Abstract
Pulmonary hypertension due to left heart disease (PH-LHD) is the most frequent manifestation of PH but lacks any approved treatment. Activin receptor type IIA-Fc fusion protein (ActRIIA-Fc) was found previously to be efficacious in experimental and human pulmonary arterial hypertension (PAH). Here we tested the hypothesis that ActRIIA-Fc improves pulmonary vascular remodeling and alleviates PH in models of PH-LHD, specifically in subtypes of heart failure with reduced ejection fraction (PH-HFrEF) and preserved ejection fraction (PH-HFpEF). Treatment with murine ActRIIA-Fc reduced cardiac remodeling and improved cardiac function in two mouse models of left heart disease without PH, confirming that this inhibitor of activin-class ligand signaling can exert cardioprotective effects in heart failure. In a mouse model of PH-HFrEF with prolonged pressure overload caused by transverse aortic constriction, ActRIIA-Fc treatment significantly reduced pulmonary vascular remodeling, pulmonary fibrosis, and pulmonary hypertension while exerting beneficial structural, functional, and histological effects on both the left and right heart. Additionally, in an obese ZSF1-SU5416 rat model of PH-HFpEF with metabolic dysregulation, therapeutic treatment with ActRIIA-Fc normalized SMAD3 overactivation in pulmonary vascular and perivascular cells, reversed pathologic pulmonary vascular and cardiac remodeling, improved pulmonary and cardiac fibrosis, alleviated PH, and produced marked functional improvements in both cardiac ventricles. Studies in vitro revealed that treatment with ActRIIA-Fc prevents an abnormal, glucose-induced, activin-mediated, migratory phenotype in human pulmonary artery smooth muscle cells, providing a mechanism by which ActRIIA-Fc could exert therapeutic effects in experimental PH-HFpEF with metabolic dysregulation. Our results demonstrate that ActRIIA-Fc broadly corrects cardiopulmonary structure and function in experimental PH-LHD, including models of PH-HFrEF and PH-HFpEF, leading to alleviation of PH under diverse pathophysiological conditions. These findings highlight the important pathogenic contributions of activin-class ligands in multiple forms of experimental PH and support ongoing clinical evaluation of human ActRIIA-Fc (sotatercept) in patients with PH-HFpEF.
In Group 2 pulmonary hypertension (PH), left heart failure can cause retrograde elevation of blood pressure that leads to dysfunction of the pulmonary arterial system and subsequent right heart failure. Activin receptor type IIA-Fc fusion protein (ActRIIA-Fc) improves cardiopulmonary remodeling and function in models of Group 2 PH through mutually reinforcing, beneficial actions at multiple sites. 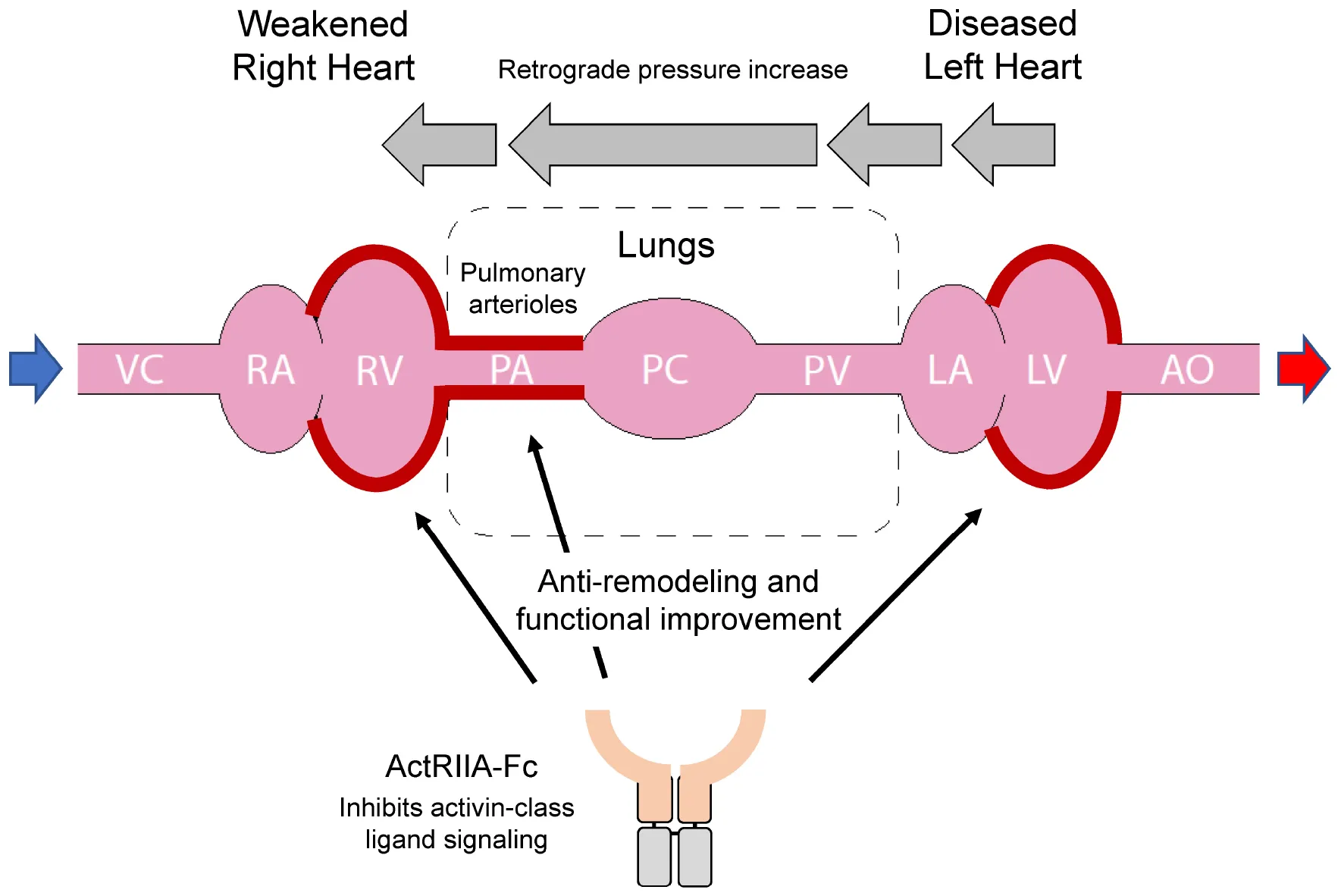
Introduction
Pulmonary hypertension (PH) accompanies a spectrum of common and rare diseases (1). The most prevalent form, PH associated with left heart disease (PH-LHD, Group 2 PH), is caused by left heart failure, including subtypes with reduced ejection fraction (PH-HFrEF) or preserved ejection fraction (PH-HFpEF) (2, 3). In both subtypes of PH-LHD, functional correlates of pulmonary vascular remodeling predict higher morbidity and mortality than in its absence (4, 5). Cardiac dysfunction associated with HFrEF and HFpEF can be treated with a range of efficacious therapies (6), including the recently-approved empagliflozin (7), but there are no approved treatments for either type of PH-LHD (2). Diverse treatments approved for pulmonary arterial hypertension (PAH, Group 1 PH) have so far been found ineffective in PH-LHD (8).
