 Yan Zhong Liu
Yan Zhong Liu Zong Xiang Li†
Zong Xiang Li† Lin Lin Zhang
Lin Lin Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 12 October 2022
Sec. Atherosclerosis and Vascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.972836
This article is part of the Research Topic Metabolic Alterations in Cardiopulmonary Vascular Dysfunction View all 8 articles
Vascular calcification (VC) is an important hallmark of cardiovascular disease, the osteo-/chondrocyte phenotype differentiation of vascular smooth muscle cells (VSMCs) is the main cause of vascular calcification. Accumulating evidence shows that mitochondrial dysfunction may ultimately be more detrimental in the VSMCs calcification. Mitochondrial participate in essential cellular functions, including energy production, metabolism, redox homeostasis regulation, intracellular calcium homeostasis, apoptosis, and signal transduction. Mitochondrial dysfunction under pathological conditions results in mitochondrial reactive oxygen species (ROS) generation and metabolic disorders, which further lead to abnormal phenotypic differentiation of VSMCs. In this review, we summarize existing studies targeting mitochondria as a treatment for VC, and focus on VSMCs, highlighting recent progress in determining the roles of mitochondrial processes in regulating the phenotype transition of VSMCs, including mitochondrial biogenesis, mitochondrial dynamics, mitophagy, mitochondrial energy metabolism, and mitochondria/ER interactions. Along these lines, the impact of mitochondrial homeostasis on VC is discussed.
Vascular calcification (VC), the ectopic deposition of calcium (Ca2+) apatite within the vascular wall, is prevalent in chronic kidney disease and diabetes mellitus and contributes to subsequent cardiovascular morbidity and mortality (1). Calcification occurs in both the intimal and medial layers of the arteries. Intimal calcification is linked to arterial obstruction and atherosclerotic plaque rupture. In contrast, medial calcification is linked to vessel stiffness, systolic hypertension, and increased pulse wave velocity leading to increased diastolic dysfunction and heart failure (2). Several meta-analyses have shown that aortic and coronary artery calcification predicts overall mortality in high-risk populations and leads to a multifold increase in cardiovascular and cerebrovascular mortality (3–5).
Calcification of the vascular medial layer, initially thought to be a passive process is actually an active, tightly regulated process driven primarily by vascular smooth muscle cells (VSMCs). VSMCs play an indispensable role in the phenotypic transition of chondroblasts and osteoblasts and lead to VC (6). The molecular mechanism underlying phenotypic transition of VSMCs is complex, mainly including elevated intracellular Ca2+ concentration (7), oxidative stress (8), and metabolic reprogramming (9). However, the regulatory mechanisms among these factors remain largely elusive. Notably, growing evidence indicates a vital role of mitochondrial in phenotypic transition of VSMCs (10–13). Mitochondrial constantly integrate signals from environment and respond accordingly to match vascular function to metabolic requirements of the organ tissue, and play a key role in energy production, redox homeostasis, Ca2+ homeostasis, and signal transduction, while mitochondrial dysfunction contributes to altered metabolism, oxidative stress, and increased apoptosis in VSMCs (14). Regardless of apoptosis or necrosis, cellular components such as Ca2+ and DNA are released after cell death, resulting in the deposition of Ca2+ and phosphorus, which may lead to VC (2). Following recent new insight in the mitochondrial function of VSMCs in VC, we believe it is time to focus on the complex role of mitochondrial homeostasis in phenotypic transition of VSMCs. In this review, we discuss the distinct phenotypes that VSMCs acquire in VC, and highlight the role of mitochondria in maintaining VSMCs function.
Vascular smooth muscle cells are the predominant cell type of blood vessels, alternating with elastic fibrous layers to form the medial layer of vascular, they can produce contraction and relaxation responses through the interaction of myosin and actin, and regulate blood pressure and blood flow distribution in various parts of the body. Unlike terminally differentiated cells such as cardiomyocytes and skeletal muscle cells, even mature VSMCs exhibit a high degree of phenotypic plasticity. VSMCs showed no significant proliferation, migration, and secretion of extracellular matrix under healthy physiological conditions, known as differentiated and contractile phenotype. In response to various pathological stimuli and biochemical regulations such as mechanical forces, vasoactive agents, changes in pyrophosphate levels, inflammatory mediators, and oxidative stress, VSMCs can re-enter the cell cycle and switch from a differentiated and contractile phenotype to dedifferentiated and synthetic phenotype, characterized by a reduction in marker of a range of cytoskeletal proteins such as smooth muscle myosin heavy chain (SM-MHC), α-SMA, and smooth muscle 22α (SM-22α). Previously, a binary model of VSMCs phenotype switching was established whereby transforming growth factor-β (TGF-β) stimulation promoted a quiescent “contractile” VSMCs phenotype, whereas platelet-derived growth factor-BB (PDGF-BB) or Angiotensin I (Ang II) stimulation triggered to the adoption of a pro-migratory, hyper-proliferative “synthetic” phenotype, which contributed to vascular diseases (15). However, the features of the contractile phenotype of VSMCs were evident, while the non-contractile phenotype displayed multiple characteristics. More recently it has become clear that under specific pathological conditions, VSMCs can maintain a spectrum of phenotypes and can express feature of osteoblasts, chondrocytes (6), macrophage-like (16, 17), foam cells (18), adipocyte-like (19), fibroblast-like (20), and pluripotent vascular stem cells (21). This plasticity of the VSMCs phenotype is associated with various vascular diseases including VC, atherosclerosis, and hypertension (Figure 1). The phenotype of VSMCs in VC was found to switch toward an osteo/chondrocyte phenotype, which is characterized by decrease in contractile protein expression and upregulation of mineralization-related markers such as runt-related transcription factor 2 (RUNX2), osterix (OSX), osteopontin (OPN), osteocalcin (OX), alkaline phosphatase (ALP), collagen type II, and type χ (22). Although the phenotypic transformation of VSMCs is thought to predate the development of vascular diseases including VC, the molecular mechanisms leading to the loss of the contractile phenotype and the upregulation of markers of the osteo/chondrocyte phenotype are at present not fully understood. There have been several recent studies assessing the beneficial role of mitochondria in maintaining redox, Ca2+ homeostasis and metabolic regulation (Table 1). Thus, targeting mitochondria to regulate VSMCs phenotypic transition provides a potential therapeutic direction for alleviating VC.
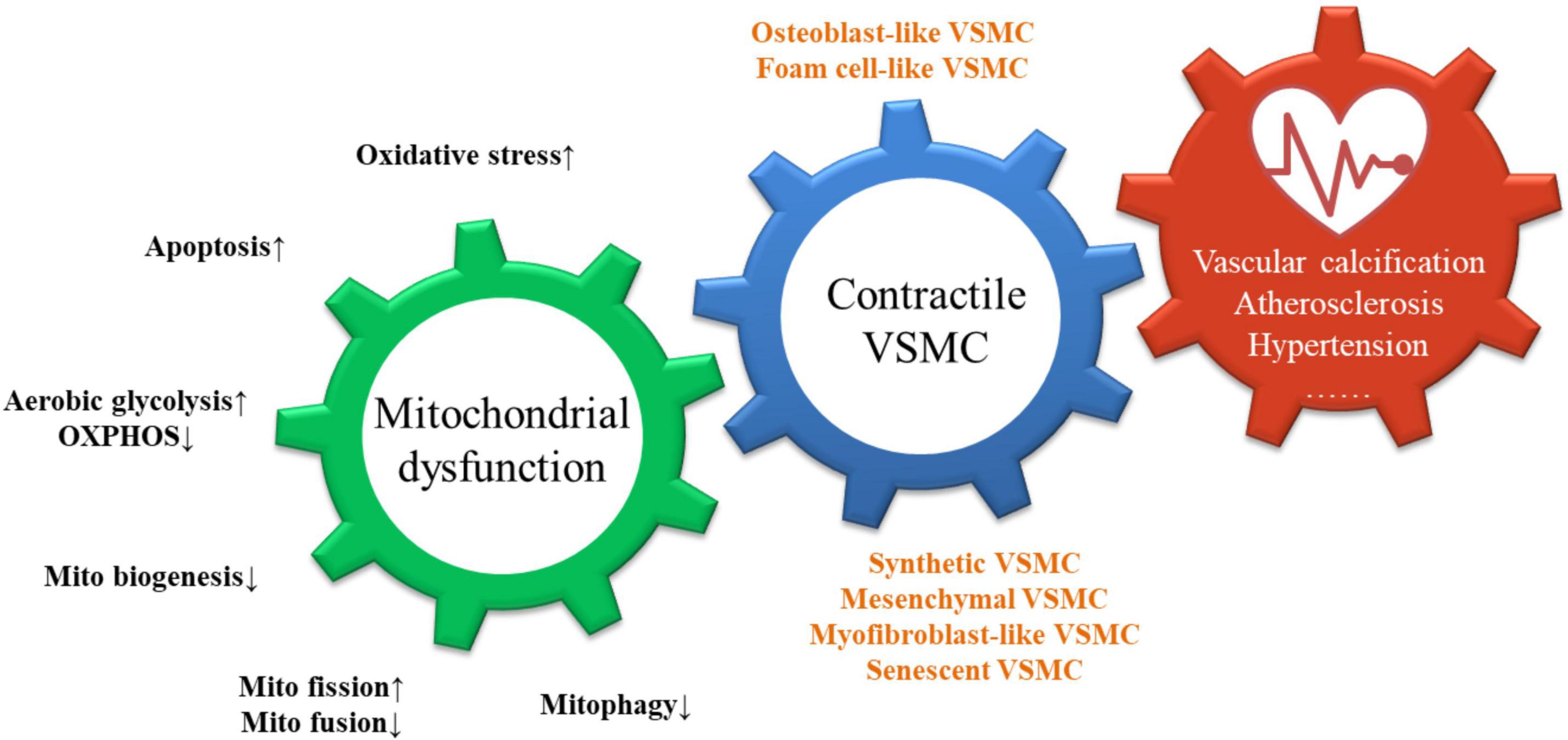
Figure 1. Consequences of mitochondrial dysfunction in VSMCs.
