 Shifeng Ning
Shifeng Ning Min Han
Min Han Li Liu
Li Liu Chengwei Liu
Chengwei Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 03 July 2023
Sec. Cardiovascular Genetics and Systems Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1109008
This article is part of the Research Topic Genomic Architecture of Cardiovascular Diseases, Volume II View all 6 articles
Objective: To characterize the cardiac phenotype associated with the novel pathogenic variant (c.1526del) of LMNA gene, which we identified in a large, six-generation family.
Methods and Results: A family tree was constructed. The clinical data of living and deceased family members were collected. DNA samples from 7 family members were analyzed for LMNA mutations using whole-exome high-throughput sequencing technology. The clinical presentation of pathogenic variant carriers was evaluated. In this six-generation family (n = 67), one member experienced sudden death at the age of 40-years-old. Three pathogenic variant carriers were identified to possess a novel heterozygous deletion mutation in LMNA gene (HGVS: NM_170707.4, c.1526del) located at exon 9 of LMNA chr1:156137145, which creates a premature translational stop signal (p.Pro509Leufs*39) in the LMNA gene and results in an mutant lamin A protein product. The main symptoms of the pathogenic variant carriers were palpitation, fatigue, and syncope, which typically occurred around 20-years-old. AV-conduction block and non-sustained ventricular tachycardia were the first signs of disease and would rapidly progress to atrial standstill around 30-years-old. Significant right atrial enlargement and bicuspid aortic valve malformation was also commonly seen in patients who carried this pathogenic variant.
Conclusion: The pathogenic variant of c.1526del p.P509Lfs*39 was a frameshift deletion located at exon 9 of LMNA chr1:156137145 and causes severe right atrial enlargement, sick sinus syndrome, atrial standstill, ventricular tachycardia, and bicuspid aortic valve malformation. Our findings expand the phenotypic spectrum of novel LMNA gene mutations.
The concept of atrial cardiomyopathy (ACM) was initially proposed a decade ago (1) and was widely accepted until recently. According to EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies, ACM is defined as “any complex of structural, architectural, contractile or electrophysiological changes affecting the atria with the potential to produce clinically-relevant manifestations”. It's classified into four classes: principally cardiomyocyte changes, principally fibrotic changes, combined cardiomyocyte-pathology/fibrosis, and primarily non-collagen infiltration (with or without cardiomyocyte changes) (2). However, this histological-based classification system has limited practical application for most patients due to the need for an invasive atrial biopsy to confirm diagnosis. ACM is characterized by a dilated atrium with various arrhythmias (mainly atrial arrhythmias), and was found to be associated with Ebstein anomaly (3), dilated cardiomyopathy (DCM) (4), myocarditis (5), amyloidosis (6) and muscular dystrophies (7). Although its prevalence is unknown, ACM was often observed in various clinical conditions, such as isolated atrial fibrillation, diabetes, aging, smoking, heart failure, valvular disease, amyloidosis and inflammatory infiltration, etc (2, 6). The ACM could be sporadic or familial, suggesting the possibility of genetic determinants in some forms of ACM. The first case of familial atrial cardiomyopathy was described by Nagle RE et al. in 1972 (8), who described a familial syndrome almost exclusively affecting the atria and atrioventricular-conducting system, with a combination of ectopic supraventricular rhythms and atrioventricular block that eventually progressed to atrial standstill. Thereafter, additional cases of ACM with hereditary components were reported and a variety of gene mutations associated with ACM were identified, including heterozygous mutations of SCN5A (9) and GJA5 (encoding connexin 40) genes (10), the gain-of-function KCNQ1 missense variant (11), homozygous mutation in the natriuretic peptide precursor A (NPPA) gene (12), nonsense mutation in the STA Gene (13), nonsense mutation in the LMNA gene (c.475G > T) (14), missense mutation in the LMNA gene (p.R335W) (15), etc. Currently, the LMNA gene, located on chromosome 1q21.1–21.2, is thought to be one of the most common disease-associated genes for familial DCM with respects to conduction system diseases (15). As of now, a total of 498 LMNA pathogenic variants have been identified and are associated with more than 15 different phenotypes (16, 17). Here we report a novel pathogenic variant of LMNA gene, discovered to be a frame-shift mutation of the LMNA gene at c.1526del, in a large six-generation family. The clinical manifestation was ACM, including giant right atrium, sick sinus syndrome, atrial standstill, left ventricular tachycardia, and bicuspid aortic valve malformation. To the best of our knowledge, this is a novel identification of a pathogenic variant of LMNA gene. Our findings facilitate improved understanding of the LMNA mutation genotype and its clinical phenotype to other known laminopathies.
This study was approved by the Institutional Ethical Committee on Human Research at the Wuhan Asia Heart Hospital. All patients gave written informed consent for participation in the study, including blood sampling, isolation of DNA, molecular genetic testing, and provision of their clinical data.
The pedigree was created based on information provided by the family members and their medical records. Clinical data from the proband and other living mutation-carriers were collected from their respective medical records. In addition, medical reports and examination results (electrocardiogram (ECG), Holter-ECG, pacemaker/implantable cardioverter-defibrillator (ICD) interrogations, echocardiography) of both living and deceased family members were collected if available.
