger etosis in monocytes. Immunol Lett. (2019) 207:36–45. doi: 10.1016/j.imlet.2019.02.001
30. Cerletti C, de Gaetano G, Lorenzet R. Platelet—leukocyte interactions: multiple links between inflammation, blood coagulation and vascular risk. Mediterr J Hematol Infect Dis. (2010) 2(3):e2010023. doi: 10.4084/mjhid.2010.023
31. Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. (2015) 107(3):321–30. doi: 10.1093/cvr/cvv147
32. Vallania F, Zisman L, Macaubas C, Hung SC, Rajasekaran N, Mason S, et al. Multicohort analysis identifies monocyte gene signatures to accurately monitor subset-specific changes in human diseases. Front Immunol. (2021) 12:659255. doi: 10.3389/fimmu.2021.659255
33. Cormican S, Griffin MD. Human monocyte subset distinctions and function: insights from gene expression analysis. Front Immunol. (2020) 11:1070. doi: 10.3389/fimmu.2020.01070
34. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. (2014) 14(6):392–404. doi: 10.1038/nri3671
35. Italiani P, Mosca E, Della Camera G, Melillo D, Migliorini P, Milanesi L, et al. Profiling the course of resolving vs. persistent inflammation in human monocytes: the role of il-1 family molecules. Front Immunol. (2020) 11:1426. doi: 10.3389/fimmu.2020.01426
36. Meierovics AI, Cowley SC. Mait cells promote inflammatory monocyte differentiation into dendritic cells during pulmonary intracellular infection. J Exp Med. (2016) 213(12):2793–809. doi: 10.1084/jem.20160637
37. Stansfield BK, Ingram DA. Clinical significance of monocyte heterogeneity. Clin Transl Med. (2015) 4:5. doi: 10.1186/s40169-014-0040-3
38. Mandel J, Casari M, Stepanyan M, Martyanov A, Deppermann C. Beyond hemostasis: platelet innate immune interactions and thromboinflammation. Int J Mol Sci. (2022) 23(7):3868. doi: 10.3390/ijms23073868
39. Newman SL, Devery-Pocius JE, Ross GD, Henson PM. Phagocytosis by human monocyte-derived macrophages. Independent function of receptors for c3b (cr1) and ic3b (cr3). Complement. (1984) 1(4):213–27. doi: 10.1159/000467840
40. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. (2010) 116(16):e74–80. doi: 10.1182/blood-2010-02-258558
41. Zawada AM, Rogacev KS, Rotter B, Winter P, Marell RR, Fliser D, et al. Supersage evidence for cd14++cd16+ monocytes as a third monocyte subset. Blood. (2011) 118(12):e50–61. doi: 10.1182/blood-2011-01-326827
42. Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. (2011) 118(5):e16–31. doi: 10.1182/blood-2010-12-326355
43. Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK, et al. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J Pathol. (2015) 185(10):2596–606. doi: 10.1016/j.ajpath.2015.06.001
44. Thomas G, Tacke R, Hedrick CC, Hanna RN. Nonclassical patrolling monocyte function in the vasculature. Arterioscler Thromb Vasc Biol. (2015) 35(6):1306–16. doi: 10.1161/atvbaha.114.304650
45. Ong SM, Hadadi E, Dang TM, Yeap WH, Tan CT, Ng TP, et al. The pro-inflammatory phenotype of the human non-classical monocyte subset is attributed to senescence. Cell Death Dis. (2018) 9(3):266. doi: 10.1038/s41419-018-0327-1
46. Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, et al. Human cd14dim monocytes patrol and sense nucleic acids and viruses via tlr7 and tlr8 receptors. Immunity. (2010) 33(3):375–86. doi: 10.1016/j.immuni.2010.08.012
47. Witten A, Martens L, Schäfer AC, Troidl C, Pankuweit S, Vlacil AK, et al. Monocyte subpopulation profiling indicates cdk6-derived cell differentiation and identifies subpopulation-specific mirna expression sets in acute and stable coronary artery disease. Sci Rep. (2022) 12(1):5589. doi: 10.1038/s41598-022-08600-7
48. Mazzone A, De Servi S, Mazzucchelli I, Fossati G, Gritti D, Canale C, et al. Increased expression of cd11b/cd18 on phagocytes in ischaemic disease: a bridge between inflammation and coagulation. Eur J Clin Invest. (1997) 27(8):648–52. doi: 10.1046/j.1365-2362.1997.1610710.x
49. Kashiwagi M, Imanishi T, Ozaki Y, Satogami K, Masuno T, Wada T, et al. Differential expression of toll-like receptor 4 and human monocyte subsets in acute myocardial infarction. Atherosclerosis. (2012) 221(1):249–53. doi: 10.1016/j.atherosclerosis.2011.12.030
50. Dopheide JF, Obst V, Doppler C, Radmacher MC, Scheer M, Radsak MP, et al. Phenotypic characterisation of pro-inflammatory monocytes and dendritic cells in peripheral arterial disease. Thromb Haemost. (2012) 108(6):1198–207. doi: 10.1160/th12-05-0327
51. Williams H, Mack CD, Li SCH, Fletcher JP, Medbury HJ. Nature versus number: monocytes in cardiovascular disease. Int J Mol Sci. (2021) 22(17):9119. doi: 10.3390/ijms22179119
52. Thaler B, Hohensinner PJ, Krychtiuk KA, Matzneller P, Koller L, Brekalo M, et al. Differential in vivo activation of monocyte subsets during low-grade inflammation through experimental endotoxemia in humans. Sci Rep. (2016) 6:30162. doi: 10.1038/srep30162
53. Xiang Y, Liang B, Zhang X, Zheng F. Lower hdl-c levels are associated with higher expressions of cd16 on monocyte subsets in coronary atherosclerosis. Int J Med Sci. (2020) 17(14):2171–9. doi: 10.7150/ijms.47998
54. Afanasieva OI, Filatova AY, Arefieva TI, Klesareva EA, Tyurina AV, Radyukhina NV, et al. The association of lipoprotein(a) and circulating monocyte subsets with severe coronary atherosclerosis. J Cardiovasc Dev Dis. (2021) 8(6):63. doi: 10.3390/jcdd8060063
55. Cappellari R, D'Anna M, Bonora BM, Rigato M, Cignarella A, Avogaro A, et al. Shift of monocyte subsets along their continuum predicts cardiovascular outcomes. Atherosclerosis. (2017) 266:95–102. doi: 10.1016/j.atherosclerosis.2017.09.032
56. Vinci R, Pedicino D, Bonanni A, D'Aiello A, Severino A, Pisano E, et al. A novel monocyte subset as a unique signature of atherosclerotic plaque rupture. Front Cell Dev Biol. (2021) 9:753223. doi: 10.3389/fcell.2021.753223
57. Gembillo G, Siligato R, Cernaro V, Satta E, Conti G, Salvo A, et al. Monocyte to hdl ratio: a novel marker of resistant hypertension in ckd patients. Int Urol Nephrol. (2022) 54(2):395–403. doi: 10.1007/s11255-021-02904-9
58. Tang X, Tan Y, Yang Y, Li M, He X, Lu Y, et al. Association of the monocyte-to-high-density lipoprotein cholesterol ratio with diabetic retinopathy. Front Cardiovasc Med. (2021) 8:707008. doi: 10.3389/fcvm.2021.707008
59. Sağ S, Yıldız A, Aydin Kaderli A, Gül BC, Bedir Ö, Ceğilli E, et al. Association of monocyte to hdl cholesterol level with contrast induced nephropathy in stemi patients treated with primary pci. Clin Chem Lab Med. (2017) 55(1):132–8. doi: 10.1515/cclm-2016-0005
60. Kaplan IG, Kaplan M, Abacioglu OO, Yavuz F, Saler T. Monocyte/hdl ratio predicts hypertensive complications. Bratisl Lek Listy. (2020) 121(2):133–6. doi: 10.4149/bll_2020_018
61. Avci A, Biricik S, Avci BS, Yesiloglu O, Sumbul HE, Icme F, et al. The new prognostic factor for pulmonary embolism: the ratio of monocyte count to hdl cholesterol. Am J Emerg Med. (2021) 46:212–6. doi: 10.1016/j.ajem.2020.07.026
62. Koupenova M, Kehrel BE, Corkrey HA, Freedman JE. Thrombosis and platelets: an update. Eur Heart J. (2017) 38(11):785–91. doi: 10.1093/eurheartj/ehw550
63. Angiolillo DJ, Capodanno D, Goto S. Platelet thrombin receptor antagonism and atherothrombosis. Eur Heart J. (2010) 31(1):17–28. doi: 10.1093/eurheartj/ehp504
64. Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR. Dynamics of leukocyte-platelet adhesion in whole blood. Blood. (1991) 78(7):1730–7. doi: 10.1182/blood.V78.7.1730.1730
65. Moore KL, Stults NL, Diaz S, Smith DF, Cummings RD, Varki A, et al. Identification of a specific glycoprotein ligand for p-selectin (cd62) on myeloid cells. J Cell Biol. (1992) 118(2):445–56. doi: 10.1083/jcb.118.2.445
66. Furie B, Furie BC. Role of platelet p-selectin and microparticle psgl-1 in thrombus formation. Trends Mol Med. (2004) 10(4):171–8. doi: 10.1016/j.molmed.2004.02.008