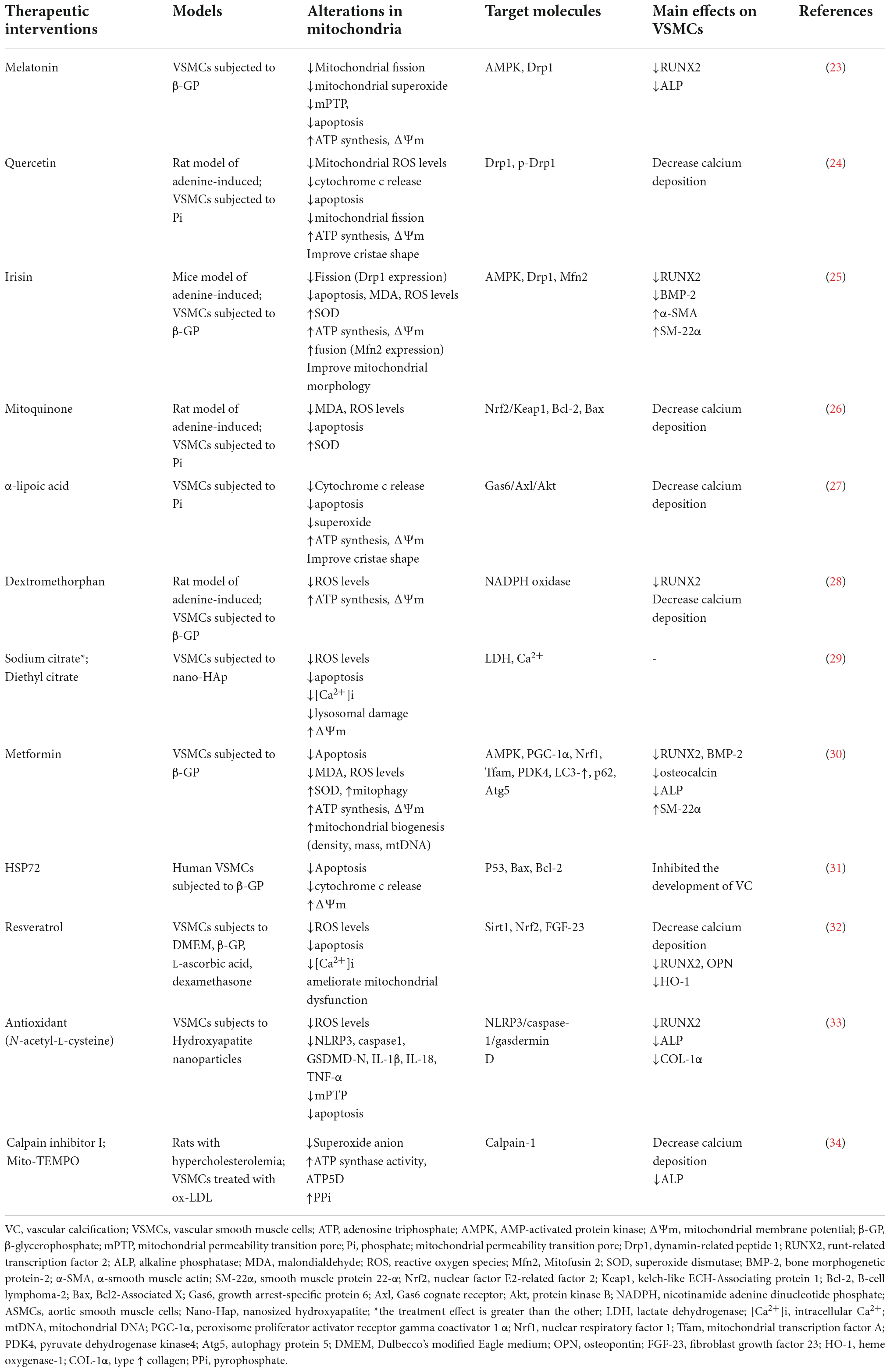
Table 1. Summary of targeted mitochondria in the treatment of vascular calcification.
As shown, mitochondrial dysfunction is manifested by an increase in oxidative stress, apoptosis, aerobic glycolysis, mitochondrial fission, and a decrease in OXPHOS, mitochondrial biosynthesis, mitochondrial fusion, and mitophagy. These triggers interact and act synergistically to promote the loss of the contractile phenotype of VSMCs and the transition to osteoblast-like, foam-cell-like phenotypes, leading to the development of vascular diseases including vascular calcification, atherosclerosis, and hypertension.
Mitochondria perform diverse yet interconnected functions, producing ATP and many biosynthetic intermediates to meet the needs of the cell’s metabolic capacity and also contribute to the cellular stress response. Under appropriate conditions, in response to oxidative stress, mitochondrial dysfunction, and an increase in the energy requirements of the cells, cells can stimulate functional mitochondrial proliferation via mitochondrial biogenesis to re-establish redox, energy metabolism, and counter-inflammatory milieu homeostasis to rejuvenate the number and size of mitochondria and prevent cell death (35). Thus, mitochondrial biogenesis is defined as the process via which cells increase their mitochondrial mass (36). If this process is blocked, not only the cell energy regulation will be out of control, but also due to mitochondrial dysfunction and activation of apoptosis, mitochondria will produce excessive reactive oxygen species, resulting in oxidative stress damage and calcium concentration imbalance, which in turn induces the osteogenic differentiation and calcification of VSMCs (37). Correct mitochondrial biogenesis is very complex and requires the coordinated synthesis of mitochondrial DNA (mtDNA) encoded protein, import of proteins encoded by the nuclear genome and synthesized on cytosolic ribosomes, assembly of the dual genetic origin derived proteins, as well as mtDNA replication (38, 39).
Mitochondrial biogenesis is regulated mainly at the level of transcription. The peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α, the most well-known, is thought to act as a master regulator of mitochondrial biogenesis that controls the optimal mitochondrial content by interacting with various transcription factors, such as peroxisome proliferator-activated receptors (PPARs), mitochondrial transcription factor A (TFAM) (40), estrogen-related receptor (ERR) (41), nuclear respiratory factors (Nrfs), and uncoupling protein 2 (UCP2) (42). PGC-1α cooperatively activates the transcription of Nrf1/2, which in turn regulate the transcription of TFAM. TFAM translocates to the mitochondrial matrix and stimulates mitochondrial DNA replication and mitochondrial gene expression. β-Glycerophosphate (β-GP) a widely used inducer of VC leads to phenotypic transition of VSMCs. Recent evidence shows that in a model of β-GP-induced calcification in VSMCs, a significant reduction in mitochondrial density is displayed, accompanied by mtDNA content, cellular ATP concentration, and expression of mitochondrial biogenesis-related genes such as PGC-1α, Nrf1, and Tfam decreased, suggesting impairment of mitochondrial biogenesis (30). AMP-activated protein kinase (AMPK) and sirtuin1 (Sirt1) are two energy sensors that directly affect PGC-1α activity through phosphorylation and deacetylation, respectively, acting as a crucial link in the maintenance of mitochondrial homeostasis. Pharmacological agents such as metformin or resveratrol can restore mitochondrial biogenesis-related gene expression by activating the AMPK/PGC-1α, Sirt1/Nrf2 signaling pathway, thereby counteracting the osteogenic phenotype transformation of VSMCs. Interestingly, mitophagy is required for metformin-induced mitochondrial biogenesis in VSMCs (30), this suggests that mitophagy and mitochondrial biogenesis are coordinated and that proper mitochondrial quality control is critical for the maintenance of cell function. Meanwhile, decreased expression of PGC-1α and Nrf1/2 in VSMCs proliferation and migration is a common feature of vascular diseases such as atherosclerosis and vascular calcification (32, 43). As the key regulator of mitochondrial biogenesis, activation of Sirt1 can alleviate VSMCs mineralization by upregulating the expression of the calcification inhibitors OPN and osteoprotegerin (OPG), Sirt1 directly acts on the RUNX2 level in hyperglycemic conditions by deacetylation of the RUNX2 promoter (44). During mitochondrial biogenesis, Sirt1 participates inside the nucleus in the induction of PGC-1α that is pivotal to orchestrate the activation of a broad set of transcription factors and nuclear hormone receptors enhancing the expression of the nucleus-encoded mitochondrial genes. In addition to regulating transcription of nucleus-encoded mitochondrial genes, Sirt1 also present as free protein in mitochondria with PGC-1α, and interacts with Tfam. In mitochondria, Sirt1 and PGC-1α form several multiprotein complexs such as PGC-1α/SIRT1, TFAM/PGC-1α, and TFAM/SIRT1. Furthermore, both PGC-1α and Sirt1 reasonably participate with Tfam in the formation of higher molecular weight multiprotein complex, which could correspond to macro-complexes involved in mtDNA transactions (45).
Mitochondria as highly dynamic organelles can be known as isolated organelles or joined to form larger dynamic networks, they can also be distributed unevenly in the cytosol to meet local energy demands of the cell, this changeable and adaptable nature of mitochondria involving their morphology and subcellular distributions is collectively called mitochondrial dynamics (46). Mitochondrial fusion is the union of two mitochondria resulting in one mitochondrion, it requires three large GTP-hydrolyzing enzymes of the dynamin superfamily: Mitofusion1 (Mfn1), Mfn2, Optic atrophy 1 (Opa1). Mfn1, Mfn2 are located on the mitochondrial outer membrane and are required for outer membrane fusion, and Opa1 is associated with the inner membrane, Cells lacking Opa1 were found to mitochondrial outer membrane fusion, but such events did not progress to inner membrane fusion (47, 48). Mitochondrial fission is the division of one mitochondrion into two smaller mitochondria. The critical mediator of mitochondrial fission is Dynamin-related protein 1 (Drp1), a large GTPase, that is recruited to the mitochondrial outer membrane via a collection of receptor proteins, including Mitochondrial fission factor (Mff), Mitochondrial dynamics protein of 49 kDa and 51 kDa (MiD49, MiD50), and Fission protein 1 (Fis1). Once on mitochondria, Drp1 assembles around the tubule and constricts it in a GTP-dependent manner to mediate scission (49). Mitochondrial fission was reported to contribute to mitochondrial apoptosis and was also found to be a prerequisite for mitophagy, while mitochondrial fusion is associated with increased mitochondrial metabolism (50).
The nature of the mitochondrial dynamic network depends on the proper balance between mitochondrial fusion and fission, which are important for biological processes such as maintenance of mitochondrial function, metabolic control, apoptosis, and senescence (51, 52). Fragmented mitochondria clustered in VSMCs often occurs in most vascular diseases, increased mitochondrial fission could result in increased fragmented mitochondria that contribute to the apoptosis-resistant, hyper-proliferative mitochondrial phenotype seen in rat model of spontaneous hypertension (53), pulmonary arterial hypertension (PAH) (54), and atherosclerosis (AS) (55). It is characterized by up-regulated expression of fission proteins (Drp1, Fis1) and down-regulated expression of fusion proteins (Mfn2, Opa1). Drp1 dephosphorylated at serine 637 and phosphorylation increased at serine 616 in PDGF- and Ang II-stimulated VSMCs, and promoted mitochondrial binding of Drp1 to activate mitochondrial fission (56, 57). The mechanism may be that PDGF induces Ca2+ release in the endoplasmic reticulum (ER) and triggers extracellular Ca2+ influx, and increased intracellular Ca2+ ([Ca2+]i) concentration can promote mitochondrial fission through extracellular regulated protein kinase (ERK1/2)-mediated phosphorylation of Drp1 in the cytoplasm (58, 59). In contrast, upregulation of Mfn2 in hyper-proliferative VSMCs exhibits highly anti-proliferative effects on VSMCs in vivo and in vitro (60), Mfn2 can inactivate the ERK1/2 cascade by blocking the Ras downstream pathway and ultimately arresting the cell cycle in G0/G1 phases (61). However, rat VSMCs treated with H2O2 induced upregulation of Mfn2 and caspase-3, and caspase-9, showing mitochondria-mediated apoptosis, suggesting that Mfn2 regulates VSMC homeostasis and viability (62). Robert et al. found that in isolated resistance arteries from Opa1± mice, Opa1 haploinsufficiency leads to high mitochondrial fission, mitochondrial cristae disturbance and significantly increased ROS production in VSMC, and upregulation of Mfn1 but not of Mfn2 to compensate for the defect in mitochondrial fusion control caused by Opa1 deficiency (63). In addition, excessive mitochondrial fission exacerbates oxidative stress and disordered energy supply responsible for VSMCs dedifferentiation, directly playing a role in VC. It has been reported that Drp1 expression was significantly increased in human arterial smooth muscle cells compared to non-calcified regions, accompanied by more fragmented mitochondria (64). In a inorganic phosphate (Pi)-induced VSMCs calcification model, increased expression and phosphorylation of Drp1 leads to mitochondrial fragmentation, ROS overproduction, and mitochondrial cristae disorder-mediated apoptosis directly promotes VC (24). Oxidative stress-induced VSMCs matrix mineralization, mitochondrial dysfunction, and ALP activity were attenuated by Drp1 inhibitor.
