
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 10 May 2023
Sec. Atherosclerosis and Vascular Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1099069
 Ramesh Chennupati1*†
Ramesh Chennupati1*† Isabella Solga1,†
Isabella Solga1,† Patricia Wischmann1,†Paul Dahlmann1
Patricia Wischmann1,†Paul Dahlmann1 Feyza Gül Celik1Daniela Pacht1Aslıhan Şahin1Vithya Yogathasan1
Feyza Gül Celik1Daniela Pacht1Aslıhan Şahin1Vithya Yogathasan1 Mohammad Rabiul Hosen2
Mohammad Rabiul Hosen2 Norbert Gerdes1,3Malte Kelm1,3
Norbert Gerdes1,3Malte Kelm1,3 Christian Jung1,3*
Christian Jung1,3*
Background: In acute myocardial infarction and heart failure, anemia is associated with adverse clinical outcomes. Endothelial dysfunction (ED) is characterized by attenuated nitric oxide (NO)-mediated relaxation responses which is poorly studied in chronic anemia (CA). We hypothesized that CA is associated with ED due to increased oxidative stress in the endothelium.
Methods: CA was induced by repeated blood withdrawal in male C57BL/6J mice. Flow-Mediated Dilation (FMD) responses were assessed in CA mice using ultrasound-guided femoral transient ischemia model. Tissue organ bath was used to assess vascular responsiveness of aortic rings from CA mice, and in aortic rings incubated with red blood cells (RBCs) from anemic patients. In the aortic rings from anemic mice, the role of arginases was assessed using either an arginase inhibitor (Nor-NOHA) or genetic ablation of arginase 1 in the endothelium. Inflammatory changes in plasma of CA mice were examined by ELISA. Expression of endothelial NO synthase (eNOS), inducible NO synthase (iNOS), myeloperoxidase (MPO), 3-Nitrotyrosine levels, and 4-Hydroxynonenal (4-HNE) were assessed either by Western blotting or immunohistochemistry. The role of reactive oxygen species (ROS) in ED was assessed in the anemic mice either supplemented with N-Acetyl cysteine (NAC) or by in vitro pharmacological inhibition of MPO.
Results: The FMD responses were diminished with a correlation to the duration of anemia. Aortic rings from CA mice showed reduced NO-dependent relaxation compared to non-anemic mice. RBCs from anemic patients attenuated NO-dependent relaxation responses in murine aortic rings compared to non-anemic controls. CA results in increased plasma VCAM-1, ICAM-1 levels, and an increased iNOS expression in aortic vascular smooth muscle cells. Arginases inhibition or arginase1 deletion did not improve ED in anemic mice. Increased expression of MPO and 4-HNE observed in endothelial cells of aortic sections from CA mice. NAC supplementation or inhibition of MPO improved relaxation responses in CA mice.
Conclusion: Chronic anemia is associated with progressive endothelial dysfunction evidenced by activation of the endothelium mediated by systemic inflammation, increased iNOS activity, and ROS production in the arterial wall. ROS scavenger (NAC) supplementation or MPO inhibition are potential therapeutic options to reverse the devastating endothelial dysfunction in chronic anemia.
Endothelial dysfunction (ED) is central to a plethora of cardiovascular diseases (CVD). ED is defined by the inability to maintain vascular homeostasis due to decreased nitric oxide (NO) production (1, 2). NO is a major vasodilator and anti-inflammatory signalling molecule, which plays an important role in vascular tone and blood pressure regulation. Decreased NO bioavailability disrupts the non-thrombogenic intimal surface, which promotes adhesion and aggregation of platelets and monocytes to the activated endothelial surface (3). The bioavailability of NO mainly depends on the availability of eNOS' substrate, arginine, and coupling state of eNOS. Arginases are catabolizing enzymes of arginine, which share the common substrate with eNOS, and are known to limit NO bioavailability (4, 5). Both isoforms of arginase (1 and 2) are expressed in endothelial cells of mice and humans (6). Endothelial arginase 1 is known to contribute to ED in coronary arteries of diabetic patients (7), and animal models of hypertension, ischemia/reperfusion, and aging (8, 9). In addition, endothelial arginase 2 upregulation and associated NO dysregulation have been reported in diabetes, aging (10), and hyperlipidemia (11). Inhibition of arginases led to increased NO-bioavailability and consequently improved the endothelium-dependent relaxation responses in different disease states (8).
The bioavailability of NO depends on the coupling state of eNOS, which relies on several factors such as oxidative depletion of tetrahydrobiopterin (BH4), S-glutathionylation, reactive oxygen, and nitrogen species (12). Concomitantly, uncoupled eNOS in the NO generation system leads to the production of superoxide anions (O2 −) rather than NO (13). Thus, superoxide anions are not only associated with reduced NO production but also enhance pre-existing oxidative stress. Additionally, peroxynitrite is known to cause depletion of cofactor BH4, which leads to uncoupling of eNOS and thus, reduced NO-bioavailability (14). Recent studies also showed that myeloperoxidase (MPO), which is upregulated in endothelial cells during inflammation, limits NO-bioavailability by producing reactive oxidants such as hypochlorous acid (15).
Anemia is frequently observed in patients with CVD such as acute and chronic coronary syndromes as well as heart failure. Approximately 30 to 40% of patients with acute myocardial infarction (AMI) have evident anemia upon admission (16), or during hospitalization (17). It is well-studied that anemia alone or in combination with other morbid conditions leads to poor prognosis in AMI (18). These studies have concluded that dysfunctional RBCs in anemia influence the severity of CVD. We recently showed that acute blood-loss anemia transiently increases FMD due to an acute increase in shear stress and cardiac output. Simultaneously acute anemia induces RBC dysfunction by reducing NO bioavailability, increasing ROS formation, reducing membrane integrity, and increasing NO scavenging by free plasma hemoglobin (19). However, it is unclear whether the complications related to chronic anemia are restricted to RBC dysfunction or also associated with systemic alteration in endothelial cells that may result in ED.
In this study, we evaluated the systemic ED in a newly established murine blood-loss chronic anemia model. Additionally, we also investigated whether the RBCs from anemic patients affect the endothelial function using co-incubation with aortic rings followed by endothelial function analysis. To our knowledge, this is the first study to evaluate systemic ED in a blood-loss anemia murine model.
All procedures were approved and performed in accordance with the guidelines of LANUV (Landesamt für Natur, Umwelt- und Verbraucherschutz Nordrhein-Westfalen, Germany). Mice were treated by following the European Convention for the protection of Vertebrate Animals used for Experimental and other Scientific Purposes (Council of Europe Treaty Series No. 123). The approved permits for the animal experiments are 84-02.04. 2020.A073 and 84-02.04.2018.A234. The C57Bl/6J (wild type, WT) mice were obtained from Janvier Labs (Saint-Berthevin Cedex, France). Mice with endothelial ablation of arginase 1 (ARG1) were generated by crossing Arg1fl/fl (C57BL/6-Arg1tm1Pmu/J, Jackson laboratory) mice with tamoxifen-inducible Cdh5-creERT2 [Tg(Cdh5-cre/ERT2)1Rha, MGI:3848982] mice which were kindly provided by Prof Dr E. Lammert (Heinrich-Heine-University of Düsseldorf). To induce the recombination, the mice were injected with 50 mg/Kg body weight tamoxifen (Sigma) for 5 consecutive days. The resulting mice that lack ARG1 in their endothelium Arg1fl/fl/Cdh5-cre/ERT2-pos are named as EC-Arg1-KO in the manuscript. The respective Cre-negative littermates were used as controls. All mice were housed in standard cages (constant room temperature and humidity, 12hr light/dark cycles) and had free access to standard pelleted chow and tap water.
To induce anemia, WT, EC-Arg1-KO and respective control mice were divided into two groups, a sham group without anemia and a group with induced chronic anemia. To induce chronic anemia, a repetitive mild blood withdrawal (by <20 g/L changes in Hb) from the facial vein was performed every third day for a time period of six weeks. At the end of the 6th week mice which showed hemoglobin levels < 10 g/L, are considered for experiments. The hematological parameters after anemia induction are represented in Supplementary Figure S1.
