 Huili Zhang
Huili Zhang Qi Zhan1
Qi Zhan1 Biao Huang
Biao Huang Yigang Wang
Yigang Wang

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 19 August 2022
Sec. Cardiovascular Therapeutics
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.952755
Gene therapy has revolutionized the field of medicine, offering new hope for those with common and rare diseases. For nearly three decades, adeno-associated virus (AAV) has shown significant therapeutic benefits in multiple clinical trials, mainly due to its unique replication defects and non-pathogenicity in humans. In the field of cardiovascular disease (CVD), compared with non-viral vectors, lentiviruses, poxviruses, and adenovirus vectors, AAV possesses several advantages, including high security, low immunogenicity, sustainable and stable exogenous gene expression etc., which makes AAV one of the most promising candidates for the treatment of many genetic disorders and hereditary diseases. In this review, we evaluate the current information on the immune responses, transport pathways, and mechanisms of action associated with AAV-based CVD gene therapies and further explore potential optimization strategies to improve the efficiency of AAV transduction for the improved safety and efficiency of CVD treatment. In conclusion, AAV-mediated gene therapy has great potential for development in the cardiovascular system.
According to data provided by The Global Burden of Diseases, Injuries, and Risk Factors Study in 2019, cardiovascular disease (CVD) is one of the leading causes of deaths, accounting for 18.5 million deaths, i.e., about one third of all mortalities worldwide, making it a major public health issue, with China having the largest number of cardiovascular disease deaths (1). This suggests that, in the future, providing both CVD prevention and treatment interventions in the context of sustainable infrastructure development may play important roles in global CVD control.
Since the successful engraftment of β-galactosidase-expressing endothelial cells into denuded iliofemoral arteries in 1989, gene therapy has quickly emerged as a candidate for the treatment of CVD (2). In the 1970s, Friedmann and Roblin formally proposed the concept of gene therapy (3): a strategy to treat diseases by means of non-viral or viral vectors that introduces therapeutic exogenous genes into target cells and tissues to supplement or correct defective genes through in vitro and in vivo techniques (4, 5). However, due to barriers to the effective delivery of gene fragments or gene-editing tools in the body, the development of gene therapy was slow until scientists discovered that some viruses seemed to be able to overcome these barriers. Virus surface proteins effectively recognize cell receptors and carry genetic material into the host cell, where it is continuously produced by the host's cell factories, making viruses ideal delivery vehicles for transgenes (6–8). Therefore, the application of viral vectors has ushered in key advancements in the conceptual exploration and clinical application of gene therapy.
Adeno-associated virus (AAV) vectors have attracted widespread attention because of their unique advantages in effectively, persistently, and safely expressing foreign genes in host cells with low immunogenicity (9). Over the past 30 years, recombinant (r) AAV products have been successfully adopted for the clinical treatment of a variety of diseases, including cancers (10), ophthalmic diseases (11), blood diseases, neurological diseases, and immune system diseases among others (12). To date, three drugs based on rAAV vectors have been approved for marketing. The first rAAV-based drug, Glybera (Alipogene Tiparvovec, AT), which was approved by the European Union, is an AAV1 vector drug that delivers DNA encoding functional lipoprotein lipase (LPL) to skeletal muscle for patients with LPL deficiency due to gene mutations (13), but it was taken off the market soon after its release because the price was unexpectedly high. In 2017, Luxturna was the first AAV-based ophthalmic gene therapy drug approved by the FDA and is used for the symptomatic treatment of retinal dystrophy associated with double-copy RPE65 mutations (14). In 2019, Zolgensma was the second AAV-based drug approved by the FDA and utilizes AAV9 expressing a functional SMN1 transgene to treat spinal muscular atrophy type I (15).
Together, conditions of the cardiovascular system can broadly be classified into two main categories: heart diseases and vascular diseases. It is encouraging that the development of modern molecular biology has prompted more thorough research on CVD, focusing on the overexpression, knockdown, and knockout of key pathogenic genes. Such genetic manipulation requires advances in technology or transmitters, hence the emergence of viral vectors that can act as delivery vehicles may lead to new insights. From the vast sea of viruses, preceding research has screened out the three most commonly used virus tools for CVD, namely, HIV-1, adenovirus, and parvovirus (e.g., AAV), which have the following common characteristics: (1) high infection efficiency; (2) low cytotoxicity; and (3) strong expression effects (8). Among them, rAAV vectors have become the best choice for gene therapy of CVD due to their unique advantages.
In recent years, with the development of high-throughput genetic engineering, cell therapy, and immunotherapy, research on CVD has become increasingly in-depth. Currently, many clinical studies have shown the impressive benefits of AAV-based therapies, 18 studies of rAAVs-based CVD gene therapies have been documented (from https://clinicaltrials.gov/; keywords, adeno associated virus/interventional studies/CVD). We provide a detailed overview of these preclinical and clinical studies, systematically introduce the optimized strategies being used to develop AAV as gene delivery vectors with improved specific targeting capabilities, and discuss the prospects of and potential research into using this vector system in the rapidly evolving field of CVD treatment.
AAV is an icosahedral parvovirus that cannot replicate without the aid of host cell machinery or helper viruses. It was first described as a contaminating component of purified adenovirus preparations in the mid-1960s and was soon discovered in human tissues (16). AAV is one of the most actively developed vectors for cell and gene therapy applications and has contributed to encouraging results in clinical research into a variety of human diseases. AAV is a 4.7 Kbp single-stranded DNA virus whose genome consists of Rep and Cap genes flanked by inverted terminal repeats (ITRs), each consisting of 145 bases (4, 10, 17). Two promoters, p5 and P19, on the left side of the AAV genome transcribe mRNAs of different lengths and generate four different capsid proteins containing overlapping sequences (Rep78, Rep68, Rep52, and Rep40) by selective shearing (4, 18). The capsid genes encode three capsid proteins (VP1, VP2, VP3) and assembly activator protein (AAP) through the alternative splicing of different start codons (19), and the proportion of the three structural proteins in a mature virion is approximately 1:1:10. The viral capsid serves as the vehicle for viral gene delivery (Figure 1) (12). In general, replication and efficient infection only occur with the involvement of helper adenoviruses or herpes simplex virus. Interestingly, AAV can establish latency in the absence of helper viruses. In the latent state, Rep mediates the activation of AAVS1-specific DNA synthesis at human chromosome 19q13.4qter and the formation of integrated complexes in vitro and in vivo (20, 21).