Mdivi-1, the Drp1 selective inhibitor, is widely used as a pharmacological intervention to inhibit mitochondrial fission process. The treatment of Mdivi-1 can significantly reversed Drp1-induced mitochondrial fission and ROS generation both in vivo and in vitro (57). In diabetic vascular injury, the imbalance of redox homeostasis leads to the accumulation of ROS and the increase of NADPH oxidase activity, resulting in oxidative stress, which directly leads to the proliferation of VSMCs and further leads to excessive mitochondrial fission. Mdivi-1 can inhibit the expression of Drp1 and the synthesis of ROS in VSMCs under high-glucose conditions, and up-regulate the expression of Mfn2, inhibiting the proliferation of VSMCs (65). Mdivi-1 also effectively reduce the characteristics of osteogenic phenotype transformation such as RUNX2 and type I collagen secretion in the calcification model of VSMCs (64). However, in different VSMCs lesion models, the intervention scheme of Mdivi-1 concentration on mitochondria has not been unified (57, 64), too high Mdivi-1 concentration (50–100 μM) can inhibit the activity of mitochondrial complex I in vitro (66), which may be a point that needs to be paid attention to in future research. Recent evidence suggests that the effects of melatonin in β-GP-treated VSMCs were similar to those of Mdivi1. Melatonin reduces VSMC calcium deposition and ALP activity through the AMPK/Drp1 pathway, and mitochondrial fragmentation and superoxide levels are improved, with a concomitant reduction in apoptosis (23). Furthermore, the myokine irisin and antioxidant Quercetin also alleviate VC progression by inhibiting mitochondrial fission through the Drp1 pathway (24, 25). The plant extract salidroside can down-regulate Drp1 and up-regulate Mfn2 in a dose-dependent manner to inhibit mitochondrial fission and restore mitochondrial network homeostasis, reduce ROS levels, and reverse high glucose-induced VSMCs proliferation (67).
In sum, these all results suggest that reducing mitochondrial fission can limit phenotypic transition of VSMCs in VC and rise the possibility that properly manipulating mitochondrial dynamic may be protective in vascular diseases.
The impaired or superfluous mitochondria are degraded by a selective form of autophagy, known as mitophagy. As a common component of mitochondrial homeostasis control processes, mitochondrial fission selectively separates aged or damaged mitochondria from the normal mitochondrial pool, which is then targeted by the mitophagy system and transported to lysosomes for degradation and Recycling. Besides general autophagy, mitophagy also plays a role in VC. Recent studies have shown that mitophagy plays a protective role against VC by reversing mitochondrial dysfunction, oxidative stress, and apoptosis (68). Defective mitophagy promotes VSMC apoptosis, and restoration of mitophagy flux can prevent cell death (69).
In mammalian cells, two independent pathways mediate mitophagy. One pathway is mitophagy receptor-mediated and the other is ubiquitin-dependent-mediated (70). Two proteins on the outer membrane of mammalian cells are functionally similar to yeast Atg32 and are suggested to be the two major receptors that mediate mitophagy. BCL2 interacting protein 3 like (BNIP3L, or was called NIX) was originally identified as a mitophagy receptor required for autophagic clearance during erythrocyte maturation. It was recently found that NIX carries a potential LC3-interacting region (LIR) that induces specific mitophagy by binding to the autophagy mediator LC3 (71). The BCL2 interacting protein 3 (BNIP3), a homolog of NIX, was found to mediate lactate-induced osteogenic phenotype transition and calcium deposition in VSMCs. Lactic acid inhibits BNIP3 expression and blocks the binding of LC3-II and BNIP3, reduces mitophagy flux, and causes the backlog of damaged mitochondria, oxidative stress, and mitochondrial permeability transition pore (mPTP)-mediated apoptosis. Overexpression of BNIP3 attenuates high levels of oxidative stress and VC, to some extent (68). FUNDC1 is another mitophagy receptor localized to the outer mitochondrial membrane. FUNDC1 was reported to be involved in Ang-II-induced VSMCV proliferation and migration, which was further exacerbated when FUNDC1 expression was inhibited (72).
Ubiquitin-dependent mitophagy mediated by PTEN-induced putative kinase protein1 (PINK1) and Parkin is one of the most-studied mitophagy mechanisms in mammalian cells. Under normal conditions, PINK1 enters the mitochondria via translocases at the outer and inner mitochondrial membranes and is cleaved by mitochondrial matrix enzymes and proteases when crossing the inner mitochondrial membrane, causing it to retrograde into the cytosol and be rapidly degraded by the proteasome (73). However, When mitochondria are damaged, a decrease in mitochondrial membrane potential (MMP) and mitochondrial depolarization inhibit PINK1 degradation, which leads to the accumulation of PINK1 on the impaired mitochondria outer membrane, phosphorylating ubiquitin attached to mitochondrial outer membrane proteins and recruiting Parkin from the cytosol to the mitochondria, inducing the occurrence of mitophagy (50). Notably, PINK1 is expressed at extremely low levels during normal mitochondrial function, and endogenous PINK1 can only be detected at the mitochondrial outer membrane under specific mitochondrial stress conditions. Thus, PINK1 acts as a mitochondrial damage sensor by accumulating on the surface of mitochondria when there is impaired import (74). The study by He et al. showed that PINK1/Parkin expression was significantly up-regulated in apelin-13-induced human aortic VSMCs proliferation in vitro and atherosclerotic lesions of ApoE-/- mice in vivo (55). And these results were confirmed by another study (75). These evidences suggest that the PINK1/Parkin regulatory axis plays an important role in the VSMCs hyper-proliferative phenotype. In addition, deficiency of PINK1/Parkin resulted in increased caspase activity and the number of apoptotic cells following oxidized-LDL (ox-LDL) treated human VSMCs, after up-regulation of PINK1, mitophagy flux was increased, and the number of apoptotic cells induced by ox-LDL was significantly reduced (76). Interestingly, no enhancement of mitophagy flux was found upon overexpression of Parkin alone, likely indicating that Parkin functions in two distinct mitochondrial pathways to promote cell survival. One pathway requires PINK1 to initiate mitophagy, and the other pathway is Parkin-dependent ubiquitination and proteasomal degradation of pro-apoptotic Bax (76). Besides, the regulation of general autophagy by Sirt1 is well-known, and there is evidence that Sirt1 plays a protective role in cardiac ischemia-reperfusion and bone metabolism disorders through PINK1/Parkin-mediated mitophagy (77, 78). In VC, the decrease of Sirt1 is an important mechanism causing the osteogenic phenotype transition of VSMC (44), but there is currently no direct evidence that Sirt1 regulates VSMC mitophagy, thereby alleviating VC. Metformin can affect mitochondrial biogenesis by increasing mitophagy, thereby protecting against β-GP-induced osteogenic phenotype transformation of VSMCs. All together, mitophagy acted as a mitochondrial homeostasis regulatory mechanism to protect VSMCs from apoptosis induced by factors such as oxidative and metabolic stress, thereby affecting VC.
Under harmful environmental stimuli, the excessive production of mtROS induces the transcription of osteogenic genes (such as Msx2, Runx2) and osteogenic differentiation through JNK or NF-κB pathways; The decrease in ATP/ADP ratio and membrane potential caused by the decrease in mitochondrial quality control activates the caspase-9/3 cascade by promoting the release of cytochrome c, which ultimately leads to VC (Figure 2).
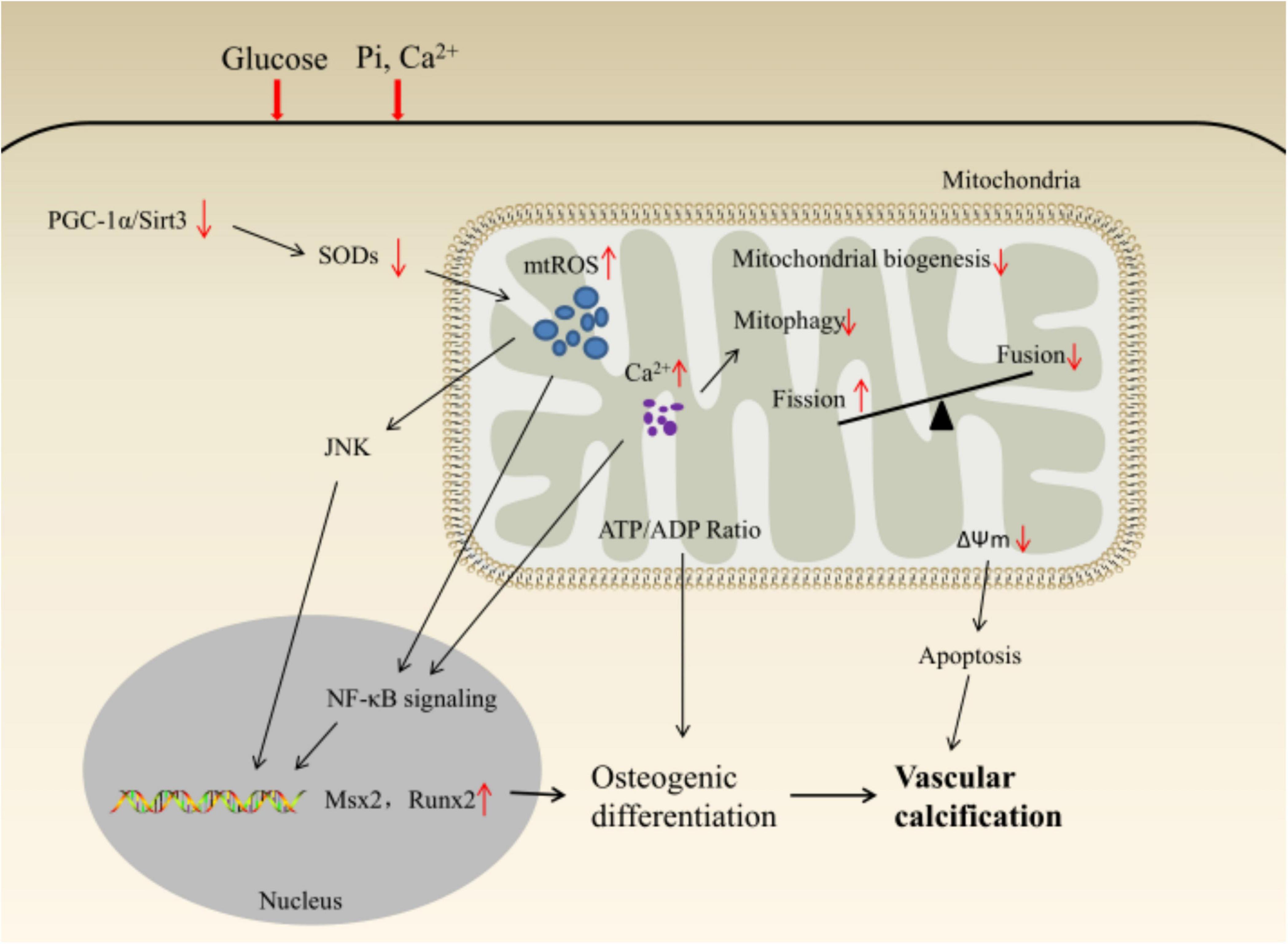
Figure 2. Schematic illustration of mitochondrial homeostasis and VSMCs phenotypic transition.
Evidence has shown that oxidative stress markers increased with renal function deterioration from the early stages of CKD in children and adults, accompanied by vascular dysfunction (79, 80). Persistent oxidative stress and ROS-induced signaling are key features of cardiovascular disease and are major drivers of VSMCs osteogenic phenotype transformation and VC (81, 82). Mechanistically, H2O2 could induce VSMC osteogenic differentiation by activating the PI3K/AKT/Runx2 signaling axis and inhibiting the expression of VSMC contractile phenotype markers in vitro, and up-regulated the expression of ALP (83, 84). In vivo and in vitro studies have shown that the increase of MMP leads to ROS production and increases IKKβ phosphorylation and IκBα degradation in a time-dependent manner, thereby inducing the phenotypic transition of VSMC to osteoblast-like phenotype by activating NF-κB and/or Elk-1/serum response factor (SRF) signaling pathways (85). In addition, NF-kB can inhibit the expression of calcification inhibitor ankylosis protein homolog (ANKH) through RNA-destabilizing factor tristetraprolin (TTP)-dependent, and enhance the activity of ALP, then reducing the secretion of PPi and aggravating VC (86). Oxidative stress-dependent ER stress can activate the PERK/eIF2a-ATF4-CHOP signaling pathway to induce the phenotype switch of VSMCs (87), and inhibition of ER stress can restore the contractile phenotype of calcified VSMCs (88). As by-products of mitochondrial oxidative phosphorylation (OXPHOS), mitochondria are the main source of intracellular ROS production (89). Under physiological conditions, the balance between ROS generation and ROS scavenging is highly controlled. Depending on the context, regulated ROS could trigger a variety of cellular responses, including signaling pathways involved in cell survival, initiation coordinated activation of mitochondrial fission and mitophagy to clear abnormal mitochondria (90). The imbalance between ROS production and antioxidant defense mechanisms is responsible for oxidative stress. In the diabetic state, both factors coexist, with reduced endogenous antioxidant defense efficiency and increased reactive oxygen species production both key mechanisms leading to the development of diabetic vascular complications (91). Advanced glycation end products (AGEs) interact with their major receptor RAGE to enhance intracellular oxidative stress levels through NADPH oxidase (NOXs) and mitochondrial pathways (92), inducing VSMCs osteogenic genes expression. The interaction between mitochondria and NOX-derived O2– constitutes a feed-forward cycle in which NOX2 increases mtROS production through reverse electron transfer. The increased production of mtROS further activates cytoplasmic NOXs, enhancing the production of cellular O2–, creating a vicious circle (93). When overexpressing mitochondrial NOX4, mice VSMCs showed decreased expression levels of TFAM and SOD2, indicating mitochondrial biogenesis and antioxidant dysfunction, which may increase ROS levels in a feed-forward manner and induce VSMC apoptosis, Aging, and aortic calcification (94). AGEs-induced upregulation of OPN, OCN, and Runx2 was inhibited by silencing NOX4, and osteogenesis and calcium deposition in VSMCs were reversed (95).
