 Katongo H. Mutengo
Katongo H. Mutengo Sepiso K. Masenga
Sepiso K. Masenga Naome Mwesigwa
Naome Mwesigwa Kaushik P. Patel
Kaushik P. Patel Annet Kirabo
Annet Kirabo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 25 August 2022
Sec. General Cardiovascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.968184
This article is part of the Research Topic Gaps, Priorities and Advances in Cardiovascular Medicine in Africa View all 7 articles
Hypertension is a risk factor for end organ damage and death and is more common in persons with HIV compared to the general population. Several mechanisms have been studied in the pathogenesis of hypertension. Current evidence suggests that the epithelial sodium channel (ENaC) plays a key role in regulating blood pressure through the transport of sodium and water across membranes in the kidney tubules, resulting in retention of sodium and water and an altered fluid balance. However, there is scarcity of information that elucidates the role of ENaC in HIV as it relates to increasing the risk for development or pathogenesis of hypertension. This review summarized the evidence to date implicating a potential role for altered ENaC activity in contributing to hypertension in patients with HIV.
The epithelial sodium channel (ENaC) is a membrane-bound ion channel that transports sodium [Na+] ions and is abundantly expressed in various epithelial cells (1, 2). It plays a critical role in blood pressure and volume control, classically through its regulation of sodium reabsorption in the distal nephron (3). It is known that the role of ENaC in blood pressure regulation is mainly controlled by the activation of the renin-angiotensin-aldosterone system (4–6). However, other extrarenal factors have also been implicated in ENaC activation including the reactive oxygen species (ROS) (6, 7).
ROS are produced as a by-product of normal aerobic metabolism, and levels are regulated by critical antioxidant enzymes, which include superoxide dismutase, the glutathione peroxidases, catalase, and glucose-6-phosphate dehydrogenase (8). Under regulated conditions, the ROS are essential for immune signaling pathways and are incorporated into immune responses (9, 10). However, excess ROS are detrimental to vascular health and are produced when there is increased activity of reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, the main enzyme responsible for generating superoxide from oxygen (8, 11). Other ROS-generating enzymes include xanthine oxidases, and cyclooxygenases (12). Importantly, both human immunodeficiency virus (HIV) and hypertension are associated with an increase in ROS generation (13–15), and therefore potentially contribute to the upregulation of ENaC. While clear and well-documented evidence supports the role of ENaC in the pathogenesis of salt-sensitive hypertension (6, 16, 17), it is not clear if there is a potential synergism between HIV and ENaC, despite recent evidence from our research indicating a high prevalence of salt-sensitivity in people with HIV (PWH) who had hypertension (18). Further studies to elucidate the link between HIV, salt sensitivity and the additive role that ENaC may play remains to be explored.
The major risk factor for cardiovascular diseases (CVDs) is hypertension and sub-Sahara Africa has a weighty synchronization of hypertension and HIV (19–22), making it a prime site for this area of research. Easy access to combined antiretroviral therapy (cART) as well as newer innovations in HIV programs has seen HIV transition from a once fatal disease to a chronic and manageable condition. Even regions hardest hit by the epidemic such as Eastern and Southern Africa; which account for 54% of the global HIV burden, have observed a decline in new HIV infections by 28% and deaths by 44% between 2010 and 2018 (23). However, these positive strides have also seen a progression of non-infectious chronic complications such as CVDs (24–26). HIV has been known to confer about 1.5 to 2-fold increased relative risk to CVDs such as myocardial infarction, heart failure and stroke (24, 25, 27, 28), with hypertension being a consistent underlying risk factor. The association between HIV-related factors and traditional risk factors for atherosclerosis, also associated with hypertension, has been implicated as causing a higher risk of CVDs in PWH (29–32). Therefore, major research efforts need to be made to cope with this rising burden of CVDs. This review provides evidence linking a possible role for activation of ENaC during HIV infection in contributing to hypertension.
The epithelial Na+ channel (ENaC; Figure 1) is a membrane-bound ion channel that is selectively permeable to Na+ ions and consists of three main subunits called α, β, and γ with a fourth (δ) that is analogous in function to the α subunit found in some tissues where it acts in place of α (1, 2, 33, 34).
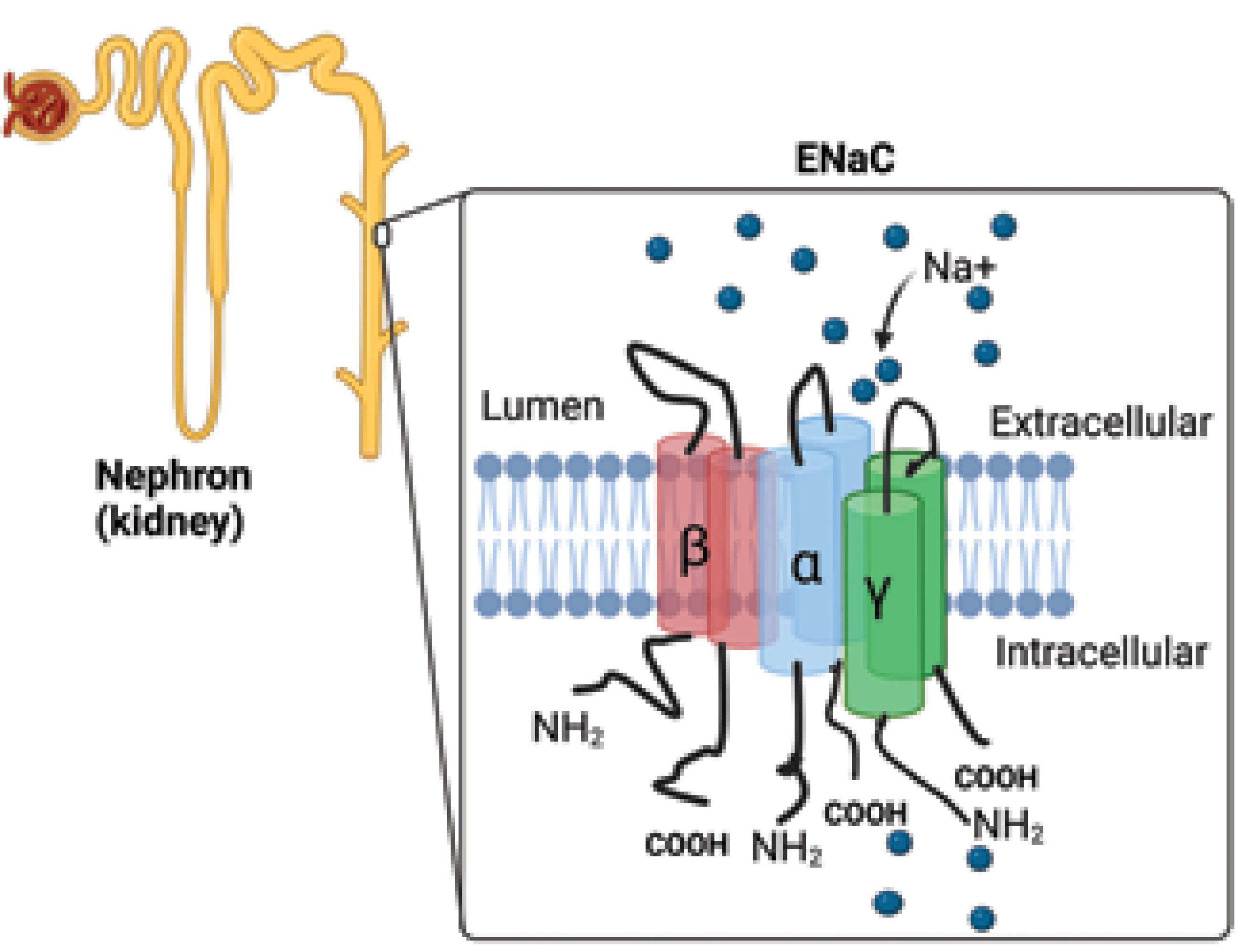
Figure 1. Epithelial sodium channel. The epithelial Na+ channel (ENaC) is responsible for the reabsorption of sodium ions by the epithelia lining the distal part of the kidney tubule (collecting duct). The ENaC consists of three main subunits called α, β, and γ. NH2, amino terminal group; COOH, carboxyl group, Na+, sodium ions.
There are four major ENaC regulatory mechanisms: The first is Na+ self-inhibition in which Na+ can either induce an allosteric change in ENaC or slowly inhibit the channel over time (35–37). This type of regulation enables Na+ reabsorption to be controlled via concentration of Na+ in the distal nephron. ENaC regulation also occurs via post-translational proteolytic cleavage, resulting in the abandonment of inhibitory pathways, making the channel more likely to remain open (38–40). Lipid signaling molecules also play a role in both increasing and decreasing the opening probability of ENaC via both binding to cationic sequences and inhibitory lipids (41, 42). Finally, aldosterone may also help regulate ENaC regulation in several different ways. Angiotensin II (Ang) II also influences this process by promoting secretion of the key ENaC regulator, aldosterone from the adrenal zona glomerulosa (4). The activation and expression of serum-and-glucocorticoid-induced kinase (SGK1) through the influence of aldosterone contributes to the increased expression and opening probability of ENaC at the plasma membrane (43–45).
ENaC’s role in the kidney, where it acts as a control mechanism for Na+ and K+, has been extensively studied (5, 6, 39, 45, 46). It is typically expressed in the aldosterone-sensitive distal nephron (ASDN) of epithelial cells. In Na+ control, it acts as a rate-limiting step for transepithelial Na+ reabsorption, total body net Na+ content, fluid volume, and blood pressure in the nephron collecting duct (47). It is also critical to the secretion of K+ in ASDN (48). Elevation of serum potassium is considered to be one of the most important agonist for aldosterone secretion (49). Aldosterone, as aforementioned, acts through the mineralocorticoid receptor to modulate changes in blood volume and sodium balance by increasing ENaC abundance and activity (4, 50). Opening of the ENaC channels results in a lumen to intracellular Na+ flux, which is facilitated by the negative electrical potential and the steep Na+ concentration gradient across the apical membrane. Na+ is then actively transported across the basolateral membrane by the Na+/K + ATPase in exchange for potassium, and potassium then exits the apical membrane K+ channels (51). Hyperkalemia and hypokalemia are common in HIV, although their impact on renal salt handling and ENaC remains unexplored. For example, commonly used drugs such as Amphotericin B to treat cryptococcal meningitis in HIV, chronic diarrhea, AIDS-associated enteropathies as well as detectable HIV mRNA levels were associated with hypokalemia (52–54). Conversely, hyperkalemia has been seen with the use of high-dose trimethoprim-sulfamethoxazole or intravenous pentamidine for pneumocystis jirovecii pneumonia attributed to an amiloride-like mechanism by blocking apical membrane sodium channels in the mammalian distal nephron (55–57). On the other hand, a study exploring the spectrum of electrolyte abnormalities in black African people living with human immunodeficiency virus and diabetes mellitus (DM) found that comorbidity of HIV and DM increased the chance of hypokalemia by 97% for every unit increase in DM duration attributed to a greater degree of insulin resistance, as both conditions progress (58). On the contrary, the non-HIV infected in this study had a 10% increased chance of hyperkalemia for every unit increase in DM attributed to dysautonomia in long- standing DM which predisposes to hyporeninaemic hypo-aldosteronism and associated hyperkalaemia (58). In vitro data suggest that individuals infected with HIV have alterations in transcellular K+ transport. Caramelo et al. (59) demonstrated that L-arginine–induced, rapid-onset hyperkalemia, was only present in the HIV infected and not the healthy controls suggesting that HIV-positive individuals have a systemic derangement in transmembrane K+ transport that is coincident with inhibited aldosterone response. Although inhibition of aldosterone has traditionally been thought to downregulate ENaC function, it has been postulated that ENaC activity is high in some instances when there are no significant changes in aldosterone, termed aldosterone-independent ENaC activation, where Vasopressin is implicated in the increased activity (60). However, it remains unclear whether disruption of transmembrane K+ transport with inhibited aldosterone response found in HIV (59) can affect ENaC function and activation.
In addition, extrarenal factors such as immune activation through the generation of ROS also upregulate the ENaC (7). ENaCs have also been found to be expressed in immune cells such as the dendric cells (DCs), which contribute to the pathogenesis of salt-sensitive hypertension (61, 62). It is worth noting that DCs are also the main site of HIV seeding, altering their function (63, 64). Whether this contributes to the upregulation of ENaC in these cells remains to be determined.
While studies on the impact of blood pressure changes and ENaC function in HIV are lacking, PWH have been widely reported to have a higher prevalence of hypertension than the general population (65, 66). A study by Masenga et al. (18) showed that PWH who were hypertensive, also demonstrated a high salt sensitivity compared to those who were normotensive and HIV-negative individuals. Because hypertension is driven by dysregulated stimulation of the RAAS (46, 67, 68), the process that also upregulates ENaC function (4–6), an increase in blood pressure can be viewed as a driver or a consequence of increased ENaC activity. Genetic variations in blood pressure responses have been extensively studied in the general population, revealing that individuals with rare variants of RAAS pathway were 1.55 times more likely to have salt-sensitivity compared to non-carriers, suggesting an association between RAAS and Na+ transporters (69). Although the effects of HIV and cART contribute to oxidative stress which contributes to RAAS activation (9, 70–73), investigating whether specific genetic variations contribute to the upregulation of ENaC in PWH is warrantable.