After obtaining the written informed consent, peripheral blood samples were collected from the proband and some members of the 3rd–6th generations (n = 24). The genomic DNA was extracted using the TIANamp Blood DNA Kit (TIANGEN, Beijing, China) according to the manufacturer's standard protocol. Genetic testing was performed by the Beijing Berry Hekang Medical Laboratory using high-throughput whole-exome sequencing technology and the Verita Trekker variant detection system. The sequencing results were analyzed using the Enliven variant site annotation interpretation system, which had been independently developed by Berry Gene Data.
The proband was a 41-year-old woman who was born to a consanguineous family and presented to our hospital with the chief complaints of worsening palpitation and syncope for 2 weeks. A diagnosis of non-sustained ventricular tachycardia (VT) was made at a local hospital, where she was successfully cardioverted with intravenous amiodarone. She also complained of worsening dyspnea during physical activity, fatigue, and intermittent edema of both lower extremities for the past 13 years. She denied paroxysmal nocturnal dyspnea and a history of hypertension or diabetes. Past medical history included pacemaker implantation (Medtronic Sensia SEDRL1) 5 years ago due to a slow heart rate (3rd-degree AV conduction block according to medical records). Per medical records regarding the permanent pacemaker implantation, 3,830 electrodes placed either in the right atrial septum, the lateral wall or near the coronary sinus orifice could not be captured by 10 V pacing, suggesting possible atrial standstill. She also had noticeable right atrial enlargement and severe tricuspid regurgitation (TR). An ECG 8 years ago revealed a slow heart rate (40 bpm) with a junctional escape rhythm. Family history is remarkable for paternal sudden cardiac death (SCD) at the age of 40 and a sister who underwent pacemaker implantation due to sick sinus syndrome at 32-years-old at a local hospital. The proband's cousin is noted to have a very similar, although unspecified, heart disease diagnosed at the age of 40 and pacemaker implantation. Upon admission, physical examination revealed blood pressure (112/58mmHg), heart rate (71 beats/min), respiratory rate (18/min) and oxygen saturation level (99% at room air) were all within normal limits. Auscultation of the heart revealed a grade 3–4/6 systolic murmur heard in the subxiphoid area. The ECG showed a pacing rhythm of 711beats/min (Figure 1A). Cardiac enlargement, particularly right atrial dilation was seen on chest x-Ray (CXR) (Figure 1B). Transthoracic echocardiography (TTE) revealed extreme enlargement of the right atrium (RA, 109 × 77.3 mm), an enlarged right ventricular (RV) chamber (42 mm), severe TR, mild enlargement of left atrium (LA, 45 mm), left ventricular (LV) chamber (53 mm), and mild mitral regurgitation (MR) (Figure 1C). The left ventricular systolic function (LVEF) was within normal range (54%). A bicuspid aortic valve malformation (type 1A) was noticed on TTE examination without significant stenosis (forward flow velocity 1.5 m/s) (Figure 1D). The extremely large RA was confirmed via computed tomography angiography (CTA) (Figure 1E). Labwork indicated CBC, plasma electrolytes, troponin levels, several inflammatory and tumor markers, and renal function were within normal range, but slightly elevated N-terminal B-type natriuretic peptide (NT-pro-BNP, 417.7 pg/ml) and hepatic dysfunction. Coronary heart disease was ruled out by CTA (Figures 2A–C), and amyloidosis cardiomyopathy was excluded via protein electrophoresis (Figures 2D,E) and biopsy of abdominal fat (Congo Red stain) (Figures 2F,G). During hospitalization, the patient experienced a recurrent episode of syncope and ECG confirmed the diagnosis of VT responsive to intravenous amiodarone (Figures 3A,B). Given the patient's family history and suspicion of inherited cardiomyopathy, genetic testing of the patient and select family members was subsequently ordered.
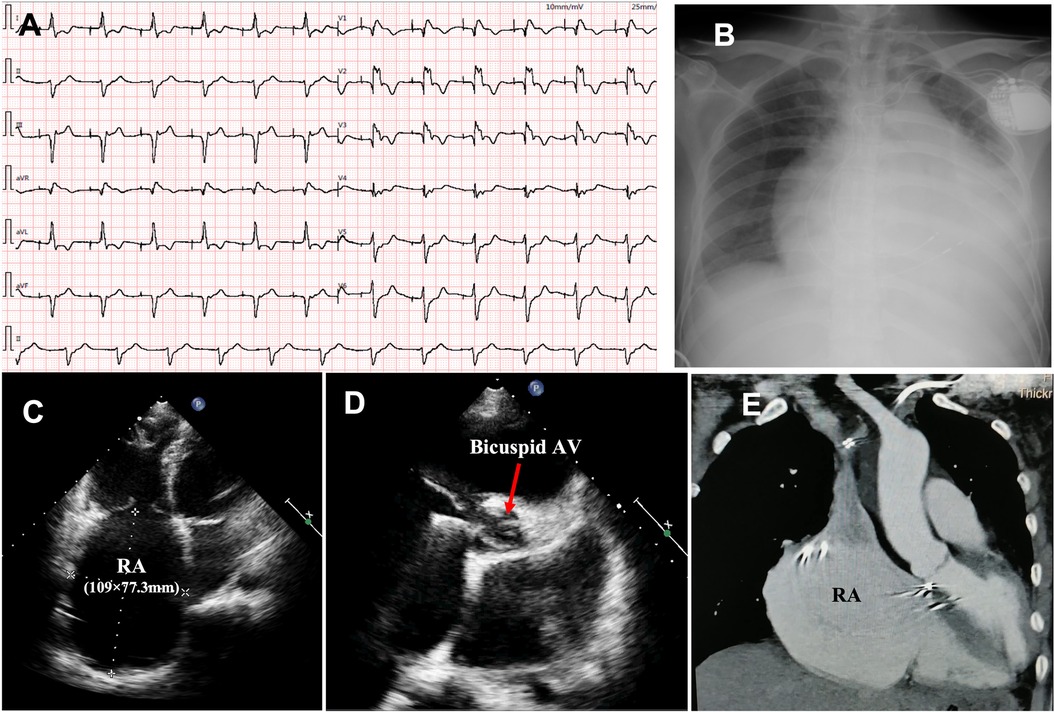
Figure 1. Upon admission, ECG showed pacing rhythm at 71 beats/min) (A); chest x-ray showed cardiac enlargement (B); TTE revealed an extremely enlarged RA (C) and bicuspid aortic valve malformation (D); the enlarged RA was confirmed by CTA (E). ECG, electrocardiogram; CXR, chest x-ray; TTE, transthoracic echocardiography; CTA, computed tomography angiography; RA, right atrium; AV, aortic valve.