ROS-targeted antioxidant therapy proved to be a viable treatment in an in vivo model of VC (96). Evidence suggests that the NADPH oxidase inhibitor dextromethorphan significantly inhibits the increase in ROS production and improves mitochondrial activity, thereby reducing aortic calcification in rats with chronic kidney disease (28). The mitochondria-targeted antioxidant mitoquinone attenuates vascular calcification by inhibiting oxidative stress and apoptosis in VSMCs through the Keap1/Nrf2 pathway (26). Mitochondrial antioxidants (mito-TEMPO) and calpain-1 inhibitors significantly inhibited superoxide anion production in aortic mitochondria, and corrected PPi metabolic disorder, while reducing ALP expression (34). The natural antioxidant α-lipoic acid inhibits VSMC apoptosis in vitro and in vivo, protects mitochondrial function through antioxidant capacity and restores the Gas6/Axl/Akt survival pathway (27). In addition, mitochondrial uncoupling proteins (U) are considered to be important antioxidants that reduce oxidative stress and oxidative damage by maintaining ROS homeostasis. Zhou et al. showed that activation of peroxisome proliferator-activated receptorγ (PPARγ) could inhibit ROS-induced VSMC proliferation and migration by upregulating UCP2 expression (97). Antioxidants (MnTMPyP, SOD1/2) and mitochondrial respiratory chain inhibitors (rotenone, CCCP, UCP2) can attenuate Pi-induced ROS generation and calcium deposition (85). Pi is an important substrate for ATP synthesis in the process of OXPHOS and participates in various biological functions such as signal transduction and energy metabolism. Apurinic/pyrimidine endonuclease 1/redox factor-1 (APE1/Ref-1) is a multifunctional protein that plays a pleiotropic role in the control of cellular responses to oxidative stress, Gene silencing of APE1/Ref-1 expression reduces basal DNA repair functions and redox activity and is involved in Pi-induced VC, APE1/Ref-1 overexpression significantly inhibits Pi-induced mtROS production, apoptosis, and calcification of VSMCs in vitro and in vivo (98). It should be noted that although anti-oxidants can all reduce superoxide levels, there are differences in their effect on vascular calcification, which may suggest that scavenging free radicals in vivo alone is not enough and that its production may need to be inhibited at the source (96).
Metabolic disturbance of Ca2+ is a common phenomenon in diabetes and CKD (99, 100). The increase in cyclic Ca2+ is a risk factor for vascular disease and mortality. Serum Ca2+ has increased by approximately 0.1 mmol/L, the risk ratio of death is 1.13 (101). The source, amplitude, frequency, and duration of Ca2+ signaling are important determinants of VSMCs phenotypic transition (102, 103). [Ca2+]i is mediated by extracellular Ca2+ influx and intracellular Ca2+ release. High Pi induces persistent depolarization of the plasma membrane through PiT-1 and -2, the latter activates voltage-gated Ca2+ channels (VGCC) leading to Ca2+ influx. Mitochondria are the important hub for intracellular Ca2+ signaling networks. Mitochondrial Ca2+ uptake plays a key role in the normal physiological functions of cells, including ATP production, ROS generation, autophagy, rectification of intracytoplasmic calcium signaling, and regulation of cell death (104, 105).
Ca2+ uptake by mitochondria is a process that relies on a gated mitochondrial Ca2+ uniporter (MCU) complex. MCU is a low-affinity, high-selectivity Ca2+ channel that localizes to the inner mitochondrial membrane (IMM) and is responsible for mitochondrial Ca2+ uptake (106). Recent evidence suggests that MCU-dependent mitochondrial Ca2+ overload leads to overproduction of mtROS (37), which promotes Drp1-dependent mitochondrial fission and mitophagy, thereby inducing VSMC proliferation, and inhibition of MCU-dependent mitochondrial Ca2+ uptake might ameliorate or reverse abnormal VSMC proliferation (58, 107). In addition, mitochondrial Ca2+ activates Ca2+-sensitive dehydrogenases in the tricarboxylic acid cycle such as pyruvate dehydrogenase, glycerol phosphate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase (108) and downregulating NAD+/NADH ratio and NAD+-dependent Sirt3 activity further inhibits SOD2 activity, mediating activation of the JNK pathway to increase mtROS production (106), the increased mtROS leads to the opening of mitochondrial permeability transition pore (mPTP), which promotes the release of apoptotic factors such as cytochrome c into the cytoplasm through ruptured mitochondria, and finally triggers cell death (104, 109). Activating the AMPK/PGC-1α/sirtuin-3 (Sirt3) pathway can inhibit MCU-induced mitochondrial Ca2+ overload, alleviate mtROS accumulation, stabilize mitochondrial membranes, and promote mitochondrial fusion to improve mitochondrial quality control (110).
In addition, due to the limitation of MCU, mitochondria can only take up Ca2+ at high concentrations of Ca2+, so Ca2+ is most likely to be directly transferred from ER to mitochondria through mitochondria-associated ER membranes (MAMs) and control ATP production and apoptosis (111). ER is the main intracellular organelle for the rapid and specific release of Ca2+, known as the Ca2+ reservoir (112). Accumulating evidence suggests that Ca2+ release from the ER is critical for mitochondrial dysfunction and increased mtROS (113, 114). Ca2+ transport between ER and mitochondria is mainly co-regulated by ER-mitochondria encounter structure (ERMES), Mfn2, inositol-1,4,5-triphosphate receptor (IP3R)/Grp75/voltage-dependent anion channel (VDAC), and MCU channels (115). ER release of Ca2+ is mainly mediated by IP3R, and then VDAC (located in the mitochondria outer membrane) is responsible for translocation from the mitochondrial outer membrane to the MCU. It was reported that histamine-stimulated IP3R mediates Ca2+ release from the endoplasmic reticulum to mitochondria, and the increased mitochondrial calcium concentration can enhance oxidative phosphorylation by activating four mitochondrial dehydrogenases, improving the metabolic efficiency of VSMCs, and modulate the phenotypic transition of VSMCs (116), and PKA/Mfn2 can regulate the connection between mitochondria and ER, the overexpression of Mfn2 can increase the functional coupling between the two organelles, which is conducive to the rapid flow of mitochondrial Ca2+ (116). Xia et al. found that nanosized hydroxyapatite (nano-HAp) can increase mitochondrial Ca2+ uptake in VSMCs, which in turn leads to excessive ROS production and oxidation of mtDNA (ox-mtDNA), and the release of more ox-mtDNA from mitochondria to the cytoplasm activates NLRP3/caspase-1/gasdermin D and induce VC by secreting inflammatory factors, antioxidants can effectively reverse this process and alleviate the progress of VC (33). In addition, the anticoagulants sodium citrate and diethyl citrate can also effectively reduce cell necrosis and lysosomal damage from [Ca2+]i overload induced by nano-HAp, avoiding further VC from cell death (29).
Metabolic reprogramming, considered to alter the activity of selected energy-producing or biosynthetic pathways, is a key regulator of VSMCs phenotypic transition and signal transduction processes (117, 118). Unusually, even though OXPHOS is much more efficient than glycolysis to generate ATP, a substantial proportion of ATP in VSMCs still originates from glycolysis rather than OXPHOS (118). The metabolism of VSMCs is characterized by a significant production of lactate even under fully oxygenated conditions. About 30% of the ATP supply in VSMC is derived from aerobic glycolysis, and 80–90% of the glycolytic flux contributes to lactate production (118, 119). VSMCs exhibit higher glucose uptake and aerobic glycolytic capacity during the phenotypic transition to maximize the energy supply required by cells (9, 120, 121). This increase in aerobic glycolysis resembles the Warburg effect in tumor cells, is an important feature of the transformation of multiple cellular phenotypes (122), and provides a potential target for clinical intervention. Although the recent view is contrary to Warburg’s original contention that increased aerobic glycolysis is not a consequence of ATP compensation of dysfunctional or damaged mitochondria (123). However, mitochondria’s healthy and larger presence enables them to compete with the LDH reaction for the intermediates of glycolysis (pyruvate and NADH), thereby minimizing lactate accumulation (124).
The expressions of GLUT-1 and glycolytic enzymes HK2 and PKM2 are up-regulated under calcifying conditions. Under the catalysis of LDHA, pyruvate is more inclined to generate lactate, the end product of glycolysis. Excessive lactate exposure not only leads to the imbalance of intracellular redox homeostasis and apoptosis of VSMCs, but also reduces the expression of biogenesis factors such as PGC-1α, Nrf1, and TFAM. Meanwhile, lactate impairs mitochondrial function through pathways such as mPTP opening rate and mitophagy. In addition, PDK4 is elevated under calcifying conditions, which inhibits OXPHOS. On the other hand, it promotes SMAD1/5/8 and SMAD4 to form heteromeric complexes and enter the nucleus, thereby regulating the transcription of osteogenic genes such as Runx2 and BMP-2 (Figure 3).
