 Qing Liu
Qing Liu Suyao Li
Suyao Li Yuran Qiu3
Yuran Qiu3 Francisco J. Rios
Francisco J. Rios Zhiguo Zou
Zhiguo Zou Rhian M. Touyz
Rhian M. Touyz
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 03 February 2023
Sec. General Cardiovascular Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1002438
This article is part of the Research TopicReceptors in Cardiovascular Diseases: Mechanisms, diagnosis and treatmentView all 5 articles
Receptor tyrosine kinases (RTKs) are a class of membrane spanning cell-surface receptors that transmit extracellular signals through the membrane to trigger diverse intracellular signaling through tyrosine kinases (TKs), and play important role in cancer development. Therapeutic approaches targeting RTKs such as vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR), and platelet-derived growth factor receptor (PDGFR), and TKs, such as c-Src, ABL, JAK, are widely used to treat human cancers. Despite favorable benefits in cancer treatment that prolong survival, these tyrosine kinase inhibitors (TKIs) and monoclonal antibodies targeting RTKs are also accompanied by adverse effects, including cardiovascular toxicity. Mechanisms underlying TKI-induced cardiovascular toxicity remain unclear. The transient receptor potential melastatin-subfamily member 7 (TRPM7) is a ubiquitously expressed chanzyme consisting of a membrane-based ion channel and intracellular α-kinase. TRPM7 is a cation channel that regulates transmembrane Mg2+ and Ca2+ and is involved in a variety of (patho)physiological processes in the cardiovascular system, contributing to hypertension, cardiac fibrosis, inflammation, and atrial arrhythmias. Of importance, we and others demonstrated significant cross-talk between TRPM7, RTKs, and TK signaling in different cell types including vascular smooth muscle cells (VSMCs), which might be a link between TKIs and their cardiovascular effects. In this review, we summarize the implications of RTK inhibitors (RTKIs) and TKIs in cardiovascular toxicities during anti-cancer treatment, with a focus on the potential role of TRPM7/Mg2+ as a mediator of RTKI/TKI-induced cardiovascular toxicity. We also describe the important role of TRPM7 in cancer development and cardiovascular diseases, and the interaction between TRPM7 and RTKs, providing insights for possible mechanisms underlying cardiovascular disease in cancer patients treated with RTKI/TKIs.
Receptor tyrosine kinases (RTKs) are a class of membrane-spanning cell-surface receptors that transmit extracellular signals through the membrane to trigger diverse intracellular signaling (1). In humans, 58 RTKs have been described that fall into 20 subfamilies (2).They all share a highly conserved structure, comprising a ligand-binding extracellular domain, an alpha-helical transmembrane domain and the tyrosine kinase domain (TKD) (3). Canonically, binding of ligands to RTKs induces dimerization and/or oligomerization of extracellular domains, resulting in activation of TKDs via trans-autophosphorylation and subsequent recruitment and activation of downstream signaling proteins. The activation of RTKs leads to phosphorylation and activation of numerous tyrosine kinases such as Abl, c-Src, Ras, PI-3K, JAK, and ALK, which regulatecellular processes, such as cell migration, differentiation, apoptosis, contraction, metabolism and survival (4). Activation of RTKs and TKs is critically involved in abnormal cell growth in cancer.
Abnormal expression and overactivation of RTKs, including vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR), and platelet-derived growth factor receptor (PDGFR) are associated with tumor invasion, metastasis, and tumor angiogenesis (5). Inhibiting these RTKs and their downstream signaling pathways reduce tumorigenesis and over the past 20 years there has been enormous interest in developing RTKI/TKIs as anti-cancer drugs (5). Despite favorable anti-cancer benefits, and prologed survival, TKIs are also accompanied by a profile of cardiovascular toxicities including hypertension, heart failure (HF) and arrhythmias (6). Understanding mechanisms of these side effects would improve the management of TKI-related cardiovascular toxicity and the clinical outcome.
The transient receptor potential melastatin-subfamily member 7 (TRPM7) possesses both ion channel and enzymatic functions. TRPM7 channel is permeable to divalent cations such as Zn2+, Mg2+ and Ca2+, and the α-kinase domain phosphorylates downstream substrates including annexin-1, eukaryotic elongation factor 2 (eEF2)'s cognate kinase (eEF2K), phospholipase Cγ2 (PLCγ2), myosin IIA, SMAD2, tropomodulin 1, myelin basic protein (MBP), cAMP response element binding protein (CREB), and RhoA (7–15). TRPM7 plays an important role in the cardiovascular system, regulating cardiac and vascular ion homeostasis, vascular smooth muscle cell function, vascular morphology and cardiac function. Abnormal TRPM7 activity has been implicated in hypertension, cardiac fibrosis, inflammation and atrial fibrillation (AF) (16–19), with TRPM7 downregulation promoting cardiovascular injury. On the other hand, aberrant expression of TRPM7 has been identified in various tumors, suggesting its significant involvement in tumorigenesis and cancer development (20–28).
In this review, we summarize the implications of RTK and TK inhibition in human cancers, with a focus on their cardiovascular toxicities. We also describe the important role of TRPM7 in cancer development and cardiovascular diseases, and the interaction between TRPM7 and RTKs, providing insights for possible mechanisms whereby anti-cancer drugs targeting RTKs induce cardiovascular toxicity.
Under physiologic conditions, RTK activity is tightly controlled. However, dysregulated signaling through RTKs promotes an imbalance between cell proliferation and cell death, which is implicated in cancer development. Mechanisms underlying the aberrant activation of RTKs are associated with overexpression (29), mutations (30), chromosomal rearrangement (31), autocrine activation (32), and RTK interaction with other kinases, proteins, and signaling molecules (5) (Figure 1).
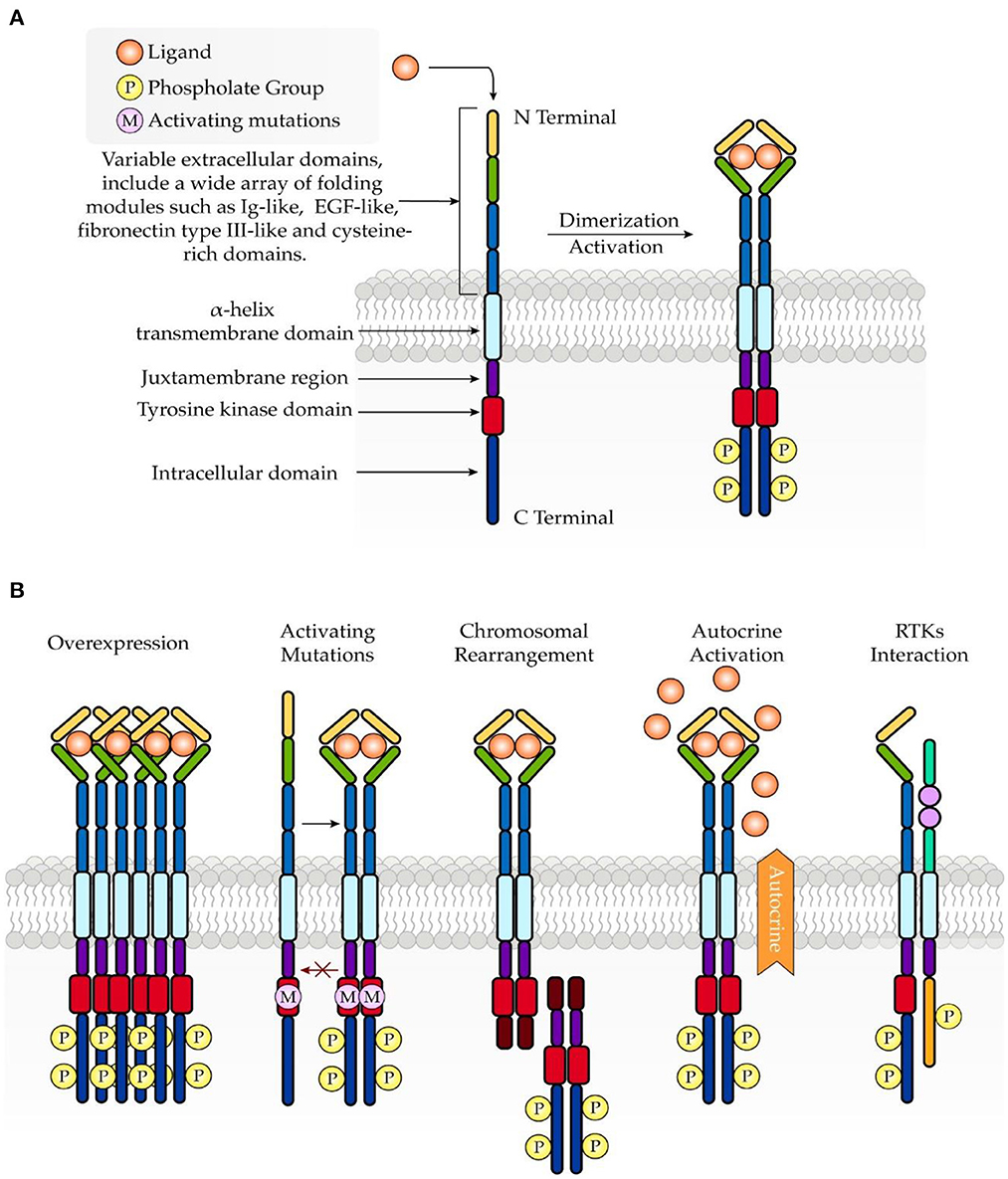
Figure 1. Schematic structure of RTKs and common mechanisms involved in oncogenesis. (A) RTKs consist of an extracellular ligand-binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain. Upon ligand binding, RTKs undergo dimerization, which further leads to activation of the intracellular tyrosine kinase domain. (B) Abnormal activation of RTKs in turmor growth is associated with overexpression, mutations, chromosomal rearrangement, autocrine activation, and RTK interaction.
TKIs are a class of pharmacologic agents that block multiple signal transduction pathways (33). In 2001, the US Food and Drug Administration (FDA) approved the first TKI imatinib, which targets the breakpoint cluster region protein- Abelson murine leukemia viral oncogene homolog (BCR-ABL) oncogene, and brought a revolutionary success to the treatment of chronic myeloid leukemia (CML) (34). To date, over 50 TKIs have been approved by the FDA. Major TKIs and their indications are listed in Table 1.
