
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med., 07 September 2022
Sec. Cardiovascular Metabolism
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.997013
This article is part of the Research TopicMyocardium Regeneration and CardioprotectionView all 6 articles
 Sean Brennan1*Simona Esposito1,2Muhammad I. M. Abdelaziz1Christopher A. Martin2Samir Makwana2Mark W. Sims2Iain B. Squire2,3
Sean Brennan1*Simona Esposito1,2Muhammad I. M. Abdelaziz1Christopher A. Martin2Samir Makwana2Mark W. Sims2Iain B. Squire2,3 Parveen Sharma1Amy E. Chadwick4
Parveen Sharma1Amy E. Chadwick4 Richard D. Rainbow1*
Richard D. Rainbow1*Hyperglycaemia at the time of myocardial infarction has an adverse effect on prognosis irrespective of a prior diagnosis of diabetes, suggesting glucose is the damaging factor. In ex vivo models of ischaemia, we demonstrated that deleterious effects of acutely elevated glucose are PKCα/β-dependent, and providing PKCα/β are inhibited, elevated glucose confers cardioprotection. Short pre-treatments with high glucose were used to investigate time-dependent glucose cardiotoxicity, with PKCα/β inhibition investigated as a potential mechanism to reverse the toxicity. Freshly isolated non-diabetic rat cardiomyocytes were exposed to elevated glucose to investigate the time-dependence toxic effects. High glucose challenge for >7.5 min was cardiotoxic, proarrhythmic and lead to contractile failure, whilst cardiomyocytes exposed to metabolic inhibition following 5-min high glucose, displayed a time-dependent protection lasting ∼15 min. This protection was further enhanced with PKCα/β inhibition. Cardioprotection was measured as a delay in contractile failure and KATP channel activation, improved contractile and Ca2+ transient recovery and increased cell survival. Finally, the effects of pre-ischaemic treatment with high glucose in a whole-heart coronary ligation protocol, where protection was evident with PKCα/β inhibition. Selective PKCα/β inhibition enhances protection suggesting glycaemic control with PKC inhibition as a potential cardioprotective therapeutics in myocardial infarction and elective cardiac surgery.
Elevated blood glucose at the time of acute myocardial infarction (AMI) has been shown to worsen prognosis irrespective of a prior diagnosis of diabetes (1–4). Elevated admission blood glucose, fasting glucose and average glucose levels during hospitalization have all been associated with increased mortality in acute coronary syndrome (ACS) patients (4–6). Combined, these data indicate that elevated glucose over a short time frame can worsen ACS prognosis and may explain why rapid glucose lowering treatment can be beneficial. Consequently, The American Heart Association (7), European Cardiovascular Society (8), and The National Institute for Health and Care Excellence (NICE, United Kingdom) (9) all recommend careful monitoring and limiting of both hyper- and hypoglycaemia in AMI patients.
Our, and others, previous findings show a damaging effect of enhanced glycolytic metabolism, via acutely elevated extracellular glucose, on cellular function in otherwise physiological conditions and abolition of cardioprotection against simulated ischemia, through activation of protein kinase C (PKC) α and β isoforms (10–17). The clinically negative impact of acute hyperglycaemia during ACS is, therefore, supported by non-diabetic cellular, whole tissue ex vivo, in vivo, and clinical data (2, 9). The 2021 NICE guidelines for acute coronary syndromes recommend further research in optimal management of hyperglycaemia in ACS. In this manuscript we investigate the time-dependence of the damaging effect of enhanced glycolytic metabolism and whether inhibition of protein kinase C (PKC) α and β isoforms can reduce the onset of these damaging effects.
Whilst glucose-insulin-potassium infusions have been a mainstay of hospital treatments for myocardial infarction, the benefits of this have not been always clear (18–21). The most effective use of this treatment appears to be as a cardioprotective pre-treatment in elective surgery (22–24). We have previously shown that glucose has acute cardiotoxicity, in this context referring to an increased proarrhythmicity with enhanced cell death and infarct size in cell and animal models (14, 17, 25). This is not, however, the complete story, where the duration of exposure to an elevated glucose has significant ramifications for cellular outcome. Our data suggests there is a “temporal cliff-edge” between elevated glucose being protective and toxic to cardiomyocytes.
Here, we show that exposure to elevated glucose for >7.5 min decreased contractile recovery following a period of metabolic inhibition, suggesting that this triggers a period of enhanced glycolytic metabolism that is potentially cardiotoxic. If the duration of high glucose challenge was less than 5 min, it did not appear to cause cardiotoxicity and enhanced cell survival in a metabolic inhibition protocol, indicating cardioprotection. These data led to the hypothesis that brief (<5 min) challenges of elevated glucose would impart cardioprotection, analogous to the brief ischaemic intervals used in ischaemic preconditioning and as previously reported with ethanol pre-treatment (26, 27). Conversely, longer duration of glucose perfusion imparted toxicity, suggesting a tipping point where the damaging PKCαβ-dependent toxicity dominates. Finally, selective PKCαβ inhibition abolished the longer duration toxicity of elevated glucose and further improved cardioprotection.
All methods were carried out in accordance with relevant guidelines and regulations. Adult male Wistar rats (300–400 g, 12–14 weeks old, n = 94, bred in-house at the University of Leicester), were killed by concussion and cervical dislocation. Animals were maintained in 1,800 cm2 Tecniplast cages, using BED01/8 dates and corn cob for bedding, housed in individually ventilated caging (specific-pathogen-free). The number of cage companions for the animals of 300–400 g is 5 based on a calculation of 350 cm2 being required for each rat of that size. The care and death of the animals confirmed to the requirements of the UK Animals (Scientific Procedures) Act 1986 (SI 2012/3039). All procedures were approved by the University of Leicester animal welfare ethical review board (AWERB_2018_44). The ARRIVE guidelines for reporting experiments involving animals have been followed in this study (28).
All perfusing solutions were based on a modified Tyrode’s solution used in our previous publications (14, 17, 25). The solution contained (in mM), 135 NaCl, 5 KCl, 0.33 NaH2PO4, 5 Na-Pyruvate, 10 HEPES, 1 MgCl2, and 2 CaCl2 (pH 7.4) and is referred to as normal Tyrode’s solution (NT). For all experiments, NT with 5 mM glucose was considered as the control glucose concentration as it is within the physiological fasting range (NICE guideline PH38). This is a lower concentration of glucose than is traditionally used for these kind of experiments, often 11.1 mM, however, our previous data suggests alterations in functional parameters in cells at 10 mM glucose; a concentration would be considered as hyperglycaemic in vivo (14). Clinically, hyperglycaemia is defined as a blood glucose concentration above 11 mM (NICE guideline [CG130]). To investigate the effects of raised glucose, NT solutions containing 20 mM glucose were used, and mannitol (5 mM glucose with 15 mM mannitol) was used to ensure all normoglycaemic Tyrode’s solutions were osmotically balanced. This 20 mM glucose solution is referred to as “high glucose” for simplicity in the text. This glucose concentration, although well into the pathophysiological range, has been shown to be relevant to real clinical scenarios during MI (3, 17).
Nominally Ca2+ free solution used during the isolation of cardiomyocytes was as outlined above with no added Ca2+. Substrate-free 2 mM Ca2+ Tyrode’s solution (SFT) contained (in mM), 140 NaCl, 5 KCl, 0.33 NaH2PO4, 10 HEPES, 20 mM sucrose, 1 MgCl2, and 2 CaCl2 (pH 7.4). The pipette solution for whole cell electrophysiological recording contained (in mM), 30 KOH, 5 EGTA, 110 KCl, 10 HEPES, 1 MgCl2, 0.61 CaCl2, 1 ATP, 0.1 ADP, and 0.1 GTP (pH 7.2). Pipette solution for cell attached patch recording contained (in mM), 140 KCl, 10 HEPES, 1 CaCl2, and 0.5 MgCl2 (pH 7.4).
Adult male Wistar rats (300–350 g) were killed by concussion and cervical dislocation. The protocol for the isolation of cardiomyocytes was as described previously (14, 17, 25, 29). Briefly, the heart was rapidly excised and placed into cold nominally Ca2+-free NT solution. The heart was then cannulated via the aorta on Langendorff apparatus and warmed (37°C) Ca2+-free NT was perfused in a retrograde manner via the aorta to clear residual blood. Following 6 min of perfusion, the solution was exchanged, and the heart perfused with Ca2+-free NT containing 0.66 mg/ml collagenase (Type II, Worthington’s, Lakewood, NJ, United States), 0.5 mg/ml protease (type XIV 15% Ca2+, Sigma-Aldrich, Gillingham, United Kingdom) and 1.93 mg/ml BSA prepared from factor V albumin (Sigma-Aldrich, Gillingham, United Kingdom) for 4–7 min until identification of rod-shaped cardiomyocytes in the perfusate. The solution was then exchanged for nominally Ca2+-free solution for 2 min, the heart cut down and the tissue washed three times with 2 mM Ca2+-containing NT. Cardiomyocytes were then mechanically dispersed from the tissue in a shaking water bath. Typically, this method yielded 70–90% rod-shaped cardiomyocytes and were stored in NT at room temperature in 60 mm culture dishes on a rocking platform to prevent pelleting of the cells. Cells isolated with this method are viable for up to 18 h following isolation with no detriment to contractility. Cells were typically used within 9 h for this study.
The human ventricular cell line AC16 cells (Sigma-Aldrich, Gillingham, United Kingdom) were maintained and cultured in low glucose (5 mM) Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma-Aldrich, Gillingham, United Kingdom) supplemented with 10% foetal bovine serum (FBS) (Sigma-Aldrich, Gillingham, United Kingdom), 4 mM glutamine (Sigma-Aldrich, Gillingham, United Kingdom) and penicillin (100 IU)/streptomycin (100 mg/ml) (Sigma-Aldrich, Gillingham, United Kingdom) at 37°C under a humidified 5% CO2 atmosphere. Cells were used up to passage 20.
Cells were seeded at 10,000 cells/ml on standard 96-well tissue culture plate. The growth media was removed from the wells, cells were washed twice with Hank’s Balanced Salt Solution (HBSS) and replaced with high glucose (20 mM) DMEM with above supplements for 5, 15, or 30 min. Following glucose incubation, media was removed and saved for evaluation and AC16 cells were lysed in somatic cell ATP releasing reagent (Sigma-Aldrich, Gillingham, United Kingdom). Aliquots of the same lysate were then used to determine the ATP and protein level of the each well using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Southampton, United Kingdom) and BCA protein assay (Pierce, Thermofisher, Altrincham, United Kingdom), respectively, according to manufacturer’s guidelines. The lactate dehydrogenase (LDH) content of the lysate and media from the same well were measured according to the instructions of Cytotoxicity Detection Kit (Roche Diagnostics, Welwyn Garden City, United Kingdom) (30).
Cardiomyocytes were perfused at a rate of 5 ml/min at 32 ± 2°C and were stimulated to contract via electric field stimulation (EFS) at a rate of 1 Hz Digitimer DSXX (Welwyn Garden City, United Kingdom). The term “contractile cells” is used to indicate the percentage, or number, of rod-shaped cardiomyocytes contracting in rhythm with the EFS. Contractile function was observed via a JVC CCTV camera (JVC, Yokohama, Kanagawa, Japan) and recorded to DVD Panasonic DVD recorder (Panasonic, Kadoma, Osaka, Japan). Cells were perfused for 5 min with NT solution followed by 2.5–20 min perfusion with 20 mM glucose NT as indicated in Figure 1. The percentage of rod-shaped cardiomyocytes were counted every 30 s and used to form the time course (Figure 1A). The number of asynchronous contractions, contractions that occur in addition to the EFS and last for longer than 2 s in duration, were also counted. These asynchronous events could lead to the cell failing to contract as displayed in Figures 1A,B (14).
