 Benzhen Wang
Benzhen Wang Guangsong Shan
Guangsong Shan Zhen Bing
Zhen Bing Qi Zhang2
Qi Zhang2 Zipu Li
Zipu Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cardiovasc. Med., 15 March 2023
Sec. Pediatric Cardiology
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1083188
This article is part of the Research TopicCase Reports in Pediatric Cardiology: 2022View all 31 articles
Dilated cardiomyopathy (DCM) is one of the leading causes of heart failure in children with diverse clinical characteristics. To date, DCM with a giant atrium as the first manifestation is rare and has not been reported in previous literature. We report a case of a male infant born with a significantly enlarged right atrium. Due to worsened clinical symptoms and the risk of arrhythmias and thrombosis, we performed the surgical reduction of the right atrium. Unfortunately, DCM and a progressive re-enlargement of the right atrium appeared during midterm follow-up. The mother's echocardiogram also suggested DCM, and the patient was eventually considered for a diagnosis of familial DCM. This case may expand the clinical spectrum of DCM and reminds us of the importance of good follow-up of children with idiopathic dilatation of the right atrium.
Dilated cardiomyopathy (DCM) is one of the leading causes of heart failure in children with complex etiologies and diverse clinical manifestations (1). Marked enlargement of the right atrium (RA) as the initial presentation of DCM has not been reported. Herein, we report an infant born with a giant right atrium and developed DCM during midterm follow-up.
We report the case of a male infant born from cesarean section after a normal pregnancy, with an Apgar score of 10/10 at 1st and 5th minutes, respectively. His mother was a 28-year-old, healthy, Gesta 2 para 2 woman who was presented for an obstetric ultrasound at 30 weeks of gestational age. An enlargement of the right atrium was noted. No other fetal anatomic abnormalities were detected. After birth, the case was admitted to the cardiac intensive care unit at the Qingdao Women and Children's Hospital. Family medical history was normal.
Heart rate, respiratory rate, blood pressure, and oxygen saturation on room air were normal on admission. No cyanosis, heart murmurs, wheezing, or hepatomegaly were present. Blood gas analysis, routine test, and liver and renal function were normal. The patient's and his mother's erythrocyte sedimentation rate, antinuclear antibodies, and extractable nuclear antigens were negative. N-terminal pro-brain natriuretic peptide reached 2,035 pg/mL (reference ranges <125 pg/mL). Transthoracic echocardiogram (TTE) showed a giant RA (50 mm × 48 mm) with normal right and left ventricular volumes, a 15 mm secundum atrial septal defect (ASD), the tricuspid annulus was 14 mm, slight tricuspid regurgitation, a pulmonary arterial systolic pressure (PASP) of 45 mmHg, and left ventricular ejection fraction (LVEF) of 65% (Figure 1A). The chest x-ray showed an enlarged heart, and the cardiothoracic ratio (CTR) reached about 0.80 (Figure 1B). Electrocardiograph showed sinus tachycardia, right axis deviation, and right bundle branch block (Figure 1C). Computed tomography angiography indicated abnormalities similar to TTE. After extensive discussion, the initial diagnosis we considered was idiopathic dilatation of the right atrium (IDRA). The patient was discharged and followed up regularly every month in the clinic.