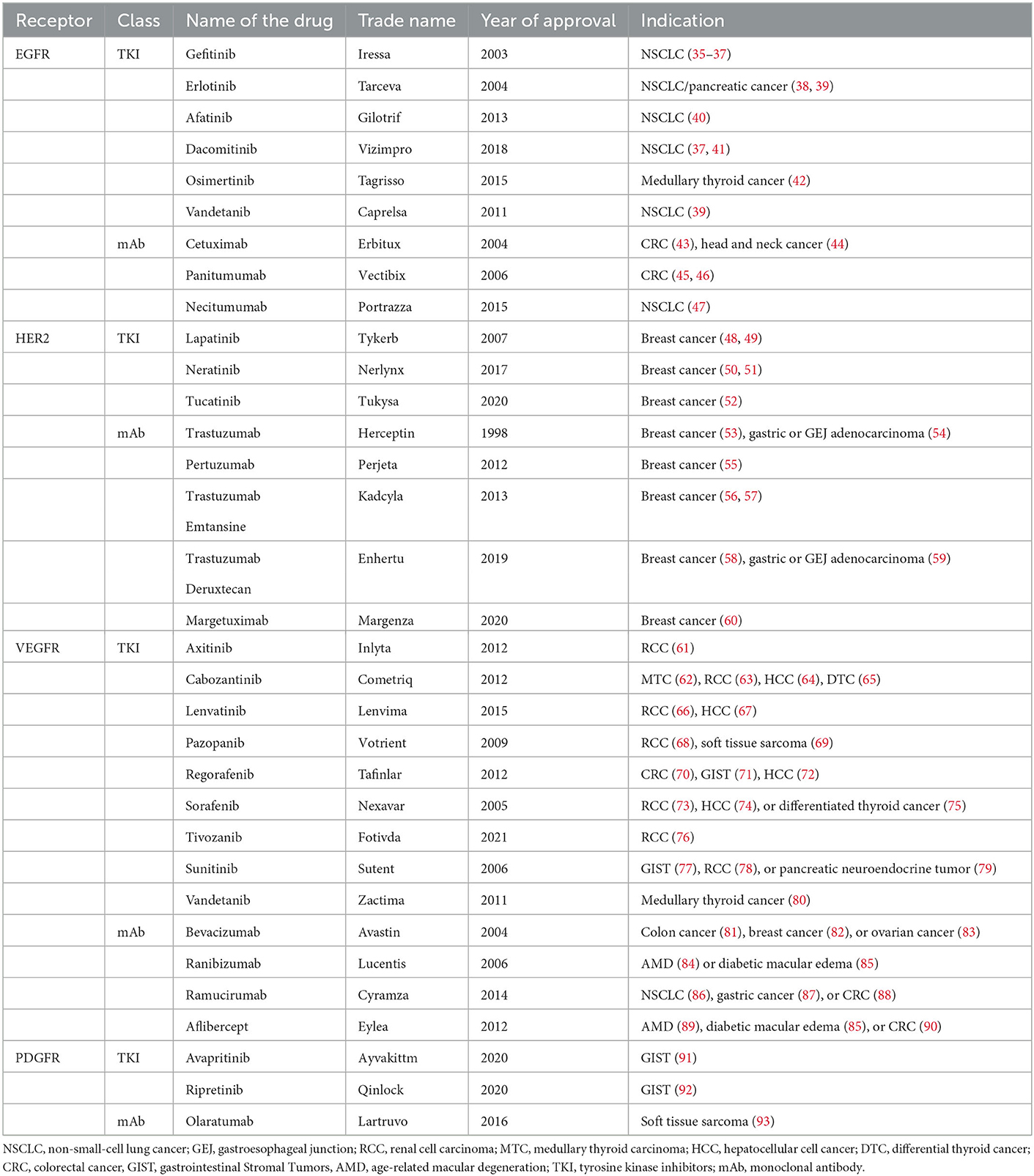
Table 1. TKIs approved by FDA in cancer treatment.
VEGF signaling plays a critical role in angiogenesis, cell proliferation and survival. Following advances in knowledge about the role of angiogenesis in promoting tumor growth (94, 95), multiple clinical trials demonstrated that VEGFR inhibitors yield incremental improvements in outcomes for a variety of solid tumors. However, the increasing use of these agents is also associated with a wide spectrum of side effects, most frequently related to cardiovascular toxicity, which might be linked to direct effects of VEGF inhibitors on the vasculature (96).
Hypertension is considered as the main cardiovascular side effect of VEGFR-TKIs. Almost every trial reports treatment-induced blood pressure elevation and up to 80% of patients develop hypertension, either de novo or worsening of previously controlled hypertension (97). As outlined in a meta-analysis including 77 VEGF inhibitors, severe hypertension occurred in 7.4%, cardiac dysfunction in 2.3%, arterial thromboembolism in 1.8%, and cardiac ischemic in 1.7% of patients, and there was no significant difference in cardiovascular risk between anti-VEGF monoclonal antibody and TKIs (98). Molecular mechanisms underlying the development of hypertension during VEGFR-TKI therapy remain unclear. However, many studies have shown that VEGFR inhibitors reduce the level of vasodilators, including nitric oxide (NO) and PGI2, which are crucial in the development of hypertension. In patients treated with VEGFR-TKI, plasma levels of NO and its metabolites are decreased, but return to baseline following withdrawal (99). Also, VEGFR-TKI therapy is associated with an elevated level of endothelin-1 (ET-1, a potent vasoconstrictor) (100). ET-1 receptor antagonists have been shown to be effective to treat VEGFR-TKI-associated hypertension in a pre-clinical study suggesting the involvement of ET-1 in VEGFR-TKI-related hypertension (101).
VEGFR inhibition is known to cause both arterial thrombosis event (ATE), particularly cardiac ischemia, and venous thromboembolism (VTE). The risk of ATE associated with anti-VEGF TKIs is greater than that of VTE, with an incidence generally <3% (102, 103). A meta-analysis of 19 randomized controlled trials including 9,711 patients treated with anti-VEGF TKIs showed a significantly increased risk of developing ATE when compared with controls (OR 2.26, 95% CI: 1.38–3.68, p = 0.001), with cardiac ischemia/infarction (67.4%) as the most common events for ATE (104). However, another meta-analysis of 7,441 patients from 17 phrase II/III trials reported no difference in the relative risk of VTE for anti-VEGF TKIs compared with controls (105). Several mechanisms have been proposed to account for the thromboembolic events of anti-VEGF therapy. Apart from the facilitation of endothelial cell proliferation and survival, VEGF activity is crucially involved in the maintenance of vascular integrity (106, 107). Hence, blockage of VEGF signaling impairs the integrity and regenerative capacity of endothelial cells, subsequently leading to thrombosis. Moreover, decreased level of PGI2 and NO related to anti-VEGF therapy creates a procoagulant environment in the vessel wall, predisposing patients to thromboembolic events (106).
Meta-analysis of trials of VEGFR-TKIs (sunitinib, sorafenib, pazopanib, axitinib, vandetanib, cabozantinib, ponatinib and regorafenib) including 10,647 patients demonstrated a pooled incidence of asymptomatic HF of 2.4% and symptomatic HF of 1.2%. Notably, there was no apparent difference in the risk of cardiovascular toxicity between the relatively specific VEGFR-TKIs (e.g., axitinib) and those targeting against a broader range of tyrosine kinases (e.g., sunitinib, sorafenib, and pazopanib) (108). Mechanisms underlying VEGFR-TKI-associated HF appear to be highly relevant to the cardiac afterload increased by endothelial dysfunction and hypertension (109). Mitochondrial dysfunction and cytochrome C-induced apoptosis might also be important, which are caused by the on-target VEGF signaling inhibition of the PI3K-AKT pathway (110). Moreover, inhibition of not only angiogenesis but also other off-targets, such as PDGFR and the adenosine monophosphate-activated protein kinase (AMPK), might be implicated in potential mechanisms that lead to HF (6).
The incidence of QT prolongation associated with VEGFR-TKIs varies widely among individual drugs. Vandetanib has the highest incidence and the most significant prolongation, with up to 8% of patients exhibiting a corrected QT (QTc) interval duration of >500 ms (111). Meta-analysis of VEGFR-TKI related clinical trials demonstrated an incidence of 4.4% of all-grade QT prolongation when compared to non-TKI therapy (112). The exact mechanism of QT prolongation from anti-VEGF TKIs is unclear, while it has been hypothesized that these TKIs might interact with the human ether-a-go-go-related gene (hERG) potassium channels, predisposing to QT prolongation (113). Of major significance, hypomagnesemia, possibly due to abnormal TRPM7 activity, is a major cause of long QT syndrome (114).
EGFR is a cell surface protein that binds to epidermal growth factor, thus inducing receptor dimerization and tyrosine autophosphorylation leading to cell proliferation. Mutations in this gene have been associated with a number of cancers, including adenocarcinoma of the lung (115), glioblastoma and epithelial tumors of the head and neck (116). Based on the FDA Adverse Events Reporting System (FAERS), a public database that contains nearly 17 million (adverse event) AE reports, EGFR TKIs have been associated with HF and cardiac arrhythmias such as atrial fibrillation and QT prolongation (117). In a retrospective cohort study of 123 patients with advanced non-small cell lung cancer (NSCLC) who received Osimertinib, the first third-generation EGFR-TKI, the incidence of cardiac AEs was 4.9% (118).
A meta-analysis of 10 randomized clinical trials involving 12,000 patients treated by trastuzumab, a monoclonal antibody targeting HER2 (the family member of EGFR), indicated that the incidence of asymptomatic decline of left ventricular ejection fraction (LVEF, normal range 50–70%) and symptomatic HF was 7.5 and 1.9%, respectively (119). A case-control study of 53 patients receiving HER2-targeted therapy further found that an LVEF <55% at any surveillance timepoint was associated with higher risk for HF, suggesting that baseline cardiac function might be an important factor that determines the cardiac outcome (120). Additionally, an increasing number of case reports have demonstrated significant HF induced by Osimertinib in patients with lung cancer, a problem that could be complicated by the coincidence of QT prolongation (121–123). In animal studies, Threadgill et al. found that wild-type mice displayed cardiac dysfunction and increased cardiac fibrosis after 3-month exposure to EGFR-TKIs (124). Intriguingly, female mice exhibited increased cardiac adverse effects, suggesting that sex might influence the susceptibility to TKI-mediated toxicity (124). Furthermore, taking advantages of the myoblast cell line H9c2 and in vivo rat cardiomyocytes, Korashy et al. showed that Gefitinib, a selective inhibitor of EGFR, induces cardiovascular toxicity through modulating the cardiac PTEN/AKT/FoxO3a pathway and the formation of CYP1A1-mediated reactive metabolites (125).
In a retrospective study based on the world health organization (WHO) pharmacovigilance database VigiBase, among 98,765 adverse reactions reported with NSCLC-targeted therapies including EGFR-TKIs, 1,783 (1.8%) were cardiac arrhythmias (126). The most frequently reported cardiac arrhythmia associated with EGFR-TKIs is QT prolongation. Rociletinib, a third-generation EGFR-TKI targeting common EGFR-activating mutations, was found to increase the risk of corrected QT prolongation compared to chemotherapy (6.7 vs. 0%) in patients with advanced or metastatic NSCLC (127). Case reports showed that Osimertinib induced QT prolongation in patients with lung cancer, while discontinuation of the drug led to the alleviation of prolonged QT interval (121, 123, 128). The scenario is further complicated by the concurrence of HF, while whether there is a casual link remains unclear (121, 123). These studies also recommend that careful monitoring of electrocardiogram (ECG) and serum potassium, a cation that importantly affects QT interval, is required in neoplastic patients receiving EGFR-TKIs therapy (128). It is also worth noting that several clinical studies failed to provide evidence supporting any correlation between anti-EGFR therapy and cardiac abnormalities including QT prolongation, and thus future adequately powered clinical trials are still required (129, 130).
PDGFR signaling pathway plays a critical role in promoting cardiomyocyte proliferation and heart regeneration (131). Various TKIs that inhibit VEGFR/PDGFR were shown to induce significant cell death in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) (132). Additionally, in an animal model of myocardial infarction, PDGF gene transfer was able to improve left ventricular function (133). In considering the potential cardioprotective effect of PDGF, it is not surprising that anti-PDGFR therapy in cancer patients was associated with cardiac AEs. Sunitinib, a TKI with multiple targets including PDGFR, was reported to induce blood pressure elevation and LVEF reduction (134). Similarly, Dasatinib, a multitargeted TKI, was shown to prolong ventricular effective refractory period and impair left ventricular mechanical function in dogs at a low-dose administration (135). Moreover, in a recent Japanese cohort study, cardiotoxic AEs including congestive HF, pericardial effusion and QT prolongation were frequently reported in patients with chronic myeloid leukemia (CML) and gastrointestinal stromal tumor (GIST) (136). However, findings of these studies should be interpreted carefully, since the observed cardiac effects might be attributed to other targets of the treatment, while available evidence regarding PDGFR-selective TKIs still lacks.