Mutations in ENaC result in human hypertension or hypotension, connecting it to the regulation of blood pressure (74). Beyond its role in the kidney, ENaC has been implicated in the interplay between hypertension, salt sensitivity, and the immune system (17, 61). The immune system’s response to hypertension may be a function of self-tolerance loss. In this case, hypertension would be considered an autoimmune disease. There is also evidence to suggest that the immune system plays an important role in salt sensitivity of blood pressure (SSBP). SSBP is defined as changes in blood pressure (BP) which mirror changes in dietary salt intake/depletion (75). The immune system’s link to hypertension and salt-sensitivity is based on the activity of antigen-presenting cells (APCs), particularly macrophages, DCs, and B cells. In response to hypertensive stimuli, such as norepinephrine and dietary salt, these APCs enter the vasculature and kidney along with T lymphocytes where they converge in aiding Na+ retention and vasoconstriction leading to the elevation of blood pressure and consequent end-organ damage (44, 62, 76–78). Coincidentally, APCs like monocytes stand out as the most abundant immune cell type found in atherosclerotic plaques (79, 80). There are three distinct types of human monocytes based on their expression of cluster of differentiation-14 (CD14) and CD16 soluble surface markers: classical with little or no CD16 (CD14++CD16-), intermediate with expression of both CD14 and CD16 (CD14++CD16 +), and non-classical with low CD14 (CD14lowCD16++) monocytes (79, 81). The classical monocytes are rapidly recruited to invade the inflamed tissue during injury or inflammation and contribute to immunological responses. Such a response comprises of recognizing and removing microorganisms and dying cells while non-classical monocytes patrol the endothelium as part of innate surveillance (82). The intermediate monocytes are recruited at a later stage of inflammation and involved in pro-inflammatory response (82). Interestingly high levels of circulating monocytes with CD16 and CD14 have been observed during HIV-infection (83–85). These immune cells are key drivers of atherosclerotic processes leading to CVDs (85–88). In fact, soluble CD14 has been found to be elevated in PWH with hypertension (89), although the contribution of ENaC to these pathoimmune responses in PWH remain to be studied. Furthermore, Loperena et al. (90) observed a direct relationship between 48-h uniaxial hypertensive stretching of endothelial cells and the transformation of classical monocytes (CD14++CD16-) into intermediate monocytes (CD14++CD16+). Loperana et al. further demonstrated that these CD14++CD16+ monocytes produced significant pro-inflammatory cytokines (interlukin-6 [IL-6], interlukin-1β [IL-1β], interlukin-23 [IL-23], and tumor necrosis factor α [TNF-α]) that drove T-cell proliferation in the human volunteers. This study excluded PWH and was therefore unable to make comparisons in this group, but a hypothesis can be postulated on the basis of a loose association with other studies showing an elevation of the monocyte-soluble marker CD14 in hypertensive PWH (89) and with upregulation of ENaC in DCs, which are simply activated monocytes (61), and increased CD14++CD16+ markers with high salt concentration (91).
Additionally, recent studies have identified a connection between EnaC, hypertension, and innate immunity. An exploration of Na+-dependent EnaC activity in DCs and its signaling mechanisms has shown that, as Na+ concentration increases extracellularly, both NADPH-oxidase activation and superoxide generation also increase (91). This leads to isolevuglandins (IsoLG)-adduct proteins, which are formed from lipid peroxidation of arachidonic acid and are extremely immunoreactive. IsoLG-adduct proteins produced in monocyte-derived DCs can lead to immune tolerance depletion in salt-induced hypertension (62, 92). As IsoLG-adduct proteins increase in an EnaC-dependent mechanism through elevated extracellular high salt conditions, the surface expression of CD83 and CD16, and the B7 ligands CD80 and CD86, increases in a process important for the maturation of DCs and antigen presentation (91, 92). On the other hand, contrasting effects of HIV-1 on DC phenotype and/or function have been reported. While some studies have reported an upregulation of the markers CD80, CD83, and CD86 upon exposure to HIV-1 or its proteins (93), others reported no effect (94), creating a controversy as to whether EnaC activation can also be variable, as the aforementioned markers are also associated with EnaC activation.
SGK-1, an intracellular salt-sensing kinase, plays a role in blood pressure modulation and EnaC regulation in the distal convoluted tubule (61). In APCs, SGK-1 controls the increased expression of the α- and γ-subunits of EnaC as mediation in salt-sensitive hypertension. This regulation results in the production of IsoLG-adducts and interleukin-1β (IL-1β) as well as the activation of T cells (61). SGK1 activity increases autoimmunity as it restricts the function of Treg cells and reciprocally regulates the development of T helper 17/regulatory T cell (Th17/Treg) balance. More Th17 means more IL-17 and therefore contribute to the development of hypertension (95, 96). There is also evidence of a possible connection between IL-1β and salt-sensitive hypertension (97, 98). According to both animal and human studies, cytokines like IL-1β generate a pro-inflammatory state which may result in heightened blood pressure due to variation in renal, endothelial, and immune responses. In mice specifically, the pharmacological inhibition of IL-1β, targeted antibody treatment of IL-1β, and genetic deletion have all proven to lead to decreased blood pressure (99–101). However, it remains unclear whether the pro-inflammatory state induced by IL-1β is necessarily a good indicator of resultant hypertension in PWH, especially since HIV-1 infection in human monocytes efficiently induce IL-1β expression and inflammasome activation (102). This inflammation is also exacerbated by high salt concentration which has been shown to aid in the production of IL-1β via blood mononuclear cells and the EnaC-dependent production of IL-1β in DCs (91, 103). A summary of the possible interaction between HIV, EnaC, ROS and activated T cells in the evolution of hypertension are presented in Figure 2.
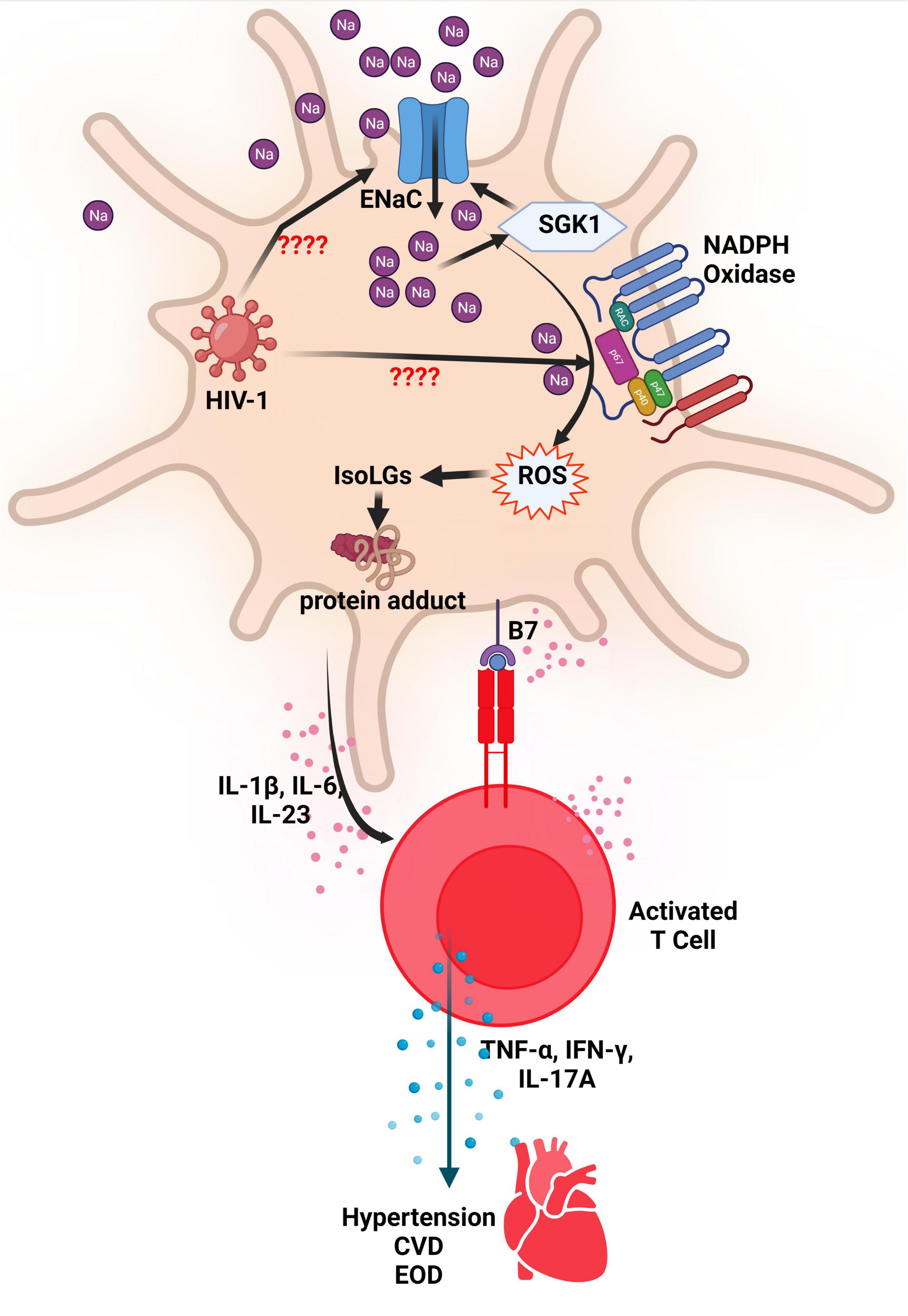
Figure 2. ENaC and NADPH oxidase activation in DCs resulting in hypertension. The increased activity of the endothelial sodium channel (ENaC) leads to an increase in Na+ ions that activates the enzyme NADPH Oxidase. The increased activity of NADPH Oxidase results in production of reactive oxygen species (ROS) which leads to formation of Isolevuglandins (IsoLGs). IsoLGs adduct to intracellular proteins and become neoantigens that are presented to T cells to activate them. Activated T cells produce inflammatory cytokines that contribute to development or progression of hypertension, cardiovascular disease (CVD) and end organ damage (EOD). It is thought that HIV-1 activates ENaC and NADPH in antigen presenting cells resulting in similar effects. SGK1, serum-and-glucocorticoid-induced kinase; NADPH, Reduced nicotinamide adenine dinucleotide phosphate; Na, sodium; HIV-1, human immunodeficiency virus type 1; IL, interleukin; TNF-α, tumor necrosis factor alpha; IFN-γ, interferon gamma; CVD, cardiovascular disease; EOD, end organ damage.
Evidence now strongly suggests that vascular EnaC contributes to the pathogenesis of arterial hypertension propagated by endothelial dysfunction (104). Vascular endothelium is a single layer of cells that forms the first barrier in the blood vessel lumen. In larger vessels, this layer is in contact with vascular smooth muscle, collagen and elastic fibers (105). The endothelium is a biologically active cell layer that produces mechanical sensors and transducers which maintain vascular homeostasis, vascular tone, and vascular integrity (105, 106). Normal endothelial function requires the regulated balance in these mechano-sensors and transducers. This balance is a function of a number of endothelium-derived relaxation factors such as vasodilator prostaglandins, nitric oxide (NO), endothelium-dependent hyperpolarizing factors, and endothelium-derived contraction factors (107–109). However, NO appears to be the main contributor to vascular homeostasis (110, 111).
There is an inverse relationship between NO availability and the generation of ROS. Excess ROS is detrimental to vascular endothelium and production is increased in situations of reduced NO, the major factor in vascular homeostasis (110–113). The reduction in the production of NO occurs mainly through two processes. The first is the increased activity of NAD(P)H oxidase, an enzyme responsible for the generating superoxide from oxygen. The superoxide reacts with NO to form peroxynitrite (OONO-), thereby oxidatively inactivating the NO (11). Second is the uncoupling of the oxidoreductase enzyme endothelial NO synthase (eNOS). This means that eNOS is converted from an enzyme that oxidizes L-arginine to NO into one that reduces molecular oxygen to a superoxide anion which is one of the main ROS (11, 114). The increase in these endothelial ROS induces lipid oxidation, leading to the production of oxidized low-density lipoprotein (LDL) cholesterol (115, 116). Oxidized LDL is further internalized in the endothelium by binding to a transmembrane glycoprotein, lectin-like oxidized low-density lipoprotein (LDL) receptor-1 (LOX-1) (117). Excess sub-endothelial deposition of oxidized LDL activates the endothelium to increase expression of cellular adhesion molecules that stimulate platelet aggregation, increased attachment and migration of inflammatory cells into the intima, such as monocytes which transform into lipid-laden macrophages called foam cells (106, 117, 118). There is also increased production of vasoconstrictors and proliferation and migration of vascular smooth muscle cells and deposition of collagen (117). ROS also induce direct toxicity to endothelial cells leading to apoptosis and inflammation further repeating the vicious cycle of the ROS generation (106). Eventually, these processes cause stiffness and narrowing of blood vessels; an atherosclerotic process which leads to hypertension. The common traditional risk factors for CVDs such as obesity, hypertension, diabetes mellitus, smoking, aging, and hypercholesterolemia can further increase the possibility of the generation of ROS (119–122). These risk factors are heightened in HIV-infection and trigger several pathological pathways that cause endothelial dysfunction and subsequent atherosclerosis (14, 70, 71). ROS also stimulate EnaC activity (6, 7), and possibly can play an additive or multiplicative role in the onset of hypertension in PWH.
A study by Jia et al. found that EnaC activation mediates endothelial permeability, which in turn promotes macrophage infiltration, M1 polarization, and oxidative stress leading to cardiac fibrosis and diastolic dysfunction in female mice with dietary obesity (44). In human studies, there is increased expression of the EnaC in neutrophils from hypertensive patients suggesting an inflammatory role (123). Upregulation of the EnaC has been linked to stiffening of the cortical actin cytoskeleton in endothelial cells including those in the coronary vessels, impaired endothelial NO release, increased oxidative stress, increased vascular permeability, and immune activation (124). Incidentally, similar mechanisms are induced by HIV (14, 15, 28, 125), further addressing the possibility of why PWH are at a higher risk of developing CVDs. These factors can lead to modulation of vascular tone and stimulation of tissue remodeling, leading to heart failure with preserved systolic function (HfpEF) (124).