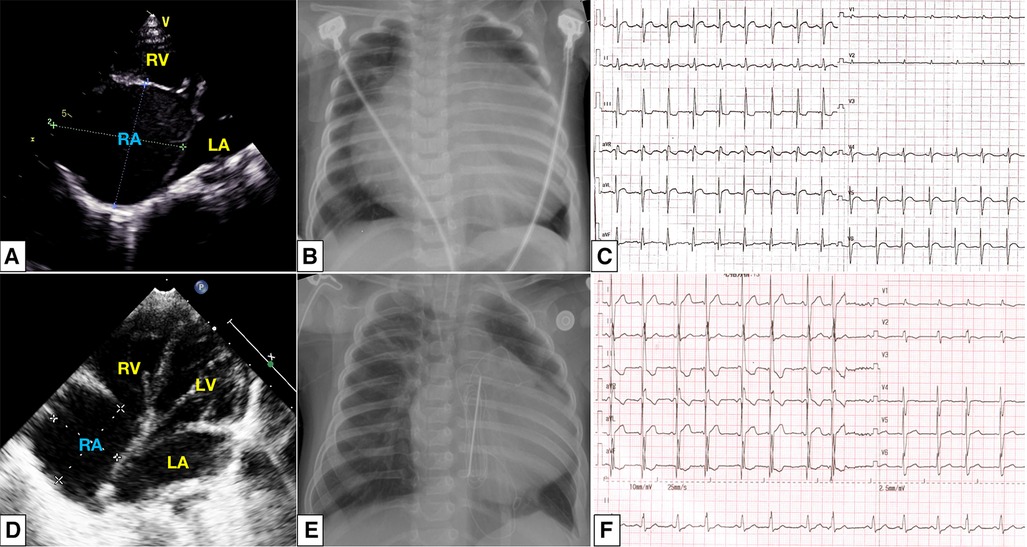
Figure 1. At initial presentation: a dilated right atrium was noted in apical four-chamber view (A); chest radiography showed cardiomegaly with a CTR of 80% (B); electrocardiograph showed sinus tachycardia, right axis deviation, and incomplete right bundle branch block (C). After surgical reduction, a slightly enlarged right atrium and intact atrial septum (D), CTR decreased to 0.61 (E), and the atrial premature beat was detected (F). CTR, cardiothoracic ratio;
Clinical symptoms worsened during the patient's follow-up, such as feeding difficulty, shortness of breath, and growth retardation. Considering the possibility of compression caused by the enlarged RA and the risk of arrhythmias and thrombosis, we performed the surgical reduction of RA at 3 months with consent obtained from the parents. Transesophageal echocardiography and intraoperative findings confirmed the diagnosis of giant RA and ASD. Resection of extensive RA wall and atrioseptopexy were performed. At surgery, biopsies of the RA wall, right ventricle (RV) and left atrium (LA) revealed unspecific inflammatory cell infiltration and various degrees of focal myocardial hypertrophy and degeneration (Figures 2A–2C). Transmission electron microscopy (TEM) demonstrated damage to myofibrils and mitochondria, with a blurring of mitochondrial cristae (Figures 2D–2F). The patient recovered sufficiently post-operation. Repeat TTE showed a slightly enlarged RA, intact atrial septum, and normal LVEF (Figure 1D). Chest x-ray revealed a decreased CTR as 0.61 (Figure 1E). At 1-year follow-up, there was a progressive enlargement of the RA (Figures 3A,B), and the patient remained asymptomatic except for growth retardation. Unexpectedly, TTE 2 years after epilepsy surgery demonstrated an enlarged left ventricle (LV) with a left ventricular end-diastolic diameter (LVEDD) of 40 mm, a giant RA (52 mm × 39 mm), a normal PASP of 20 mmHg, and reduced LVEF of 55% (Figures 3C,D). Anti-heart failure therapy was administered, including captopril, diuretics, metoprolol, and acetylsalicylic acid. The patient had a good condition at 5 years post-operation. The size of the RA and LV did not change significantly, and the LVEF was about 50% (Figures 3E,F). Cardiac magnetic resonance (CMR) showed cardiomegaly at the expense of the right atrium with a decreased LVEF of 45% and no typical manifestations of ventricular myocardial fibrosis (Figure 4).

Figure 2. H&E staining showing the pathological changes in myocardial tissues of the right atrium (A), right ventricle (B), and left atrium (C). TEM shows damage to myofibrils and mitochondria in the right atrium (D), right ventricle (E), and left atrium (F). Scar bar: 1 µm. Magnification: ×25,000 and ×20,000. TEM, transmission electron microscopy.