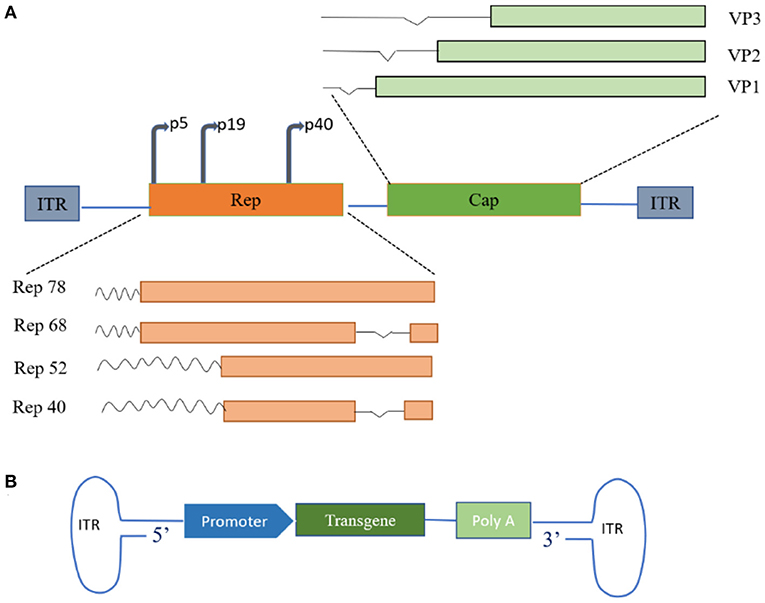
Figure 1. Schematic of adeno-associated virus (AAVs) genome structure. (A) Wild-type AAV genome structure. The wild-type AAV comprises a small, unenveloped, single-stranded DNA genome of approximately 4.7 Kb, encoding replication (Rep) and capsid (Cap) genes. Rep and Cap protein products are assembled into 60-polymeric viral capsids under suitable conditions. (B) Recombinant AAV genome structure. Therapeutic transgenes replace parts of the genome encoding proteins of the viral capsid. ITR, inverted terminal repeat; VP, capsid proteins.
The rAAV vectors currently in use worldwide are constructed by replacing a section of the viral genome between the two ITRs with a genetic payload of interest, and rAAVs have shown some unique advantages in clinical trials compared to wild-type AAVs (14, 19, 22). The development of rAAV vectors has received overwhelming enthusiasm from the research community. It is generally believed that the key to gene therapy is the unification of specificity and controllability. AAV has a simple structure, and 13 human and primate AAV serotypes and hundreds of AAV mutants have been described (23–25). In addition, different serotypes of AAV can infect diverse cell types, depending on the receptors on the cell surface targeted by AAV, and these serotypes and their associated receptors also determine the transport routes taken by the different AAV serotypes within the cell (Table 1). With the development of high-throughput biotechnology, an increasing variety of specific AAV serotypes are being developed, and this has largely driven the application of AAV as agents to fight human disease.
The viral capsid protein significantly affects the affinity of the AAV vector for a given tissue. In vitro studies support evidence that the transduction of AAV to cells requires AAV capsid protein interactions with primary receptors and co-receptors on the target cell surface. AAV enters the target cell, initiates a cascade of reactions, and finally, integrates DNA into the target cell chromosome (without helper virus) or otherwise persistently expresses viral genes via additional methods (with helper virus present) (4, 16, 23, 26, 27). In conclusion, during natural infection, AAV relies on its capsid proteins attaching to glycans expressed on the surface of target cells, such as heparan sulfate (AAV2, 3, 6, 13), sialic acid (AAV1,4,5,6), laminin receptor (AAV8), and galactose (AAV9) (16, 22). Subsequently, the virus undergoes internalization and intracellular transport through endocytosis, then escapes from the endosome under the control of the capsid protein while completing nuclear import, releasing the viral genome. Once the DNA is released from the capsid, the single-stranded genome is immediately synthesized and converted into double-stranded DNA (Figure 2) (4, 16, 22, 26).
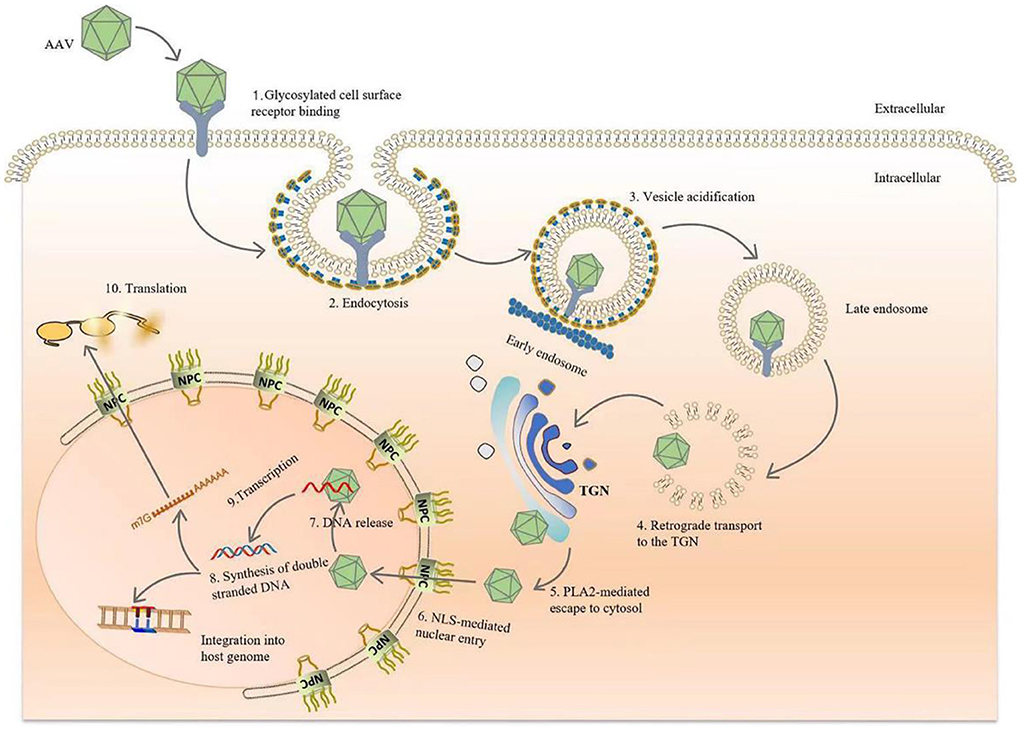
Figure 2. Diagram of AAV transduction mechanism. AAV is bound by glycosylated receptors on the surface of host cells, initiating endocytosis, after which it is rapidly transported to the reverse Golgi apparatus (TGN). After arriving at the TGN, the conformations of VP1 and VP2 change due to the acidification of endocytic vesicles in the lower intracellular pH environment, and the phospholipase A2 domain of the capsid species is exposed. After endosomal escape, AAV crosses the Golgi membrane and enters the nucleus via a nuclear pour complex (NPC) guided by the nuclear localization sequence (NLS). Once in the nucleus, DNA is released from the capsid, and host cell DNA polymerase is used to rapidly initiate second-strand synthesis. Finally, viral genomes persistently express the transgenes. Vector genomes also integrate into the host genome to establish latency, although the probability of this event occurring is extremely low.