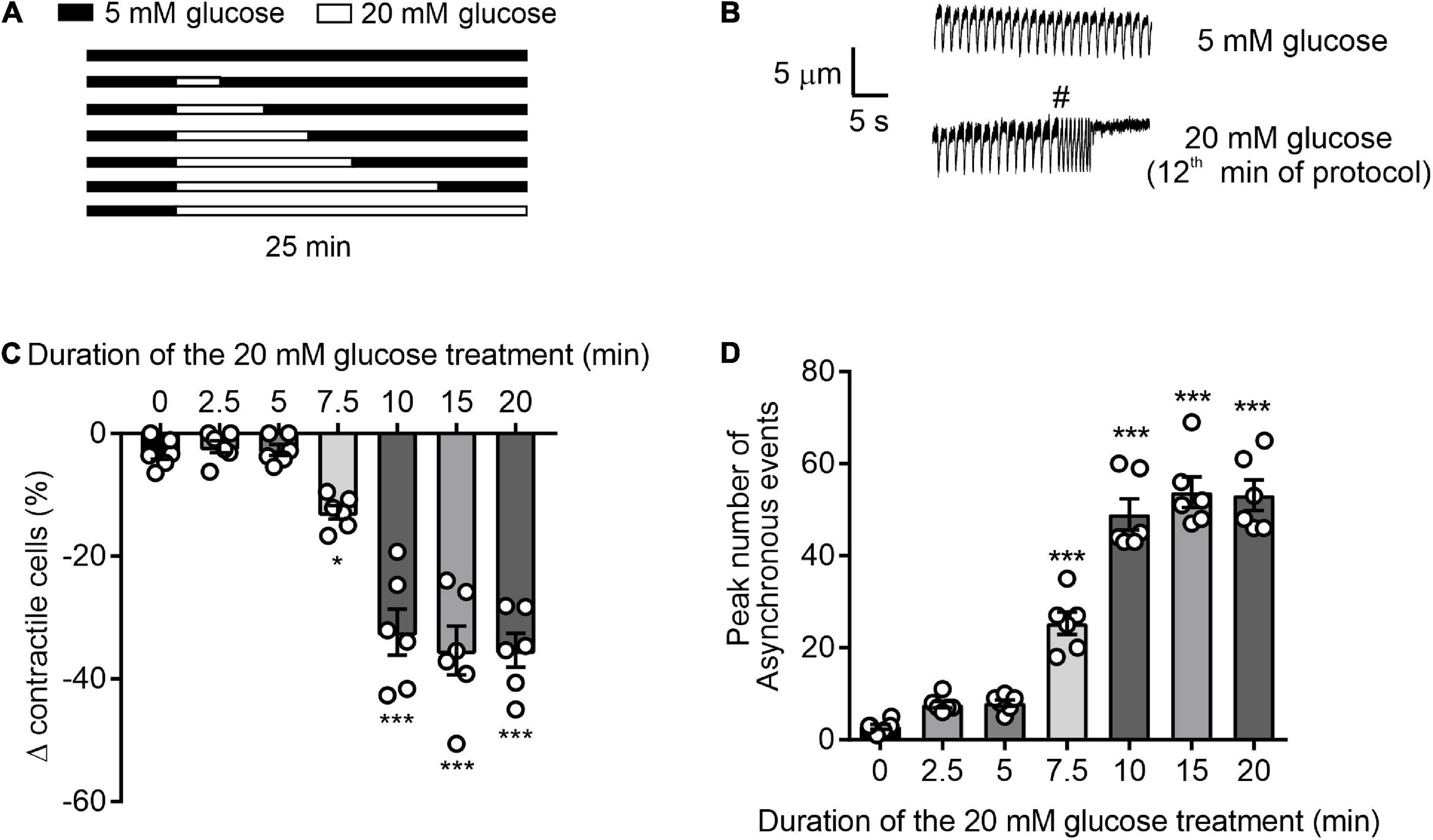
Figure 1. Time course of glucose-induced dysregulation of cardiomyocyte contractile function. (A) Illustration of the experimental design used in this figure. (B) Example traces showing cardiomyocyte contractions, initiated by 1 Hz EFS, in the presence of 5 or 20 mM glucose. Cardiomyocytes that contracted outside of the 1 Hz EFS, as demonstrated by # in panel B, or failed to contract were not defined as contractile cells. Therefore, only cardiomyocytes that contracted in time with 1 Hz EFS were counted as contractile cells. (C) Bar chart showing the mean percentage change in contractile cells (*p < 0.05, ***p < 0.001. One Way ANOVA with Holm-Sidak post-test). (D) Bar chart showing the mean number of arrhythmic episodes in each data set. ***p < 0.001 One-Way ANOVA with Holm-Sidak post-test. n = 6 experiments from six animals (>98 cells for each data set).
Using the same apparatus as above, cardiomyocytes were subjected to a metabolic inhibition and reperfusion (MI/R) protocol which consisted of 3 min perfusion with NT, 7 min of perfusion with a substrate-free metabolic inhibition Tyrode’s solution (SFT-MI) and 10 min washout with EFS at 1 Hz throughout. SFT-MI contained 2 mM Na-cyanide and 1 mM iodoacetic acid to block oxidative phosphorylation and glycolysis, respectively. The time to contractile failure, contractile recovery, and cell survival (as measured using Trypan blue exclusion) were noted for each cell. A contractile recovery of between 25 and 30% was indicative of a normal contractile recovery for control cardiomyocytes with anything above 45% contractile recovery representing cardioprotection (25). Modifications to this protocol, such as pre-treatment and changes to the washout protocol, are indicated in the figures and text.
Metabolic inhibition-activated currents were recorded in both whole cell and cell-attached patch configurations. Whole cell recordings were made from isolated cardiomyocytes using an Axopatch 200B amplifier, digitised using a Digidata 1440 and recorded and analysed using pCLAMP10.7 software (RRID:SCR_011323, Axon instruments, Scientifica, Uckfield, United Kingdom). Cardiomyocytes were perfused with NT solution at 5 ml/min at 32 ± 2°C with the membrane voltage held at 0 mV to inactivate all voltage-gated currents and to avoid the inward rectifier (IK1) current. Application of SFT-MI solution caused an activation of a metabolic inhibition-sensitive outward current attributed to the cardiac KATP current. The time to activation of this current was recorded and compared to that in cell pre-treated with a 20 mM glucose NT pre-treatment. Cardiac KATP (Kir6.2/SUR2A) current activation was also measured directly using cell-attached patch recording, with KATP being readily identified by its single channel current. Cell attached patches of membrane were held at 40 mV (an equivalent of ∼−110 mV from the sum of the pipette holding potential and the approximated resting membrane potential of −75 mV). At this potential, assuming a Kir6.2/SUR2A single channel conductance of around 70–80 pS, the single channel current should be ∼10 pA for each level of opening.
Fura-2 measurements were made from cardiomyocytes as described previously (14, 17). Briefly, cardiomyocytes were loaded with 5 μM Fura-2 AM for 20 min at room temperature, then allowed to settle for a further 10 min in a heated perfusion chamber before being washed with NT solution prior to the start of the experiment. Cardiomyocytes were perfused at 5 ml/min at 32 ± 2°C and stimulated to contract at 1 Hz using electric field stimulation (EFS). Fluorescence was excited at alternately at 340 and 380 nm via a monochromator (PTI) and the emission collected above 520 nm using a Roper Cascade CCD camera (Photometrics, Tuscan, AZ, United States). Images were recorded using EasyRatioPro software (PTI, Birmingham, NJ, United States) and the data presented as the 340:380 ratio (Figure 2F), or a change in ratio (Figure 2G).
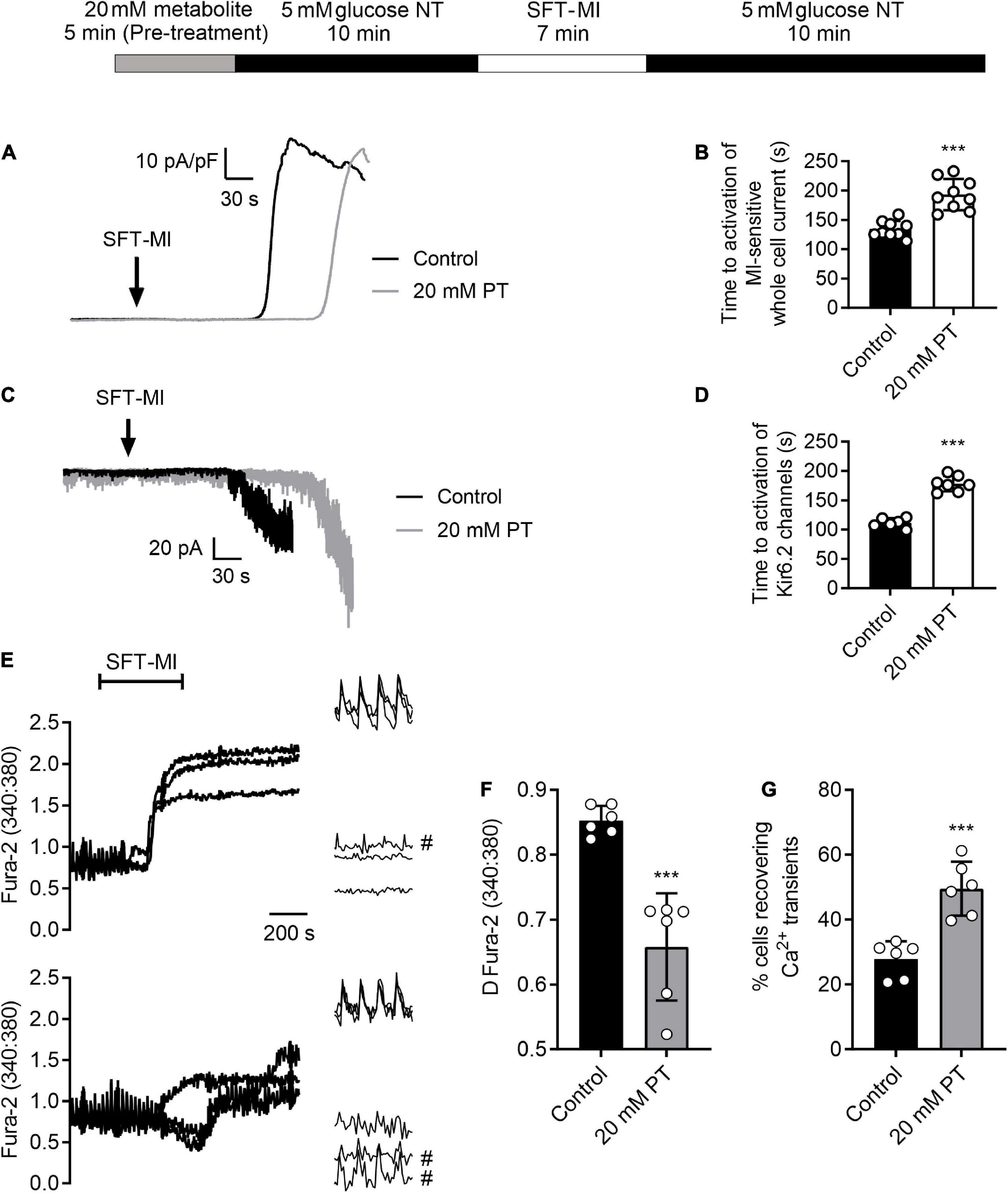
Figure 2. High glucose pre-treatment delays activation of the metabolic inhibition-sensitive KATP current and limits calcium overload during metabolic inhibition. (A) Example traces of whole-cell current recorded at 0 mV showing the activation of a metabolically-sensitive outward K+ current. (B) Bar chart showing the mean time to activation of this metabolic inhibition-sensitive current in control conditions (perfused for 15 min with NT prior to SFT-MI) or with 5 min high glucose pre-treatment. Mean time to activation was significantly delayed following 20 mM PT [***P > 0.001, Unpaired T-test, n = 9 (>14) for each group]. (C) Example traces of cell attached recording of sarcolemmal Kir6.2/SUR2A (IKATP) current activation by metabolic inhibition, identified by its single channel conductance. (D) Bar chart showing the mean time to activation of Kir6.2/SUR2A channels with a burst duration longer than 250 ms. [***P > 0.001, Unpaired T-test, n = > 6 (>10) for each group]. (E) Example traces and expanded transients, recorded at the beginning and end of the protocol, from three cells in control conditions (perfused for 15 mins prior to SFT-MI with NT solution) and three cells in a 20 mM glucose pre-treated group (perfused for 5 min with 20 mM glucose NT followed by 10 min washout with NT). Cells recovering transients are marked with #. Cardiomyocytes were stimulated to contract using 1 Hz EFS and perfused at 34 ± 2°C. Fura-2 ratio images were taken once every 5 s to minimise photo bleaching. Transients were recorded at a rate of 12 ratios per second (24 images). (F) The mean change in Fura-2 ratio at the end of the 10 min simulated reperfusion period showing a reduction in Ca2+ accumulation in the 20 mM glucose pre-treated group compared to control conditions [***P < 0.001, unpaired T-test, n = 6 (>84 cells) per group]. (G) Mean percentage of cells recovering Ca2+ transients at the end of the simulated reperfusion period showing an increase following 20 mM glucose pre-treatment [***P < 0.001, unpaired T-test, n = 6 (>84 cells) per group].