A key pathogenic factor in PH-LHD is progressive pulmonary vascular remodeling. In left heart disease, elevated pressure can be transmitted in a retrograde manner from the left atrium to the right ventricle (RV) independent of secondary pulmonary vascular remodeling (9). This results in a PH-LHD subtype termed passive, or isolated, post-capillary PH (IpcPH-LHD) (2, 10). A second subgroup of more severely affected patients with precapillary vascular remodeling and combined post- and precapillary PH (CpcPH-LHD) displays pulmonary pathophysiology resembling that in PAH (Group 1 PH) (2, 5, 9, 10). In this case, PH impairs RV function and in the absence of effective treatments eventually causes death by right heart failure. Although features of metabolic syndrome such as higher body mass index, elevated hemoglobin A1c levels, and diabetes are considered risk factors for PH-HFpEF (11–13), the potential influence of metabolic syndrome on pulmonary vascular remodeling in left heart disease is largely unexplored.
Activin receptor signaling is implicated in left heart failure and PAH vascular pathology, raising the possibility that this pathway contributes to PH-LHD pathogenesis. Imbalanced signaling by the transforming growth factor-β (TGF-β) superfamily contributes extensively to pathologic vascular remodeling in PAH, with overactive, pro-proliferative SMAD2/3 signaling occurring along with deficient, antiproliferative SMAD1/5/8 signaling (14). The activin-class ligands activin A, growth differentiation factor 8 (GDF8), and GDF11—prominent activators of SMAD2/3-pathway signaling—are conspicuously upregulated in small pulmonary arteries in both experimental and human PAH (15). Importantly, sequestration of activin-class ligands with an Fc-fusion protein incorporating the extracellular domain of activin receptor type IIA (ActRIIA-Fc) exerts antiproliferative and inflammation-suppressing effects in the lung vasculature, reverses pulmonary vascular remodeling, and reduces PH in experimental PAH, thus exhibiting disease-modifying activity not observed with vasodilator-based therapy (15, 16). Additional evidence implicates activin receptor signaling in pathologic RV remodeling associated with experimental PAH (16) and left heart failure associated with models of systemic pressure overload, aging, ischemia, and acute ischemia-reperfusion injury (17–20). Therapeutic use of ActRIIA-Fc (sotatercept) provides clinically meaningful improvement in patients with PAH, even in those receiving multiple background therapies (21), underscoring the strong pathogenic roles for activin receptor signaling in PAH progression.
Based on these extensive clinical and preclinical observations, we hypothesized that sequestration of activin-class ligands with ActRIIA-Fc will alleviate experimental PH-LHD. Our results in models of PH-HFrEF and PH-HFpEF are the first to indicate that activin receptor signaling plays a critical role in pulmonary vascular remodeling in experimental PH-LHD. The ability of ActRIIA-Fc to reverse pathologic remodeling in the left heart, right heart, and pulmonary vasculature in experimental PH-LHD suggests that multi-ligand sequestration with this agent could be a promising therapeutic approach to treat Group 2 PH.
Materials and methods
Animal studies
All animal experiments were approved by the Institutional Animal Care and Use Committee at Acceleron Pharma Inc., a subsidiary of Merck & Co., Inc., Rahway, NJ, USA and performed in accordance with the guidelines from the NIH Guide for the Care and Use of Laboratory Animals. Male C57BL/6 mice (10 weeks old, Jackson Laboratory) were used for TAC and MI models as described (18, 22), and male Balb/c mice (10 weeks old, Jackson Laboratory) were used for the prolonged TAC model to establish PH. Male obese ZSF1 rats (8 and 23 weeks old) and their lean littermates (Charles River, Wilmington, MA, USA) were used for the PH-HFpEF study. PH was established by a single subcutaneous injection of a vascular endothelial growth factor receptor antagonist, Sugen 5416 (SU5416, 100 mg/kg; Cayman), suspended in CMC buffer (0.5% sodium carboxymethyl cellulose, 0.4% polysorbate 80, 0.9% sodium chloride, and 0.9% benzyl alcohol) (23, 24). Animals were euthanized in all experiments by heart and lung removal en bloc according to AVMA guidelines.
Hemodynamic measurements
Animals were anesthetized with 3–4% isoflurane and placed on controlled heating pads. Right ventricular systolic pressure (RVSP) was measured by advancing a 2F curve tip pressure transducer catheter (SPR-513, Millar Instruments) into the right ventricle (RV) via the right jugular vein under 1.5–2% isoflurane anesthesia. En-bloc heart and lungs were collected, and lungs were perfused with physiological saline via the right ventricular outflow tract to flush blood cells from the pulmonary circulation. RV hypertrophy was assessed by calculating Fulton’s index, the weight ratio of the RV free wall to the combined left ventricle (LV) + septum [RV/(LV + S)].
Histopathology and immunohistochemistry
After perfusion, the right bronchus was ligated and the right lung lobes were dissected and snap frozen for biochemical analysis. The left lung lobe was inflated with a formalin-agarose mixture [0.5% w/v low melting agarose in 1% neutral buffered formalin (NBF)] at a constant pressure of 20 cm H2O. The inflated lungs were fixed in 10% NBF for 48 h (16). The left lung lobe was blocked, embedded in paraffin, and sectioned. Formalin-fixed, paraffin-embedded lung sections were stained with hematoxylin and eosin (H&E) and Masson’s trichome for histological analysis. Immunohistochemical staining was performed using antibodies against phospho-SMAD3 (pSMAD3) (cat# ab52903, Abcam), activin A (cat# PA5-47004, ThermoFisher), and GDF11 (cat# NBP2-57399, Novus). Dual immunofluorescence staining was performed using combinations of antibodies against pSMAD3 (cat# ab52903, Abcam) and smooth muscle α-actin (cat# A5228, Sigma) or CD31 (cat# ab182981, Abcam). DAPI (4′,6-diamidino-2-phenylindole) was used to identify cell nuclei.