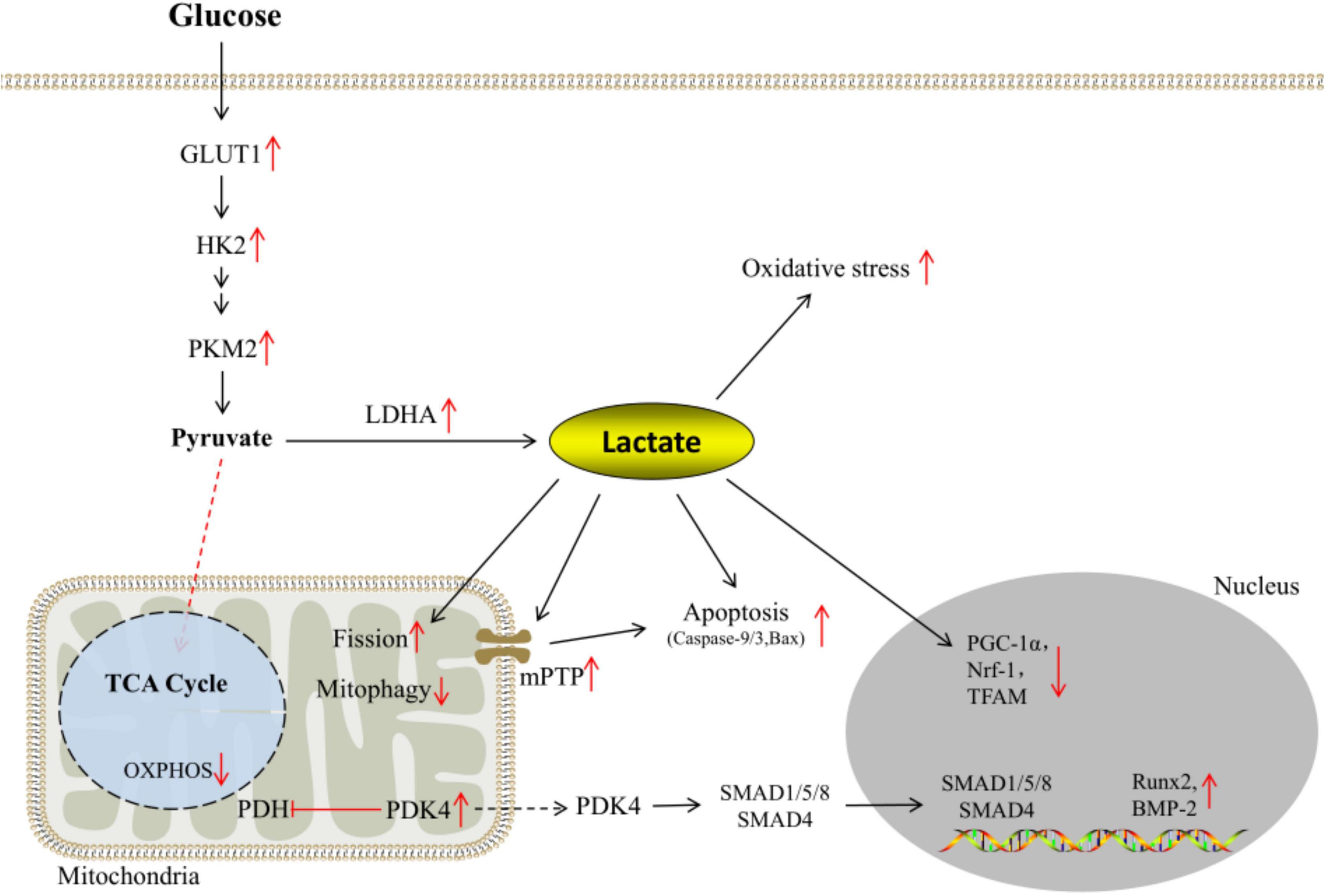
Figure 3. Reprogramming of glucose metabolism under calcifying conditions.
Glucose metabolism is critical for VSMCs phenotype regulation. Glucose uptake and lactate production increased during VSMCs calcification, suggesting an increased proportion of glycolysis in VSMCs glucose metabolism (9, 125). Multiple studies have demonstrated elevated blood lactate levels in diabetic patients (126, 127). Although we are currently unable to establish a direct link between blood lactate and diabetic VSMCs calcification, blood lactate concentrations can provide valuable insights into overall metabolic health (128). Mechanistically, lactate promotes Drp1-mediated mitochondrial fission and BNIP3-mediated mitophagy impairment by activating the nuclear receptor subfamily 4 group A member 1 (NR4A1)/DNA-dependent protein kinase, catalytic subunit (DNA-PKcs)/p53 pathway, ultimately leading to apoptosis and accelerate VSMCs calcification (11). In addition, lactate is closely linked to mitochondrial dysfunction and intracellular oxidative stress (68). Glucose uptake by cells is primarily through glucose transporters, followed by the conversion of glucose to pyruvate by the combined action of key rate-limiting enzymes such as hexokinase 2 (HK2) and pyruvate kinase in glycolysis. Pyruvate is then catalyzed by the pyruvate dehydrogenase complex for irreversible oxidative decarboxylation into acetyl-CoA, which enters the tricarboxylic acid cycle, or is catalyzed by lactate dehydrogenase to lactate. Glucose transporter1 (GLUT-1) is the major isoform in VSMCs. In β-GP-induced VC, the expression of GLUT-1 increased in a time-dependent manner and contributed to the proliferation of VSMCs (129, 130). Excessive GLUT-1 in humans results in a 44% increase in intracellular glucose concentration, similar to a diabetic environment (125, 131), while high intracellular glucose concentrations increase the expression of osteogenic transcription factors such as BMP-2, OPN, collagen type I, and ALP (125, 132, 133). In addition, the xxGLUT-1 polymorphism genotypes are associated with VC in early-stage kidney patients (125).
Hypoxia triggers VSMCs calcification and osteogenic differentiation in a HIF-1-dependent and mitochondrial ROS-dependent manner (134–136). As a key effector of hypoxia response, the hypoxia-inducible factor-1α (HIF-1α) can affect the transcription and activity of related metabolic enzymes and regulate the phenotypic transformation of VSMCs. In hypoxia, hydroxylation of HIF-1α is inhibited, and the accumulated HIF-1α binds to HIF-1 β to form a dimer, which in turn generates nuclear translocation and binds to specific DNA sequences of target genes (hypoxia response elements), and activate transcription of target genes (137). In human carotid smooth muscle, growth factors such as PDGF activate HIF-1 through PI3K/Akt/GSK3b pathway, hyperpolarized MMP by promoting HK2 translocation to mitochondria, inhibits OXPHOS of glucose, contributing to the increased of VSMCs glycolysis flux (138). In addition, activation of signal transducer and activator of transcription 3 (STAT3) could be a key event in the upregulation of HK2 activity and glycolysis in VSMCs, and inhibition of the STAT3/HK2 signaling axis can reduce glycolysis and inhibit VSMCs proliferation and migration (139). Pyruvate kinase isoform M2 (PKM2) is a crucial rate-limiting enzyme during glycolysis that accelerates VSMC proliferation and migration (140). In the Warburg effect, PKM2 not only inhibits mitochondrial Ca2+ levels, leading to the conversion of glucose metabolism from OXPHOS to aerobic glycolysis, but also promotes HIF-1α-mediated transcription of multiple metabolism-related genes such as lactate dehydrogenase (LDH)-A1 and GLUT-1 through nuclear translocation, ensuring that lactate levels are elevated (141). Pyruvate dehydrogenase kinase 4 (PDK4) is a mitochondrial matrix enzyme that negatively regulates the activity of the pyruvate dehydrogenase complex (PDC) through phosphorylation, thus contributing to mitochondrial function and glucose utilization in VSMCs (142). A growing number of studies have shown that the expression of PDK4 and aerobic glycolysis are increased in VSMCs calcification models induced by Pi or AGEs (143, 144). Recent evidences suggest that elevated PDK4 expression affects cellular metabolism and accelerates VSMCs calcification in at least the following ways: (1) PDK4 overexpression impairs mitochondrial function, manifested by decreased mitochondrial aerobic rate, ATP production, maximal respiration, and ROS-mediated apoptosis, resulting in decreased mitochondrial tricarboxylic acid cycle activity promotes the conversion of pyruvate to lactate production (30, 145); (2) PDK4 regulates the downstream signaling pathway of BMP2 and activate SMAD1/5/8 through PDK4 phosphorylation, resulting in phosphorylation SMADs translocating to the nucleus and promoting the transcription of osteogenic genes (146); (3) PDK4 increases in an AGEs/HIF-1α-dependent manner and promotes autophagy during VSMCs calcification (147), which may be a compensatory mechanism for cellular self-protection. PDK4 inhibition can decrease AGEs-induced ALP activity, Runx2 activation, and calcium deposition content and significantly downregulate lactate production (144, 148). Evidence suggests that interventions such as metformin and zinc supplementation can inhibit the osteogenic phenotype transition and calcification of VSMCs by reducing PDK4 expression levels (30, 149).
In VSMCs, mitochondrial dysfunction can lead to oxidative stress, and metabolic transformation, triggering a series of cascade reactions. Therefore, targeting mitochondria is an attractive option in VC therapy. However, existing measures such as mitochondrial antioxidants have not yet shown satisfactory results in clinical trials. Zhang et al. believe that mitochondrial dysfunction is a dynamic network of changes, rather than damage to individual mitochondria (14). Meng et al. believe that the redox state of organelles is different and should be accurately detected, rather than non-specific treatment (150), how accurately assessing mitochondrial damage and regulating the balance of mitochondrial homeostasis is the key to future pharmacological research. In addition, lifestyle interventions such as exercise (14), fasting (151), and calorie restriction can improve mitochondrial function and balance redox homeostasis from the perspective of integrated physiology (152), which has broad application prospects.
Endoplasmic reticulum and mitochondria are physically and functionally closely linked, affecting the ROS generation, metabolism, and survival of cells via the regulation of ions such as Ca2+ and magnesium (Mg2+). Mg2+ is the second most abundant divalent cation in cells. As a natural antagonist of Ca2+, Mg2+ participates in the regulation of Ca2+ homeostasis competitively. In addition, Mg2+ acts as a cofactor for more than 300 metabolic responses and affects cardiovascular disease by regulating mitochondrial function, metabolism, oxidative stress, and inflammation (153). Daw et al. demonstrated that lactate acts as a signaling molecule to trigger dynamic changes in Mg2+ between the ER and mitochondria, thereby enhancing mitochondrial bioenergetics (154). Mg2+ supplementation has shown therapeutic potential in models of pulmonary arterial hypertension (155), but there is currently a lack of evidence in VC.
In conclusion, mitochondria play critical roles in cellular physiology, including metabolic flexibility, energy production, maintenance of redox homeostasis, calcium homeostasis processing, and cell death. Little is currently known about the interactions between mitochondria and other organelles and the mediating role of mitochondria between ions and metabolism, exploring the role of mitochondria in the phenotypic transition of VSMCs may provide clues for the precise prevention and treatment of VC.
DW, YPL, and YZL conceived and designed the research. YZL, ZXL, and LLZ collected the manuscript data and designed the tables and images. YZL and ZXL wrote the manuscript. DW and YPL supervised the study and revised the manuscript. All authors contributed to the article and approved the submitted version.
We gratefully acknowledged the financial support by Natural Science Foundation of Fujian Province (2017J01627) and China Postdoctoral Science Foundation (2021M700782).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Timmis A, Townsend N, Gale C, Grobbee R, Maniadakis N, Flather M, et al. European society of cardiology: cardiovascular disease statistics 2017. Eur Heart J. (2018) 39:508–79. doi: 10.1093/eurheartj/ehx628
2. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
3. Niu Q, Hong Y, Lee CH, Men C, Zhao H, Zuo L. Abdominal aortic calcification can predict all-cause mortality and CV events in dialysis patients: a systematic review and meta-analysis. PLoS One. (2018) 13:e0204526. doi: 10.1371/journal.pone.0204526
4. Leow K, Szulc P, Schousboe JT, Kiel DP, Lewis JR. Prognostic value of abdominal aortic calcification: a systematic review and meta-analysis of observational studies. J Am Heart Assoc. (2021) 10:e017205.
5. Leow K, Szulc P, Schousboe J, Kiel D, Teixeira A, Sheikh H, et al. Abdominal aortic calcification and cardiovascular events: a systematic review and meta-analysis of observational studies. Calcified Tissue Int. (2019) 28(Suppl. 4):S371–2.
6. Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, et al. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res. (2009) 104:733–41. doi: 10.1161/circresaha.108.183053
7. Rodenbeck SD, Zarse CA, McKenney-Drake ML, Bruning RS, Sturek M, Chen NX, et al. Intracellular calcium increases in vascular smooth muscle cells with progression of chronic kidney disease in a rat model. Nephrol Dial Transplant. (2017) 32:450–8. doi: 10.1093/ndt/gfw274
8. Badran A, Nasser SA, Mesmar J, El-Yazbi AF, Bitto A, Fardoun MM, et al. Reactive oxygen species: modulators of phenotypic switch of vascular smooth muscle cells. Int J Mol Sci. (2020) 21:8764. doi: 10.3390/ijms21228764
9. Rashdan N, Macrae VJH. Investigating the role of aerobic glycolysis in arterial calcification. Heart. (2017) 103(Suppl. 5):A144–5. doi: 10.1136/heartjnl-2017-311726.221
10. Tsai YT, Yeh HY, Chao CT, Chiang CK. Superoxide dismutase 2 (SOD2) in vascular calcification: a focus on vascular smooth muscle cells, calcification pathogenesis, and therapeutic strategies. Oxid Med Cell Longev. (2021) 2021:6675548. doi: 10.1155/2021/6675548
11. Zhu Y, Han XQ, Sun XJ, Yang R, Ma WQ, Liu NF. Lactate accelerates vascular calcification through NR4A1-regulated mitochondrial fission and BNIP3-related mitophagy. Apoptosis. (2020) 25:321–40. doi: 10.1007/s10495-020-01592-7
12. Huynh DTN, Heo KS. Role of mitochondrial dynamics and mitophagy of vascular smooth muscle cell proliferation and migration in progression of atherosclerosis. Arch Pharm Res. (2021) 44:1051–61. doi: 10.1007/s12272-021-01360-4
13. Jia Y, Wang M, Mao C, Yu F, Wang Y, Xiao R, et al. COMP-prohibitin 2 interaction maintains mitochondrial homeostasis and controls smooth muscle cell identity. Cell Death Dis. (2018) 9:676. doi: 10.1038/s41419-018-0703-x
14. Zhang X, Gao F. Exercise improves vascular health: role of mitochondria. Free Radic Biol Med. (2021) 177:347–59. doi: 10.1016/j.freeradbiomed.2021.11.002
15. Maguire EM, Xiao Q. Noncoding RNAs in vascular smooth muscle cell function and neointimal hyperplasia. FEBS J. (2020) 287:5260–83. doi: 10.1111/febs.15357
16. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-Cell RNA-Seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. (2018) 122:1661–74. doi: 10.1161/circresaha.117.312509
17. Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J, et al. Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switching and potential therapeutic targets for atherosclerosis in mouse and human. Circulation. (2020) 142:2060–75. doi: 10.1161/circulationaha.120.048378
18. Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Jaw JE, et al. Smooth muscle cells contribute the majority of foam cells in ApoE (Apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol. (2019) 39:876–87. doi: 10.1161/atvbaha.119.312434
19. Berry DC, Jiang Y, Graff JM. Mouse strains to study cold-inducible beige progenitors and beige adipocyte formation and function. Nat Commun. (2016) 7:10184. doi: 10.1038/ncomms10184
20. Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. (2019) 25:1280–9. doi: 10.1038/s41591-019-0512-5
21. Dobnikar L, Taylor AL, Chappell J, Oldach P, Harman JL, Oerton E, et al. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat Commun. (2018) 9:4567. doi: 10.1038/s41467-018-06891-x
22. Quaglino D, Boraldi F, Lofaro FD. The biology of vascular calcification. Int Rev Cell Mol Biol. (2020) 354:261–353. doi: 10.1016/bs.ircmb.2020.02.007
23. Chen WR, Zhou YJ, Sha Y, Wu XP, Yang JQ, Liu F. Melatonin attenuates vascular calcification by inhibiting mitochondria fission via an AMPK/Drp1 signalling pathway. J Cell Mol Med. (2020) 24:6043–54. doi: 10.1111/jcmm.15157
24. Cui L, Li Z, Chang X, Cong G, Hao L. Quercetin attenuates vascular calcification by inhibiting oxidative stress and mitochondrial fission. Vascul Pharmacol. (2017) 88:21–9. doi: 10.1016/j.vph.2016.11.006
25. Wang PW, Pang Q, Zhou T, Song XY, Pan YJ, Jia LP, et al. Irisin alleviates vascular calcification by inhibiting VSMC osteoblastic transformation and mitochondria dysfunction via AMPK/Drp1 signaling pathway in chronic kidney disease. Atherosclerosis. (2022) 346:36–45. doi: 10.1016/j.atherosclerosis.2022.02.007
26. Cui L, Zhou Q, Zheng X, Sun B, Zhao S. Mitoquinone attenuates vascular calcification by suppressing oxidative stress and reducing apoptosis of vascular smooth muscle cells via the Keap1/Nrf2 pathway. Free Radic Biol Med. (2020) 161:23–31. doi: 10.1016/j.freeradbiomed.2020.09.028
27. Kim H, Kim HJ, Lee K, Kim JM, Kim HS, Kim JR, et al. α-Lipoic acid attenuates vascular calcification via reversal of mitochondrial function and restoration of Gas6/Axl/Akt survival pathway. J Cell Mol Med. (2012) 16:273–86. doi: 10.1111/j.1582-4934.2011.01294.x
28. Liu ES, Chen NC, Jao TM, Chen CL. Dextromethorphan reduces oxidative stress and inhibits uremic artery calcification. Int J Mol Sci. (2021) 22:12277. doi: 10.3390/ijms222212277
29. Zhang CY, Sun XY, Ouyang JM, Gui BS. Diethyl citrate and sodium citrate reduce the cytotoxic effects of nanosized hydroxyapatite crystals on mouse vascular smooth muscle cells. Int J Nanomed. (2017) 12:8511–25. doi: 10.2147/ijn.S145386
30. Ma WQ, Sun XJ, Wang Y, Zhu Y, Han XQ, Liu NF. Restoring mitochondrial biogenesis with metformin attenuates β-GP-induced phenotypic transformation of VSMCs into an osteogenic phenotype via inhibition of PDK4/oxidative stress-mediated apoptosis. Mol Cell Endocrinol. (2019) 479:39–53. doi: 10.1016/j.mce.2018.08.012
31. Chung C, Lim K, Lu T. Arterial calcification can be inhibited via maintained mitochondrial homeostasis in chronic kidney disease. Circulation. (2013) 128(Suppl. 22):A18849.
32. Zhang P, Li Y, Du Y, Li G, Wang L, Zhou F. Resveratrol ameliorated vascular calcification by regulating sirt-1 and Nrf2. Transplant Proc. (2016) 48:3378–86. doi: 10.1016/j.transproceed.2016.10.023
33. Xia Y, Li B, Zhang F, Wu Q, Wen S, Jiang N, et al. Hydroxyapatite nanoparticles promote mitochondrial-based pyroptosis via activating calcium homeostasis and redox imbalance in vascular smooth muscle cells. Nanotechnology. (2022) 33:275101. doi: 10.1088/1361-6528/ac61ca
34. Tang F, Chan E, Lu M, Zhang X, Dai C, Mei M, et al. Calpain-1 mediated disorder of pyrophosphate metabolism contributes to vascular calcification induced by oxLDL. PLoS One. (2015) 106:e0129128. doi: 10.1371/journal.pone.0129128
35. Piantadosi CA, Suliman HB. Redox regulation of mitochondrial biogenesis. Free Radic Biol Med. (2012) 53:2043–53. doi: 10.1016/j.freeradbiomed.2012.09.014
36. Valero T. Mitochondrial biogenesis: pharmacological approaches. Curr Pharm Des. (2014) 20:5507–9. doi: 10.2174/138161282035140911142118
37. Nguyen NT, Nguyen TT, Da Ly D, Xia JB, Qi XF, Lee IK, et al. Oxidative stress by Ca(2+) overload is critical for phosphate-induced vascular calcification. Am J Physiol Heart Circ Physiol. (2020) 319:H1302–12. doi: 10.1152/ajpheart.00305.2020
38. Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. (2010) 47:69–84. doi: 10.1042/bse0470069
39. Wenz T. Regulation of mitochondrial biogenesis and PGC-1α under cellular stress. Mitochondrion. (2013) 13:134–42. doi: 10.1016/j.mito.2013.01.006
40. Sun L, Zhao M, Yu XJ, Wang H, He X, Liu JK, et al. Cardioprotection by acetylcholine: a novel mechanism via mitochondrial biogenesis and function involving the PGC-1α pathway. J Cell Physiol. (2013) 228:1238–48. doi: 10.1002/jcp.24277
41. Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, et al. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci USA. (2004) 101:6472–7. doi: 10.1073/pnas.0308686101
42. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. (1999) 98:115–24. doi: 10.1016/s0092-8674(00)80611-x
43. Chong H, Wei Z, Na M, Sun G, Zheng S, Zhu X, et al. The PGC-1α/NRF1/miR-378a axis protects vascular smooth muscle cells from FFA-induced proliferation, migration and inflammation in atherosclerosis. Atherosclerosis. (2020) 297:136–45. doi: 10.1016/j.atherosclerosis.2020.02.001
44. Lu CL, Liao MT, Hou YC, Fang YW, Zheng CM, Liu WC, et al. Sirtuin-1 and its relevance in vascular calcification. Int J Mol Sci. (2020) 21:1593. doi: 10.3390/ijms21051593
45. Aquilano K, Vigilanza P, Baldelli S, Pagliei B, Rotilio G, Ciriolo MR. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis. J Biol Chem. (2010) 285:21590–9. doi: 10.1074/jbc.M109.070169
46. Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. (2020) 214:204–24. doi: 10.1038/s41580-020-0210-7
47. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. (2020) 15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711
48. Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. (2014) 19:630–41. doi: 10.1016/j.cmet.2014.03.011
49. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. (2018) 62:341–60. doi: 10.1042/ebc20170104
50. Ma K, Chen G, Li W, Kepp O, Zhu Y, Chen Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front Cell Dev Biol. (2020) 8:467. doi: 10.3389/fcell.2020.00467
51. Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. (2016) 21:379–87. doi: 10.1083/jcb.201511036
52. Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. (2007) 8:870–9. doi: 10.1038/nrm2275
53. Zhang X, Chen W, Li J, Qi S, Hong S, Wang Y, et al. Involvement of mitochondrial fission in calcium sensing receptor-mediated vascular smooth muscle cells proliferation during hypertension. Biochem Biophys Res Commun. (2018) 495:454–60. doi: 10.1016/j.bbrc.2017.11.048
54. Chen KH, Dasgupta A, Lin J, Potus F, Bonnet S, Iremonger J, et al. Epigenetic dysregulation of the dynamin-related protein 1 binding partners MiD49 and MiD51 increases mitotic mitochondrial fission and promotes pulmonary arterial hypertension: mechanistic and therapeutic implications. Circulation. (2018) 13:287–304. doi: 10.1161/circulationaha.117.031258
55. He L, Zhou Q, Huang Z, Xu J, Zhou H, Lv D, et al. PINK1/Parkin-mediated mitophagy promotes apelin-13-induced vascular smooth muscle cell proliferation by AMPKα and exacerbates atherosclerotic lesions. J Cell Physiol. (2019) 23:8668–82. doi: 10.1002/jcp.27527
56. Ikeda Y, Iwabayashi M, Sasaki Y, Akasaki Y, Higuchi K, Miyata M, et al. Renin-angiotensin signal develops arterial senescence and atherosclerosis via modulation of mitochondrial dynamics. Hypertension. (2017) 70:A440. doi: 10.1161/hyp.70.suppl_1.p440
57. Deng Y, Li S, Chen Z, Wang W, Geng B, Cai J. Mdivi-1, a mitochondrial fission inhibitor, reduces angiotensin-II- induced hypertension by mediating VSMC phenotypic switch. Biomed Pharmacother. (2021) 140:111689. doi: 10.1016/j.biopha.2021.111689