When AAV successfully infects a cell, it determines the final location of its genes based on the presence or absence of helper viruses. In the absence of helper viruses, AAV is nearly incapable of replication, gene expression is suppressed, and the AAV genome integrates into a 4 kb region of chromosome 19q13.4, establishing a latent infection (20, 21). With a helper virus, AAVs can undergo nucleic acid replication, viral gene expression, virion production, and ultimately, virulent infection (14, 32). Interestingly, rAAV rarely integrates DNA into the genome of the target cell, and thus largely avoids the likelihood of inadvertent genome insertion (15).
The transduction efficiency of AAV is critical for AAV-based gene therapy, and several approaches used to improve this efficiency have emerged. Treatment of AAV with the metal ions cobalt and zinc increased the transduction efficiency of the AAV vector 10-fold in a dose-dependent manner (33). Most worthy of mention are the modification and engineering of the AAV capsid and the remodeling strategy of the promoter, which will be highlighted later.
The gene delivery approaches of AAV in CVD can be mainly summarized as follows. The most common is systemic intravenous delivery, which is simple but relatively unspecific. The second is intramyocardial delivery, which has the advantages of strong specificity and no endothelial barrier but causes greater damage (34). Anterograde/retrograde coronary injection and pericardial injection have also been selected as routes of AAV delivery for CVD (34). Intravenous and coronary injections are commonly used as delivery methods in clinical studies because maximizing patient safety is the primary clinical concern (34, 35).
Currently, AAV is one of the most promising vector platforms for in vivo gene transfer due to its unique advantages. However, immune recognition of AAVs is a potential challenge to its therapeutic efficacy. Early preclinical and clinical trials have demonstrated the high prevalence of neutralizing antibodies (NAbs) in the global population due to the natural exposure of humans and non-human primates to wild-type AAV (23, 36–38). These pre-existing human host immune responses potentially inhibit the efficiency of rAAV gene therapy. To date, various strategies have been developed to address the challenges raised by NAbs, all with encouraging success. Chemical modification of capsids alter the surface of AAV to facilitate their evasion of immunological detection (39). The administration of empty capsids, which act as bait to neutralize NAbs, leads to AAV vectors carrying exogenous payloads infecting target tissues efficiently and accurately (40, 41). IgG-cleaving enzyme modification by IdeS or IdeZ also offers a potential way to rescue gene transfer of the AAV vector by removing pre-existing anti-AAV antibodies and prevent AAV neutralization in vivo (42, 43). In non-human primate experiments, the transient removal of pre-existing AAV vector NAbs in vivo using plasmapheresis allowed the sustained expression of high levels of AAV-vector-delivered transgenes (44, 45). Notably, Orlowski et al. used apheresis and AAV9 particles coupled to agarose beads to selectively deplete pre-existing carrier-bound antibodies in vivo. Strategically, this depletion of anti-AAV antibodies was specific, and the immunoadsorption technique greatly increased the AAV transduction rate in the heart and liver of animals (46). Taken together, these strategies circumvent the deleterious effects of anti-AAV antibodies to a certain extent and can significantly improve and enhance the gene therapy efficacy of rAAVs.
The rAAV-mediated gene management of patients with refractory or relapsing CVD has been the subject of extensive research. Although efficacy has been demonstrated in preclinical studies and in phase 1 and 2 trials, there is no clear evidence of efficacy in large randomized placebo-controlled trials. What is worrying, however, is that, in a recent clinical study, 9 AAV-cFVIII-treated hemophilia A dogs continued to express coagulation factors for 10 years of follow-up. Moreover, the therapeutic gene segment carried by AAV was partially integrated into the host cell chromosome, especially in the vicinity of genes related to cell growth, which has the potential to induce cancer, although the gene integration rate was extremely low (47). In addition, a European study found that AAV DNA was detectable in 21% of patients, and the main viral subtypes were AAV2 and AAV2/13 (28). This study shatters the myth that AAV viral vectors are absolutely safe. A 46-nt liver-specific enhancer-promoter element in the wild-type AAV2 genome has been reported to be associated with the dysregulation of human hepatocellular carcinoma (HCC) driver genes (48). Schäffer et al. integrated AAV2/13 into the genomes of Thai and Mongolian HCC patients and found that they did not induce tumorigenesis (49). Overall, the very few potential gene-integration risks of AAV do not invalidate the advantages of AAV (50, 51). This is because AAV remains, mainly in its episomal form, in non-dividing mature cells and is still considered a safe vector choice compared to retroviruses that integrate into genomes.
Different serotypes of AAV have diverse affinity for tissues or target organs. The difference between the 13 native serotypes and hundreds of wild-type variants lies in the sequence variation of the Cap gene, which encodes individual capsid proteins, resulting in the variable infection efficiency, tissue affinity, and expression times of the AAV serotypes in different cells and tissues (14, 26, 40). Among these serotypes, AAV1, 6, 8, and 9 are considered the most favorable candidates for specific cardiac transduction (52–55). AAV9 has the best cardiac transduction specificity and can be expressed stably and with high efficiency. AAV1, 5, 8, and 9 have good transduction specificity for blood vessels. In recent years, new methods to screen and obtain more targeted rAAV vectors have been implemented, such as point mutation of capsid proteins to obtain AAV variants and the introduction of new peptides into capsid proteins. A reversible RNA switch hammerhead ribozyme was reported to efficiently regulate transgene expression in vivo without involving immunogenic non-self proteins, broadening the application dimensions of AAV gene therapy (56). Anc80L65, a synthetic AAV vector, can effectively achieve cardiac gene transfer and more stable expression after myocardial administration compared to AAV9, providing a new therapeutic strategy for CVD patients (57). Endothelial cells play a central role in the treatment of CVD. In a recent publication by Bozoglu et al., a significantly improved endothelial transduction rate was achieved with an rAAV coated with endothelial-affine peptides and polyamidoamine (PAMAMs) (58). Another novel strategy is to build a diverse in vivo targeting library by constructing AAV variants with Cap gene codon mutations, resulting in emergent synthetic variants with enhanced properties that can circumvent A20 mAb neutralizing effects (59). These improvements complement the deficiencies in AAV vectors and highlight the potential for the rational selection of AAV serotypes and capsid optimization in therapeutic strategies for CVD.
In addition to identifying the most efficient myocardial-tissue-specific serotypes, another major factor to be considered is promoter selection. Promoters can be classified as constitutive (broad-spectrum type), tissue-restricted, and inducible, according to their expression patterns (60, 61). rAAV-based CVD gene therapy uses more constitutive promoters and specific gene expression regulatory elements to enable the expression of exogenous genes in specific tissue sites, which is more suitable for the therapeutic indication (10, 60, 62, 63). Many researchers have demonstrated these tissue-restricted promoters' utility and ability to enhance the specific expression of transgenes in the heart and vascular system; these include myocardium-specific promoters showing cardiac-selective expression and vascular-smooth-muscle-cell-specific and endothelial-cell-specific promoters.