67. Lindmark E, Tenno T, Siegbahn A. Role of platelet p-selectin and cd40 ligand in the induction of monocytic tissue factor expression. Arterioscler Thromb Vasc Biol. (2000) 20(10):2322–8. doi: 10.1161/01.atv.20.10.2322
68. Martins P, van Gils JM, Mol A, Hordijk PL, Zwaginga JJ. Platelet binding to monocytes increases the adhesive properties of monocytes by up-regulating the expression and functionality of β(1) and β(2) integrins. J Leukoc Biol. (2006) 79(3):499–507. doi: 10.1189/jlb.0605318
69. Schulz C, von Brühl ML, Barocke V, Cullen P, Mayer K, Okrojek R, et al. Emmprin (cd147/basigin) mediates platelet-monocyte interactions in vivo and augments monocyte recruitment to the vascular wall. J Thromb Haemost. (2011) 9(5):1007–19. doi: 10.1111/j.1538-7836.2011.04235.x
70. Schmidt R, Bültmann A, Fischel S, Gillitzer A, Cullen P, Walch A, et al. Extracellular matrix metalloproteinase inducer (cd147) is a novel receptor on platelets, activates platelets, and augments nuclear factor kappab-dependent inflammation in monocytes. Circ Res. (2008) 102(3):302–9. doi: 10.1161/circresaha.107.157990
71. Chimen M, Evryviadou A, Box CL, Harrison MJ, Hazeldine J, Dib LH, et al. Appropriation of gpibα from platelet-derived extracellular vesicles supports monocyte recruitment in systemic inflammation. Haematologica. (2020) 105(5):1248–61. doi: 10.3324/haematol.2018.215145
72. von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, et al. Heterophilic interactions of platelet factor 4 and rantes promote monocyte arrest on endothelium. Blood. (2005) 105(3):924–30. doi: 10.1182/blood-2004-06-2475
73. Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: a transcellular delivery system for rantes promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol. (2005) 25(7):1512–8. doi: 10.1161/01.Atv.0000170133.43608.37
74. Kasper B, Brandt E, Brandau S, Petersen F. Platelet factor 4 (cxc chemokine ligand 4) differentially regulates respiratory burst, survival, and cytokine expression of human monocytes by using distinct signaling pathways. J Immunol. (2007) 179(4):2584–91. doi: 10.4049/jimmunol.179.4.2584
75. Kasper B, Winoto-Morbach S, Mittelstädt J, Brandt E, Schütze S, Petersen F. Cxcl4-induced monocyte survival, cytokine expression, and oxygen radical formation is regulated by sphingosine kinase 1. Eur J Immunol. (2010) 40(4):1162–73. doi: 10.1002/eji.200939703
76. Gleissner CA, Shaked I, Erbel C, Böckler D, Katus HA, Ley K. Cxcl4 downregulates the atheroprotective hemoglobin receptor cd163 in human macrophages. Circ Res. (2010) 106(1):203–11. doi: 10.1161/circresaha.109.199505
77. Gleissner CA. Macrophage phenotype modulation by cxcl4 in atherosclerosis. Front Physiol. (2012) 3(1):1–7. doi: 10.3389/fphys.2012.00001
78. Gawaz M, Neumann FJ, Dickfeld T, Koch W, Laugwitz KL, Adelsberger H, et al. Activated platelets induce monocyte chemotactic protein-1 secretion and surface expression of intercellular adhesion molecule-1 on endothelial cells. Circulation. (1998) 98(12):1164–71. doi: 10.1161/01.cir.98.12.1164
79. Santos JCD, Cruz MS, Bortolin RH, Oliveira KM, Araújo JNG, Duarte VHR, et al. Relationship between circulating vcam-1, icam-1, e-selectin and mmp9 and the extent of coronary lesions. Clinics (Sao Paulo). (2018) 73:e203. doi: 10.6061/clinics/2018/e203
80. Zuchtriegel G, Uhl B, Hessenauer ME, Kurz AR, Rehberg M, Lauber K, et al. Spatiotemporal expression dynamics of selectins govern the sequential extravasation of neutrophils and monocytes in the acute inflammatory response. Arterioscler Thromb Vasc Biol. (2015) 35(4):899–910. doi: 10.1161/atvbaha.114.305143
81. Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, et al. E-selectin engages psgl-1 and cd44 through a common signaling pathway to induce integrin alphalbeta2-mediated slow leukocyte rolling. Blood. (2010) 116(3):485–94. doi: 10.1182/blood-2009-12-259556
82. Huo Y, Ley K. Adhesion molecules and atherogenesis. Acta Physiol Scand. (2001) 173(1):35–43. doi: 10.1046/j.1365-201X.2001.00882.x
83. Alon R, Kassner PD, Carr MW, Finger EB, Hemler ME, Springer TA. The integrin vla-4 supports tethering and rolling in flow on vcam-1. J Cell Biol. (1995) 128(6):1243–53. doi: 10.1083/jcb.128.6.1243
84. Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA, Gimbrone MA Jr, et al. Mcp-1 and il-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. (1999) 398(6729):718–23. doi: 10.1038/19546
85. Luscinskas FW, Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA, et al. C-c and c-x-c chemokines trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Ann N Y Acad Sci. (2000) 902:288–93. doi: 10.1111/j.1749-6632.2000.tb06324.x
86. Meisel SR, Shapiro H, Radnay J, Neuman Y, Khaskia AR, Gruener N, et al. Increased expression of neutrophil and monocyte adhesion molecules lfa-1 and mac-1 and their ligand icam-1 and vla-4 throughout the acute phase of myocardial infarction: possible implications for leukocyte aggregation and microvascular plugging. J Am Coll Cardiol. (1998) 31(1):120–5. doi: 10.1016/s0735-1097(97)00424-5
87. Watanabe T, Fan J. Atherosclerosis and inflammation mononuclear cell recruitment and adhesion molecules with reference to the implication of icam-1/lfa-1 pathway in atherogenesis. Int J Cardiol. (1998) 66(Suppl 1):S45–53; discussion S5. doi: 10.1016/S0167-5273(98)00147-8
88. Badimon L, Peña E, Arderiu G, Padró T, Slevin M, Vilahur G, et al. C-reactive protein in atherothrombosis and angiogenesis. Front Immunol. (2018) 9:430. doi: 10.3389/fimmu.2018.00430
89. Owens AP III, Mackman N. Sources of tissue factor that contribute to thrombosis after rupture of an atherosclerotic plaque. Thromb Res. (2012) 129(Suppl 2):S30–3. doi: 10.1016/j.thromres.2012.02.026
90. Shantsila E, Lip GY. The role of monocytes in thrombotic disorders. Insights from tissue factor, monocyte-platelet aggregates and novel mechanisms. Thromb Haemost. (2009) 102(5):916–24. doi: 10.1160/th09-01-0023
91. Neumann FJ, Ott I, Marx N, Luther T, Kenngott S, Gawaz M, et al. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler Thromb Vasc Biol. (1997) 17(12):3399–405. doi: 10.1161/01.atv.17.12.3399
92. Osnes LT, Westvik AB, Joø GB, Okkenhaug C, Kierulf P. Inhibition of IL-1 induced tissue factor (TF) synthesis and procoagulant activity (PCA) in purified human monocytes by IL-4, IL-10 and IL-13. Cytokine. (1996) 8(11):822–7. doi: 10.1006/cyto.1996.0110
93. Stojkovic S, Thulin Å, Hell L, Thaler B, Rauscher S, Baumgartner J, et al. IL-33 stimulates the release of procoagulant microvesicles from human monocytes and differentially increases tissue factor in human monocyte subsets. Thromb Haemost. (2017) 117(7):1379–90. doi: 10.1160/th16-10-0784
94. Schecter AD, Rollins BJ, Zhang YJ, Charo IF, Fallon JT, Rossikhina M, et al. Tissue factor is induced by monocyte chemoattractant protein-1 in human aortic smooth muscle and THP-1 cells. J Biol Chem. (1997) 272(45):28568–73. doi: 10.1074/jbc.272.45.28568
95. Ernofsson M, Siegbahn A. Platelet-derived growth factor-BB and monocyte chemotactic protein-1 induce human peripheral blood monocytes to express tissue factor. Thromb Res. (1996) 83(4):307–20. doi: 10.1016/0049-3848(96)00139-9
96. Leipner C, Grün K, Müller A, Buchdunger E, Borsi L, Kosmehl H, et al. Imatinib mesylate attenuates fibrosis in coxsackievirus b3-induced chronic myocarditis. Cardiovasc Res. (2008) 79(1):118–26. doi: 10.1093/cvr/cvn063
97. Poitevin S, Cochery-Nouvellon E, Dupont A, Nguyen P. Monocyte IL-10 produced in response to lipopolysaccharide modulates thrombin generation by inhibiting tissue factor expression and release of active tissue factor-bound microparticles. Thromb Haemost. (2007) 97(4):598–607. doi: 10.1160/TH06-11-0622