While the cause of hypertension in HIV is multifaceted, it can generally be accepted that ROS play a crucial role in the onset and sustenance of hypertension. Reduction in NO release is the hallmark of endothelial dysfunction which triggers a cascade of events leading to hypertension and this reduction is enhanced by high Na+ concentration (126). A study on the effects of HIV on the pulmonary vasculature showed that HIV causes endothelial damage and mediator-related vasoconstriction through stimulation by the envelope gp 120, including direct release and effects of endothelin-1 (vasoconstrictor), interlukin-6 and TNF-α (127). The inflammatory mediators, IL-6 and TNF-α, which induce mediator-related vasoconstriction, also contribute to formation of ROS, which are important for the upregulation of ENaC (6, 7, 128). Upregulation of ENaC further leads to a cycle of impaired endothelial NO release, increased oxidative stress, increased vascular permeability, and immune activation which contribute to atherosclerosis and hypertension (124, 128). Increasing NO bioavailibity is important for smooth muscle relaxation and overall vascular health (110, 111). However, oxidative processes reduces NO bioavailability (11, 129), and these can be induced by both HIV and ENaC upregulation. HIV therefore likely plays a synergistic role in the development of hypertension through its possible indirect role in the vascular ENaC activation.
Non-invasive measurements such as flow-mediated dilatation (FMD) of the brachial artery have been used as surrogates to determine endothelium-dependent vasorelaxation (129). A study by Oliviero et al. showed that FMD is impaired in naive untreated PWH versus healthy participants (130), indicating inadequate release of vasodilators, including nitric oxide, which cause dilation in a healthy artery (131). Oliviero et al. further found a strong inverse correlation between FMD values and HIV mRNA levels which favored the direct role for early stage of viral infection in vascular dysfunction independent of cART and metabolic factors. On the contrary, another study looking at FMD reported no significant changes in the HIV-treatment naïve, HIV on cART and healthy participants, despite high levels of inflammatory markers in the HIV-treatment naïve (132). Although generally, cART has been linked to the development atherosclerotic changes mostly contributed by ROS generated as a result of lipid abnormalities (72, 133–136). With scarcity of data linking the ENaC to cardiac pathologies and HIV, more studies are warranted to examine this connection.
Glycoprotein 120 (gp120) is a critical protein in the binding of the HIV to cell surface receptors of target cell. It is expressed on the outer layer on the virus, in free form in the body fluids of infected people, as well as the virus infected cells (137). Gp120 is critical for virus infection, as the protein is necessary for binding to specific cell surface receptor CD4, mainly expressed on CD4+ T cells, and the coreceptor [e.g., chemokine receptor C-C Motif Chemokine Receptor 5 (CCR5) or C-X-C chemokine receptor type 4 (CXCR4)] (138). The molecular and cellular mechanisms through which gp120 induce vascular injury is multifaceted. Studies have demonstrated that gp120 acts synergistically with HIV nuclear protein trans-activator of transcription (Tat) to stimulate release of endothelial microvesicles which further confer pathological effects on endothelial cells by inducing inflammation, oxidative stress, and senescence as well as enhancing susceptibility to apoptosis (15). This HIV glycoprotein also acts as an agonist to proinflammatory cytokines such as TNF- α (139), endothelial monocyte-activating polypeptide II (140) and interleukin-6 and interleukin-8 (15), as well as induce apoptotic signaling transduction pathways through the p38 mitogen-activated protein kinase (MAPK) (141). All these pathways contribute to ROS generation, endothelial apoptosis, platelet aggregation and formation of atheroma and are also known to affect ENaC and thus may interact during HIV to develop hypertension (6, 7).
On the other hand, negative regulatory factor (Nef) is the most frequently transcribed HIV accessory protein in the early stages of viral infection. It is critical for maintaining virus production and progression to acquired immune deficiency syndrome (AIDS) (142). Nef has been found to have the ability to downregulate CD4 receptors and major histocompatibility complex class 1 (MCH 1), which benefits HIV by interfering with viral recognition and destruction of infected cells by cytotoxic T-cells (142, 143). Nef has been shown to persist in the blood of ART-treated PWH and cause ROS generation, release of monocyte attractant protein-1 (MCP-1), and consequent apoptosis of endothelial cells (144, 145). On the other hand, these are similar processes found with the upregulation of ENaC (17) and may interact during HIV.
Both Nef and Tat have also been shown to induce NADPH oxidases (NOX) as well as immense enhancement of their responses to a variety of stimuli (146, 147). Seven distinct NOX isoforms have been identified, of which NOX1, 2, 4, and 5 have been characterized in the cardiovascular system (148). However, NOX4 is the most abundant NOX isoform in the renal tubular segments, including the ASDN, although significant amounts have also been found in the pulmonary vasculature and microglia (148–151). Animal studies have demonstrated that Tat augments NOX4, facilitating the production of ROS (151, 152). The NOX4-dependent pathway regulates many factors essential for proper sodium handling in the distal nephron (149). A study by Cowley et al. showed that Dahl salt-sensitive rats lacking NOX4 have lower blood pressure and renal injury when placed on a high salt diet than wild type controls (153). This was supported by another study by Pavlov et al. (149) which showed that NOX4 deletion in Dahl salt-sensitive rat challenged with a high salt diet attenuated high salt-induced upregulation of ENaC activity in the cortical collecting ducts. Pavlov et al. further showed that H2O2 upregulated ENaC activity, and H2O2 production was reduced in both the renal cortex and medulla in salt-sensitive rats with deleted NOX4 who were fed a high salt diet, suggesting that ENaC upregulation in the context of renal injury might result from overstimulation of the NOX4-dependent pathway and increased ROS generation (149). With evidence indicating influence of HIV proteins on NOX and generation of ROS, it can be hypothesized that HIV may have an effect on the renal ENaC salt handling and salt sensitive hypertension. However, as data is still scarce in this area, the relationship between HIV and ART on salt handling through ENaC remains unknown.
Metabolic risks such as weight gain, dyslipidemia and hyperglycemia, which are part of the metabolic syndrome, have been identified in patients on cART which can alter normal endothelial function (72, 134, 135, 154, 155). Sax et al. showed that weight gain was greater in more recent trials and with the use of newer integrase strand transfer inhibitor (INSTIs) such as dolutegravir than protease inhibitors (135). Sax et al. further showed that demographic factors associated with weight gain included lower CD4 cell count, higher HIV type 1 RNA, no injection drug use, female sex, and black race. Several observational studies have also indicated significant weight gain in PWH on INSTIs with some indicating a predisposition toward black race and female sex (72, 156, 157). Excessive weight gain, particularly central obesity is associated with vascular endothelial dysfunction. Healthy perivascular adipose tissue (PVAT) ensures normal endothelial vasodilation function, however, enhanced oxidative stress and pro-cytokine generation found with obesity-associated PVAT is associated with reduced NO availability promoting the aforementioned cascade of events leading to atherosclerosis (133). Obesity is also associated with elevated aldosterone and insulin levels, both of which stimulates ENaC to conserve sodium and are thought to be a likely cause of refractory hypertension in this population (51).
High levels of insulin also stimulate the renin-angiotensin-aldosterone system (RAAS) resulting in increased peripheral vascular resistance and hypertension (158). RAAS is crucial for blood pressure regulation as well as the pathogenesis of cardiovascular diseases (159, 160). The RAAS involves the secretion of renin, an enzyme secreted by the juxtaglomerular cells of the kidneys in response to various stimuli such as decreased afferent arteriolar perfusion, decreased sodium content in the macula densa and sympathetic activation (68). Renin activates the conversion of angiotensinogen to angiotensin I, which is further cleaved to into Ang II by the angiotensin converting enzyme (ACE) (68). Ang II acts on the angiotensin receptor I (AT1R) and induces deleterious effects such as vasoconstriction, sympathetic activation, reduction in NO synthesis, inflammation and oxidative stress; processes leading to the pathogenesis of hypertension (46, 67, 159, 160). The effects of Ang II are counteracted by another enzyme angiotensin converting enzyme II (ACE 2), which acts on the angiotensin receptor II (AT2R), although these receptors are more restricted in tissue expression and affinity in adults (161). The AT2R acts as the catalytic site for the ACE 2 which results in conversion of Ang II to angiotensin-1,7 (Ang-1,7) (161). Ang-1,7 causes vasodilatation, inhibits oxidative cellular damage by blocking NADPH-oxidase, and stimulates NO production via nitric synthase pathways (162). All of these cellular processes have a protective effect on the endothelium. However, there is limited data on whether HIV and obesity cause further downregulation of Ang-1,7 although both HIV and obesity have been shown to cause upregulation of Ang II via RAAS, potentially contributing to hypertension (73). As previously mentioned, Ang II promotes secretion of the key ENaC regulator, aldosterone, from the adrenal zona glomerulosa, which is essential for the pathogenesis of hypertension (4).
A study by Srinivasa et al. showed that HIV-infected subjects on cART with excess visceral adipose tissue had significantly higher RAAS activation than HIV-infected subjects without excess visceral adipose tissue (73). This study further showed that HIV-infected subjects with excess visceral adipose tissue had increased aldosterone and plasma renin activity compared to the HIV-infected subjects without excess visceral adipose tissue (73), suggesting the potential role of dysregulated exogenous aldosterone production and RAAS activation from adipose tissue (163, 164). In addition, aldosterone stimulates both the innate and adaptive immune systems leading to macrophages and T cells accumulation in the kidneys, heart, and vasculature which contribute to the development of hypertension and other cardiometabolic diseases (165). The persistence of immune activation in HIV (144, 166) and the metabolic toxicity associated with cART (72, 134, 135, 154, 155) may potentially play an additive or multiplicative role on the effects of aldosterone and ENaC activation, and therefore increase the risk of hypertension in PWH. Given the widespread use of dolutegravir, it is worth prospectively investigating the impact of weight gain on ENaC function in PWH.
Several animal studies have shown that adipose cells in small arteries demonstrate increased expression of endothelin-1, endothelin type A and endothelin type B receptors, and TNF- α, which augment adrenergic vasoconstriction while impairing tonic NO release (167–170). In addition, high glucose levels lead to enhanced non-enzymatic glycation, sorbitol-myo-inositol-mediated changes, redox potential alterations, and the activation of protein kinase C which induces oxidative stress (171). The low-density lipoproteins also activate intracellular pathways to increase local and systemic inflammation, monocyte adhesion, endothelial cell dysfunction and apoptosis, and smooth muscle cell proliferation, leading to foam cell formation and genesis of atherosclerotic plaque (172, 173). The effect of these various metabolic risks occurs in a synergic fashion with HIV (27, 174–178), and also found with salt-sensitivity and ENaC activation (16, 61, 91), (Figure 3) suggesting an imperative need to investigate the contribution of Na+ and ENaC activity, and their interaction with anti-retroviral therapy in PWH.
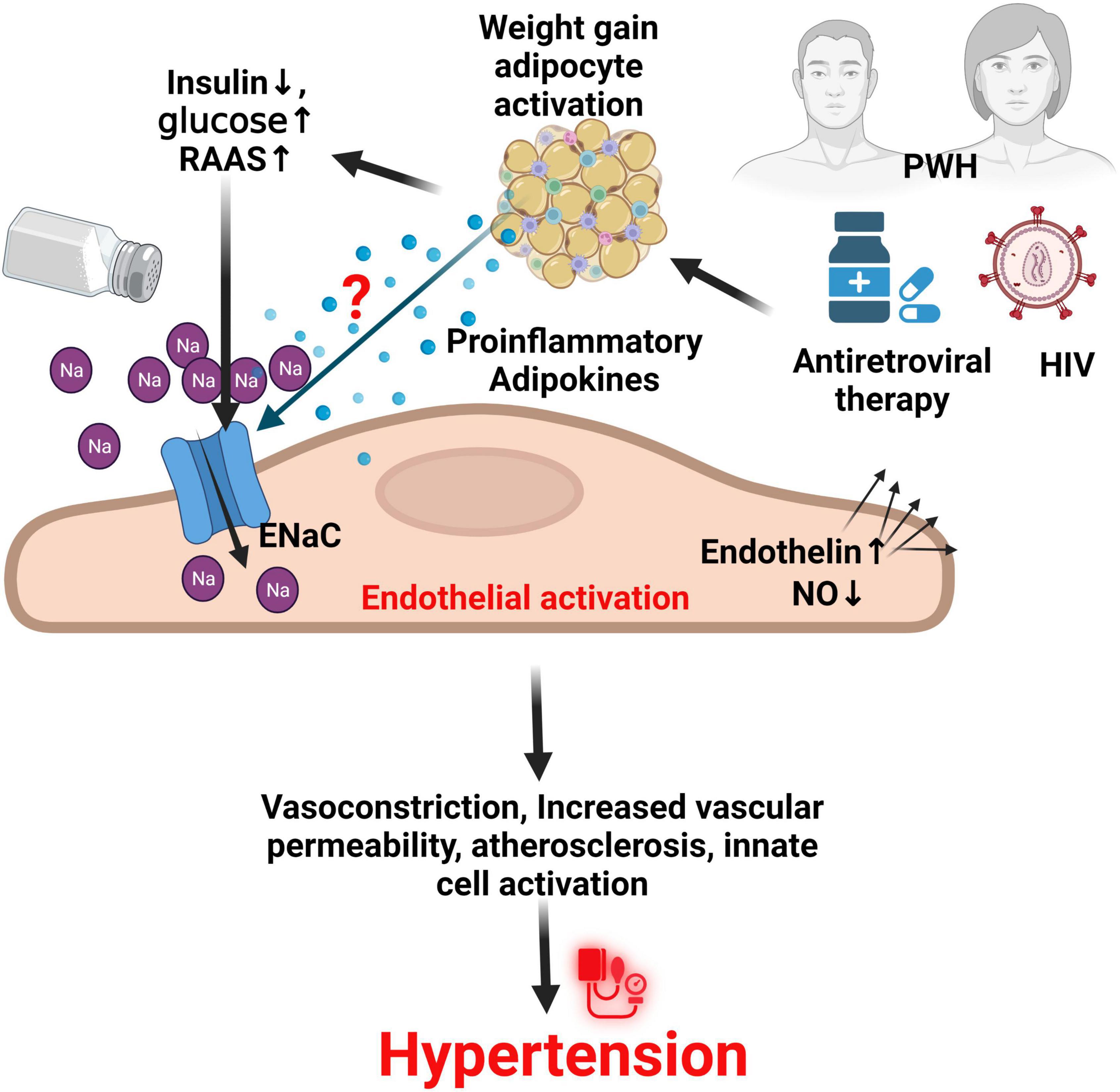
Figure 3. ENaC in endothelial cells and the role of HIV and ART in hypertension. It is thought that HIV infection and use of antiretroviral therapy especially dolutegravir, synergizes with other factors such as increased glucose, reduced insulin action and production, and activation of the renin angiotensin aldosterone system (RAAS) through unknown mechanisms to increase the activity of the endothelial sodium channel (ENaC) on activated endothelial cells. This results in several effects that contribute to hypertension. NO, nitric oxide; PWH, persons with HIV; Na, sodium; HIV, human immunodeficiency virus.
