 ChangXin Sun1,2†
ChangXin Sun1,2† ZePing Wang1,2†
ZePing Wang1,2† LanQing Hu2
LanQing Hu2 XiaoNan Zhang2
XiaoNan Zhang2 JiYe Chen2
JiYe Chen2 ZongLiang Yu2
ZongLiang Yu2 LongTao Liu2*
LongTao Liu2* Min Wu3*
Min Wu3*- 1Beijing University of Chinese Medicine, Beijing, China
- 2Xiyuan Hospital, China Academy of Chinese Medical Sciences, Beijing, China
- 3Guang'anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
Increasing researches have considered gut microbiota as a new “metabolic organ,” which mediates the occurrence and development of metabolic diseases. In addition, the liver is an important organ of lipid metabolism, and abnormal lipid metabolism can cause the elevation of blood lipids. Among them, elevated low-density lipoprotein cholesterol (LDL-C) is related with ectopic lipid deposition and metabolic diseases, and statins are widely used to lower LDL-C. In recent years, the gut microbiota has been shown to mediate statins efficacy, both in animals and humans. The effect of statins on microbiota abundance has been deeply explored, and the pathways through which statins reduce the LDL-C levels by affecting the abundance of microbiota have gradually been explored. In this review, we discussed the interaction between gut microbiota and cholesterol metabolism, especially the cholesterol-lowering effect of statins mediated by gut microbiota, via AMPK-PPARγ-SREBP1C/2, FXR and PXR-related, and LPS-TLR4-Myd88 pathways, which may help to explain the individual differences in statins efficacy.
Introduction
Blood lipids come from two sources. Exogenous fat is absorbed through the intestine and then decomposed into lipid particles, and endogenous lipids particles are generated in liver. All lipids particles can only be transported in the blood by binding to the corresponding apolipoprotein. Dyslipidemia mainly includes hypertriglyceridemia, low high-density lipoprotein cholesterol (HDL-C), high low-density lipoprotein cholesterol (LDL-C), and combined hyperlipidemia. In addition, high level of LDL-C can cause ectopic lipid deposition and associated metabolic diseases, such as non-alcoholic fatty liver disease (1), hypertension (2), and atherosclerosis (AS) (3). Among these, stroke and ischemic heart disease, which are AS-related diseases, are main causes of premature death among the Chinese population (4). Therefore, as the most effective drug against AS, statins remain an important choice especially for those whose LDL-C cannot be sufficiently reduced through modification of diet and living habits (5). However, elder patients with other comorbidities are more prone to the side effects of statins and face increased risks of musculoskeletal symptoms, diabetes, and hemorrhagic stroke (5, 6), which largely limits the use of statins.
Gut microbiota regulates several diseases, including liver diseases (7), neuropsychiatric disorders (8), and metabolic diseases, including type 2 diabetes, dyslipidemia, and obesity (9), mainly by regulating energy homeostasis, glucose metabolism, lipid metabolism, and other metabolic processes (10). As a critical part of the gut-liver axis, gut microbiota performs several functions in the metabolism of short-chain fatty acid (SCFA), bile acid (BA), trimethylamine-N-oxide (TMAO), and lipopolysaccharides (LPS), and has a significant impact on immunological, metabolic, structural, and neurological landscapes in the human body (11). Presently, the specific effects of lipid-lowering drugs, such as statins, on the gut microbiota remain unclear (12). This review elucidates how various statins affect the gut microbiota and LDL-C metabolism through the gut-liver axis to discover their potential therapeutic implications and explain why the efficacy of statins varies in different populations.
Microbial effects on cholesterol metabolism
Gut microbiota, which is influenced by diet and living environment, is individually unique and highly related to cholesterol metabolism (13). Commonly, BAs and other active substances synthesized by the liver are secreted into the upper part of the small intestine, and affect the abundance of gut microbiota and microbiota-derived metabolites (MDMs) in jejunum, ileum and colon. Similarly, based on the mesenteric vein-portal vein system, metabolites in the gut are absorbed and transported directly to the liver through the gut mucosa and then transformed into metabolic raw materials and regulatory molecules. SCFA (14), secondary bile acid (SBA) (15), TMAO (16) and LPS (17) are main MDMs that can significantly regulate the cholesterol metabolism and even reduce LDL-C through the gut-liver axis. Thus, the gut microbiota and metabolites play key roles in regulating LDL-C levels.
The SCFAs, BAs, TMAO, and LPS are major gut microbiota metabolites that broadly affect the human cholesterol metabolism. SCFAs mainly include acetate, propionate, and butyrate, which are produced by the human gut microbiota through the fermentation of dietary fiber and amino acids obtained via hydrolysis using various pathways (18). The human gut microbiota includes the following genera: Lactobacillus, Eubacterium, Faecalis, Bifidobacterium, Clostridium, Bacteroides and Butyrimionas (19–22). Lactobacillus (23–25) and Bifidobacterium (26, 27). Increase in Lactobacillus and Bifidobacterium are positively correlated with the total SCFAs (28). Additionally, most members of the Prevotellaceae, Clostridiaceae, Ruminococcaceae, Lactobacillaceae, and Lachnospiraceae families are putative butyrate producers and own anti-inflammatory properties (29). Clostridiaceae and Lachnospiraceae are also acetate producers. Moreover, the Erysipelotrichaceae family is primarily responsible for the production of acetate and butyrate, based on metatranscriptome profiles (30). Studies have revealed that the Coriobacteriaceae family positively correlates with the serum LDL-C levels in mice fed with a high fat diet (HFD) (31).
BAs mainly exist as primary BAs, chenodeoxycholic acid (CDCA), and cholic acid (CA) before fermentation by gut microbiota, whereas the SBAs, deoxycholic acid (DCA), and lithocholic acid (LCA) are generated by Clostridium and some strains of Lachnospiraceae and Peptostreptococcaceae families in the gut; DCA and LCA are the main products (32). The Clostridium (33–35) and Lachnospiraceae (35) share the same trend of change with LDL-C levels. An experiment revealed that the percentage of Gram-positive bacteria, especially Clostridium, is significantly increased by HFD, which results in the production of DCA in the feces. Suppressing Gram-positive bacteria, including Clostridium, with antibiotics decreased the fecal DCA in this experiment (36). Lactobacillus can increase the relative proportion of intestinal Clostridium and the production of SBAs (DCA and LCA) in rats fed in a high-cholesterol diet (37). Moreover, Lactobacillus decreased the absorption of BAs and increased the excretion of BAs in feces (38). A significant correlation exists between the increase in Bifidobacterium and the decrease in fecal LCA (39). Low abundance of Bacteroidetes and high abundance of Erysipelotrichaceae indicate excessive SBAs in feces (40).
Methanogenic bacteria have been reported to deplete both trimethylamine (TMA) and TMAO (41, 42), and a randomized controlled trial showed that probiotic supplementation (such as Lactobacillus and Bifidobacterium) significantly increased the TMAO levels (43). Naghipour et al. revealed the production of TMA is related to three biochemical pathways. Based on genetic testing, comparing the genes involved in the biochemical pathways, it has been confirmed that the Bacteroides lack the related genes, whereas these genes exist in the Firmicutes, Proteobacteria and Actinobacteria phyla (44). Among them, reduction in Firmicutes (45), Proteobacteria and Actinobacteria (46) is often accompanied by reduction in the LDL-C levels. Falony et al. (47) suggested that Bacteroidetes did not produce TMA, and Wang et al. (48) observed that Bacteroides were negatively correlated with the plasma TMAO levels. However, a previous research revealed that TMA and TMAO levels were associated with an increased Firmicutes/Bacteroidetes ratio (49), which was consistent with the above findings.
LPS is specific to Gram-negative bacteria. At the phylum level, Bacteroidetes and Proteobacteria are the two most dominant Gram-negative bacteria in the gut microbiota. Although the relative abundance of Bacteroidetes decreased significantly after the HFD intervention, the relative abundance of Proteobacteria was observed to increase (50). In addition, the absolute abundance of the gut microbiota doubled by HFD intervention (51), which indicated that although the relative abundance of Bacteroidetes was reduced, the total number may not have changed significantly. We observed that SCFAs, BAS, TMAO, and LPS are related to the ability of the gut microbiota to regulate the LDL-C levels via metabolites (Table 1). However, correlative research reports are insufficient, and long-term studies are still needed to determine the impact of gut microbiota on SCFAs, BAs, TMAO, and LPS.

Table 1. Relationship between different microbiota and its metabolites.
Points of contact between gut microbiota and cholesterol metabolism
SCFAs
Different SCFAs play different roles in the body; acetate's concentration is the highest in the blood, propionate helps in liver gluconeogenesis, and butyrate is the main energy source for the colon cells (52). Earlier experiments have demonstrated a relationship between SCFAs and LDL-C (53, 54). In addition, SCFAs significantly reduced the cholesterol levels in the plasma and liver of male rats (55), and the proliferation of bacteria that produce SCFAs can effectively reduce the blood cholesterol in hamsters with hypercholesterolemia (56). A HFD reduced the formation of SCFAs, whereas supplementing fermentable dietary fiber affected the SCFAs and reduced the plasma cholesterol levels (57).