98. del Conde I, Nabi F, Tonda R, Thiagarajan P, López JA, Kleiman NS. Effect of P-selectin on phosphatidylserine exposure and surface-dependent thrombin generation on monocytes. Arterioscler Thromb Vasc Biol. (2005) 25(5):1065–70. doi: 10.1161/01.ATV.0000159094.17235.9b
99. Hollerbach A, Müller-Calleja N, Canisius A, Orning C, Lackner KJ. Induction of tissue factor expression by anti-β2-glycoprotein I is mediated by tumor necrosis factor α. J Thromb Thrombolysis. (2020) 49(2):228–34. doi: 10.1007/s11239-019-01970-2
100. Xie H, Zhou H, Wang H, Chen D, Xia L, Wang T, et al. Anti-β(2)GPI/β(2)GPI induced TF and TNF-α expression in monocytes involving both TLR4/MyD88 and TLR4/TRIF signaling pathways. Mol Immunol. (2013) 53(3):246–54. doi: 10.1016/j.molimm.2012.08.012
101. López-Pedrera C, Buendía P, Cuadrado MJ, Siendones E, Aguirre MA, Barbarroja N, et al. Antiphospholipid antibodies from patients with the antiphospholipid syndrome induce monocyte tissue factor expression through the simultaneous activation of NF-kappaB/rel proteins via the p38 mitogen-activated protein kinase pathway, and of the MEK-1/ERK pathway. Arthritis Rheum. (2006) 54(1):301–11. doi: 10.1002/art.21549
102. Saito M, Makino Y, Inoue K, Watanabe Y, Hoshi O, Kubota T. Anti-DNA antibodies cross-reactive with β(2)-glycoprotein I induce monocyte tissue factor through the TLR9 pathway. Immunol Med. (2021) 44(2):124–35. doi: 10.1080/25785826.2020.1796285
103. Mészáros K, Aberle S, Dedrick R, Machovich R, Horwitz A, Birr C, et al. Monocyte tissue factor induction by lipopolysaccharide (LPS): dependence on LPS-binding protein and CD14, and inhibition by a recombinant fragment of bactericidal/permeability-increasing protein. Blood. (1994) 83(9):2516–25. doi: 10.1182/blood.V83.9.2516.2516
104. Langer F, Spath B, Fischer C, Stolz M, Ayuk FA, Kröger N, et al. Rapid activation of monocyte tissue factor by antithymocyte globulin is dependent on complement and protein disulfide isomerase. Blood. (2013) 121(12):2324–35. doi: 10.1182/blood-2012-10-460493
105. Scalise V, Sanguinetti C, Neri T, Cianchetti S, Lai M, Carnicelli V, et al. PCSK9 induces tissue factor expression by activation of TLR4/NFkB signaling. Int J Mol Sci. (2021) 22(23):12640. doi: 10.3390/ijms222312640
106. Khajuria A, Houston DS. Induction of monocyte tissue factor expression by homocysteine: a possible mechanism for thrombosis. Blood. (2000) 96(3):966–72. doi: 10.1182/blood.V96.3.966
107. Kawata J, Aoki M, Ishimaru Y, Ono T, Sagara K, Narahara S, et al. Mechanism of tissue factor production by monocytes stimulated with neutrophil elastase. Blood Cells Mol Dis. (2015) 54(2):206–9. doi: 10.1016/j.bcmd.2014.10.005
108. Yamamoto Y, Morita T, Tanaka T, Ikeda K, Kikuchi H, Oguri G, et al. Amiodarone inhibits tissue factor expression in monocytic THP-1 cells. Eur J Pharmacol. (2013) 701(1–3):14–9. doi: 10.1016/j.ejphar.2012.11.064
109. Khan MM, Douglas SD, Benton TD. Substance P-neurokinin-1 receptor interaction upregulates monocyte tissue factor. J Neuroimmunol. (2012) 242(1–2):1–8. doi: 10.1016/j.jneuroim.2011.10.012
110. Kotla S, Singh NK, Kirchhofer D, Rao GN. Heterodimers of the transcriptional factors NFATc3 and FosB mediate tissue factor expression for 15(S)-hydroxyeicosatetraenoic acid-induced monocyte trafficking. J Biol Chem. (2017) 292(36):14885–901. doi: 10.1074/jbc.M117.804344
111. Stephens AC, Rivers RP. Suppression of human monocyte tissue factor synthesis by antisense oligodeoxynucleotide. Thromb Res. (1997) 85(5):387–98. doi: 10.1016/s0049-3848(97)00027-3
112. Ferro D, Basili S, Alessandri C, Cara D, Violi F. Inhibition of tissue-factor-mediated thrombin generation by simvastatin. Atherosclerosis. (2000) 149(1):111–6. doi: 10.1016/s0021-9150(99)00291-9
113. Collier MEW, Ambrose AR, Goodall AH. Does hsa-miR-223-3p from platelet-derived extracellular vesicles regulate tissue factor expression in monocytic cells? Platelets. (2022) 33(7):1031–42. doi: 10.1080/09537104.2022.2027903
114. Vatsyayan R, Kothari H, Pendurthi UR, Rao LV. 4-hydroxy-2-nonenal enhances tissue factor activity in human monocytic cells via p38 mitogen-activated protein kinase activation-dependent phosphatidylserine exposure. Arterioscler Thromb Vasc Biol. (2013) 33(7):1601–11. doi: 10.1161/atvbaha.113.300972
115. Corseaux D, Meurice T, Six I, Rugeri L, Ezekowitz MD, Rouvier P, et al. Basic fibroblast growth factor increases tissue factor expression in circulating monocytes and in vascular wall. Circulation. (2000) 101(16):2000–6. doi: 10.1161/01.cir.101.16.2000
116. Lv B, Tang Y, Chen F, Xiao X. Vasoactive intestinal peptide and pituary adenylate cyclase-activating polypeptide inhibit tissue factor expression in monocyte in vitro and in vivo. Shock. (2009) 31(2):185–91. doi: 10.1097/SHK.0b013e31817d423a
117. Gould TJ, Lysov Z, Swystun LL, Dwivedi DJ, Zarychanski R, Fox-Robichaud AE, et al. Extracellular histones increase tissue factor activity and enhance thrombin generation by human blood monocytes. Shock. (2016) 46(6):655–62. doi: 10.1097/shk.0000000000000680
118. Ichikawa K, Yoshinari M, Iwase M, Wakisaka M, Doi Y, Iino K, et al. Advanced glycosylation end products induced tissue factor expression in human monocyte-like U937 cells and increased tissue factor expression in monocytes from diabetic patients. Atherosclerosis. (1998) 136(2):281–7. doi: 10.1016/s0021-9150(97)00221-9
119. Sanguinetti C, Scalise V, Neri T, Celi A, Susini V, Franzini M, et al. Binding of gamma-glutamyl transferase to TLR4 signalling allows tissue factor activation in monocytes. Int J Mol Sci. (2022) 23(20):12207. doi: 10.3390/ijms232012207
120. Gerrits AJ, Koekman CA, Yildirim C, Nieuwland R, Akkerman JW. Insulin inhibits tissue factor expression in monocytes. J Thromb Haemost. (2009) 7(1):198–205. doi: 10.1111/j.1538-7836.2008.03206.x
121. Dong R, Chen W, Feng W, Xia C, Hu D, Zhang Y, et al. Exogenous bradykinin inhibits tissue factor induction and deep vein thrombosis via activating the eNOS/phosphoinositide 3-kinase/Akt signaling pathway. Cell Physiol Biochem. (2015) 37(4):1592–606. doi: 10.1159/000438526
122. Amadio P, Baldassarre D, Sandrini L, Weksler BB, Tremoli E, Barbieri SS. Effect of cigarette smoke on monocyte procoagulant activity: focus on platelet-derived brain-derived neurotrophic factor (BDNF). Platelets. (2017) 28(1):60–5. doi: 10.1080/09537104.2016.1203403
123. Sovershaev TA, Egorina EM, Unruh D, Bogdanov VY, Hansen JB, Sovershaev MA. BMP-7 induces TF expression in human monocytes by increasing F3 transcriptional activity. Thromb Res. (2015) 135(2):398–403. doi: 10.1016/j.thromres.2014.11.031
124. Barstad RM, Hamers MJ, Stephens RW, Sakariassen KS. Retinoic acid reduces induction of monocyte tissue factor and tissue factor/factor VIIa-dependent arterial thrombus formation. Blood. (1995) 86(1):212–8. doi: 10.1182/blood.V86.1.212.bloodjournal861212
125. Kasthuri RS, Glover SL, Jonas W, McEachron T, Pawlinski R, Arepally GM, et al. PF4/heparin-antibody complex induces monocyte tissue factor expression and release of tissue factor positive microparticles by activation of FcγRI. Blood. (2012) 119(22):5285–93. doi: 10.1182/blood-2011-06-359430
126. Kim JE, Kim KJ, Ahn W, Han KS, Kim HK. Local anesthetics inhibit tissue factor expression in activated monocytes via inhibition of tissue factor mRNA synthesis. Clin Appl Thromb Hemost. (2011) 17(6):E4–9. doi: 10.1177/1076029610378500
127. Cirillo P, Ziviello F, Pellegrino G, Conte S, Cimmino G, Giaquinto A, et al. The adipokine apelin-13 induces expression of prothrombotic tissue factor. Thromb Haemost. (2015) 113(2):363–72. doi: 10.1160/th14-05-0451