The cardiovascular selective promoters currently developed include the cardiac troponin T promoter (cTnT) (64–67), atrial natriuretic factor (ANF) promoter (68), cardiac troponin T promoter (TNT4) (68), cardiac myocyte-specific cardiac troponin T (MYC) promoter (69), cardiac sodium-calcium exchanger (NCX1), and cardiac myosin light chain 2v (MLC-2v) promoter (70, 71), as well as the endothelial-specific Tie-2 promoter (72–74). Indeed, the rational selection of specific promoters is an alternative strategy to facilitate the development of in vivo gene delivery using rAAV-based therapies.
HF, the leading cause of death from cardiovascular diseases, is a severe progressive disease that damages the heart and its function and irreversibly worsens without any therapeutic intervention (75, 76). When the heart's ejection capacity decreases, the body first activates a variety of compensatory mechanisms to maintain the heart's function at relatively normal levels (77), and these compensatory mechanisms play key roles in the development of HF. The pathophysiological changes are quite complex and can be summarized as three aspects: abnormal hemodynamics, the activation of neurohumoral regulation mechanisms, and ventricular remodeling (54, 77–80). Therefore, therapeutics based on the above three aspects are key to the treatment of HF.
Understanding the pathological mechanisms and natural history of HF is the basis for achieving desired treatment outcomes. According to a large number of randomized controlled trials (Table 2), interventions that improve left ventricular systolic function are the main treatment modalities. In recent years, a better understanding of the pathogenesis of HF has led to a flurry of clinical trials. A prevalent clinical strategy for rAAV gene therapy in patients with severe HF is left ventricular function improvement and remodeling by driving the sarcoplasmic reticulum calcium ATPase (SERCA2a) pump to restoring calcium cycling and reduce ventricular rhythmic abnormalities in HF patients (103–105). SERCA2a strictly regulates myocardial contractility and mainly controls the transport of cytosolic calcium ions to the sarcoplasmic reticulum of cardiomyocytes (106, 107). In large animal models, AAV2/1-mediated hSERCA2a gene delivery via percutaneous circulation maximized SERCA2a expression and duration of exposure, significantly improved left ventricular function in a sheep model of HF, and showed no adverse outcomes for myocardial fibrosis or myocardial necrosis (108). While in CUPID phase 1/2a clinical trial, the safety of AAV1/SERCA2a for advanced HF was confirmed to be good, and the body function of most patients was improved; although, due to pre-existing NAbs in the body, two patients failed to improve (109). This also implies that it is urgent that we develop rAAV gene therapy that circumvents NAbs. With the exception of levosimendan (a Ca-sensitizer), treatment of HF patients focuses on reducing afterload, modification of cardiac remodeling, and other mechanisms.

Table 2. Preclinical application of AAV in cardiovascular disease.
In rodent models, activation of the calcineurin-NFAT signaling pathway controls the hypertrophic growth of the myocardium, ultimately leading to heart failure. Remes et al. used AAV9 to express decoy oligodeoxynucleotides (dODN) to neutralize NFATc1-c4 in cardiomyocytes and reduce NFAT transcriptional activity, thereby preserving cardiac function during stress overload in mouse models of severe HF (82). Tafazzin (TAZ) is a mitochondrial protein associated with the inner mitochondrial membrane and is required for normal cardiolipin biosynthesis. Mutations in the TAZ gene cause Barth syndrome, but AAV9-hTZA gene therapy successfully reversed novel dysfunction in a cardio-specific Taz-knockout mouse model (81). A recent study demonstrated that AAV-JP2NT entry into the myocardium attenuated HF development and stress-induced transcriptional remodeling (110).
Cardiomyopathy is a heterogeneous disease that results in systolic and diastolic dysfunction of the heart due to pathological changes to the myocardium (111). Most cardiomyopathies are nonhereditary, e.g., alcoholic cardiomyopathy, chemotherapy-related cardiomyopathy, infection-induced cardiomyopathy, and secondary cardiomyopathy linked to autoimmune disease. However, in most epidemiological studies, the genetic-linked diversity of cardiomyopathy types, such as hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), dilated cardiomyopathy (DCM) and diabetic cardiomyopathy (DbCM), is still a complex issue (55, 102). Existing evidence implies that most myocardial diseases are caused by related pathogenic gene mutations. These genes are mainly involved in encoding cell structure and related functional proteins, and most are genes encoding sarcomeric proteins that have significant effects on cardiac function. In addition, several genes encoding cytoskeleton components and related connexins were also identified (111–113). Available data suggest that gene variants encoding sarcomere proteins may individually or collectively affect cardiac function, even if they do not cause myocardial disease.
Repairing and improving the systolic and diastolic functions of the heart is a long-term effective strategy against cardiomyopathy, and multiple gene therapies have been used to improve cardiac capacity, including cardiac regeneration, improving skeletal muscle function, and reducing cardiac hypertrophy (97, 99–101, 111). The current findings show that rAAV9 reversed the effects of HCM caused by the D166V mutation by delivering S15D-RLC into the hearts of humanized transgenic HCM-D166V mice, significantly improving intact cardiac function. Interestingly, rAAV9-S15D-RLC therapy enhanced systolic and diastolic function, but did not alter functioning WT-RLC (55). Mutations in the LaminA gene cause DCM, and delivery of Dominantly Negative acting Sun1 miniprotein (DNSUN1) using AAV9 significantly improved the progression of DCM and delayed HF, resulting in at least a five-fold extension in lifespan in mice that develop LaminA-induced DCM. This is because DNSUN1 protects cardiomyocytes from contraction-induced stress by disrupting SUN1 to disrupt the SUN-KASH LINK complex (96). The retro-orbital delivery of AAV9-cTnT-Klf5, an important transcription factor Krüppel-like factor 5 (KLF5), during cardiovascular remodeling directly binds the NADPH oxidase 4 (NOX4) promoter, and induction of NOX4 expression leads to oxidative stress and promotes the accumulation of myocardial ceramides, leading to DbCM (65). This suggests that the pharmacological or genetic inhibition of KLF5 may alleviate oxidative stress and improve myocardial function in diabetic mice. Combinations of gene therapy and antioxidants are promising new ideas for DbCM treatment. Martinez et al. investigated the effects of the systemic delivery of muscle A-kinase anchor protein β (mAKAPβ) in mouse models of myocardial infarction. The systemic administration of AAV9sc.shmAKAP (a novel self-complementary serotype 9 AAV expressing short-hairpin RNA of mAKAPβ under the control of a cardiomyocyte-specific promoter) inhibited mAKAPβ expression in cardiomyocytes and promoted the long-term stable recovery of the left ventricular ejection fraction, providing conceptual support for mAKAPβ being a therapeutic target for ischemic cardiomyopathy (114).