A Previous research revealed that oral acetate (58, 59), propionate (60), and butyrate supplementation (61, 62) can improve the expression of lipid metabolism genes in mice with a HFD. Examples include the upregulation of cyclic adenosine monophosphate-responsive element binding protein-regulated transcription coactivator (CRTC) 2 and phosphorylated acetyl-CoA carboxylase (p-ACC), or downregulation of peroxisome proliferator-activated receptor (PPAR) γ, acetyl-CoA carboxylase (ACC), and sterol regulatory element binding protein 1 (SREBP1) (63). Among these, PPARγ is considered as the central medium for SCFA to play a beneficial role (64). The knockout of PPARγ in the liver completely abolished the effect of SCFAs, indicating that SCFAs act through related pathways in the liver. SCFAs acetate, propionate, and butyrate were the main pathways that inhibited PPARγ expression and activated the AMP-activated protein kinase (AMPK) pathway in this experiment (64). Activating the AMPK signaling pathway inhibited the expression of SREBP1c and SREBP2 directly (65). The SREBP plays a role in activating ACC and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) (66), among which HMGCR is the key enzyme for cholesterol synthesis. Thus, a decrease in SREBP expression is often accompanied by a decrease in plasma lipoprotein and cholesterol levels (67). Suppressing the AMPK-SREBP signaling pathway regulates cholesterol metabolism and prevents high LDL-C levels, which is beneficial for lowering LDL-C levels in serum and reducing liver lipid accumulation (66).
Among the free fatty acid receptors (FFAR), FFAR 2 and 3 are generally considered as SCFA receptors; however, studies have revealed that propionate and butyrate are not significantly associated with FFAR 3 (68). Acetate, propionate, and butyrate can activate FFAR 2 in the intestine and promote the secretion and intestinal peristalsis of peptide YY (PYY) and glucagon-like peptide 1 (GLP-1). Further studies have shown that HFD can reduce the expression of GLP-1 receptors in the liver, while supplementation with butyrate, a GLP-1 sensitizer, upregulates the levels of GLP-1 receptors in the liver, which stimulates AMPK phosphorylation (69). In general, the SCFAs-FFAR 2 signaling pathway is beneficial for improving cholesterol metabolism and reducing LDL-C levels; however, there is no current literature to explain the relationship between GLP-1 and the AMPK signaling pathway.
Studies have discovered that increased SCFAs induce the upregulation of cytochrome p-450 enzyme cholesterol 7α-hydroxylase (CYP7A1) (70) and 27α-hydroxylase (CYP27A1) (71) in the liver. Upregulation of CYP7A1 promotes cholesterol converted into BAs (72), thereby increasing cholesterol consumption and lowering LDL-C levels. Dietary acetate supplementation does not increase CYP7A1 mRNA levels (59), but increased propionate (53) and butyrate (54) levels can lead to the upregulation of CYP7A1, promote cholesterol catabolism, and reduce plasma LDL-C levels. Therefore, CYP7A1 is also a pathway for SCFAs to regulate cholesterol metabolism and LDL-C levels. In summary, SCFAs are closely related to the regulation of LDL-C via AMPK, FFAR 2, and CYP7A1-related signaling pathways, which are the main signaling pathways through which SCFAs regulate lipid metabolism and improve high LDL-C levels in the liver and intestine.
Bile acids
BAs are important components of bile and are stored and concentrated in the gallbladder. BAs flow into the intestine under the control of filling and emptying of the gallbladder. Approximately 5% of BAs cannot be absorbed by hepatocytes through the hepatoenteric circulation and are excreted back into the bile (73). The liver maintains the stability of the BA pool by synthesizing BAs, which is the main catabolic pathway that consumes cholesterol in the body and is the only quantitative significant cholesterol catabolic mechanism (74). The nuclear receptor farnesoid X receptor (FXR) and cell surface receptor G protein-coupled bile acid receptor 5 (TGR5) are the key receptors required by Bas in maintaining systemic cholesterol levels and energy metabolism. The FXR and TGR5 signaling pathways regulate the liver BA synthesis and intestinal BA excretion via the FXR activation of TGR5 in the enterocytes (75).
CYP7A1 and CYP27A1 are involved in the first step of the convert of cholesterol into BAs (76); however, CYP7A1 is also a rate-limiting enzyme in the entire conversion process. CYP7A1 is simultaneously inhibited by an FXR-small heterodimer partner (SHP), which is the downstream target of FXR in the liver, whereas FXR in enterocytes inhibits CYP7A1 activation in hepatocytes by secreting fibroblast growth factor 19 (FGF19) and binding to fibroblast growth factor receptor 4 (FGFR4) on the hepatocyte membrane (77). The expression level of FGF19 was increased 16-fold in the ileum of rabbits fed in a high-cholesterol diet, and CYP7A1 decreased by 75% in the liver (78). A study by Jones revealed that the total cholesterol concentration in the liver after CYP7A1 knockout in mice was significantly higher than that in non-knockout mice (79). CDCA, CA, DCA, and LCA are all FXR agonists that can activate the FXR-CYP7A1 signaling pathway (80), which inhibits cholesterol consumption and causes abnormal cholesterol levels. Animal studies have confirmed that dietary supplementation with Lactobacillus can reduce the relative abundance of Clostridium, LDL-C/HDL-C, and the production of DCA and LCA in the intestines of rats that were fed in a high-cholesterol diet (37). In summary, changing Clostridium and Lactobacillus can regulate the production of LCA, DCA, and cholesterol consumption.
TGR5 can be activated by CDCA, CA, DCA, and LCA to promote the secretion and release of PYY and GLP-1 (81). Compared with normal mice, the expression levels of CYP7A1 and CYP27A1 increased in mice whose the TGR 5 gene was knocked out, indicating that TGR5 is involved in inhibiting the expression of CYP7A1 and CYP27A1 (82). However, there were also differences in the lipid-lowering effects of TGR5 between the female and male mice. LDL-C levels reduced significantly only in female mice whose TGR5 gene was knocked out.
Pregnane X receptor (PXR) is activated by LCA hepatoxic BAs to prevent cholestatic liver disease; however, it is not affected by CDCA, CA, or DCA (83). In addition, PXR can inhibit CYP7A1 transcription through SHP, which is a key activator of the activation of CYP7A1 gene in the hepatocytes (84). Additionally, PXR activation leads to the upregulation of SREBP2 and HMGCR and broadly induces cholesterol synthesis both in mice experiments and clinical researches (85). Kim et al. (86) discovered that PXR was related to a decrease in Lactobacillus and Bifidobacterium levels and an increase in the ratio of Firmicutes/Bacteroides. As a part of the gut-liver axis, BAs regulate the cholestatic metabolism via a multi-pathway process involving FXR, TGR5, and PXR, which are closely related to the LDL-C levels.
Trimethylamine-N-oxide
TMA, which forms TMAO in the liver, is generated by the action of the gut microbiota by using dietary precursors such as choline, choline-containing compounds, betaine, and L-carnitine (87). Among these, choline is closely related to high TG and low HDL-C levels (88). Part of the TMA produced by the gut microbiota is fermented to produce methane by the gut microbiota, and the rest is absorbed into the liver (41).
TMAO is formed by flavin monooxygenase (FMO), which is expressed in the liver. Among the five members of the FMO family, only FMO1 and FMO3 can oxidize TMA to TMAO in the liver, especially FMO3 (89). FMO3 significantly decreases the expression of genes involved in de novo lipogenesis. In high-cholesterol diet mice, FMO3 knockout can affect the biliary lipid secretion and slow down cholesterol absorption in the intestine (90). Studies have revealed that the excretion of BAs indirectly induced by TMAO is a potential target for cholesterol-lowering treatments and is an independent risk factor. TMAO could cause an increase in serum LDL-C concentration and a decrease in the relative abundance of CYP7A1 in the hepatocytes; however, there was no significant change in the relative abundance of CYP27A1 (91–93). In addition, the expression of FXR (encoded by Nr1h4) and SHP (encoded by Nr0b2) were significantly upregulated by TMAO, which indicated that the FXR-SHN pathway is regulated by TMAO via CYP7A1 and cholesterol consumption (93). In mice not interfered with the gut microbiota, dietary supplementation with TMAO or precursors of TMA reduced in vivo reverse cholesterol transport (94). Antibiotics can reduce the production of TMAO by inhibiting the gut bacteria associated with TMAO (95). Moreover, further research has shown that oral broad-spectrum antibiotics can completely offset the effects of supplementing TMA precursors and reversing cholesterol transport (94).
LPS
LPS, which is composed of lipids and polysaccharides, is a unique chemical component of the outer wall of Gram-negative bacteria. Generally, LPS is difficult to shed from the cell wall and sloughs off when bacteria die. LPS can enter the circulatory system after translocating by disrupting the intestinal epithelial barrier and increasing epithelial permeability.
In a meta-analysis of the gut microbiota and inflammatory markers, modulation of the gut microbiota reduced the LPS levels by 22–28% (96). Another meta-analysis showed that the translocation of LPS was directly related to the concentration of LDL-C, which was more pronounced in smokers or obese individuals (97, 98). LPS promotes hepatic proprotein convertase subtilisin/kexin-9 (PCSK9) synthesis that suppresses cholesterol consumption by binding to LDL-C receptors on cell membranes (17, 99). The mechanism by which LPS increases the synthesis of PCSK9 in the liver may be related to the phosphatidylinositol-3-kinase (PI3K)-Akt signaling pathway; further studies are required in this regard (100).