TRPM7 human gene is located on the long arm of chromosome 15, consisting of 19 exons and encoding a 1,863 amino acid protein with a molecular weight of 210 kDa (137). The basic structure of TRPM7 consists of N-terminal hydrophobic region (H1) and four Melastatin Homologous Regions (MHR), six transmembrane segments, and C-terminal transient receptor potential (TRP) region followed by the coiled coil (CC) domain connecting loop, serine/threonine-rich domains and an α-type kinase domain (Figure 2) (4).
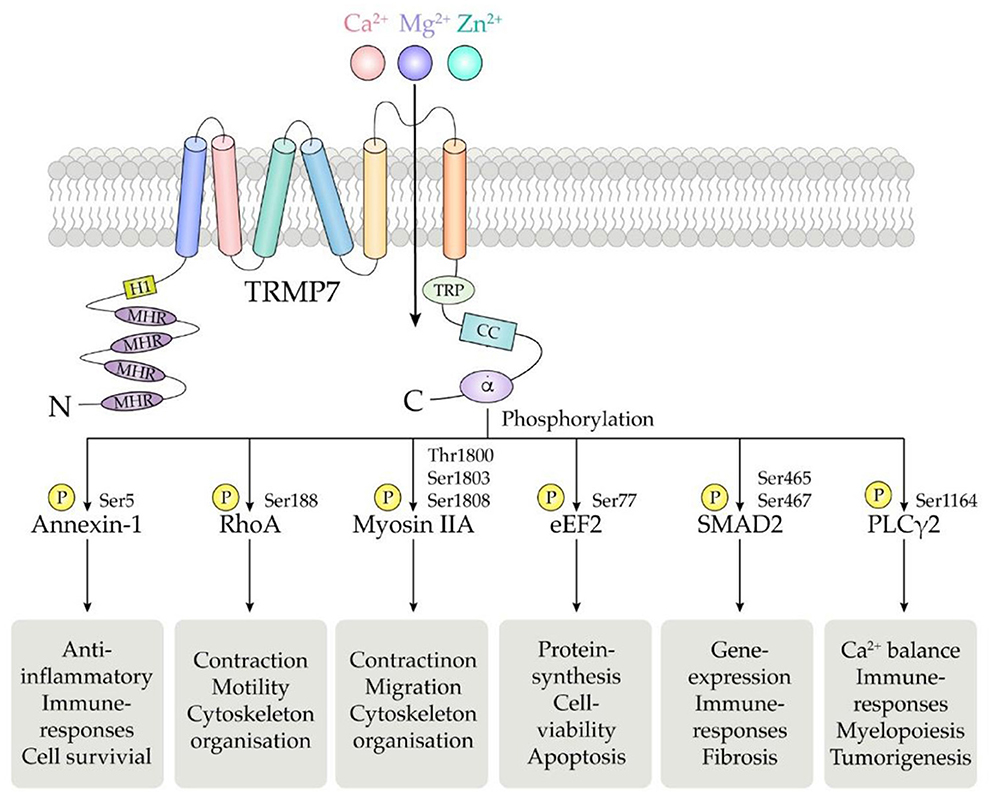
Figure 2. Schematic structure of the TRPM7 chanzyme. It consists of six transmembrane segments with the channel pore formed between segment 5 and 6. TRPM7 channel is permeable to divalent cations including Mg2+, Ca2+, and Zn2+.The α-kinase domain is able to phosphorylate diverse downstream substrates such as annexin-1, RhoA, myosin IIA, eEF2, SMAD2, and PLCγ2, at specific phosphorylation residues.
TRPM7 expression has been confirmed in the most abundant cell types of the cardiovascular system including VSMCs, endothelial cells (ECs) and cardiomyocytes (16, 138, 139). In 2005, we were amongst the first to identify and characterize TRPM7 in VSMCs, where the chanzyme acts as a functionally important regulator of Mg2+ homeostasis and cell growth (16). We recently further showed that TRPM7 is membrane-bound in VSMCs, mediating Mg2+ and calcium (Ca2+) influx and exerting effects on cell migration and proliferation (17). TRPM7 was found to be a contributor to the development of a proliferative phenotype of VSMCs trigged by angiotensin II (Ang II), and vascular calcification induced by phosphate (140, 141). In ECs, Inoue and Xiong discovered the TRPM7-like outward rectifying currents by whole-cell patch-clamp experiments (138). The physiological function of TRPM7 in ECs is more likely to associate with its Mg2+ permeability, since silencing TRPM7 mimics the effects of Mg2+ deficiency on cell behavior and Mg2+ regulates endothelial barrier functions through TRPM7 (142, 143). Additionally, endothelial functions such as cell adhesion and tube formation are negatively regulated by TRPM7 with mechanisms involving the extracellular signal-regulated kinase (ERK1/2) pathway (144). TRPM7 and its sister homolog TRPM6 are also observed in cardiomyocytes from all chamber walls of human hearts (139). Cardiac TRPM7 influences cardiac action potentials in a Mg2+-sensitive manner, while TRPM7 deletion in embryonic myocardium disrupts cardiac automaticity via the regulation of Hcn4 expression (145, 146).
TRPM7 activity is regulated by various vasoactive agents such as Ang II, aldosterone, bradykinin and C-type natriuretic peptide (CNP) (16, 147–149). Ang II enhances TRPM7 protein expression through Ang II type 1 receptor-mediated ERK1/2 signaling, which contributes to phenotypic change and proliferation of VSMCs (140). Chronic treatment with aldosterone upregulates the plasma membrane expression of TRPM7 in HEK cells, a process occurring via a mineralocorticoid receptor (MR)-dependent genomic signaling cascade involving serum- and glucocorticoid-regulated kinase 1 (SGK1) and a functional TRPM7 α-kinase domain (148). Bradykinin, a known vasodilator, was found to mediate the expression of TRPM7 and its kinase substrate annexin-1 in VSMC via molecular mechanisms involving phospholipase C (PLC), protein kinase C (PKC) and c-Src (149). Furthermore, C-type natriuretic peptide (CNP), a peptide produced by the vascular endothelium, has recently been shown to affect TRPM7-mediated Ca2+ entry in chondrocytes and stimulate bone growth via activating natriuretic peptide receptor 2 (NPR2) (147).
Growing evidence indicates an impoartnt role for TRPM7 in cardiac development (146). TRPM7 plays an indispensable role for myocardial proliferation during early cardiogenesis, as TRPM7 deletion before embryonic day 9 led to congestive heart failure and death by embryonic day 11.5 (150). These findings highlight the importance of TRPM7 in the integrity and function of the cardiovascular system.
Cardiac fibrosis is defined as the excessive accumulation of fibrillar extracellular matrix in the cardiac interstitium, a pathological process contributing to various heart diseases including HF, myocardial infarction, dilated and ischemic cardiomyopathies and arrhythmias (151). We demonstrated that TRPM7 deficiency is associated with cardiac dysfunction, inflammation and fibrosis in mice by Mg2+ dependent effects (17). Intriguingly, TRPM7 was observed to contribute to cardiac fibrosis induced by Ang II and hydrogen peroxide (H2O2) (152, 153). In these studies, 2-APB, a pharmacological non-specific inhibitor of TRPM7, was shown to attenuate cardiac fibrosis via effects on cardiac fibroblasts (152, 153). Given the important role of TRPM7 played in cardiac fibrosis, several studies have investigated TRPM7 as the potential therapeutic target. Tang et al. showed that Astragaloside IV, an important constituent of traditional Chinese medicine, inhibits cardiac fibrosis through modulating TRPM7 (154). Moreover, it was also shown that sacubitril, a drug well-known for treating heart failure, ameliorates cardiac fibrosis by acting on fibroblasts and cardiomyocytes via inhibiting TRPM7 channel (155).
Hypertension is a well-known major risk factor for cardiovascular diseases and exerts negative effects on healthy longevity (156). Direct evidence supporting TRPM7 involvement in the development of hypertension was first demonstrated in Ang II-induced hypertension, where the development of hypertension was amplified in TRPM7-deficent mice. This was associated with impaired endothelial function and amplified cardiac remodeling and left ventricular dysfunction (18). On the other hand, it was shown that TRPM7 inhibition by pharmacologic agents reduced hypertension induced by leptin. (157). Leptin receptor and TRPM7 colocalized in glomus cells of carotid bodies, where leptin regulates blood pressure through acting on TRPM7 (157). Furthermore, calpain, the substrate of TRPM7 kinase, might also play an important role in the development of hypertension. Calpain acts as downstream mediators in Ang II-induced cardiovascular remodeling, while calpastatin, a calpain-specific inhibitor, was able to prevent Ang II-dependent cardiac hypertrophy and perivascular inflammation (158). In addition, TRPM7 might contribute to hypertension via its Mg2+ permeability. Despite inconsistent findings to support the correlation between serum Mg2+ and hypertension, Mg2+ supplementation at a dose of 368 mg/d for 3 months has been shown to lower blood pressure in adults (159–161).
In the last decade, increasing evidence have established a link between TRPM7 and atrial fibrillation (AF), which is most commonly sustained arrhythmia and a major cause of morbidity and mortality (19, 162–164). Elevated TRPM7 expression has been observed in atrial myocytes and peripheral blood from patients with AF (162, 164). Yue et al. further showed that TRPM7 is the major Ca2+ permeable channel in human atrial fibroblasts, which might contribute to atrial fibrosis in human AF (19). These findings suggest that TRPM7 is associated with the pathogenesis of AF, which might be related to its cation permeability property. TRPM7 is highly expressed in sinoatrial node, where it influences diastolic membrane depolarization and automaticity, suggesting a possible role of TPRM7 in sick sinus syndrome and atrioventricular block (146). TRPM7 might also be implicated in ventricular arrhythmias, since cardiac TRPM7 deletion in mice is associated with a high risk of developing cardiomyopathy, characterized by impaired repolarization and ventricular arrhythmias (150). Importantly, attention has also been drawn to the involvement of TRPM7 in ischemic heart disease (IHD), since increased expression of TRPM7 and TRPM6 was observed in cardiac biopsies from patients with IHD (139). Consistently, in a murine model of myocardial infarction, TRPM7 expression was remarkably upregulated in cardiac fibroblasts, accompanied by enhanced Ca2+ influx (165).
Intracellular Mg2+, which is regulated by TRPM7 cation channel activation, is an essential cation and second messenger involved in tyrosine kinase signaling and regulation of RTKs (166). Mechanisms whereby Mg2+ regulates the activation of RTKs are elusive, however the current concept of “two metal catalysis” shows the requirement of two Mg2+ ions for proper kinase phosphorylation. In physiologic conditions, the ligand-receptor interaction induces binding of the first Mg2+ ion to the kinase, which allows the binding of the second Mg2+ leading to a proper signal propagation of the TK. This complex signaling pathway is deficient in conditions of Mg2+ deprivation (167). Moreover, affinity analysis showed that the binding-free energies of ATP to target enzymes are lower in the presence of Mg2+ ions than those in the absence, suggesting that Mg2+ enhances the binding affinities of ATP to the protein kinases (168) (Figure 3).