Many patients with vascular disease still respond poorly to traditional drug treatments, and current medical therapies are not suitable for all CVD patients. Over the past 20 years, some promising CVD therapy developments have been seen, ranging from plasmid therapy to viral-mediated gene therapy. In particular, studies targeting major complications in CVD, including metabolic and genetic-related diseases such as hyperglycemia, hypercholesterolemia, and atherosclerosis, have yielded encouraging results (115). In recent sophisticated analyses conducted in randomized controlled trials, overexpression of the acquired functional variant proprotein convertase subtilisin/kexin type 9 (PCSK9) induced atherosclerosis in mice. In addition, targeting the low-density lipoprotein receptor (LDLR) with an integrated AAV-CRISPR vector and liver-directed destruction of LDLR was demonstrated to be a viable and reliable alternative way to study atherosclerosis (115, 116). AAV-directed LDLR gene therapy reduced plasma cholesterol levels in some patients in two clinical trials (from https://clinicaltrials.gov/; NCT02651675 and NCT04148001). Therefore, some patients with dyslipidemia or even atherosclerosis may benefit from AAV-mediated LDLR therapy and improvements in their apolipoprotein levels. Conversely, lowering serum low-density lipoprotein cholesterol in hyperlipidemia animal models was an effective strategy for reducing atherosclerotic CVD. The role of SIRT6 was also demonstrated by a preclinical study that involved creating a carotid plaque mouse model. In short, SIRT6 inhibits HIF-1α degradation to stimulate angiogenesis-related signaling pathways, revealing the cardiovascular risk associated with SIRT6 expression and suggesting that SIRT6 knockout is a feasible route to inhibit the development of atherosclerosis (117).
In brief, rAAV gene therapies provide the new opportunities for CVD patients, targeted therapy for refractory diseases is likely to have a far-reaching impact, which also implies that the optimization of viral capsid protein sequences and promoters will be a promising research direction in future preclinical trials. These strategies may better reap the potential benefits of rAAV gene therapy and more adequately address the distress and troubles of CVD patients.
HZ designed, conceived, and wrote the manuscript. QZ reviewed the literature and supervised the manuscript. BH provided suggestions for manuscript revision. YW participated in the revision and polishing of the manuscript. XW participated in the revision and funding. All authors contributed to the article and approved the submitted version.
This study was supported by Hangzhou Science and Technology Bureau (20201203B44), Public Welfare Technology Project of Zhejiang Province (LGF21H160033), and the Grant for 521 talent project of ZSTU.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Roth GA, Mensah GA, Fuster V. The global burden of cardiovascular diseases and risks: a compass for global action. Journal of the American College of Cardiology. (2020) 76:2980–1. doi: 10.1016/j.jacc.2020.11.021
2. Nabel Elizabeth G, Plautz G, Boyce Frederick M, Stanley James C, Nabel Gary J. Recombinant Gene Expression in vivo Within Endothelial Cells of the Arterial Wall. Science. (1989) 244:1342–4. doi: 10.1126/science.2499928
3. Friedmann T, Roblin R. Gene therapy for human genetic disease? Science. (1972) 175:949–55. doi: 10.1126/science.175.4025.949
4. Li C, Samulski JR. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet. (2020) 21:255–72. doi: 10.1038/s41576-019-0205-4
5. Ferrari G, Thrasher AJ, Aiuti A. Gene therapy using haematopoietic stem and progenitor cells. Nature Reviews Genetics. (2021) 22:216–34. doi: 10.1038/s41576-020-00298-5
6. Weklak D, Pembaur D, Koukou G, Jönsson F, Hagedorn C, Kreppel F. Genetic and chemical capsid modifications of adenovirus vectors to modulate vector–host interactions. Viruses. (2021) 13.doi: 10.3390/v13071300
7. Nectow AR, Nestler JE. Viral tools for neuroscience. Nat Rev Neurosci. (2020) 21:669–81. doi: 10.1038/s41583-020-00382-z
8. Robbins PD, Tahara H, Ghivizzani CS. Viral vectors for gene therapy. Trends Biotechnol. (1998) 16:35–40. doi: 10.1016/S0167-7799(97)01137-2
9. Venditti CP. Safety questions for AAV gene therapy. Nat Biotechnol. (2021) 39:24–26. doi: 10.1038/s41587-020-00756-9
10. Santiago-Ortiz JL, Schaffer VD. Adeno-associated virus (AAV) vectors in cancer gene therapy. J Controlled Release. (2016) 240:287–301. doi: 10.1016/j.jconrel.2016.01.001
11. Gordon K, Del Medico A, Sander I, Kumar A, Hamad B. Gene therapies in ophthalmic disease. Nat Rev Drug Discov. (2019) 18:415–6. doi: 10.1038/d41573-018-00016-1
12. Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, Goodspeed K, Gray SJ, Kay CN, et al. Tremblay: current clinical applications of in vivo gene therapy with AAVs. Mol Therapy. (2021) 29:464–88. doi: 10.1016/j.ymthe.2020.12.007
13. Miller N. Glybera and the future of gene therapy in the European Union. Nat Rev Drug Discov. (2012) 11:419. doi: 10.1038/nrd3572-c1
14. Bucher K, Rodríguez-Bocanegra E, Dauletbekov D, Fischer DM. Immune responses to retinal gene therapy using adeno-associated viral vectors – Implications for treatment success and safety. Prog Retin. (2021) 83:100915. doi: 10.1016/j.preteyeres.2020.100915
15. Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. (2019) 18:358–78. doi: 10.1038/s41573-019-0012-9
16. Hacker UT, Bentler M, Kaniowska D, Morgan M, Büning H. Towards clinical implementation of adeno-associated virus (aav) vectors for cancer gene therapy: current status and future perspectives. Cancers. (2020) 12:1889. doi: 10.3390/cancers12071889
17. El Andari D. Grimm: production, processing, and characterization of synthetic aav gene therapy vectors. Biotechnol J. (2021) 16:2000025. doi: 10.1002/biot.202000025