In few studies, the HMGCR mRNA levels in the liver increased after 4h of LPS treatment, which increased the hepatic cholesterol synthesis and serum cholesterol levels (101). In vitro studies by Ye et al. revealed that exposure to LPS resulted in the overexpression of SREBP2 and SREBP cleavage-activating protein (SCAP) (102). SCAP is required to activate SREBP1/2 in the hepatocytes, and the knockout of SCAP negatively regulated SREBP1/2 expression and cholesterol synthesis (103). Studies have demonstrated that LPS-induced inflammatory response leads to the downregulation of FXR, SHP, and CYP7A1, resulting in downregulation of LDL-C clearance and cholesterol consumption in the long term (104, 105). In addition, LPS inhibited the expression of liver receptor homolog (LRH) that can positively regulate CYP7A1, which may explain the relationship between CYP7A1, FXR and SHP (105).
Toll-like receptor 4 (TLR4) is considered as a potential receptor for LPS, and activates by an adaptor protein, such as myeloid differentiation factor 88 (Myd88). Prenatal exposure of mice to LPS (intraperitoneal injection) resulted in significantly elevated serum LDL-C levels and high LDL-C levels in the offspring. Cao et al. (2019) suggested more pronounce serum LDL-C changes in TLR2 knockout offspring mice and indicated that this may be due to a compensatory increase in TLR4 caused by TLR2 deficiency, manifested by an increased expression of TLR4 and Myd88 overactivity. Thus, the presence of LPS led to elevated serum LDL-C levels by activating TLR4 (106). Studies have discovered that the TLR4-Myd88 signaling pathway is related to lipid metabolism, as shown by the fact that the lipid-lowering effects of multiple lipid-lowering drugs are eliminated in Myd88 knockout mice (107). Elevated LPS in hepatocytes promoted the expression of SREBP2 by activating TLR4-Myd88 and the relative inflammatory response pathway (JNK/c-Jun) (108, 109).
Statins-related potential pathways regulating cholesterol metabolism
Statins and gut microbiota
Statins are well known as the most commonly used cholesterol-lowering drugs, which can directly reduce plasma cholesterol levels, especially LDL-C (110), by inhibiting cholesterol synthesis and HMGCR. The cholesterol-lowering effect of statins is closely related to the gut microbiota and can be impaired for the imbalance if gut microbiota (12, 51). In addition, different statins, such as atorvastatin, simvastatin, and rosuvastatin, have different effects on different microbiota (111) and thereby affect MDM (Table 2).

Table 2. The effect of some statins on the gut microbiota.
Compared to HFD mice that received atorvastatin, LDL-C levels were significantly higher in HFD mice that received atorvastatin and antibiotics. In this experiment, antibiotic-treated mice and mice without antibiotics showed different responses to statins, and LDL-lowering effect varied. LDL-C was lower in HFD mice that only received atorvastatin, whereas the results of HFD mice treated with atorvastatin and antibiotics were insignificant compared to untreated HFD mice (51). The diversity of gut microbiota changes after statin treatment (12), and is possibly related to a decrease in the incidence of gut microbiota imbalance (117). Sun et al. have revealed that hyperlipidemic patients with atorvastatin sensitivity manifested a higher diversity of gut microbiota, such as greater number of Lactobacillus, Eubacterium, Faecalibacterium, Bifidobacterium and fewer Clostridium genera (112).
Animal experiments have confirmed that the effect of simvastatin in reducing of serum LDL-C is affected by the gut microbiota. In a control experiment of simvastatin and antibiotics combined with simvastatin in HFD mice, serum LDL-C levels were significantly reduced in HFD mice that were treated with simvastatin, whereas serum LDL-C levels were higher in mice that were treated with antibiotics and simvastatin (118). A previous study showed that simvastatin changed the composition of gut microbiota in rats fed with HFD and caused an elevation in Ruminococcaceae and Lactobacillus levels (113). Another study showed that the relative abundance of Firmicutes and the portion of Ruminococcaceae decreased, while Bacteroidetes increased when followed simvastatin therapy (114).
Nolan et al. have suggested that the intervention of rosuvastatin in mice with HFD resulted in a significant increase in the Lachnospiraceae and Erysipelotrichaceae families and a significant decrease in Proteobacteria phylum, Coriobacteriaceae, Akkermansiaceae family, and Akkermansia (115). However, a clinical study revealed that the Firmicutes and Fusobacterium phyla and Ruminococcaceae, Clostridiaceae, Lachnospiraceae, Coriobacteriaceae and Erysipelotrichaceae families were negatively correlated with the LDL-lowering effect of rosuvastatin, while the Bacteroidaceae family had a positive correlation (116). The results of Lachnospiraceae and Erysipelotrichaceae families were opposite in the two experiments, and the changes in the rest of the gut microbiota in the two experiments were consistent. The application of rosuvastatin reduced the Firmicutes/Bacteroidetes ratio and increased the abundance of Bacteroides and Butyricimonas simultaneously (22). We found that statins can regulate cholesterol metabolism through MDM, and the major gut microbiota affected by statins (Figure 1).
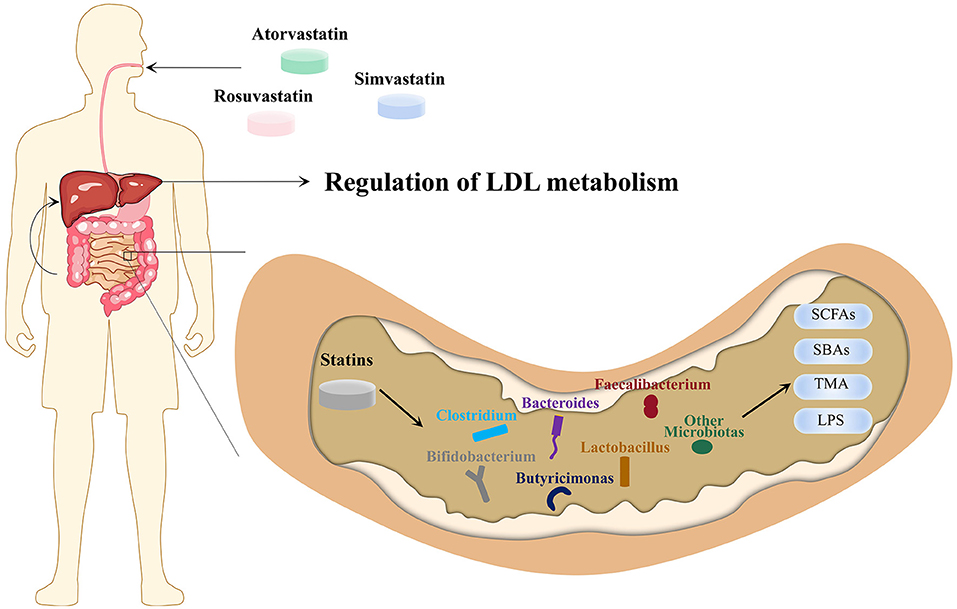
Figure 1. Microbiota-driven, LDL-lowering effect of statins. Atorvastatin, simvastatin and rosuvastatin enter the intestine via the upper gastrointestinal tract through oral administration and affect the absolute and relative abundance of gut microbiota, which in turn affects SCFAs, SBAs, TMA, LPS and other MDMs. The MDMs are transported to the liver through the portal venous system, ultimately achieving the purpose of regulating cholesterol metabolism and LDL-C levels. LPS, lipopolysaccharides; MDMs, microbiota-derived metabolites; LDL-C, low-density lipoprotein cholesterol; SBAs, secondary bile acid; SCFAs, short-chain fatty acid; TMA, trimethylamine.
AMPK-PPARγ-SREBP1C/2
Regarding SCFA, there are more gut microbiota that produce SCFA in patients with hyperlipidemia who are sensitive to atorvastatin (112). The application of atorvastatin reduced the Firmicutes/Bacteroidetes ratio to a level close to that of normal mice (22), among which the abundance of butyrate-producing Firmicutes (such as the Faecalibacterium) had been increased by statins (119). Similarly butyrate-producing Bacteroidetes (including the Bacteroides and Butyricimonas) that were treated with atorvastatin and simvastatin also increased (22). However, Khan et al. observed that patients with high cholesterol levels had significantly higher levels of Firmicutes than those in healthy patients. The increase in the proportion of Faecalibacterium and Bifidobacterium in patients with high cholesterol treated with atorvastatin was consistent with the results of previous studies (120).
Hu et al. observed that the PPARγ and HMGCR protein expression decreased in the liver of HFD rats after atorvastatin intervention (121). The expression of HMGCR in mice treated with antibiotics was twice as high as that in mice without any special intervention (64), suggesting that HMGCR expression is partly influenced by the gut microbiota. The SREBP family is activated in low-cholesterol situation, in which SREBP2 plays a major role (102, 122). Therefore, it is challenging to observe changes in SREBP without being affected by cholesterol. Zimmermann et al. discovered that atorvastatin intervention increased the average levels of SREBP2 expression in the liver of HFD mice, and the effect of atorvastatin was reversed in antibiotic-treated HFD mice (51). This suggests that antibiotics attenuate the SREBP2 response to a low-cholesterol state by eliminating the positive effect of gut microbiota-mediated lipid-lowering action of atorvastatin.