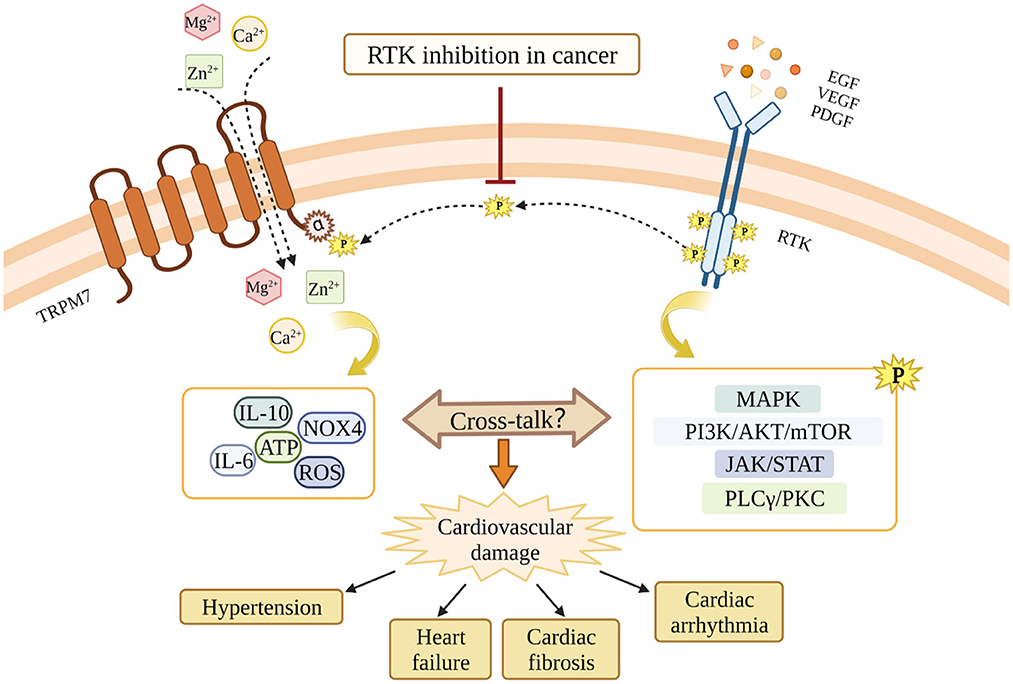
Figure 3. Potential cross-talk between RTKs and TRPM7 and the potential involvement in RTKs inhibition-related cardiovascular toxicity. RTKs inhibition blocks the interaction between RTKs and TRPM7, and leads to TRPM7 dysfunction. Through mechanisms involving TRPM7, RTKs inhibition might cause cardiovascular damages in cancer patients. The MAPK pathway, the PI3K/AKT/mTOR pathway, the JAK/STAT pathway, and the PLCγ/PKC pathway are downstream signaling cascades activated by RTKs. The dashed line indicates the possibility that RTKs and TRPM7 interact. This still awaits confirmation.
In renal distal convoluted tubule (DCT), EGF increases TRPM6 activity and surface expression, while in cancer patients receiving anti-EGFR treatment, serum level of Mg2+ was significantly decreased (169–171). In a similar fashion, EGF through binding to EGFR and activating downstream signaling, regulates cell membrane proteins expression and currents of TRPM7 (21, 172). PDGF was shown to dramatically enhance the gene and protein expression of TRPM7 in a time-dependent manner in hepatic stellate cells (HSCs), and PDGF-induced cell proliferation was prevented by TRPM7 inhibition (173). Consistently, Xu et al. found that carvacrol, a pharmacological inhibitor of TPRM7, attenuated the activation and proliferation of HSCs induced by PDGF through TRPM7-meidated cellular signaling involving the mitogen-activated protein kinases (MAPK) (174). In a human osteoblast cell line, PDGF upregulated TRPM7 expression after 4 h of treatment, an effect that could be sustained for a 24 h-period (175). The regulation of TRPM7 by PDGF was further shown to importantly modulate Mg2+ influx, and proliferation and migration of human osteoblasts (175). Moreover, nerve growth factor (NGF) via its receptor TrkA, a family member of RTKs, regulated the outward-rectifying TRPM7-like currents in hippocampal neurons through a phospholipase C (PLC)-dependent manner (176). Taken together, these studies have highlighted a critical regulation of TRPM7 by the RTKs signaling, which is associated with important biological effects in various cell types.
It is worth noting that most evidence connecting RTKs and TRPM7 were from non-cardiovascular studies. The direct evidence demonstrating a link between TKIs and TPRM7 in the cardiovascular system was from our previous study and this link was primarily functional (177). We showed that in VSMCs, EGF promotes Mg2+ influx through the TRPM7 channel and consequently regulates VSMCs function and vascular morphology. Of importance, the effects of EGF on TRPM7 and VSMCs were attenuated by gefitinib, a TKI that exclusively targets EGFR. In addition, we also found that in VSMCs from TRPM7 kinase-deficient mice (TRPM7+/Δkinase) and aortic tissues from TRPM7-kinase dead (TRPM7R/R) mice, EGFR expression and EGFR phosphorylation (Y845) were reduced, respectively, supporting a significant cross-talk (177). Moreover, our study demonstrated that EGF/EGFR was able to mediate the kinase activity of TRPM7 in VSMCs, due to the observation that EGF enhanced TRPM7 phosphorylation at Ser 1511 (177).
Experimental treatment with the EGFR inhibitor erlotinib causes hypomagnesemia that gradually increases after 3 weeks of treatment, suggesting a cumulative chronic effects on the Mg2+ handling by kidneys and intestines (178). Oxidative stress is another important mechanism that might be responsible for reduced activity of Mg2+ channels observed after EGFR inhibition. Erlotinib activated Nox4, a NADPH oxidase highly expressed in the cardiovascular system that generates H2O2 (179), and it was previously demonstrated that H2O2 cause inhibition of TRPM6 and TRPM7 activities and subsequently reduction in Mg2+ (180, 181). However, it is still elusive whether these important mechanisms are observed in cells from the vasculature.
Vascular dysfunction in hypertension resembles the vascular phenotype in aging, with hypertension being defined as a condition of premature vascular aging (182). Cellular and experimental studies demonstrated that TRPM7-deficiency is associated with increased molecular markers of senescence, including p16, WRN and phosphorylation of P66Shc (183). Importantly, expression of these markers is increased in experimental hypertension, induced by infusion with Ang II or aldosterone, chronic kidney ischemia and pulmonary arterial hypertension (182). Important pathologic implications of the TRPM7-Mg2+ axis deficiency is associated with cardiovascular inflammation by mechanisms dependent on macrophage infiltration to cardiac tissues leading to increased galectin-3, IL-6, IL-10, phosphor-P66Shc and reduced phosphor-Stat3 leading to cardiac fibrosis and diastolic dysfunction (17). Additionally, TRPM7 is downregulated in VSMC from PAH patients and in experimental models, effects that were exacerbated by waixenicin A, a TRPM7 inhibitor. Intracellular mechanisms involved MEK/ERK pathway (184).
Another important factor that should be taken into consideration is that TRPM7 is highly permeable to Zn2+. Hence, downregulation of TRPM7 induced by TKI might affect intracellular concentration of Zn2+. Clinical investigations showed reduced serum Zn2+ as adverse effect of EGFR inhibition and Zn2+ supplementation was able to reduce dermatitis these patients (185). Of importance, Zn2+ is a potent antioxidant and its deficiency is associated with mitochondrial and endoplasmic reticulum (ER) stresses, increased ROS production and dysregulation of cellular metabolism (186). Clinically, Zn2+ deficiency is associated with high incidence of cardiovascular diseases, including hypertension and diabetes (187). Mechanisms underlying these effects are still elusive and might involve oxidative stress and inflammatory response.
Mg2+ deficiency is directly associated with risk for diabetes development. Physiologically, Mg2+ is a co-factor of the ATPase that limits the opening of the ATP-sensitive potassium channels, leading to increase in calcium influx and results in insulin release. Therefore, low Mg2+ reduces ATPase activity and consequent hyperactivity of the ATP-sensitive potassium channels (KATP), inhibiting Ca2+ influx resulting in defective insulin secretion (188). Furthermore, hypomagnesemia interferes with optimal binding of insulin to receptor and because IRS are TK receptor, reduced in Mg2+ interferes with intracellular signaling pathways mediated by IRS1 and IRS2 activation (167, 189). Of importance, a randomized clinical trial involving 1,122 subjects showed that hypomagnesemia is associated with the development of impaired glucose tolerance and type 2 diabetes (190).
It should be highlighted that while it is clear that there is important cross-talk between RTK/TK and TRPM7 at the functional level, direct interacting p-tyr sites between RTKs/TKs and TRPM7 still awaits confirmation. However, there is growing evidence that both TRPM6 and TRPM7 possesses phosphorylation sites, which potentially may link to the RTK/TK pathway (14, 191). Moreover, adaptor proteins may act as links between the systems.
RTKs such as VEGFR, EGFR and PDGFR are important therapeutic targets for human cancers, however, inhibitors of RTKs and TKs are also accompanied by a profile of adverse cardiovascular effects through unclear mechanisms. Since the cross-talk between RTKs and TRPM7 exists in multiple cell types, and TRPM7 is critically involved in both cancers and cardiovascular diseases, we suggest that TRPM7 might play a role in the cardiovascular toxicity associated with RTKI/TKIs (Figure 3). TRPM7 is ubiquitously expressed and while it is an important regulator of intracellular Mg2+, it also influences intarcellular Ca2+ homeostasis and downstream kinases and substartes that may also play a role in TRPM7-RTK/TK crosstalk. Unraveling these TRPM7-dependent processes might provide greater insights into the molecular mechanisms that underlie cardiovascular disease in patients treated with RTKI/TKIs.
QL and SL wrote the manuscript. YQ and JZ provided support for the tables and figures. FR, ZZ, and RT provided guidance and corrected the manuscript. All authors contributed to the article and approved the submitted version.