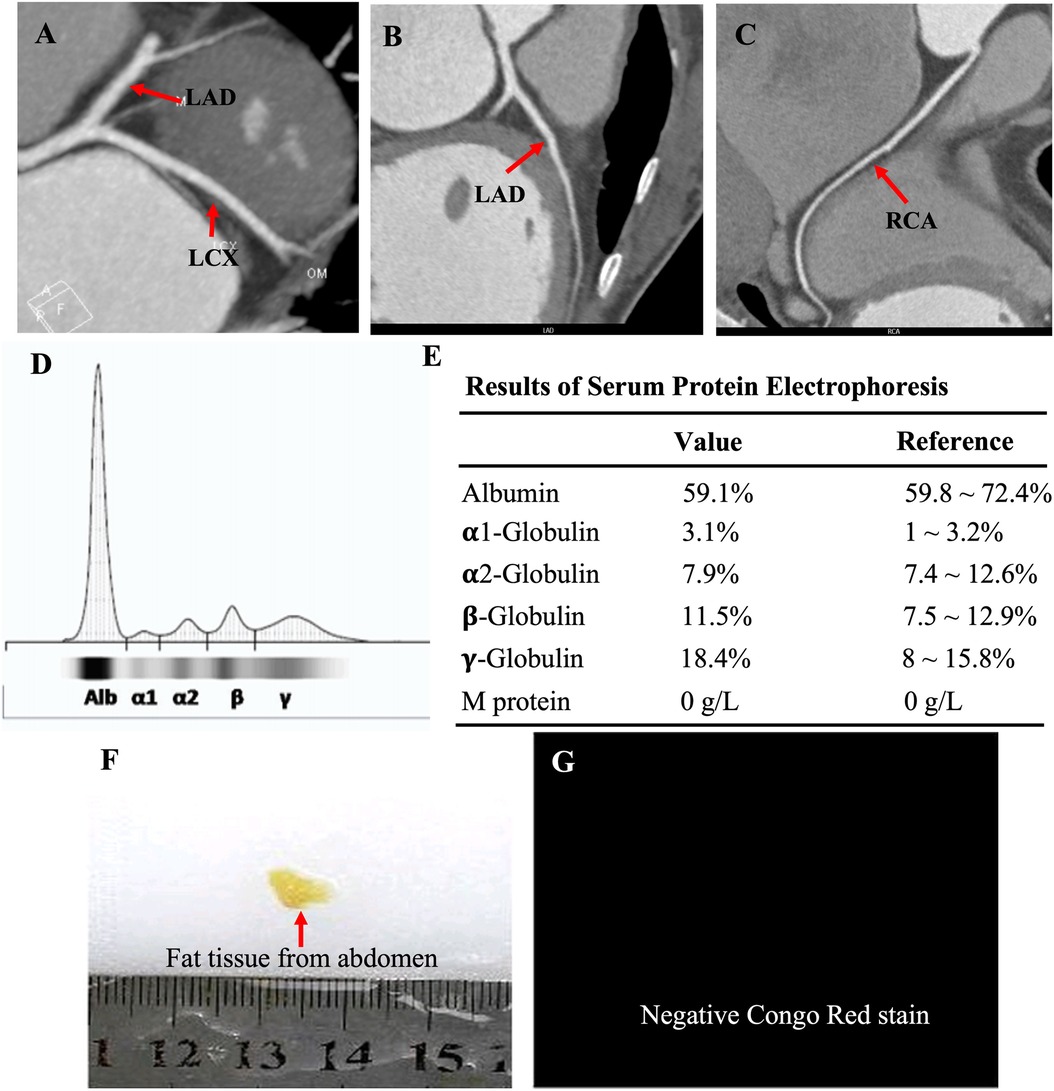
Figure 2. CTA showed normal coronary arteries, including the LAD, LCX and RCA (A–C); protein electrophoresis showed normal range of α1-, α2-, β-, γ-globulin and M-protein (D,E); biopsy of abdominal fat showed a negative Congo Red stain (F,G), ruling out potential amyloid cardiomyopathy.