In another experiment, the feces of HFD mice tended to have less acetate and butyrate and more propionate. Acetate and propionate levels increased significantly after simvastatin intervention, whereas butyrate levels did not change significantly. Similarly, the expression of HMGCR and SREBP1c were lower after simvastatin intervention in the liver and were closer to the levels in normal rats (113). Gu et al. also found that lipid metabolism-related genes SREBP1c and ACC1 in the liver were reduced after simvastatin treatment (123). SREBP and PCSK9 are closely related. Both SREBP1c and SREBP2 in fasting mice promoted the expression of hepatic PCSK9, which elevated serum PCSK9 levels (124). Thus, the lipid-lowering effect of simvastatin mediated by gut microbiota may be related to the reduction in SREBP1c levels.
In brief, atorvastatin can significantly regulate the serum LDL-C level in model animals by regulating the PPARγ signaling pathway and HMGCR gene expression.
FXR and PXR signaling pathways
Atorvastatin treatment of hypercholesterolemic patients can selectively regulate the relative abundance of several functionally dominant gut microbiota. Clostridium is an important microbiota involved in producing SBAs and increasing SBA-mediated dyslipidemia induced by HFD. Reducing Clostridium can inhibit the production of SBAs, prevent CYP7A1 from being inhibited by FXR and PXR, and ultimately increase cholesterol consumption. Statins can be also used for Clostridium infections in the intestine (27). Moreover, the relative abundance of Bifidobacterium, which can reduce LCA, was decreased in hypercholesterolemic patients (120).
A previous study showed that the CYP7A1 induction by atorvastatin appears to be due to suppressed FXR signaling in the liver and intestine (125). Simvastatin promoted CYP7A1 expression in the liver of HFD mice; however, the effect of simvastatin was impaired by antibiotics. In addition, simvastatin significantly reduced serum CDCA and DCA levels in HFD mice but had no significant effect on CA and LCA levels. Antibiotics can cause an increase in CDCA and a decrease in LCA, which may be related to the unbalanced gut microbiota in the production of LCA (118). In experiments with the combined intervention of atorvastatin and antibiotics, average levels of hepatic FXR expression in control mice, HFD mice, and atorvastatin-treated mice were lower than in mice given the same intervention and antibiotics (51). This confirms the influence of gut microbiota on the lipid-lowering effect of statins. FXR-SHP-CYP7A1 signaling pathway is one of the lipid-lowering pathways of atorvastatin. Moreover, FXR can activate PXR through PPARα (126). Li et al. observed that reducing secondary BAs such as DCA and LCA can regulate cholesterol metabolism by inhibiting the FXR-SHP pathway and reducing the expression of SREBP1c through the FXR-PXR pathway (127).
Compared with HFD rats, simvastatin could intervention resulted in a significant decrease in LDL-C levels and a significant increase in total BAs excretion, which led to the promotion of cholesterol consumption in rats that were not fed an HFD. Zhang et al. revealed that simvastatin intervention could antagonize the inhibitory effect of HFD on the expression of CYP7A1 in the liver tissue of rats (113). In an experiment investigating PXR and BAs excretion, administration of atorvastatin lowered BAs levels in the intestine, but was of no use in PXR knockout mice (128). The PXR-SHP-CYP7A1 signaling pathway showed a new potential in increasing BA synthesis and promoting cholesterol consumption. In addition, Catry et al. discovered that simvastatin increased the expression of SREBP2 in the intestinal tract of mice and indicated that enterocyte SREBP2 overexpression was related to the expression of cholesterol excretion genes (129). This may explain how simvastatin increase the excretion of BAs.
LPS-TLR4-Myd88
Gram-positive Firmicutes bacteria and Gram-negative Bacteroidetes bacteria account for 80% of the normal intestinal tract microbiota, and can exceed to 90% through an HFD (22). A previous study showed that the ileum was injured after exposure to low doses of LPS to the small intestine (130). LPS disrupted intestinal intercellular tight junction through a TLR4-dependent intracellular mechanism, resulting in increased intestinal permeability (131). However, we are yet to determine whether LPS damage to the gut affects the FXR and PXR pathways in enterocytes, thereby affecting the regulation of cholesterol by the gut microbiota. We observed that LPS translocation causes abnormal cholesterol metabolism and elevated serum LDL-C levels via TLR4-Myd88 in hepatocytes (106–109), which can be prevented by statins. Moreover, TLR4-Myd88 can activate both ERK and JNK signaling pathways in mice hepatocytes, thereby inhibiting CYP7A1 expression (132). ERK and JNK were important components of mitogen-activated protein kinases (MAPK) signaling pathway and were closely related to glucose and lipid metabolism (133). Repression of CYP7A1 by ERK and JNK is abolished when the Myd88 is disappeared in mice (132).
Statins can suppress TLR4 expression on cell membranes, including hepatocytes and enterocytes, modulate the TLR4-Myd88 signaling pathway, and prevent disease progression (134, 135). The TLR4-Myd88 pathway is restricted by SCFAs, and statins have been previously found to increase the SCFA-producing bacteria. Oral administration of butyrate reduces TLR4 and Myd88 expression in the mouse liver and repairs the damaged intestinal barrier (136). In another animal study, serum LPS levels were significantly decreased, and TLR4 expression in the liver and adipose tissue was significantly decreased after butyrate administration (137). SCFAs reduced the TLR4-Myd88 pathway activation by reducing LPS levels; however, whether SCFAs can directly affect the expression of TLR4 and Myd88 remains unclear. We summarize the effects of statins administration on cholesterol metabolism via gut microbiota and MDM that include SCFAs, BAs, TMAO, and LPS (Figure 2).
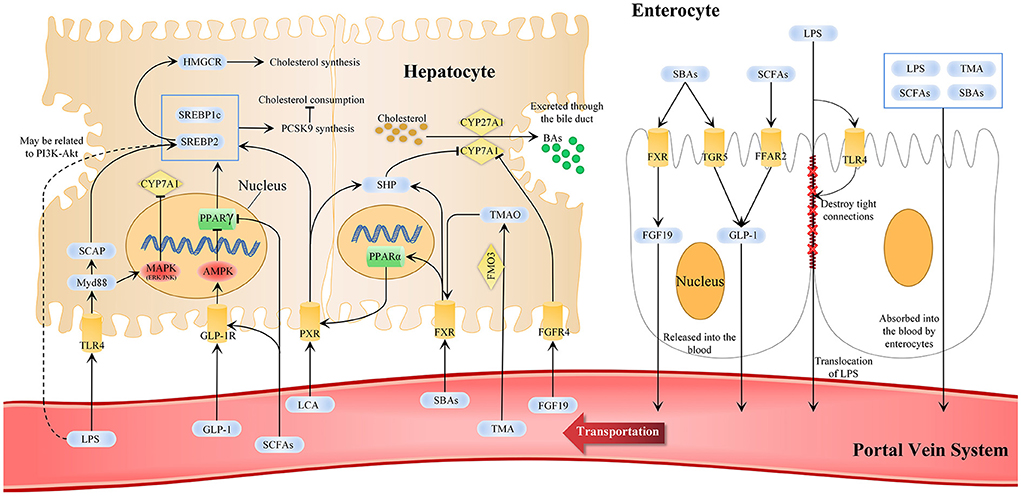
Figure 2. Statins-related microbiota metabolism and pathways in cholesterol metabolisms. In cholesterol metabolism, changes in gut microbiota composition and its metabolites are markers for regulating the gut-liver axis and can reflect the effect of statins. The liver-gut axis can be used as one of the ways statins affect cholesterol metabolism and can explain the why the efficacy of statins differs individually. SCFAs, SBAs, TMA, LPS, and other MDMs are changed after statins administration. SCFAs and SBAs can activate enterocytes to secrete FGF19 and GLP-1 into the blood, and others are absorbed by enterocytes into the blood circulation and finally absorbed in the liver through the portal vein system. This affects the AMPK-PPARγ-SREBP1C/2, FXR and TGR5 signaling pathways and LPS-TLR4-Myd88 and PI3K-Akt in hepatocytes to promote BAs excretion, reduce cholesterol synthesis and promote cholesterol consumption. AMPK, AMP-activated protein kinase; CYP7A1, cytochrome p-450 enzyme cholesterol 7α-hydroxylase; CYP27A1, cytochrome p-450 enzyme cholesterol 27α-hydroxylase; FXR, farnesoid X receptor; FGF 19, fibroblast growth factor 19; FGFR4, fibroblast growth factor receptor 4; GLP-1, glucagon-like peptide 1; GLP-1R, GLP-1 receptor; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; LPS, lipopolysaccharides; MAPK, mitogen-activated protein kinases; MDMs, microbiota-derived metabolites; Myd88, myeloid differentiation factor 88; PI3K-Akt, the phosphatidylinositol-3-kinase; PPAR, peroxisome proliferator-activated receptor γ; SBAs, secondary bile acid; SCAP, SREBP cleavage-activating protein; SCFAs, short-chain fatty acid; SHP, FXR-small heterodimer partner; SREBP, sterol regulatory element binding protein; TGR5, cell surface receptor G protein-coupled bile acid receptor 5; TLR4, Toll-like receptor 4; TMA, trimethylamine; TMAO, trimethylamine-N-oxide.