It is worth noting that gender differences have been noted in human immunodeficiency virus (HIV) infection in different geographic areas. In 2020, women aged 15 years and older accounted for at least 56% of HIV infections in sub-Saharan Africa (SSA), while in western and central Europe and North America, the same age group accounted for only 20% (179). Although the epidemiological problem of hypertension in HIV-infected adults is well studied (65, 66, 180), gender influences on the pathogenesis of hypertension in PHW are not as well studied, despite the geographical diversity of HIV infection.
Existing studies have shown that the prevalence of hypertension is generally higher in men than women, regardless of age, although severity in women increases markedly with age (181–183). Hypertension in men is mainly attributed to low socioeconomic status and behavioral factors such as high alcohol consumption and smoking, while advancing age and hormonal imbalances from the onset of menopause and combined oral contraceptives pills (COCs) appear to be main contributors in women (181, 183). In general, women have higher CD8+ T cell activation, higher interferon-stimulated genes (which are both protective in HIV progression and accompanying comorbidities) and the protective effect of estrogen on the development of hypertension appear to be what protects women living with HIV from developing hypertension earlier than men (184). Animal studies have also shown that estrogen reduces ENaC function and blood pressure (185), providing further evidence to explain postmenopausal hypertension. More studies are therefore needed to show whether this effect extends to PWH.
Gender differences have been identified in Na+ storage between muscle and skin which may be one of the factors underlying sex-specific susceptibility. For example, a study by Wang et al. showed that men have higher levels of Na+ in their skin than in muscle, while women accumulate Na+ to greater levels in their muscle (186). Increased Na+ accumulation in the skin compared to muscle is associated with increased risk for the development and pathogenesis of hypertension (69, 150). Interestingly, Barbaro et al. demonstrated that monocytes from humans with high skin Na+ exhibited increased IsoLG-adduct accumulation and CD83 expression (91), thought to lead to immune tolerance depletion in salt-induced hypertension (62, 92). In the previous section, it was stated that soluble markers such as CD14 were found to be elevated in PWH with hypertension, with upregulation of ENaC, and with high-salt concentration (61, 89, 91). Siedner et al. found that HIV-infected women on long-term ART had significantly elevated levels of soluble CD14 and soluble CD163 compared to HIV-infected men (187). Evidence from the general population also indicates that women exhibit significant changes in blood pressure in response to high-salt consumption compared to men (188, 189). This has been attributed to factors such as failure to suppress aldosterone following high-salt load (190), a key mechanism leading to protection from salt-sensitive hypertension (191, 192). However, further studies are needed to investigate the contribution of salt-sensitivity and ENaC to hypertension along with gender differences in PWH.
There are also gender differences in factors affecting the gut microbiota in the development of hypertension. A study of over 200, 000 participants characterizing gender-specific dietary patterns showed that women were 2.4 times more likely to consume too much sugar, 1.4 times more likely to consume too much fat, and 1.4 times as likely to consume fiber intake below the recommendations (193). Excess sugar and fat, and less fiber has a tendency to tip the gut microbiota toward dysbiosis (194), which has been shown to cause increase in blood pressure in animal studies (195, 196). High dietary sodium consumption may contribute to gut dysbiosis by altering the gut microbial composition, and inducing an inflammatory response which alters the gut anatomy (197), and is associated with increased immunogenic IsoLG-adduct formation responsible for salt-sensitivity and upregulation of ENaC (61, 198), and women have been reported to be more sensitive to salt (188, 189). The group by McDonough et al. reported that in mice, sodium channels in the kidney demonstrate a sex-specific adaptation to high-salt diet that ultimately preserve electrolyte homeostasis (199). Several sodium transporters along the nephron (Na+/H+exchanger 3 (NHE3), bumetanide-sensitive Na+-K+-2Cl cotransporter (NKCC2), Na+-Cl– cotransporter (NCC), and the ENaC) play specific roles in the ultimate maintenance of electrolyte homeostasis and changes in the activity of one may affect the expression and activity of the other transporters (200–202). ENaC particularly mediates the final step in sodium reabsorption, making it an important regulator of sodium and blood pressure (165). Sex-specific differences in the expression of sodium channels along the nephron were observed when mice were fed high salt (4% sodium chloride), but the fold changes were similar along the distal nephron and collecting duct where ENaC is located (200–202). However, compared to other transporters, inhibition of ENaC resulted in increased excretion of sodium and water in male rats compared to females suggesting that the response of ENaC in males may be more pronounced than in females (165). There is also some evidence of patho-immune gender-specific mechanisms in the development of hypertension involving IL-17 secretion, NO and Ang II (67, 203–205). A study by Zimmerman et al. showed that infusion on Ang II in Male Sprague-Dawley rats produced a greater increase in the number of renal Th17 cells (206), and Th17 cells have been associated with Ang II-induced increases in BP, salt-induced increase in SGK1 and ENaC activation (77, 96). It has also been shown that HIV promotes the production of renin by CD4 T-cells, which is a source of endogenous RAAS activation (207), however, sex differences in the HIV population need further studies.
A summary of potential sex differences contributing to hypertension are shown in Figure 4. However, a number of questions remain unanswered regarding the sexes in terms of interaction between ENaC activity, HIV infection, ROS and antiviral therapy and hypertension.
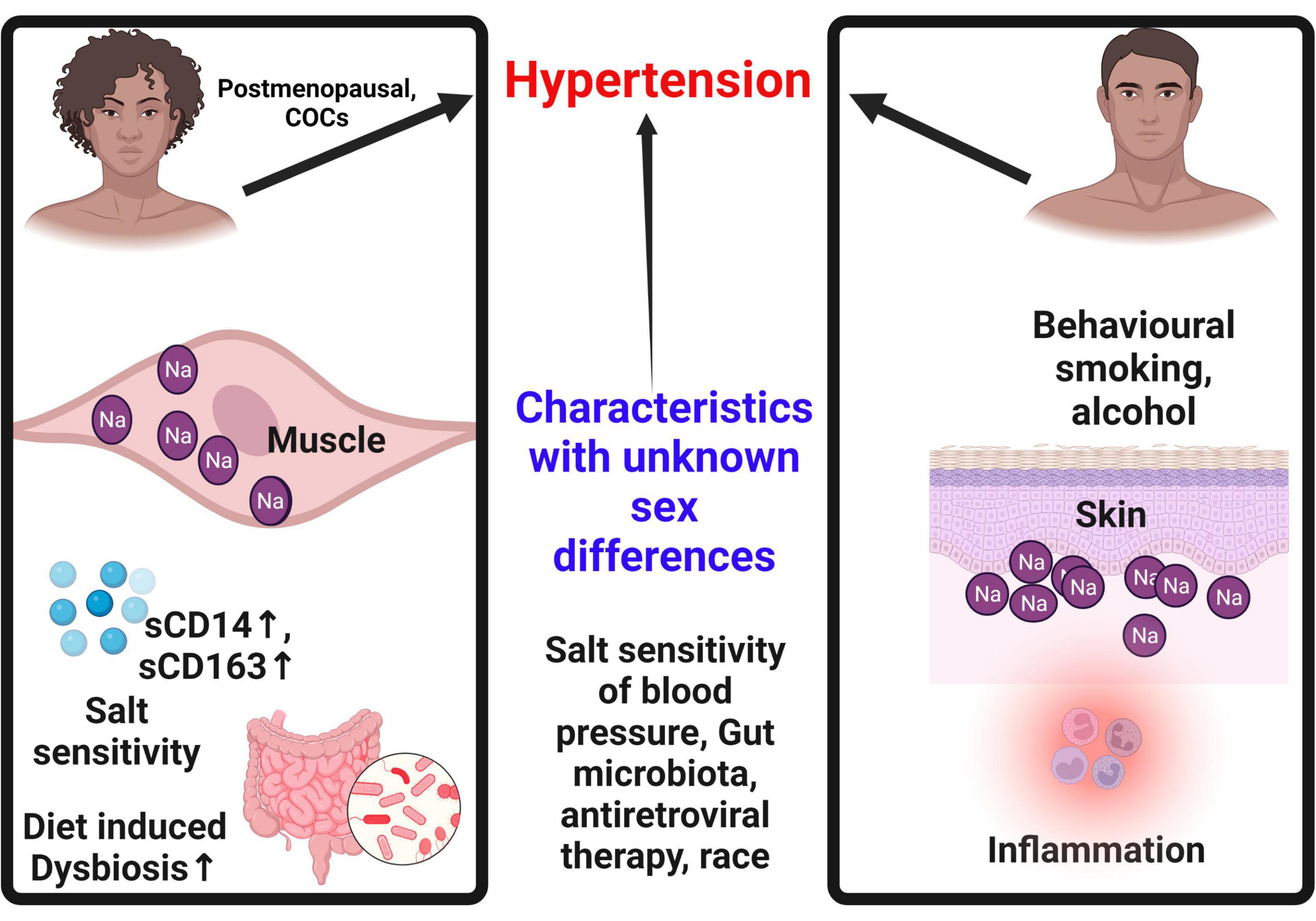
Figure 4. Sex differences contributing to hypertension. Sex specific characteristics contribute to hypertension. In females, hypertension is only common after menopause. Females tend to store more sodium in the muscle than in the skin, a concept that may be protective when compared to males. Males on the other hand tend to store more sodium in the skin eliciting an inflammatory cascade that contributes to hypertension. Females are more susceptible to salt sensitivity and diet induced dysbiosis that contribute to hypertension. Use of combined oral contraceptives is a predictor of hypertension in females. Some behavioral patterns common in men such as alcohol and smoking increase the risk of hypertension in males. COCs, combined oral contraceptives; Na, sodium; sCD14, soluble cluster of differentiation 14; sCD163, soluble cluster of differentiation 163.
The protean external and internal gender-specific influences in the pathogenesis of hypertension, should prompt the transition from putative approaches to the development and integration of gender-specific interventions for the prevention of CVD risk factors such as hypertension in PWH. There still remain a paucity of data on the contribution of gender to the pathogenesis of hypertension and role of ENaC in HIV-infected individuals.
Evidence has indicated a favorable effect on blood pressure reduction with ENaC blockers such as amiloride, which inhibits the endothelial amiloride sensitive subunits of the ENaC, as well as aldosterone blockers such as spironolactone (208, 209). However, as the hallmark of endothelial dysfunction is driven by reduction in NO release and increase generation of ROS, which can occur in the setting of HIV, potential therapies such as antioxidant therapy can be considered in the HIV population in the control of ENaC driven hypertension. Studies have indicated that PWH, including those on cART have high levels of total plasma peroxides and oxidative stress index compared to the control group (210). Preclinical studies have demonstrated that therapies which improves NO bioavailability, such as Lazaroids (inhibitors of lipid membrane peroxidation and appear to function as oxygen free radical scavengers) have been shown to reduce blood pressure in animal models (211). Additionally, vitamin D has also been shown to reduce renin-angiotensin-aldosterone system (RAAS) activity as well as regulating vascular oxidative stress and can be considered as a potential therapy for hypertension, especially that PWH appear to have lower vitamin D- levels compared to controls (212–214). However, due to the multifaceted nature of hypertension etiology in humans, results on the use antioxidants as potential treatment for hypertension remain conflicting (215). Nonetheless, a consideration for anti-hypertensive drugs that also exert antioxidant effects such as nebivolol, carvedilol, and celiprolol is an alternative option (215). More studies are required to explore the potential for antioxidant therapy to treat hypertension.
The multifactorial link between HIV and CVDs is well established. Hypertension, an important driver in the development of most CVD events, requires more robust studies. Salt-sensitive hypertension has already been demonstrated to be highly prevalent in PWH, but molecular studies such as the role of ENaC are still scarce in regions most affected by both HIV and CVDs, such as the sub-Sahara Africa. This review aimed to highlight the pathogenesis surrounding salt-sensitivity and ENaC and their contribution to the pathogenesis of hypertension in PWH, with the hope that more can be invested in preventing the onset and progression of CVDs in PWH.
• ENaC regulates blood pressure.
• Hypertension is more common in persons with HIV compared to persons without HIV.
• In HIV, the activity of ENaC is likely increased resulting in increased risk for the development of hypertension in persons with HIV.
• Some sex differences in hypertension are explained by the role of ENaC. However, the mechanisms still remain to be explored.