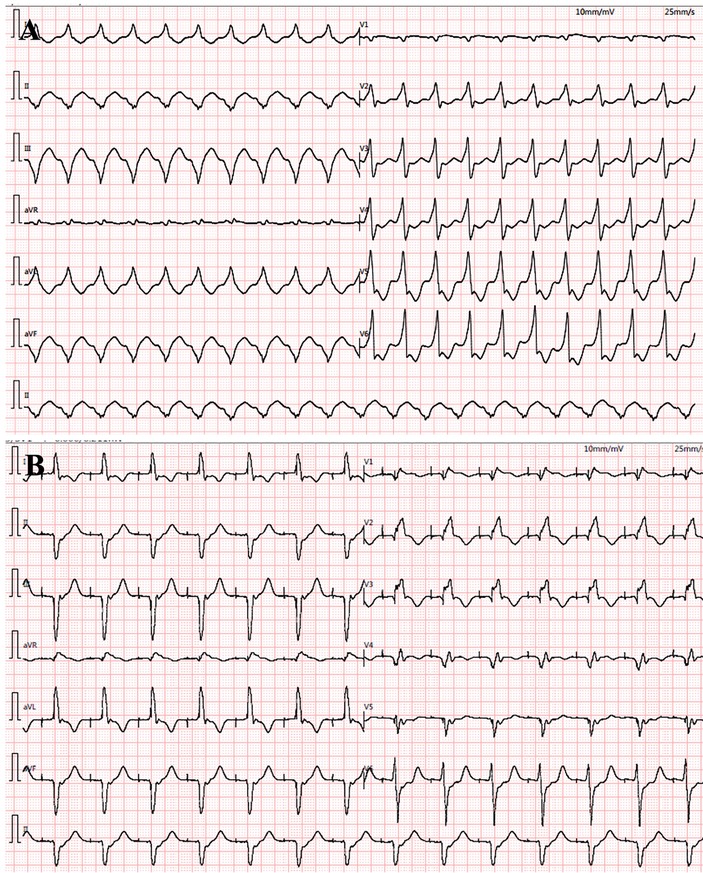
Figure 3. During hospitalization, ventricular tachycardia (VT) reoccurred (A) and was cardioverted with intravenous amiodarone treatment (B).
The constructed family tree included 67 family members spanning over six generations (Figure 4A). Since medical records and samples were unavailable for the 1st and 2nd generations, blood or other genetic samples were only acquired from some members of the 3rd–6th generations. Although genetic samples were obtained from 24 family members, only 7 (III-8, IV-3, V-2, V-3, V-4, V-5, VI-1) eventually completed genetic testing due to cost and other reasons. In total, 1 deceased (III-4) and 1 very sick due to “heart disease” (III-6) at an early age and presumed mutation-carriers, as well as 3 genetically verified mutation-carriers (IV-3, V-2, V-4) were identified (Figure 4B). Genetic testing revealed that the proband carried a frameshift deletion in the LMNA gene at c.1526del (p.P509Lfs*39) in exon 9 on chromosome 1 (HGVS: NM_170707.4, c.1526del: p.P509Lfs*39), which caused changes in the open reading frame of the gene and resulted in loss-of-function of lamin A/C. This variant was not found in the China Genome Database (https://db.cngb.org), Human Exon Database (ExAC, http://exac.broadinstitute.org), Reference Population Thousand Genomes (1000G) (http://browser.1000genomes.org) or Population Genome Mutation Frequency Database (gnomAD, https://gnomad.broadinstitute.org). According to the American Society for Medical Genetics and Genomics (ACMG) guidelines (18) and Recommendations from the working group of Clingen Sequence Variant Interpretation (SVI) on the application of the guideline standards (19, 20), this c.1526del mutation of LMNA gene was a novel pathogenic variant with strong evidence for pathogenicity (PVS1 + PM2). Among the 7 family members who underwent genetic testing, two of the proband's sisters were found to be carrying the same pathogenic variant.