58. Wang L, Yu T, Lee H, O’Brien DK, Sesaki H, Yoon Y. Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc Res. (2015) 106:272–83. doi: 10.1093/cvr/cvv005
59. Yu T, Jhun BS, Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Signal. (2011) 14:425–37. doi: 10.1089/ars.2010.3284
60. Zhou W, Chen KH, Cao W, Zeng J, Liao H, Zhao L, et al. Mutation of the protein kinase a phosphorylation site influences the anti-proliferative activity of mitofusin 2. Atherosclerosis. (2010) 211:216–23. doi: 10.1016/j.atherosclerosis.2010.02.012
61. Li D, Li X, Guan Y, Guo X. Mitofusin-2-mediated tethering of mitochondria and endoplasmic reticulum promotes cell cycle arrest of vascular smooth muscle cells in G0/G1 phase. Acta Biochim Biophys Sin (Shanghai). (2015) 47:441–50. doi: 10.1093/abbs/gmv035
62. Guo X, Chen KH, Guo Y, Liao H, Tang J, Xiao RP. Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ Res. (2007) 101:1113–22. doi: 10.1161/circresaha.107.157644
63. Robert P, Nguyen PMC, Richard A, Grenier C, Chevrollier A, Munier M, et al. Protective role of the mitochondrial fusion protein OPA1 in hypertension. FASEB J. (2021) 35:e21678. doi: 10.1096/fj.202000238RRR
64. Rogers MA, Maldonado N, Hutcheson JD, Goettsch C, Goto S, Yamada I, et al. Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ Res. (2017) 121:220–33. doi: 10.1161/circresaha.116.310293
65. Maimaitijiang A, Zhuang X, Jiang X, Li Y. Dynamin-related protein inhibitor downregulates reactive oxygen species levels to indirectly suppress high glucose-induced hyperproliferation of vascular smooth muscle cells. Biochem Biophys Res Commun. (2016) 471:474–8. doi: 10.1016/j.bbrc.2016.02.051
66. Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, et al. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell. (2017) 40:583–94.e6. doi: 10.1016/j.devcel.2017.02.020
67. Zhuang X, Maimaitijiang A, Li Y, Shi H, Jiang X. Salidroside inhibits high-glucose induced proliferation of vascular smooth muscle cells via inhibiting mitochondrial fission and oxidative stress. Exp Ther Med. (2017) 14:515–24. doi: 10.3892/etm.2017.4541
68. Zhu Y, Ji JJ, Yang R, Han XQ, Sun XJ, Ma WQ, et al. Lactate accelerates calcification in VSMCs through suppression of BNIP3-mediated mitophagy. Cell Signal. (2019) 58:53–64. doi: 10.1016/j.cellsig.2019.03.006
69. Moulis M, Grousset E, Faccini J, Richetin K, Thomas G, Vindis C. The multifunctional sorting protein PACS-2 controls mitophagosome formation in human vascular smooth muscle cells through mitochondria-ER contact sites. Cells. (2019) 8:638. doi: 10.3390/cells8060638
70. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. (2018) 20:1013–22. doi: 10.1038/s41556-018-0176-2
71. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. (2010) 11:45–51. doi: 10.1038/embor.2009.256
72. Wang X, Han X, Liu N. FUNDC1 mediated mitophagy alleviates rat aortic smooth muscle cell proliferation, migration induced by angiotensin II. Atherosclerosis. (2017) 263:e132. doi: 10.1016/j.atherosclerosis.2017.06.420
73. Sekine S, Youle RJ. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. (2018) 16:2. doi: 10.1186/s12915-017-0470-7
74. Bingol B, Sheng M. Mechanisms of mitophagy: PINK1, parkin, USP30 and beyond. Free Radic Biol Med. (2016) 100:210–22. doi: 10.1016/j.freeradbiomed.2016.04.015
75. Nahapetyan H, Faccini J, Mucher E, Grazide M-H, Elbaz M, Martinet W, et al. Defective autophagy in vascular smooth muscle cells promotes an unstable atherosclerotic plaque phenotype and increased expression of mitophagy markers in Apo E-/- mice. Atherosclerosis. (2017) 263:e5. doi: 10.1016/j.atherosclerosis.2017.06.043
76. Swiader A, Nahapetyan H, Faccini J, D’Angelo R, Mucher E, Elbaz M, et al. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget. (2016) 7:28821–35. doi: 10.18632/oncotarget.8936
77. Das S, Mitrovsky G, Vasanthi HR, Das DK. Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid Med Cell Longev. (2014) 2014:345105. doi: 10.1155/2014/345105
78. Wang S, Deng Z, Ma Y, Jin J, Qi F, Li S, et al. The role of autophagy and mitophagy in bone metabolic disorders. Int J Biol Sci. (2020) 16:2675–91. doi: 10.7150/ijbs.46627
79. Chien SJ, Lin IC, Hsu CN, Lo MH, Tain YL. Homocysteine and arginine-to-asymmetric dimethylarginine ratio associated with blood pressure abnormalities in children with early chronic kidney disease. Circ J. (2015) 79:2031–7. doi: 10.1253/circj.CJ-15-0412
80. Kotur-Stevuljeviæ J, Peco-Antiæ A, Spasiæ S, Stefanoviæ A, Paripoviæ D, Kostiæ M, et al. Hyperlipidemia, oxidative stress, and intima media thickness in children with chronic kidney disease. Pediatr Nephrol. (2013) 28:295–303. doi: 10.1007/s00467-012-2323-5
81. Byon CH, Heath JM, Chen Y. Redox signaling in cardiovascular pathophysiology: a focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol. (2016) 9:244–53. doi: 10.1016/j.redox.2016.08.015
82. Hu CT, Shao YD, Liu YZ, Xiao X, Cheng ZB, Qu SL, et al. Oxidative stress in vascular calcification. Clin Chim Acta. (2021) 519:101–10. doi: 10.1016/j.cca.2021.04.012
83. Liu L, Liu ZZ, Chen H, Zhang GJ, Kong YH, Kang XX. Oxidized low-density lipoprotein and β-glycerophosphate synergistically induce endothelial progenitor cell ossification. Acta Pharmacol Sin. (2011) 32:1491–7. doi: 10.1038/aps.2011.128
84. Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. (2008) 283:15319–27. doi: 10.1074/jbc.M800021200
85. Zhao MM, Xu MJ, Cai Y, Zhao G, Guan Y, Kong W, et al. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. (2011) 79:1071–9. doi: 10.1038/ki.2011.18
86. Zhao G, Xu MJ, Zhao MM, Dai XY, Kong W, Wilson GM, et al. Activation of nuclear factor-kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int. (2012) 82:34–44. doi: 10.1038/ki.2012.40
87. Zhang R, Jiang M, Zhang J, Qiu Y, Li D, Li S, et al. Regulation of the cerebrovascular smooth muscle cell phenotype by mitochondrial oxidative injury and endoplasmic reticulum stress in simulated microgravity rats via the PERK-eIF2α-ATF4-CHOP pathway. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165799. doi: 10.1016/j.bbadis.2020.165799
88. Duan XH, Chang JR, Zhang J, Zhang BH, Li YL, Teng X, et al. Activating transcription factor 4 is involved in endoplasmic reticulum stress-mediated apoptosis contributing to vascular calcification. Apoptosis. (2013) 18:1132–44. doi: 10.1007/s10495-013-0861-3
89. Dröse S, Brandt U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J Biol Chem. (2008) 283:21649–54. doi: 10.1074/jbc.M803236200
90. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
91. Matough FA, Budin SB, Hamid ZA, Alwahaibi N, Mohamed J. The role of oxidative stress and antioxidants in diabetic complications. Sultan Qaboos Univ Med J. (2012) 12:5–18. doi: 10.12816/0003082
92. Basta G, Lazzerini G, Del Turco S, Ratto GM, Schmidt AM, De Caterina R. At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products. Arterioscler Thromb Vasc Biol. (2005) 25:1401–7. doi: 10.1161/01.ATV.0000167522.48370.5e
93. Dikalov SI, Nazarewicz RR, Bikineyeva A, Hilenski L, Lassègue B, Griendling KK, et al. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid Redox Signal. (2014) 20:281–94. doi: 10.1089/ars.2012.4918
94. Canugovi C, Stevenson MD, Vendrov AE, Hayami T, Robidoux J, Xiao H, et al. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. (2019) 26:101288. doi: 10.1016/j.redox.2019.101288
95. Tada Y, Yano S, Yamaguchi T, Okazaki K, Ogawa N, Morita M, et al. Advanced glycation end products-induced vascular calcification is mediated by oxidative stress: functional roles of NAD(P)H-oxidase. Horm Metab Res. (2013) 45:267–72. doi: 10.1055/s-0032-1329965
96. Brodeur MR, Bouvet C, Bouchard S, Moreau S, Leblond J, Deblois D, et al. Reduction of advanced-glycation end products levels and inhibition of RAGE signaling decreases rat vascular calcification induced by diabetes. PLoS One. (2014) 9:e85922. doi: 10.1371/journal.pone.0085922
97. Zhou Y, Zhang MJ, Li BH, Chen L, Pi Y, Yin YW, et al. PPARγ inhibits VSMC proliferation and migration via attenuating oxidative stress through upregulating UCP2. PLoS One. (2016) 11:e0154720. doi: 10.1371/journal.pone.0154720
98. Lee KM, Lee EO, Lee YR, Joo HK, Park MS, Kim CS, et al. APE1/Ref-1 inhibits phosphate-induced calcification and osteoblastic phenotype changes in vascular smooth muscle cells. Int J Mol Sci. (2017) 18:2053. doi: 10.3390/ijms18102053
99. Yang H, Chen XY, Kuang SJ, Zhou MY, Zhang L, Zeng Z, et al. Abnormal Ca(2+) handling contributes to the impairment of aortic smooth muscle contractility in Zucker diabetic fatty rats. J Mol Cell Cardiol. (2020) 141:82–92. doi: 10.1016/j.yjmcc.2020.03.009
100. Nilsson J, Nilsson LM, Chen YW, Molkentin JD, Erlinge D, Gomez MF. High glucose activates nuclear factor of activated T cells in native vascular smooth muscle. Arterioscler Thromb Vasc Biol. (2006) 26:794–800. doi: 10.1161/01.Atv.0000209513.00765.13
101. Reid IR, Birstow SM, Bolland MJ. Calcium and cardiovascular disease. Endocrinol Metab (Seoul). (2017) 32:339–49. doi: 10.3803/EnM.2017.32.3.339
102. Kudryavtseva O, Aalkjaer C, Matchkov VV. Vascular smooth muscle cell phenotype is defined by Ca2+-dependent transcription factors. FEBS J. (2013) 280:5488–99. doi: 10.1111/febs.12414
103. House SJ, Potier M, Bisaillon J, Singer HA, Trebak M. The non-excitable smooth muscle: calcium signaling and phenotypic switching during vascular disease. Pflugers Arch. (2008) 456:769–85. doi: 10.1007/s00424-008-0491-8
104. Wang Y, Li X, Zhao F. MCU-dependent mROS generation regulates cell metabolism and cell death modulated by the AMPK/PGC-1α/SIRT3 signaling pathway. Front Med (Lausanne). (2021) 8:674986. doi: 10.3389/fmed.2021.674986
105. Phadwal K, Feng D, Zhu D, MacRae VE. Autophagy as a novel therapeutic target in vascular calcification. Pharmacol Ther. (2020) 206:107430. doi: 10.1016/j.pharmthera.2019.107430
106. Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y, et al. MCU-dependent mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene. (2017) 36:5897–909. doi: 10.1038/onc.2017.167
107. Chen Z, Zhou Q, Chen J, Yang Y, Chen W, Mao H, et al. MCU-dependent mitochondrial calcium uptake-induced mitophagy contributes to apelin-13-stimulated VSMCs proliferation. Vascul Pharmacol. (2022) 144:106979. doi: 10.1016/j.vph.2022.106979
108. Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. (2009) 1787:1309–16. doi: 10.1016/j.bbabio.2009.01.005
109. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. (2004) 287:C817–33. doi: 10.1152/ajpcell.00139.2004