• Use of antiretroviral therapy likely modulates ENaC function through yet unknown mechanisms.
KM conceptualized the study and wrote the draft manuscript. SM and KM wrote different sections of the manuscript. SM created all the figures. KP and NM edited and reviewed the manuscript. AK edited and reviewed the manuscript, conceptualized the framework and finalized the manuscript as well as obtained funding for the manuscript. All authors contributed to article reviews, edited and approved the final version of this manuscript.
This work was supported by the Fogarty International Center of the National Institutes of Health grants 2D43TW009744 (KM), D43 TW009744 and D43 TW009337 (SM), R01DK114663, R01DK129311 and endowed McIntyre Professorship (KP), R03HL155041, and R01HL144941 (AK). The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Noreng S, Bharadwaj A, Posert R, Yoshioka C, Baconguis I. Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife. (2018) 7:e39340. doi: 10.7554/eLife.39340
2. Giraldez T, Afonso-Oramas D, Cruz-Muros I, Garcia-Marin V, Pagel P, González-Hernández T, et al. Cloning and functional expression of a new epithelial sodium channel delta subunit isoform differentially expressed in neurons of the human and monkey telencephalon. J Neurochem. (2007) 102:1304–15. doi: 10.1111/j.1471-4159.2007.04622.x
3. Bhalla V, Hallows KR. Mechanisms of ENaC regulation and clinical implications. J Am Soc Nephrol. (2008) 19:1845–54. doi: 10.1681/ASN.2008020225
4. Zaika O, Mamenko M, Staruschenko A, Pochynyuk O. Direct activation of ENaC by angiotensin II: Recent advances and new insights. Curr Hypertens Rep. (2013) 15:17–24. doi: 10.1007/s11906-012-0316-1
5. Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, Pochynyuk O. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem. (2012) 287:660–71. doi: 10.1074/jbc.M111.298919
6. Pavlov TS, Staruschenko A. Involvement of ENaC in the development of salt-sensitive hypertension. Am J Physiol Renal Physiol. (2017) 313:F135–40. doi: 10.1152/ajprenal.00427.2016
7. Ilatovskaya DV, Pavlov TS, Levchenko V, Staruschenko A. ROS production as a common mechanism of ENaC regulation by EGF, insulin, and IGF-1. Am J Physiol Cell Physiol. (2013) 304:C102–11. doi: 10.1152/ajpcell.00231.2012
8. Dan Dunn J, Alvarez LAJ, Zhang X, Soldati T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. (2015) 6:472–85. doi: 10.1016/j.redox.2015.09.005
9. Yang Y, Bazhin AV, Werner J, Karakhanova S. Reactive oxygen species in the immune system. Int Rev Immunol. (2013) 32:249–70. doi: 10.3109/08830185.2012.755176
10. Rosales C. Neutrophils at the crossroads of innate and adaptive immunity. J Leukoc Biol. (2020) 108:377–96. doi: 10.1002/JLB.4MIR0220-574RR
11. Loscalzo J. Oxidative stress in endothelial cell dysfunction and thrombosis. Pathophysiol Haemost Thromb. (2002) 32:359–60. doi: 10.1159/000073600
12. Poznyak AV, Grechko AV, Orekhova VA, Khotina V, Ivanova EA, Orekhov AN. NADPH oxidases and their role in atherosclerosis. Biomedicines. (2020) 8:206. doi: 10.3390/biomedicines8070206
13. Lassègue B, Griendling KK. Reactive oxygen species in hypertension*: An update. Am J Hypertens. (2004) 17:852–60. doi: 10.1016/j.amjhyper.2004.02.004
14. Marincowitz C, Genis A, Goswami N, De Boever P, Nawrot TS, Strijdom H. Vascular endothelial dysfunction in the wake of HIV and ART. FEBS J. (2019) 286:1256–70. doi: 10.1111/febs.14657
15. Hijmans JG, Stockelman K, Levy M, Madden Brewster L, Bammert TD, Greiner JJ, et al. Effects of HIV-1 gp120 and TAT-derived microvesicles on endothelial cell function. J Appl Physiol. (2019) 126:1242–9. doi: 10.1152/japplphysiol.01048.2018
16. Mutchler SM, Kirabo A, Kleyman TR. Epithelial sodium channel and salt-sensitive hypertension. Hypertension. (2021) 77:759–67. doi: 10.1161/HYPERTENSIONAHA.120.14481
17. Pitzer AL, Van Beusecum JP, Kleyman TR, Kirabo A. ENaC in salt-sensitive hypertension: Kidney and beyond. Curr Hypertens Rep. (2020) 22:69. doi: 10.1007/s11906-020-01067-9
18. Masenga SK, Kirabo A, Hamooya BM, Nzala S, Kwenda G, Heimburger DC, et al. HIV-positive demonstrate more salt sensitivity and nocturnal non-dipping blood pressure than HIV-negative individuals. Clin Hypertens. (2021) 27:2. doi: 10.1186/s40885-020-00160-0
19. Bygrave H, Golob L, Wilkinson L, Roberts T, Grimsrud A. Let’s talk chronic disease: Can differentiated service delivery address the syndemics of HIV, hypertension and diabetes? Curr Opin HIV AIDS. (2020) 15:256–60. doi: 10.1097/COH.0000000000000629
20. Gouda HN, Charlson F, Sorsdahl K, Ahmadzada S, Ferrari AJ, Erskine H, et al. Burden of non-communicable diseases in sub-Saharan Africa, 1990–2017: Results from the Global Burden of Disease Study 2017. Lancet Glob Heal. (2019) 7:e1375–87. doi: 10.1016/S2214-109X(19)30374-2
21. Todowede OO, Mianda SZ, Sartorius B. Prevalence of metabolic syndrome among HIV-positive and HIV-negative populations in sub-Saharan Africa – A systematic review and meta-analysis 11 Medical and Health Sciences 1117 Public Health and Health Services. Syst Rev. (2019) 8:1–17. doi: 10.1186/s13643-018-0927-y
22. Global Burden of Disease Report [GBDR]. Global burden of disease (GBD 2019) | Institute for health metrics and evaluation. (2019). Available online at: http://www.healthdata.org/gbd/2019 (accessed November 6, 2021).
23. United Nations Programme on HIV/AIDS [UNAIDS]. UNAIDS Data 2018. Geneva: UNAIDS (2018). p. 1–376.
24. Triant VA. Cardiovascular disease and HIV infection. Curr HIV/AIDS Rep. (2013) 10:199–206. doi: 10.1007/s11904-013-0168-6
25. So-Armah K, Freiberg MS. HIV and cardiovascular disease: Update on clinical events, special populations, and novel biomarkers. Curr HIV/AIDS Rep. (2018) 15:233–44. doi: 10.1007/s11904-018-0400-5
26. Kengne AP, Ntsekhe M. Challenges of cardiovascular disease risk evaluation in people living with HIV infection. Circulation. (2018) 137:2215–7. doi: 10.1161/CIRCULATIONAHA.118.033913
27. Kaplan-Lewis E, Aberg JA, Lee M. Atherosclerotic cardiovascular disease and anti-retroviral therapy. Curr HIV/AIDS Rep. (2016) 13:297–308. doi: 10.1007/s11904-016-0331-y
28. Nou E, Lo J, Grinspoon SK. Inflammation, immune activation, and cardiovascular disease in HIV. AIDS. (2016) 30:1495–509. doi: 10.1097/QAD.0000000000001109
29. Bittencourt MS. Estimating cardiovascular risk in hiv-infected patients. Arq Bras Cardiol. (2020) 114:76–7. doi: 10.36660/abc.20190747
30. Masenga SK, Hamooya BM, Nzala S, Kwenda G, Heimburger DC, Mutale W, et al. Patho-immune mechanisms of hypertension in HIV: A systematic and thematic review. Curr Hypertens Rep. (2019) 21:1–19. doi: 10.1007/s11906-019-0956-5
31. Noubiap JJN, Nansseu JRN, Bigna JJR, Jingi AM, Kengne AP. Prevalence and incidence of dyslipidaemia among adults in Africa: A systematic review and meta-analysis protocol. BMJ Open. (2015) 5:e007404. doi: 10.1136/bmjopen-2014-007404
32. Ahmed D, Roy D, Cassol E. Examining relationships between metabolism and persistent inflammation in HIV patients on antiretroviral therapy. Mediators Inflamm. (2018) 2018:6238978. doi: 10.1155/2018/6238978
33. Gonzales EB, Kawate T, Gouaux E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature. (2009) 460:599–604. doi: 10.1038/nature08218
34. Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature (1994) 367:463–7. doi: 10.1038/367463a0
35. Kleyman TR, Kashlan OB, Hughey RP. Epithelial Na + channel regulation by extracellular and intracellular factors. Annu Rev Physiol. (2018) 80:263–81. doi: 10.1146/annurev-physiol-021317-121143
36. Kashlan OB, Blobner BM, Zuzek Z, Tolino M, Kleyman TR. Na+ inhibits the epithelial Na+ channel by binding to a site in an extracellular acidic cleft*. J Biol Chem. (2015) 290:568–76. doi: 10.1074/jbc.M114.606152
37. Anantharam A, Tian Y, Palmer LG. Open probability of the epithelial sodium channel is regulated by intracellular sodium. J Physiol. (2006) 574(Pt 2):333. doi: 10.1113/jphysiol.2006.109173
38. Kleyman TR, Satlin LM, Hallows KR. Opening lines of communication in the distal nephron. J Clin Invest. (2013) 123:4139–41. doi: 10.1172/JCI71944
39. Sheng S, Carattino MD, Bruns JB, Hughey RP, Kleyman TR. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am J Physiol Ren Physiol. (2006) 290:1488–96. doi: 10.1152/ajprenal.00439.2005
40. Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM, et al. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the gamma-subunit. J Biol Chem. (2007) 282:6153–60. doi: 10.1074/jbc.M610636200
41. Wei Y, Lin DH, Kemp R, Yaddanapudi GSS, Nasjletti A, Falck JR, et al. Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J Gen Physiol. (2004) 124:719. doi: 10.1085/jgp.200409140
42. Sun P, Antoun J, Lin DH, Yue P, Gotlinger KH, Capdevila J, et al. Cyp2c44-epoxygenase is essential for preventing the renal sodium absorption during increasing dietary potassium (K)-intake. Hypertension. (2012) 59:339. doi: 10.1161/HYPERTENSIONAHA.111.178475
43. Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. (2006) 86:1151–78. doi: 10.1152/physrev.00050.2005
44. Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, et al. A novel role for type 1 angiotensin receptors on T lymphocytes to limit target organ damage in hypertension. Circ Res. (2012) 110:1604. doi: 10.1161/CIRCRESAHA.111.261768
45. Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PMT, Kohan DE. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol. (2015) 10:135. doi: 10.2215/CJN.05760513
46. Mishra S, Ingole S, Jain R. Salt sensitivity and its implication in clinical practice. Indian Heart J. (2018) 70:556–64. doi: 10.1016/j.ihj.2017.10.006
47. Morla L, Edwards A, Crambert G. New insights into sodium transport regulation in the distal nephron: Role of G-protein coupled receptors. World J Biol Chem. (2016) 7:44. doi: 10.4331/wjbc.v7.i1.44
48. Kleyman TR, Carattino MD, Hughey RP. ENaC at the cutting edge: Regulation of epithelial sodium channels by proteases. J Biol Chem. (2009) 284:20447. doi: 10.1074/jbc.R800083200
49. Arroyo JP, Ronzaud C, Lagnaz D, Staub O, Gamba G. Aldosterone paradox: Differential regulation of ion transport in distal nephron. Physiology (Bethesda). (2011) 26:115–23. doi: 10.1152/physiol.00049.2010
50. Soundararajan R, Pearce D, Ziera T. The role of the ENaC-regulatory complex in aldosterone-mediated sodium transport. Mol Cell Endocrinol. (2011) 350:242–7. doi: 10.1016/j.mce.2011.11.003
51. Bubien JK. Epithelial Na+ channel (ENaC), hormones, and hypertension. J Biol Chem. (2010) 285:23527–31. doi: 10.1074/jbc.R109.025049
52. Bernardo JF, Murakami S, Branch RA, Sabra R. Potassium depletion potentiates amphotericin-B-induced toxicity to renal tubules. Nephron. (1995) 70:235–41. doi: 10.1159/000188590
53. Musso CG, Belloso WH, Glassock RJ. Water, electrolytes, and acid-base alterations in human immunodeficiency virus infected patients. World J Nephrol. (2016) 5:33–42. doi: 10.5527/wjn.v5.i1.33
54. Garza Tovar OA, Pérez AAM, Pérez MEG, Robledo IU, Galarza FFG, Márquez FCL. Serum electrolytes and renal alterations in HIV-seropositive Mexican subjects. Medicine (Baltimore). (2021) 100:e26016. doi: 10.1097/MD.0000000000026016
55. Velázquez H, Perazella MA, Wright FS, Ellison DH. Renal mechanism of trimethoprim-induced hyperkalemia. Ann Intern Med. (1993) 119:296–301. doi: 10.7326/0003-4819-119-4-199308150-00008
56. Greenberg S, Reiser IW, Chou SY. Hyperkalemia with high-dose trimethoprim-sulfamethoxazole therapy. Am J Kidney Dis. (1993) 22:603–6. doi: 10.1016/S0272-6386(12)80937-1
57. Kleyman TR, Roberts C, Ling BN. A mechanism for pentamidine-induced hyperkalemia: Inhibition of distal nephron sodium transport. Ann Intern Med. (1995) 122:103–6. doi: 10.7326/0003-4819-122-2-199501150-00004
58. Pillay P, Pillay S, McHunu N. The spectrum of electrolyte abnormalities in black African people living with human immunodeficiency virus and diabetes mellitus at Edendale Hospital, Pietermaritzburg, South Africa. South Afr J HIV Med. (2020) 21:1095. doi: 10.4102/sajhivmed.v21i1.1095
59. Caramelo C, Bello E, Ruiz E, Rovira A, Gazapo RM, Alcazar JM, et al. Hyperkalemia in patients infected with the human immunodeficiency virus: Involvement of a systemic mechanism. Kidney Int. (1999) 56:198–205. doi: 10.1046/j.1523-1755.1999.00530.x
60. Stockand JD. The role of the epithelial Na + channel (enac) in high AVP but low aldosterone states. Front Physiol. (2012) 3:304. doi: 10.3389/fphys.2012.00304
61. Van Beusecum JP, Barbaro NR, McDowell Z, Aden LA, Xiao L, Pandey AK, et al. High salt activates CD11c + antigen-presenting cells via SGK (Serum Glucocorticoid Kinase) 1 to promote renal inflammation and salt-sensitive hypertension. Hypertens (Dallas, Tex 1979). (2019) 74:555–63. doi: 10.1161/HYPERTENSIONAHA.119.12761
62. Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. (2017) 21:1009–20. doi: 10.1016/j.celrep.2017.10.002
63. Kristoff J, Rinaldo CR, Mailliard RB. Role of dendritic cells in exposing latent HIV-1 for the kill. Viruses. (2020) 12:37. doi: 10.3390/v12010037
64. Koh WH, Lopez P, Ajibola O, Parvarchian R, Mohammad U, Hnatiuk R, et al. HIV-Captured DCs regulate T Cell migration and cell-cell contact dynamics to enhance viral spread. iScience. (2020) 23:101427. doi: 10.1016/j.isci.2020.101427
65. Fahme SA, Bloomfield GS, Peck R. Hypertension in HIV-Infected Adults. Hypertension. (2018) 72:44–55. doi: 10.1161/HYPERTENSIONAHA.118.10893
66. Davis K, Perez-Guzman P, Hoyer A, Brinks R, Gregg E, Althoff KN, et al. Association between HIV infection and hypertension: A global systematic review and meta-analysis of cross-sectional studies. BMC Med. (2021) 19:105. doi: 10.1186/s12916-021-01978-7
67. Giani JF, Janjulia T, Kamat N, Seth DM, Blackwell WLB, Shah KH, et al. Renal angiotensin-converting enzyme is essential for the hypertension induced by nitric oxide synthesis inhibition. J Am Soc Nephrol. (2014) 25:2752–63. doi: 10.1681/ASN.2013091030
68. Fountain JH, Lappin SL. Physiology, renin angiotensin system - StatPearls - NCBI bookshelf. Treasure Island, FL: StatPearls (2021).