RT was supported by grants from the Leducq Foundation, Dr. Phil Gold Chair, McGill University and European Commission, and Canada Research Chair Program of the Canadian Institutes of Health Research. ZZ was sponsored by Shanghai Pujiang Program (21PJD039).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Butti R, Das S, Gunasekaran VP, Yadav AS, Kumar D, Kundu GC. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol Cancer. (2018) 17:34. doi: 10.1186/s12943-018-0797-x
2. Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. (2000) 19:5548–57. doi: 10.1038/sj.onc.1203957
3. Hubbard SR. Structural analysis of receptor tyrosine kinases. Prog Biophys Mol Biol. (1999) 71:343–58. doi: 10.1016/S0079-6107(98)00047-9
4. Zou ZG, Rios FJ, Montezano AC, Touyz RM. TRPM7, magnesium, and signaling. Int J Mol Sci. (2019) 20:1877. doi: 10.3390/ijms20081877
5. Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer. (2018) 17:58. doi: 10.1186/s12943-018-0782-4
6. Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. (2007) 7:332–44. doi: 10.1038/nrc2106
7. Dorovkov MV, Ryazanov AG. Phosphorylation of annexin I by TRPM7 channel-kinase. J Biol Chem. (2004) 279:50643–6. doi: 10.1074/jbc.C400441200
8. Perraud AL, Zhao X, Ryazanov AG, Schmitz C. The channel-kinase TRPM7 regulates phosphorylation of the translational factor eEF2 via eEF2-k. Cell Signal. (2011) 23:586–93. doi: 10.1016/j.cellsig.2010.11.011
9. Clark K, Middelbeek J, Lasonder E, Dulyaninova NG, Morrice NA, Ryazanov AG, et al. TRPM7 regulates myosin IIA filament stability and protein localization by heavy chain phosphorylation. J Mol Biol. (2008) 378:790–803. doi: 10.1016/j.jmb.2008.02.057
10. Deason-Towne F, Perraud AL, Schmitz C. Identification of Ser/Thr phosphorylation sites in the C2-domain of phospholipase C gamma2 (PLCgamma2) using TRPM7-kinase. Cell Signal. (2012) 24:2070–5. doi: 10.1016/j.cellsig.2012.06.015
11. Romagnani A, Vettore V, Rezzonico-Jost T, Hampe S, Rottoli E, Nadolni W, et al. TRPM7 kinase activity is essential for T cell colonization and alloreactivity in the gut. Nat Commun. (2017) 8:1917. doi: 10.1038/s41467-017-01960-z
12. Voringer S, Schreyer L, Nadolni W, Meier MA, Woerther K, Mittermeier C, et al. Inhibition of TRPM7 blocks MRTF/SRF-dependent transcriptional and tumorigenic activity. Oncogene. (2020) 39:2328–44. doi: 10.1038/s41388-019-1140-8
13. Ogata K, Tsumuraya T, Oka K, Shin M, Okamoto F, Kajiya H, et al. The crucial role of the TRPM7 kinase domain in the early stage of amelogenesis. Sci Rep. (2017) 7:18099. doi: 10.1038/s41598-017-18291-0
14. Cai N, Bai Z, Nanda V, Runnels LW. Mass spectrometric analysis of TRPM6 and TRPM7 phosphorylation reveals regulatory mechanisms of the channel-kinases. Sci Rep. (2017) 7:42739. doi: 10.1038/srep42739
15. Dorovkov MV, Beznosov SN, Shah S, Kotlianskaia L, Kostiukova AS. Effect of mutations imitating the phosphorylation by TRPM7 kinase on the function of the N-terminal domain of tropomodulin. Biofizika. (2008) 53:943–9. doi: 10.1134/S0006350908060055
16. He Y, Yao G, Savoia C, Touyz RM. Transient receptor potential melastatin 7 ion channels regulate magnesium homeostasis in vascular smooth muscle cells: role of angiotensin II. Circ Res. (2005) 96:207–15. doi: 10.1161/01.RES.0000152967.88472.3e
17. Rios FJ, Zou ZG, Harvey AP, Harvey KY, Nosalski R, Anyfanti P, et al. Chanzyme TRPM7 protects against cardiovascular inflammation and fibrosis. Cardiovasc Res. (2020) 116:721–35. doi: 10.1093/cvr/cvz164
18. Antunes TT, Callera GE, He Y, Yogi A, Ryazanov AG, Ryazanova LV, et al. Transient receptor potential melastatin 7 cation channel kinase: new player in angiotensin II-induced hypertension. Hypertension. (2016) 67:763–73. doi: 10.1161/HYPERTENSIONAHA.115.07021
19. Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, et al. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. (2010) 106:992–1003. doi: 10.1161/CIRCRESAHA.109.206771
20. Yee NS, Zhou W, Liang IC. Transient receptor potential ion channel Trpm7 regulates exocrine pancreatic epithelial proliferation by Mg2+-sensitive Socs3a signaling in development and cancer. Dis Model Mech. (2011) 4:240–54. doi: 10.1242/dmm.004564
21. Gao H, Chen X, Du X, Guan B, Liu Y, Zhang H, et al. enhances the migration of cancer cells by up-regulation of TRPM7. Cell Calcium. (2011) 50:559–68. doi: 10.1016/j.ceca.2011.09.003
22. Sun Y, Sukumaran P, Varma A, Derry S, Sahmoun AE, Singh BB. Cholesterol-induced activation of TRPM7 regulates cell proliferation, migration, and viability of human prostate cells. Biochim Biophys Acta. (2014) 1843:1839–50. doi: 10.1016/j.bbamcr.2014.04.019
23. Gao S-L, Kong C-Z, Zhang Z, Li Z-L, Bi J-B, Liu X-K. TRPM7 is overexpressed in bladder cancer and promotes proliferation, migration, invasion and tumor growth. Oncol Rep. (2017) 38:1967–76. doi: 10.3892/or.2017.5883
24. Guilbert A, Gautier M, Dhennin-Duthille I, Haren N, Sevestre H, Ouadid-Ahidouch H. Evidence that TRPM7 is required for breast cancer cell proliferation. Am J Physiol Cell Physiol. (2009) 297:C493–502. doi: 10.1152/ajpcell.00624.2008
25. Chen W-L, Barszczyk A, Turlova E, Deurloo M, Liu B, Yang BB, et al. Inhibition of TRPM7 by carvacrol suppresses glioblastoma cell proliferation, migration and invasion. Oncotarget. (2015) 6:16321–40. doi: 10.18632/oncotarget.3872
26. Auwercx J, Kischel P, Lefebvre T, Jonckheere N, Vanlaeys A, Guenin S, et al. TRPM7 modulates human pancreatic stellate cell activation. Cells. (2022) 11:2255. doi: 10.3390/cells11142255
27. Liu H, Dilger JP, Lin J, A. pan-cancer-bioinformatic-based literature review of TRPM7 in cancers. Pharmacol Ther. (2022) 240:108302. doi: 10.1016/j.pharmthera.2022.108302
28. Meng S, Alanazi R, Ji D, Bandura J, Luo ZW, Fleig A, et al. Role of TRPM7 kinase in cancer. Cell Calcium. (2021) 96:102400. doi: 10.1016/j.ceca.2021.102400
29. Basu A, Contreras AG, Datta D, Flynn E, Zeng L, Cohen HT, et al. Overexpression of vascular endothelial growth factor and the development of post-transplantation cancer. Cancer Res. (2008) 68:5689–98. doi: 10.1158/0008-5472.CAN-07-6603
30. Harrison PT, Vyse S, Huang PH. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin Cancer Biol. (2020) 61:167–79. doi: 10.1016/j.semcancer.2019.09.015
31. Kolomietz E, Al-Maghrabi J, Brennan S, Karaskova J, Minkin S, Lipton J, et al. Primary chromosomal rearrangements of leukemia are frequently accompanied by extensive submicroscopic deletions and may lead to altered prognosis. Blood. (2001) 97:3581–8. doi: 10.1182/blood.V97.11.3581
32. Kentsis A, Reed C, Rice KL, Sanda T, Rodig SJ, Tholouli E, et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat Med. (2012) 18:1118–22. doi: 10.1038/nm.2819
33. Thomson RJ, Moshirfar M, Ronquillo Y. Tyrosine Kinase Inhibitors. Treasure Island, FL: StatPearls (2021).
34. Cohen MH, Williams G, Johnson JR, Duan J, Gobburu J, Rahman A, et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res. (2002) 8:935–42.
35. Muhsin M, Graham J, Kirkpatrick P. Gefitinib. Nat Rev Drug Discov. (2003) 2:515–6. doi: 10.1038/nrd1136
36. Kris MG, Natale RB, Herbst RS, Lynch TJ, Prager D, Belani CP, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. (2003) 290:2149–58. doi: 10.1001/jama.290.16.2149
37. Mok TS, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Improvement in overall survival in a randomized study that compared dacomitinib with gefitinib in patients with advanced non-small-cell lung cancer and EGFR-activating mutations. J Clin Oncol. (2018) 36:2244−50. doi: 10.1200/JCO.2018.78.7994
38. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. (2007) 25:1960–6. doi: 10.1200/JCO.2006.07.9525
39. Natale RB, Thongprasert S, Greco FA, Thomas M, Tsai CM, Sunpaweravong P, et al. Phase III trial of vandetanib compared with erlotinib in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol. (2011) 29:1059–66. doi: 10.1200/JCO.2010.28.5981
40. de Marinis F, Catania C, Passaro A. Afatinib in NSCLC harbouring EGFR mutations. Lancet Oncol. (2014) 15:e148–e9. doi: 10.1016/S1470-2045(13)70402-9
41. Addeo A. Dacomitinib in NSCLC: a positive trial with little clinical impact. Lancet Oncol. (2018) 19:e4. doi: 10.1016/S1470-2045(17)30923-3
42. Tsubata Y, Watanabe K, Saito R, Nakamura A, Yoshioka H, Morita M, et al. Osimertinib in poor performance status patients with T790M-positive advanced non-small-cell lung cancer after progression of first- and second-generation EGFR-TKI treatments (NEJ032B). Int J Clin Oncol. (2022) 27:112–20. doi: 10.1007/s10147-021-02043-2
43. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. (2004) 351:337–45. doi: 10.1056/NEJMoa033025
44. Guntinas-Lichius O. Cetuximab in head and neck cancer. N Engl J Med. (2008) 359:2725; author reply 6. doi: 10.1056/NEJMc082096
45. Saltz L, Easley C, Kirkpatrick P. Panitumumab. Nat Rev Drug Discov. (2006) 5:987–8. doi: 10.1038/nrd2204
46. Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. (2007) 25:1658–64. doi: 10.1200/JCO.2006.08.1620
47. Thatcher N, Hirsch FR, Luft AV, Szczesna A, Ciuleanu TE, Dediu M, et al. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): an open-label, randomised, controlled phase 3 trial. Lancet Oncol. (2015) 16:763–74. doi: 10.1016/S1470-2045(15)00021-2
48. Moy B, Kirkpatrick P, Kar S, Goss P. Lapatinib. Nat Rev Drug Discov. (2007) 6:431–2. doi: 10.1038/nrd2332
49. Gomez HL, Doval DC, Chavez MA, Ang PCS, Aziz Z, Nag S, et al. Efficacy and safety of lapatinib as first-line therapy for ErbB2-amplified locally advanced or metastatic breast cancer. J Clin Oncol. (2008) 26:2999–3005. doi: 10.1200/JCO.2007.14.0590
50. Neratinib Approved for HER2+ breast cancer. Cancer Discov. (2017) 7:OF1. doi: 10.1158/2159-8290.CD-NB2017-110
51. Park JW, Liu MC, Yee D, Yau C, van 't Veer LJ, Symmans WF, et al. Adaptive randomization of neratinib in early breast cancer. N Engl J Med. (2016) 375:11–22. doi: 10.1056/NEJMoa1513750
52. Murthy RK, Loi S, Okines A, Paplomata E, Hamilton E, Hurvitz SA, et al. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N Engl J Med. (2020) 382:597–609. doi: 10.1056/NEJMoa1914609
53. Barnes DM, Miles DW. Response of metastatic breast cancer to trastuzumab? Lancet. (2000) 355:160–1. doi: 10.1016/S0140-6736(99)00430-4
54. Bang Y-J, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. (2010) 376:687–97. doi: 10.1016/S0140-6736(10)61121-X
55. Baselga J, Cortés J, Kim S-B, Im S-A, Hegg R, Im Y-H, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. (2012) 366:109–19. doi: 10.1056/NEJMoa1113216
56. Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. (2012) 367:1783–91. doi: 10.1056/NEJMoa1209124
57. Krop IE, Kim S-B, González-Martín A, LoRusso PM, Ferrero J-M, Smitt M, et al. Trastuzumab emtansine versus treatment of physician's choice for pretreated HER2-positive advanced breast cancer (TH3RESA): a randomised, open-label, phase 3 trial. Lancet Oncol. (2014) 15:689–99. doi: 10.1016/S1470-2045(14)70178-0