18. Weger S, Hammer E, Heilbronn R. Differential contribution of adeno-associated virus type 2 Rep protein expression and nucleic acid elements to inhibition of adenoviral replication in cis and in trans. J Virol. (2014) 88:14126–37. doi: 10.1128/JVI.02350-14
19. Maurer AC, Pacouret S, Cepeda Diaz AK, Blake J, Andres-Mateos E, Vandenberghe HL. The assembly-activating protein promotes stability and interactions between aav's viral proteins to nucleate capsid assembly. Cell Reports. (2018) 23:1817–30. doi: 10.1016/j.celrep.2018.04.026
20. Hamilton H, Gomos J, Berns KI, Falck-Pedersen EE. Adeno-associated virus site-specific integration and AAVS1 disruption. J Virol. (2004) 78:7874–82. doi: 10.1128/JVI.78.15.7874-7882.2004
21. Xiang Q, Huang L, Guo S, Chen F, Zha X, Chen B, et al. Liu: A method mediated AAVS1 recombination with Rep mRNA and homologous arms. Acta Biochimica et Biophysica Sinica. (2012) 44:1015–22. doi: 10.1093/abbs/gms076
22. Dhungel BP, Bailey CG, Rasko J. Journey to the center of the cell: tracing the path of AAV transduction. Trends Mol Med. (2021) 27:172–84. doi: 10.1016/j.molmed.2020.09.010
23. Verdera HC, Kuranda K, Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol Ther. (2020) 28:723–46. doi: 10.1016/j.ymthe.2019.12.010
24. Ronzitti G, Gross D-A F. Mingozzi: human immune responses to adeno-associated virus (AAV) vectors. Front Immunol. (2020) 11:670. doi: 10.3389/fimmu.2020.00670
25. Wu Z, Asokan A, Samulski JR. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Molecular Therapy. (2006) 14:316–27. doi: 10.1016/j.ymthe.2006.05.009
26. Zengel J, Carette EJ. Chapter Two - Structural and cellular biology of adeno-associated virus attachment and entry. In: Ed Kielian MTC. Mettenleiter, Roossinck MJ. Advances in Virus Research. Academic Press. (2020). doi: 10.1016/bs.aivir.2020.01.002
27. Pillay S, Meyer NL, Puschnik AS, Davulcu O, Diep J, Ishikawa Y, et al. Carette: An essential receptor for adeno-associated virus infection. Nature. (2016) 530:108–12. doi: 10.1038/nature16465
28. T. La Bella Imbeaud S, Peneau C, Mami I, Datta S, Bayard Q, et al. Adeno-associated virus in the liver: natural history and consequences in tumour development. Gut. (2020) 69:737. doi: 10.1136/gutjnl-2019-318281
29. Mietzsch M, Jose A, Chipman P, Bhattacharya N, Daneshparvar N, McKenna R, et al. Agbandje-mckenna: completion of the aav structural atlas: serotype capsid structures reveals clade-specific features. Viruses. (2021) 13. doi: 10.3390/v13010101
30. Zincarelli C, Soltys S, Rengo G, Rabinowitz EJ. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. (2008) 16:1073–80. doi: 10.1038/mt.2008.76
31. Liu Y, Fortmann SD, Shen J, Wielechowski E, Tretiakova A, Yoo S, et al. Campochiaro: AAV8-antiVEGFfab Ocular Gene Transfer for Neovascular Age-Related Macular Degeneration. Mol Ther. (2018) 26:542–9. doi: 10.1016/j.ymthe.2017.12.002
32. Hüser D, Gogol-Döring A, Chen W, Heilbronn R. Adeno-associated virus type 2 wild-type and vector-mediated genomic integration profiles of human diploid fibroblasts analyzed by third-generation PacBio DNA sequencing. J Virol. (2014) 88:11253–63. doi: 10.1128/JVI.01356-14
33. Rambhai HK, Ashby FJ, 3rd, Qing K, Srivastava A. Role of Essential Metal Ions in AAV Vector-Mediated Transduction. Mol Ther. (2020) 18:159–66. doi: 10.1016/j.omtm.2020.05.019
34. Ishikawa K, Tilemann L, Fish K, Hajjar JR. Gene delivery methods in cardiac gene therapy. J Gene Med. (2011) 13:566–72. doi: 10.1002/jgm.1609
35. Katz MG, Swain JD, Tomasulo CE, Sumaroka M, Fargnoli A, Bridges RC. Current strategies for myocardial gene delivery. J Mol Cell Cardiol. (2011) 50:766–76. doi: 10.1016/j.yjmcc.2010.09.003
36. Colella P, Ronzitti G, Mingozzi F. Emerging issues in aav-mediated in vivo gene therapy. Mol Ther. (2017) 8:87–104. doi: 10.1016/j.omtm.2017.11.007
37. Mingozzi F, High AK. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. (2013) 122:23–36. doi: 10.1182/blood-2013-01-306647
38. Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Pogoda JM, et al. Zsebo: Prevalence of AAV1 neutralizing antibodies and consequences for a clinical trial of gene transfer for advanced heart failure. Gene Ther. (2016) 23:313–9. doi: 10.1038/gt.2015.109
39. György B, Fitzpatrick Z, Crommentuijn MHW, Mu D, Maguire AC. Naturally enveloped AAV vectors for shielding neutralizing antibodies and robust gene delivery in vivo. Biomaterials. (2014) 35:7598–609. doi: 10.1016/j.biomaterials.2014.05.032
40. Pei X, Earley LF, He Y, Chen X, Hall NE, Samulski RJ, et al. Efficient capsid antigen presentation from adeno-associated virus empty virions in vivo. Front Immunol. (2018) 9:844. doi: 10.3389/fimmu.2018.00844
41. Mingozzi F, Anguela XM, Pavani G, Chen Y, Davidson RJ, Hui DJ, et al. High: Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci Translat Med. (2013) 5:194ra92–ra92. doi: 10.1126/scitranslmed.3005795
42. Elmore ZC, Oh DK, Simon KE, Fanous MM A. Asokan: Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. JCI insight. (2020) 5:e139881. doi: 10.1172/jci.insight.139881
43. Leborgne C, Barbon E, Alexander JM, Hanby H, Delignat S, Cohen DM, et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat Med. (2020) 26:1096–101. doi: 10.1038/s41591-020-0911-7
44. Chicoine LG, Montgomery CL, Bremer WG, Shontz KM, Griffin DA, Heller KN, et al. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol Ther. (2014) 22:338–47. doi: 10.1038/mt.2013.244
45. Bertin B, Veron P, Leborgne C, Deschamps J-Y, Moullec S, Fromes Y, et al. Capsid-specific removal of circulating antibodies to adeno-associated virus vectors. Sci Rep. (2020) 10:864. doi: 10.1038/s41598-020-57893-z
46. Orlowski A, Katz MG, Gubara SM, Fargnoli AS, Fish KM, Weber T. Successful transduction with aav vectors after selective depletion of anti-aav antibodies by immunoadsorption. Mol Ther. (2020) 16:192–203. doi: 10.1016/j.omtm.2020.01.004