128. Williams JC, Klein TW, Goldberger BA, Sleasman JW, Mackman N, Goodenow MM. Δ(9)-tetrahydrocannabinol (THC) enhances lipopolysaccharide-stimulated tissue factor in human monocytes and monocyte-derived microvesicles. J Inflamm (Lond). (2015) 12:39. doi: 10.1186/s12950-015-0084-1
129. Sovershaev MA, Egorina EM, Hansen JB, Østerud B, Pacher P, Stasch JP, et al. Soluble guanylate cyclase agonists inhibit expression and procoagulant activity of tissue factor. Arterioscler Thromb Vasc Biol. (2009) 29(10):1578–86. doi: 10.1161/atvbaha.109.192690
130. Gadomska G, Ziołkowska K, Boinska J, Filipiak J, Rość D. Activation of tf-dependent blood coagulation pathway and vegf-a in patients with essential thrombocythemia. Medicina (Kaunas). (2019) 55(2):54. doi: 10.3390/medicina55020054
131. Millington-Burgess SL, Harper MT. Cytosolic and mitochondrial Ca(2+) signaling in procoagulant platelets. Platelets. (2021) 32(7):855–62. doi: 10.1080/09537104.2021.1881951
132. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209(4):819–35. doi: 10.1084/jem.20112322
133. Sadiku P, Walmsley SR. Hypoxia and the regulation of myeloid cell metabolic imprinting: consequences for the inflammatory response. EMBO Rep. (2019) 20(5):e47388. doi: 10.15252/embr.201847388
134. Pang X, Wang Y, Liu M. M1-macrophage polarization is upregulated in deep vein thrombosis and contributes to the upregulation of adhesion molecules. Hum Immunol. (2019) 80(10):883–9. doi: 10.1016/j.humimm.2019.05.001
135. Ichiki M, Sakai Y, Nango M, Nakamura K, Matsui H, Cho H, et al. Experimental venous thrombi: mri characteristics with histopathological correlation. Br J Radiol. (2012) 85(1012):331–8. doi: 10.1259/bjr/37592039
136. Kimball AS, Obi AT, Luke CE, Dowling AR, Cai Q, Adili R, et al. Ly6clo monocyte/macrophages are essential for thrombus resolution in a murine model of venous thrombosis. Thromb Haemost. (2020) 120(2):289–99. doi: 10.1055/s-0039-3400959
137. Henke PK, Wakefield T. Thrombus resolution and vein wall injury: dependence on chemokines and leukocytes. Thromb Res. (2009) 123(Suppl 4):S72–8. doi: 10.1016/s0049-3848(09)70148-3
138. McGuinness CL, Humphries J, Waltham M, Burnand KG, Collins M, Smith A. Recruitment of labelled monocytes by experimental venous thrombi. Thromb Haemost. (2001) 85(6):1018–24. doi: 10.1055/s-0037-1615957
139. Mosevoll KA, Johansen S, Wendelbo Ø, Nepstad I, Bruserud Ø, Reikvam H. Cytokines, adhesion molecules, and matrix metalloproteases as predisposing, diagnostic, and prognostic factors in venous thrombosis. Front Med (Lausanne). (2018) 5:147. doi: 10.3389/fmed.2018.00147
140. Grinnell F. Fibronectin and wound healing. J Cell Biochem. (1984) 26(2):107–16. doi: 10.1002/jcb.240260206
141. Madlener M, Parks WC, Werner S. Matrix metalloproteinases (mmps) and their physiological inhibitors (timps) are differentially expressed during excisional skin wound repair. Exp Cell Res. (1998) 242(1):201–10. doi: 10.1006/excr.1998.4049
142. Gillitzer R, Goebeler M. Chemokines in cutaneous wound healing. J Leukoc Biol. (2001) 69(4):513–21. doi: 10.1189/jlb.69.4.513
143. Mukhopadhyay S, Johnson TA, Duru N, Buzza MS, Pawar NR, Sarkar R, et al. Fibrinolysis and inflammation in venous thrombus resolution. Front Immunol. (2019) 10:1348. doi: 10.3389/fimmu.2019.01348
144. Wakefield TW, Strieter RM, Wilke CA, Kadell AM, Wrobleski SK, Burdick MD, et al. Venous thrombosis-associated inflammation and attenuation with neutralizing antibodies to cytokines and adhesion molecules. Arterioscler Thromb Vasc Biol. (1995) 15(2):258–68. doi: 10.1161/01.atv.15.2.258
145. Varma MR, Varga AJ, Knipp BS, Sukheepod P, Upchurch GR, Kunkel SL, et al. Neutropenia impairs venous thrombosis resolution in the rat. J Vasc Surg. (2003) 38(5):1090–8. doi: 10.1016/s0741-5214(03)00431-2
146. Sharma S, Hofbauer TM, Ondracek AS, Chausheva S, Alimohammadi A, Artner T, et al. Neutrophil extracellular traps promote fibrous vascular occlusions in chronic thrombosis. Blood. (2021) 137(8):1104–16. doi: 10.1182/blood.2020005861
147. Wakefield TW, Myers DD, Henke PK. Mechanisms of venous thrombosis and resolution. Arterioscler Thromb Vasc Biol. (2008) 28(3):387–91. doi: 10.1161/atvbaha.108.162289
148. Waltham M, Burnand KG, Collins M, McGuinness CL, Singh I, Smith A. Vascular endothelial growth factor enhances venous thrombus recanalisation and organisation. Thromb Haemost. (2003) 89(1):169–76. doi: 10.1055/s-0037-1613556
149. Szade A, Grochot-Przeczek A, Florczyk U, Jozkowicz A, Dulak J. Cellular and molecular mechanisms of inflammation-induced angiogenesis. IUBMB Life. (2015) 67(3):145–59. doi: 10.1002/iub.1358
150. Moldovan NI, Goldschmidt-Clermont PJ, Parker-Thornburg J, Shapiro SD, Kolattukudy PE. Contribution of monocytes/macrophages to compensatory neovascularization: the drilling of metalloelastase-positive tunnels in ischemic myocardium. Circ Res. (2000) 87(5):378–84. doi: 10.1161/01.res.87.5.378
151. Anghelina M, Krishnan P, Moldovan L, Moldovan NI. Monocytes/macrophages cooperate with progenitor cells during neovascularization and tissue repair: conversion of cell columns into fibrovascular bundles. Am J Pathol. (2006) 168(2):529–41. doi: 10.2353/ajpath.2006.050255
152. Nguyen KP, McGilvray KC, Puttlitz CM, Mukhopadhyay S, Chabasse C, Sarkar R. Matrix metalloproteinase 9 (mmp-9) regulates vein wall biomechanics in murine thrombus resolution. PLoS One. (2015) 10(9):e0139145. doi: 10.1371/journal.pone.0139145
153. Nosaka M, Ishida Y, Kimura A, Kuninaka Y, Taruya A, Furuta M, et al. Contribution of the tnf-α (tumor necrosis factor-α)-tnf-rp55 (tumor necrosis factor receptor p55) axis in the resolution of venous thrombus. Arterioscler Thromb Vasc Biol. (2018) 38(11):2638–50. doi: 10.1161/atvbaha.118.311194
154. Downing LJ, Strieter RM, Kadell AM, Wilke CA, Austin JC, Hare BD, et al. Il-10 regulates thrombus-induced vein wall inflammation and thrombosis. J Immunol. (1998) 161(3):1471–6. doi: 10.4049/jimmunol.161.3.1471
155. Henke PK, Wakefield TW, Kadell AM, Linn MJ, Varma MR, Sarkar M, et al. Interleukin-8 administration enhances venous thrombosis resolution in a rat model. J Surg Res. (2001) 99(1):84–91. doi: 10.1006/jsre.2001.6122
156. Nosaka M, Ishida Y, Kimura A, Kuninaka Y, Taruya A, Ozaki M, et al. Crucial involvement of il-6 in thrombus resolution in mice via macrophage recruitment and the induction of proteolytic enzymes. Front Immunol. (2019) 10:3150. doi: 10.3389/fimmu.2019.03150
157. Wojcik BM, Wrobleski SK, Hawley AE, Wakefield TW, Myers DD Jr, Diaz JA. Interleukin-6: a potential target for post-thrombotic syndrome. Ann Vasc Surg. (2011) 25(2):229–39. doi: 10.1016/j.avsg.2010.09.003
158. Purdy M, Obi A, Myers D, Wakefield T. P- and e-selectin in venous thrombosis and non-venous pathologies. J Thromb Haemost. (2022) 20(5):1056–66. doi: 10.1111/jth.15689
159. Miyazaki C, Shimizu K, Nagasawa Y, Chiba T, Sakuma K, Aimoto M, et al. Effects of enhanced intracranial pressure on blood pressure and the cardio-ankle vascular index in rabbits. J Atheroscler Thromb. (2021) 28(11):1241–9. doi: 10.5551/jat.59451
160. Olsen F. Correlation between infiltration of mononuclear cells and production of connective tissue in acute hypertensive vascular disease. Acta Pathol Microbiol Scand A. (1971) 79(1):15–21. doi: 10.1111/j.1699-0463.1971.tb00507.x
161. Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, et al. Dc isoketal-modified proteins activate t cells and promote hypertension. J Clin Invest. (2014) 124(10):4642–56. doi: 10.1172/jci74084
162. Ishibashi M, Hiasa K, Zhao Q, Inoue S, Ohtani K, Kitamoto S, et al. Critical role of monocyte chemoattractant protein-1 receptor ccr2 on monocytes in hypertension-induced vascular inflammation and remodeling. Circ Res. (2004) 94(9):1203–10. doi: 10.1161/01.Res.0000126924.23467.A3
163. Wenzel P. Monocytes as immune targets in arterial hypertension. Br J Pharmacol. (2019) 176(12):1966–77. doi: 10.1111/bph.14389
164. Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, et al. Lysozyme m-positive monocytes mediate angiotensin ii-induced arterial hypertension and vascular dysfunction. Circulation. (2011) 124(12):1370–81. doi: 10.1161/circulationaha.111.034470
165. Lu X, Crowley SD. Inflammation in salt-sensitive hypertension and renal damage. Curr Hypertens Rep. (2018) 20(12):103. doi: 10.1007/s11906-018-0903-x
166. Stumpf C, Raaz D, Klinghammer L, Schneider M, Schmieder RE, Garlichs CD, et al. Platelet cd40 contributes to enhanced monocyte chemoattractant protein 1 levels in patients with resistant hypertension. Eur J Clin Invest. (2016) 46(6):564–71. doi: 10.1111/eci.12635
167. Mikolajczyk TP, Szczepaniak P, Vidler F, Maffia P, Graham GJ, Guzik TJ. Role of inflammatory chemokines in hypertension. Pharmacol Ther. (2021) 223:107799. doi: 10.1016/j.pharmthera.2020.107799
168. Imiela AM, Siedliński M, Dobrowolski P, Pręgowska-Chwała B, Kabat M, Oliveira Silva RN, et al. Altered monocytic phenotypes are linked to a hypertension form: a clinical observational study. Kardiol Pol. (2022) 80(3):346–9. doi: 10.33963/KP.a2022.0036
169. Artigues C, Richard V, Roussel C, Lallemand F, Henry JP, Thuillez C. Increased endothelium–monocyte interactions in salt-sensitive hypertension: effect of l-arginine. J Cardiovasc Pharmacol. (2000) 35(3):468–73. doi: 10.1097/00005344-200003000-00018
170. Blasi ER, Rocha R, Rudolph AE, Blomme EA, Polly ML, McMahon EG. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int. (2003) 63(5):1791–800. doi: 10.1046/j.1523-1755.2003.00929.x
171. Mervaala EM, Müller DN, Park JK, Schmidt F, Löhn M, Breu V, et al. Monocyte infiltration and adhesion molecules in a rat model of high human renin hypertension. Hypertension. (1999) 33(1 Pt 2):389–95. doi: 10.1161/01.hyp.33.1.389
172. Schweigert O, Adler J, Längst N, Aïssi D, Duque Escobar J, Tong T, et al. Crip1 expression in monocytes related to hypertension. Clin Sci (Lond). (2021) 135(7):911–24. doi: 10.1042/cs20201372
173. Loperena R, Van Beusecum JP, Itani HA, Engel N, Laroumanie F, Xiao L, et al. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: roles of stat3, interleukin 6 and hydrogen peroxide. Cardiovasc Res. (2018) 114(11):1547–63. doi: 10.1093/cvr/cvy112
174. Petri M. Antiphospholipid syndrome. Transl Res. (2020) 225:70–81. doi: 10.1016/j.trsl.2020.04.006
175. Yasuda S, Bohgaki M, Atsumi T, Koike T. Pathogenesis of antiphospholipid antibodies: impairment of fibrinolysis and monocyte activation via the p38 mitogen-activated protein kinase pathway. Immunobiology. (2005) 210(10):775–80. doi: 10.1016/j.imbio.2005.10.009
176. Corban MT, Duarte-Garcia A, McBane RD, Matteson EL, Lerman LO, Lerman A. Antiphospholipid syndrome: role of vascular endothelial cells and implications for risk stratification and targeted therapeutics. J Am Coll Cardiol. (2017) 69(18):2317–30. doi: 10.1016/j.jacc.2017.02.058
177. Matsuura E, Kobayashi K, Matsunami Y, Lopez LR. The immunology of atherothrombosis in the antiphospholipid syndrome: antigen presentation and lipid intracellular accumulation. Autoimmun Rev. (2009) 8(6):500–5. doi: 10.1016/j.autrev.2008.12.018
178. Harper BE, Wills R, Pierangeli SS. Pathophysiological mechanisms in antiphospholipid syndrome. Int J Clin Rheumtol. (2011) 6(2):157–71. doi: 10.2217/ijr.11.9
179. Breen KA, Sanchez K, Kirkman N, Seed PT, Parmar K, Moore GW, et al. Endothelial and platelet microparticles in patients with antiphospholipid antibodies. Thromb Res. (2015) 135(2):368–74. doi: 10.1016/j.thromres.2014.11.027
180. Cho CS, Cho ML, Chen PP, Min SY, Hwang SY, Park KS, et al. Antiphospholipid antibodies induce monocyte chemoattractant protein-1 in endothelial cells. J Immunol. (2002) 168(8):4209–15. doi: 10.4049/jimmunol.168.8.4209
181. Virachith S, Saito M, Watanabe Y, Inoue K, Hoshi O, Kubota T. Anti-β(2)-glycoprotein i antibody with DNA binding activity enters living monocytes via cell surface DNA and induces tissue factor expression. Clin Exp Immunol. (2019) 195(2):167–78. doi: 10.1111/cei.13229
182. Saito M, Makino Y, Inoue K, Watanabe Y, Hoshi O, Kubota T. Anti-DNA antibodies cross-reactive with β(2)-glycoprotein i induce monocyte tissue factor through the tlr9 pathway. Immunol Med. (2021) 44(2):124–35. doi: 10.1080/25785826.2020.1796285
183. Kolyada A, Barrios DA, Beglova N. Dimerized domain v of beta2-glycoprotein i is sufficient to upregulate procoagulant activity in pma-treated u937 monocytes and require intact residues in two phospholipid-binding loops. Antibodies (Basel). (2017) 6(2):8. doi: 10.3390/antib6020008
184. Oku K, Atsumi T, Bohgaki M, Amengual O, Kataoka H, Horita T, et al. Complement activation in patients with primary antiphospholipid syndrome. Ann Rheum Dis. (2009) 68(6):1030–5. doi: 10.1136/ard.2008.090670
185. López-Pedrera C, Cuadrado MJ, Herández V, Buendïa P, Aguirre MA, Barbarroja N, et al. Proteomic analysis in monocytes of antiphospholipid syndrome patients: deregulation of proteins related to the development of thrombosis. Arthritis Rheum. (2008) 58(9):2835–44. doi: 10.1002/art.23756
186. Naranjo L, Stojanovich L, Djokovic A, Andreoli L, Tincani A, Maślińska M, et al. Circulating immune-complexes of igg/igm bound to b2-glycoprotein-i associated with complement consumption and thrombocytopenia in antiphospholipid syndrome. Front Immunol. (2022) 13:957201. doi: 10.3389/fimmu.2022.957201
187. Zhou Z, You Y, Wang F, Sun Y, Teng J, Liu H, et al. Urine proteomics differentiate primary thrombotic antiphospholipid syndrome from obstetric antiphospholipid syndrome. Front Immunol. (2021) 12:702425. doi: 10.3389/fimmu.2021.702425
188. Zhang G, Cai Q, Zhou H, He C, Chen Y, Zhang P, et al. Oxldl/β2gpi/anti-β2gpi ab complex induces inflammatory activation via the tlr4/nf-κb pathway in huvecs. Mol Med Rep. (2021) 23(2):148. doi: 10.3892/mmr.2020.11787
189. Zhang X, Xie Y, Zhou H, Xu Y, Liu J, Xie H, et al. Involvement of tlr4 in oxidized ldl/β2gpi/anti-β2gpi-induced transformation of macrophages to foam cells. J Atheroscler Thromb. (2014) 21(11):1140–51. doi: 10.5551/jat.24372
190. Pérez-Sánchez L, Patiño-Trives AM, Aguirre-Zamorano M, Luque-Tévar M, Ábalos-Aguilera MC, la Rosa I A-d, et al. Characterization of antiphospholipid syndrome atherothrombotic risk by unsupervised integrated transcriptomic analyses. Arterioscler Thromb Vasc Biol. (2021) 41(2):865–77. doi: 10.1161/atvbaha.120.315346
191. Juárez-Vicuña Y, Guzmán-Martín CA, Martínez-Martínez LA, Hernández-Díazcouder A, Huesca-Gómez C, Gamboa R, et al. Mir-19b-3p and mir-20a-5p are associated with the levels of antiphospholipid antibodies in patients with antiphospholipid syndrome. Rheumatol Int. (2021) 41(7):1329–35. doi: 10.1007/s00296-021-04864-w
192. Manganelli V, Truglia S, Capozzi A, Alessandri C, Riitano G, Spinelli FR, et al. Alarmin hmgb1 and soluble rage as new tools to evaluate the risk stratification in patients with the antiphospholipid syndrome. Front Immunol. (2019) 10:460. doi: 10.3389/fimmu.2019.00460
193. Štok U, Štucin N, Blokar E, Ambrožič A, Sodin-Šemrl S, Čučnik S, et al. Antiphospholipid antibody syndrome-associated increased surface expression of vla4 integrin on human monocytes. Biomedicines. (2022) 10(10):2341. doi: 10.3390/biomedicines10102341
194. Martirosyan A, Petrek M, Navratilova Z, Blbulyan A, Boyajyan A, Manukyan G. Differential regulation of proinflammatory mediators following lps- and atp-induced activation of monocytes from patients with antiphospholipid syndrome. Biomed Res Int. (2015) 2015:292851. doi: 10.1155/2015/292851
195. Blbulyan A, Martirosyan A, Petrek M, Navratilova Z, Manukyan G. Antiphospholipid syndrome and monocytes: new aspects. Georgian Med News. (2017) 268–269:12–7.