Morphological analyses
Wall thickness of pulmonary arteries was measured in H&E-stained sections with HALO software. Wall thickness was determined by the formula [outer diameter (OD) – inner diameter (ID)]/OD × 100% as described previously (25). Approximately 30 vessels with OD < 100 μm were randomly selected, and the outer and inner diameters of the vessels were annotated to calculate wall thickness.
Echocardiography
Animals were anesthetized with 3–4% isoflurane and maintained at 1.5–2% isoflurane during echocardiography. A Vevo 3100 imaging system with MX201 scanhead (VisualSonics, Toronto, ON, Canada) was used to perform echocardiography for measurement of pulmonary. Pulmonary artery acceleration time (PAAT), tricuspid annular plane systolic excursion (TAPSE), isovolumic relaxation time (IVRT), and mitral inflow velocity (E) and mitral annular velocity (E’), from which the E/E’ ratio was derived. Briefly, rats were placed supine on a heated platform and allowed to breathe spontaneously. The right ventricular outflow tract was visualized using a modified parasternal long axis view. PAAT was measured as pulmonary artery blood flow time from start to peak velocity from the pulse wave doppler tracings recorded in the lumen of the main pulmonary artery distal to the pulmonary valve. TAPSE was obtained from the apical four-chamber view directing the M-mode doppler beam through the lateral annulus of the tricuspid valve plane. E, the maximal transmittal flow velocity during early inflow, was recorded using the apical four chamber view. E’, the peak mitral annular velocity during early filling, was obtained by tissue doppler imaging by placing the pulsed waved doppler at the septal corner of the mitral annulus. Using the recordings of transmitral flow velocities, we measured IVRT from the time interval between aortic valve closure and mitral valve opening. For each parameter, measurements from three individual heartbeats per animal were collected and averaged.
Cell culture
Cardiomyocytes derived from human induced pluripotent stem cells (iPSCs, ATCC-ACS-1021) were cultured as instructed. For cardiomyocyte differentiation in vitro, a STEMdiff ventricular cardiomyocyte differentiation kit (STEMCELL Technologies, cat# 05010) was used according to the manufacturer’s instructions. Cardiomyocytes were cultured for 15 days in maintenance medium (STEMCELL Technologies, cat# 05020) to promote maturation. To induce hypertrophy, cardiomyocytes were treated with 10 nM of endothelin 1 (ET-1, Sigma-Aldrich, cat#E7764) for 24 h. Human pulmonary artery smooth muscle cells (PASMCs) were purchased (ATCC, cat# PCS-100-023) and cultured in vascular cell basal medium (ATCC, cat# PCS-100-030) supplemented with a vascular smooth muscle cell growth kit (ATCC, cat# PCS-100-042) in 5% CO2 at 37°C. Cells from passages 4–7 were used for experiments. Sotagliflozin (Selleckchem, cat# S8103) was used as described for each experiment.
Western blotting
Cells were lysed in RIPA buffer (Sigma, cat# R0278) on ice for 20 min and centrifuged at 15,000 rpm for 15 min at 4°C. Supernatants were quantified by BCA (Thermo Fisher, cat# A53225), and 30–40 μg of protein per sample was used for gel electrophoresis on a 4–15% gel (Bio-Rad, cat# 4568085) and transferred onto nitrocellulose membranes at 250 mA for 90 min. Tissue samples were pulverized and homogenized in RIPA buffer with small pulses for 3 min at 4°C. Homogenates were kept on ice for 20 min, and protein quantification was performed by BCA using 20–30 μg of protein per sample. Blots were probed with antibodies against SGLT1 (Cell Signaling Technologies, cat# 5042), pERK1/2 (Cell Signaling Technologies, cat# 9101S), ERK (Cell Signaling Technologies, cat# 4695S), pJNK (Cell Signaling Technologies, cat# 4668S), JNK (Cell Signaling Technologies, cat# 9252S), and GLUT1 (Cell Signaling Technologies, cat# 12939S).
Real time RT-PCR
Pulverized samples (10–20 mg) were homogenized in QIAzol Lysis Reagent, and total RNA extraction was performed with an RNeasy Plus Mini Kit (Qiagen, cat# 74034) according to the manufacturer’s instructions. RNA concentration was measured with a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and 0.5–1 μM of RNA was used for cDNA syntheses. Taqman probes (Thermo Fisher Scientific) were used for quantitative real-time RT-PCR (qRT-PCR). At least four technical replicates were used for each group.
Cellular migration assay
Human PASMCs (5 × 104) were seeded in a Boyden chamber (Sigma-Aldrich, cat# ECM508) in vascular cell basal medium supplemented with a vascular smooth muscle cell growth kit and incubated overnight. The next day, medium in the lower chamber was replaced with basal medium alone or medium containing 25 mM glucose while the upper chamber contained only basal medium. Inhibitors were added to both chambers, but activin A was added only to the lower chamber. Experimental values were measured at the end of a 72 h incubation period.
Assay for activin A
Human PASMCs (1.5 × 105) were seeded in 24-well plates in vascular cell basal medium supplemented with a vascular smooth muscle cell growth kit. When cells reached 60–70% confluency, they were incubated with glucose-deficient medium for 6 h. Medium was then collected (defined as time zero), and cells were treated for 24 or 48 h with medium containing either 5 or 25 mM glucose. Medium samples were collected at each time point, and activin A concentrations in samples were measured with an ELISA kit (R&D Systems, cat# DAC00B) according to the manufacturer’s instructions.
Statistical analysis
Data are presented as means ± standard error of the mean (SEM). Comparisons between groups were analyzed using either Student’s t-test or ANOVA with Dunnett’s or Tukey’s post-hoc test for multiple comparisons. Differences were considered significant at P < 0.05.