Supplementation of NAC: Male (WT) mice, which were 10–11 week-old, were induced with chronic anemia. After two weeks of anemia induction, anemic and respective sham mice were given 1% (w/v) NAC (Sigma) through drinking water for 4 weeks. After 6 weeks of anemia, mice were sacrificed and tissues were collected.
The 10–11 week-old C57BL/6J male mice were used for the assessment of flow-mediated dilation (FMD) responses. The measurements were performed before, and after 2-, 4-, 6, -weeks of anemia induction. FMD responses were assessed using the Vevo 2,100 high-resolution ultrasound scanner using a 30–70 MHz linear transducer (Visual Sonics Inc., Toronto, Canada) as described previously (20). Mice were kept under 1.5%–2% isoflurane anesthesia, the transducer was placed using a stereotactic holder and adjusted manually to visualise the external iliac artery. A vascular occluder (8 mm diameter, Harvard Apparatus, Boston, MA, USA) was placed around the lower limb (20). Baseline images of the vessel were first recorded, the cuff was inflated to 200 mmHg, and pressure was kept constant for 5 min (Druckkalibriergerät KAL 84, Halstrup Walcher, Kirchzarten, Germany); then the cuff was released to assess FMD. The upstream diameter of the vessel was determined every 30 s both during inflation and deflation of the cuff. Changes in vessel diameter were quantified as the percentage of baseline (%) = [diameter (max)/diameter (baseline)] × 100.
Krebs-Ringer bicarbonate-buffered salt solution (KRB) contained (in mM): 118.5 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25.0 NaHCO3 and 5.5 glucose. The KRB solution was continuously aerated with 95% O2/5% CO2 and maintained at 37°C. Indomethacin (INDO; Sigma Aldrich) was dissolved in ethanol. Acetylcholine (ACH), phenylephrine (PHE), NG-nitro-arginine methyl ester (L-NAME) and sodium nitroprusside (SNP; all Sigma Aldrich) were dissolved in KRB solution. Myeloperoxidase inhibitor (AZD5904) was purchased from PromoCell and dissolved in dimethyl sulfoxide (DMSO).
Animals were euthanized under deep isoflurane anesthesia (4.5%). The thoracic aorta was dissected free from perivascular adipose tissue, and 2 mm size aortic rings were mounted in an organ bath equipped with force transducers (Hugo Sachs Electronic, HARVARD apparatus GmbH, March-Hugstetten Germany) or wire-myograph system (Danish Myotechnology, Aarhus, Denmark). Arterial segments were distended stepwise to 1 gr force or 9.8 mN. All aortic segments were allowed 40 min incubation prior to experiments and stimulated with depolarizing 60 mM KCl as described previously (21). Prior to experiment, endothelial integrity was checked by assessing relaxation responses to ACH (10 µM) in PHE (10 µM) pre-contracted arteries. The aortic segments were pre-contracted and segments which showed less than 80% relaxation responses were excluded from experiments.
Initially, a concentration-response curve (CRC) for PHE (0.001–10 μM) was constructed. During the contraction induced by 10 µM PHE, an ACH CRC (1 nM–10 μM) was generated. Next, experiments were repeated in the presence of the cyclooxygenase inhibitor INDO (10 µM) and in the presence of both INDO and the NOS inhibitor L-NAME (100 µM) to assess the contribution of NO to arterial relaxation.
During contraction of the aortic rings with PHE (10 µM) in the presence of INDO (10 µM) and L-NAME (100 µM), the relaxing effects of the NO donor SNP (10 nM–10 µM) were recorded.
The protocol is based on previous studies (22) with adjustments for murine aortic rings. Briefly, thoracic aortas from 10 to 11 week-old mice were dissected and placed in cold KRB buffer. Anemic and non-anemic patient blood was collected in heparin-coated tubes, and 40% hematocrit (Hct) was prepared with KRB buffer. Aortic segments were transferred to the 40% Hct containing KRB buffer and placed in an incubator for 6 h at 37°C. We previously (unpublished data) assessed the best suitable time for co-incubation experiments based on the viability and maximum responses to agonists (PHE, ACH) of aortic rings. After 6 h of incubation, aortic segments were mounted in the wire-myograph system and endothelial-dependent relaxation responses were assessed. Approval numbers for patient samples collection are 5481R, 2018–14, and 2018–47 issued by clinical ethics committee, Universitätsklinikum Düsseldorf, Germany. All patients were given their written consent according to Helsinki ethical principles.
The thoracic aortas were thawed and homogenized using a mixer mill (MM400 Retsch Haan, Germany) and a brief sonification in a radioimmunoprecipitation assay (RIPA) buffer. For further processing, the protein concentration was determined with the DC™ Protein Assay Kit (Bio-Rad, Feldkirchen, Germany). The samples were denatured and loaded on a 4%–12% Bis-Tris gradient gel QPAGE™ (Smobio, Hsinchu City, Taiwan). Proteins were then transferred onto nitrocellulose membranes and a total protein staining was performed by using Revert Total Protein Stains (LI-COR Biosciences GmbH, Bad Homburg, Germany). The unspecific sites were blocked using intercept TBS Protein-Free Blocking Buffer (LI-COR) for 60 min at RT. Next, the membrane was incubated with either iNOS polyclonal antibody (1:1,000 in blocking buffer; ThermoFisher Scientific, Schwerte, Germany) or 3-Nitrotyrosine monoclonal antibody (1:1,000 in blocking buffer; Santa Cruz Biotechnology, Inc., Heidelberg, Germany) for overnight at 4°C. Afterwards, the membranes were incubated with corresponding secondary antibodies (IRDye® 700/800CW secondary antibodies; LI-COR) for 1 h at room temperature. The membranes were visualized with the Odyssey® Fc Imaging System (LI-COR).
Thoracic aortas from anemic and respective sham mice were collected, fixed in formaldehyde (4%) for 2 h, and treated with sucrose (30%) overnight. Next, the aortic segments were embedded in Tissue-Tek O.C.T and frozen until further use. Tissue sections (4 µm) were incubated overnight at 4°C with rat anti-mouse CD31 [1:100 in blocking solution (2.5% BSA); BD Biosciences, Heidelberg, Germany] and either with mouse anti-eNOS/NOS Type III (1:100 in 2.5% BSA; BD Biosciences), mouse anti-iNOS (1:100 in 2,5% BSA; Abcam), rabbit anti-4HNE (1:200 in 2,5% BSA; Abcam) or rabbit anti-myeloperoxidase (1:200 in 2,5% BSA; antibody.com) antibodies. Followed by incubating with respective anti-Mouse Alexa Fluor 555 (1:1,000 in 2.5% BSA; Thermo Fisher), goat anti-rat Alexa Fluor 488 (1:1,000 in 2.5% BSA; ThermoFisher) or anti-Rabbit Alexa Fluor 555 (1:1,000 in 2.5% BSA; ThermoFisher) secondary antibodies. Additionally, autofluorescence was quenched using Vector® TrueVIEW® Autofluorescence Quenching Kit (BIOZOL Vectorlabs, Eching, Germany). The stained tissue sections were mounted with the ProLong™ Diamond Antifade Mountant mounting medium (ThermoFisher) and imaged with a Leica DM6 M microscope.
The blood was collected from different experimental groups, plasma was prepared and snap-frozen. Plasma samples were stored at −80°C until further use. Mouse ICAM-1/CD54, VCAM-1/CD106, ELISA kits were purchased from R&D Systems (Bio-Techne GmbH, Wiesbaden Germany). Additionally, mouse haptoglobin, free hemoglobin, and transferrin plasma levels were assessed by using ELISA kits from Abcam. Mouse soluble transferrin receptor, ferritin and erythropoietin ELISA kits were obtained from MyBioSource (BIOZOL, Eching, Germany). All assays were performed according to the manufacturer's instructions. The plasma inflammatory markers were analyzed using a multiplex system (Bioplex, BioLegend).