196. Martirosyan A, Petrek M, Kishore A, Manukyan G. Immunomodulatory effects of therapeutic plasma exchange on monocytes in antiphospholipid syndrome. Exp Ther Med. (2016) 12(2):1189–95. doi: 10.3892/etm.2016.3441
197. Arora S, Nair S, Prabhu R, Avanthika C, Jhaveri S, Samayam S, et al. Role of direct oral anticoagulation agents as thromboprophylaxis in antiphospholipid syndrome. Cureus. (2021) 13(10):e19009. doi: 10.7759/cureus.19009
198. Poels K, van Leent MMT, Reiche ME, Kusters PJH, Huveneers S, de Winther MPJ, et al. Antibody-mediated inhibition of ctla4 aggravates atherosclerotic plaque inflammation and progression in hyperlipidemic mice. Cells. (2020) 9(9):1987. doi: 10.3390/cells9091987
199. Libby P. The changing landscape of atherosclerosis. Nature. (2021) 592(7855):524–33. doi: 10.1038/s41586-021-03392-8
200. Steinberg D. The ldl modification hypothesis of atherogenesis: an update. J Lipid Res. (2009) 50(Suppl):S376–81. doi: 10.1194/jlr.R800087-JLR200
201. Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, et al. Combined inhibition of ccl2, cx3cr1, and ccr5 abrogates ly6c(hi) and ly6c(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. (2008) 117(13):1649–57. doi: 10.1161/circulationaha.107.745091
202. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. (2012) 12(2):223–32. doi: 10.1016/j.chom.2012.06.006
203. Flores-Gomez D, Bekkering S, Netea MG, Riksen NP. Trained immunity in atherosclerotic cardiovascular disease. Arterioscler Thromb Vasc Biol. (2021) 41(1):62–9. doi: 10.1161/atvbaha.120.314216
204. Edgar L, Akbar N, Braithwaite AT, Krausgruber T, Gallart-Ayala H, Bailey J, et al. Hyperglycemia induces trained immunity in macrophages and their precursors and promotes atherosclerosis. Circulation. (2021) 144(12):961–82. doi: 10.1161/circulationaha.120.046464
205. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. Mtor- and hif-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345(6204):1250684. doi: 10.1126/science.1250684
206. Keating ST, Groh L, Thiem K, Bekkering S, Li Y, Matzaraki V, et al. Rewiring of glucose metabolism defines trained immunity induced by oxidized low-density lipoprotein. J Mol Med (Berl). (2020) 98(6):819–31. doi: 10.1007/s00109-020-01915-w
207. Sohrabi Y, Lagache SMM, Schnack L, Godfrey R, Kahles F, Bruemmer D, et al. Mtor-dependent oxidative stress regulates oxldl-induced trained innate immunity in human monocytes. Front Immunol. (2018) 9:3155. doi: 10.3389/fimmu.2018.03155
208. Groh LA, Verel DE, van der Heijden C, Matzaraki V, Moorlag S, de Bree LC, et al. Immune modulatory effects of progesterone on oxldl-induced trained immunity in monocytes. J Leukoc Biol. (2022) 112(2):279–88. doi: 10.1002/jlb.3ab1220-846r
209. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34(8):1731–8. doi: 10.1161/atvbaha.114.303887
210. Wang F, Zhang Z, Fang A, Jin Q, Fang D, Liu Y, et al. Macrophage foam cell-targeting immunization attenuates atherosclerosis. Front Immunol. (2018) 9:3127. doi: 10.3389/fimmu.2018.03127
211. Clarke MC, Bennett MR. Cause or consequence: what does macrophage apoptosis do in atherosclerosis? Arterioscler Thromb Vasc Biol. (2009) 29(2):153–5. doi: 10.1161/atvbaha.108.179903
212. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. (2014) 114(12):1852–66. doi: 10.1161/circresaha.114.302721
213. Xiang Y, Liang B, Zhang X, Qiu X, Deng Q, Yu L, et al. Atheroprotective mechanism by which folic acid regulates monocyte subsets and function through DNA methylation. Clin Epigenetics. (2022) 14(1):32. doi: 10.1186/s13148-022-01248-0
214. Vrijenhoek JE, Pasterkamp G, Moll FL, de Borst GJ, Bots ML, Catanzariti L, et al. Extracellular vesicle-derived cd14 is independently associated with the extent of cardiovascular disease burden in patients with manifest vascular disease. Eur J Prev Cardiol. (2015) 22(4):451–7. doi: 10.1177/2047487313518478
215. Oggero S, de Gaetano M, Marcone S, Fitzsimons S, Pinto AL, Ikramova D, et al. Extracellular vesicles from monocyte/platelet aggregates modulate human atherosclerotic plaque reactivity. J Extracell Vesicles. (2021) 10(6):12084. doi: 10.1002/jev2.12084
216. Baig S, Parvaresh Rizi E, Shabeer M, Chhay V, Mok SF, Loh TP, et al. Metabolic gene expression profile in circulating mononuclear cells reflects obesity-associated metabolic inflexibility. Nutr Metab (Lond). (2016) 13:74. doi: 10.1186/s12986-016-0135-5
217. Leite F, Lima M, Marino F, Cosentino M, Ribeiro L. Dopaminergic receptors and tyrosine hydroxylase expression in peripheral blood mononuclear cells: a distinct pattern in central obesity. PLoS One. (2016) 11(1):e0147483. doi: 10.1371/journal.pone.0147483
218. Karam BS, Chavez-Moreno A, Koh W, Akar JG, Akar FG. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc Diabetol. (2017) 16(1):120. doi: 10.1186/s12933-017-0604-9
219. Milasan A, Tessandier N, Tan S, Brisson A, Boilard E, Martel C. Extracellular vesicles are present in mouse lymph and their level differs in atherosclerosis. J Extracell Vesicles. (2016) 5:31427. doi: 10.3402/jev.v5.31427
220. Goichot B, Grunebaum L, Desprez D, Vinzio S, Meyer L, Schlienger JL, et al. Circulating procoagulant microparticles in obesity. Diabetes Metab. (2006) 32(1):82–5. doi: 10.1016/s1262-3636(07)70251-3
221. Lin FJ, Tseng WK, Yin WH, Yeh HI, Chen JW, Wu CC. Residual risk factors to predict major adverse cardiovascular events in atherosclerotic cardiovascular disease patients with and without diabetes mellitus. Sci Rep. (2017) 7(1):9179. doi: 10.1038/s41598-017-08741-0
222. Dasu MR, Devaraj S, Jialal I. High glucose induces il-1beta expression in human monocytes: mechanistic insights. Am J Physiol Endocrinol Metab. (2007) 293(1):E337–46. doi: 10.1152/ajpendo.00718.2006
223. Omoto S, Nomura S, Shouzu A, Nishikawa M, Fukuhara S, Iwasaka T. Detection of monocyte-derived microparticles in patients with type ii diabetes mellitus. Diabetologia. (2002) 45(4):550–5. doi: 10.1007/s00125-001-0772-7
224. Nomura S. Dynamic role of microparticles in type 2 diabetes mellitus. Curr Diabetes Rev. (2009) 5(4):245–51. doi: 10.2174/157339909789804404
225. Bessler WK, Kim G, Hudson FZ, Mund JA, Mali R, Menon K, et al. Nf1+/− monocytes/macrophages induce neointima formation via ccr2 activation. Hum Mol Genet. (2016) 25(6):1129–39. doi: 10.1093/hmg/ddv635
226. Nomura S, Inami N, Shouzu A, Urase F, Maeda Y. Correlation and association between plasma platelet-, monocyte- and endothelial cell-derived microparticles in hypertensive patients with type 2 diabetes mellitus. Platelets. (2009) 20(6):406–14. doi: 10.1080/09537100903114545
227. Nomura S, Kanazawa S, Fukuhara S. Effects of efonidipine on platelet and monocyte activation markers in hypertensive patients with and without type 2 diabetes mellitus. J Hum Hypertens. (2002) 16(8):539–47. doi: 10.1038/sj.jhh.1001447
228. Coenen KR, Gruen ML, Lee-Young RS, Puglisi MJ, Wasserman DH, Hasty AH. Impact of macrophage toll-like receptor 4 deficiency on macrophage infiltration into adipose tissue and the artery wall in mice. Diabetologia. (2009) 52(2):318–28. doi: 10.1007/s00125-008-1221-7
229. Hosseinkhani B, Kuypers S, van den Akker NMS, Molin DGM, Michiels L. Extracellular vesicles work as a functional inflammatory mediator between vascular endothelial cells and immune cells. Front Immunol. (2018) 9:1789. doi: 10.3389/fimmu.2018.01789