Results
ActRIIA-Fc improves cardiac structure, function, and histology and reduces cardiomyocyte injury markers in models of left heart failure
We evaluated ActRIIA-Fc effects in mouse models of left heart failure caused by sustained pressure overload or myocardial infarction because activin receptor signaling has been implicated in multiple forms of left heart disease (17–20). LV pressure overload induced in mice by transverse aortic constriction (TAC) for 3 weeks led to detrimental changes in cardiac structure, function, and histology (Figure 1). Treatment with a murine form of ActRIIA-Fc (RAP-011) exerted significant cardioprotective effects on these parameters when compared with vehicle treatment (Figure 1). This result is consistent with previously reported beneficial effects of activin receptor-like kinase 4 (ALK4) haploinsufficiency on cardiac hypertrophy, dysfunction, and fibrosis in a similar mouse model of LV pressure overload (17). Treatment with RAP-011 also produced beneficial effects in a mouse model of myocardial infarction (Supplementary Figure 1), consistent with previously reported benefits of ActRIIB ligand inhibition under comparable conditions (20). These results confirm cardioprotective effects of ActRIIA-Fc similar to those observed with other methods of activin-signaling inhibition in experimental heart failure.
FIGURE 1
To determine whether ActRIIA-Fc exerts cardioprotective effects through regulation of cardiomyocyte signaling, we evaluated effects of a human form of ActRIIA-Fc (ACE-011) on injury markers in cultured cardiomyocytes derived from human induced pluripotent stem cells (iPSCs) (26). Treatment with endothelin-1 (ET-1) induced cellular injury in these cardiomyocytes, including increased NPPB expression and a shift in the relative abundance of myosin heavy chain isoforms from α to β (increased MYH7:MYH6 ratio) (Supplementary Figure 2). Co-treatment with ACE-011 significantly limited ET-1–induced NPPB expression and partially normalized the MYH7:MYH6 ratio in these cells (Supplementary Figure 2). Together, the above results indicate that ActRIIA-Fc exerts cardioprotective effects in multiple mouse models of left heart failure, consistent with previous studies of activin signaling pathway inhibition, and imply that activin-class ligands act directly on cardiomyocytes to drive pathogenic cellular activity in these contexts.
ActRIIA-Fc improves cardiopulmonary function and alleviates PH in a mouse model of PH-HFrEF
To determine whether ActRIIA-Fc alleviates PH caused by LHD, we evaluated effects of RAP-011 treatment in a TAC-PH mouse model of PH-HFrEF in which prolonged TAC causes PH (Figure 2A) (27). Compared with sham controls, this extended TAC intervention produced elevated pressures in the left atrium and induced PH (Figures 2B–D), hemodynamic effects associated with pathologic pulmonary vascular remodeling (Figures 2E, F) and pulmonary fibrosis (Figures 2G, H). Treatment with RAP-011 beginning 2 weeks after the start of TAC significantly improved each of these cardiopulmonary parameters as compared with vehicle treatment (Figures 2B–H). These results indicate that RAP-011 alleviates PH arising from sustained LV pressure overload.
FIGURE 2
We then investigated whether alleviation of PH in this model by treatment with RAP-011 was accompanied by beneficial effects on heart structure. Treatment with RAP-011 biweekly for 6 weeks beginning 2 weeks after TAC onset provided significant protection against adverse cardiac remodeling when compared with vehicle-treated mice (Figure 3). Specifically, RAP-011 treatment reduced cardiac hypertrophy (Figures 3A–C), improved LV contractility (Figures 3D, E) and LV diastolic function (Figures 3F, G), and reduced LV fibrosis (Figures 3H, I) compared with these measures in vehicle-treated controls. RAP-011 treatment also conferred significant protection against RV remodeling in this model (Supplementary Figure 3). Together, these findings establish that treatment with ActRIIA-Fc alleviates PH and protects against pathologic cardiopulmonary remodeling in this model of PH-HFrEF.
FIGURE 3
ActRIIA-Fc reduces pulmonary remodeling and improves cardiopulmonary function in the obese ZSF1-Su rat model of PH-HFpEF
We next tested whether therapeutic treatment with ActRIIA-Fc would exert favorable cardiopulmonary effects in a rat model of PH-HFpEF. In this two-hit model (23), obese ZSF1 rats develop PH-HFpEF after a single high dose of SU5416 (Su), an inhibitor of vascular endothelial growth factor receptor-2 used to induce pulmonary endothelial injury. Treatment with SU5416 alone did not induce pulmonary hypertension or diastolic dysfunction in lean rats (Supplementary Figure 4), while ZSF1 rats in the absence of SU5416 exhibited diastolic dysfunction without pulmonary hypertension (Supplementary Figure 4). Obese ZSF1-Su rats have been shown to recapitulate hemodynamic features and clinical outcomes of patients with PH-HFpEF, notably including CpcPH-HFpEF. As one aspect of our evaluation in this model, we compared ActRIIA-Fc effects with those of sildenafil, a representative phosphodiesterase type 5 (PDE5) inhibitor, because therapeutic agents in this class have been assessed in patients with HFpEF (28, 29). Preclinically, sildenafil therapy produces modest improvements in systemic hypertension and LV stiffness in obese ZSF1 rats without PH (30), which model disease features observed in some patients with HFpEF.
To mimic a clinical stage in which disease has progressed substantially before onset of treatment, we initiated treatment with either RAP-011 or vehicle 6 weeks after administration of SU5416, at which time RV dysfunction is prominent, and continued treatment biweekly for 8 weeks (Figure 4A). As determined by right heart catheterization and echocardiography, obese ZSF1-Su rats treated with vehicle exhibited significantly increased RV systolic pressure (RVSP) and reduced pulmonary artery acceleration time (PAAT) (Figure 4B and Supplementary Figure 5). Therapeutic treatment with RAP-011 fully reversed these changes to values observed in lean controls, whereas sildenafil treatment improved RVSP by 19% and PAAT by 36%. In addition, treatment with RAP-011, but not sildenafil, fully normalized RV hypertrophy (Fulton index) and tricuspid annular plane systolic excursion (TAPSE) (Figure 4B and Supplementary Figure 5). Histologic analysis confirmed the presence of pulmonary vascular remodeling in obese ZSF1-Su rats, as described previously. Therapeutic treatment with RAP-011 restored pulmonary vessel structure to conditions observed in lean controls (Figures 4C, D). Finally, elevated levels of pulmonary fibrosis present in obese ZSF1-Su rats were partially reversed by therapeutic treatment with RAP-011 (Figures 4E, F). Together, these results indicate that therapeutic treatment with ActRIIA-Fc reduces fibrosis, reverses RV and pulmonary vascular remodeling, improves RV function, and alleviates PH in the obese ZSF1-Su model of PH-HFpEF.