After 6 weeks of anemia induction, the thoracic aorta was isolated from anemic and respective sham mice, and snap-frozen until further use. The aortas were thawed and homogenized in a lysis buffer using a mixer mill (MM400 Retsch). RNA was isolated from tissue lysates using RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. Reverse transcription was performed following the manufacturer's instructions using Superscript™ IV VILO™ Master Mix (Invitrogen). The real-time PCR was performed using the QuantStudio-7 Flex (ThermoFisher). The TaqMan probes (ThermoFisher) used for real-time PCR were glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Mm03302249_g1), MPO (Mm00447886_m1) and iNOS (Mm00440502_m1,). Relative expression levels were obtained by normalization with GAPDH.
All concentration response curves (CRCs) for contractile stimuli are expressed as absolute values. Relaxing responses were expressed as a percentage reduction of the level of contraction. Individual CRCs were fitted to a non-linear sigmoid regression curve (Graphpad Prism 8.0). Data normality was checked with D'Agostino Person test. The normally distributed data for multiple groups were analysed with two-way analysis of variance (ANOVA) with Bonferroni's post-hoc test and significance was calculated for all the concentration–response curves. Comparisons between two groups were performed with unpaired t-test or Mann–Whitney test depending on the normality. Sensitivity (pEC50), and maximal effect (Emax) are shown as means ± SEM.
To assess the effect of CA on endothelial function in vivo, we determined flow-mediated dilation (FMD) responses before induction of anemia (basal) and after 2-, 4-, 6- weeks of anemia. The average FMD responses significantly abrogated after 4- and 6-weeks (6 ± 1% and 5 ± 2%, respectively) of anemia induction compared to before induction of anemia (13 ± 2%) (Figures 1A,B) indicating the progressive ED in CA. Furthermore, we assessed the endothelium-dependent and independent relaxation responses in the isolated aortic rings using an organ bath. The CA mice showed a reduced relaxation (Emax 68 ± 4%) response to acetylcholine (ACH) compared to sham mice (Emax 84 ± 4%) (Figure 2A; Table 1). Furthermore, in the presence of NOS inhibitor (L-NAME, 100 µM), relaxation responses were completely inhibited in both anemic and sham groups (Figure 2B; Table 1), which shows that the relaxation responses are entirely mediated by NO. In addition, the relaxation responses to exogenous NO donor [Sodium nitroprusside (SNP)] are similar in CA mice compared to the sham group suggesting that smooth muscle sensitivity to NO is unaltered in CA mice compared to sham mice (Figure 2C; Table 1). Taken together, CA mice showed reduced endothelium-dependent relaxation responses. These results demonstrate the altered endothelial function in CA.
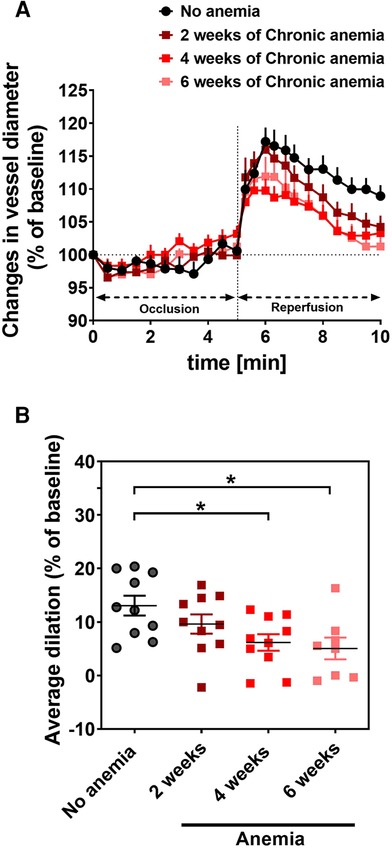
Figure 1. Chronic anemia is associated with altered flow-mediated dilation (FMD) responses. (A) Changes in vessel diameter (% ratio of baseline diameter) before (black circles), 2 (dark red squares), 4 (red squares), and 6 weeks (light red squares) after anemia induction. The occlusion and releasing phase of the cuff (reperfusion) are indicated in the panel. (B) Average FMD in the reperfusion phase (6–10 min) was normalized to baseline. Values are shown as the means ± SEM (n = 10 per group).*, p ≤ 0.05. One-Way ANOVA with Sidak's post-hoc test was used to compare average FMD between baseline and respective week.
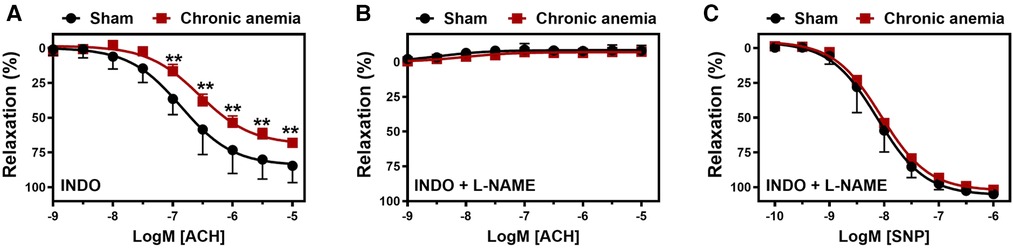
Figure 2. Chronic anemia is associated with reduced endothelial-dependent relaxation responses. Aortic rings were isolated from sham (black circles) and chronic anemic (dark red squares) mice. Aortic segments were pre-contracted with phenylephrine (10 µM) and relaxation responses to acetylcholine (ACH, 1 nM–10 µM) were measured using organ bath. (A) relaxation in the presence of indomethacin (10 µM, COX inhibitor). (B) relaxation in the presence of indomethacin and L-NAME (100 µM, NOS inhibitor). (C) Relaxation responses to sodium nitroprusside (SNP, 10 nM-10 µM) in the presence of indomethacin and L-NAME. All values are mean values ± SEM (n = 8–10 per group). **, p ≤ 0.01. CRCs were analysed by Two-Way ANOVA and Bonferroni ‘s post-hoc test to compare sham and chronic anemic groups.
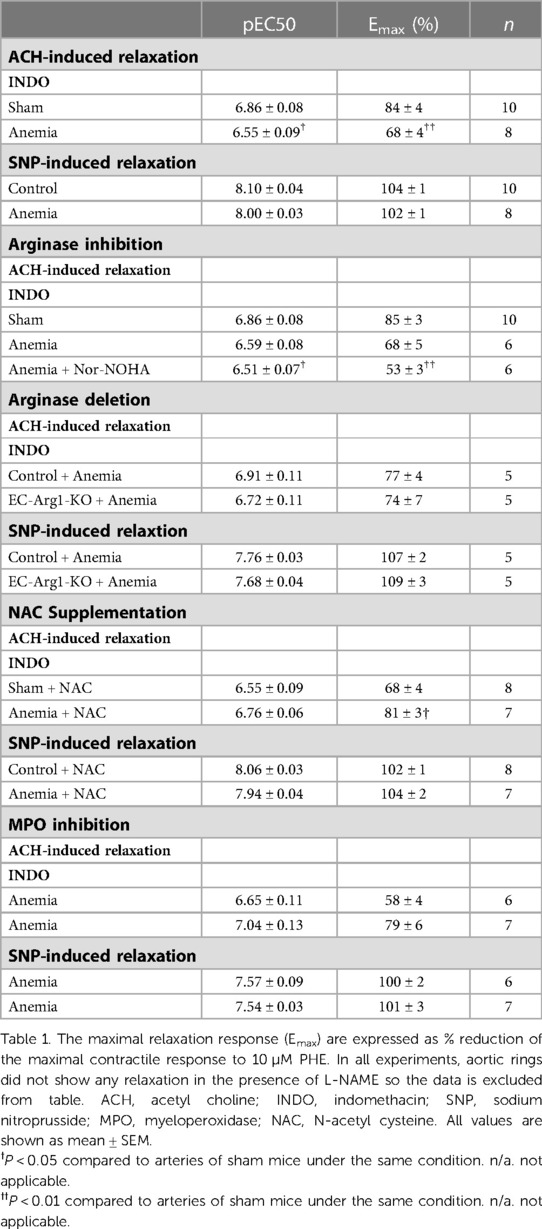
Table 1. Effect of anemia on endothelial-dependent and -independent relaxation responses.