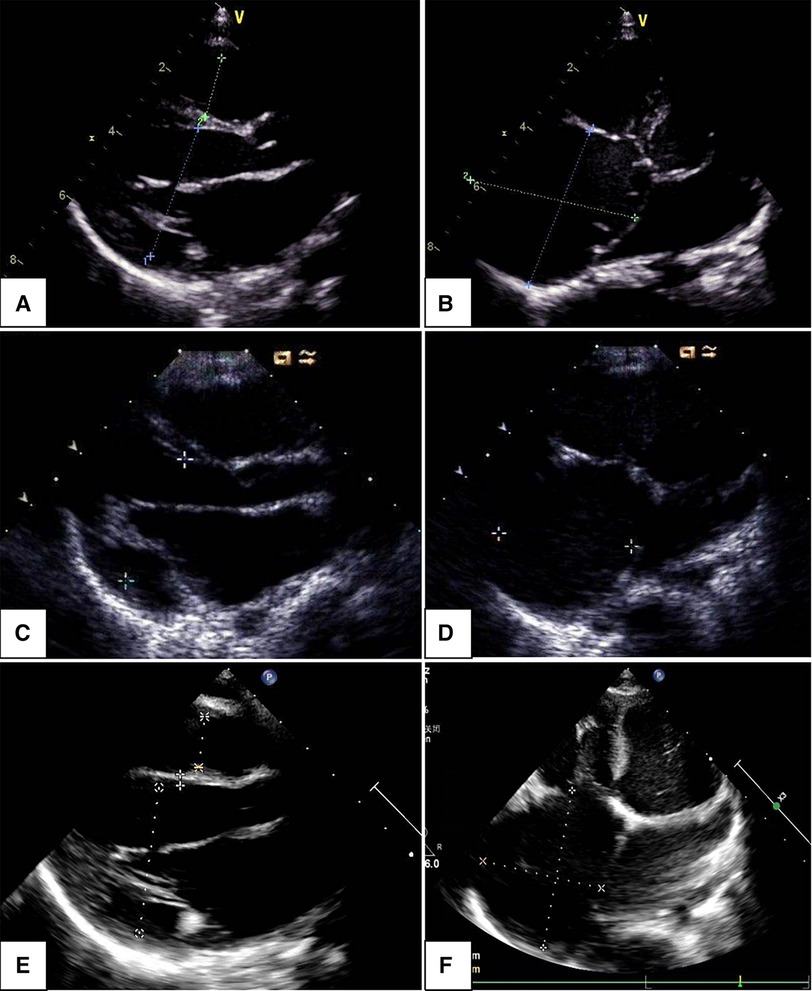
Figure 3. Changes in echocardiogram with follow-up time: parasternal long axis view indicated left ventricular end-diastolic dimension increased within 1, 2, and 5 years (A,C,E); apical four-chamber view showed an enlarged right atrium, the changes were not significant within 1, 2, and 5 years (B,D,F).
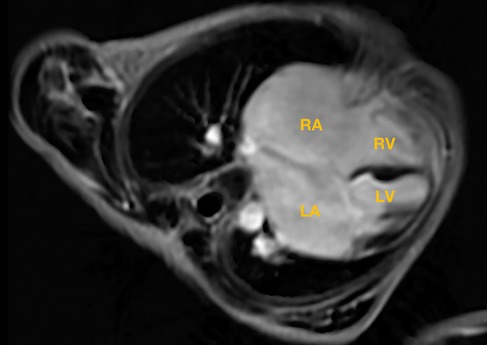
Figure 4. Cardiac magnetic resonance showed an enlarged right atrium, left atrium, and left ventricle; no typical manifestations of ventricular myocardial fibrosis were detected. RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle.
A genetic study showed a heterozygous missense Pkp2 variant (c.2380T > G; OMIM: 609040; SCV 002576312) related to arrhythmogenic right ventricular cardiomyopathy (ARVC) (Supplementary material S1). Further Sanger sequencing indicated that his mother presented the same variant in Pkp2; the variant was classified as a variant of uncertain significance according to the American College of Medical Genetics and Genomics (ACMG) guidelines. Although his mother was asymptomatic, her echocardiography showed an enlarged LA and LV (LVEDD 55 mm), mild to moderate mitral regurgitation, and reduced LVEF (48%) without other cardiac structural defects. Then, the mother was diagnosed with DCM, and the diagnosis of familial DCM was established based on the patient's and his mother's features.