FIGURE 4
ActRIIA-Fc improves remodeling and function of both left and right heart in obese ZSF1-Su rats
Based on the beneficial cardiac effects of ActRIIA-Fc observed in the above experiments, we investigated cardiac effects of ActRIIA-Fc treatment more extensively in the PH-HFpEF model. As before, we initiated treatment with either ActRIIA-Fc (RAP-011) or vehicle 6 weeks after administration of SU5416 and continued treatment biweekly for 8 weeks (weeks 14–22, Figure 4A).
As determined by right heart catheterization and echocardiography, LV and RV dysfunction and abnormal remodeling were prominent in diseased rats before treatment initiation at week 14 (Figures 5A–C). Follow-up echocardiographic analysis at week 22 revealed further disease progression of all evaluated parameters in vehicle-treated, obese ZSF1-Su rats, compared with assessments made either at baseline (week 14) or in lean controls (week 22) (Figures 5A–C). Therapeutic treatment of obese ZSF1-Su rats with RAP-011 normalized LV mass and produced significant improvements in two measures of LV diastolic dysfunction, E/E’ [the ratio of mitral inflow velocity (E) to mitral annular velocity (E’)] and isovolumic relaxation time (IVRT) (Figure 5A). RAP-011 treatment also significantly improved RV remodeling [RV free-wall thickness (RVFWT)] and RV function [RV fractional area change (RVFAC) and TAPSE] (Figure 5B), which confirmed the result for TAPSE above (Supplementary Figure 5). In addition, RAP-011 treatment in obese ZSF1-Su rats significantly improved the myocardial performance index (MPI), a measure of global cardiac function (Figure 5C). Finally, RAP-011 treatment significantly reduced fibrosis in both the LV and RV compared with vehicle-treated controls (Figure 5D and Supplementary Figure 6). These results provide compelling evidence that beneficial effects of ActRIIA-Fc treatment on PH are accompanied by reversal of pathologic cardiac remodeling as well as functional improvements in both ventricles in this model of PH-HFpEF.
FIGURE 5
ActRIIA-Fc inhibits canonical and non-canonical TGF-β superfamily signaling in the lungs of obese ZSF1-Su rats
We next investigated signaling pathways whose activity is influenced by ActRIIA-Fc in the lungs of obese ZSF1-Su rats. Compared with lean controls, obese ZSF1-Su rats displayed elevated activation of SMAD3 in pulmonary perivascular cells as determined by immunostaining for phospho-SMAD3 (pSMAD3) (Figures 6A, B). In obese ZSF1-Su rats, therapeutic treatment with RAP-011 normalized pSMAD3 immunostaining in these perivascular cells (Figures 6A, B), which were shown separately by dual immunofluorescence staining to include vascular smooth muscle cells and endothelial cells (Supplementary Figures 7, 8). Consistent with disease-related overactivation of SMAD3 in the pulmonary vasculature, perivascular regions of ZSF-Su rat lung were found to contain increased immunostaining for activin A and GDF11 (Supplementary Figure 9), prominent activators of SMAD2/3 signaling previously implicated in PAH (15). Obese ZSF1-Su rats treated with vehicle exhibited elevated activation of the p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) pathways in lung as determined by protein blot analysis of phospho–extracellular-signal-regulated kinase (pERK) and pJNK levels in whole lung lysate (Figure 6C). In obese ZSF1-Su rats, RAP-011 treatment reduced pERK and pJNK levels, whereas total ERK and total JNK expression remained unchanged (Figure 6C). These results indicate that lung tissue in obese ZSF1-Su rats displays elevated activation of pathways associated with TGF-β superfamily canonical (pSMAD3) and non-canonical (pERK and pJNK) signaling and that therapeutic treatment with ActRIIA-Fc inhibits the activation of both types.
FIGURE 6
ActRIIA-Fc inhibits glucose-induced release of activin and migration of human PASMCs mediated by the SGLT pathway
To explore mechanisms by which ActRIIA-Fc improves pulmonary vascular remodeling in obese ZSF1-Su rats, we examined the activity of hPASMCs in vitro under elevated glucose conditions to model metabolic comorbidities observed in patients with PH-HFpEF. We observed that glucose treatment produced a significant, time-dependent increase in activin A release by hPASMCs, with pronounced increases in extracellular activin A concentrations occurring at 24 h of exposure and increases of more than 10-fold at 48 h (Figure 7A). Sotagliflozin, a dual inhibitor of sodium-glucose cotransporters (SGLT), significantly reduced activin A release by hPASMCs under elevated glucose conditions (Figure 7B), implicating this transporter type in glucose-induced release of activin A. Treatment of cultured hPASMCs with ACE-011 completely blocked glucose-induced release of activin A (Figure 7C).
FIGURE 7
We next investigated migratory activity of hPASMCs in vitro using a Boyden chamber migration assay. Treatment with activin A significantly stimulated migration of hPASMCs, and this migratory effect was blocked completely by co-treatment with ACE-011 (Figure 7D). Treatment with elevated glucose levels increased migration of hPASMCs beyond rates observed with normal physiologic glucose levels, and this effect was completely blocked by either SGLT inhibitor or ACE-011 (Figure 7E). These results implicate a novel mechanism contributing to aberrant vascular remodeling in which glucose-induced release of activin A by PASMCs triggers their transition to an abnormal migratory phenotype, thereby promoting maladaptive pulmonary vascular modeling in the context of metabolic disorder.
Finally, we explored whether SGLT is similarly dysregulated in lungs of obese ZSF1-Su rats in vivo. Compared with lean controls, obese ZSF1-Su rats treated with vehicle exhibited increased pulmonary expression of SGLT1 protein and reduced expression of glucose transporter GLUT1 (Figure 7F). Treatment of obese ZSF1-Su rats with RAP-011 normalized SGLT1 protein expression in lung tissue without detectable changes in GLUT1 protein expression (Figure 7F). As summarized schematically in Figure 7G, our concordant findings from in vitro and in vivo models implicate SGLT, activin A, and activin receptor-mediated signaling—both canonical and non-canonical—as key components in a PASMC-mediated process by which hyperglycemia promotes aberrant pulmonary vascular remodeling and PH in PH-HFpEF. These results also provide mechanistic insight into ActRIIA-Fc alleviation of PH associated with metabolic comorbidities in this preclinical model of HFpEF.