The protocol for generation of an infarct using coronary ligation was previously described (17). Briefly, the heart was rapidly excised, placed in cold NT solution and then mounted on Langendorff apparatus via the aorta, perfused in a retrograde fashion and allowed to stabilise for 1 h and solutions bubbled with 100% O2 throughout. Any hearts that were not contractile (>60 bpm) following 1 h stabilisation or fell off of the canula at any part of the protocol, were excluded (2 of 29 hearts). The left anterior descending (LAD) coronary artery was ligated (40 min) to cause ischaemia using 5-0 USP braided silk suture and two pipette tips to form a reversible knot around the artery. The knot was then removed to start the 3-h reperfusion phase with the suture remaining in place to allow for re-ligation at the end of reperfusion. During all phases, temperature was maintained at 37°C by submerging the heart in Tyrode’s solution using a heated water jacket. Following re-ligation at the end of the protocol, Evans Blue dye (1% in Tyrode’s solution) was perfused through the heart, staining the areas unaffected by the ligation. The heart was then cut down from the Langendorff canula, wrapped in parafilm to prevent freeze-drying, and frozen for 1 h at −20°C. The heart was then cut into eight slices using a scalpel blade and stained in a Na2HPO4/NaH2PO4 solution (∼2:1 ratio of 0.1 M stock solutions until pH 7.4) with 10 mg/ml 2,3,5-triphenyltetrazolium chloride (Sigma Aldrich) to identify area at risk (AAR) and infarcted area (IA, white in appearance), respectively. To determine the AAR and IA, each slice was scanned on both sides and weighed. AAR and IA were calculated for each of the slices from the heart using ImageJ (RRID:SCR_003070). The AAR, IA and unaffected area sizes from ImageJ were then used to calculate the percentage infarct of the AAR by weight. Data were analysed by three researchers, including the experimenter. The two additional researchers were blinded to the conditions, and the mean of all three analyses were used for each heart.
Tat-linked PKC inhibitor peptides were used at a concentration of 100 nM as described previously (14, 25, 31). Cardiomyocytes were preincubated with PKCα and β-specific Tat-linked inhibitor peptides for 15 min prior to the protocol outlined in each figure. PKCα and β were also pharmacologically inhibited in isolated cardiomyocytes using Gö6976 (300 nM: Tocris, Bristol, United Kingdom) and LY379196 (300 nM; a kind gift from Eli Lilly, IND, United States) which were perfused with the NT solutions as indicated in the figures. Equivalent concentrations of vehicle (DMSO) had no effect on cardiomyocyte function.
All contractile function data was analysed in Microsoft Excel 2019 (RRID:SCR_016137). All patch clamp data was analysed in pCLAMP 10.7 (RRID:SCR_011323)1 software and Microsoft Excel 2019. Ca2+ fluorescence measurements were analysed using Winfluor V4.0.2 (J Dempster, University of Strathclyde)2 and Microsoft Excel 2019. Whole heart data was analysed using ImageJ V1.533 and Microsoft Excel 2019. All graphs were made in GraphPad Prism V9.3 (RRID:SCR_002798)4 and statistical analysis performed in Prism 9.3 as indicated in the text. Data is reported as n = animals (cells) and bar charts presented with mean and standard deviation in solid bars with the individual mean per animal superimposed as circles. Data for AC16 cells is reported as n = experiments. Each experiment was the mean of >12 replicates for each condition. The statistical test(s), including post-tests, used for each data set are described in each figure legend along with the n number and the P value where appropriate.
To determine the duration of 20 mM glucose NT (high glucose) perfusion required to negatively impact contractile function, cardiomyocytes were exposed to high glucose for time intervals between 2.5 and 20 mins (Figure 1). In accordance with our previous findings (14), durations of high glucose perfusion ≥ 7.5 min reduced the proportion of rod-shaped cardiomyocytes maintaining contractile function to EFS and increased the number of asynchronous, ectopic contractions (Figures 1A–C). In cells with high glucose perfused for ≤5 min, there was no significant change in the percentage of contractile cardiomyocytes, or in the number of ectopic contractions. These data show that in isolated cardiomyocytes, exposure to high glucose for >7.5 min negatively impacts contractile function. Conversely, brief exposures to high levels of metabolic substrate, in the form of high glucose (5 min), did not alter contractile function.
A body of clinical work reports that elevated glucose at admission for ACS, irrespective of diabetic status, is deleterious to patient outcome (2, 3, 32–34). Furthermore, others have reported that active management of hyperglycaemia with insulin can improve patient outcomes (18–21). It was hypothesised that short (≤5 min) exposure to high glucose, to increase levels of glycolytic substrate, would impart cardioprotection against metabolic inhibition, akin to the short burst of ischaemia used to impart cardioprotection with ischaemic preconditioning (IPC).
To investigate whether a short (5 min) high glucose challenge could impart cardioprotection against ischemia to isolated cardiomyocytes, the metabolic inhibition protocol was adapted to include a period of high glucose pre-treatment followed by a 5 min washout period with 5 mM glucose, as indicated in Figure 3. For this experiment, three durations of pre-treatment were used, 5, 10, and 15 min prior to the metabolic inhibition. With a 5 min high glucose pre-treatment there was a delay in the time to contractile failure (Figure 3C), increase in the percentage of cells recovering their contractile function (Figure 3D) and an increase in cell survival (Figure 3E); these are hallmarks of various established cardioprotective stimuli such as IPC and adenosine (25). With IPC, a delay in the time to contractile failure of ∼60 s is accompanied by increases of contractile recovery of ∼30% and cell survival of ∼15% are routinely recorded (25). A 10 min pre-treatment reduced the number of rod-shaped cardiomyocytes contractile at the start of the protocol (Figure 3A) but did not alter the other parameters measured (Figures 3B–D). Finally, a 15 min pre-treatment with high glucose was cardiotoxic as indicated by a reduced number of contractile cells (Figure 3A), shortened the time to contractile failure (Figure 3B) and fewer cardiomyocytes that recovered contractile function (Figure 3C). Together, these data suggest that a 5 min challenge with high glucose can impart cardioprotection against metabolic inhibition, however, longer exposures result in high glucose-dependent cardiotoxicity. These data suggest that prolonged exposure to enhanced glycolytic metabolite is cardiotoxic. Although this may be paradoxical to what might be expected, however, Supplementary Figure 1 shows that prolonged exposure to high glucose decreases ATP production in AC16 cardiomyocytes. Crucially, shorter duration of high glucose did not reduce ATP production. This could provide an explanation as to why prolonged exposure resulted in poor outcome in the simulated ischemia experiments.
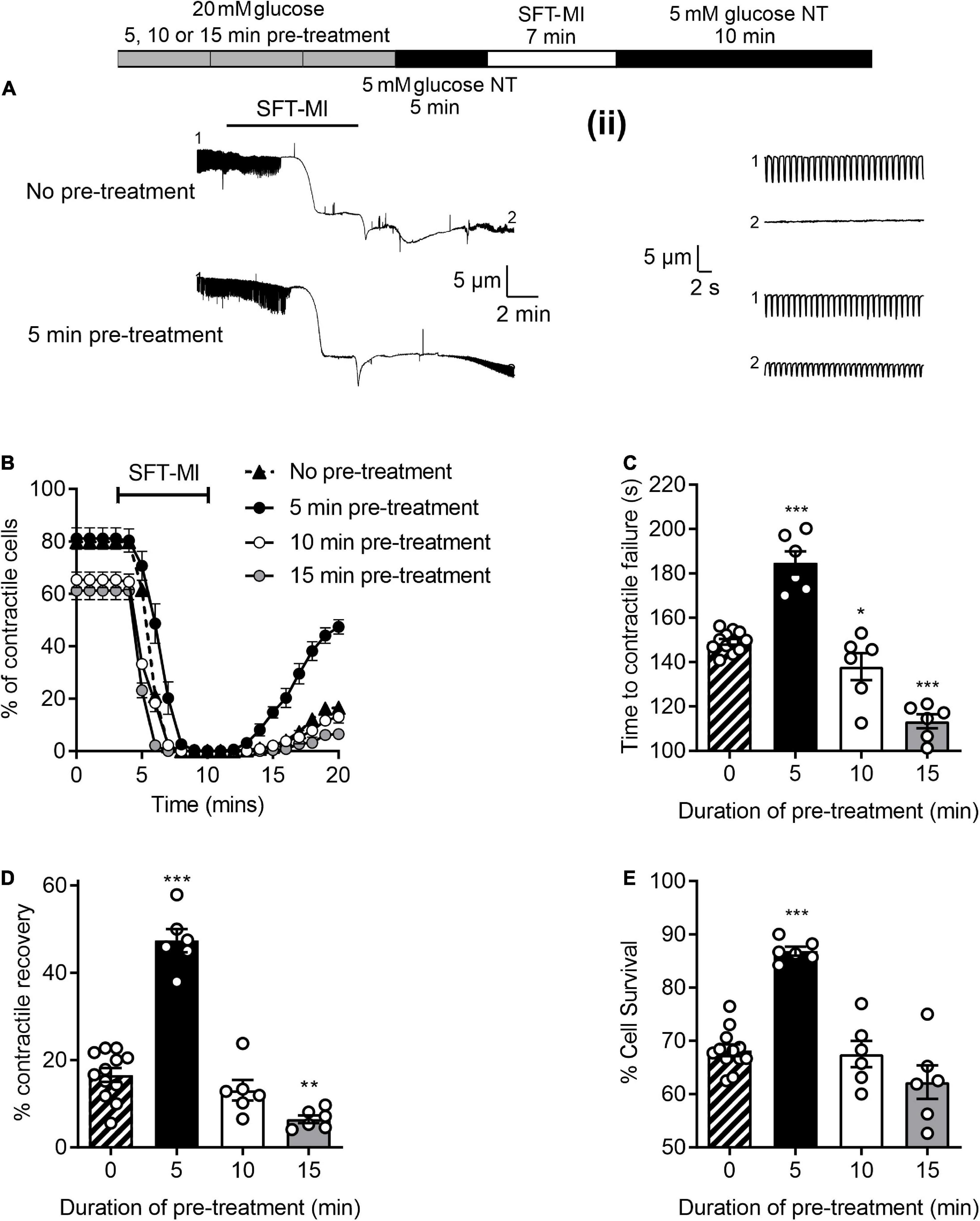
Figure 3. A short pre-treatment with elevated glucose imparts a cardioprotection, but longer durations reveal glucose-dependent toxicity. (A) Example video edge detection measurements from a cell in control (5 mM glucose) conditions and a cell following 5 min of pre-treatment with glucose prior to the MI/R protocol. (ii) Expanded traces showing 30 s of contractions from the beginning (1) and end (2) of the traces in panel (A). (B) Time course showing the number of rod-shaped contractile cells from 3 min before the start of the metabolic inhibition in control (no pre-treatment), or following a 5, 10, or 15 min pre-treatment with 20 mM glucose NT. Bar charts showing (C) the mean time to contractile failure in metabolic inhibition, (D) the mean percentage of contractile recovery at the end of the MI/R protocol and (E) the mean percentage cell survival at the end of the MI/R protocol [**P < 0.01, ***P < 0.001, One-Way ANOVA with Holm-Sidak post-test, n = 6 (>131) cells for each group].