Arginase 1 activity is known to be enhanced in various pathological states and is also known to limit NO bioavailability (4, 23). We assessed the relaxation responses after arginase inhibition in anemic mice. The aortic segments from anemic mice treated with Nor-NOHA (non-specific inhibitor of Arginase 1 & 2) did not show any improvement in the relaxation responses (Emax 53 ± 3%) compared to untreated (Emax 68 ± 5%) aortic segments (Figure 3A; Table 1). In addition, endothelial-dependent relaxation responses are not improved in anemic EC-Arg1-KO (Emax 74 ± 6%) and respective anemic control mice (Emax 77 ± 4%) compared to the sham mice (Emax 91 ± 5%) (Figure 3B). These relaxation responses are mediated by NO (Figure 3C; Table 1). The endothelium-independent relaxation to SNP was comparable in both groups (Figure 3D; Table 1). Taken together, these results exclude the potential role of arginases in ED associated with anemia.
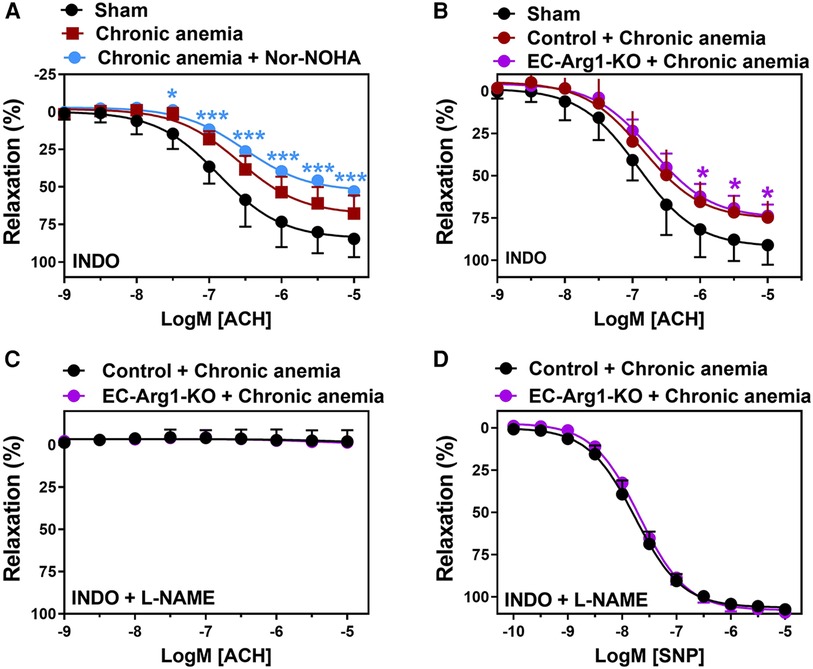
Figure 3. Inhibition of arginases or arginase 1 ablation does not improve endothelium- dependent relaxation responses in anemic mice. (A) Aortic rings were isolated from sham (black circles) and chronic anemic mice and incubated in the presence (blue circles) or absence (red squares) of Nor-NOHA (arginase inhibitor, 1 µM). Relaxation responses in the presence of indomethacin (10 µM, COX inhibitor) were measured in phenylephrine (10 µM) pre-contracted arteries. (B–D) Aortic rings were isolated from chronic anemic control (dark red circles) and EC-Arg1-KO mice (purple circles). All aortic segments were pre-contracted with phenylephrine (10 µM) and relaxation responses to acetylcholine (1 nM–10 µM) were measured using organ bath. (B) Relaxation in the presence of indomethacin (10 µM). (C) Relaxation in the presence of indomethacin and L-NAME (100 µM, NOS inhibitor). (D) Relaxation responses to sodium nitroprusside (SNP, 10 nM-10 µM) in the presence of indomethacin and L-NAME. All values are mean values ± SEM (n = 6–10 per group). *, p ≤ 0.05; ***, p ≤ 0.001. CRCs were analysed by Two-Way ANOVA and Bonferroni's post-hoc test to compare anemic Nor-NOHA treated or anemic EC-Arg1-KO with sham mice.
To evaluate whether the reduced relaxation responses in CA are also associated with decreased eNOS abundance, we analyzed the protein expression of eNOS in aortic lysates from anemic and respective sham mice. Interestingly, the eNOS levels are similar in CA mice and sham mice (Figure 4A, Supplementary Figure S5). In addition, immunohistochemical analyses also demonstrated similar patterns of eNOS expression in CA and sham mice (Figure 4B). We further assessed the levels of iNOS, a signature molecule for vascular inflammation. The aortic lysates from CA mice showed enhanced iNOS expression compared to sham mice (Figure 4C, Supplementary Figure S5). The immunohistochemistry analysis showed that iNOS expression is increased in vascular smooth muscle cells of CA mice compared to sham mice (Figure 4D).
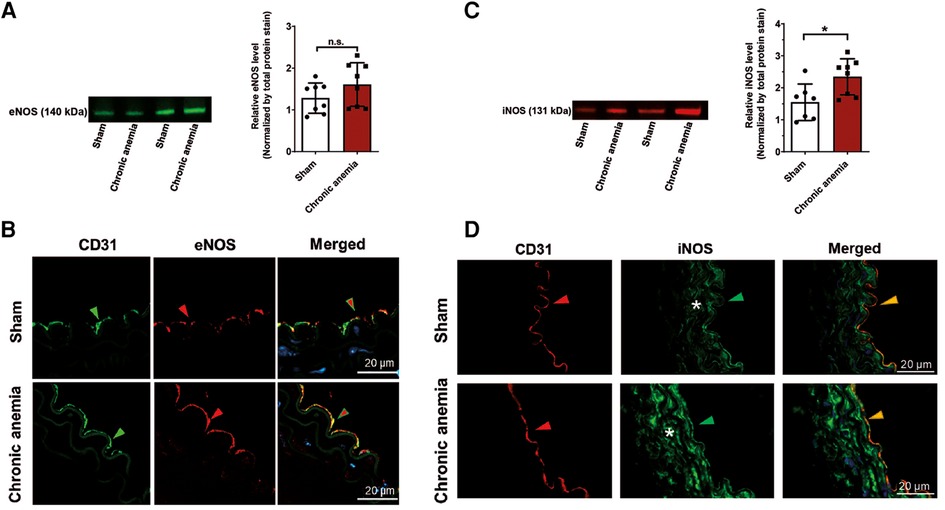
Figure 4. Expression of iNOS is increased in chronic anemic mice. (A,C) Representative Western blot images showing protein expression levels and quantification of eNOS (A) and iNOS (C) in the thoracic aortas isolated from sham and chronic anemic mice. Values are shown as means ± SEM (n = 7–8 per group). *, p ≤ 0.05. Unpaired t-test (A) or Mann–Whitney test (C) was used to compare between two groups. (B,D) Representative immunohistochemistry images showing the expression of CD31, eNOS, or iNOS. DAPI (blue) staining is used to detect the nucleus. Arrowheads are in the luminal side pointing towards the respective staining. Asterisk represents the smooth muscle area. Results are representative pictures of 3 (sham) and 3 (chronic anemic) independent experiments.
To investigate whether CA is associated with enhanced endothelial activation, we assessed plasma ICAM-1 and VCAM-1 level. The results showed that both ICAM-1 and VCAM-1 levels were increased in CA mice compared to sham mice (Figure 5A). Strikingly, we also observed increased plasma ICAM-1 and VCAM-1 levels in chronic coronary syndrome (CCS) patients with anemia compared to non-anemic patients (Figure 5B). Additionally, assessment of the systemic inflammatory markers demonstrated that IL6 and IL17A were progressively increased with duration of anemia (Supplementary Figure S2) demonstrating systemic inflammation in CA mice. To assess whether anemia leads to increased ROS generation in the aorta, we quantified generation of ROS-derived tyrosine adducts in the aortic lysates from anemic mice and control mice. Indeed, CA mice showed an enhanced 3-Nitrotyrosine levels in the aorta (Figure 5C). These results demonstrate that anemia results in the activation of endothelium and increased ROS production. Immunohistochemistry assessment of 4-hydroxynonenal (product of lipid peroxidation) in the aorta revealed an enhanced ROS activity in the CA mice (Figure 5D).