Discussion
There is major unmet need for patients with PH-LHD. PH is highly prevalent in patients with LHD and is associated with a worse prognosis than LHD without PH (2, 5). Elevated left atrial pressure due to LHD causes pulmonary venous congestion and increased hydrostatic pressure in pulmonary capillaries, thereby leading to IpcPH-LHD and CpcPH-LHD (2). Despite shared features in the pathophysiology of CpcPH-LHD and PAH (11), including pre-capillary pulmonary vascular remodeling, vasodilators used to treat PAH are ineffective in patients with PH-LHD and can even be harmful (8, 31). In the present study, we find that therapeutic treatment with ActRIIA-Fc exerts reverse remodeling effects in the pulmonary vasculature, left heart, and right heart in rats with experimental CpcPH-HFpEF, whereas the vasodilator sildenafil does not, consistent with previous clinical results (32). Here we also find that ActRIIA-Fc exerts protective cardiopulmonary effects in an established model of a second major type of PH-LHD, CpcPH-HFrEF.
Pulmonary vascular remodeling is a key pathologic feature in patients with PH and is associated with diminished pulmonary arterial compliance, impaired RV–pulmonary arterial coupling, reduced diffusing capacity of the lungs, abnormal RV remodeling, and higher patient morbidity or mortality (2, 9, 10, 33). We recently identified unbalanced signaling by the SMAD pathways—with important contributions by activin-class ligands—in pathogenic vascular remodeling in lung that underlies human and experimental PAH (15). In the present study, we similarly found evidence of increased expression of activin A and GDF11 in the pulmonary vasculature in two experimental models of PH-LHD. Several cell types are implicated in pulmonary vascular remodeling, including endothelial cells, vascular SMCs (VSMCs), adventitial fibroblasts, and immune cells (15, 16, 34). Here we focused our analysis in vitro on pulmonary VSMCs because they are a nearly universal component of vascular remodeling (35) and contribute to progressive pre-capillary remodeling in CpcPH-HFpEF (3, 9), as confirmed in our histological assessment of obese ZSF1-Su rats. Unlike terminally differentiated skeletal or cardiac muscle cells, VSMCs retain remarkable plasticity in adulthood, which enables vessel growth and adaptive remodeling but also contributes broadly to cardiovascular pathologies (36).
The lung has historically not been considered an organ prominently afflicted by glucose dysregulation in diabetes (37). However, our studies using hPASMCs reveal an activin-mediated mechanism by which hyperglycemia causes an abnormal phenotypic shift in hPASMCs that could contribute to pulmonary vascular remodeling and PH in experimental PH-HFpEF. Despite the resemblance between CpcPH-HFpEF and PAH with regard to vascular remodeling, the glucose-associated aspect of this mechanism differs from other mechanistic elements identified so far for activin signaling and pulmonary vascular remodeling in the context of experimental PAH (15, 16, 38).
In our model, activin A produced by hPASMCs acts in an autocrine or paracrine manner to mediate a glucose-inducible shift in these cells to an abnormal migratory phenotype (Figure 7). We speculate that activin A secreted by PASMCs in vivo could potentially also affect the activity of other cell types in the pulmonary vascular microenvironment. Our results indicate that glucose induction of activin A release by hPASMCs is partly mediated by SGLT. Additionally, our results indicate that SGLT1 protein is upregulated in the lungs of obese ZSF1-Su rats in the setting of metabolic syndrome, and ActRIIA-Fc treatment normalizes these SGLT1 levels. Thus, beneficial effects of ActRIIA-Fc treatment on PASMCs under these conditions arise not only from sequestration of extracellular activin A and potentially other activin-class ligands—with consequent inhibition of canonical and non-canonical activin receptor-mediated signaling—but also from downregulation of SGLT1 protein by an indirect mechanism yet to be determined. Although many studies have demonstrated adverse effects of hyperglycemia on VSMC or endothelial cell function (39), surprisingly few have examined the severity of PH in patients with diabetes, especially for PH types other than PAH (40). However, clinical studies have identified activin A as a prominent marker of cardiovascular pathology, including increased arterial intima–media thickness, in the context of metabolic syndrome (41–44).
In the present study, ActRIIA-Fc also exerted protective cardiopulmonary effects in experimental PH-HFrEF. As underscored here by the beneficial effects of ActRIIA-Fc in models of left heart disease without PH, activin receptor signaling has been implicated previously as an important contributor to pathologic cardiac remodeling in multiple types of left heart failure. This aspect of ActRIIA-Fc activity resembles cardioprotective effects of ALK4 haplodeficiency and effects of other activin receptor pathway inhibitors (ActRIIB-Fc and a dual-specific antibody against ActRIIA and ActRIIB) in models of left ventricular failure associated with aging or systemic pressure overload (17, 18). The roles of activin pathway signaling in cardiac muscle, especially by activin A and GDF11, have been controversial (18). However, recent studies consistently implicate activin receptor–mediated SMAD2/3 signaling as a partially compensatory pathway that becomes maladaptive in heart failure and ischemia-reperfusion injury (17–20). Elevated levels of circulating activin A in patients support a role for this ligand in abnormal myocardial remodeling, diabetic cardiomyopathy, HFrEF, and HFpEF (42, 44–46). Interestingly, activin A, GDF8, and GDF11 induce similar but non-identical pathologic profiles in left ventricular cardiomyocytes (18). We speculate that multi-ligand inhibition might therefore advantageously prevent overlapping as well as distinct activities of activin-class ligands, which could be mutually reinforcing in the therapeutic treatment of cardiovascular disease.
Our results obtained with complementary experimental models display robust concordance and support the translatability of our findings. In this study, we focused on pulmonary VSMCs due in part to their important roles in vascular muscularization and remodeling. However, evidence strongly suggests that other pulmonary vascular cell types contribute to PH and are regulated extensively by activin pathway signaling (14–16, 38). It will therefore be important to investigate the contributions of these signaling mechanisms to dysregulated endothelial cell, fibroblast, and immune cell activities implicated in PH-HFpEF. Immune cells and inflammatory processes play major roles in PH (14, 47–49), and recent evidence indicates that ActRIIA-Fc suppresses inflammation as one component of its multi-factorial mechanism of action in experimental PAH (16). An ongoing phase 2 study is evaluating sotatercept, a human analog of ActRIIA-Fc, in patients with CpcPH-HFpEF.
Statements
Data availability statement
The original contributions presented in this study are included in this article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee at Acceleron Pharma Inc., a subsidiary of Merck & Co., Inc., Rahway, NJ, USA and performed in accordance with the guidelines from the NIH Guide for the Care and Use of Laboratory Animals.