It was hypothesised that the ischemic protection afforded to cells via a 5 min enhanced glycolytic load would decline by increasing the time interval between the high glucose challenge and metabolic inhibition. To investigate this, various time intervals (2–20 min) between high glucose pre-treatment and metabolic insult were included in the metabolic inhibition protocol, as indicated in the top of Figure 4. There was a marked delay in the time to contractile failure, an increase in contractile recovery and cell survival following a 5 min high glucose challenge with a 2-, 5-, or 10-min gap between the pre-treatment and the metabolic inhibition, however, this protection was largely lost by 15 min and completely lost with a 20 min gap (Figure 4). These data suggest a time interval-dependent, high glucose induced, cardioprotection. In further figures in this study, a 10 min gap between high glucose challenge and metabolic inhibition was used, as the protection afforded was significantly greater than control conditions but not at the potential upper limit of the protection (as seen with a 2- or 5-min gap).
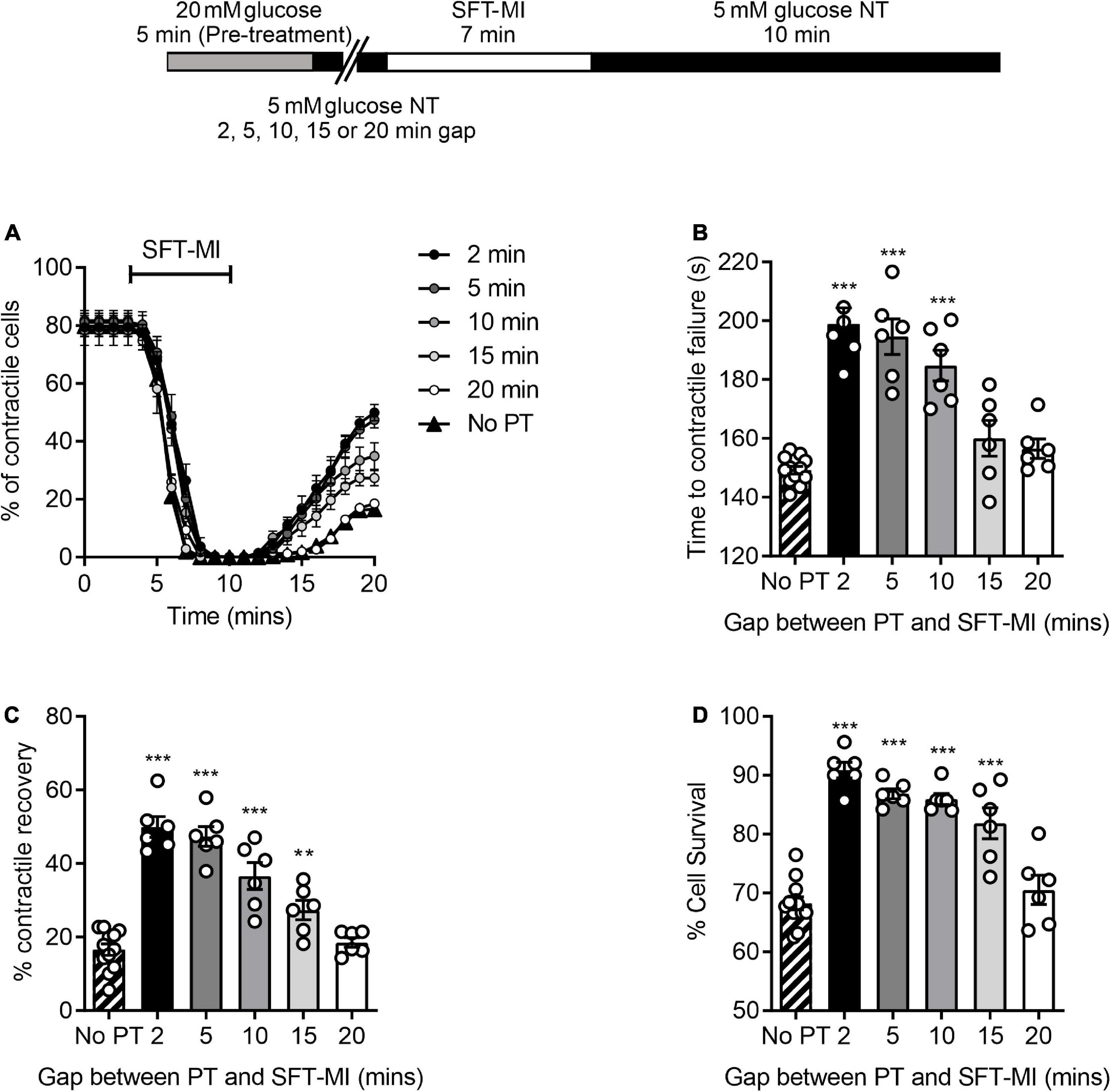
Figure 4. A 5 min pre-treatment with 20 mM glucose imparts a short-lived cardioprotection. (A) Time course showing the percentage of contractile cardiomyocytes throughout the metabolic inhibition protocol following a 5 min high glucose pre-treatment and a variable gap between the pre-treatment and metabolic insult. (B) Mean time to contractile failure, (C) mean percentage contractile recovery and (D) mean percentage of cell survival [**p < 0.01, ***p < 0.001, One-Way ANOVA with Holm-Sidak post-test. n = 6 experiments from six animals (>86 cells for each data set)].
To establish whether this protection was also triggered by alterations of glycolytic metabolism, the protocol was carried out with challenges with non-metabolised and metabolised sugars. A pre-treatment with 20 mM L-glucose, a non-metabolised enantiomer of D-glucose, showed no difference in the time to contractile failure, the contractile recovery or the cell survival compared to no pre-treatment control. This contrasted with the 20 mM D-glucose pre-treatment where there was a significant increase in all three measurements of cardioprotection (Figure 5). In addition, exchanging the 20 mM glucose pre-treatment for a 15 mM fructose with 5 mM glucose also imparted a cardioprotected-like phenotype, as did increasing pyruvate to 10 mM, with a significant increase in the time to contractile failure, increased contractile recovery and cell survival (Figure 5). These data suggest that enhanced glycolytic metabolism is responsible for the protective effect of a short period of high glucose pre-treatment.
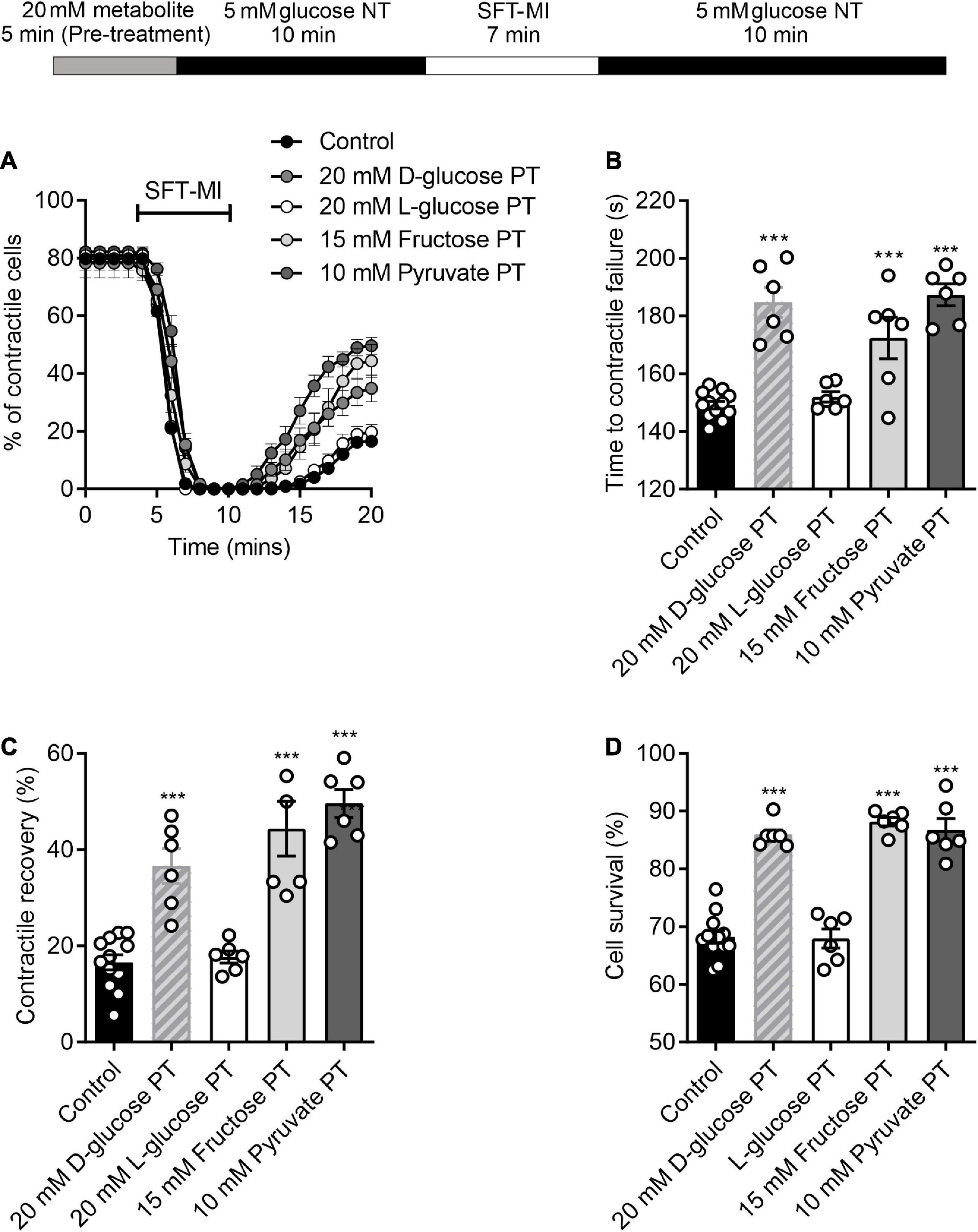
Figure 5. Protection afforded by high glucose pre-treatment can be mimicked with other glycolytic metabolites. (A) Example time course showing the percentage of contractile cells during the MI/R protocol following 5 min pre-treatment with 20 mM glucose, 15 mM fructose, 10 mM pyruvate, or 20 mM L-glucose. (B) Bar chart showing mean time to contractile failure. All pre-treatments show a significant delay in the time to contractile failure, except for L-glucose, which is a non-metabolised enantiomer [**P < 0.01, ***P < 0.001, One-Way ANOVA with Holm-Sidak post-test, n = 6 (>98 cells) for each group]. Bar charts showing (C) the percentage contractile recovery and (D) cell survival at the end of the MI/R protocol. In each case there is a significant increase in contractile recovery, except for the L-glucose pre-treated group [***P < 0.001, One-Way ANOVA with Holm-Sidak post-test, n = 6 (>102 cells) for each group].
Our previous reports show that cardioprotective stimuli delays ATP depletion and thereby delays the time to downstream activation of the sarcolemmal cardiac KATP current (25). Comparable to cardioprotective stimuli, a 5 min high glucose pre-treatment delayed the time to activation of the metabolic inhibition-activated whole-cell current (Figures 2A,B) and Kir6.2/SUR2A sarcolemmal KATP current, measured using cell-attached recording (Figures 2C,D), in keeping with a hypothesized delayed ATP depletion.
Additionally, cardioprotective stimuli reduce the accumulation of intracellular Ca2+ during metabolic inhibition, and into reperfusion (25, 35–38). Fluorescence imaging was used to determine whether elevation of intracellular Ca2+ during metabolic inhibition and reperfusion can be attenuated with high glucose pre-treatment used as a cardioprotective stimuli. Figure 2E shows example traces in control conditions and following high glucose pre-treatment, showing a significant reduction in the end Fura-2 ratio and the change in Fura-2 ratio following pre-treatment (Figure 2F). Similar to the contractile recovery data in Figure 5 and Supplementary Figure 1, the recovery of Ca2+ transients at the end of the recording period were also increased in 20 mM glucose pre-treated cardiomyocytes (Figure 2G). Combined, these data show that short high glucose challenges display characteristics of established cardioprotective stimuli such as IPC.