58. Modi S, Saura C, Yamashita T, Park YH, Kim S-B, Tamura K, et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N Engl J Med. (2020) 382:610–21. doi: 10.1056/NEJMoa1914510
59. Shitara K, Bang Y-J, Iwasa S, Sugimoto N, Ryu M-H, Sakai D, et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N Engl J Med. (2020) 382:2419–30. doi: 10.1056/NEJMoa2004413
60. “Super Trastuzumab” extends PFS in late-line breast cancer. Cancer Discov. (2019) 9:OF5. doi: 10.1158/2159-8290.CD-NB2019-069
61. Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. (2011) 378:1931–9. doi: 10.1016/S0140-6736(11)61613-9
62. Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. (2011) 29:2660–6. doi: 10.1200/JCO.2010.32.4145
63. Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med. (2015) 373:1814–23. doi: 10.1056/NEJMoa1510016
64. Abou-Alfa GK, Meyer T, Cheng A-L, El-Khoueiry AB, Rimassa L, Ryoo B-Y, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med. (2018) 379:54–63. doi: 10.1056/NEJMoa1717002
65. Brose MS, Robinson B, Sherman SI, Krajewska J, Lin C-C, Vaisman F, et al. Cabozantinib for radioiodine-refractory differentiated thyroid cancer (COSMIC-311): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. (2021) 22:1126–38. doi: 10.1016/S1470-2045(21)00332-6
66. Hindié E. Lenvatinib plus pembrolizumab for renal cell carcinoma. N Engl J Med. (2021) 385:287. doi: 10.1056/NEJMc2107518
67. Kudo M, Finn RS, Qin S, Han K-H, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. (2018) 391:1163–73. doi: 10.1016/S0140-6736(18)30207-1
68. Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. (2010) 28:1061–8. doi: 10.1200/JCO.2009.23.9764
69. van der Graaf WTA, Blay J-Y, Chawla SP, Kim D-W, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. (2012) 379:1879–86. doi: 10.1016/S0140-6736(12)60651-5
70. Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. (2013) 381:303–12. doi: 10.1016/S0140-6736(12)61900-X
71. Kang Y-K, George S, Jones RL, Rutkowski P, Shen L, Mir O, et al. Avapritinib versus regorafenib in locally advanced unresectable or metastatic GI stromal tumor: a randomized, open-label phase III study. J Clin Oncol. (2021) 39:3128–39. doi: 10.1200/JCO.21.00217
72. Bruix J, Qin S, Merle P, Granito A, Huang Y-H, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2017) 389:56–66. doi: 10.1016/S0140-6736(16)32453-9
73. Rini BI, Pal SK, Escudier BJ, Atkins MB, Hutson TE, Porta C, et al. Tivozanib versus sorafenib in patients with advanced renal cell carcinoma (TIVO-3): a phase 3, multicentre, randomised, controlled, open-label study. Lancet Oncol. (2020) 21:95–104. doi: 10.1016/S1470-2045(19)30735-1
74. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. (2008) 359:378–90. doi: 10.1056/NEJMoa0708857
75. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. (2014) 384:319–28. doi: 10.1016/S0140-6736(14)60421-9
76. Chang E, Weinstock C, Zhang L, Fiero MH, Zhao M, Zahalka E, et al. FDA approval summary: tivozanib for relapsed or refractory renal cell carcinoma. Clin Cancer Res. (2022) 28:441–5. doi: 10.1158/1078-0432.CCR-21-2334
77. Joensuu H. Sunitinib for imatinib-resistant GIST. Lancet. (2006) 368:1303–4. doi: 10.1016/S0140-6736(06)69489-0
78. Motzer RJ, Ravaud A, Patard J-J, Pandha HS, George DJ, Patel A, et al. Adjuvant sunitinib for high-risk renal cell carcinoma after nephrectomy: subgroup analyses and updated overall survival results. Eur Urol. (2018) 73:62–8. doi: 10.1016/j.eururo.2017.09.008
79. Kulke MH, Lenz H-J, Meropol NJ, Posey J, Ryan DP, Picus J, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. (2008) 26:3403–10. doi: 10.1200/JCO.2007.15.9020
80. Wells SA, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. (2012) 30:134–41. doi: 10.1200/JCO.2011.35.5040
81. Boisen MK, Johansen JS, Dehlendorff C, Larsen JS, Østerlind K, Hansen J, et al. Primary tumor location and bevacizumab effectiveness in patients with metastatic colorectal cancer. Ann Oncol. (2013) 24:2554–9. doi: 10.1093/annonc/mdt253
82. von Minckwitz G, Eidtmann H, Rezai M, Fasching PA, Tesch H, Eggemann H, et al. Neoadjuvant chemotherapy and bevacizumab for HER2-negative breast cancer. N Engl J Med. (2012) 366:299–309. doi: 10.1056/NEJMoa1111065
83. Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. (2019) 381:2416–28. doi: 10.1056/NEJMoa1911361
84. Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. (2006) 355:1419–31. doi: 10.1056/NEJMoa054481
85. Wells JA, Glassman AR, Ayala AR, Jampol LM, Aiello LP, Antoszyk AN, et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N Engl J Med. (2015) 372:1193–203. doi: 10.1056/NEJMoa1414264
86. Ramucirumab extends survival in advanced NSCLC. Cancer Discov. (2014) 4:OF6. doi: 10.1158/2159-8290.CD-NB2014-087
87. Wilke H, Muro K, Van Cutsem E, Oh S-C, Bodoky G, Shimada Y, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. (2014) 15:1224–35. doi: 10.1016/S1470-2045(14)70420-6
88. Tabernero J, Yoshino T, Cohn AL, Obermannova R, Bodoky G, Garcia-Carbonero R, et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. (2015) 16:499–508. doi: 10.1016/S1470-2045(15)70127-0
89. Stewart MW, Grippon S, Kirkpatrick P. Aflibercept. Nat Rev Drug Discov. (2012) 11:269–70. doi: 10.1038/nrd3700
90. Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausová J, Macarulla T, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. (2012) 30:3499–506. doi: 10.1200/JCO.2012.42.8201
91. Avapritinib Approved for GIST Subgroup. Cancer Discov. (2020) 10:334. doi: 10.1158/2159-8290.CD-NB2020-003
92. Villanueva MT. Ripretinib turns off the switch in GIST. Nat Rev Drug Discov. (2019) 18:499. doi: 10.1038/d41573-019-00099-4
93. Olaratumab approved for soft-tissue sarcoma. Cancer Discov. (2016) 6:1297. doi: 10.1158/2159-8290.CD-NB2016-141
94. Liu G, Chen T, Ding Z, Wang Y, Wei Y, Wei X. Inhibition of FGF-FGFR and VEGF-VEGFR signalling in cancer treatment. Cell Prolif. (2021) 54:e13009. doi: 10.1111/cpr.13009
95. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol. (2018) 9:978. doi: 10.3389/fimmu.2018.00978
96. Sundararajan S, Kumar A, Poongkunran M, Kannan A, Vogelzang NJ. Cardiovascular adverse effects of targeted antiangiogenic drugs: mechanisms and management. Fut Oncol. (2016) 12:1067–80. doi: 10.2217/fon.16.4
97. Small HY, Montezano AC, Rios FJ, Savoia C, Touyz RM. Hypertension due to antiangiogenic cancer therapy with vascular endothelial growth factor inhibitors: understanding and managing a new syndrome. Can J Cardiol. (2014) 30:534–43. doi: 10.1016/j.cjca.2014.02.011
98. Abdel-Qadir H, Ethier JL, Lee DS, Thavendiranathan P, Amir E. Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: a systematic review and meta-analysis. Cancer Treat Rev. (2017) 53:120–7. doi: 10.1016/j.ctrv.2016.12.002
99. Robinson ES, Khankin EV, Choueiri TK, Dhawan MS, Rogers MJ, Karumanchi SA, et al. Suppression of the nitric oxide pathway in metastatic renal cell carcinoma patients receiving vascular endothelial growth factor-signaling inhibitors. Hypertension. (2010) 56:1131–6. doi: 10.1161/HYPERTENSIONAHA.110.160481
100. de Jesus-Gonzalez N, Robinson E, Penchev R, von Mehren M, Heinrich MC, Tap W, et al. Regorafenib induces rapid and reversible changes in plasma nitric oxide and endothelin-1. Am J Hypertens. (2012) 25:1118–23. doi: 10.1038/ajh.2012.97
101. Kappers MH, de Beer VJ, Zhou Z, Danser AH, Sleijfer S, Duncker DJ, et al. Sunitinib-induced systemic vasoconstriction in swine is endothelin mediated and does not involve nitric oxide or oxidative stress. Hypertension. (2012) 59:151–7. doi: 10.1161/HYPERTENSIONAHA.111.182220
102. Eisen T, Sternberg CN, Robert C, Mulders P, Pyle L, Zbinden S, et al. Targeted therapies for renal cell carcinoma: review of adverse event management strategies. J Natl Cancer Inst. (2012) 104:93–113. doi: 10.1093/jnci/djr511
103. Herrmann J, Lerman A. An update on cardio-oncology. Trends Cardiovasc Med. (2014) 24:285–95. doi: 10.1016/j.tcm.2014.07.003
104. Qi WX, Shen Z, Tang LN, Yao Y. Risk of arterial thromboembolic events with vascular endothelial growth factor receptor tyrosine kinase inhibitors: an up-to-date meta-analysis. Crit Rev Oncol Hematol. (2014) 92:71–82. doi: 10.1016/j.critrevonc.2014.04.004
105. Sonpavde G, Je Y, Schutz F, Galsky MD, Paluri R, Rosenberg JE, et al. Venous thromboembolic events with vascular endothelial growth factor receptor tyrosine kinase inhibitors: a systematic review and meta-analysis of randomized clinical trials. Crit Rev Oncol Hematol. (2013) 87:80–9. doi: 10.1016/j.critrevonc.2012.12.006
106. Ferroni P, Formica V, Roselli M, Guadagni F. Thromboembolic events in patients treated with anti-angiogenic drugs. Curr Vasc Pharmacol. (2010) 8:102–13. doi: 10.2174/157016110790226660
107. Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer. (2007) 96:1788–95. doi: 10.1038/sj.bjc.6603813
108. Ghatalia P, Morgan CJ, Je Y, Nguyen PL, Trinh QD, Choueiri TK, et al. Congestive heart failure with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Crit Rev Oncol Hematol. (2015) 94:228–37. doi: 10.1016/j.critrevonc.2014.12.008
109. Dobbin SJH, Cameron AC, Petrie MC, Jones RJ, Touyz RM, Lang NN. Toxicity of cancer therapy: what the cardiologist needs to know about angiogenesis inhibitors. Heart. (2018) 104:1995–2002. doi: 10.1136/heartjnl-2018-313726
110. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. (2005) 115:2108–18. doi: 10.1172/JCI24682
111. Zamorano JL, Lancellotti P, Rodriguez Muñoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur Heart J. (2016) 37:2768–801. doi: 10.1093/eurheartj/ehw211
112. Ghatalia P, Je Y, Kaymakcalan MD, Sonpavde G, Choueiri TK. QTc interval prolongation with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Br J Cancer. (2015) 112:296–305. doi: 10.1038/bjc.2014.564
113. Strevel EL, Ing DJ, Siu LL. Molecularly targeted oncology therapeutics and prolongation of the QT interval. J Clin Oncol. (2007) 25:3362–71. doi: 10.1200/JCO.2006.09.6925
114. TeBay C, Hill AP, Windley MJ. Metabolic and electrolyte abnormalities as risk factors in drug-induced long QT syndrome. Biophys Rev. (2022) 14:353–67. doi: 10.1007/s12551-022-00929-7
115. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. (2007) 7:169–81. doi: 10.1038/nrc2088
116. Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. (2011) 11:9–22. doi: 10.1038/nrc2982
117. Anand K, Ensor J, Trachtenberg B, Bernicker EH. Osimertinib-induced cardiotoxicity: a retrospective review of the FDA adverse events reporting system (FAERS). JACC Cardiooncol. (2019) 1:172–8. doi: 10.1016/j.jaccao.2019.10.006
118. Kunimasa K, Kamada R, Oka T, Oboshi M, Kimura M, Inoue T, et al. Cardiac adverse events in EGFR-mutated non-small cell lung cancer treated with osimertinib. JACC CardioOncol. (2020) 2:1–10. doi: 10.1016/j.jaccao.2020.02.003
119. Hahn VS, Zhang KW, Sun L, Narayan V, Lenihan DJ, Ky B. Heart failure with targeted cancer therapies: mechanisms and cardioprotection. Circ Res. (2021) 128:1576–93. doi: 10.1161/CIRCRESAHA.121.318223