Author contributions
SJ, EA, and GL planned the research. SJ and JL performed in vivo experiments. EA, JL, and YD performed in vitro experiments. SJ, EA, JL, YD, and GL analyzed data. PA, RK, and GL provided guidance on experimental designs and data analysis. SJ, EA, SB, MA, and GL wrote the manuscript, which was reviewed and approved for submission by all authors.
Acknowledgments
We thank Jonathan Lu for discussion and clinical input. Research described in this manuscript was funded by Acceleron Pharma Inc., a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
Conflict of interest
All authors were employed by Acceleron Pharma Inc., a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1064290/full#supplementary-material
References
1.
HoeperMHumbertMSouzaRIdreesMKawutSSliwa-HahnleKet alA global view of pulmonary hypertension.Lancet Respir Med. (2016) 4:306–22.
2.
AdlerJGerhardtFWissmüllerMAdlerCBaldusSRosenkranzS. Pulmonary hypertension associated with left-sided heart failure.Curr Opin Cardiol. (2020) 35:610–9.
3.
GuazziMGhioSAdirY. Pulmonary hypertension in HFpEF and HFrEF: JACC review topic of the week.J Am Coll Cardiol. (2020) 76:1102–11. 10.1016/j.jacc.2020.06.069
4.
RosenkranzSGibbsJWachterRDe MarcoTVonk-NoordegraafAVachiéryJ. Left ventricular heart failure and pulmonary hypertension.Eur Heart J. (2015) 37:942–54.
5.
RosenkranzSLangIBlindtRBondermanDBruchLDillerGet alPulmonary hypertension associated with left heart disease: updated recommendations of the Cologne Consensus Conference 2018.Int J Cardiol. (2018) 272:53–62. 10.1016/j.ijcard.2018.08.080
6.
MurphySIbrahimNJanuzziJJr.Heart failure with reduced ejection fraction: a review.JAMA. (2020) 324:488–504.
7.
AnkerSButlerJFilippatosGFerreiraJBocchiEBöhmMet alEmpagliflozin in heart failure with a preserved ejection fraction.N Engl J Med. (2021) 385:1451–61.
8.
LteifCAtayaADuarteJ. Therapeutic challenges and emerging treatment targets for pulmonary hypertension in left heart disease.J Am Heart Assoc. (2021) 10:e020633.
9.
FayyazAEdwardsWMaleszewskiJKonikEDuBrockHBorlaugBet alGlobal pulmonary vascular remodeling in pulmonary hypertension associated with heart failure and preserved or reduced ejection fraction.Circulation. (2018) 137:1796–810.
10.
LevineASimonMGladwinM. Pulmonary vascular disease in the setting of heart failure with preserved ejection fraction.Trends Cardiovasc Med. (2019) 29:207–17.
11.
AssadTHemnesALarkinEGlazerAXuMWellsQet alClinical and biological insights into combined post- and pre-capillary pulmonary hypertension.J Am Coll Cardiol. (2016) 68:2525–36. 10.1016/j.jacc.2016.09.942
12.
RanchouxBNadeauVBourgeoisAProvencherSTremblayÉOmuraJet alMetabolic syndrome exacerbates pulmonary hypertension due to left heart disease.Circ Res. (2019) 125:449–66.
13.
TrammellAHemnesATsengVShahAPhillipsLHartC. Influence of body weight and diabetes mellitus in patients with pulmonary hypertension.Am J Cardiol. (2020) 134:130–7.
14.
AndrePJoshiSBriscoeSAlexanderMLiGKumarR. Therapeutic approaches for treating pulmonary arterial hypertension by correcting imbalanced TGF-β superfamily signaling.Front Med. (2022) 8:814222. 10.3389/fmed.2021.814222
15.
YungLYangPJoshiSAugurZKimSBocoboGet alACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension.Sci Transl Med. (2020) 12:eaaz5660. 10.1126/scitranslmed.aaz5660
16.
JoshiSLiuJBloomTKaraca AtabayEKuoTLeeMet alSotatercept analog suppresses inflammation to reverse experimental pulmonary arterial hypertension.Sci Rep. (2022) 12:7803. 10.1038/s41598-022-11435-x
17.
LiCChenYWangQHouJWangHWangYet alPartial inhibition of activin receptor-like kinase 4 attenuates pressure overload-induced cardiac fibrosis and improves cardiac function.J Hypertens. (2016) 34:1766–77.
18.
RohJHobsonRChaudhariVQuinteroPYeriABensonMet alActivin type II receptor signaling in cardiac aging and heart failure.Sci Transl Med. (2019) 11:eaau8680. 10.1126/scitranslmed.aau8680
19.
MaggaJVainioLKilpioTHulmiJTaponenSLinRet alSystemic blockade of ACVR2B ligands protects myocardium from acute ischemia-reperfusion injury.Mol Ther. (2019) 27:600–10. 10.1016/j.ymthe.2019.01.013
20.
CastilleroEAkashiHNajjarMJiRBrandstetterLWangCet alActivin type II receptor ligand signaling inhibition after experimental ischemic heart failure attenuates cardiac remodeling and prevents fibrosis.Am J Physiol Heart Circ Physiol. (2020) 318:H378–90. 10.1152/ajpheart.00302.2019
21.
HumbertMMcLaughlinVGibbsJGomberg-MaitlandMHoeperMPrestonIet alSotatercept for the treatment of pulmonary arterial hypertension.N Engl J Med. (2021) 384:1204–15.
22.
ParajuliNYuanYZhengXBedjaDCaiZ. Phosphatase PTEN is critically involved in post-myocardial infarction remodeling through the Akt/interleukin-10 signaling pathway.Basic Res Cardiol. (2012) 107:248. 10.1007/s00395-012-0248-6
23.
LaiYTabimaDDubeJHughanKVanderpoolRGoncharovDet alSIRT3-AMP-activated protein kinase activation by nitrite and metformin improves hyperglycemia and normalizes pulmonary hypertension associated with heart failure with preserved ejection fraction.Circulation. (2016) 133:717–31.
24.
AbeKTobaMAlzoubiAItoMFaganKCoolCet alFormation of plexiform lesions in experimental severe pulmonary arterial hypertension.Circulation. (2010) 121:2747–54.
25.
OkaMKaroorVHommaNNagaokaTSakaoEGolembeskiSet alDehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension.Cardiovasc Res. (2007) 74:377–87. 10.1016/j.cardiores.2007.01.021
26.