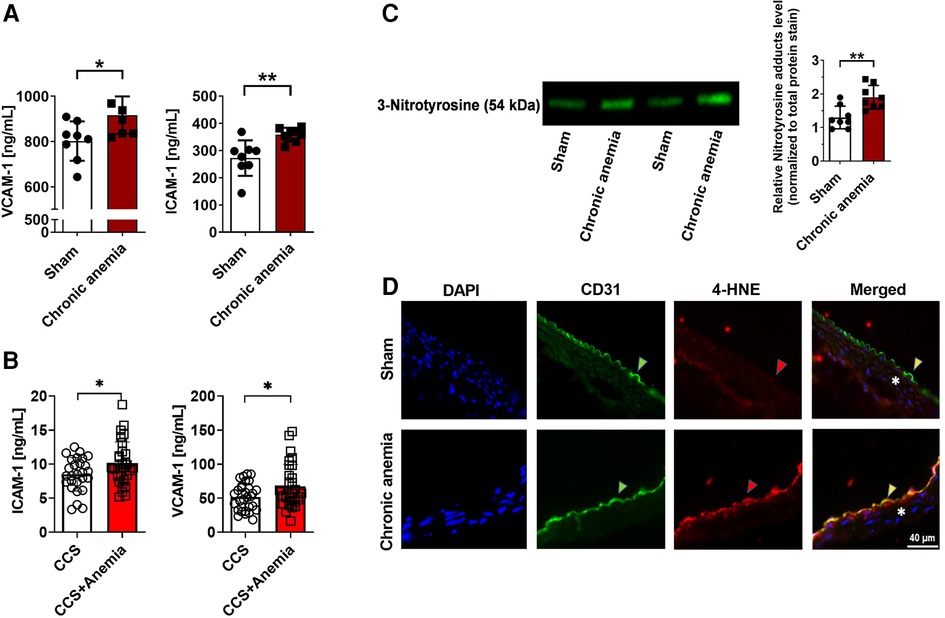
Figure 5. Chronic anemia is associated with increased inflammation and ROS formation. (A) Plasma VCAM-1 and ICAM-1 levels in sham (white bars), and chronic (dark red bar) anemic mice. (B) Plasma VCAM-1 and ICAM-1 levels in CCS patients without anemia (white bars) and with anemia (red bar). (C) Expression of nitrotyrosine protein adducts in the aortic lysates from sham (white bar), and anemic mice (red bar). Values are shown as means ± SEM. *, p ≤ 0.05; **, p ≤ 0.01. Unpaired t-test was used to compare between two groups. (D) Representative immunohistochemistry images of aortic sections stained with endothelial cell marker CD-31 (anti-CD31), and also for 4-HNE (anti-4-HNE). DAPI (blue) staining is used to detect the nucleus. Arrowheads are in the luminal side pointing towards the respective staining. Asterisk represents the smooth muscle area. Results are representative pictures of 5 (sham) and 5 (chronic anemic) independent experiments.
We assessed whether the ROS scavenger (NAC) improves the endothelial function and thus ED in CA mice. Aortic rings isolated from CA mice which were supplemented with NAC showed significantly improved endothelium-dependent relaxation responses (Emax 81 ± 3%) compared to untreated CA mice (Emax 68 ± 4%) (Figure 6A; Table 1). These relaxation responses are entirely attributed to NO (Figure 6B). The endothelium-independent relaxation to SNP was preserved in both groups (Figure 6C). A recent study showed that MPO expression is enhanced in ED as a consequence of vascular inflammation (15). Interestingly, the aortic lysates from CA mice also showed an increased expression of MPO compared to sham mice (Figure 6D, Supplementary Figure S5). MPO expression was also enhanced in the aortic endothelium of chronic anemic mice (Supplementary Figure S3). In addition, aortic rings treated with MPO inhibitor (AZD5904, 10 µM) showed significantly improved the relaxation responses in CA mice (Figures 6E–G; Table 1). These results conclude the potential role of ROS in anemia-associated ED.
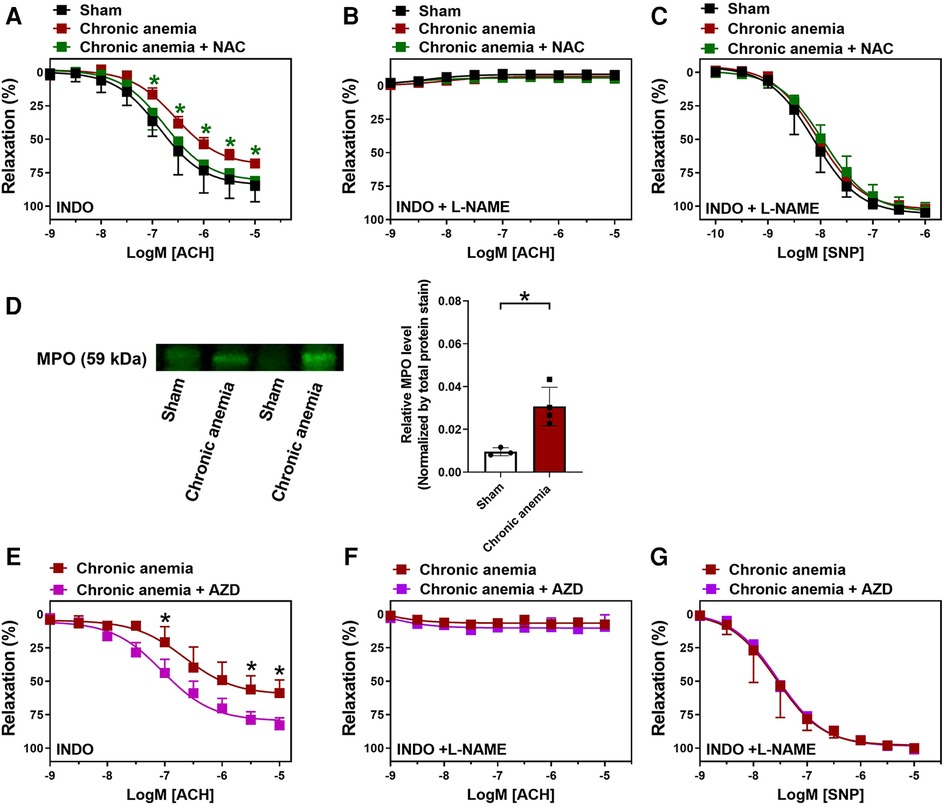
Figure 6. N-Acetyl cysteine (NAC) treatment or MPO inhibition improves the endothelium-dependent relaxation responses in chronic anemic mice. Aortic rings were isolated from sham (black circles), and chronic anemic mice with (green squares) and without (dark red squares) NAC supplementation. Aortic segments were pre-contracted with phenylephrine (10 µM) and relaxation responses to acetylcholine (1 nM–10 µM) were measured using an organ bath. (A) Relaxation in the presence of indomethacin (10 µM, COX inhibitor). (B) Relaxation in the presence of indomethacin and L-NAME (100 µM, NOS inhibitor). (C) Relaxation responses to sodium nitroprusside (SNP, 10 nM-10 µM) in the presence of indomethacin and L-NAME. (D) Protein level expression of MPO in the aortic lysates isolated from sham (white bar), and chronic anemic mice (red bar). In a different experimental setup (E–G), aortic rings from chronic anemic mice were incubated in the presence (pink squares) or absence (dark red squares) of myeloperoxidase inhibitor (AZD5904, 10 µM) and the relaxation responses were assessed to acetylcholine (E,F) and SNP. (G) All values are mean values ± SEM (n = 7–8 per group). *, p ≤ 0.05. CRCs were analysed by Two-Way ANOVA and Bonferroni's post-hoc test to compare treated (NAC or AZD) anemic group of mice with non-treated anemic mice. To compare MPO expression (D) Mann–Whitney test was used.