110. Gao P, Jiang Y, Wu H, Sun F, Li Y, He H, et al. Inhibition of mitochondrial calcium overload by SIRT3 prevents obesity- or age-related whitening of brown adipose tissue. Diabetes. (2020) 69:165–80. doi: 10.2337/db19-0526
111. Gao P, Yan Z, Zhu Z. Mitochondria-associated endoplasmic reticulum membranes in cardiovascular diseases. Front Cell Dev Biol. (2020) 8:604240. doi: 10.3389/fcell.2020.604240
112. Carreras-Sureda A, Pihán P, Hetz C. Calcium signaling at the endoplasmic reticulum: fine-tuning stress responses. Cell Calcium. (2018) 70:24–31. doi: 10.1016/j.ceca.2017.08.004
113. Hamilton S, Terentyeva R, Martin B, Perger F, Li J, Stepanov A, et al. Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res Cardiol. (2020) 115:38. doi: 10.1007/s00395-020-0797-z
114. Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, Lin YH, Hausenloy DJ. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine. (2020) 57:102884. doi: 10.1016/j.ebiom.2020.102884
115. Alevriadou BR, Patel A, Noble M, Ghosh S, Gohil VM, Stathopulos PB, et al. Molecular nature and physiological role of the mitochondrial calcium uniporter channel. Am J Physiol Cell Physiol. (2021) 320:C465–82. doi: 10.1152/ajpcell.00502.2020
116. Morales PE, Torres G, Sotomayor-Flores C, Peña-Oyarzún D, Rivera-Mejías P, Paredes F, et al. GLP-1 promotes mitochondrial metabolism in vascular smooth muscle cells by enhancing endoplasmic reticulum-mitochondria coupling. Biochem Biophys Res Commun. (2014) 446:410–6. doi: 10.1016/j.bbrc.2014.03.004
117. Metallo CM, Vander Heiden MG. Metabolism strikes back: metabolic flux regulates cell signaling. Genes Dev. (2010) 24:2717–22. doi: 10.1101/gad.2010510
118. Shi J, Yang Y, Cheng A, Xu G, He F. Metabolism of vascular smooth muscle cells in vascular diseases. Am J Physiol Heart Circ Physiol. (2020) 319:H613–31. doi: 10.1152/ajpheart.00220.2020
119. Barron JT, Parrillo JE. Production of lactic acid and energy metabolism in vascular smooth muscle: effect of dichloroacetate. Am J Physiol. (1995) 268(Pt 2):H713–9. doi: 10.1152/ajpheart.1995.268.2.H713
120. Werle M, Kreuzer J, Höfele J, Elsässer A, Ackermann C, Katus HA, et al. Metabolic control analysis of the Warburg-effect in proliferating vascular smooth muscle cells. J Biomed Sci. (2005) 12:827–34. doi: 10.1007/s11373-005-9010-5
121. Salabei JK, Hill BG. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol. (2013) 1:542–51. doi: 10.1016/j.redox.2013.10.011
122. Lin X, Xiao Z, Chen T, Liang SH, Guo H. Glucose metabolism on tumor plasticity, diagnosis, and treatment. Front Oncol. (2020) 10:317. doi: 10.3389/fonc.2020.00317
123. Vaupel P, Multhoff G. Revisiting the Warburg effect: historical dogma versus current understanding. J Physiol. (2021) 599:1745–57. doi: 10.1113/jp278810
124. Glancy B, Kane DA, Kavazis AN, Goodwin ML, Willis WT, Gladden LB. Mitochondrial lactate metabolism: history and implications for exercise and disease. J Physiol. (2021) 599:863–88. doi: 10.1113/jp278930
125. Rufino M, Hernández D, Barrios Y, Salido E. The GLUT-1 XbaI gene polymorphism is associated with vascular calcifications in nondiabetic uremic patients. Nephron Clin Pract. (2008) 108:c182–7. doi: 10.1159/000118940
126. Ishitobi M, Hosaka T, Morita N, Kondo K, Murashima T, Kitahara A, et al. Serum lactate levels are associated with serum alanine aminotransferase and total bilirubin levels in patients with type 2 diabetes mellitus: a cross-sectional study. Diabetes Res Clin Pract. (2019) 149:1–8. doi: 10.1016/j.diabres.2019.01.028
127. Wu M, Huang X, Yan D, Pan H, Li F, Ren M, et al. Interactions among endotoxin, uric acid, and lactate in relation to the risk of type 2 diabetes: a population-based study. J Diabetes. (2020) 12:605–15. doi: 10.1111/1753-0407.13039
128. Broskey NT, Zou K, Dohm GL, Houmard JA. Plasma lactate as a marker for metabolic health. Exerc Sport Sci Rev. (2020) 48:119–24. doi: 10.1249/jes.0000000000000220
129. Vesely ED, Heilig CW, Brosius FC III. GLUT1-induced cFLIP expression promotes proliferation and prevents apoptosis in vascular smooth muscle cells. Am J Physiol Cell Physiol. (2009) 297:C759–65. doi: 10.1152/ajpcell.00213.2009
130. Idelevich A, Rais Y, Monsonego-Ornan E. Bone Gla protein increases HIF-1alpha-dependent glucose metabolism and induces cartilage and vascular calcification. Arterioscler Thromb Vasc Biol. (2011) 31:e55–71. doi: 10.1161/atvbaha.111.230904
131. Zhou J, Deo BK, Hosoya K, Terasaki T, Obrosova IG, Brosius FC III, et al. Increased JNK phosphorylation and oxidative stress in response to increased glucose flux through increased GLUT1 expression in rat retinal endothelial cells. Invest Ophthalmol Vis Sci. (2005) 46:3403–10. doi: 10.1167/iovs.04-1064
132. Chen NX, Duan D, O’Neill KD, Moe SM. High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol Dial Transplant. (2006) 21:3435–42. doi: 10.1093/ndt/gfl429
133. Moe SM, Duan D, Doehle BP, O’Neill KD, Chen NX. Uremia induces the osteoblast differentiation factor Cbfa1 in human blood vessels. Kidney Int. (2003) 63:1003–11. doi: 10.1046/j.1523-1755.2003.00820.x
134. Mokas S, Larivière R, Lamalice L, Gobeil S, Cornfield DN, Agharazii M, et al. Hypoxia-inducible factor-1 plays a role in phosphate-induced vascular smooth muscle cell calcification. Kidney Int. (2016) 90:598–609. doi: 10.1016/j.kint.2016.05.020
135. Balogh E, Tóth A, Méhes G, Trencsényi G, Paragh G, Jeney V. Hypoxia triggers osteochondrogenic differentiation of vascular smooth muscle cells in an HIF-1 (Hypoxia-Inducible Factor 1)-dependent and reactive oxygen species-dependent manner. Arterioscler Thromb Vasc Biol. (2019) 39:1088–99. doi: 10.1161/atvbaha.119.312509
136. Tóth A, Csiki DM, Nagy B Jr., Balogh E, Lente G, Ababneh H, et al. Daprodustat accelerates high phosphate-induced calcification through the activation of HIF-1 signaling. Front Pharmacol. (2022) 13:798053. doi: 10.3389/fphar.2022.798053
137. Thompson CB. Into thin air: how we sense and respond to hypoxia. Cell. (2016) 167:9–11. doi: 10.1016/j.cell.2016.08.036
138. Lambert CM, Roy M, Robitaille GA, Richard DE, Bonnet S. HIF-1 inhibition decreases systemic vascular remodelling diseases by promoting apoptosis through a hexokinase 2-dependent mechanism. Cardiovasc Res. (2010) 88:196–204. doi: 10.1093/cvr/cvq152
139. Heiss EH, Schachner D, Donati M, Grojer CS, Dirsch VM. Increased aerobic glycolysis is important for the motility of activated VSMC and inhibited by indirubin-3’-monoxime. Vascul Pharmacol. (2016) 83:47–56. doi: 10.1016/j.vph.2016.05.002
140. Zhao X, Tan F, Cao X, Cao Z, Li B, Shen Z, et al. PKM2-dependent glycolysis promotes the proliferation and migration of vascular smooth muscle cells during atherosclerosis. Acta Biochim Biophys Sin (Shanghai). (2020) 52:9–17. doi: 10.1093/abbs/gmz135
141. Lee YB, Min JK, Kim JG, Cap KC, Islam R, Hossain AJ, et al. Multiple functions of pyruvate kinase M2 in various cell types. J Cell Physiol. (2022) 237:128–48. doi: 10.1002/jcp.30536
142. Ma WQ, Sun XJ, Zhu Y, Liu NF. PDK4 promotes vascular calcification by interfering with autophagic activity and metabolic reprogramming. Cell Death Dis. (2020) 11:991. doi: 10.1038/s41419-020-03162-w
143. Rashdan NA, Sim AM, Cui L, Phadwal K, Roberts FL, Carter R, et al. Osteocalcin regulates arterial calcification via altered Wnt signaling and glucose metabolism. J Bone Miner Res. (2020) 35:357–67. doi: 10.1002/jbmr.3888
144. Zhu Y, Ma WQ, Han XQ, Wang Y, Wang X, Liu NF. Advanced glycation end products accelerate calcification in VSMCs through HIF-1α/PDK4 activation and suppress glucose metabolism. Sci Rep. (2018) 8:13730. doi: 10.1038/s41598-018-31877-6
145. Leem J, Lee IK. Mechanisms of vascular calcification: the pivotal role of pyruvate dehydrogenase kinase 4. Endocrinol Metab (Seoul). (2016) 31:52–61. doi: 10.3803/EnM.2016.31.1.52
146. Lee SJ, Jeong JY, Oh CJ, Park S, Kim JY, Kim HJ, et al. Pyruvate dehydrogenase kinase 4 promotes vascular calcification via SMAD1/5/8 phosphorylation. Sci Rep. (2015) 5:16577. doi: 10.1038/srep16577
147. Yang R, Zhu Y, Wang Y, Ma W, Han X, Wang X, et al. HIF-1α/PDK4/autophagy pathway protects against advanced glycation end-products induced vascular smooth muscle cell calcification. Biochem Biophys Res Commun. (2019) 517:470–6. doi: 10.1016/j.bbrc.2019.07.102
148. Ma WQ, Han XQ, Wang Y, Wang X, Zhu Y, Liu NF. Nε-carboxymethyl-lysine promotes calcium deposition in VSMCs via intracellular oxidative stress-induced PDK4 activation and alters glucose metabolism. Oncotarget. (2017) 8:112841–112854. doi: 10.18632/oncotarget.22835
149. Nagy A, Pethõ D, Gáll T, Zavaczki E, Nyitrai M, Posta J, et al. Zinc inhibits HIF-prolyl hydroxylase inhibitor-aggravated VSMC calcification induced by high phosphate. Front Physiol. (2019) 10:1584. doi: 10.3389/fphys.2019.01584
150. Meng J, Lv Z, Zhang Y, Wang Y, Qiao X, Sun C, et al. Precision redox: the key for antioxidant pharmacology. Antioxid Redox Signal. (2021) 34:1069–82. doi: 10.1089/ars.2020.8212
151. Mehrabani S, Bagherniya M, Askari G, Read MI, Sahebkar A. The effect of fasting or calorie restriction on mitophagy induction: a literature review. J Cachexia Sarcopenia Muscle. (2020) 11:1447–58. doi: 10.1002/jcsm.12611
152. Savencu CE, Linţa A, Farcaş G, Bînã AM, Creţu OM, Maliţa DC, et al. Impact of dietary restriction regimens on mitochondria, heart, and endothelial function: a brief overview. Front Physiol. (2021) 12:768383. doi: 10.3389/fphys.2021.768383
153. Liu M, Dudley SC Jr. Magnesium, oxidative stress, inflammation, and cardiovascular disease. Antioxidants (Basel). (2020) 9:907. doi: 10.3390/antiox9100907
154. Daw CC, Ramachandran K, Enslow BT, Maity S, Bursic B, Novello MJ, et al. Lactate elicits ER-mitochondrial Mg(2+) dynamics to integrate cellular metabolism. Cell. (2020) 183:474–89.e17. doi: 10.1016/j.cell.2020.08.049
Keywords: vascular calcification, vascular smooth muscle cell (VSMC), mitochondria, phenotypic switch, cardiovascular disease
Citation: Liu YZ, Li ZX, Zhang LL, Wang D and Liu YP (2022) Phenotypic plasticity of vascular smooth muscle cells in vascular calcification: Role of mitochondria. Front. Cardiovasc. Med. 9:972836. doi: 10.3389/fcvm.2022.972836
Received: 19 June 2022; Accepted: 20 September 2022;
Published: 12 October 2022.
Edited by:
Olga Tura-Ceide, Hospital Clinic of Barcelona, SpainReviewed by:
Joon-Young Park, Baylor University, United StatesCopyright © 2022 Liu, Li, Zhang, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Wang, d2FuZ3hpYW9kYW5kYW4yMDEzQDE2My5jb20=; Yi Ping Liu, eXBsaXUxOTY2QDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.