Conclusion
This review aims to describe the effect of statins on serum LDL-C levels via the gut microbiota and to summarize the underlying mechanisms. Therefore, we focused on evidence describing statin alterations in gut microbiota abundance and their effects on gut MDM based on laboratory models and human data. As most studies have demonstrated, the LDL-C lowering effect of statins as direct inhibitors of HMGCR is potentially affected by the gut microbiota. In conclusion, scientific studies based on laboratory models have revealed that statins can alter the pathways associated with gut MDM, which in turn regulate the cholesterol metabolism and LDL-C levels. In particular, statins can modulate the abundance of gut microbiota producing SCFAs, SBAs, TMAO, and LPS, which in turn affect the AMPK-SREBP, FXR, PXR, FFAR2, and TLR4-Myd88 signaling pathways. Unfortunately, clinical trails on the effect of statins on serum LDL-C levels via the gut-liver axis are limited. However, preclinical studies have shown positive roles for the effects of statins on high LDL-C levels via the MDMs and gut liver axis, and it is fair to assume similar effects on the human gut and liver. Further clinical trials and experimental studies are required to better elucidate whether statins can effectively lower LDL-C levels through the gut microbiota.
Author contributions
CS, MW, and LL designed and directed the manuscript. CS and ZW wrote the manuscript. LH and XZ revised the manuscript. JC searched the literature. ZY designed the illustrations and tables. All the authors approved the manuscript for publication, read, and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81973689 and 82074254) and the Natural Science Foundation of Beijing Municipality (No. 7202176).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Katsiki N, Mikhailidis DP, Mantzoros CS. Non-alcoholic fatty liver disease and dyslipidemia: an update. Metabolism. (2016) 65:1109–23. doi: 10.1016/j.metabol.2016.05.003
2. Ke C, Zhu X, Zhang Y, Shen Y. Metabolomic characterization of hypertension and dyslipidemia. Metabolomics. (2018) 14:117. doi: 10.1007/s11306-018-1408-y
3. Aboonabi A, Meyer RR, Singh I. The association between metabolic syndrome components and the development of atherosclerosis. J Hum Hypertens. (2019) 33:844–55. doi: 10.1038/s41371-019-0273-0
4. Zhou M, Wang H, Zeng X, Yin P, Zhu J, Chen W, et al. Mortality, morbidity, and risk factors in China and its provinces, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. (2019) 394:1145–58. doi: 10.1016/S0140-6736(19)30427-1
5. Horodinschi RN, Stanescu AMA, Bratu OG, Pantea Stoian A, Radavoi DG, Diaconu CC. Treatment with Statins in Elderly Patients. Medicina (Kaunas). (2019) 55:721. doi: 10.3390/medicina55110721
6. Pinal-Fernandez I, Casal-Dominguez M, Mammen AL. Statins: pros and cons. Med Clin (Barc). (2018) 150:398–402. doi: 10.1016/j.medcli.2017.11.030
7. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol. (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
8. Mancini A, Campagna F, Amodio P, Tuohy KM. Gut : liver : brain axis: the microbial challenge in the hepatic encephalopathy. Food Funct. (2018) 9:1373–88. doi: 10.1039/C7FO01528C
9. Martinez KB, Leone V, Chang EB. Western diets, gut dysbiosis, and metabolic diseases: are they linked? Gut Microbes. (2017) 8:130–42. doi: 10.1080/19490976.2016.1270811
10. Sonnenburg JL, Bäckhed F. Diet-microbiota interactions as moderators of human metabolism. Nature. (2016) 535:56–64. doi: 10.1038/nature18846
11. Adak A, Khan MR. An insight into gut microbiota and its functionalities. Cell Mol Life Sci. (2019) 76:473–93. doi: 10.1007/s00018-018-2943-4
12. Dias AM, Cordeiro G, Estevinho MM, Veiga R, Figueira L, Reina-Couto M, et al. Gut bacterial microbiome composition and statin intake-A systematic review. Pharmacol Res Perspect. (2020) 8:e00601. doi: 10.1002/prp2.601
13. Vourakis M, Mayer G, Rousseau G. The role of gut microbiota on cholesterol metabolism in atherosclerosis. Int J Mol Sci. (2021) 22:8074. doi: 10.3390/ijms22158074
14. Deng M, Qu F, Chen L, Liu C, Zhang M, Ren F, et al. SCFAs alleviated steatosis and inflammation in mice with NASH induced by MCD. J Endocrinol. (2020) 245:425–37. doi: 10.1530/JOE-20-0018
15. Di Ciaula A, Garruti G, Lunardi Baccetto R, Molina-Molina E, Bonfrate L, Wang DQ, et al. Bile acid physiology. Ann Hepatol. (2017) 16(Suppl. 1: s3-105.):s4–s14. doi: 10.5604/01.3001.0010.5493
16. Pathak P, Helsley RN, Brown AL, Buffa JA, Choucair I, Nemet I, et al. Small molecule inhibition of gut microbial choline trimethylamine lyase activity alters host cholesterol and bile acid metabolism. Am J Physiol Heart Circ Physiol. (2020) 318:H1474–h86. doi: 10.1152/ajpheart.00584.2019
17. Feminò R, Feminò G, Cavezzi A, Troiani E. PCSK9 inhibition, LDL and lipopolysaccharides: a complex and “dangerous” relationship. Int Angiol. (2021) 40:248–60. doi: 10.23736/S0392-9590.21.04632-0
18. Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. (2017) 19:29–41. doi: 10.1111/1462-2920.13589
19. Lee J, d'Aigle J, Atadja L, Quaicoe V, Honarpisheh P, Ganesh BP, et al. Gut Microbiota-Derived Short-Chain Fatty Acids Promote Poststroke Recovery in Aged Mice. Circ Res. (2020) 127:453–65. doi: 10.1161/CIRCRESAHA.119.316448
20. Chang Y, Chen Y, Zhou Q, Wang C, Chen L, Di W, et al. Short-chain fatty acids accompanying changes in the gut microbiome contribute to the development of hypertension in patients with preeclampsia. Clin Sci (Lond). (2020) 134:289–302. doi: 10.1042/CS20191253
21. Zhou L, Zhang M, Wang Y, Dorfman RG, Liu H, Yu T, et al. Faecalibacterium prausnitzii Produces Butyrate to Maintain Th17/Treg Balance and to Ameliorate Colorectal Colitis by Inhibiting Histone Deacetylase 1. Inflamm Bowel Dis. (2018) 24:1926–40. doi: 10.1093/ibd/izy182
22. Kim J, Lee H, An J, Song Y, Lee CK, Kim K, et al. Alterations in gut microbiota by statin therapy and possible intermediate effects on hyperglycemia and hyperlipidemia. Front Microbiol. (2019) 10:1947. doi: 10.3389/fmicb.2019.01947
23. Yang M, Zheng J, Zong X, Yang X, Zhang Y, Man C, et al. Preventive effect and molecular mechanism of lactobacillus rhamnosus JL1 on food-borne obesity in mice. Nutrients. (2021) 13:3989. doi: 10.3390/nu13113989
24. Reamtong O, Thiangtrongjit T, Kosoltanapiwat N, Panbangred W, Prangthip P. Potential benefits of L. acidophilus in dyslipidemic rats. Sci Rep. (2021) 11:6115. doi: 10.1038/s41598-021-85427-8
25. Wu Y, Li X, Tan F, Zhou X, Mu J, Zhao X. Lactobacillus fermentum CQPC07 attenuates obesity, inflammation and dyslipidemia by modulating the antioxidant capacity and lipid metabolism in high-fat diet induced obese mice. J Inflamm (Lond). (2021) 18:5. doi: 10.1186/s12950-021-00272-w
26. Bordoni A, Amaretti A, Leonardi A, Boschetti E, Danesi F, Matteuzzi D, et al. Cholesterol-lowering probiotics: in vitro selection and in vivo testing of bifidobacteria. Appl Microbiol Biotechnol. (2013) 97:8273–81. doi: 10.1007/s00253-013-5088-2
27. Wang K, Yu X, Li Y, Guo Y, Ge L, Pu F, et al. Bifidobacterium bifidum TMC3115 can characteristically influence glucose and lipid profile and intestinal microbiota in the middle-aged and elderly. Probiotics Antimicrob Proteins. (2019) 11:1182–94. doi: 10.1007/s12602-018-9441-8
28. Zhu W, Lin K, Li K, Deng X, Li C. Reshaped fecal gut microbiota composition by the intake of high molecular weight persimmon tannin in normal and high-cholesterol diet-fed rats. Food Funct. (2018) 9:541–51. doi: 10.1039/C7FO00995J
29. Esquivel-Elizondo S, Ilhan ZE, Garcia-Peña EI, Krajmalnik-Brown R. Insights into butyrate production in a controlled fermentation system via gene predictions. mSystems. (2017) 2:e00051–17. doi: 10.1128/mSystems.00051-17