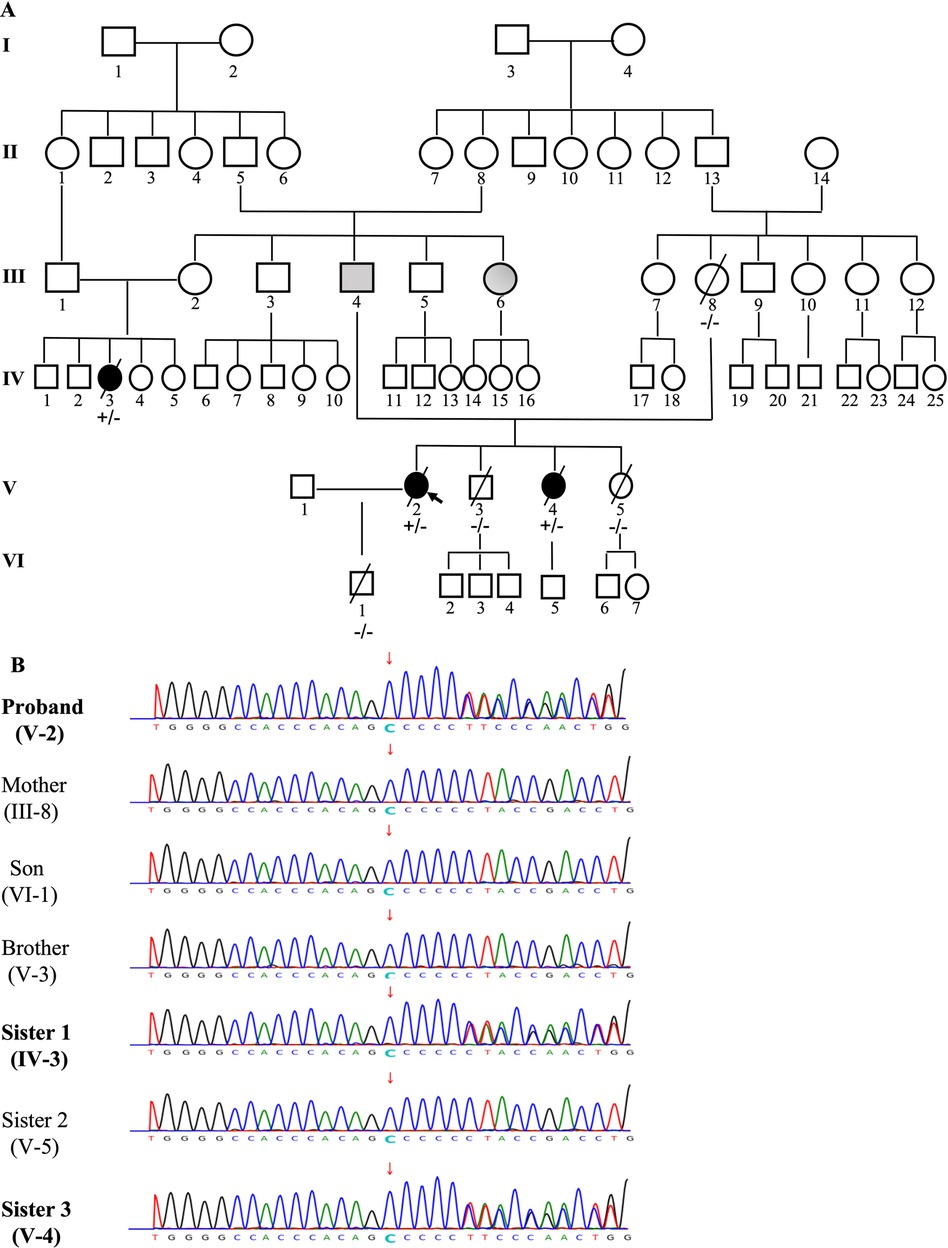
Figure 4. The pedigree of the family (A) and genetic testing results (B). The pedigree of this family was constructed based on a questionnaire on health history, medical records, and/or blood samples of available members that included 67 family members over six generations. Individuals are indicated by generation and pedigree number. Males and females are shown with squares and circles, respectively. Black circles/squares are affected (3 members), gray is presumedly affected (2 members), white is unaffected. Arrow indicates the proband (A). Genetic testing was performed in 7 members of the family (III-8, IV-3, V-2, V-3, V-4, V-5, VI-1). A pathogenic variant c.1526del: p.P509Lfs*39, a frameshift deletion in the LMNA gene in exon 9 on chromosome 1 was identified in 3 members ( IV-3, V-2, V-4), which causes changes in the open reading frame of the gene and results in loss-of-function of Lamin A/C (B).
The patients carrying the c.1526del pathogenic variant shared some similar clinical characteristics, including clinical symptoms, ECG and TTE findings. ECG revealed a slow arrhythmia, AV-conduction block rapidly progressing to atrial standstill, and non-sustained ventricular tachycardia. TTE showed right-sided cardiac enlargement and extreme enlargement of the right atrium. Left side of the heart was less involved. A bicuspid aortic valve malformation (type A) was also typically noted on TTE examination (Table 1).
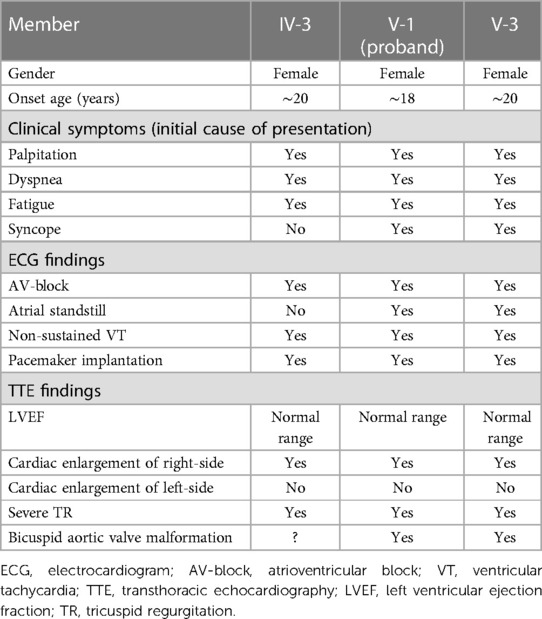
Table 1. Clinical characteristics of the patients carrying pathogenic variant of c.1526del: p.P509Lfs*39.