We further evaluated whether the dysfunctional RBCs impair the endothelial-dependent relaxation responses in anemia. Aortic rings from WT mice were incubated with RBCs (Hct 40%) isolated from CCS patients with and without anemia for 6 h at 37°C. The respective patients’ characteristics are summarized in Supplementary Table S1. In a separate set of experiments, aortic rings from WT mice were incubated with RBCs (Hct 40%) from anemic mice and sham mice. Evaluation of vascular function using wire-myograph demonstrated that aortic segments incubated with RBCs from anemic CCS patients show a reduced ACH-induced relaxation response (Emax 65 ± 5%; Figure 7A) compared to aortic segments incubated with RBCs of non-anemic CCS patients (Emax 82 ± 5%; Figure 7A). Inhibition of NOS by L-NAME demonstrated that these relaxation responses are mediated by NO (Figure 7B). Endothelium-independent relaxation responses to SNP were similar in both groups (Figure 7C). We also observed altered endothelium-dependent and independent relaxation responses in the aortic rings incubated with anemic mice aortic RBCs (Supplementary Figure S4). In addition, elevated expression of 4-HNE was observed in the aortic rings incubated with RBCs from anemic patients compared to aortic rings incubated with non-anemic patients (Figure 7D). Taken altogether, these results demonstrate that RBCs from anemic patients induce ROS production in the endothelium resulting in endothelial dysfunction due to altered NO-mediated relaxation responses.
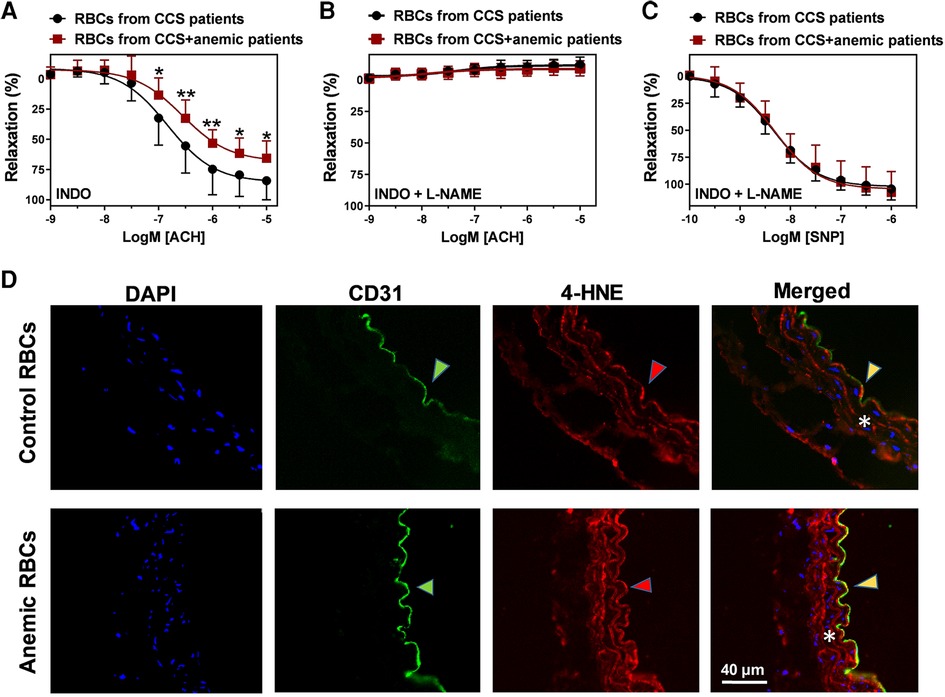
Figure 7. RBCs from anemic patients induce endothelial dysfunction in murine aortic rings. RBCs were isolated from chronic coronary syndrome (CCS) patients with anemia (red squares) and without anemia (black circles). Haematocrit (40%) was prepared in a KRB buffer. Aortic rings were incubated with haematocrit for 6 h at 37°C, mounted in a wire-myograph and pre-contracted with phenylephrine (10 µM). (A) Relaxation responses to acetylcholine (1 nM–10 µM) in the presence of indomethacin (10 µM, COX inhibitor). (B) Relaxation responses to acetylcholine in the presence of L-NAME (100 µM, NOS inhibitor) and indomethacin. (C) Endothelium-independent relaxation responses to (SNP, 10 nM-10 µM, NO donor). Values are shown as means ± SEM. *, p ≤ 0.05; **, p ≤ 0.01. CRCs were analysed by Two-Way ANOVA and Bonferroni's post-hoc test. Control group (stable CCS), n = 10; anemic group (CCS + Anemia), n = 12. (D) Thoracic aortas isolated from WT mice and incubated for 6 h at 37°C with RBCs from anemic or control patients. Representative immunohistochemistry images of aortic sections stained with endothelial cell marker CD-31 (anti-CD31), and 4-HNE (anti- 4-HNE). DAPI (blue) staining is used to detect the nucleus. Arrowheads are in the luminal side pointing towards the respective staining. Asterisk represents the smooth muscle area. Results are representative pictures of 5 (sham) and 5 (chronic anemic) independent experiments.
In this study, we evaluated the effects of chronic anemia on ED in an established blood loss anemia mouse model with a hypothesis that CA alters endothelial function and lead to ED. The major findings of the study are that (1) anemic mice show impaired in vivo FMD responses, and abrogated endothelial NO-dependent relaxation responses in vitro, (2) the abrogated endothelial-dependent relaxation responses in CA are not attributed to enhanced arginase activity, (3) eNOS expression is preserved whereas enhanced iNOS expression is observed in vascular smooth muscle cells of CA mice, (4) CA is associated with increased endothelial activation and ROS production, (5) scavenging of ROS improved the endothelial function in CA mice, (6) RBCs from anemic patients induce ED in murine aortic rings.
We used a blood-loss CA mouse model to assess ED. In this model, we draw blood < 15% of the circulating volume/day on every third day for six weeks. We replaced the blood loss volume with 350 µl NaCl (0.9%) or saline to avoid any volume changes. This resulted in moderate anemia with increased plasma erythropoietin and soluble transferrin receptor levels. We previously demonstrated that arterial lactate concentration and the distribution of Evans blue dye remained within the normal range in this model indicating no signs of hemorrhage or rheological volume changes (19).
Decreased NO-dependent relaxation responses are a hallmark of ED (24, 25). Severe anemia in hemoglobinopathies such as sickle cell disease or β-thalassemia is associated with ED in both humans (26) and in respective mouse models (27, 28). Our moderate blood loss CA mouse model showed decreased NO-dependent relaxation responses similar to mouse models of hemoglobinopathies. However, the ED in hemoglobinopathies is majorly attributed to increased inflammation and hemolysis, which are mildly elevated in our CA model (Supplementary Figure S2). To prove that RBCs from anemic patients induce ED, we performed co-incubation experiments by incubating RBCs from anemic patients with mouse thoracic aortic rings. We used an adapted incubation protocol as described previously (22). In line with our hypothesis, the aortic rings incubated with RBCs of anemic patients showed reduced NO-dependent relaxation responses, which indicate that RBCs induce ED. The exact signalling mechanism of how anemic RBCs induce ED is not known. We believe that the altered secretome of RBCs might contribute to this. In addition, a recent study showed the possible role of RBC-derived extracellular vesicles in anemia-associated ED (29). We recently showed that acute (3 days) blood loss anemia is associated with a transient increase in FMD responses because of compensatory increases in cardiac output associated with increased shear stress at the endothelial surface (19). Here we demonstrate unequivocally that the acute response to anemia is compromised by progressive decrease in FMD responses in CA mice correlating with the duration of anemia. In addition, inflammatory markers such as IL6 and IL-17A were unchanged in a subacute anemic mouse model, however, these markers are significantly increased in chronic anemia. Based on our previous publication (19) and the present research work, we summarized the vascular phenotypic differences between acute and chronic anemic mouse models in Supplementary Figure S6. The differences between the two models might be explained by acute and chronic adaptation of the vascular system to anemia. We believe that in acute anemia the vascular system undergoes compensatory changes to improve the endothelial function, especially at peripheral level. However, these compensatory effects are compromised over the time.