47. Nguyen GN, Everett JK, Kafle S, Roche AM, Raymond HE, Leiby J, et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat Biotechnol. (2021) 39:47–55. doi: 10.1038/s41587-020-0741-7
48. Logan GJ, Dane AP, Hallwirth CV, Smyth CM, Wilkie EE, Amaya AK, et al. Identification of liver-specific enhancer–promoter activity in the 3′ untranslated region of the wild-type AAV2 genome. Nat Genet. (2017) 49:1267–73. doi: 10.1038/ng.3893
49. Schäffer AA, Dominguez DA, Chapman LM, Gertz EM, Budhu A, Forgues M, et al. Integration of adeno-associated virus (AAV) into the genomes of most Thai and Mongolian liver cancer patients does not induce oncogenesis. BMC Genomics. (2021) 22:814. doi: 10.1186/s12864-021-08098-9
50. Spector LP, Tiffany M, Ferraro NM, Abell NS, Montgomery SB, Kay AM. Evaluating the genomic parameters governing raav-mediated homologous recombination. Mol Ther. (2021) 29:1028–1046. doi: 10.1016/j.ymthe.2020.11.025
51. McCarty DM, Young SM, Samulski JR. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annual Rev Genet. (2004) 38:819–45. doi: 10.1146/annurev.genet.37.110801.143717
52. Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. (2005) 23:321–8. doi: 10.1038/nbt1073
53. Su H, Huang Y, Takagawa J, Barcena A, Arakawa-Hoyt J, Ye J, et al. Kan: AAV Serotype-1 mediates early onset of gene expression in mouse hearts and results in better therapeutic effect. Gene Therapy. (2006) 13:1495–502. doi: 10.1038/sj.gt.3302787
54. Prakoso D, Tate M, Blasio MJD, Ritchie HR. Adeno-associated viral (AAV) vector-mediated therapeutics for diabetic cardiomyopathy - current and future perspectives. Clini Sci (London, England). (2021) 135:1369–87. doi: 10.1042/CS20210052
55. Yadav S, Yuan C-C, Kazmierczak K, Liang J, Huang W, Takeuchi LM, et al. Szczesna-Cordary: Therapeutic potential of AAV9-S15D-RLC gene delivery in humanized MYL2 mouse model of HCM. J Mol Med. (Berlin, Germany). (2019) 97:1033–47. doi: 10.1007/s00109-019-01791-z
56. Zhong G, Wang H, He W, Li Y, Mou H, Tickner ZJ, et al. A reversible RNA on-switch that controls gene expression of AAV-delivered therapeutics in vivo. Nat Biotechnol. (2020) 38:169–75. doi: 10.1038/s41587-019-0357-y
57. Katz MG, Hadas Y, Bailey RA, Fazal S, Vincek A, Madjarova SJ, et al. Eliyahu: Efficient cardiac gene transfer and early-onset expression of a synthetic adeno-associated viral vector, Anc80L65, after intramyocardial administration. J Thoracic Cardiovas Surg. (2021). S0022-5223(21)00915-6. doi: 10.1016/j.jtcvs.2021.05.050
58. Bozoglu T, Lee S, Ziegler T, Jurisch V, Maas S, Baehr A, et al. Endothelial retargeting of AAV9 in vivo. Adv Sci. (Weinheim, Baden-Wurttemberg, Germany). (2022) 9:e2103867–e2103867. doi: 10.1002/advs.202103867
59. Ogden PJ, Kelsic ED, Sinai S, Church MG. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science (New York, NY.). (2019) 366:1139–1143. doi: 10.1126/science.aaw2900
60. Pacak CA, J B. Byrne: AAV vectors for cardiac gene transfer: experimental tools and clinical opportunities. Mol Ther. (2011) 19:1582–90. doi: 10.1038/mt.2011.124
61. Barth AS, Kizana E, Smith RR, Terrovitis J, Dong P, Leppo MK, et al. Lentiviral vectors bearing the cardiac promoter of the Na+-Ca2+ exchanger report cardiogenic differentiation in stem cells. Mol Ther. (2008) 16:957–964. doi: 10.1038/mt.2008.30
62. Powell SK, Samulski RJ, McCown JT. AAV capsid-promoter interactions determine cns cell-selective gene expression in vivo. Mol Ther. (2020) 28:1373–80. doi: 10.1016/j.ymthe.2020.03.007
63. Breton C, Furmanak T, Avitto AN, Smith MK, Latshaw C, Yan H, et al. Increasing the specificity of AAV-based gene editing through self-targeting and short-promoter strategies. Mol Ther. (2021) 29:1047–56. doi: 10.1016/j.ymthe.2020.12.028
64. Bezzerides VJ, Caballero A, Wang S, Ai Y, Hylind RJ, Lu F, et al. Gene therapy for catecholaminergic polymorphic ventricular tachycardia by inhibition of Ca(2+)/calmodulin-dependent kinase. Circulation. (2019) 140:405–19. doi: 10.1161/CIRCULATIONAHA.118.038514
65. Kyriazis ID, Hoffman M, Gaignebet L, Lucchese AM, Markopoulou E, Palioura D, et al. KLF5 Is Induced by FOXO1 and causes oxidative stress and diabetic cardiomyopathy. Circulat Res. (2021) 128:335–57. doi: 10.1161/CIRCRESAHA.120.316738
66. Konkalmatt PR, Beyers RJ, O'Connor DM, Xu Y, Seaman ME, A B. French: Cardiac-selective expression of extracellular superoxide dismutase after systemic injection of adeno-associated virus 9 protects the heart against post-myocardial infarction left ventricular remodeling. Circulation. Cardiovascular imaging. (2013) 6:478–86. doi: 10.1161/CIRCIMAGING.112.000320
67. Prasad KMR, Xu Y, Yang Z, Acton ST, French AB. Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther. (2011) 18:43–52. doi: 10.1038/gt.2010.105
68. Ni L, Scott L, Jr Campbell HM, Pan X, Alsina KM, et al. Atrial-specific gene delivery using an adeno-associated viral vector. Circulat Res. (2019) 124:256–62. doi: 10.1161/CIRCRESAHA.118.313811
69. Li J, Tan Y, Passariello CL, Martinez EC, Kritzer MD, Li X, et al. Signalosome-regulated serum response factor phosphorylation determining myocyte growth in width versus length as a therapeutic target for heart failure. Circulation. (2020) 142:2138–54. doi: 10.1161/CIRCULATIONAHA.119.044805
70. Su H, Joho S, Huang Y, Barcena A, Arakawa-Hoyt J, Grossman W, et al. Adeno-associated viral vector delivers cardiac-specific and hypoxia-inducible VEGF expression in ischemic mouse hearts. Proc Natl Acad Sci U.S.A. (2004) 101:16280–5. doi: 10.1073/pnas.0407449101
71. Franz WM, Breves D, Klingel K, Brem G, Hofschneider PH R. Kandolf: Heart-specific targeting of firefly luciferase by the myosin light chain-2 promoter and developmental regulation in transgenic mice. Circ Res. (1993) 73:629–38. doi: 10.1161/01.RES.73.4.629
72. Karnani P K. Kairemo: Targeting endothelial growth with monoclonal antibodies against Tie-1 kinase in mouse models. Clin Cancer Res. (2003) 9:3821s−6s.