DCM is a frequent cause of heart failure and sudden cardiac death worldwide. It is now well established the significance of genetic etiology with currently more than 40 DCM genes implicated. Among them mutations on TTN gene (encoding titin which provides structure, flexibility, and stability to sarcomeres) was the largest contributor, following by LMNA gene, SCN5A and DES gene (21, 22). The LMNA gene is composed of 12 exons and encodes intermediate filament proteins lamin A and C via alternative splicing of exon 10 (23, 24). The lamin A/C proteins polymerize to form a scaffold called the nuclear lamina (NL) at the nuclear periphery, which consists of a long α-helical domain flanked by globular amino-terminal (head) and carboxy-terminal (tail) domains. Lamins first dimerize using their α-helical rod domain, then polymerize in a polar head-to-tail manner, forming protofilaments. Between three and four protofilaments associate laterally to form an intermediate filament (25, 26). Its expression pattern is not solely confined to the atria as it also exists in skeletal muscle and ventricular myocardium (23, 24). Lamin A/C possesses a key function in the stability of the nuclear envelope, chromatin organization, and DNA-repair (25). It plays a major role in transmitting mechanical signals to the nuclear core, transportation through the nuclear envelope, and regulation of transcription (27, 28). LMNA gene mutations typically present in an autosomal dominant manner and can result in a variety of phenotypes such as lipodystrophy, muscular disease, neuropathy, progeria, and cardiomyopathy (29, 30). The cardiac phenotype includes dilated cardiomyopathy, heart failure, progressive atrioventricular block (AVB), atrial fibrillation (AF), ventricular tachycardia and fibrillation (VT/VF), and SCD (16, 17, 25, 30). The most common manifestation of the cardiac phenotype is disturbance in the electrical conducting system presenting as atrioventricular block (AV-block), and both atrial and ventricular arrhythmias (31, 32). A meta-analysis by van Berlo JH et al. (31) showed that initial ECG findings in patients with the LMNA gene mutation appeared as low amplitude P-waves and PR-intervals, and 92% of patients had developed arrhythmias after age 30% and 28% received pacemaker therapy. The cardiac phenotype of the LMNA mutation can present as predominantly right-sided cardiomyopathy (15, 33). Zhang et al. (15) identified a pathogenic variant which was a missense variant in rod 2B domain (c.1003C > T p.R335W) of LMNA gene in a large Chinese family where affected members expressed clinical findings such as atrial enlargement, atrial arrhythmia, sick sinus syndrome and AV-block.
LMNA-related cardiomyopathy accounts for 5%–10% of familial DCM (31) and the clinical outcome is considerably worse for patients harboring LMNA mutations compared to those without (34, 35). Although the precise pathophysiological processes underlying the occurrence and progression of arrhythmia in patients carrying the pathogenic variant of LMNA gene remains unknown, the following mechanisms may be involved (1) decreased spontaneous action potential beat frequency, decreased pacemaker current (I-f) density, (2) prolonged action potential duration, increased L-type calcium current density, (3) delayed post-depolarizations (DADs), arrhythmias, and increased heart rate variability, (4) DADs, arrhythmias, and cessation of spontaneous discharges caused by beta-adrenergic stimulation and rapid pacing (36). Given the extremely poor prognosis of cardiomyopathy-causing in LMNA pathogenic variant carriers, early identification of affected family members is imperative. Genetic testing allows for both early identification and diagnosis of high-risk family members. If a pathogenic variant is identified in an asymptomatic individual, regular clinical cardiovascular screening (e.g., ECG) is recommended to detect the first signs of disease, which can then be managed through early intervention. If family members are not found to carry the mutation, they can be diagnosed as unaffected and do not require serial follow-up.
In this study, we identified a novel pathogenic variant in LMNA gene which has not been reported in literature thus far. This is a c.1526del (a single nucleotide deletion) located in exon 9 on chromosome 1 (HGVS: NM_170707.4, c.1526del: p.P509Lfs*39), causing a change in the open reading frame of the gene and resulting in loss-of-function of lamin A/C. The clinical manifestation of the affected members presented similarly to the pathogenic variant reported by Zhang et al. (15), but clinical symptoms of the c.1526del pathogenic variant presented earlier in life (∼20 years old). Although the patient's symptoms were nonspecific, auxiliary examination provided clues to warrant further investigation. ECG findings presented as gradual worsening of electrical conduction due to sinus bradycardia, sick sinus syndrome, AV-block, and atrial/ventricular arrhythmia progressing to eventual atrial electrical quiescence. A giant right atrium was a significant finding associated with this pathogenic variant and accompanied by RV enlargement and bicuspid aortic valve malformation. All affected members required eventual pacemaker implantation, with some possibly needing ICD implantation due to VT. Therefore, genetic screening of family members is necessary once familial inheritance is suspected as it could provide early diagnosis and treatment for the affected family members. Actually, a same site mutation of c.1526 (c.1526dup, p.Asn509fs) has been reported in ClinVar, however different from the pathogenic variant we identified which was a novel deletion mutation and presented as cardiac phenotype, this variants was seen in Charcot-Marie-Tooth disease type 2, cardiovascular phenotype was uncertain.