69. Kelly TN, Li C, Hixson JE, Gu D, Rao DC, Huang J, et al. Resequencing study identifies rare renin-angiotensin-aldosterone system variants associated with blood pressure salt-sensitivity: The GenSalt study. Am J Hypertens. (2017) 30:495–501. doi: 10.1093/ajh/hpx004
70. Cui HL, Ditiatkovski M, Kesani R, Bobryshev YV, Liu Y, Geyer M, et al. HIV protein Nef causes dyslipidemia and formation of foam cells in mouse models of atherosclerosis. FASEB J. (2014) 28:2828–39. doi: 10.1096/fj.13-246876
71. Peck RN, Kingery JR. Immunology of hypertension in people with HIV. J Am Heart Assoc. (2020) 9:e015725. doi: 10.1161/JAHA.120.015725
72. Norwood J, Turner M, Bofill C, Rebeiro P, Shepherd B, Bebawy S, et al. Brief report: Weight gain in persons with HIV switched from efavirenz-based to integrase strand transfer inhibitor-based regimens. J Acquir Immune Defic Syndr. (2017) 76:527–31. doi: 10.1097/QAI.0000000000001525
73. Srinivasa S, Fitch KV, Wong K, Torriani M, Mayhew C, Stanley T, et al. RAAS activation is associated with visceral adiposity and insulin resistance among HIV-infected patients. J Clin Endocrinol Metab. (2015) 100:2873–82. doi: 10.1210/jc.2015-1461
74. Enslow BT, Stockand JD, Berman JM. Liddle’s syndrome mechanisms, diagnosis and management. Integr Blood Press Control. (2019) 12:13. doi: 10.2147/IBPC.S188869
75. Elijovich F, Weinberger MH, Anderson CAM, Appel LJ, Bursztyn M, Cook NR, et al. Salt sensitivity of blood pressure. Hypertension. (2016) 68:e7–46. doi: 10.1161/HYP.0000000000000047
76. Harrison DG, Vinh A, Lob H, Madhur MS. Role of the adaptive immune system in hypertension. Curr Opin Pharmacol. (2010) 10:203–7. doi: 10.1016/j.coph.2010.01.006
77. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertens (Dallas, Tex 1979). (2010) 55:500–7. doi: 10.1161/HYPERTENSIONAHA.109.145094
78. Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, et al. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am J Physiol Ren Physiol. (2008) 295:515–24. doi: 10.1152/ajprenal.00527.2007
79. Rogacev KS, Cremers B, Zawada AM, Seiler S, Binder N, Ege P, et al. CD14++CD16+ monocytes independently predict cardiovascular events: A cohort study of 951 patients referred for elective coronary angiography. J Am Coll Cardiol. (2012) 60:1512–20. doi: 10.1016/j.jacc.2012.07.019
80. Berg KE, Ljungcrantz I, Andersson L, Bryngelsson C, Hedblad B, Fredrikson GN, et al. Elevated CD14++CD16- monocytes predict cardiovascular events. Circ Cardiovasc Genet. (2012) 5:122–31. doi: 10.1161/CIRCGENETICS.111.960385
81. Belge K-U, Dayyani F, Horelt A, Siedlar M, Frankenberger M, Frankenberger B, et al. The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J Immunol. (2002) 168:3536–42. doi: 10.4049/jimmunol.168.7.3536
82. Sprangers S, Vries TJD, Everts V. Monocyte heterogeneity: Consequences for monocyte-derived immune cells. J Immunol Res. (2016) 2016:1475435. doi: 10.1155/2016/1475435
83. Liang H, Duan Z, Li D, Li D, Wang Z, Ren L, et al. Higher levels of circulating monocyte-platelet aggregates are correlated with viremia and increased sCD163 levels in HIV-1 infection. Cell Mol Immunol. (2015) 12:435–43. doi: 10.1038/cmi.2014.66
84. Chen P, Su B, Zhang T, Zhu X, Xia W, Fu Y, et al. Perturbations of monocyte subsets and their association with T helper cell differentiation in acute and chronic HIV-1-infected patients. Front Immunol. (2017) 8:272. doi: 10.3389/fimmu.2017.00272
85. Luo L, Han Y, Song X, Zhu T, Zeng Y, Li T. CD16-expressing monocytes correlate with arterial stiffness in HIV-infected ART-naïve men. HIV Clin Trials. (2018) 19:39–45. doi: 10.1080/15284336.2018.1437863
86. McKibben RA, Margolick JB, Grinspoon S, Li X, Palella FJ, Kingsley LA, et al. Elevated levels of monocyte activation markers are associated with subclinical atherosclerosis in men with and those without HIV infection. J Infect Dis. (2015) 211:1219–28. doi: 10.1093/infdis/jiu594
87. Pereyra F, Lo J, Triant VA, Wei J, Buzon MJ, Fitch KV, et al. Increased coronary atherosclerosis and immune activation in HIV-1 elite controllers. AIDS. (2012) 26:2409–12. doi: 10.1097/QAD.0b013e32835a9950
88. Bengtsson E, Hultman K, Edsfeldt A, Persson A, Nitulescu M, Nilsson J, et al. CD163+ macrophages are associated with a vulnerable plaque phenotype in human carotid plaques. Sci Rep. (2020) 10:14362. doi: 10.1038/s41598-020-71110-x
89. Manner IW, Baekken M, Kvale D, Oektedalen O, Pedersen M, Nielsen SD, et al. Markers of microbial translocation predict hypertension in HIV-infected individuals. HIV Med. (2013) 14:354–61. doi: 10.1111/hiv.12015
90. Loperena R, Van Beusecum JP, Itani HA, Engel N, Laroumanie F, Xiao L, et al. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: Roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc Res. (2018) 114:1547–63. doi: 10.1093/cvr/cvy112
91. Ruggeri Barbaro N, Van Beusecum J, Xiao L, Do Carmo L, Pitzer A, Loperena R, et al. Sodium activates human monocytes via the NADPH oxidase and isolevuglandin formation. Cardiovasc Res. (2021) 117:1358–71. doi: 10.1093/cvr/cvaa207
92. Kirabo A, Fontana V, De Faria APC, Loperena R, Galindo CL, Wu J, et al. DC isoketal-modified proteins activate T cells and promotehypertension. J Clin Invest. (2014) 124:4642. doi: 10.1172/JCI74084
93. Fantuzzi L, Purificato C, Donato K, Belardelli F, Gessani S. Human immunodeficiency virus Type 1 gp120 induces abnormal maturation and functional alterations of dendritic cells: A novel mechanism for AIDS pathogenesis. J Virol. (2004) 78:9763–72. doi: 10.1128/JVI.78.18.9763-9772.2004
94. Nasi A, Amu S, Göthlin M, Jansson M, Nagy N, Chiodi F, et al. Dendritic cell response to HIV-1 is controlled by differentiation programs in the cells and strain-specific properties of the virus. Front Immunol. (2017) 8:244. doi: 10.3389/fimmu.2017.00244
95. Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, et al. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. (2011) 57:469–76. doi: 10.1161/HYPERTENSIONAHA.110.162941
96. Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L, et al. A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI insight. (2017) 2:e92801. doi: 10.1172/jci.insight.92801
97. Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, et al. Interleukin-1 augments salt retention in angiotensin II-induced hypertension via nitric oxide-dependent regulation of the NKCC2 sodium co-transporter. Cell Metab. (2016) 23:360. doi: 10.1016/j.cmet.2015.11.013
98. Yang Y, Liu X, Liu Y, Fu H, Gao Y, Liu X, et al. The development of salt-sensitive hypertension regulated by PSGL-1 gene in mice. Cell Biosci. (2018) 8:20. doi: 10.1186/s13578-018-0218-2
99. Ling YH, Krishnan SM, Chan CT, Diep H, Ferens D, Chin-Dusting J, et al. Anakinra reduces blood pressure and renal fibrosis in one kidney/DOCA/salt-induced hypertension. Pharmacol Res. (2017) 116:77–86. doi: 10.1016/j.phrs.2016.12.015
100. Chamberlain J, Francis S, Brookes Z, Shaw G, Graham D, Alp NJ, et al. Interleukin-1 regulates multiple atherogenic mechanisms in response to fat feeding. PLoS One. (2009) 4:e5073. doi: 10.1371/journal.pone.0005073
101. Bhaskar V, Yin J, Mirza AM, Phan D, Vanegas S, Issafras H, et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis. (2011) 216:313–20. doi: 10.1016/j.atherosclerosis.2011.02.026
102. Guo H, Gao J, Taxman DJ, Ting JPY, Su L. HIV-1 infection induces interleukin-1β production via TLR8 protein-dependent and NLRP3 inflammasome mechanisms in human monocytes. J Biol Chem. (2014) 289:21716–26. doi: 10.1074/jbc.M114.566620
103. Shapiro L, Dinarello C. Hyperosmotic stress as a stimulant for proinflammatory cytokine production. | Semantic Scholar. (2022). Available online at: https://www.semanticscholar.org/paper/Hyperosmotic-stress-as-a-stimulant-for-cytokine-Shapiro-Dinarello/0c83e3ecd4912e971f542e509db4fdef0e590f39 (accessed May 11, 2022).