Our previous publications (14, 16, 17), and others (13, 39) all suggest a link to protein kinase C (PKC) in the damaging effects of elevated glucose in cardiomyocytes. In particular, we have shown that glucose activates PKCα and β to increase cellular excitability leading to aberrant Ca2+ release events and changes in action potential morphology (14) which causes more damage following an ischaemic challenge (17). The experiments outlined in Figure 1 showed that increasing durations of high glucose perfusion increased the arrhythmic events seen in cardiomyocytes. This pro-arrhythmicity, seemingly triggered by a high glucose challenge, was largely reversed following pre-treatment with either 300 nM Gö6976 (PKCα and β inhibitor) or 100 nM LY379196 (PKCβ inhibitor), further confirming our previous findings of an involvement of PKCα and β (Figures 6A–D) (14, 17).
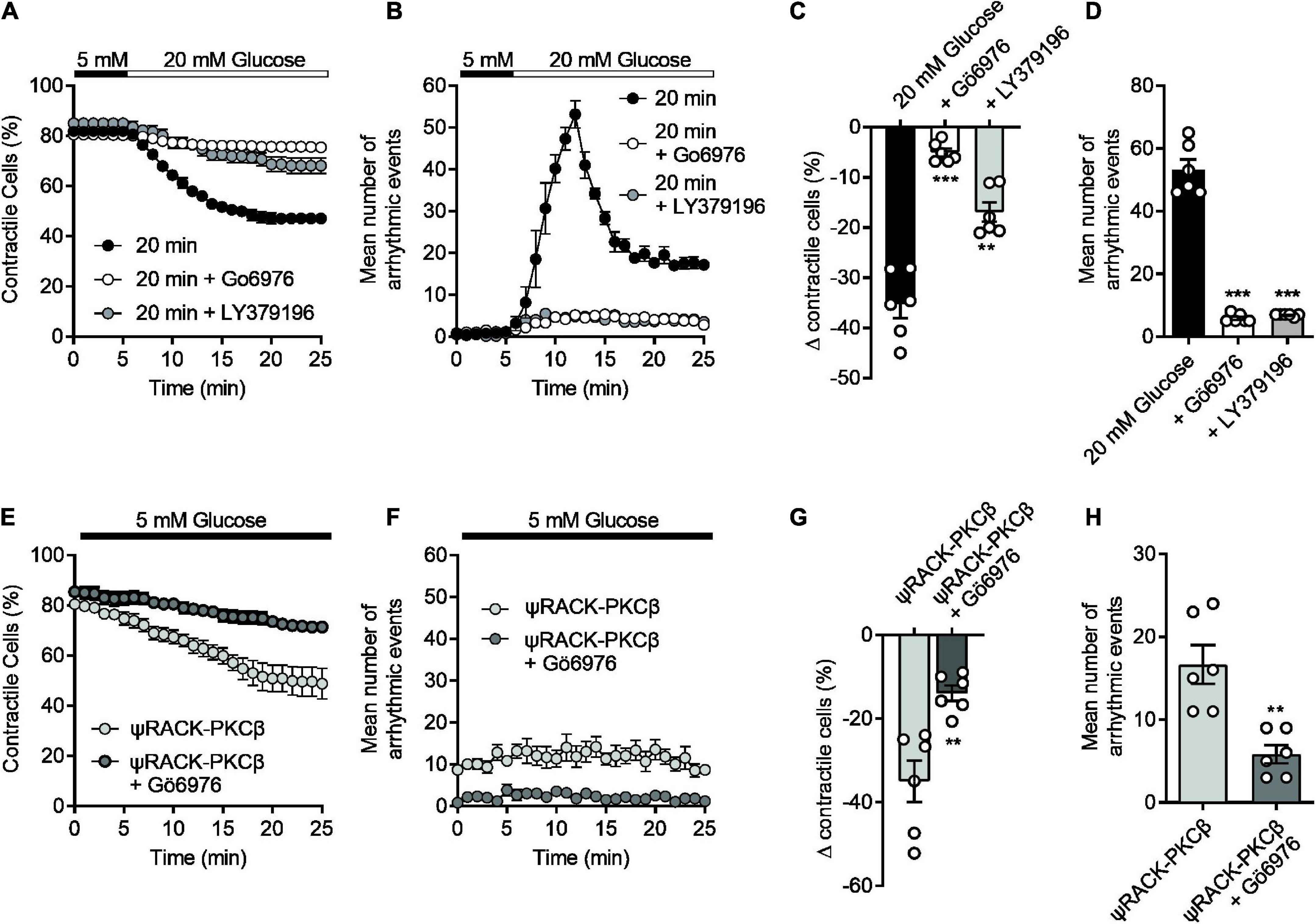
Figure 6. The high glucose-induced disruption of contractile function in isolated cardiomyocytes can be reversed with selective PKCα/β inhibition and mimicked with a PKCβ activator. (A) Time course showing the loss of contractile cardiomyocytes over a 20 min exposure to 20 mM glucose in the absence (black circles) and presence (white circles, Gö6976, grey circles, LY379196) of PKCα/β inhibitors. (B) Time course showing the number of contractions outside of the 1 Hz EFS in the cardiomyocytes recorded in panel (A). (C) The mean change in contractile cells at the end of the protocol and (D) the mean peak number of arrhythmic evens in each condition (***P < 0.001, **P < 0.01, One Way ANOVA with Holm-Sidak post-test, n = 6 experiments from six animals, >123 cells for each group). (E) Time course showing the loss of contractile function in cardiomyocytes pre-incubated for 15 min with Tat-ψRACK-PKCβ-activating peptide in the absence (light grey) and presence (dark grey) Gö6976 in 5 mM glucose. (F) Time course showing the number of contractions outside of the 1 Hz EFS in the cardiomyocytes recorded in panel (E). (G) The mean change in contractile cells at the end of the protocol and (H) the mean peak number of arrhythmic evens in each condition [**P < 0.0026 (G), **P < 0.0018 (H), unpaired t-test, n = 6 experiments from six animals, >105 cells for each group].
In cardiomyocytes pre-incubated with a cell permeant, tat-peptide linked, ψRACK-PKCβ-specific activating peptide, there was a similar increase in cardiac cell pro-arrhythmicity, in the absence of high glucose, that was reversed by treatment with Gö6976 (Figures 6E–H). Furthermore, use of the PKCβ activating peptide in metabolic inhibition experiments also reduced contractile recovery and cell survival analogous to treatment with high glucose, and was restored to control levels with selective PKCα/β inhibition (Supplementary Figure 2). These data suggest that we can mimic the damaging effects of a high glucose challenge with specific PKCβ activation.
Although there were no obvious deleterious effects of a 5 min pre-treatment with 20 mM glucose (Figures 1A,C), it is clear from the data in Figures 1, 3, and our previous publications (11, 14, 16, 17), that deleterious effects occur when pre-treatments longer than 7.5 min are used. Given our previous findings, various selective PKCαβ inhibitors were used in combination with a 5 min high glucose challenge to determine whether a larger degree of cardioprotection against metabolic inhibition could be observed. Either small-molecule inhibitors, Go6976 (PKCα and β, 300 nM), LY379196 (PKCβ, 100 nM), or cell permeant Tat-peptide linked PKCα and β inhibitor peptides (50 nM of each peptide) were included in the high glucose pre-treatment. The PKC inhibitors had no effect on the outcome of the MI/R protocol in the absence of a high glucose stimulus (Supplementary Figure 3). In all cases where PKCα and/or β were inhibited along with the high glucose pre-treatment, contractile recovery was increased compared to high glucose pre-treatment alone, indicative of greater cardioprotection (Figure 7). There was, however, no further delay in the time to contractile failure or further increase in the percentage of cells surviving the MI/R protocol (Figure 7). In keeping with our previous findings, selective PKCα/β inhibition did not affect the tested parameters in 5 mM glucose with no high glucose challenge (Figure 7).
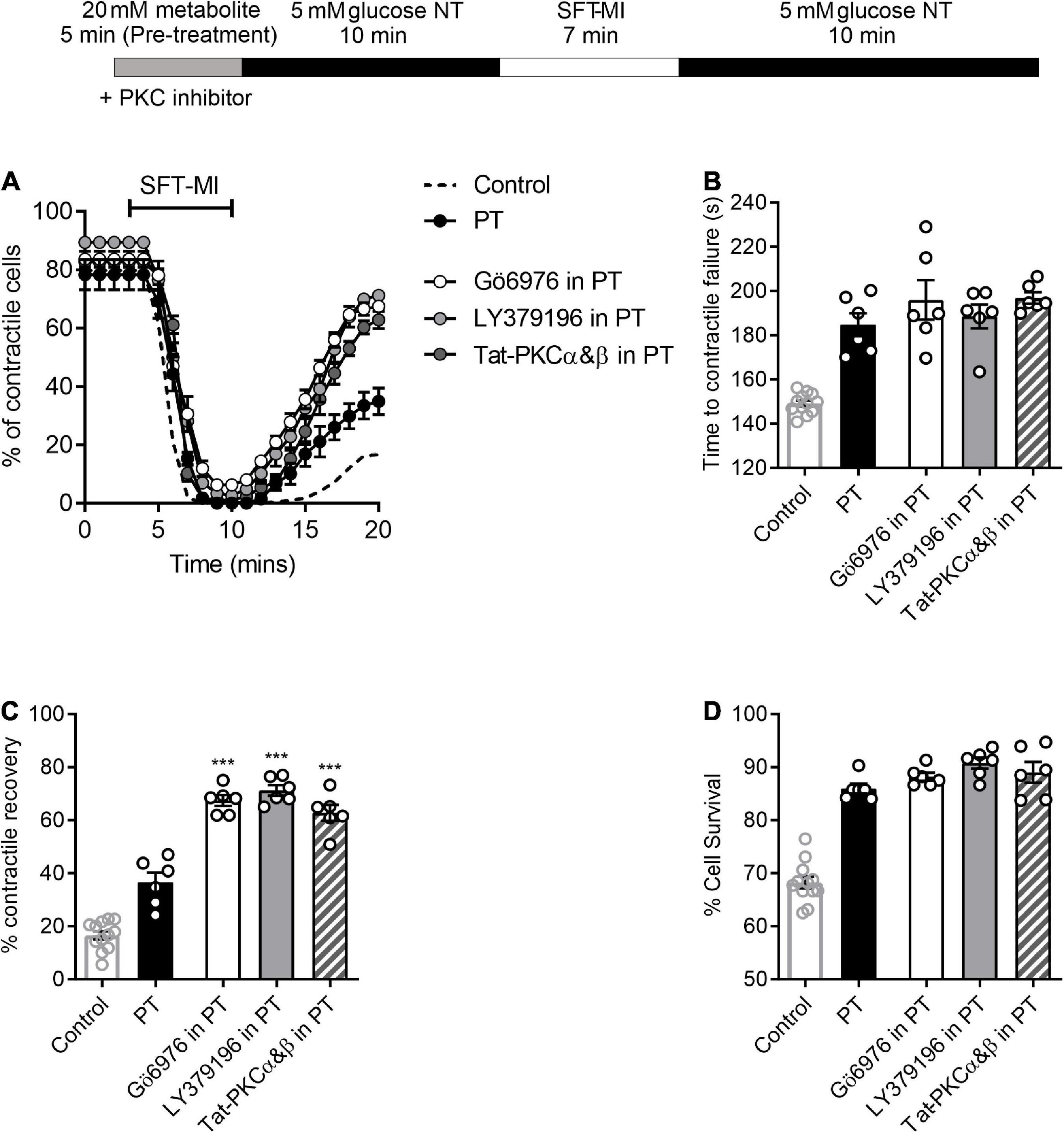
Figure 7. PKCα and/or β-selective inhibitors enhance the protected phenotype imparted by short high glucose pre-treatment. (A) Time course showing the percentage of contractile cardiomyocyte throughout the metabolic inhibition protocol following high glucose pre-treatment with or without PKC inhibitors, as indicated. (B) Bar chart showing the mean time to contractile failure, (C) mean percentage contractile recovery and (D) mean percentage of cell survival following metabolic inhibition in the absence and presence of selective PKC inhibition and with or without pre-treatment [***p < 0.001, One Way ANOVA with Holm-Sidak post-test. n = > 6 animals (>83 cells) for each data set. Each data set is compared to PT in the absence of PKC inhibitor. Control (no PT) data shown for comparison].