In summary, this study describes a novel cardiac phenotype caused by a newly identified pathogenic variant in LMNA gene (c.1526del p.P509Lfs*39) in a large family, where affected family members were clinically characterized by a giant right atrium, sick sinus syndrome, atrial/ventricular arrhythmia with progressive loss of atrial electric activity to atrial standstill, and potential accompanying bicuspid aortic valve malformation. This study expands the phenotype of the pathogenic variant in LMNA gene and also provides genetic information regarding atrial cardiomyopathy. To the best of our knowledge, this is a novel pathogenic variant first identified by our research team.
This study carries several limitations. First, medical records and samples were unavailable in the 1st and 2nd generations, and information from members of the 3rd–6th generation may not be entirely accurate. Second, genetic testing was not performed on all living family members due to cost and difficulty with travel, so the actual number of affected members could not be identified. Lastly, as this novel pathogenic variant in LMNA gene was determined to be autosomal dominant, there is a possibility that the proband's pathogenic variant of LMNA gene may come from her father. Unfortunately, the proband's father passed at an early age so his gene mutation status could not be verified.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author’s.
The studies involving human participants were reviewed and approved by the Institutional Ethical Committee on Human Research at the Wuhan Asia Heart Hospital. The patients/participants provided their written informed consent to participate in this study.
SN, MH, RQ, XH and ZX contributed to patient diagnosis, treatment, and follow-up. SN and LL drafted this manuscript. LL and CL revised the final version of the manuscript. All authors agreed to be accountable for the content of the work. All authors contributed to the article and approved the submitted version.
We would like to thank Xuan He for the diligent assistance in preparing select materials.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Dong M, Liu T, Li G. Atrial cardiomyopathy—a not yet classified cardiomyopathy? Int J Cardiol. (2011) 151:394–6. doi: 10.1016/j.ijcard.2011.07.003
2. Goette A, Kalman JM, Aguinaga L, Akar J, Cabrera JA, Chen SA, et al. EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: definition, characterization, and clinical implication. EP Eur. (2016) 18:1455–90. doi: 10.1093/europace/euw161
3. Piérard LA, Henrard L, Demoulin JC. Persistent atrial standstill in familial Ebstein’s anomaly. Br Heart J. (1985) 53:594–7. doi: 10.1136/hrt.53.6.594
4. Fazelifar AF, Arya A, Haghjoo M, Sadr-Ameli MA. Familial atrial standstill in association with dilated cardiomyopathy. Pacing Clin Electrophysiol. (2005) 28:1005–8. doi: 10.1111/j.1540-8159.2005.00198.x
5. Habara M, Fujieda H, Nakamura Y. Images in cardiology: atrial myocarditis: a possible cause of idiopathic enlargement of bilateral atria. Heart. (2006) 92:842. doi: 10.1136/hrt.2005.077420
6. Maeda S, Tanaka T, Hayashi T. Familial atrial standstill caused by amyloidosis. Br Heart J. (1988) 59:498–500. doi: 10.1136/hrt.59.4.498
7. Hong JS, Ki CS, Kim JW, Suh YL, Kim JS, Baek KK, et al. Cardiac dysarrhythmias, cardiomyopathy and muscular dystrophy in patients with Emery-Dreifuss muscular dystrophy and limb-girdle muscular dystrophy type 1B. J Korean Med Sci. (2005) 20:283–90. doi: 10.3346/jkms.2005.20.2.283
8. Nagle RE, Smith B, Williams DO. Familial atrial cardiomyopathy with heart block. Br Heart J. (1972) 34:205. doi: 10.1136/hrt.34.3.244
9. Tan RB, Gando I, Bu L, Cecchin F, Coetzee W. A homozygous SCN5A mutation associated with atrial standstill and sudden death. Pacing Clin Electrophysiol. (2018) 21:1–7. doi: 10.1111/pace.13386
10. Groenewegen WA, Firouzi M, Bezzina CR, Vliex S, van Langen IM, Sandkuijl L, et al. A cardiac sodium channel mutation cosegregates with a rare connexin 40 genotype in familial atrial standstill. Circ Res. (2003) 92:14–22. doi: 10.1161/01.RES.0000050585.07097.D7
11. Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. (2003) 299:251–4. doi: 10.1126/science.1077771
12. Disertori M, Quintarelli S, Grasso M, Pilotto A, Narula N, Favalli V, et al. Autosomal recessive atrial dilated cardiomyopathy with standstill evolution associated with mutation of natriuretic peptide precursor A. Circ Cardiovasc Genet. (2013) 6:27–36. doi: 10.1161/CIRCGENETICS.112.963520
13. Sakata K, Shimizu M, Ino H, Yamaguchi M, Terai H, Fujino N, et al. High incidence of sudden cardiac death with conduction disturbances and atrial cardiomyopathy caused by a nonsense mutation in the STA gene. Circulation. (2005) 111(25):3352–8. doi: 10.1161/CIRCULATIONAHA.104.527184