104. Kusche-Vihrog K, Jeggle P, Oberleithner H. The role of ENaC in vascular endothelium. Pflügers Arch Eur J Physiol. (2013) 466:851–9. doi: 10.1007/s00424-013-1356-3
105. Fu BM, Tarbell JM. Mechano-sensing and transduction by endothelial surface glycocalyx: Composition, structure, and function. Wiley Interdiscip Rev Syst Biol Med. (2013) 5:381–90. doi: 10.1002/wsbm.1211
106. Jebari-Benslaiman S, Galicia-García U, Larrea-Sebal A, Olaetxea JR, Alloza I, Vandenbroeck K, et al. Pathophysiology of atherosclerosis. Int J Mol Sci. (2022) 23:3346. doi: 10.3390/ijms23063346
107. Godo S, Shimokawa H. Endothelial functions. Arterioscler Thromb Vasc Biol. (2017) 37:e108–14. doi: 10.1161/ATVBAHA.117.309813
108. Shimokawa H. 2014 Williams Harvey lecture: Importance of coronary vasomotion abnormalities-from bench to bedside. Eur Heart J. (2014) 35:3180–93. doi: 10.1093/eurheartj/ehu427
109. Ohashi J, Sawada A, Nakajima S, Noda K, Takaki A, Shimokawa H. Mechanisms for enhanced endothelium-derived hyperpolarizing factor-mediated responses in microvessels in mice. Circ J. (2012) 76:1768–79. doi: 10.1253/circj.CJ-12-0197
110. Cyr AR, Huckaby LV, Shiva SS, Zuckerbraun BS. Nitric oxide and endothelial dysfunction. Crit Care Clin. (2020) 36:307. doi: 10.1016/j.ccc.2019.12.009
111. Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. (2018) 100:1–19. doi: 10.1016/j.vph.2017.05.005
112. Bohlen HG. Nitric oxide and the cardiovascular system. Compr Physiol. (2015) 5:803–28. doi: 10.1002/cphy.c140052
113. Mutchler SM, Straub AC. Compartmentalized nitric oxide signaling in the resistance vasculature. Nitric Oxide. (2015) 49:8. doi: 10.1016/j.niox.2015.05.003
114. Tan BL, Norhaizan ME, Liew WPP. Nutrients and oxidative stress: Friend or foe? Oxid Med Cell Longev. (2018) 2018:9719584. doi: 10.1155/2018/9719584
115. Kruth HS, Huang W, Ishii I, Zhang WY. Macrophage foam cell formation with native low density lipoprotein. J Biol Chem. (2002) 277:34573–80. doi: 10.1074/jbc.M205059200
116. Carnevale R, Bartimoccia S, Nocella C, Di Santo S, Loffredo L, Illuminati G, et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis. (2014) 237:108–16. doi: 10.1016/j.atherosclerosis.2014.08.041
117. Kattoor AJ, Goel A, Mehta JL. LOX-1: Regulation, signaling and its role in atherosclerosis. Antioxidants (Basel, Switzerland). (2019) 8:218. doi: 10.3390/antiox8070218
118. Ropraz P, Imhof BA, Matthes T, Wehrle-Haller B, Sidibé A. Simultaneous study of the recruitment of monocyte subpopulations under flow in vitro. J Vis Exp. (2018) 2018:e58509. doi: 10.3791/58509
119. Panth N, Paudel KR, Parajuli K. Reactive oxygen species: A key hallmark of cardiovascular disease. Adv Med. (2016) 2016:1–12. doi: 10.1155/2016/9152732
120. Lakier JB. Smoking and cardiovascular disease. Am J Med. (1992) 93:8S–12S. doi: 10.1016/0002-9343(92)90620-Q
121. Gulati A, Dalal J, Padmanabhan TNC, Jain P, Patil S, Vasnawala H. Lipitension: Interplay between dyslipidemia and hypertension. Indian J Endocrinol Metab. (2012) 16:240. doi: 10.4103/2230-8210.93742
122. Dikalov SI, Nazarewicz RR. Angiotensin II-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal. (2013) 19:1085–94. doi: 10.1089/ars.2012.4604
123. Reus-Chavarría E, Martínez-Vieyra I, Salinas-Nolasco C, Chávez-Piña AE, Méndez-Méndez JV, López-Villegas EO, et al. Enhanced expression of the Epithelial Sodium Channel in neutrophils from hypertensive patients. Biochim Biophys Acta Biomembr. (2019) 1861:387–402. doi: 10.1016/j.bbamem.2018.11.003
124. Hill MA, Jaisser F, Sowers JR. Role of the vascular endothelial sodium channel activation in the genesis of pathologically increased cardiovascular stiffness. Cardiovasc Res. (2022) 118:130–40. doi: 10.1093/cvr/cvaa326
125. Louboutin JP, Strayer DS. Blood-brain barrier abnormalities caused by HIV-1 gp120: Mechanistic and therapeutic implications. ScientificWorldJournal. (2012) 2012:482575. doi: 10.1100/2012/482575
126. Oberleithner H, Riethmüller C, Schillers H, MacGregor GA, De Wardener HE, Hausberg M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci U S A. (2007) 104:16281–6. doi: 10.1073/pnas.0707791104
127. Pellicelli AM, Barbaro G, Palmieri F, Girardi E, D’Ambrosio C, Rianda A, et al. Primary pulmonary hypertension in HIV patients: A systematic review. Angiology. (2001) 52:31–41. doi: 10.1177/000331970105200105
128. Tanase DM, Gosav EM, Radu S, Ouatu A, Rezus C, Ciocoiu M, et al. Arterial hypertension and interleukins: Potential therapeutic target or future diagnostic marker? Int J Hypertens. (2019) 2019:3159283. doi: 10.1155/2019/3159283
129. Kovacs L, Kress TC, Belin de Chantemèle EJ. HIV, combination antiretroviral therapy, and vascular diseases in men and women. JACC Basic to Transl Sci. (2022) 7:410–21. doi: 10.1016/j.jacbts.2021.10.017
130. Oliviero U, Bonadies G, Apuzzi V, Foggia M, Bosso G, Nappa S, et al. Human immunodeficiency virus per se exerts atherogenic effects. Atherosclerosis. (2009) 204:586–9. doi: 10.1016/j.atherosclerosis.2008.10.012
131. Weissgerber TL. Flow-mediated dilation: Can new approaches provide greater mechanistic insight into vascular dysfunction in preeclampsia and other diseases? Curr Hypertens Rep. (2014) 16:487. doi: 10.1007/s11906-014-0487-z
132. Dysangco A, Liu Z, Stein JH, Dubé MP, Gupta SK. HIV infection, antiretroviral therapy, and measures of endothelial function, inflammation, metabolism, and oxidative stress. PLoS One. (2017) 12:e0183511. doi: 10.1371/journal.pone.0183511
133. Virdis A. Endothelial dysfunction in obesity: Role of inflammation. High Blood Press Cardiovasc Prev. (2016) 23:83–5. doi: 10.1007/s40292-016-0133-8
134. Hirigo AT, Gutema S, Eifa A, Ketema W. Experience of dolutegravir-based antiretroviral treatment and risks of diabetes mellitus. SAGE Open Med Case Rep. (2022) 10:2050313X2210794. doi: 10.1177/2050313X221079444
135. Sax PE, Erlandson KM, Lake JE, McComsey GA, Orkin C, Esser S, et al. Weight gain following initiation of antiretroviral therapy: Risk factors in randomized comparative clinical trials. Clin Infect Dis. (2020) 71:1379–89. doi: 10.1093/cid/ciz999
136. Waters DD, Hsue PY. Lipid abnormalities in persons living with HIV infection. Can J Cardiol. (2019) 35:249–59. doi: 10.1016/j.cjca.2018.11.005
137. Habeshaw JA, Dalgleish AG, Bountiff L, Newell AL, Wilks D, Walker LC, et al. AIDS pathogenesis: HIV envelope and its interaction with cell proteins. Immunol Today. (1990) 11:418–25. doi: 10.1016/0167-5699(90)90162-3
138. Chen B. Molecular mechanism of HIV-1 entry. Trends Microbiol. (2019) 27:878–91. doi: 10.1016/j.tim.2019.06.002
139. Alves NMP, de Moura RR, Bernardo LC, Agrelli A, de Oliveira ASLE, da Silva NP, et al. In silico analysis of molecular interactions between HIV-1 glycoprotein gp120 and TNF receptors. Infect Genet Evol. (2021) 92:104837. doi: 10.1016/j.meegid.2021.104837
140. Green LA, Yi R, Petrusca D, Wang T, Elghouche A, Gupta SK, et al. HIV envelope protein gp120-induced apoptosis in lung microvascular endothelial cells by concerted upregulation of EMAP II and its receptor, CXCR3. Am J Physiol Lung Cell Mol Physiol. (2014) 306:L372–82. doi: 10.1152/ajplung.00193.2013
141. Gao R, Fang Q, Zhang X, Xu Q, Ye H, Guo W, et al. R5 HIV-1 gp120 Activates p38 MAPK to induce rat cardiomyocyte injury by the CCR5 coreceptor. Pathobiology. (2019) 86:274–84. doi: 10.1159/000502238
142. Pereira EA, daSilva LLP. HIV-1 Nef: Taking control of protein trafficking. Traffic. (2016) 17:976–96. doi: 10.1111/tra.12412
143. Pawlak EN, Dikeakos JD. HIV-1 Nef: A master manipulator of the membrane trafficking machinery mediating immune evasion. Biochim Biophys Acta. (2015) 1850:733–41. doi: 10.1016/j.bbagen.2015.01.003
144. Chelvanambi S, Bogatcheva NV, Bednorz M, Agarwal S, Maier B, Alves NJ, et al. HIV-Nef protein persists in the lungs of aviremic patients with HIV and induces endothelial cell death. Am J Respir Cell Mol Biol. (2019) 60:357–66. doi: 10.1165/rcmb.2018-0089OC
145. Wang T, Green LA, Gupta SK, Kim C, Wang L, Almodovar S, et al. Transfer of intracellular HIV Nef to endothelium causes endothelial dysfunction. PLoS One. (2014) 9:e91063. doi: 10.1371/journal.pone.0091063
146. Vilhardt F, Plastre O, Sawada M, Suzuki K, Wiznerowicz M, Kiyokawa E, et al. The HIV-1 Nef protein and phagocyte NADPH oxidase activation. J Biol Chem. (2002) 277:42136–43. doi: 10.1074/jbc.M200862200
147. Agarwal S, Sharma H, Chen L, Dhillon NK. NADPH oxidase-mediated endothelial injury in HIV- and opioid-induced pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. (2020) 318:L1097–108. doi: 10.1152/ajplung.00480.2019
148. Gray SP, Shah AM, Smyrnias I. NADPH oxidase 4 and its role in the cardiovascular system. Vasc Biol. (2019) 1:H59–66. doi: 10.1530/VB-19-0014
149. Pavlov TS, Palygin O, Isaeva E, Levchenko V, Khedr S, Blass G, et al. NOX4-dependent regulation of ENaC in hypertension and diabetic kidney disease. FASEB J. (2020) 34:13396–408. doi: 10.1096/fj.202000966RR
150. Barman SA, Chen F, Su Y, Dimitropoulou C, Wang Y, Catravas JD, et al. NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler Thromb Vasc Biol. (2014) 34:1704–15. doi: 10.1161/ATVBAHA.114.303848
151. Jadhav VS, Krause KH, Singh SK. HIV-1 Tat C modulates NOX2 and NOX4 expressions through miR-17 in a human microglial cell line. J Neurochem. (2014) 131:803–15. doi: 10.1111/jnc.12933
152. Ivanov AV, Valuev-Elliston VT, Ivanova ON, Kochetkov SN, Starodubova ES, Bartosch B, et al. Oxidative stress during HIV infection: Mechanisms and consequences. Oxid Med Cell Longev. (2016) 2016:8910396. doi: 10.1155/2016/8910396
153. Cowley AW, Yang C, Zheleznova NN, Staruschenko A, Kurth T, Rein L, et al. Evidence of the importance of Nox4 in production of hypertension in dahl salt-sensitive rats. Hypertens (Dallas, Tex 1979). (2016) 67:440–50. doi: 10.1161/HYPERTENSIONAHA.115.06280
154. Hamooya BM, Musonda P, Mutale W, Masenga SK, Halwiindi H, Mutengo KH, et al. Prevalence of low high-density lipoprotein among young adults receiving antiretroviral therapy in Zambia: An opportunity to consider non-communicable diseases in resource-limited settings. PLoS One. (2021) 16:e0247004. doi: 10.1371/journal.pone.0247004
155. Masenga SK, Elijovich F, Koethe JR, Hamooya BM, Heimburger DC, Munsaka SM, et al. Hypertension and metabolic syndrome in persons with HIV. Curr Hypertens Rep. (2020) 22:78. doi: 10.1007/s11906-020-01089-3
156. Bakal DR, Coelho LE, Luz PM, Clark JL, De Boni RB, Cardoso SW, et al. Obesity following ART initiation is common and influenced by both traditional and HIV-/ART-specific risk factors. J Antimicrob Chemother. (2018) 73:2177–85. doi: 10.1093/jac/dky145
157. Esber AL, Chang D, Iroezindu M, Bahemana E, Kibuuka H, Owuoth J, et al. Weight gain during the dolutegravir transition in the African Cohort Study. J Int AIDS Soc. (2022) 25:e25899. doi: 10.1002/jia2.25899
158. Tanaka M. Improving obesity and blood pressure. Hypertens Res. (2019) 43:79–89. doi: 10.1038/s41440-019-0348-x
159. Jia G, Aroor AR, Hill MA, Sowers JR. Role of renin-angiotensin-aldosterone system activation in promoting cardiovascular fibrosis and stiffness. Hypertension. (2018) 72:537–48. doi: 10.1161/HYPERTENSIONAHA.118.11065
160. Patel S, Rauf A, Khan H, Abu-Izneid T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed Pharmacother. (2017) 94:317–25. doi: 10.1016/j.biopha.2017.07.091
161. Levy BI. How to explain the differences between renin angiotensin system modulators. Am J Hypertens. (2005) 18:134S–41S. doi: 10.1016/j.amjhyper.2005.05.005
162. Mendoza Torres E, Oyarzún A, Mondaca Ruff D, Azocar A, Chiong M, Castro PF, et al. ACE2 and vasoactive peptides: Novel players in cardiovascular/renal remodeling and hypertension. Ther Adv Cardiovasc Dis. (2015) 9:217–37. doi: 10.1177/1753944715597623
163. Briones AM, Cat AND, Callera GE, Yogi A, Burger D, He Y, et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: Implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertens (Dallas, Tex 1979). (2012) 59:1069–78. doi: 10.1161/HYPERTENSIONAHA.111.190223
164. Huby AC, Antonova G, Groenendyk J, Gomez-Sanchez CE, Bollag WB, Filosa JA, et al. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation. (2015) 132:2134–45. doi: 10.1161/CIRCULATIONAHA.115.018226
165. Ferreira NS, Tostes RC, Paradis P, Schiffrin EL. Aldosterone, inflammation, immune system, and hypertension. Am J Hypertens. (2021) 34:15–27. doi: 10.1093/ajh/hpaa137
166. Novelli S, Lécuroux C, Goujard C, Reynes J, Villemant A, Blum L, et al. Persistence of monocyte activation under treatment in people followed since acute HIV-1 infection relative to participants at high or low risk of HIV infection. eBioMedicine. (2020) 62:103129. doi: 10.1016/j.ebiom.2020.103129
167. Sánchez A, Martínez P, Muñoz M, Benedito S, García-Sacristán A, Hernández M, et al. Endothelin-1 contributes to endothelial dysfunction and enhanced vasoconstriction through augmented superoxide production in penile arteries from insulin-resistant obese rats: Role of ETA and ETB receptors. Br J Pharmacol. (2014) 171:5682–95. doi: 10.1111/bph.12870
168. Virdis A, Masi S, Colucci R, Chiriacò M, Uliana M, Puxeddu I, et al. Microvascular endothelial dysfunction in patients with obesity. Curr Hypertens Rep. (2019) 21:32. doi: 10.1007/s11906-019-0930-2
169. Kleniewska P, Goraca A. Influence of endothelin 1 receptor blockers and a nitric oxide synthase inhibitor on reactive oxygen species formation in rat lungs. Physiol Res. (2016) 65:789–98. doi: 10.33549/physiolres.933263
170. Xia N, Horke S, Habermeier A, Closs EI, Reifenberg G, Gericke A, et al. Uncoupling of endothelial nitric oxide synthase in perivascular adipose tissue of diet-induced obese mice. Arterioscler Thromb Vasc Biol. (2016) 36:78–85. doi: 10.1161/ATVBAHA.115.306263
171. Knapp M, Tu X, Wu R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol Sin. (2019) 40:1. doi: 10.1038/s41401-018-0042-6
172. Hurtubise J, McLellan K, Durr K, Onasanya O, Nwabuko D, Ndisang JF. The different facets of dyslipidemia and hypertension in atherosclerosis. Curr Atheroscler Rep. (2016) 18:82. doi: 10.1007/s11883-016-0632-z
173. Helkin A, Stein JJ, Lin S, Siddiqui S, Maier KG, Gahtan V. Dyslipidemia Part 1 – review of lipid metabolism and vascular cell physiology. Vasc Endovascular Surg. (2016) 50:107–18. doi: 10.1177/1538574416628654
174. Hsue PY, Scherzer R, Hunt PW, Schnell A, Bolger AF, Kalapus SC, et al. Carotid intima-media thickness progression in HIV-infected adults occurs preferentially at the carotid bifurcation and is predicted by inflammation. J Am Heart Assoc. (2012) 1:jah3–000422. doi: 10.1161/JAHA.111.000422
175. Beires MT, Silva-Pinto A, Santos AC, Madureira AJ, Pereira J, Carvalho D, et al. Visceral adipose tissue and carotid intima-media thickness in HIV-infected patients undergoing cART: A prospective cohort study. BMC Infect Dis. (2018) 18:32. doi: 10.1186/s12879-017-2884-9
176. Bellinati PQ, Alfieri DF, Flauzino T, Junior PFG, Rossi DJ, Breganó JW, et al. Association of lower adiponectin plasma levels, increased age and smoking with subclinical atherosclerosis in patients with HIV-1 infection. Curr HIV Res. (2020) 18:292–306. doi: 10.2174/1570162X18666200609114741
177. Parrinello CM, Landay AL, Hodis HN, Gange SJ, Norris PJ, Young M, et al. Association of subclinical atherosclerosis with lipid levels amongst antiretroviral-treated and untreated HIV-infected women in the Women’s Interagency HIV study. Atherosclerosis. (2012) 225:408–11. doi: 10.1016/j.atherosclerosis.2012.09.035
178. Ruderman SA, Crane HM, Nance RM, Whitney BM, Harding BN, Mayer KH, et al. Brief report: Weight gain following ART initiation in ART-Naïve people living with HIV in the current treatment era. J Acquir Immune Defic Syndr. (2021) 86:339–43. doi: 10.1097/QAI.0000000000002556