Protein kinase Cε is conventionally thought to be part of the cardioprotective pathway, where selective inhibition of this isoform removes protection afforded by many protective stimuli (25, 40, 41). 20 mM D-glucose-induced protection was unaffected by selective PKCε inhibition with a cell permeant Tat peptide-linked PKCε inhibitor peptide with either a 5- or 10-min gap between glucose and metabolic inhibition (Supplementary Figure 4). This peptide has previously been shown, in our hands, to abolish cardioprotection afforded by a number of stimuli (14, 25). These findings suggest that the short pre-treatment with 20 mM glucose was not causing protection by activation of PKCε, therefore making this distinct from other conventional cardioprotective interventions.
Having demonstrated that selective PKCα/β inhibition enhanced the protective effects of a short pre-treatment with high glucose in isolated cells, a similar protocol was used in the whole heart in a left anterior descending (LAD) coronary artery ligation model. A 5-min pre-treatment with glucose, with or without Gö6976, had no significant effect on the mean infarct size [34.1 ± 1.3, 31.5 ± 1.9, and 33.4 ± 3% in control (n = 11), 5 min pre-treatment (n = 4) and 5 min pre-treatment with 300 nM Gö6976 (n = 4)]. This may be because different durations of substrate perfusion are required for whole organ systems such as the heart compared to experiments on isolated cardiomyocytes, which allow almost instant changes in solution to be performed. The duration of the high glucose pre-treatment was increased to 15 min, which caused a detectable increase the infarct size [from 34.1 ± 1.3% in control conditions to 41 ± 2.3% (n = 6) following 15 min pre-treatment with high glucose (Figures 8A,B)]. Co-treatment with Gö6976, however, markedly reduced the infarct following high glucose challenge from 34.1 ± 1.3 to 15.6 ± 1.7%. This reduction in infarct size was not seen in hearts treated with Gö6976 in the absence of a high glucose pre-treatment (33.1 ± 2.3%, n = 4; Figure 8B). These data show that in whole-heart ex vivo experiments, a high glucose challenge for 15-min increased infarct size that was reversed with PKCα/β inhibition, further demonstrating a PKC-dependence to the modulation.
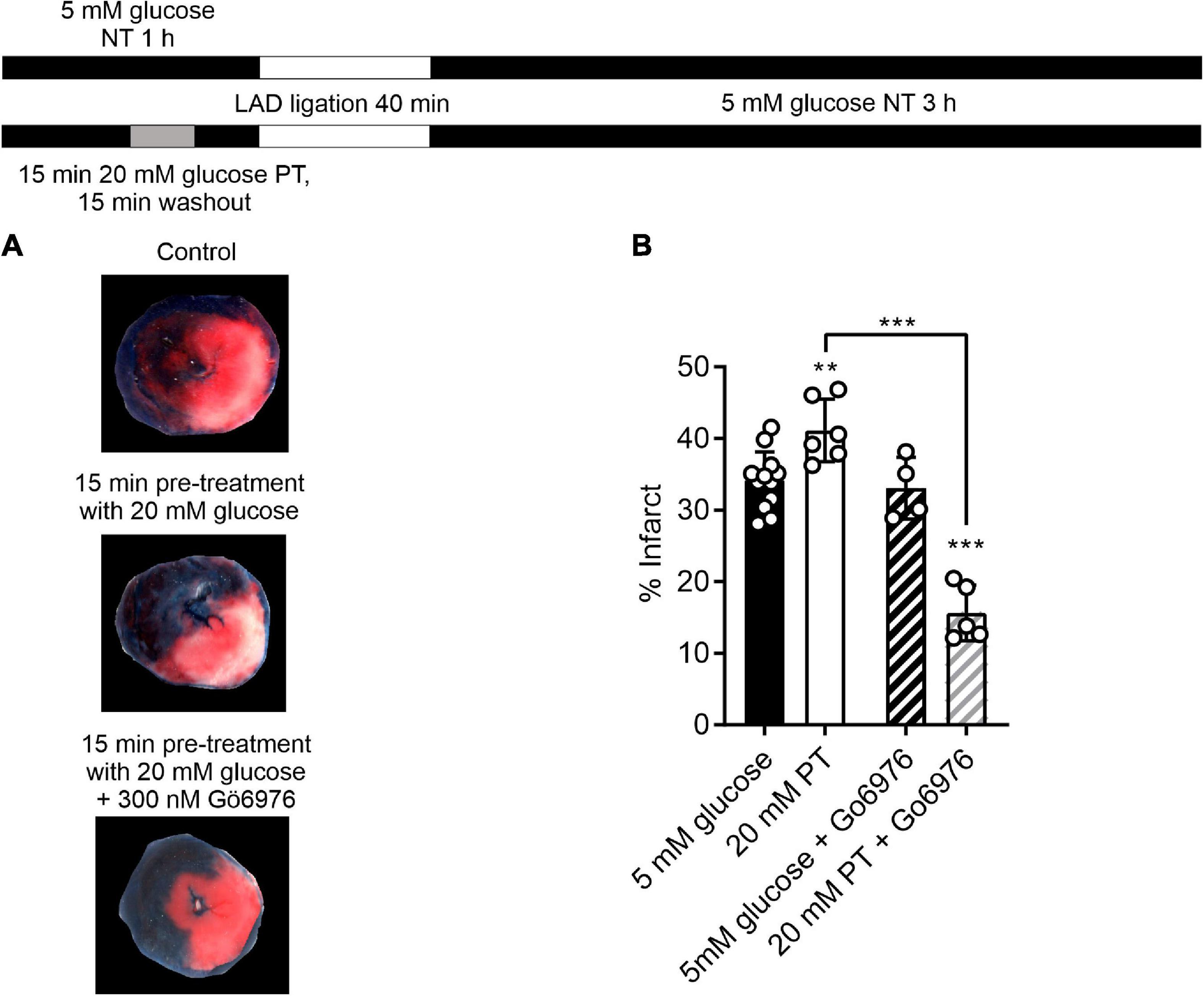
Figure 8. Selective inhibition of PKCα/β with Gö6976 reveals a 20 mM glucose pre-treatment dependent reduction in infarct size. (A) Example images from a control heart, heart following an amended protocol using a 15-min pre-treatment with 20 mM glucose and a heart following 15-min pre-treatment with 20 mM glucose in the presence of 300 nM Gö6976. The experimental protocol is shown above. (B) Bar chart showing the mean percentage infarct by weight of the different groups. Circles show data from individual hearts (**P < 0.01 and ***P < 0.001 Two-way ANOVA with Tukey’s post-test, n = 12, 6, 4, and 5 hearts for control, 20 mM PT, 5 mM glucose with Gö6976 and 20 mM PT with Gö6976 groups). There was no difference in the effectiveness of coronary blockade in the 4 groups measured as the percentage area at risk (41.9 ± 4.5, 43.2 ± 4.5, 39.4 ± 2.3, and 38.9 ± 2.6% (mean ± S.D., n = 12, 6, 4, and 5 hearts for control, 20 mM PT, 5 mM glucose with Gö6976 and 20 mM PT with Gö6976 groups, Two-way ANOVA).
This study demonstrates that a short 5-min challenge with increased extracellular metabolic substrate imparted a cardioprotective phenotype to cardiomyocytes that was short-lived in these experimental models but gave a protection that was as substantive as well-established methods of cardioprotection, such as ischaemic preconditioning (14, 25). The protection afforded by the elevated metabolic substrate was further enhanced by co-treatment with selective PKCα and/or β inhibition, similar to our previous findings (11, 14, 17, 25). These findings suggest that we should be giving consideration of the damage that glucose could be doing not just in terms of weeks, but rather over the duration of minutes.
Evidence supports this with Kir6.2 openings and whole cell current. Although short burst of glucose did not increase ATP levels in cells (Supplementary Figure 1), the time for Kir6.2 opening was delayed thereby demonstrating ATP was preserved for longer; a hallmark of established cardioprotective stimuli, such as IPC (25). We suggest that the 5 min pre-treatment used in the cellular section of this study was not long enough to fully activate the deleterious, PKCα/β-dependent cardiotoxicity.
This current report results demonstrates a critical time-dependent effect, whereby glucose is cardiotoxic to cardiomyocytes with greater than 7.5 min exposure but protective with less than 5 min, suggesting a temporal switch between protection and toxicity. Although more clinical research is needed to determine whether this role translates to the clinical scenario, it does fit with the clinical data reporting elevated admission glucose in ACS patients is associated with poor outcome. Such time-dependence does, however, offer a plausible explanation to the conflicting reports on the benefit of insulin infusion to reduce glucose in ACS patients (42–44), together with the conflicting reports of benefit with glucose-inulin-potassium infusion (18–21). It is plausible that the benefit of this therapy may be time-dependent and that the control of glycaemia in patients in more effective with an early intervention. A greater appreciation of this time-dependent effect is also required for basic research, where acute increases of extracellular glucose has been reported to be cardiotoxic to cardiomyocytes by our lab (14, 17), and others (13, 15, 45–51). Furthermore, we have also demonstrated this in whole heart ex vivo measurements of infarct size both in this and our previous manuscript (17).
The concentration of glucose used in this study, 5 mM as our “normoglycaemic”, is no unique amongst researchers, however, 11.1 mM is often used and 25 mM may be used in culture media. Although these elevated concentrations are used elsewhere to compensate for other metabolic substrates that might be found in vivo, our studies have previously shown that even 10 mM glucose gives rise to differences in cardiac cell function and response to conditioning stimuli (14). We see no detriment in cellular or whole heart recordings using 5 mM glucose as our normoglycaemic control, and advocate this concentration as an appropriate physiologically normal glucose (14, 17, 25).
The complex relationship between glucose, time and myocardial damage has also been reported with other metabolites. Non-obesogenic mice fed a high-fat diet exhibit increased susceptibility to myocardial reperfusion injury (49), however, switching back to a normal diet before cardiac insult reduced cardiac vulnerability (48). Ethanol has also been reported to have a direct cardioprotective effect on the I/R challenged myocardium (26), and similar to our glucose work described here, ethanol can prevent its own protection if it is not washed out or sufficiently metabolized prior to the onset of ischemia (27). Combined, these reports suggest that acute exposure to elevated levels of metabolite, somewhat paradoxically, result in greater damage during metabolic inhibition. Although the reasons behind this require further research, we suggest that, for glucose at least, the damage be in part due to the build-up of metabolites that are forced into diacylglycerol synthesis, which then cause aberrant activation of DAG-sensitive PKC isoforms (13, 52, 53).
A noticeable difference that we can report between the ethanol-dependent and glucose-dependent cardioprotection is the PKC isoforms that are involved. Both ethanol and arachidonic acid (AA) have been shown to activate PKCε (26, 46, 54, 55), activation of which is well established to impart cardioprotection (56, 57). However, the use of a Tat-PKCε inhibitor peptide did not alter the cardioprotective effect of glucose pre-treatment (Supplementary Figure 4). The use of selective PKCα/β inhibitors further enhanced the cardioprotection imparted by glucose pre-treatment, indicating PKCα/β activation within minutes can cause deleterious effect to cell function (Figure 7).
Previous experiments using acute hyperglycaemia in animal models has shown an increased infarct size and reduced functional recovery (14, 44, 58, 59), therefore it is difficult to imagine a scenario where clinicians would briefly introduce hyperglycaemia as a cardioprotective therapy. There is a growing body of clinical evidence that demonstrates that glucose, irrespective of diabetic status, is associated with poor outcome for patients with myocardial infarction (3, 8, 34, 51, 60, 61). This damaging outcome combined with the large proportion of patients that have elevated glucose, perhaps though stress-induced hyperglycaemia (3, 17, 62), outlines the need to prevent the harmful effects of glucose in ACS. What is compelling, and evidenced in our previous publications (14, 16, 17), is that acute elevations of glucose coupled with PKCαβ inhibition mask the damaging effects of glucose, and so reveals a glucose metabolism-dependent protective effect. This was evident in both isolated cell models, and in whole heart coronary ligation experiments. These findings raise the possibility that PKCα/β inhibition may provide a novel therapy to confer cardioprotection in ACS patients with hyperglycaemia, whilst preventing the risk of causing hypoglycaemia by glucose lowering interventions. Indeed, PKC inhibitors of the isoforms identified to play a role in glucose-induced cardiac dysregulation are already in use in clinical trials. The PKCβ inhibitor, Ruboxistaurin, an LY379196 analogue, is currently in phase I/II evaluation for treatment of heart failure (NCT02769611), however, is listed in this documentation as a PKCα inhibitor. In addition, various, pre-clinical studies have suggested PKCβ inhibition as a potential therapeutic target in heart failure via several mechanisms (63). Inhibition of PKCβ at the point of ischaemic damage may reduce the glucose-dependent exacerbation of this injury and also help attenuate the development of heart failure.