120. Yu AF, Moskowitz CS, Chuy KL, Yang J, Dang CT, Liu JE, et al. Cardiotoxicity surveillance and risk of heart failure during HER2 targeted therapy. JACC CardioOncol. (2020) 2:166–75. doi: 10.1016/j.jaccao.2020.03.002
121. Bardaro F, Stirpe E. Osimertinib induced cardiac failure and QT-prolongation in a patient with advanced pulmonary adenocarcinoma. J Oncol Pharm Pract. (2022) 28:989–94. doi: 10.1177/10781552211073823
122. Piper-Vallillo AJ, Costa DB, Sabe MA, Asnani A. Heart failure associated with the epidermal growth factor receptor inhibitor osimertinib. JACC CardioOncol. (2020) 2:119–22. doi: 10.1016/j.jaccao.2020.01.003
123. Ikebe S, Amiya R, Minami S, Ihara S, Higuchi Y, Komuta K. Osimertinib-induced cardiac failure with QT prolongation and torsade de pointes in a patient with advanced pulmonary adenocarcinoma. Int Cancer Conf J. (2021) 10:68–71. doi: 10.1007/s13691-020-00450-2
124. Barrick CJ Yu M, Chao HH, Threadgill DW. Chronic pharmacologic inhibition of EGFR leads to cardiac dysfunction in C57BL/6J mice. Toxicol Appl Pharmacol. (2008) 228:315–25. doi: 10.1016/j.taap.2007.12.012
125. Alhoshani A, Alanazi FE, Alotaibi MR, Attwa MW, Kadi AA, Aldhfyan A, et al. EGFR inhibitor gefitinib induces cardiotoxicity through the modulation of cardiac PTEN/Akt/FoxO3a pathway and reactive metabolites formation: in vivo and in vitro rat studies. Chem Res Toxicol. (2020) 33:1719–28. doi: 10.1021/acs.chemrestox.0c00005
126. Waliany S, Zhu H, Wakelee H, Padda SK, Das M, Ramchandran K, et al. Pharmacovigilance analysis of cardiac toxicities associated with targeted therapies for metastatic NSCLC. J Thorac Oncol. (2021) 16:2029–39. doi: 10.1016/j.jtho.2021.07.030
127. Yang JC, Reckamp KL, Kim YC, Novello S, Smit EF, Lee JS, et al. Efficacy and safety of rociletinib versus chemotherapy in patients with EGFR-mutated NSCLC: the results of TIGER-3, a phase 3 randomized study. JTO Clin Res Rep. (2021) 2:100114. doi: 10.1016/j.jtocrr.2020.100114
128. Kondo M, Kisanuki M, Kokawa Y, Gohara S, Kawano O, Kagiyama S, et al. Case report: QT prolongation and abortive sudden death observed in an 85-year-old female patient with advanced lung cancer treated with tyrosine kinase inhibitor osimertinib. Front Cardiovasc Med. (2021) 8:655808. doi: 10.3389/fcvm.2021.655808
129. Munasinghe WP, Mittapalli RK Li H, Hoffman DM, Holen KD, Menon RM, et al. Evaluation of the effect of the EGFR antibody-drug conjugate ABT-414 on QT interval prolongation in patients with advanced solid tumors likely to over-express EGFR. Cancer Chemother Pharmacol. (2017) 79:915–22. doi: 10.1007/s00280-017-3284-y
130. Jang SB, Kim KB, Sim S, Cho BC, Ahn MJ, Han JY, et al. Cardiac safety assessment of lazertinib: findings from patients with EGFR mutation-positive advanced NSCLC and preclinical studies. JTO Clin Res Rep. (2021) 2:100224. doi: 10.1016/j.jtocrr.2021.100224
131. Yue Z, Chen J, Lian H, Pei J, Li Y, Chen X, et al. PDGFR-beta signaling regulates cardiomyocyte proliferation and myocardial regeneration. Cell Rep. (2019) 28:966–78 e4. doi: 10.1016/j.celrep.2019.06.065
132. Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med. (2017) 9:eaaf2584. doi: 10.1126/scitranslmed.aaf2584
133. Rashid FN, Clayton ZE, Ogawa M, Perdomo J, Hume RD, Kizana E, et al. Platelet derived growth factor-A (Pdgf-a) gene transfer modulates scar composition and improves left ventricular function after myocardial infarction. Int J Cardiol. (2021) 341:24–30. doi: 10.1016/j.ijcard.2021.07.021
134. Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. (2007) 370:2011–9. doi: 10.1016/S0140-6736(07)61865-0
135. Izumi-Nakaseko H, Fujiyoshi M, Hagiwara-Nagasawa M, Goto A, Chiba K, Kambayashi R, et al. Dasatinib can impair left ventricular mechanical function but may lack proarrhythmic effect: a proposal of non-clinical guidance for predicting clinical cardiovascular adverse events of tyrosine kinase inhibitors. Cardiovasc Toxicol. (2020) 20:58–70. doi: 10.1007/s12012-019-09538-5
136. Motokawa T, Ikeda S, Ueno Y, Eguchi M, Minami T, Kawano H, et al. Comparison of dasatinib- and imatinib-related cardiotoxic adverse events in Japanese patients with chronic myeloid leukemia and gastrointestinal stromal tumor. Circ Rep. (2022) 4:1–8. doi: 10.1253/circrep.CR-21-0140
137. Yee NS, Kazi AA, Yee RK. Cellular and developmental biology of TRPM7 channel-kinase: implicated roles in cancer. Cells. (2014) 3:751–77. doi: 10.3390/cells3030751
138. Inoue K, Xiong ZG. Silencing TRPM7 promotes growth/proliferation and nitric oxide production of vascular endothelial cells via the ERK pathway. Cardiovasc Res. (2009) 83:547–57. doi: 10.1093/cvr/cvp153
139. Andriule I, Pangonyte D, Almanaityte M, Patamsyte V, Kupryte M, Karciauskas D, et al. Evidence for the expression of TRPM6 and TRPM7 in cardiomyocytes from all four chamber walls of the human heart. Sci Rep. (2021) 11:15445. doi: 10.1038/s41598-021-94856-4
140. Zhang Z, Wang M, Fan XH, Chen JH, Guan YY, Tang YB. Upregulation of TRPM7 channels by angiotensin II triggers phenotypic switching of vascular smooth muscle cells of ascending aorta. Circ Res. (2012) 111:1137–46. doi: 10.1161/CIRCRESAHA.112.273755
141. Lee CT, Ng HY, Kuo WH, Tain YL, Leung FF, Lee YT. The role of TRPM7 in vascular calcification: comparison between phosphate and uremic toxin. Life Sci. (2020) 260:118280. doi: 10.1016/j.lfs.2020.118280
142. Baldoli E, Maier JA. Silencing TRPM7 mimics the effects of magnesium deficiency in human microvascular endothelial cells. Angiogenesis. (2012) 15:47–57. doi: 10.1007/s10456-011-9242-0
143. Zhu D, You J, Zhao N, Xu H. Magnesium regulates endothelial barrier functions through TRPM7, MagT1, and S1P1. Advanced science. (2019) 6:1901166. doi: 10.1002/advs.201901166
144. Zeng Z, Inoue K, Sun H, Leng T, Feng X, Zhu L, et al. TRPM7 regulates vascular endothelial cell adhesion and tube formation. Am J Physiol Cell Physiol. (2015) 308:C308–18. doi: 10.1152/ajpcell.00275.2013
145. Gwanyanya A, Andriule I, Istrate BM, Easmin F, Mubagwa K, Macianskiene R. Modulation of the cardiac myocyte action potential by the magnesium-sensitive TRPM6 and TRPM7-like current. Int J Mol Sci. (2021) 22:8744. doi: 10.3390/ijms22168744
146. Sah R, Mesirca P, Van den Boogert M, Rosen J, Mably J, Mangoni ME, et al. Ion channel-kinase TRPM7 is required for maintaining cardiac automaticity. Proc Natl Acad Sci USA. (2013) 110:E3037–46. doi: 10.1073/pnas.1311865110
147. Miyazaki Y, Ichimura A, Kitayama R, Okamoto N, Yasue T, Liu F, et al. C-type natriuretic peptide facilitates autonomic Ca(2+) entry in growth plate chondrocytes for stimulating bone growth. Elife. (2022) 11:e71931. doi: 10.7554/eLife.71931.sa2
148. Valinsky WC, Jolly A, Miquel P, Touyz RM, Shrier A. Aldosterone upregulates transient receptor potential melastatin 7 (TRPM7). J Biol Chem. (2016) 291:20163–72. doi: 10.1074/jbc.M116.735175
149. Callera GE, He Y, Yogi A, Montezano AC, Paravicini T, Yao G, et al. Regulation of the novel Mg2+ transporter transient receptor potential melastatin 7 (TRPM7) cation channel by bradykinin in vascular smooth muscle cells. J Hypertens. (2009) 27:155–66. doi: 10.1097/HJH.0b013e3283190582
150. Sah R, Mesirca P, Mason X, Gibson W, Bates-Withers C, Van den Boogert M, et al. Timing of myocardial trpm7 deletion during cardiogenesis variably disrupts adult ventricular function, conduction, and repolarization. Circulation. (2013) 128:101–14. doi: 10.1161/CIRCULATIONAHA.112.000768
151. Hu F, Li M, Han F, Zhang Q, Zeng Y, Zhang W, et al. Role of TRPM7 in cardiac fibrosis: a potential therapeutic target (review). Exp Ther Med. (2021) 21:173. doi: 10.3892/etm.2020.9604
152. Yu Y, Chen S, Xiao C, Jia Y, Guo J, Jiang J, et al. TRPM7 is involved in angiotensin II induced cardiac fibrosis development by mediating calcium and magnesium influx. Cell Calcium. (2014) 55:252–60. doi: 10.1016/j.ceca.2014.02.019
153. Guo JL Yu Y, Jia YY, Ma YZ, Zhang BY, Liu PQ, et al. Transient receptor potential melastatin 7 (TRPM7) contributes to H2O2-induced cardiac fibrosis via mediating Ca(2+) influx and extracellular signal-regulated kinase 1/2 (ERK1/2) activation in cardiac fibroblasts. J Pharmacol Sci. (2014) 125:184–92. doi: 10.1254/jphs.13224FP
154. Lu J, Wang QY, Zhou Y, Lu XC, Liu YH, Wu Y, et al. Astragaloside against cardiac fibrosis by inhibiting TRPM7 channel. Phytomedicine. (2017) 30:10–7. doi: 10.1016/j.phymed.2017.04.002
155. Jia T, Wang X, Tang Y, Yu W, Li C, Cui S, et al. Sacubitril ameliorates cardiac fibrosis through inhibiting TRPM7 channel. Front Cell Dev Biol. (2021) 9:760035. doi: 10.3389/fcell.2021.760035
156. Reges O, Ning H, Wilkins JT, Wu CO, Tian X, Domanski MJ, et al. Association of cumulative systolic blood pressure with long-term risk of cardiovascular disease and healthy longevity: findings from the lifetime risk pooling project cohorts. Hypertension. (2021) 77:347–56. doi: 10.1161/HYPERTENSIONAHA.120.15650
157. Shin MK, Eraso CC, Mu YP, Gu C, Yeung BHY, Kim LJ, et al. Leptin induces hypertension acting on transient receptor potential melastatin 7 channel in the carotid body. Circ Res. (2019) 125:989–1002. doi: 10.1161/CIRCRESAHA.119.315338
158. Letavernier E, Perez J, Bellocq A, Mesnard L, de Castro Keller A, Haymann JP, et al. Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ Res. (2008) 102:720–8. doi: 10.1161/CIRCRESAHA.107.160077
159. Correa S, Guerra-Torres XE, Waikar SS, Mc Causland FR. Serum magnesium, blood pressure, and risk of hypertension and chronic kidney disease progression in the CRIC study. Hypertension. (2021) 78:1771–80. doi: 10.1161/HYPERTENSIONAHA.121.17694
160. Khan AM, Sullivan L, McCabe E, Levy D, Vasan RS, Wang TJ. Lack of association between serum magnesium and the risks of hypertension and cardiovascular disease. Am Heart J. (2010) 160:715–20. doi: 10.1016/j.ahj.2010.06.036
161. Zhang X, Li Y, Del Gobbo LC, Rosanoff A, Wang J, Zhang W, et al. Effects of magnesium supplementation on blood pressure: a meta-analysis of randomized double-blind placebo-controlled trials. Hypertension. (2016) 68:324–33. doi: 10.1161/HYPERTENSIONAHA.116.07664
162. Zhang YH, Sun HY, Chen KH, Du XL, Liu B, Cheng LC, et al. Evidence for functional expression of TRPM7 channels in human atrial myocytes. Basic Res Cardiol. (2012) 107:282. doi: 10.1007/s00395-012-0282-4
163. Macianskiene R, Almanaityte M, Jekabsone A, Mubagwa K. Modulation of human cardiac TRPM7 current by extracellular acidic pH depends upon extracellular concentrations of divalent cations. PLoS ONE. (2017) 12:e0170923. doi: 10.1371/journal.pone.0170923
164. Duzen IV, Yavuz F, Vuruskan E, Saracoglu E, Poyraz F, Goksuluk H, et al. Leukocyte TRP channel gene expressions in patients with non-valvular atrial fibrillation. Sci Rep. (2017) 7:9272. doi: 10.1038/s41598-017-10039-0
165. Li MJ, Zhou YM, Tang YH, Cao F. Increased expression of transient receptor potential melastatin 7 in mouse cardiac fibroblasts post myocardial infarction. Zhonghua Xin Xue Guan Bing Za Zhi. (2008) 36:641–5.