30. Hugenholtz F, Davids M, Schwarz J, Müller M, Tomé D, Schaap P, et al. Metatranscriptome analysis of the microbial fermentation of dietary milk proteins in the murine gut. PLoS ONE. (2018) 13:e0194066. doi: 10.1371/journal.pone.0194066
31. Mu H, Zhou Q, Yang R, Zeng J, Li X, Zhang R, et al. Naringin attenuates high fat diet induced non-alcoholic fatty liver disease and gut bacterial dysbiosis in mice. Front Microbiol. (2020) 11:585066. doi: 10.3389/fmicb.2020.585066
32. Wang Q, Hao C, Yao W, Zhu D, Lu H, Li L, et al. Intestinal flora imbalance affects bile acid metabolism and is associated with gallstone formation. BMC Gastroenterol. (2020) 20:59. doi: 10.1186/s12876-020-01195-1
33. Chiu HF, Huang YC, Lu YY, Han YC, Shen YC, Golovinskaia O, et al. Regulatory/modulatory effect of prune essence concentrate on intestinal function and blood lipids. Pharm Biol. (2017) 55:974–9. doi: 10.1080/13880209.2017.1285323
34. Shi N, Zhang S, Silverman G, Li M, Cai J, Niu H. Protective effect of hydroxychloroquine on rheumatoid arthritis-associated atherosclerosis. Animal Model Exp Med. (2019) 2:98–106. doi: 10.1002/ame2.12065
35. Li R, Yao Y, Gao P, Bu S. The therapeutic efficacy of curcumin vs. Metformin in modulating the gut microbiota in NAFLD rats: a comparative study. Front Microbiol. (2020) 11:555293. doi: 10.3389/fmicb.2020.555293
36. Wang L, Gong Z, Zhang X, Zhu F, Liu Y, Jin C, et al. Gut microbial bile acid metabolite skews macrophage polarization and contributes to high-fat diet-induced colonic inflammation. Gut Microbes. (2020) 12:1–20. doi: 10.1080/19490976.2020.1819155
37. Hu X, Wang T, Li W, Jin F, Wang L. Effects of NS Lactobacillus strains on lipid metabolism of rats fed a high-cholesterol diet. Lipids Health Dis. (2013) 12:67. doi: 10.1186/1476-511X-12-67
38. Palaniyandi SA, Damodharan K, Suh JW, Yang SH. Probiotic Characterization of Cholesterol-Lowering Lactobacillus fermentum MJM60397. Probiotics Antimicrob Proteins. (2020) 12:1161–72. doi: 10.1007/s12602-019-09585-y
39. Okazaki Y, Sitanggang NV, Sato S, Ohnishi N, Inoue J, Iguchi T, et al. Burdock fermented by Aspergillus awamori elevates cecal Bifidobacterium, and reduces fecal deoxycholic acid and adipose tissue weight in rats fed a high-fat diet. Biosci Biotechnol Biochem. (2013) 77:53–7. doi: 10.1271/bbb.120551
40. Kamp KJ, Cain KC, Utleg A, Burr RL, Raftery D, Luna RA, et al. Bile acids and microbiome among individuals with irritable bowel syndrome and healthy volunteers. Biol Res Nurs. (2021) 23:65–74. doi: 10.1177/1099800420941255
41. Brugère JF, Borrel G, Gaci N, Tottey W, O'Toole PW, Malpuech-Brugère C. Archaebiotics: proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut Microbes. (2014) 5:5–10. doi: 10.4161/gmic.26749
42. Dridi B, Fardeau ML, Ollivier B, Raoult D, Drancourt M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int J Syst Evol Microbiol. (2012) 62(Pt 8):1902–7. doi: 10.1099/ijs.0.033712-0
43. Chen S, Jiang PP Yu D, Liao GC, Wu SL, Fang AP, et al. Effects of probiotic supplementation on serum trimethylamine-N-oxide level and gut microbiota composition in young males: a double-blinded randomized controlled trial. Eur J Nutr. (2021) 60:747–58. doi: 10.1007/s00394-020-02278-1
44. Naghipour S, Cox AJ, Peart JN, Du Toit EF, Headrick JP. Trimethylamine N-oxide: heart of the microbiota-CVD nexus? Nutr Res Rev. (2021) 34:125–46. doi: 10.1017/S0954422420000177
45. He WS, Li L, Rui J, Li J, Sun Y, Cui D, et al. Tomato seed oil attenuates hyperlipidemia and modulates gut microbiota in C57BL/6J mice. Food Funct. (2020) 11:4275–90. doi: 10.1039/D0FO00133C
46. Zhu L, Wang J, Ding X, Bai S, Zeng Q, Xuan Y, et al. Serum trimethylamine-N-oxide and gut microbiome alterations are associated with cholesterol deposition in the liver of laying hens fed with rapeseed meal. Anim Nutr. (2021) 7:1258–70. doi: 10.1016/j.aninu.2021.02.008
47. Falony G, Vieira-Silva S, Raes J. Microbiology meets big data: the case of gut microbiota-derived trimethylamine. Annu Rev Microbiol. (2015) 69:305–21. doi: 10.1146/annurev-micro-091014-104422
48. Wang X, Li X, Dong Y. Vitamin D decreases plasma trimethylamine-n-oxide level in mice by regulating gut microbiota. Biomed Res Int. (2020) 2020:9896743. doi: 10.1155/2020/9896743
49. Cho CE, Taesuwan S, Malysheva OV, Bender E, Tulchinsky NF, Yan J, et al. Trimethylamine-N-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: a randomized controlled trial. Mol Nutr Food Res. (2017) 61:201600324. doi: 10.1002/mnfr.201770016
50. Singh RP, Halaka DA, Hayouka Z, Tirosh O. High-fat diet induced alteration of mice microbiota and the functional ability to utilize fructooligosaccharide for ethanol production. Front Cell Infect Microbiol. (2020) 10:376. doi: 10.3389/fcimb.2020.00376
51. Zimmermann F, Roessler J, Schmidt D, Jasina A, Schumann P, Gast M, et al. Impact of the gut microbiota on atorvastatin mediated effects on blood lipids. J Clin Med. (2020) 9:1596. doi: 10.3390/jcm9051596
52. Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. (2016) 7:189–200. doi: 10.1080/19490976.2015.1134082
53. Tong LT, Zhong K, Liu L, Qiu J, Guo L, Zhou X, et al. Effects of dietary wheat bran arabinoxylans on cholesterol metabolism of hypercholesterolemic hamsters. Carbohydr Polym. (2014) 112:1–5. doi: 10.1016/j.carbpol.2014.05.061
54. Du Y, Li X, Su C, Xi M, Zhang X, Jiang Z, et al. Butyrate protects against high-fat diet-induced atherosclerosis via up-regulating ABCA1 expression in apolipoprotein E-deficiency mice. Br J Pharmacol. (2020) 177:1754–72. doi: 10.1111/bph.14933
55. Shah S, Fillier T, Pham TH, Thomas R, Cheema SK. Intraperitoneal administration of short-chain fatty acids improves lipid metabolism of long-evans rats in a sex-specific manner. Nutrients. (2021) 13:892. doi: 10.3390/nu13030892
56. Hao W, He Z, Zhu H, Liu J, Kwek E, Zhao Y, et al. Sea buckthorn seed oil reduces blood cholesterol and modulates gut microbiota. Food Funct. (2019) 10:5669–81. doi: 10.1039/C9FO01232J
57. Jakobsdottir G, Xu J, Molin G, Ahrné S, Nyman M. High-fat diet reduces the formation of butyrate, but increases succinate, inflammation, liver fat and cholesterol in rats, while dietary fibre counteracts these effects. PLoS ONE. (2013) 8:e80476. doi: 10.1371/journal.pone.0080476
58. Olaniyi KS, Owolabi MN, Atuma CL, Agunbiade TB, Alabi BY. Acetate rescues defective brain-adipose metabolic network in obese Wistar rats by modulation of peroxisome proliferator-activated receptor-γ. Sci Rep. (2021) 11:18967. doi: 10.1038/s41598-021-98605-5
59. Fushimi T, Suruga K, Oshima Y, Fukiharu M, Tsukamoto Y, Goda T. Dietary acetic acid reduces serum cholesterol and triacylglycerols in rats fed a cholesterol-rich diet. Br J Nutr. (2006) 95:916–24. doi: 10.1079/BJN20061740
60. Song B, Zhong YZ, Zheng CB, Li FN, Duan YH, Deng JP. Propionate alleviates high-fat diet-induced lipid dysmetabolism by modulating gut microbiota in mice. J Appl Microbiol. (2019) 127:1546–55. doi: 10.1111/jam.14389
61. Aguilar EC, da Silva JF, Navia-Pelaez JM, Leonel AJ, Lopes LG, Menezes-Garcia Z, et al. Sodium butyrate modulates adipocyte expansion, adipogenesis, and insulin receptor signaling by upregulation of PPAR-γ in obese Apo E knockout mice. Nutrition. (2018) 47:75–82. doi: 10.1016/j.nut.2017.10.007
62. Yu C, Liu S, Chen L, Shen J, Niu Y, Wang T, et al. Effect of exercise and butyrate supplementation on microbiota composition and lipid metabolism. J Endocrinol. (2019) 243:125–35. doi: 10.1530/JOE-19-0122
63. Fu C, Liu L, Li F. Acetate alters the process of lipid metabolism in rabbits. Animal. (2018) 12:1895–902. doi: 10.1017/S1751731117003275
64. den Besten G, Bleeker A, Gerding A, van Eunen K, Havinga R, van Dijk TH, et al. Short-chain fatty acids protect against high-fat diet-induced obesity via a PPARγ-dependent switch from lipogenesis to fat oxidation. Diabetes. (2015) 64:2398–408. doi: 10.2337/db14-1213
65. Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. (2011) 13:376–88. doi: 10.1016/j.cmet.2011.03.009
66. Han HJ, Song X, Yadav D, Hwang MS, Lee JH, Lee CH, et al. Ulmus macrocarpa Hance modulates lipid metabolism in hyperlipidemia via activation of AMPK pathway. PLoS ONE. (2019) 14:e0217112. doi: 10.1371/journal.pone.0217112
67. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. (2002) 109:1125–31. doi: 10.1172/JCI0215593
68. Lin HV, Frassetto A, Kowalik EJ Jr, Nawrocki AR, Lu MM, Kosinski JR, et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE. (2012) 7:e35240. doi: 10.1371/journal.pone.0035240
69. Zhou D, Chen YW, Zhao ZH, Yang RX, Xin FZ, Liu XL, et al. Sodium butyrate reduces high-fat diet-induced non-alcoholic steatohepatitis through upregulation of hepatic GLP-1R expression. Exp Mol Med. (2018) 50:1–12. doi: 10.1038/s12276-018-0183-1
70. Hamada Y, Goto M, Nishimura G, Nagasaki H, Seino Y, Kamiya H, et al. The alpha-glucosidase inhibitor miglitol increases hepatic CYP7A1 activity in association with altered short-chain fatty acid production in the gut of obese diabetic mice. Metabol Open. (2020) 5:100024. doi: 10.1016/j.metop.2020.100024
71. Zhang Z, Fan S, Huang D, Xiong T, Nie S, Xie M. Polysaccharides from fermented Asparagus officinalis with Lactobacillus plantarum NCU116 alleviated liver injury via modulation of glutathione homeostasis, bile acid metabolism, and SCFA production. Food Funct. (2020) 11:7681–95. doi: 10.1039/D0FO01435D
72. Chambers KF, Day PE, Aboufarrag HT, Kroon PA. Polyphenol effects on cholesterol metabolism via bile acid biosynthesis, CYP7A1: a review. Nutrients. (2019) 11:2588. doi: 10.3390/nu11112588
73. Housset C, Chrétien Y, Debray D, Chignard N. Functions of the gallbladder. Compr Physiol. (2016) 6:1549–77. doi: 10.1002/cphy.c150050
74. Wang Y, Ding WX Li T. Cholesterol and bile acid-mediated regulation of autophagy in fatty liver diseases and atherosclerosis. Biochim Biophys Acta Mol Cell Biol Lipids. (2018) 1863:726–33. doi: 10.1016/j.bbalip.2018.04.005
75. Chiang JYL, Ferrell JM. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am J Physiol Gastrointest Liver Physiol. (2020) 318:G554–g73. doi: 10.1152/ajpgi.00223.2019
76. Rizzolo D, Buckley K, Kong B, Zhan L, Shen J, Stofan M, et al. Bile acid homeostasis in a cholesterol 7α-hydroxylase and sterol 27-hydroxylase double knockout mouse model. Hepatology. (2019) 70:389–402. doi: 10.1002/hep.30612
77. Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol. (2018) 15:111–28. doi: 10.1038/nrgastro.2017.119
78. Shang Q, Guo GL, Honda A, Saumoy M, Salen G, Xu G. FGF15/19 protein levels in the portal blood do not reflect changes in the ileal FGF15/19 or hepatic CYP7A1 mRNA levels. J Lipid Res. (2013) 54:2606–14. doi: 10.1194/jlr.M034827
79. Jones RD, Lopez AM, Tong EY, Posey KS, Chuang JC, Repa JJ, et al. Impact of physiological levels of chenodeoxycholic acid supplementation on intestinal and hepatic bile acid and cholesterol metabolism in Cyp7a1-deficient mice. Steroids. (2015) 93:87–95. doi: 10.1016/j.steroids.2014.11.002
80. Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. (2002) 296:1313–6. doi: 10.1126/science.1070477
81. Kuhre RE, Wewer Albrechtsen NJ, Larsen O, Jepsen SL, Balk-Møller E, Andersen DB, et al. Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol Metab. (2018) 11:84–95. doi: 10.1016/j.molmet.2018.03.007
82. Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, Yang S, et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J. (2006) 398:423–30. doi: 10.1042/BJ20060537
83. Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. (2001) 98:3369–74. doi: 10.1073/pnas.051551698
84. Bhalla S, Ozalp C, Fang S, Xiang L, Kemper JK. Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1alpha. Functional implications in hepatic cholesterol and glucose metabolism. J Biol Chem. (2004) 279:45139–47. doi: 10.1074/jbc.M405423200
85. Karpale M, Käräjämäki AJ, Kummu O, Gylling H, Hyötyläinen T, Orešič M, et al. Activation of pregnane X receptor induces atherogenic lipids and PCSK9 by a SREBP2-mediated mechanism. Br J Pharmacol. (2021) 178:2461–81. doi: 10.1111/bph.15433
86. Kim S, Choi S, Dutta M, Asubonteng JO, Polunas M, Goedken M, et al. Pregnane X receptor exacerbates nonalcoholic fatty liver disease accompanied by obesity- and inflammation-prone gut microbiome signature. Biochem Pharmacol. (2021) 193:114698. doi: 10.1016/j.bcp.2021.114698
87. Zeisel SH, Warrier M. Trimethylamine N-oxide, the microbiome, and heart and kidney disease. Annu Rev Nutr. (2017) 37:157–81. doi: 10.1146/annurev-nutr-071816-064732
88. Fu BC, Hullar MAJ, Randolph TW, Franke AA, Monroe KR, Cheng I, et al. Associations of plasma trimethylamine N-oxide, choline, carnitine, and betaine with inflammatory and cardiometabolic risk biomarkers and the fecal microbiome in the Multiethnic Cohort Adiposity Phenotype Study. Am J Clin Nutr. (2020) 111:1226–34. doi: 10.1093/ajcn/nqaa015
89. Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. (2013) 17:49–60. doi: 10.1016/j.cmet.2012.12.011
90. Warrier M, Shih DM, Burrows AC, Ferguson D, Gromovsky AD, Brown AL, et al. The TMAO-generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep. (2015) 10:326–38. doi: 10.1016/j.celrep.2014.12.036
91. Charach G, Rabinovich A, Argov O, Weintraub M, Rabinovich P. The role of bile acid excretion in atherosclerotic coronary artery disease. Int J Vasc Med. (2012) 2012:949672. doi: 10.1155/2012/949672
92. Tan X, Liu Y, Long J, Chen S, Liao G, Wu S, et al. Trimethylamine N-oxide aggravates liver steatosis through modulation of bile acid metabolism and inhibition of farnesoid x receptor signaling in nonalcoholic fatty liver disease. Mol Nutr Food Res. (2019) 63:e1900257. doi: 10.1002/mnfr.201900257
93. Ding L, Chang M, Guo Y, Zhang L, Xue C, Yanagita T, et al. Trimethylamine-N-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis. (2018) 17:286. doi: 10.1186/s12944-018-0939-6
94. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. (2013) 19:576–85. doi: 10.1038/nm.3145
95. Wu WK, Chen CC, Liu PY, Panyod S, Liao BY, Chen PC, et al. Identification of TMAO-producer phenotype and host-diet-gut dysbiosis by carnitine challenge test in human and germ-free mice. Gut. (2019) 68:1439–49. doi: 10.1136/gutjnl-2018-317155
96. Barengolts E, Gut microbiota. prebiotics, probiotics, and synbiotics in management of obesity and prediabetes: review of randomized controlled trials. Endocr Pract. (2016) 22:1224–34. doi: 10.4158/EP151157.RA
97. Määttä AM, Salminen A, Pietiäinen M, Leskelä J, Palviainen T, Sattler W, et al. Endotoxemia is associated with an adverse metabolic profile. Innate Immun. (2021) 27:3–14. doi: 10.1177/1753425920971702
98. Jin C, Flavell RA. Innate sensors of pathogen and stress: linking inflammation to obesity. J Allergy Clin Immunol. (2013) 132:287–94. doi: 10.1016/j.jaci.2013.06.022
99. Melendez QM, Krishnaji ST, Wooten CJ, Lopez D. Hypercholesterolemia: the role of PCSK9. Arch Biochem Biophys. (2017) 625–26:39–53. doi: 10.1016/j.abb.2017.06.001
100. Wu YR, Li L, Sun XC, Wang J, Ma CY, Zhang Y, et al. Diallyl disulfide improves lipid metabolism by inhibiting PCSK9 expression and increasing LDL uptake via PI3K/Akt-SREBP2 pathway in HepG2 cells. Nutr Metab Cardiovasc Dis. (2021) 31:322–32. doi: 10.1016/j.numecd.2020.08.012
101. Feingold KR, Hardardottir I, Memon R, Krul EJ, Moser AH, Taylor JM, et al. Effect of endotoxin on cholesterol biosynthesis and distribution in serum lipoproteins in Syrian hamsters. J Lipid Res. (1993) 34:2147–58. doi: 10.1016/S0022-2275(20)35355-4