Giant RA is considered to be of congenital origin in the absence of conditions such as congenital heart disease, pulmonary arterial hypertension, or tricuspid valve disease. IDRA is the most common disease mentioned in recent years due to a marked enlarged right atrium, a congenital anomaly with unknown etiology reported in fetuses, infants, children, and adults (2–4). Most patients are asymptomatic, and the most common symptoms are palpitations, dyspnea, and syncope caused by atrial tachyarrhythmias (2). The diagnosis of IDRA is usually established with fetal echocardiography and transthoracic echocardiography, and CT or CMR may be beneficial for a definitive diagnosis (2–4). According to prenatal and postnatal imaging features, IDRA should be considered as the primary disorder in our case. It is important to note that IDRA may be accompanied by congenital heart defects consisting of ASD (5). Our case also has a large secundum ASD, which could have contributed to the enlargement of RA. However, the recurrence of atrial enlargement after surgical repair of ASD suggests that the effect of ASD on giant RA is limited. The postnatal management of asymptomatic patients with IDRA remains controversial. Indications for surgical reduction of RA in children with IDRA have included right atrial thrombus and atrial arrhythmias, as well as others, such as progressive dilatation of the RA, concern about airway compression, and severe tricuspid insufficiency suspected as Ebstein's anomaly (6). So far, many studies suggest that the long-term outcomes of patients post-surgery are also contradictory. Although many cases demonstrate that surgery may indeed relieve symptoms associated with IDRA, some children still suffer the recurrent attack of giant RA (4, 6). Unfortunately, the situation of our case also suggests that the surgical indications for children with IDRA should be more strictly controlled.
In patients with cardiomyopathy, huge atria are commonly seen in restrictive cardiomyopathy (RCM), leading to a significant atrial pressure increase due to limited ventricular diastolic function and progressive atrial enlargement (7). The morphological definition of RCM is based on imaging features demonstrating non-hypertrophied, non-dilated ventricles, with marked bi-atrial enlargement (7). Nevertheless, the findings are not consistent with the characteristics of RCM in this case but with the features of DCM, such as LV dilation and poor cardiac systolic function.
Endomyocardial biopsy (EMB) is the gold standard method for diagnosing acute or chronic inflammatory heart diseases and enables the identification of the underlying etiology of cardiac inflammation (8, 9). Although there are no structural or functional changes in the heart chambers except RA in the early stage of the disease in our case, it is worth noting that the histological findings of damage to LA and RA were present. On the other hand, early involvement of the LV cannot be ruled out despite the absence of an LV biopsy. With the evidence of inflammatory cell infiltration, the probability of inflammatory disorders in the myocardium should still be considered (10).
The diagnosis of familial DCM is confirmed when two or more first-degree relatives have “idiopathic” DCM and/or unexplained death at a young age, and there could be an underlying genetic etiology (11). The pathogenesis of genetically mediated DCM has been associated with genes that encode components of the cytoskeleton, sarcomeric proteins, and nuclear envelope proteins (12). Recently, Pkp2 gene, encoding components of desmosomes, has also been associated with different inherited cardiac conditions, including ARVC and Brugada syndrome (13). Few studies have suggested that Pkp2 can also lead to DCM and left ventricular non-compaction (LVNC) (14). However, these associations do not prove causality between Pkp2 and DCM in our case, and further research should be conducted on the variant we detected.
In summary, DCM with a giant atrium as the first manifestation is an uncommon complex condition. Although the etiology remains unclear, a key factor should contribute to this unique phenotype. This case also reminds us of the importance of good follow-up of children with IDRA, including those after surgery. Further follow-up is needed for this case and his mother to evaluate the prognosis of this phenotype in DCM.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the ethics committee of Qingdao Women and Children's Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
BW, GS, and ZB acquired the clinical data. QX performed the cardiac surgery. QZ performed the radiological analyses. BW and GS interpreted the data and drafted the manuscript. ZL contributed to the clinical design and study concept, and revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1083188/full#supplementary-material.