166. Li FY, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature. (2011) 475:471–6. doi: 10.1038/nature10246
167. Jacobsen DM, Bao ZQ, O'Brien P, Brooks CL 3rd, Young MA. Price to be paid for two-metal catalysis: magnesium ions that accelerate chemistry unavoidably limit product release from a protein kinase. J Am Chem Soc. (2012) 134:15357–70. doi: 10.1021/ja304419t
168. Yu L, Xu L, Xu M, Wan B, Yu L, Huang Q. Role of Mg2+ ions in protein kinase phosphorylation: insights from molecular dynamics simulations of ATP-kinase complexes. Mol Simul. (2011) 37:1143–50. doi: 10.1080/08927022.2011.561430
169. Thebault S, Alexander RT, Tiel Groenestege WM, Hoenderop JG, Bindels RJ. EGF increases TRPM6 activity and surface expression. J Am Soc Nephrol. (2009) 20:78–85. doi: 10.1681/ASN.2008030327
170. Muallem S, Moe OW. When EGF is offside, magnesium is wasted. J Clin Invest. (2007) 117:2086–9. doi: 10.1172/JCI33004
171. Hsieh MC, Wu CF, Chen CW, Shi CS, Huang WS, Kuan FC. Hypomagnesemia and clinical benefits of anti-EGFR monoclonal antibodies in wild-type KRAS metastatic colorectal cancer: a systematic review and meta-analysis. Sci Rep. (2018) 8:2047. doi: 10.1038/s41598-018-19835-8
172. Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol. (2002) 4:329–36. doi: 10.1038/ncb781
173. Fang L, Zhan S, Huang C, Cheng X, Lv X, Si H, et al. TRPM7 channel regulates PDGF-BB-induced proliferation of hepatic stellate cells via PI3K and ERK pathways. Toxicol Appl Pharmacol. (2013) 272:713–25. doi: 10.1016/j.taap.2013.08.009
174. Cai S, Wu L, Yuan S, Liu G, Wang Y, Fang L, et al. Carvacrol alleviates liver fibrosis by inhibiting TRPM7 and modulating the MAPK signaling pathway. Eur J Pharmacol. (2021) 898:173982. doi: 10.1016/j.ejphar.2021.173982
175. Abed E, Moreau R. Importance of melastatin-like transient receptor potential 7 and magnesium in the stimulation of osteoblast proliferation and migration by platelet-derived growth factor. Am J Physiol Cell Physiol. (2009) 297:C360–8. doi: 10.1152/ajpcell.00614.2008
176. Tian SL, Jiang H, Zeng Y, Li LL, Shi J. NGF-induced reduction of an outward-rectifying TRPM7-like current in rat CA1 hippocampal neurons. Neurosci Lett. (2007) 419:93–8. doi: 10.1016/j.neulet.2007.04.020
177. Zou ZG, Rios FJ, Neves KB, Alves-Lopes R, Ling J, Baillie GS, et al. Epidermal growth factor signaling through transient receptor potential melastatin 7 cation channel regulates vascular smooth muscle cell function. Clin Sci. (2020) 134:2019–35. doi: 10.1042/CS20200827
178. Mak IT, Kramer JH, Chmielinska JJ, Spurney CF, Weglicki WB, EGFR-TKI. erlotinib, causes hypomagnesemia, oxidative stress, and cardiac dysfunction: attenuation by NK-1 receptor blockade. J Cardiovasc Pharmacol. (2015) 65:54–61. doi: 10.1097/FJC.0000000000000163
179. Orcutt KP, Parsons AD, Sibenaller ZA, Scarbrough PM, Zhu Y, Sobhakumari A, et al. Erlotinib-mediated inhibition of EGFR signaling induces metabolic oxidative stress through NOX4. Cancer Res. (2011) 71:3932–40. doi: 10.1158/0008-5472.CAN-10-3425
180. Cao G, Lee KP, van der Wijst J, de Graaf M, van der Kemp A, Bindels RJ, et al. Methionine sulfoxide reductase B1 (MsrB1) recovers TRPM6 channel activity during oxidative stress. J Biol Chem. (2010) 285:26081–7. doi: 10.1074/jbc.M110.103655
181. Inoue H, Murayama T, Tashiro M, Sakurai T, Konishi M. Mg(2+)- and ATP-dependent inhibition of transient receptor potential melastatin 7 by oxidative stress. Free Radic Biol Med. (2014) 72:257–66. doi: 10.1016/j.freeradbiomed.2014.04.015
182. Harvey A, Montezano AC, Lopes RA, Rios F, Touyz RM. Vascular fibrosis in aging and hypertension: molecular mechanisms and clinical implications. Can J Cardiol. (2016) 32:659–68. doi: 10.1016/j.cjca.2016.02.070
183. Yee NS, Zhou W, Lee M, Yee RK. Targeted silencing of TRPM7 ion channel induces replicative senescence and produces enhanced cytotoxicity with gemcitabine in pancreatic adenocarcinoma. Cancer Lett. (2012) 318:99–105. doi: 10.1016/j.canlet.2011.12.007
184. Xing J, Wang M, Hong J, Gao Y, Liu Y, Gu H, et al. TRPM7 channel inhibition exacerbates pulmonary arterial hypertension through MEK/ERK pathway. Aging. (2019) 11:4050–65. doi: 10.18632/aging.102036
185. Tohyama M, Sakaguchi C, Nishina T, Hyodo I. Possible involvement of zinc deficiency in epidermal growth factor receptor inhibitor-induced xerotic dermatitis. J Dermatol. (2021) 48:1579–83. doi: 10.1111/1346-8138.16049
186. Dabravolski SA, Sadykhov NK, Kartuesov AG, Borisov EE, Sukhorukov VN, Orekhov AN. Interplay between Zn(2+) homeostasis and mitochondrial functions in cardiovascular diseases and heart ageing. Int J Mol Sci. (2022) 23:6890. doi: 10.3390/ijms23136890
187. Braun LA, Ou R, Kure C, Trang A, Rosenfeldt F. Prevalence of zinc deficiency in cardiac surgery patients. Heart Lung Circ. (2018) 27:760–2. doi: 10.1016/j.hlc.2017.07.009
188. Gommers LM, Hoenderop JG, Bindels RJ, de Baaij JH. Hypomagnesemia in type 2 diabetes: a vicious circle? Diabetes. (2016) 65:3–13. doi: 10.2337/db15-1028
189. Kostov K. Effects of magnesium deficiency on mechanisms of insulin resistance in type 2 diabetes: focusing on the processes of insulin secretion and signaling. Int J Mol Sci. (2019) 20:1351. doi: 10.3390/ijms20061351
190. Guerrero-Romero F, Rascon-Pacheco RA, Rodriguez-Moran M, de la Pena JE, Wacher N. Hypomagnesaemia and risk for metabolic glucose disorders: a 10-year follow-up study. Eur J Clin Invest. (2008) 38:389–96. doi: 10.1111/j.1365-2362.2008.01957.x
Keywords: receptor tyrosine kinase, TRPM7, cardiovascular toxicities, cancer, tyrosine kinase inhibitors, magnesium, calcium, cation channel
Citation: Liu Q, Li S, Qiu Y, Zhang J, Rios FJ, Zou Z and Touyz RM (2023) Cardiovascular toxicity of tyrosine kinase inhibitors during cancer treatment: Potential involvement of TRPM7. Front. Cardiovasc. Med. 10:1002438. doi: 10.3389/fcvm.2023.1002438
Received: 25 July 2022; Accepted: 18 January 2023;
Published: 03 February 2023.
Edited by:
Lilei Yu, Wuhan University, ChinaReviewed by:
Monica Gonzalez-Magaldi, The University of Texas at Austin, United StatesCopyright © 2023 Liu, Li, Qiu, Zhang, Rios, Zou and Touyz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rhian M. Touyz,  cmhpYW4udG91eXpAbWNnaWxsLmNh; Zhiguo Zou, em91emhpZ3VvQHJlbmppLmNvbQ==
cmhpYW4udG91eXpAbWNnaWxsLmNh; Zhiguo Zou, em91emhpZ3VvQHJlbmppLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.