Enhanced arginase expression/activity was known to be one of the detrimental mechanisms for ED (30). In our study, endothelial-specific deletion of arginase 1 did not improve the relaxation responses in CA. It has been shown that arginase 2 is also highly expressed in endothelium and contributes to ED in different disease states (8). We used a classic pharmacological approach to inhibit both arginases to further evaluate the possible role of arginase 2 in CA. The aortic rings from anemic mice which were treated with Nor-NOHA did not show any improvement in the relaxation responses. These results largely exclude the potential roles of arginases in ED associated with CA.
We determined whether the reduced relaxation responses in CA are also associated with decreased eNOS expression in endothelial cells. However, eNOS expression at mRNA and protein levels was preserved in CA mice. These results are also in line with previous studies on β-thalassemia anemic mouse model (27). Of note, like in other pathologies (31, 32), the inflammatory iNOS is significantly increased in the vascular smooth muscle cells (VSMC) of CA mice. The enhanced expression of iNOS in inflammatory disease state such as sepsis is known to be detrimental. Additionally, our preliminary findings showed that interleukin IL-6 and interleukin IL-17A levels are significantly elevated in CA mice which might contribute to the enhanced iNOS expression in VSMC. The mechanistic aspects of IL-6 and IL-17A -mediated iNOS is beyond the current research focus but will be studied in the future. The increased circulatory VCAM-1 and ICAM-1 levels in CA mice further underline these findings implicating the role of anemia in inflammation and associated changes in the endothelium.
It is very well known that inflammation and oxidative stress concur with each other in vascular diseases (33). Taking this into consideration, we assessed 3-Nitrotyrosine levels in CA mice. In line with the recent study on iron deficiency mouse model (34), we also observed increased 3-Nitrotyrosine levels in the aortic lysates of CA mice indicating enhanced oxidative stress. NAC is known to be a potential ROS scavenger, which has beneficial effects on endothelial function in various CVDs (35). In our CA mouse model, supplementation of NAC completely reversed the impaired NO-dependent relaxation responses, concluding the potential role of ROS in mediating oxidative stress in CA. We also believe that the preserved eNOS expression and impaired NO-dependent relaxation responses in CA are associated with oxidative stress-mediated eNOS-uncoupling, which was also reported in other disease models (27, 36). Noteworthy, we cannot rule out some limitations of this study. Herein, we mainly focused on the effects of anemia on the endothelium. However, the exact mechanism how anemic RBCs induces ED requires further investigation. Our in vitro studies showed that MPO inhibition with AZD5904 improves endothelial function in CA mice aortic rings. However, further in vivo studies are needed to evaluate the beneficial effects of this MPO inhibitor in improving ED.
Our data suggest that chronic anemia is associated with decreased NO production due to increased inflammation and ROS production. Additionally, we also showed that RBCs from anemic patients attenuates the endothelial relaxation responses concluding the potential role of anemic RBCs in ED. We demonstrated that increased ROS production in the aortic endothelium of anemic mice, and treating the anemic mice with ROS scavenger (NAC) improved the relaxation responses in CA. In addition, MPO expression in endothelium is elevated in CA mice, and the MPO inhibitor AZD5904 also improved the relaxation responses in CA mice. Taken together, NAC supplementation and/or MPO inhibition improves endothelial function in anemia.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Clinical ethics committee, Universitätsklinikum Düsseldorf, Germany. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Landesamt für Natur, Umwelt- und Verbraucherschutz Nordrhein-Westfalen, Germany.
RC: contributed to conception, design, collection and analysis of the data. IS contributed to qRT-PCR, biochemical assays, Western Blot analysis. PD, FC, RC, VY: contributed to myograpgh and organ bath experiments. PW: contributed to FMD analysis. DP, IS, AS: contributed to immunohistochemistry. SB: contributed to anemia induction. CJ and RC: interpreted the data and drafted the manuscript. CJ, MK: obtained the funding. MH, CJ, NG, MK: critically revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the following grants: Research Commission of the Medical Faculty of Heinrich-Heine University to R.C. (No. 2020-62), and to P.W. (No. 29-2021); Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)-Grant No. 236177352- CRC1116; projects B06, B09 to M.K., C.J., and N.G. and Grant No. 397484323—CRC/TRR259; project A05 to N.G.; We acknowledge the support of the Susanne-Bunnenberg-Stiftung at the Düsseldorf Heart Center.
The authors wish to thank Stefanie Becher for her technical assistance regarding the animal experiments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1099069/full#supplementary-material.
1. Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J. (2009) 73(4):595–601. doi: 10.1253/circj.CJ-08-1169
2. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. (2000) 87(10):840–4. doi: 10.1161/01.RES.87.10.840
3. Tousoulis D, Kampoli AM, Tentolouris C, Papageorgiou N, Stefanadis C. The role of nitric oxide on endothelial function. Curr Vasc Pharmacol. (2012) 10(1):4–18. doi: 10.2174/157016112798829760
4. Romero MJ, Platt DH, Tawfik HE, Labazi M, El-Remessy AB, Bartoli M, et al. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ Res. (2008) 102(1):95–102. doi: 10.1161/CIRCRESAHA.107.155028
5. Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. (2003) 108(16):2000–6. doi: 10.1161/01.CIR.0000092948.04444.C7
6. Yang Z, Ming XF. Arginase: the emerging therapeutic target for vascular oxidative stress and inflammation. Front Immunol. (2013) 4:149. doi: 10.3389/fimmu.2013.00149
7. Beleznai T, Feher A, Spielvogel D, Lansman SL, Bagi Z. Arginase 1 contributes to diminished coronary arteriolar dilation in patients with diabetes. Am J Physiol Heart Circ Physiol. (2011) 300(3):H777–83. doi: 10.1152/ajpheart.00831.2010
8. Pernow J, Jung C. Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovasc Res. (2013) 98(3):334–43. doi: 10.1093/cvr/cvt036
9. Santhanam L, Lim HK, Lim HK, Miriel V, Brown T, Patel M, et al. Inducible no synthase dependent S-nitrosylation and activation of Arginase1 contribute to age-related endothelial dysfunction. Circ Res. (2007) 101(7):692–702. doi: 10.1161/CIRCRESAHA.107.157727
10. Shin WS, Berkowitz DE, Ryoo SW. Increased arginase ii activity contributes to endothelial dysfunction through endothelial nitric oxide synthase uncoupling in aged mice. Exp Mol Med. (2012) 44(10):594–602. doi: 10.3858/emm.2012.44.10.068
11. Ryoo S, Berkowitz DE, Lim HK. Endothelial arginase ii and atherosclerosis. Korean J Anesthesiol. (2011) 61(1):3–11. doi: 10.4097/kjae.2011.61.1.3