1. Price JF. Congestive heart failure in children. Pediatr Rev. (2019) 40:60–70. doi: 10.1542/pir.2016-0168
2. Hofmann SR, Heilmann A, Häusler HJ, Dähnert I, Kamin G, Lachmann R. Congenital idiopathic dilatation of the right atrium: antenatal appearance, postnatal management, long-term follow-up and possible pathomechanism. Fetal Diagn Ther. (2012) 32:256–61. doi: 10.1159/000338661
3. Wagner N, Kagan KO, Abele H, Hoopmann M, Kaulitz R, Hofbeck M. Fetal idiopathic dilatation of the right atrium. Ultraschall Med. (2011) 32:81–2. doi: 10.1055/s-0028-1109998
4. Walter C, Bartrons J, Gómez O, Caffarena JM, Carretero JM. Idiopathic dilatation of the right atrium: a not so benign entity. Cardiol Young. (2020) 30:919–22. doi: 10.1017/S1047951120001353
5. Venugopalan P, Jain R. Accidental detection of a giant right atrial aneurysm in an asymptomatic infant. Acta Cardiol. (2002) 57:125–7. doi: 10.2143/AC.57.2.2005384
6. Forbes K, Kantoch MJ, Divekar A, Ross D, Rebeyka IM. Management of infants with idiopathic dilatation of the right atrium and atrial tachycardia. Pediatr Cardiol. (2007) 28:289–96. doi: 10.1007/s00246-006-0012-5
7. Muchtar E, Blauwet LA, Gertz MA. Restrictive cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. (2017) 121:819–37. doi: 10.1161/CIRCRESAHA.117.310982
8. Tschöpe C, Ammirati E, Bozkurt B, Caforio ALP, Cooper LT, Felix SB, et al. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol. (2021) 18:169–93. doi: 10.1038/s41569-020-00435-x
9. Li B, Lento PA, Pan S. Inflammatory cardiomyopathy: case-based review on clinical presentation, diagnosis, and management. Cardiol Rev. (2021) 29:230–7. doi: 10.1097/CRD.0000000000000369
10. Heymans S, Eriksson U, Lehtonen J, Cooper LT. The quest for new approaches in myocarditis and inflammatory cardiomyopathy. J Am Coll Cardiol. (2016) 68:2348–64. doi: 10.1016/j.jacc.2016.09.937
11. Peters S, Johnson R, Birch S, Zentner D, Hershberger RE, Fatkin D. Familial dilated cardiomyopathy. Heart Lung Circ. (2020) 29:566–74. doi: 10.1016/j.hlc.2019.11.018
12. Khan RS, Pahl E, Dellefave-Castillo L, Rychlik K, Ing A, Yap KL, et al. Genotype and cardiac outcomes in pediatric dilated cardiomyopathy. J Am Heart Assoc. (2022) 11:e022854. doi: 10.1161/JAHA.121.022854
13. Novelli V, Malkani K, Cerrone M. Pleiotropic phenotypes associated with Pkp2 variants. Front Cardiovasc Med. (2018) 5:184. doi: 10.3389/fcvm.2018.00184
Keywords: heart failure, right atrium, cardiomyopathy, pediatrics, cardiac surgery
Citation: Wang B, Shan G, Bing Z, Zhang Q, Xing Q and Li Z (2023) Giant right atrium in a child with dilated cardiomyopathy: A case report. Front. Cardiovasc. Med. 10:1083188. doi: 10.3389/fcvm.2023.1083188
Received: 28 October 2022; Accepted: 17 February 2023;
Published: 15 March 2023.
Edited by:
Alvise Guariento, University of Toronto, CanadaReviewed by:
Junbao Du, Peking University, China© 2023 Wang, Shan, Bing, Zhang, Xing and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zipu Li YXB1cWRAc2luYS5jb20=
Specialty Section: This article was submitted to Pediatric Cardiology, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.