179. United Nations Programme on HIV/AIDS [UNAIDS]. UNAIDS data 2021. Geneva: UNAIDS (2021). p. 4–38.
180. Nduka CU, Stranges S, Sarki AM, Kimani PK, Uthman OA. Evidence of increased blood pressure and hypertension risk among people living with HIV on antiretroviral therapy: A systematic review with meta-analysis. J Hum Hypertens. (2015) 30:355–62. doi: 10.1038/jhh.2015.97
181. American College of Cardiology [ACC]. Women and hypertension: Beyond the 2017 guideline for prevention, detection, evaluation, and management of high blood pressure in adults - American college of cardiology. (2022). Available online at: https://www.acc.org/latest-in-cardiology/articles/2018/07/27/09/02/women-and-hypertension (accessed April 23, 2022).
183. Choi HM, Kim HC, Kang DR. Sex differences in hypertension prevalence and control: Analysis of the 2010-2014 Korea National Health and Nutrition Examination Survey. PLoS One. (2017) 12:e0178334. doi: 10.1371/journal.pone.0178334
184. Scully EP. Sex differences in HIV infection. Curr HIV/AIDS Rep. (2018) 15:136–46. doi: 10.1007/s11904-018-0383-2
185. Zhang X, Ge Y, Bukhari A-A-S, Zhu Q, Shen Y, Li M, et al. Estrogen negatively regulates the renal epithelial sodium channel (ENaC) by promoting Derlin-1 expression and AMPK activation. Exp Mol Med. (2019) 51:1–12. doi: 10.1038/s12276-019-0253-z
186. Wang P, Deger MS, Kang H, Ikizler TA, Titze J, Gore JC. Sex differences in sodium deposition in human muscle and skin. Magn Reson Imaging. (2017) 36:93. doi: 10.1016/j.mri.2016.10.023
187. Siedner MJ, Zanni M, Tracy RP, Kwon DS, Tsai AC, Kakuhire B, et al. The journal of infectious diseases increased systemic inflammation and gut permeability among women with treated HIV infection in rural Uganda. J Infect Dis. (2018) 218:922–6. doi: 10.1093/infdis/jiy244
188. Faulkner JL, Belin de Chantemèle EJ. Female sex, a major risk factor for salt-sensitive hypertension. Curr Hypertens Rep. (2020) 22:99. doi: 10.1007/s11906-020-01113-6
189. Chen J. Sodium sensitivity of blood pressure in Chinese populations. Curr Hypertens Rep. (2010) 12:127–34. doi: 10.1007/s11906-009-0088-4
190. Shukri MZ, Tan JW, Manosroi W, Pojoga LH, Rivera A, Williams JS, et al. Biological sex modulates the adrenal and blood pressure responses to angiotensin II. Hypertens (Dallas, Tex 1979). (2018) 71:1083–90. doi: 10.1161/HYPERTENSIONAHA.117.11087
191. Kawarazaki W, Fujita T. Aberrant Rac1-mineralocorticoid receptor pathways in salt-sensitive hypertension. Clin Exp Pharmacol Physiol. (2013) 40:929–36. doi: 10.1111/1440-1681.12177
192. Ishii M, Atarashi K, Ikeda T, Hirata Y, Igari T, Uehara Y, et al. Role of the aldosterone system in the salt-sensitivity of patients with benign essential hypertension. Jpn Heart J. (1983) 24:79–90. doi: 10.1536/ihj.24.79
193. Bennett E, Peters SAE, Woodward M. Sex differences in macronutrient intake and adherence to dietary recommendations: Findings from the UK Biobank. BMJ Open. (2018) 8:e020017. doi: 10.1136/bmjopen-2017-020017
194. Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ. (2018) 361:36–44. doi: 10.1136/bmj.k2179
195. Adnan S, Nelson JW, Ajami NJ, Venna VR, Petrosino JF, Bryan RM, et al. Alterations in the gut microbiota can elicit hypertension in rats. Physiol Genomics. (2017) 49:96–104. doi: 10.1152/physiolgenomics.00081.2016
196. Toral M, Robles-Vera I, de la Visitación N, Romero M, Sánchez M, Gómez-Guzmán M, et al. Role of the immune system in vascular function and blood pressure control induced by faecal microbiota transplantation in rats. Acta Physiol. (2019) 227:e13285. doi: 10.1111/apha.13285
197. Smiljanec K, Lennon SL. Sodium, hypertension, and the gut: Does the gut microbiota go salty? Am J Physiol - Hear Circ Physiol. (2019) 317:H1173–82. doi: 10.1152/ajpheart.00312.2019
198. Ferguson JF, Aden LA, Barbaro NR, Van Beusecum JP, Xiao L, Simons AJ, et al. High dietary salt-induced dendritic cell activation underlies microbial dysbiosis-associated hypertension. JCI Insight. (2019) 5:e126241. doi: 10.1172/jci.insight.126241
199. Torres-Pinzon DL, Ralph DL, Veiras LC, McDonough AA. Sex-specific adaptations to high-salt diet preserve electrolyte homeostasis with distinct sodium transporter profiles. Am J Physiol Cell Physiol. (2021) 321:C897–909. doi: 10.1152/ajpcell.00282.2021
200. Sparks MA, Dilmen E, Ralph DL, Rianto F, Hoang TA, Hollis A, et al. Vascular control of kidney epithelial transporters. Am J Physiol Ren Physiol. (2021) 320:F1080. doi: 10.1152/ajprenal.00084.2021
201. Nelson JW, McDonough AA, Xiang Z, Ralph DL, Robertson JA, Giani JF, et al. Local and downstream actions of proximal tubule angiotensin II signaling on Na + transporters in the mouse nephron. Am J Physiol Renal Physiol. (2021) 321:F69–81. doi: 10.1152/ajprenal.00014.2021
202. Hu R, McDonough AA, Layton AT. Sex differences in solute transport along the nephrons: Effects of Na + transport inhibition. Am J Physiol Renal Physiol. (2020) 319:F487–505. doi: 10.1152/ajprenal.00240.2020
203. Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, et al. Interleukin-17A regulates renal sodium transporters and renal injury in angiotensin II-induced hypertension. Hypertens (Dallas, Tex 1979). (2016) 68:167–74. doi: 10.1161/HYPERTENSIONAHA.116.07493
204. Hyndman KA, Mironova EV, Giani JF, Dugas C, Collins J, McDonough AA, et al. Collecting duct nitric oxide synthase 1ß activation maintains sodium homeostasis during high sodium intake through suppression of aldosterone and renal angiotensin II pathways. J Am Heart Assoc. (2017) 6:e006896. doi: 10.1161/JAHA.117.006896
205. Pei L, Solis G, Nguyen MTX, Kamat N, Magenheimer L, Zhuo M, et al. Paracellular epithelial sodium transport maximizes energy efficiency in the kidney. J Clin Invest. (2016) 126:2509–18. doi: 10.1172/JCI83942
206. Zimmerman MA, Baban B, Tipton AJ, O’Connor PM, Sullivan JC. Sex and gender differences in renal physiology: Chronic ANG II infusion induces sex-specific increases in renal T cells in Sprague-Dawley rats. Am J Physiol Ren Physiol. (2015) 308:F706. doi: 10.1152/ajprenal.00446.2014
207. Chandel N, Ayasolla K, Lan X, Rai P, Mikulak J, Husain M, et al. Renin modulates HIV replication in T cells. J Leukoc Biol. (2014) 96:601–9. doi: 10.1189/JLB.2A0414-192R
208. Oberleithner H, Ludwig T, Riethmüller C, Hillebrand U, Albermann L, Schäfer C, et al. Human endothelium: Target for aldosterone. Hypertens (Dallas, Tex 1979). (2004) 43:952–6. doi: 10.1161/01.HYP.0000123572.45556.a5
209. Pratt JH, Eckert GJ, Newman S, Ambrosius WT. Blood pressure responses to small doses of amiloride and spironolactone in normotensive subjects. Hypertension. (2001) 38:1124–9. doi: 10.1161/hy1101.095010
210. Ojong EW, Bassey IE, Nsonwu-Anyanwu AC, Ndeh FJ, Djeufouata JDM, Njongang VN, et al. Assessment of plasma antioxidant capacity and oxidative stress in HIV/AIDS patients in Calabar, Nigeria. Sci African. (2021) 14:e01017. doi: 10.1016/j.sciaf.2021.e01017
211. Vaziri ND, Ni Z, Oveisi F, Trnavsky-Hobbs DL. Effect of antioxidant therapy on blood pressure and NO synthase expression in hypertensive rats. Hypertens (Dallas, Tex 1979). (2000) 36:957–64. doi: 10.1161/01.HYP.36.6.957
212. Argacha JF, Egrise D, Pochet S, Fontaine D, Lefort A, Libert F, et al. Vitamin D deficiency-induced hypertension is associated with vascular oxidative stress and altered heart gene expression. J Cardiovasc Pharmacol. (2011) 58:65–71. doi: 10.1097/FJC.0b013e31821c832f
213. Giménez VMM, Sanz RL, Marón FJM, Ferder L, Manucha W. Vitamin D-RAAS connection: An integrative standpoint into cardiovascular and neuroinflammatory disorders. Curr Protein Pept Sci. (2020) 21:948–54. doi: 10.2174/1389203721666200606220719
214. Wang Y, Huang X, Wu Y, Li A, Tian Y, Ren M, et al. Increased risk of vitamin D deficiency among HIV-infected individuals: A systematic review and meta-analysis. Front Nutr. (2021) 8:722032. doi: 10.3389/fnut.2021.722032
Keywords: hypertension, inflammation, HIV, cardiovasclar disease, ENaC (epithelial Na+ channel)
Citation: Mutengo KH, Masenga SK, Mwesigwa N, Patel KP and Kirabo A (2022) Hypertension and human immunodeficiency virus: A paradigm for epithelial sodium channels? Front. Cardiovasc. Med. 9:968184. doi: 10.3389/fcvm.2022.968184
Received: 13 June 2022; Accepted: 10 August 2022;
Published: 25 August 2022.
Edited by:
Ruan Kruger, North-West University, South AfricaReviewed by:
Ana Paula O. Leite, Tulane University, United StatesCopyright © 2022 Mutengo, Masenga, Mwesigwa, Patel and Kirabo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annet Kirabo, QW5uZXQuS2lyYWJvQHZ1bWMub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.