12. Karbach S, Wenzel P, Waisman A, Munzel T, Daiber A. Enos uncoupling in cardiovascular diseases–the role of oxidative stress and inflammation. Curr Pharm Des. (2014) 20(22):3579–94. doi: 10.2174/13816128113196660748
13. Roe ND, Ren J. Nitric oxide synthase uncoupling: a therapeutic target in cardiovascular diseases. Vascul Pharmacol. (2012) 57(5-6):168–72. doi: 10.1016/j.vph.2012.02.004
14. Chen W, Druhan LJ, Chen CA, Hemann C, Chen YR, Berka V, et al. Peroxynitrite induces destruction of the tetrahydrobiopterin and heme in endothelial nitric oxide synthase: transition from reversible to irreversible enzyme inhibition. Biochemistry. (2010) 49(14):3129–37. doi: 10.1021/bi9016632
15. Cheng D, Talib J, Stanley CP, Rashid I, Michaelsson E, Lindstedt EL, et al. Inhibition of mpo (myeloperoxidase) attenuates endothelial dysfunction in mouse models of vascular inflammation and atherosclerosis. Arterioscler Thromb Vasc Biol. (2019) 39(7):1448–57. doi: 10.1161/ATVBAHA.119.312725
16. Anker SD, Voors A, Okonko D, Clark AL, James MK, von Haehling S, et al. Prevalence, incidence, and prognostic value of anaemia in patients after an acute myocardial infarction: data from the optimaal trial. Eur Heart J. (2009) 30(11):1331–9. doi: 10.1093/eurheartj/ehp116
17. Salisbury AC, Alexander KP, Reid KJ, Masoudi FA, Rathore SS, Wang TY, et al. Incidence, correlates, and outcomes of acute, hospital-acquired Anemia in patients with acute myocardial infarction. Circ Cardiovasc Qual Outcomes. (2010) 3(4):337–46. doi: 10.1161/CIRCOUTCOMES.110.957050
18. Moghaddam N, Wong GC, Cairns JA, Goodman SG, Perry-Arnesen M, Tocher W, et al. Association of Anemia with outcomes among st-segment-elevation myocardial infarction patients receiving primary percutaneous coronary intervention. Circ Cardiovasc Interv. (2018) 11(12):e007175. doi: 10.1161/CIRCINTERVENTIONS.118.007175
19. Wischmann P, Kuhn V, Suvorava T, Muessig JM, Fischer JW, Isakson BE, et al. Anaemia is associated with severe rbc dysfunction and a reduced circulating No pool: vascular and cardiac enos are crucial for the adaptation to anaemia. Basic Res Cardiol. (2020) 115(4):43. doi: 10.1007/s00395-020-0799-x
20. Schuler D, Sansone R, Freudenberger T, Rodriguez-Mateos A, Weber G, Momma TY, et al. Measurement of endothelium-dependent vasodilation in mice–brief report. Arterioscler Thromb Vasc Biol. (2014) 34(12):2651–7. doi: 10.1161/ATVBAHA.114.304699
21. Zhou ZY, Zhao WR, Shi WT, Xiao Y, Ma ZL, Xue JG, et al. Endothelial-dependent and independent vascular relaxation effect of tetrahydropalmatine on rat aorta. Front Pharmacol. (2019) 10:336. doi: 10.3389/fphar.2019.00336
22. Zhou Z, Mahdi A, Tratsiakovich Y, Zahoran S, Kovamees O, Nordin F, et al. Erythrocytes from patients with type 2 diabetes induce endothelial dysfunction via arginase I. J Am Coll Cardiol. (2018) 72(7):769–80. doi: 10.1016/j.jacc.2018.05.052
23. Shosha E, Xu Z, Narayanan SP, Lemtalsi T, Fouda AY, Rojas M, et al. Mechanisms of diabetes-induced endothelial cell senescence: role of arginase 1. Int J Mol Sci. (2018) 19(4):1215. doi: 10.3390/ijms19041215
24. Farah C, Michel LYM, Balligand JL. Nitric oxide signalling in cardiovascular health and disease. Nat Rev Cardiol. (2018) 15(5):292–316. doi: 10.1038/nrcardio.2017.224
25. Naseem KM. The role of nitric oxide in cardiovascular diseases. Mol Aspects Med. (2005) 26(1–2):33–65. doi: 10.1016/j.mam.2004.09.003
26. Teixeira RS, Terse-Ramos R, Ferreira TA, Machado VR, Perdiz MI, Lyra IM, et al. Associations between endothelial dysfunction and clinical and laboratory parameters in children and adolescents with sickle cell Anemia. PLoS One. (2017) 12(9):e0184076. doi: 10.1371/journal.pone.0184076
27. Stoyanova E, Trudel M, Felfly H, Lemsaddek W, Garcia D, Cloutier G. Vascular endothelial dysfunction in beta-thalassemia occurs despite increased enos expression and preserved vascular smooth muscle cell reactivity to no. PLoS One. (2012) 7(6):e38089. doi: 10.1371/journal.pone.0038089
28. Hsu LL, Champion HC, Campbell-Lee SA, Bivalacqua TJ, Manci EA, Diwan BA, et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood. (2007) 109(7):3088–98. doi: 10.1182/blood-2006-08-039438
29. Nader E, Romana M, Guillot N, Fort R, Stauffer E, Lemonne N, et al. Association between nitric oxide, oxidative stress, eryptosis, red blood cell microparticles, and vascular function in sickle cell Anemia. Front Immunol. (2020) 11:551441. doi: 10.3389/fimmu.2020.551441
30. Lucas R, Fulton D, Caldwell RW, Romero MJ. Arginase in the vascular endothelium: friend or foe? Front Immunol. (2014) 5:589. doi: 10.3389/fimmu.2014.00589
31. Di Pietro N, Di Tomo P, Di Silvestre S, Giardinelli A, Pipino C, Morabito C, et al. Increased inos activity in vascular smooth muscle cells from diabetic rats: potential role of Ca(2+)/calmodulin-dependent protein kinase ii delta 2 (camkiidelta(2)). Atherosclerosis. (2013) 226(1):88–94. doi: 10.1016/j.atherosclerosis.2012.10.062
32. Ginnan R, Guikema BJ, Halligan KE, Singer HA, Jourd'heuil D. Regulation of smooth muscle by inducible nitric oxide synthase and nadph oxidase in vascular proliferative diseases. Free Radic Biol Med. (2008) 44(7):1232–45. doi: 10.1016/j.freeradbiomed.2007.12.025
33. Steven S, Frenis K, Oelze M, Kalinovic S, Kuntic M, Bayo Jimenez MT, et al. Vascular inflammation and oxidative stress: major triggers for cardiovascular disease. Oxid Med Cell Longev. (2019) 2019:7092151. doi: 10.1155/2019/7092151
34. Inserte J, Barrabes JA, Aluja D, Otaegui I, Baneras J, Castellote L, et al. Implications of iron deficiency in stemi patients and in a murine model of myocardial infarction. JACC Basic Transl Sci. (2021) 6(7):567–80. doi: 10.1016/j.jacbts.2021.05.004
35. Khan SA, Campbell AM, Lu Y, An L, Alpert JS, Chen QM. N-Acetylcysteine for cardiac protection during coronary artery reperfusion: a systematic review and meta-analysis of randomized controlled trials. Front Cardiovasc Med. (2021) 8:752939. doi: 10.3389/fcvm.2021.752939
36. Marchesi C, Ebrahimian T, Angulo O, Paradis P, Schiffrin EL. Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertension. (2009) 54(6):1384–92. Epub 2009/10/14. doi: 10.1161/HYPERTENSIONAHA.109.138305
Keywords: anemia, endothelial dysfunction, nitric oxide, reactive oxygen species, n-acetyl cysteine, myeloperoxidase
Citation: Chennupati R, Solga I, Wischmann P, Dahlmann P, Celik FG, Pacht D, Şahin A, Yogathasan V, Hosen MR, Gerdes N, Kelm M and Jung C (2023) Chronic anemia is associated with systemic endothelial dysfunction. Front. Cardiovasc. Med. 10:1099069. doi: 10.3389/fcvm.2023.1099069
Received: 22 November 2022; Accepted: 21 April 2023;
Published: 10 May 2023.
Edited by:
Hong Jin, Karolinska Institutet (KI), SwedenReviewed by:
Aida Collado, Karolinska Institutet (KI), Sweden© 2023 Chennupati, Solga, Wischmann, Dahlmann, Celik, Pacht, Şahin, Yogathasan, Hosen, Gerdes, Kelm and Jung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ramesh Chennupati UmFtZXNoLmNoZW5udXBhdGlAbWVkLnVuaS1kdWVzc2VsZG9yZi5kZQ== Christian Jung Y2hyaXN0aWFuLmp1bmdAbWVkLnVuaS1kdWVzc2VsZG9yZi5kZQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.