Findings that acute hyperglycaemia activates PKC to cause negative effects on cardiomyocyte function, may go in some part to explain this loss of efficacy in clinical trials compared to in vitro and in vivo animal model studies. Several groups have shown that cardioprotective stimuli fail to work in elevated glucose (50, 64–66). Our previous manuscripts in this area demonstrate that although cardioprotection can be abolished by elevated glucose, selective PKCα and β inhibition restored cardioprotection (14). PKCα/β inhibition therefore may provide a route for re-investigation of a number of failed therapeutics for imparting cardioprotection.
Our data in this study was investigated in rat cardiomyocytes and whole heart using an ex vivo coronary ligation model. Further research in this area is required to determine the therapeutic potential of glucose challenge combined with selective PKC inhibition. However, the number of studies reporting deleterious effects of glucose on cellular functions and the mapping of this to the in vivo and clinical scenario is providing potentially interesting new avenues to explore. In our manuscripts, we have taken care to describe the effects of an acute treatment with high glucose and have not discussed what could happen with chronic hyperglycaemia as might be seen in a diabetic patient. The potential involvement of advanced glycation end products (AGE), ROS sympathetic drive and the polyol pathway have not been investigated in this manuscript given that they are often associated with longer term changes in chronic hyperglycaemia (52, 53, 67). Further to this, it should be noted that the experiments in this study do not include any free fatty acids in the solutions, the preferred metabolic substrate in cardiac cells (68). In our previous studies, we demonstrate that PKC was being activated not just functionally, but also showing changes in cellular distribution in response to high glucose (14, 17), but the mechanism behind this is unclear. We hypothesise that it involves de novo synthesis of diacylglycerol from a backup of glycolytic enzymes (52, 53, 69). All of our data to date has been carried out in otherwise healthy animal tissue with no known comorbidities. This is unlike the clinical scenario where a patient presenting with an AMI would be unlikely to have no other potential confounding factors. What is clear from our and others data is that glucose demonstrates a time-dependent cardiotoxicity that may well underlie damage to cardiomyocyte integrity and function in the setting of ischaemia. This suggests that the clinical association between hyperglycaemia and adverse outcome after ACS may indeed be the result of direct glucose-toxicity rather than an epiphenomenon.
In summary, we have demonstrated in this manuscript that a short treatment/challenge with high glucose, or alternative metabolite, can impart a cardioprotected-like phenotype to both isolated cells and the whole heart. Such protection by elevated glucose requires the administration to be short restored to normal physiological levels. This protection is markedly improved by the co-administration of selective PKCα and β-selective inhibitors, which then has no time dependence to it, as the deleterious effects shown in our previous manuscripts are suppressed (14, 16, 17). Given that ∼48% of ACS patients showed a blood glucose greater than 8 mM (3, 17), indicating some degree of stress-hyperglycaemia, we suggest that selective PKCα/β inhibitors should be investigated as a potential cardioprotective alternative to aggressive lowering of blood glucose. Our findings suggest that this could turn a deleterious increase in glycaemia to a novel cardioprotective intervention.
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by University of animal welfare ethical review board (AWERB_2018_44).
SB: contributed to experimental design, acquired, analysed, interpreted the data, and drafted the manuscript. SE, MA, CM, and SM: acquired, analysed, interpreted the data, and helped with manuscript revision. MS: acquired, analysed, and interpreted the data. IS, PS, and AC: conception and design of the work, critically appraised, and revised the manuscript. RR: conception and design of the work, acquired, analysed, interpreted the data, critically appraised, and revised the manuscript. All authors approved the final version of manuscript, agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All work was carried out at the Universities of Liverpool or Leicester in RR’s laboratory.
This work was supported by the van Geest Cardiovascular Research Fund, University of Leicester (SB and RR), the British Heart Foundation (PG/16/14/32039 and PG/19/18/34280) (SB and RR) and Wellcome Trust Institutional Strategic Support Fund and University of Liverpool (SB).
We thank Lorenza Francescut for expert technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.997013/full#supplementary-material
1. Kosiborod M, Rathore SS, Inzucchi SE, Masoudi FA, Wang Y, Havranek EP, et al. Admission glucose and mortality in elderly patients hospitalized with acute myocardial infarction: implications for patients with and without recognized diabetes. Circulation. (2005) 111:3078–86. doi: 10.1161/CIRCULATIONAHA.104.517839
2. Zeller M, Verges B, L’Huillier I, Brun JM, Cottin Y. Glycemia in acute coronary syndromes. Diabetes Metab. (2006) 32:2S42–7. doi: 10.1016/s1262-3636(06)70485-2
3. Squire IB, Nelson CP, Ng LL, Jones DR, Woods KL, Lambert PC. Prognostic value of admission blood glucose concentration and diabetes diagnosis on survival after acute myocardial infarction: results from 4702 index cases in routine practice. Clin Sci (Lond). (2010) 118:527–35. doi: 10.1042/CS20090322
4. Lipton JA, Barendse RJ, Van Domburg RT, Schinkel AF, Boersma H, Simoons MI, et al. Hyperglycemia at admission and during hospital stay are independent risk factors for mortality in high risk cardiac patients admitted to an intensive cardiac care unit. Eur Heart J Acute Cardiovasc Care. (2013) 2:306–13. doi: 10.1177/2048872613489304
5. Suleiman M, Hammerman H, Boulos M, Kapeliovich MR, Suleiman A, Agmon Y, et al. Fasting glucose is an important independent risk factor for 30-day mortality in patients with acute myocardial infarction: a prospective study. Circulation. (2005) 111:754–60. doi: 10.1161/01.CIR.0000155235.48601.2A
6. Aronson D, Hammerman H, Suleiman M, Markiewicz W. Usefulness of changes in fasting glucose during hospitalization to predict long-term mortality in patients with acute myocardial infarction. Am J Cardiol. (2009) 104:1013–7. doi: 10.1016/j.amjcard.2009.05.053
7. O’Gara PT, Kushner FG, Ascheim DD, Casey DE Jr, Chung MK, de Lemos JA, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American college of cardiology foundation/American heart association task force on practice guidelines. J Am Coll Cardiol. (2013) 61:e78–140. doi: 10.1016/j.jacc.2012.11.019
8. Steg PG, James SK, Atar D, Badano LP, Blomstrom-Lundqvist C, Borger MA, et al. ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J. (2012) 33:2569–619. doi: 10.1093/eurheartj/ehs215
9. Corbett SJ. NICE recommendations for the management of hyperglycaemia in acute coronary syndrome. Heart. (2012) 98:1189–91. doi: 10.1136/heartjnl-2012-302421
10. Shizukuda Y, Reyland ME, Buttrick PM. Protein kinase C-delta modulates apoptosis induced by hyperglycemia in adult ventricular myocytes. Am J Physiol Heart Circ Physiol. (2002) 282:H1625–34. doi: 10.1152/ajpheart.00783.2001
11. Rainbow RD, Hardy ME, Standen NB, Davies NW. Glucose reduces endothelin inhibition of voltage-gated potassium channels in rat arterial smooth muscle cells. J Physiol. (2006) 575(Pt 3):833–44. doi: 10.1113/jphysiol.2006.114009
12. Straub SV, Girouard H, Doetsch PE, Hannah RM, Wilkerson MK, Nelson MT. Regulation of intracerebral arteriolar tone by K(v) channels: effects of glucose and PKC. Am J Physiol Cell Physiol. (2009) 297:C788–96. doi: 10.1152/ajpcell.00148.2009
13. Wang Z, Zhang Y, Guo J, Jin K, Li J, Guo X, et al. Inhibition of protein kinase C betaII isoform rescues glucose toxicity-induced cardiomyocyte contractile dysfunction: role of mitochondria. Life Sci. (2013) 93:116–24. doi: 10.1016/j.lfs.2013.06.002
14. Sims MW, Winter J, Brennan S, Norman RI, Ng GA, Squire IB, et al. PKC-mediated toxicity of elevated glucose concentration on cardiomyocyte function. Am J Physiol Heart Circ Physiol. (2014) 307:H587–97. doi: 10.1152/ajpheart.00894.2013
15. Mapanga RF, Essop MF. Damaging effects of hyperglycemia on cardiovascular function: spotlight on glucose metabolic pathways. Am J Physiol Heart Circ Physiol. (2016) 310:H153–73. doi: 10.1152/ajpheart.00206.2015
16. Jackson R, Brennan S, Fielding P, Sims MW, Challiss RA, Adlam D, et al. Distinct and complementary roles for alpha and beta isoenzymes of PKC in mediating vasoconstrictor responses to acutely elevated glucose. Br J Pharmacol. (2016) 173:870–87. doi: 10.1111/bph.13399
17. Brennan S, Chen S, Makwana S, Martin CA, Sims MW, Alonazi ASA, et al. A novel form of glycolytic metabolism-dependent cardioprotection revealed by PKCalpha and beta inhibition. J Physiol. (2019) 597:4481–501. doi: 10.1113/JP278332
18. Ritsinger V, Malmberg K, Martensson A, Ryden L, Wedel H, Norhammar A. Intensified insulin-based glycaemic control after myocardial infarction: mortality during 20 year follow-up of the randomised diabetes mellitus insulin glucose infusion in acute myocardial infarction (DIGAMI 1) trial. Lancet Diabetes Endocrinol. (2014) 2:627–33. doi: 10.1016/S2213-8587(14)70088-9
19. Mellbin LG, Malmberg K, Norhammar A, Wedel H, Ryden L, Investigators D. The impact of glucose lowering treatment on long-term prognosis in patients with type 2 diabetes and myocardial infarction: a report from the DIGAMI 2 trial. Eur Heart J. (2008) 29:166–76. doi: 10.1093/eurheartj/ehm518
20. van der Horst IC, Ottervanger JP, van’t Hof AW, Reiffers S, Miedema K, Hoorntje JC, et al. The impact of glucose-insulin-potassium infusion in acute myocardial infarction on infarct size and left ventricular ejection fraction [ISRCTN56720616]. BMC Med. (2005) 3:9. doi: 10.1186/1741-7015-3-9
21. Jonassen AK, Aasum E, Riemersma RA, Mjos OD, Larsen TS. Glucose-insulin-potassium reduces infarct size when administered during reperfusion. Cardiovasc Drugs Ther. (2000) 14:615–23. doi: 10.1023/a:1007802630604
22. Licker M, Reynaud T, Garofano N, Sologashvili T, Diaper J, Ellenberger C. Pretreatment with glucose-insulin-potassium improves ventricular performances after coronary artery bypass surgery: a randomized controlled trial. J Clin Monit Comput. (2020) 34:29–40. doi: 10.1007/s10877-019-00280-5
23. Ellenberger C, Sologashvili T, Kreienbuhl L, Cikirikcioglu M, Diaper J, Licker M. Myocardial protection by glucose-insulin-potassium in moderate- to high-risk patients undergoing elective on-pump cardiac surgery: a randomized controlled trial. Anesth Analg. (2018) 126:1133–41. doi: 10.1213/ANE.0000000000002777