14. Yokokawa T, Ichimura S, Hijioka N, Kaneshiro T, Yoshihisa A, Kunii H, et al. Case reports of a c.475G > T, p.E159* lamin A/C mutation with a family history of conduction disorder, dilated cardiomyopathy and sudden cardiac death. BMC Cardiovasc Disord. (2019) 19(1):298. doi: 10.1186/s12872-019-01282-6
15. Zhang Y, Lin Y, Zhang Y, Wang Y, Li Z, Zhu Y, et al. Familial atrial myopathy in a large multi-generational heart-hand syndrome pedigree carrying LMNA missense variant in rod 2B domain (p.R335W). Heart Rhythm. (2022) 19(3):466–75. doi: 10.1016/j.hrthm.2021.11.022
16. Brauch KM, Chen LY, Olson TM. Comprehensive mutation scanning of LMNA in 268 patients with lone atrial fibrillation. Am J Cardiol. (2009) 103:1426–8. doi: 10.1016/j.amjcard.2009.01.354
17. Crasto S, My I, Di Pasquale E. The broad spectrum of LMNA cardiac diseases: from molecular mechanisms to clinical phenotype. Front Physiol. (2020) 11:761. doi: 10.3389/fphys.2020.00761
18. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–23. doi: 10.1038/gim.2015.30
19. Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. (2018) 39(11):1517–24. doi: 10.1002/humu.23626
20. Ghosh R, Harrison SM, Rehm HL, Plon SE, Biesecker LG, Clingen Sequence Variant Interpretation Working Group. Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum Mutat. (2018) 39(11):1525–30. doi: 10.1002/humu.23642
21. Mestroni L, Brun F, Spezzacatene A, Sinagra G, Taylor MRG. Genetic causes of dilated cardiomypothy. Prog Pediatr Cardiol. (2014) 37(1-2):13–8. doi: 10.1016/j.ppedcard.2014.10.003
22. Gerull B, Klaassen S, Brodehl A. The genetic landscape of cardiomyopathies. Genetic Causes of Cardiac Disease, 45–91. Part of the Cardiac and Vascular Biology book series. (2019). Electronic ISSN 2509-7849, Print ISSN 2509-7830.
23. Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. (1993) 268:16321–6. doi: 10.1016/S0021-9258(19)85424-8
24. Wydner KL, McNeil JA, Lin F, Worman HJ, Lawrence JB. Chromosomal assignment of human nuclear envelope protein genes LMNA, LMNB1, and LBR by fluorescencein situhybridization. Genomics. (1996) 32:474–8. doi: 10.1006/geno.1996.0146
25. Ho CY, Lammerding J. Lamins at a glance. J Cell Sci. (2012) 125:2087–93. doi: 10.1242/jcs.087288
26. Dittmer TA, Misteli T. The lamin protein family. Genome Biol. (2011) 12:222. doi: 10.1186/gb-2011-12-5-222
27. Simon DN, Wilson KL. The nucleoskeleton as a genome-associated dynamic “network of networks”. Nat Rev Mol Cell Biol. (2011) 12:695–708. doi: 10.1038/nrm3207
28. Yamada S, Ko T, Ito M, Sassa T, Nomura S, Okuma H, et al. TEAD1 Trapping by the Q353R-lamin A/C causes dilated cardiomyopathy. Sci Adv. (2023) 9(15):eade7047. doi: 10.1126/sciadv.ade7047
29. Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. (2005) 45:969–81. doi: 10.1016/j.jacc.2004.11.066
30. Brayson D, Shanahan CM. Current insights into LMNA cardiomyopathies: existing models and missing LINCs. Nucleus. (2017) 8:17–33. doi: 10.1080/19491034.2016.1260798
31. van Berlo JH, de Voogt WG, van der Kooi AJ, Peter van Tintelen J, Bonne G, Yaou RB, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med. (2005) 83:79–83. doi: 10.1007/s00109-004-0589-1
32. Peretto G, Sala S, Benedetti S, Di Resta C, Gigli L, Ferrari M, et al. Updated clinical overview on cardiac laminopathies: an electrical and mechanical disease. Nucleus. (2018) 9:380–91. doi: 10.1080/19491034.2018.1489195
33. Quarta G, Syrris P, Ashworth M, Jenkins S, Zuborne Alapi K, Morgan J, et al. Mutations in the lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. (2012) 33:1128–36. doi: 10.1093/eurheartj/ehr451
34. Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. (2008) 156:161–9. doi: 10.1016/j.ahj.2008.01.026
35. Kumar S, Baldinger SH, Gandjbakhch E, Maury P, Sellal J-M, Androulaki AFA, et al. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J Am Coll Cardiol. (2016) 68:2299–307. doi: 10.1016/j.jacc.2016.08.058
Keywords: LMNA gene mutation, pathogenic variant, atrial cardiomyopathy, right atrial enlargement, atrial standstill, bicuspid aortic valve malformation
Citation: Ning S, Han M, Qiu R, Hong X, Xia Z, Liu L and Liu C (2023) Novel pathogenic variant in LMNA gene identified in a six-generation family causing atrial cardiomyopathy and associated right atrial conduction arrhythmias. Front. Cardiovasc. Med. 10:1109008. doi: 10.3389/fcvm.2023.1109008
Received: 27 November 2022; Accepted: 14 June 2023;
Published: 3 July 2023.
Edited by:
Seitaro Nomura, The University of Tokyo, JapanReviewed by:
Maria Rosaria D’Apice, Policlinico Tor Vergata, Italy© 2023 Ning, Han, Qiu, Hong, Xia, Liu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Liu bGl1bGlid2hAMTYzLmNvbQ== Chengwei Liu d2hsY3c2MEAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.