230. Hjuler Nielsen M, Irvine H, Vedel S, Raungaard B, Beck-Nielsen H, Handberg A. Elevated atherosclerosis-related gene expression, monocyte activation and microparticle-release are related to increased lipoprotein-associated oxidative stress in familial hypercholesterolemia. PLoS One. (2015) 10(4):e0121516. doi: 10.1371/journal.pone.0121516
231. Suades R, Padró T, Alonso R, López-Miranda J, Mata P, Badimon L. Circulating cd45+/cd3+ lymphocyte-derived microparticles map lipid-rich atherosclerotic plaques in familial hypercholesterolaemia patients. Thromb Haemost. (2014) 111(1):111–21. doi: 10.1160/th13-07-0612
232. Suades R, Padró T, Alonso R, Mata P, Badimon L. High levels of tsp1+/cd142+ platelet-derived microparticles characterise young patients with high cardiovascular risk and subclinical atherosclerosis. Thromb Haemost. (2015) 114(6):1310–21. doi: 10.1160/th15-04-0325
233. Silverstein RL. Teaching an old dog new tricks: potential antiatherothrombotic use for statins. J Clin Invest. (2012) 122(2):478–81. doi: 10.1172/jci61857
234. Chan YL, Saad S, Pollock C, Oliver B, Al-Odat I, Zaky AA, et al. Impact of maternal cigarette smoke exposure on brain inflammation and oxidative stress in male mice offspring. Sci Rep. (2016) 6:25881. doi: 10.1038/srep25881
235. Li M, Yu D, Williams KJ, Liu ML. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arterioscler Thromb Vasc Biol. (2010) 30(9):1818–24. doi: 10.1161/atvbaha.110.209577
236. Papaconstantinou J. The role of signaling pathways of inflammation and oxidative stress in development of senescence and aging phenotypes in cardiovascular disease. Cells. (2019) 8(11):1383. doi: 10.3390/cells8111383
237. Garg R, Aggarwal S, Kumar R, Sharma G. Association of atherosclerosis with dyslipidemia and co-morbid conditions: a descriptive study. J Nat Sci Biol Med. (2015) 6(1):163–8. doi: 10.4103/0976-9668.149117
238. Stojkovic S, Thulin Å, Hell L, Thaler B, Rauscher S, Baumgartner J, et al. Il-33 stimulates the release of procoagulant microvesicles from human monocytes and differentially increases tissue factor in human monocyte subsets. Thromb Haemost. (2017) 117(7):1379–90. doi: 10.1160/th16-10-0784
239. Aharon A, Tamari T, Brenner B. Monocyte-derived microparticles and exosomes induce procoagulant and apoptotic effects on endothelial cells. Thromb Haemost. (2008) 100(5):878–85. doi: 10.1160/th07-11-0691
240. Dooley LM, Ahmad TB, Pandey M, Good MF, Kotiw M. Rheumatic heart disease: a review of the current status of global research activity. Autoimmun Rev. (2021) 20(2):102740. doi: 10.1016/j.autrev.2020.102740
241. Mahla H, Harlalka KK, Bhairappa S, Manjunath CN. Spectrum of left atrial thrombus in rheumatic heart disease. BMJ Case Rep. (2014) 2014:bcr2014204370. doi: 10.1136/bcr-2014-204370
242. Kumar V, Ganguly NK, Anand IS, Wahi PL. Release of oxygen free radicals by macrophages and neutrophils in patients with rheumatic fever. Eur Heart J. (1991) 12(Suppl D):163–5. doi: 10.1093/eurheartj/12.suppl_d.163
243. Passos LSA, Becker-Greene D, Braulio R, Le TD, Gelape CL, de Almeida LFR, et al. Proinflammatory matrix metalloproteinase-1 associates with mitral valve leaflet disruption following percutaneous mitral valvuloplasty. Front Cardiovasc Med. (2021) 8:804111. doi: 10.3389/fcvm.2021.804111
244. Xiao F, Zheng R, Yang D, Cao K, Zhang S, Wu B, et al. Sex-dependent aortic valve pathology in patients with rheumatic heart disease. PLoS One. (2017) 12(6):e0180230. doi: 10.1371/journal.pone.0180230
245. Feldman M, Ginsburg I. A novel aspect may explain the mechanisms of pathogenicity of rheumatic fever, a multifactorial, autoimmune, infectious and inflammatory disorder which “licks the joints and bites the heart”: a working hypothesis. Med Hypotheses. (2020) 144:110222. doi: 10.1016/j.mehy.2020.110222
246. Deng F, Chen W, Liu L, Wang LM, Chen X. The expression and molecular mechanism of m1 macrophages in rheumatic valvular disease. Zhonghua Wai Ke Za Zhi. (2012) 50(10):933–7.23302467
247. Othieno R, Abu Affan M, Okpo E. Home versus in-patient treatment for deep vein thrombosis. Cochrane Database Syst Rev. (2007) 3:Cd003076. doi: 10.1002/14651858.CD003076.pub2
248. Zhang Y, Cui J, Zhang G, Wu C, Abdel-Latif A, Smyth SS, et al. Inflammasome activation promotes venous thrombosis through pyroptosis. Blood Adv. (2021) 5(12):2619–23. doi: 10.1182/bloodadvances.2020003041
249. Momi S, Canino J, Vismara M, Galgano L, Falcinelli E, Guglielmini G, et al. Proline-rich tyrosine kinase pyk2 regulates deep vein thrombosis. Haematologica. (2022) 107(6):1374–83. doi: 10.3324/haematol.2021.279703
250. Shahneh F, Christian Probst H, Wiesmann SC, A-Gonzalez N, Ruf W, Steinbrink K, et al. Inflammatory monocyte counts determine venous blood clot formation and resolution. Arterioscler Thromb Vasc Biol. (2022) 42(2):145–55. doi: 10.1161/atvbaha.121.317176
251. Schönfelder T, Brandt M, Kossmann S, Knopp T, Münzel T, Walter U, et al. Lack of t-bet reduces monocytic interleukin-12 formation and accelerates thrombus resolution in deep vein thrombosis. Sci Rep. (2018) 8(1):3013. doi: 10.1038/s41598-018-21273-5
252. Nosaka M, Ishida Y, Kimura A, Kuninaka Y, Inui M, Mukaida N, et al. Absence of ifn-γ accelerates thrombus resolution through enhanced mmp-9 and vegf expression in mice. J Clin Invest. (2011) 121(7):2911–20. doi: 10.1172/jci40782
253. Chen YK, Jiang XM, Gong JP. Recombinant human granulocyte colony-stimulating factor enhanced the resolution of venous thrombi. J Vasc Surg. (2008) 47(5):1058–65. doi: 10.1016/j.jvs.2007.12.042
254. Humphries J, Gossage JA, Modarai B, Burnand KG, Sisson TH, Murdoch C, et al. Monocyte urokinase-type plasminogen activator up-regulation reduces thrombus size in a model of venous thrombosis. J Vasc Surg. (2009) 50(5):1127–34. doi: 10.1016/j.jvs.2009.06.047
255. Amadio P, Tarantino E, Sandrini L, Tremoli E, Barbieri SS. Prostaglandin-endoperoxide synthase-2 deletion affects the natural trafficking of annexin a2 in monocytes and favours venous thrombosis in mice. Thromb Haemost. (2017) 117(8):1486–97. doi: 10.1160/th16-12-0968
256. Samsu N. Diabetic nephropathy: challenges in pathogenesis, diagnosis, and treatment. Biomed Res Int. (2021) 2021:1497449. doi: 10.1155/2021/1497449
257. Faselis C, Katsimardou A, Imprialos K, Deligkaris P, Kallistratos M, Dimitriadis K. Microvascular complications of type 2 diabetes mellitus. Curr Vasc Pharmacol. (2020) 18(2):117–24. doi: 10.2174/1570161117666190502103733
258. Alsharidah AS. Diabetes mellitus and diabetic nephropathy: a review of the literature on hemostatic changes in coagulation and thrombosis. Blood Res. (2022) 57(2):101–5. doi: 10.5045/br.2022.2021204
259. Li HD, You YK, Shao BY, Wu WF, Wang YF, Guo JB, et al. Roles and crosstalks of macrophages in diabetic nephropathy. Front Immunol. (2022) 13:1015142. doi: 10.3389/fimmu.2022.1015142
260. Min D, Lyons JG, Bonner J, Twigg SM, Yue DK, McLennan SV. Mesangial cell-derived factors alter monocyte activation and function through inflammatory pathways: possible pathogenic role in diabetic nephropathy. Am J Physiol Renal Physiol. (2009) 297(5):F1229–37. doi: 10.1152/ajprenal.00074.2009
261. Min D, Brooks B, Wong J, Salomon R, Bao W, Harrisberg B, et al. Alterations in monocyte cd16 in association with diabetes complications. Mediators Inflamm. (2012) 2012:649083. doi: 10.1155/2012/649083
262. Devaraj S, Jialal I. Low-density lipoprotein postsecretory modification, monocyte function, and circulating adhesion molecules in type 2 diabetic patients with and without macrovascular complications: the effect of alpha-tocopherol supplementation. Circulation. (2000) 102(2):191–6. doi: 10.1161/01.cir.102.2.191