KarakikesIAmeenMTermglinchanVWuJ. Human induced pluripotent stem cell-derived cardiomyocytes.Circ Res. (2015) 117:80–8.
27.
ChenYGuoHXuDXuXWangHHuXet alLeft ventricular failure produces profound lung remodeling and pulmonary hypertension in mice.Hypertension. (2012) 59:1170–8. 10.1161/HYPERTENSIONAHA.111.186072
28.
GuazziMVicenziMArenaRGuazziM. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study.Circulation. (2011) 124:164–74.
29.
RedfieldMChenHBorlaugBSemigranMLeeKLewisGet alEffect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial.JAMA. (2013) 309:1268–77.
30.
LeiteSMoreira-CostaLCerqueiraRSousa-MendesCAngélico-GonçalvesAFontouraDet alChronic sildenafil therapy in the ZSF1 obese rat model of metabolic syndrome and heart failure with preserved ejection fraction.J Cardiovasc Pharmacol Ther. (2021) 26:690–701. 10.1177/10742484211034253
31.
LahmTDouglasIArcherSBogaardHCheslerNHaddadFet alAssessment of right ventricular function in the research setting: knowledge gaps and pathways forward.Am J Respir Crit Care Med. (2018) 198:e15–43. 10.1164/rccm.201806-1160ST
32.
BorlaugBLewisGMcNultySSemigranMLeWinterMChenHet alEffects of sildenafil on ventricular and vascular function in heart failure with preserved ejection fraction.Circ Heart Fail. (2015) 8:533–41.
33.
MaronBLeopoldJ. Emerging concepts in the molecular basis of pulmonary arterial hypertension.Circulation. (2015) 131:2079–91.
34.
FoshatMBoroumandN. The evolving classification of pulmonary hypertension.Arch Pathol Lab Med. (2016) 141:696–703.
35.
StenmarkKFridMGrahamBTuderR. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension.Cardiovasc Res. (2018) 114:551–64. 10.1093/cvr/cvy004
36.
OwensGKumarMWamhoffB. Molecular regulation of vascular smooth muscle cell differentiation in development and disease.Physiol Rev. (2004) 84:767–801.
37.
FusoLPitoccoDAntonelli-IncalziR. Diabetic lung, an underrated complication from restrictive functional pattern to pulmonary hypertension.Diabetes Metab Res Rev. (2019) 35:e3159. 10.1002/dmrr.3159
38.
RyantoGIkedaKMiyagawaKTuLGuignabertCHumbertMet alAn endothelial activin A-bone morphogenetic protein receptor type 2 link is overdriven in pulmonary hypertension.Nat Commun. (2021) 12:1720. 10.1038/s41467-021-21961-3
39.
GraySJandeleit-DahmK. The pathobiology of diabetic vascular complications—cardiovascular and kidney disease.J Mol Med. (2014) 92:441–52. 10.1007/s00109-014-1146-1
40.
WillsonCWatanabeMTsuji-HosokawaAMakinoA. Pulmonary vascular dysfunction in metabolic syndrome.J Physiol. (2019) 597:1121–41.
41.
AndersenGUelandTKnudsenEScholzHYndestadASahraouiAet alActivin A levels are associated with abnormal glucose regulation in patients with myocardial infarction: potential counteracting effects of activin A on inflammation.Diabetes. (2011) 60:1544–51. 10.2337/db10-1493
42.
ChenWGreulichSvan der MeerRRijzewijkLLambHde RoosAet alActivin A is associated with impaired myocardial glucose metabolism and left ventricular remodeling in patients with uncomplicated type 2 diabetes.Cardiovasc Diabetol. (2013) 12:150. 10.1186/1475-2840-12-150
43.
KuoCLuYHsuCChangCChouRLiuLet alIncreased activin A levels in prediabetes and association with carotid intima-media thickness: a cross-sectional analysis from I-Lan Longitudinal Aging Study.Sci Rep. (2018) 8:9957. 10.1038/s41598-018-27795-2
44.
ZellerJKrügerCLamounier-ZepterVSagSStrackCMohrMet alThe adipo-fibrokine activin A is associated with metabolic abnormalities and left ventricular diastolic dysfunction in obese patients.ESC Heart Fail. (2019) 6:362–70. 10.1002/ehf2.12409
45.
YndestadAUelandTØieEFlorholmenGHalvorsenBAttramadalHet alElevated levels of activin A in heart failure.Circulation. (2004) 109:1379–85.
46.
TsaiYChouRKuoCChangCWuCHuangPet alCirculating activin A is a surrogate for the incidence of diastolic dysfunction and heart failure in patients with preserved ejection fraction.Circ J. (2019) 83:1514–9. 10.1253/circj.CJ-18-0837
47.
GlezevaNBaughJ. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target.Heart Fail Rev. (2014) 19:681–94. 10.1007/s10741-013-9405-8
48.
DewachterLDewachterC. Inflammation in right ventricular failure: does it matter?Front Physiol. (2018) 9:1056. 10.3389/fphys.2018.01056
49.
HuertasATuLHumbertMGuignabertC. Chronic inflammation within the vascular wall in pulmonary arterial hypertension: more than a spectator.Cardiovasc Res. (2019) 116:885–93. 10.1093/cvr/cvz308
Summary
Keywords
Group 2 pulmonary hypertension (PH), pulmonary arterial hypertension, heart failure with preserved ejection fraction, heart failure with reduced ejection fraction, ActRIIA-Fc, activin, smooth muscle cell, vascular remodeling
Citation
Joshi SR, Atabay EK, Liu J, Ding Y, Briscoe SD, Alexander MJ, Andre P, Kumar R and Li G (2023) Sotatercept analog improves cardiopulmonary remodeling and pulmonary hypertension in experimental left heart failure. Front. Cardiovasc. Med. 10:1064290. doi: 10.3389/fcvm.2023.1064290
Received
08 October 2022
Accepted
19 January 2023
Published
23 February 2023
Volume
10 - 2023
Edited by
Xiaohui Li, Central South University, China
Reviewed by
Noriaki Emoto, Kobe Pharmaceutical University, Japan; Kazufumi Nakamura, Okayama University, Japan
Updates
Copyright
© 2023 Joshi, Atabay, Liu, Ding, Briscoe, Alexander, Andre, Kumar and Li.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Li, garygl@gmail.com
This article was submitted to General Cardiovascular Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.