102. Ye Q, Lei H, Fan Z, Zheng W, Zheng S. Difference in LDL receptor feedback regulation in macrophages and vascular smooth muscle cells: foam cell transformation under inflammatory stress. Inflammation. (2014) 37:555–65. doi: 10.1007/s10753-013-9769-x
103. Moon YA. The SCAP/SREBP pathway: a mediator of hepatic steatosis. Endocrinol Metab (Seoul). (2017) 32:6–10. doi: 10.3803/EnM.2017.32.1.6
104. Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. (2004) 45:1169–96. doi: 10.1194/jlr.R300019-JLR200
105. Kim MS, Shigenaga J, Moser A, Feingold K, Grunfeld C. Repression of farnesoid X receptor during the acute phase response. J Biol Chem. (2003) 278:8988–95. doi: 10.1074/jbc.M212633200
106. Cao D, Wang W, Li S, Lai W, Huang X, Zhou J, et al. TLR2-Deficiency Promotes Prenatal LPS Exposure-Induced Offspring Hyperlipidemia. Front Physiol. (2019) 10:1102. doi: 10.3389/fphys.2019.01102
107. Wang F, Hu M, Zhu H, Yang C, Xia H, Yang X, et al. MyD88 determines the protective effects of fish oil and perilla oil against metabolic disorders and inflammation in adipose tissue from mice fed a high-fat diet. Nutr Diabetes. (2021) 11:23. doi: 10.1038/s41387-021-00159-y
108. Dong G, Huang X, Wu L, Jiang S, Tan Q, Chen S. SREBF2 triggers endoplasmic reticulum stress and Bax dysregulation to promote lipopolysaccharide-induced endothelial cell injury. Cell Biol Toxicol. (2022) 38:185–201. doi: 10.1007/s10565-021-09593-1
109. Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. (2006) 124:35–46. doi: 10.1016/j.cell.2005.12.022
110. Sirtori CR. The pharmacology of statins. Pharmacol Res. (2014) 88:3–11. doi: 10.1016/j.phrs.2014.03.002
111. Ko HHT, Lareu RR, Dix BR, Hughes JD. Statins: antimicrobial resistance breakers or makers? PeerJ. (2017) 5:e3952. doi: 10.7717/peerj.3952
112. Sun B, Li L, Zhou X. Comparative analysis of the gut microbiota in distinct statin response patients in East China. J Microbiol. (2018) 56:886–92. doi: 10.1007/s12275-018-8152-x
113. Zhang Q, Fan X, Ye R, Hu Y, Zheng T, Shi R, et al. The effect of simvastatin on gut microbiota and lipid metabolism in hyperlipidemic rats induced by a high-fat diet. Front Pharmacol. (2020) 11:522. doi: 10.3389/fphar.2020.00522
114. Zhang S, Li H, Yuan L, Zhang J, Han L, Liu R, et al. Molecular characterization of gut microbiota in high-lipid diet-induced hyperlipidemic rats treated with simvastatin. Int J Mol Med. (2020) 45:1601–15. doi: 10.3892/ijmm.2020.4516
115. Nolan JA, Skuse P, Govindarajan K, Patterson E, Konstantinidou N, Casey PG, et al. The influence of rosuvastatin on the gastrointestinal microbiota and host gene expression profiles. Am J Physiol Gastrointest Liver Physiol. (2017) 312:G488–g97. doi: 10.1152/ajpgi.00149.2016
116. Liu Y, Song X, Zhou H, Zhou X, Xia Y, Dong X, et al. Gut microbiome associates with lipid-lowering effect of rosuvastatin in vivo. Front Microbiol. (2018) 9:530. doi: 10.3389/fmicb.2018.00530
117. Vieira-Silva S, Falony G, Belda E, Nielsen T, Aron-Wisnewsky J, Chakaroun R, et al. Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nature. (2020) 581:310–5. doi: 10.1038/s41586-020-2269-x
118. He X, Zheng N, He J, Liu C, Feng J, Jia W, et al. Gut microbiota modulation attenuated the hypolipidemic effect of simvastatin in high-fat/cholesterol-diet fed mice. J Proteome Res. (2017) 16:1900–10. doi: 10.1021/acs.jproteome.6b00984
119. Stadlbauer V, Engertsberger L, Komarova I, Feldbacher N, Leber B, Pichler G, et al. Dysbiosis, gut barrier dysfunction and inflammation in dementia: a pilot study. BMC Geriatr. (2020) 20:248. doi: 10.1186/s12877-020-01644-2
120. Khan TJ, Ahmed YM, Zamzami MA, Siddiqui AM, Khan I, Baothman OAS, et al. Atorvastatin treatment modulates the gut microbiota of the hypercholesterolemic patients. Omics. (2018) 22:154–63. doi: 10.1089/omi.2017.0130
121. Hu N, Chen C, Wang J, Huang J, Yao D, Li C. Atorvastatin ester regulates lipid metabolism in hyperlipidemia rats via the PPAR-signaling pathway and HMGCR expression in the liver. Int J Mol Sci. (2021) 22:11107. doi: 10.3390/ijms222011107
122. Sharpe LJ, Brown AJ. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J Biol Chem. (2013) 288:18707–15. doi: 10.1074/jbc.R113.479808
123. Gu Y, Zhang Y, Li M, Huang Z, Jiang J, Chen Y, et al. Ferulic acid ameliorates atherosclerotic injury by modulating gut microbiota and lipid metabolism. Front Pharmacol. (2021) 12:621339. doi: 10.3389/fphar.2021.621339
124. Krysa JA, Ooi TC, Proctor SD, Vine DF. Nutritional and lipid modulation of PCSK9: effects on cardiometabolic risk factors. J Nutr. (2017) 147:473–81. doi: 10.3945/jn.116.235069
125. Fu ZD, Cui JY, Klaassen CD. Atorvastatin induces bile acid-synthetic enzyme Cyp7a1 by suppressing FXR signaling in both liver and intestine in mice. J Lipid Res. (2014) 55:2576–86. doi: 10.1194/jlr.M053124
126. Halilbasic E, Claudel T, Trauner M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol. (2013) 58:155–68. doi: 10.1016/j.jhep.2012.08.002
127. Li Y, Tian Y, Cai W, Wang Q, Chang Y, Sun Y, et al. Novel ι-Carrageenan tetrasaccharide alleviates liver lipid accumulation via the bile acid-FXR-SHP/PXR pathway to regulate cholesterol conversion and fatty acid metabolism in insulin-resistant mice. J Agric Food Chem. (2021) 69:9813–21. doi: 10.1021/acs.jafc.1c04035
128. Caparrós-Martín JA, Lareu RR, Ramsay JP, Peplies J, Reen FJ, Headlam HA, et al. Statin therapy causes gut dysbiosis in mice through a PXR-dependent mechanism. Microbiome. (2017) 5:95. doi: 10.1186/s40168-017-0312-4
129. Catry E, Pachikian BD, Salazar N, Neyrinck AM, Cani PD, Delzenne NM. Ezetimibe and simvastatin modulate gut microbiota and expression of genes related to cholesterol metabolism. Life Sci. (2015) 132:77–84. doi: 10.1016/j.lfs.2015.04.004
130. Li C, Zhou HC, Nie YL, Zhao BY, Wu CC. Effects of lipopolysaccharide on T lymphocyte cell subsets and cytokine secretion in mesenteric lymph nodes of mice: Histological and molecular study. Environ Toxicol Pharmacol. (2019) 71:103214. doi: 10.1016/j.etap.2019.103214
131. Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. (2013) 182:375–87. doi: 10.1016/j.ajpath.2012.10.014
132. Lefort C, Van Hul M, Delzenne NM, Everard A, Cani PD. Hepatic MyD88 regulates liver inflammation by altering synthesis of oxysterols. Am J Physiol Endocrinol Metab. (2019) 317:E99–e108. doi: 10.1152/ajpendo.00082.2019
133. Papa S, Choy PM, Bubici C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene. (2019) 38:2223–40. doi: 10.1038/s41388-018-0582-8
134. Bahrami A, Parsamanesh N, Atkin SL, Banach M, Sahebkar A. Effect of statins on toll-like receptors: a new insight to pleiotropic effects. Pharmacol Res. (2018) 135:230–8. doi: 10.1016/j.phrs.2018.08.014
135. Rashidian A, Muhammadnejad A, Dehpour AR, Mehr SE, Akhavan MM, Shirkoohi R, et al. Atorvastatin attenuates TNBS-induced rat colitis: the involvement of the TLR4/NF-kB signaling pathway. Inflammopharmacology. (2016) 24:109–18. doi: 10.1007/s10787-016-0263-6
136. Zhou D, Pan Q, Xin FZ, Zhang RN, He CX, Chen GY, et al. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J Gastroenterol. (2017) 23:60–75. doi: 10.3748/wjg.v23.i1.60
Keywords: gut microbiota, low density lipid cholesterol, cholesterol metabolism, statins, potential pathways
Citation: Sun C, Wang Z, Hu L, Zhang X, Chen J, Yu Z, Liu L and Wu M (2022) Targets of statins intervention in LDL-C metabolism: Gut microbiota. Front. Cardiovasc. Med. 9:972603. doi: 10.3389/fcvm.2022.972603
Received: 18 June 2022; Accepted: 08 August 2022;
Published: 08 September 2022.
Edited by:
Francesco Violi, Sapienza University of Rome, ItalyCopyright © 2022 Sun, Wang, Hu, Zhang, Chen, Yu, Liu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: LongTao Liu, bGl1bG9uZ3RhbzE5NzZAMTI2LmNvbQ==; Min Wu, d3VtaW4xOTc2MjAwMEAxMjYuY29t
†These authors have contributed equally to this work and share first authorship