263. Min D, Brooks B, Wong J, Aamidor S, Seehoo R, Sutanto S, et al. Monocyte cd163 is altered in association with diabetic complications: possible protective role. J Leukoc Biol. (2016) 100(6):1375–83. doi: 10.1189/jlb.3A1015-461RR
264. Galkina E, Ley K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J Am Soc Nephrol. (2006) 17(2):368–77. doi: 10.1681/asn.2005080859
265. Chow F, Ozols E, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int. (2004) 65(1):116–28. doi: 10.1111/j.1523-1755.2004.00367.x
266. Tesch GH. Macrophages and diabetic nephropathy. Semin Nephrol. (2010) 30(3):290–301. doi: 10.1016/j.semnephrol.2010.03.007
267. Chana RS, Martin J, Rahman EU, Wheeler DC. Monocyte adhesion to mesangial matrix modulates cytokine and metalloproteinase production. Kidney Int. (2003) 63(3):889–98. doi: 10.1046/j.1523-1755.2003.00828.x
268. Wang Y, Wei Q, Liu Q, Li Z, Zhou L, Zou F, et al. Crosstalk between monocytes and renal mesangial cells via interaction of metalloproteinases and fractalkine in diabetic nephropathy. Mol Med Rep. (2013) 8(6):1817–23. doi: 10.3892/mmr.2013.1703
269. Banba N, Nakamura T, Matsumura M, Kuroda H, Hattori Y, Kasai K. Possible relationship of monocyte chemoattractant protein-1 with diabetic nephropathy. Kidney Int. (2000) 58(2):684–90. doi: 10.1046/j.1523-1755.2000.00214.x
270. Scurt FG, Menne J, Brandt S, Bernhardt A, Mertens PR, Haller H, et al. Monocyte chemoattractant protein-1 predicts the development of diabetic nephropathy. Diabetes Metab Res Rev. (2022) 38(2):e3497. doi: 10.1002/dmrr.3497
271. Haller H, Bertram A, Nadrowitz F, Menne J. Monocyte chemoattractant protein-1 and the kidney. Curr Opin Nephrol Hypertens. (2016) 25(1):42–9. doi: 10.1097/mnh.0000000000000186
272. Tesch GH. Mcp-1/ccl2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol. (2008) 294(4):F697–701. doi: 10.1152/ajprenal.00016.2008
273. Amann B, Tinzmann R, Angelkort B. Ace inhibitors improve diabetic nephropathy through suppression of renal mcp-1. Diabetes Care. (2003) 26(8):2421–5. doi: 10.2337/diacare.26.8.2421
274. Wada T, Furuichi K, Sakai N, Iwata Y, Yoshimoto K, Shimizu M, et al. Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney Int. (2000) 58(4):1492–9. doi: 10.1046/j.1523-1755.2000.00311.x
275. Boels MGS, Koudijs A, Avramut MC, Sol W, Wang G, van Oeveren-Rietdijk AM, et al. Systemic monocyte chemotactic protein-1 inhibition modifies renal macrophages and restores glomerular endothelial glycocalyx and barrier function in diabetic nephropathy. Am J Pathol. (2017) 187(11):2430–40. doi: 10.1016/j.ajpath.2017.07.020
276. Yacoub R, Campbell KN. Inhibition of ras in diabetic nephropathy. Int J Nephrol Renovasc Dis. (2015) 8:29–40. doi: 10.2147/ijnrd.S37893
277. Moratal C, Laurain A, Naïmi M, Florin T, Esnault V, Neels JG, et al. Regulation of monocytes/macrophages by the renin-angiotensin system in diabetic nephropathy: state of the art and results of a pilot study. Int J Mol Sci. (2021) 22(11):6009. doi: 10.3390/ijms22116009
278. Roscioni SS, Heerspink HJ, de Zeeuw D. The effect of raas blockade on the progression of diabetic nephropathy. Nat Rev Nephrol. (2014) 10(2):77–87. doi: 10.1038/nrneph.2013.251
279. Lv LL, Feng Y, Wu M, Wang B, Li ZL, Zhong X, et al. Exosomal mirna-19b-3p of tubular epithelial cells promotes m1 macrophage activation in kidney injury. Cell Death Differ. (2020) 27(1):210–26. doi: 10.1038/s41418-019-0349-y
280. Jiang WJ, Xu CT, Du CL, Dong JH, Xu SB, Hu BF, et al. Tubular epithelial cell-to-macrophage communication forms a negative feedback loop via extracellular vesicle transfer to promote renal inflammation and apoptosis in diabetic nephropathy. Theranostics. (2022) 12(1):324–39. doi: 10.7150/thno.63735
281. Jia Y, Chen J, Zheng Z, Tao Y, Zhang S, Zou M, et al. Tubular epithelial cell-derived extracellular vesicles induce macrophage glycolysis by stabilizing hif-1α in diabetic kidney disease. Mol Med. (2022) 28(1):95. doi: 10.1186/s10020-022-00525-1
282. Huang Q, Wu H, Wo M, Ma J, Fei X, Song Y. Monocyte-lymphocyte ratio is a valuable predictor for diabetic nephropathy in patients with type 2 diabetes. Medicine (Baltimore). (2020) 99(19):e20190. doi: 10.1097/md.0000000000020190
Keywords: monocyte, macrophage, thrombotic diseases, arterial thrombosis, intravenous thrombolysis
Citation: Han Z, Liu Q, Li H, Zhang M, You L, Lin Y, Wang K, Gou Q, Wang Z, Zhou S, Cai Y, Yuan L and Chen H (2023) The role of monocytes in thrombotic diseases: a review. Front. Cardiovasc. Med. 10:1113827. doi: 10.3389/fcvm.2023.1113827
Received: 1 December 2022; Accepted: 18 May 2023;
Published: 2 June 2023.
Edited by:
Rongli Zhang, Case Western Reserve University, United StatesReviewed by:
Dong-Eog Kim, Dongguk University, Republic of Korea© 2023 Han, Liu, Li, Zhang, You, Lin, Wang, Gou, Wang, Zhou, Cai, Yuan and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lan Yuan eXVhbmxhbkBjZHV0Y20uZWR1LmNu Haoran Chen Y2hlbmhycnJAMTYzLmNvbQ==
†These authors contributed equally to this work
Abbreviations ANTI-β2GPI, anti-beta2glycoprotein I; AT1R, angiotensin type 1 receptor; ACS, acute coronary syndrome; AD-uPA; adenovirus uPA; ANG II, angiotensin II; APS, antiphospholipid syndrome; ASCVD, atherosclerotic cardiovascular disease; β2GPI-DV, β2GPI dimerization domainV; BFGF, basic fibroblast growth factor; CAD, coronary artery disease; CCS, chronic coronary syndrome; CKD, chornic kidney disease; CMs, classical monocytes; CRIP1, cysteine-rich intestinal protein 1; CRP, c-reactive protein; CVD, cardiovascular disease; DC, dendritic cells; DN, diabetic nephropathy; DOCA, deoxycorticosterone acetate; ECM, extracellular matrix; ET, extracellular traps; ETOSIS, extracellular trap cell death; EVs, extracellular vesicles; FCN1, ficolin-1; GP, glycoprotein; HBMMs, human blood monocyte-derived macrophages; HCQ, hydroxychloroquine; HLA-DR, mhc-class II molecules; HTN, hypertension; ICAM-1, intercellular adhesion molecule-1; IGF1, insulin-like growth factor-1;IL-8, interleukin-8; IMs, intermediate monocytes; LDL, low-density lipoproteins; LF, lactoferrin; LVH, left ventricular hypertrophy; MACE, major adverse cardiovascular events; M-CSF, macrophage colony stimulating factor; MCP-1, monocyte chemotactic protein-1; MDCS, monocyte-derived dendritic cells; MMP, matrix metalloproteinases; MPA, monocyte-platelet aggregates; MPO, myeloperoxidase; MHR, monocyte to hdl ratio; MI, myocardial infarction; MLR, monocyte-lymphocyte ratio; MPs, monocytes; microparticles; MVs, microvesicles; MΦ, macrophage; MΦI, macrophage infiltration; NCMs, non-classical monocytes; NDH, newly diagnosed hypertension; NF-κB, nuclear factor κB; OCT, optical coherence tomography; OXLDL, oxidized ldl; PCM, pre-classical monocytes; PE, pulmonary embolism; PHA, primary hyperaldosteronism; PMPs, platelet microparticles; PTGS, prostag-landin-endoperoxide synthase; RAS, renin-angiotensin system; RDN, renal denervation; RHD, rheumatic heart disease; RF, rheumatic fever; RHG-CSF, recombinant human granulocyte colony-stimulating factor; ROS, reactive oxygen species; SASP, secretory phenotype; SYK, spleen tyrosine kinase; TECs, tubular epithelial cells; TOD, target organ damage; UPA, uridylyl phosphate adenosine; VCAM-I, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor; VTE, venous thromboembolism.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.