24. Zhao K, Zhang Y, Li J, Cui Q, Zhao R, Chen W, et al. Modified glucose-insulin-potassium regimen provides cardioprotection with improved tissue perfusion in patients undergoing cardiopulmonary bypass surgery. J Am Heart Assoc. (2020) 9:e012376. doi: 10.1161/JAHA.119.012376
25. Brennan S, Jackson R, Patel M, Sims MW, Hudman D, Norman RI, et al. Early opening of sarcolemmal ATP-sensitive potassium channels is not a key step in PKC-mediated cardioprotection. J Mol Cell Cardiol. (2015) 79:42–53. doi: 10.1016/j.yjmcc.2014.10.016
26. Churchill EN, Disatnik MH, Budas GR, Mochly-Rosen D. Ethanol for cardiac ischemia: the role of protein kinase C. Ther Adv Cardiovasc Dis. (2008) 2:469–83. doi: 10.1177/1753944708094735
27. Krenz M, Baines CP, Heusch G, Downey JM, Cohen MV. Acute alcohol-induced protection against infarction in rabbit hearts: differences from and similarities to ischemic preconditioning. J Mol Cell Cardiol. (2001) 33:2015–22. doi: 10.1006/jmcc.2001.1465
28. Percie du Sert N, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, et al. Reporting animal research: explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. (2020) 18:e3000411. doi: 10.1371/journal.pbio.3000411
29. Woodier J, Rainbow RD, Stewart AJ, Pitt SJ. Intracellular zinc modulates cardiac ryanodine receptor-mediated calcium release. J Biol Chem. (2015) 290:17599–610. doi: 10.1074/jbc.M115.661280
30. Kamalian L, Chadwick AE, Bayliss M, French NS, Monshouwer M, Snoeys J, et al. The utility of HepG2 cells to identify direct mitochondrial dysfunction in the absence of cell death. Toxicol Vitro. (2015) 29:732–40. doi: 10.1016/j.tiv.2015.02.011
31. Rainbow RD, Norman RI, Everitt DE, Brignell JL, Davies NW, Standen NB. Endothelin-I and angiotensin II inhibit arterial voltage-gated K+ channels through different protein kinase C isoenzymes. Cardiovasc Res. (2009) 83:493–500. doi: 10.1093/cvr/cvp143
32. Deedwania P, Kosiborod M, Barrett E, Ceriello A, Isley W, Mazzone T, et al. Hyperglycemia and acute coronary syndrome: a scientific statement from the American heart association diabetes committee of the council on nutrition, physical activity, and metabolism. Circulation. (2008) 117:1610–9. doi: 10.1161/CIRCULATIONAHA.107.188629
33. Kosiborod M, Deedwania P. An overview of glycemic control in the coronary care unit with recommendations for clinical management. J Diabetes Sci Technol. (2009) 3:1342–51. doi: 10.1177/193229680900300614
34. Gardner LS, Nguyen-Pham S, Greenslade JH, Parsonage W, D’Emden M, Than M, et al. Admission glycaemia and its association with acute coronary syndrome in emergency department patients with chest pain. Emerg Med J. (2015) 32:608–12. doi: 10.1136/emermed-2014-204046
35. Hudman D, Standen NB. Protection from the effects of metabolic inhibition and reperfusion in contracting isolated ventricular myocytes via protein kinase C activation. J Mol Cell Cardiol. (2004) 37:579–91. doi: 10.1016/j.yjmcc.2004.05.019
36. Rodrigo GC, Standen NB. Role of mitochondrial re-energization and Ca2+ influx in reperfusion injury of metabolically inhibited cardiac myocytes. Cardiovasc Res. (2005) 67:291–300. doi: 10.1016/j.cardiores.2005.03.015
37. Rodrigo GC, Samani NJ. Ischemic preconditioning of the whole heart confers protection on subsequently isolated ventricular myocytes. Am J Physiol Heart Circ Physiol. (2008) 294:H524–31. doi: 10.1152/ajpheart.00980.2007
38. Ertracht O, Malka A, Atar S, Binah O. The mitochondria as a target for cardioprotection in acute myocardial ischemia. Pharmacol Ther. (2014) 142:33–40. doi: 10.1016/j.pharmthera.2013.11.003
39. Nishikawa T, Edelstein D, Brownlee M. The missing link: a single unifying mechanism for diabetic complications. Kidney Int Suppl (2000) 77:S26–30. doi: 10.1046/j.1523-1755.2000.07705.x
40. Inagaki K, Churchill E, Mochly-Rosen D. Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res. (2006) 70:222–30. doi: 10.1016/j.cardiores.2006.02.015
41. Budas GR, Churchill EN, Mochly-Rosen D. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacol Res. (2007) 55:523–36. doi: 10.1016/j.phrs.2007.04.005
42. Weston C, Walker L, Birkhead J, National Audit of Myocardial Infarction Project, National Institute for Clinical Outcomes Research. Early impact of insulin treatment on mortality for hyperglycaemic patients without known diabetes who present with an acute coronary syndrome. Heart. (2007) 93:1542–6. doi: 10.1136/hrt.2006.108696
43. Chakrabarti AK, Singh P, Gopalakrishnan L, Kumar V, Elizabeth Doherty M, Abueg C, et al. Admission hyperglycemia and acute myocardial infarction: outcomes and potential therapies for diabetics and nondiabetics. Cardiol Res Pract. (2012) 2012:704314. doi: 10.1155/2012/704314
44. Yu Q, Zhou N, Nan Y, Zhang L, Li Y, Hao X, et al. Effective glycaemic control critically determines insulin cardioprotection against ischaemia/reperfusion injury in anaesthetized dogs. Cardiovasc Res. (2014) 103:238–47. doi: 10.1093/cvr/cvu132
45. Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. (2008) 57:696–705. doi: 10.2337/db07-1098
46. Churchill E, Budas G, Vallentin A, Koyanagi T, Mochly-Rosen D. PKC isozymes in chronic cardiac disease: possible therapeutic targets? Annu Rev Pharmacol Toxicol. (2008) 48:569–99. doi: 10.1146/annurev.pharmtox.48.121806.154902
47. Hreiche R, Plante I, David LP, Simard C, Turgeon J, Drolet B. Impact of glucose concentration on cardiac ventricular repolarization under I Kr/I Ks blocking agents. J Mol Cell Cardiol. (2009) 47:210–20. doi: 10.1016/j.yjmcc.2009.02.004
48. Littlejohns B, Lin H, Angelini GD, Halestrap AP, Suleiman MS. Switching back to normal diet following high-fat diet feeding reduces cardiac vulnerability to ischaemia and reperfusion injury. Cell Physiol Biochem. (2014) 34:1090–100. doi: 10.1159/000366323
49. Littlejohns B, Pasdois P, Duggan S, Bond AR, Heesom K, Jackson CL, et al. Hearts from mice fed a non-obesogenic high-fat diet exhibit changes in their oxidative state, calcium and mitochondria in parallel with increased susceptibility to reperfusion injury. PLoS One. (2014) 9:e100579. doi: 10.1371/journal.pone.0100579
50. Mapanga RF, Joseph D, Symington B, Garson KL, Kimar C, Kelly-Laubscher R, et al. Detrimental effects of acute hyperglycaemia on the rat heart. Acta Physiol (Oxf). (2014) 210:546–64. doi: 10.1111/apha.12184
51. Al Jumaily T, Rose’Meyer RB, Sweeny A, Jayasinghe R. Cardiac damage associated with stress hyperglycaemia and acute coronary syndrome changes according to level of presenting blood glucose. Int J Cardiol. (2015) 196:16–21. doi: 10.1016/j.ijcard.2015.05.143
52. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. (2001) 414:813–20. doi: 10.1038/414813a
53. Shah MS, Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res. (2016) 118:1808–29. doi: 10.1161/CIRCRESAHA.116.306923
54. Huang XP, Pi Y, Lokuta AJ, Greaser ML, Walker JW. Arachidonic acid stimulates protein kinase C-epsilon redistribution in heart cells. J Cell Sci. (1997) 110(Pt 14):1625–34. doi: 10.1242/jcs.110.14.1625
55. Mackay K, Mochly-Rosen D. Arachidonic acid protects neonatal rat cardiac myocytes from ischaemic injury through epsilon protein kinase C. Cardiovasc Res. (2001) 50:65–74. doi: 10.1016/s0008-6363(00)00322-9
56. Duquesnes N, Lezoualc’h F, Crozatier B. PKC-delta and PKC-epsilon: foes of the same family or strangers? J Mol Cell Cardiol. (2011) 51:665–73. doi: 10.1016/j.yjmcc.2011.07.013
57. Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov. (2012) 11:937–57. doi: 10.1038/nrd3871
58. Kersten JR, Schmeling TJ, Orth KG, Pagel PS, Warltier DC. Acute hyperglycemia abolishes ischemic preconditioning in vivo. Am J Physiol. (1998) 275:H721–5. doi: 10.1152/ajpheart.1998.275.2.H721
59. Yang Z, Tian Y, Liu Y, Hennessy S, Kron IL, French BA. Acute hyperglycemia abolishes ischemic preconditioning by inhibiting Akt phosphorylation: normalizing blood glucose before ischemia restores ischemic preconditioning. Oxid Med Cell Longev. (2013) 2013:329183. doi: 10.1155/2013/329183
60. Timmer JR, Hoekstra M, Nijsten MW, van der Horst IC, Ottervanger JP, Slingerland RJ, et al. Prognostic value of admission glycosylated hemoglobin and glucose in nondiabetic patients with ST-segment-elevation myocardial infarction treated with percutaneous coronary intervention. Circulation. (2011) 124:704–11. doi: 10.1161/CIRCULATIONAHA.110.985911
61. Fujino M, Ishihara M, Honda S, Kawakami S, Yamane T, Nagai T, et al. Impact of acute and chronic hyperglycemia on in-hospital outcomes of patients with acute myocardial infarction. Am J Cardiol. (2014) 114:1789–93. doi: 10.1016/j.amjcard.2014.09.015
62. Dungan KM, Braithwaite SS, Preiser JC. Stress hyperglycaemia. Lancet. (2009) 373:1798–807. doi: 10.1016/S0140-6736(09)60553-5
63. Ferreira JC, Brum PC, Mochly-Rosen D. betaIIPKC and epsilonPKC isozymes as potential pharmacological targets in cardiac hypertrophy and heart failure. J Mol Cell Cardiol. (2011) 51:479–84. doi: 10.1016/j.yjmcc.2010.10.020
64. Raphael J, Gozal Y, Navot N, Zuo Z. Hyperglycemia inhibits anesthetic-induced postconditioning in the rabbit heart via modulation of phosphatidylinositol-3-kinase/Akt and endothelial nitric oxide synthase signaling. J Cardiovasc Pharmacol. (2010) 55:348–57. doi: 10.1097/FJC.0b013e3181d26583
65. Kottenberg E, Thielmann M, Kleinbongard P, Frey UH, Heine T, Jakob H, et al. Myocardial protection by remote ischaemic pre-conditioning is abolished in sulphonylurea-treated diabetics undergoing coronary revascularisation. Acta Anaesthesiol Scand. (2014) 58:453–62. doi: 10.1111/aas.12278
66. Baranyai T, Nagy CT, Koncsos G, Onodi Z, Karolyi-Szabo M, Makkos A, et al. Acute hyperglycemia abolishes cardioprotection by remote ischemic perconditioning. Cardiovasc Diabetol. (2015) 14:151. doi: 10.1186/s12933-015-0313-1
67. Vlassara H, Uribarri J. Advanced glycation end products (AGE) and diabetes: cause, effect, or both? Curr Diab Rep. (2014) 14:453. doi: 10.1007/s11892-013-0453-1
68. Hendrickson SC, St Louis JD, Lowe JE, Abdel-aleem S. Free fatty acid metabolism during myocardial ischemia and reperfusion. Mol Cell Biochem. (1997) 166:85–94. doi: 10.1023/a:1006886601825
Keywords: glucose, cardiotoxicity, cardioprotection, hyperglycaemia, protein kinase C (PKC), time-dependent
Citation: Brennan S, Esposito S, Abdelaziz MIM, Martin CA, Makwana S, Sims MW, Squire IB, Sharma P, Chadwick AE and Rainbow RD (2022) Selective protein kinase C inhibition switches time-dependent glucose cardiotoxicity to cardioprotection. Front. Cardiovasc. Med. 9:997013. doi: 10.3389/fcvm.2022.997013
Received: 18 July 2022; Accepted: 15 August 2022;
Published: 07 September 2022.
Edited by:
Dongdong Sun, Fourth Military Medical University, ChinaReviewed by:
Sapna Arjun, University College London, United KingdomCopyright © 2022 Brennan, Esposito, Abdelaziz, Martin, Makwana, Sims, Squire, Sharma, Chadwick and Rainbow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sean Brennan, c2Vhbi5icmVubmFuQGxpdmVycG9vbC5hYy51aw==; Richard D. Rainbow, cmljaGFyZC5yYWluYm93QGxpdmVycG9vbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.