73. Jia W, Kong L, Kidoya H, Naito H, Muramatsu F, Hayashi Y, et al. Takakura: Indispensable role of Galectin-3 in promoting quiescence of hematopoietic stem cells. Nature Communicat. (2021) 12:2118–2118. doi: 10.1038/s41467-021-22346-2
74. Korhonen J, Lahtinen I, Halmekytö M, Alhonen L, Jänne J, Dumont D, et al. Endothelial-specific gene expression directed by the tie gene promoter in vivo. Blood. (1995) 86:1828–1835. doi: 10.1182/blood.V86.5.1828.bloodjournal8651828
75. van de Bovenkamp Wijkstra N, Oosterveer FPT, Vonk Noordegraaf A, Bogaard HJ, van Rossum AC, et al. The value of passive leg raise during right heart catheterization in diagnosing heart failure with preserved ejection fraction. Circulation. 15:e008935. doi: 10.1161/CIRCHEARTFAILURE.121.008935
76. McMurray JJV, Pfeffer AM: Heart failure. The Lancet. (2005) 365:1877–89. doi: 10.1016/S0140-6736(05)66621-4
77. Tomasoni D, Adamo M, Lombardi CM, Metra M. Highlights in heart failure. ESC heart Failure. (2019) 6:1105–27. doi: 10.1002/ehf2.12555
78. Yousefi K, Irion CI, Takeuchi LM, Ding W, Lambert G, Eisenberg T, et al. Osteopontin promotes left ventricular diastolic dysfunction through a mitochondrial pathway. J Am College Cardiol. (2019) 73:2705–18. doi: 10.1016/j.jacc.2019.02.074
79. White SJ, Nicklin SA, Büning H, Brosnan MJ, Leike K, Papadakis ED, et al. Baker: Targeted gene delivery to vascular tissue in vivo by tropism-modified adeno-associated virus vectors. Circulation. (2004) 109:513–9. doi: 10.1161/01.CIR.0000109697.68832.5D
80. Ylä-Herttuala S, Bridges C, Katz MG, Korpisalo P. Angiogenic gene therapy in cardiovascular diseases: dream or vision? Eur Heart J. (2017) 38:1365–371. doi: 10.1093/eurheartj/ehw547
81. Wang S, Li Y, Xu Y, Ma Q, Lin Z, Schlame M, et al. AAV gene therapy prevents and reverses heart failure in a murine knockout model of barth syndrome. Circulat Res. (2020) 126:1024–39. doi: 10.1161/CIRCRESAHA.119.315956
82. Remes A, Wagner AH, Schmiedel N, Heckmann M, Ruf T, Ding L, et al. AAV-mediated expression of NFAT decoy oligonucleotides protects from cardiac hypertrophy and heart failure. Basic Res cardiol. (2021) 116:38. doi: 10.1007/s00395-021-00880-w
83. Pleger ST, Shan C, Ksienzyk J, Bekeredjian R, Boekstegers P, Hinkel R, et al. Most: Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci Translational Med. (2011) 3:92ra64–ra64. doi: 10.1126/scitranslmed.3002097
84. Ishikawa K, Fish KM, Tilemann L, Rapti K, Aguero J, Santos-Gallego CG, et al. Hajjar: Cardiac I-1c overexpression with reengineered AAV improves cardiac function in swine ischemic heart failure. Mol Ther. (2014) 22:2038–45. doi: 10.1038/mt.2014.127
85. Zhou F, Fu WD, Chen L. MiRNA-182 regulates the cardiomyocyte apoptosis in heart failure. Eur Rev Med Pharmacol Sci. (2019) 23:4917–23. doi: 10.26355/eurrev_201906_18079
86. Park CS, Cha H, Kwon EJ, Jeong D, Hajjar RJ, Kranias EG, et al. AAV-mediated knock-down of HRC exacerbates transverse aorta constriction-induced heart failure. PloS ONE. (2012) 7:e43282–e43282. doi: 10.1371/journal.pone.0043282
87. Chaanine AH, Nonnenmacher M, Kohlbrenner E, Jin D, Kovacic JC, Akar FG, et al. Weber: Effect of bortezomib on the efficacy of AAV9.SERCA2a treatment to preserve cardiac function in a rat pressure-overload model of heart failure. Gene Therapy. (2014) 21:379–86. doi: 10.1038/gt.2014.7
88. Pepe M, Mamdani M, Zentilin L, Csiszar A, Qanud K, Zacchigna S, et al. Intramyocardial VEGF-B167 gene delivery delays the progression towards congestive failure in dogs with pacing-induced dilated cardiomyopathy. Circulation Res. (2010) 106:1893–903. doi: 10.1161/CIRCRESAHA.110.220855
89. Karam S, Margaria JP, Bourcier A, Mika D, Varin A, Bedioune I, et al. Vandecasteele: cardiac overexpression of pde4b blunts β-adrenergic response and maladaptive remodeling in heart failure. Circulation. (2020) 142:161–74. doi: 10.1161/CIRCULATIONAHA.119.042573
90. Mathiyalagan P, Adamiak M, Mayourian J, Sassi Y, Liang Y, Agarwal N, et al. Sahoo: FTO-dependent N(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation. (2019) 139:518–32. doi: 10.1161/CIRCULATIONAHA.118.033794
91. Rengo G, Lymperopoulos A, Zincarelli C, Donniacuo M, Soltys S, Rabinowitz JE, et al. Koch: Myocardial adeno-associated virus serotype 6-betaARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation. (2009) 119:89–98. doi: 10.1161/CIRCULATIONAHA.108.803999
92. Weeks KL, Gao X, Du X-J, Boey EJH, Matsumoto A, Bernardo BC, et al. Phosphoinositide 3-Kinase p110α Is a master regulator of exercise-induced cardioprotection and pi3k gene therapy rescues cardiac dysfunction. Circulation. (2012) 5:523–34. doi: 10.1161/CIRCHEARTFAILURE.112.966622
93. Bianca Bernardo C, Kate Weeks L, Pongsukwechkul T, Gao X, Kiriazis H, Cemerlang N, et al. Gene delivery of medium chain acyl-coenzyme A dehydrogenase induces physiological cardiac hypertrophy and protects against pathological remodelling. Clini Sci. (2018) 132:381–397. doi: 10.1042/CS20171269
94. Yan H, Wang H, Zhu X, Huang J, Li Y, Zhou K, et al. Adeno-associated virus-mediated delivery of anti-miR-199a tough decoys attenuates cardiac hypertrophy by targeting PGC-1alpha. Mol Ther. (2020) 23:406–17. doi: 10.1016/j.omtn.2020.11.007
95. Zhong P, Peng J, Liu T, Ding H. AAV9-mediated cardiac CNTF overexpression exacerbated adverse cardiac remodeling in streptozotocin-induced type 1 diabetic models. Cardiovascular Toxicol. (2022) 22:88–96. doi: 10.1007/s12012-021-09706-6
96. Chai RJ, Werner H, Li PY, Lee YL, Nyein KT, Solovei I, et al. Stewart: Disrupting the LINC complex by AAV mediated gene transduction prevents progression of Lamin induced cardiomyopathy. Nature Communicat. (2021) 12:4722. doi: 10.1038/s41467-021-24849-4
97. Wu Y, Zhou L, Liu H, Duan R, Zhou H, Zhang F, et al. Chen: LRP6 downregulation promotes cardiomyocyte proliferation and heart regeneration. Cell Res. (2021) 31:450–62. doi: 10.1038/s41422-020-00411-7
98. Gedicke-Hornung C, Behrens-Gawlik V, Reischmann S, Geertz B, Stimpel D, Weinberger F, et al. Carrier: Rescue of cardiomyopathy through U7snRNA-mediated exon skipping in Mybpc3-targeted knock-in mice. EMBO Molecular Med. (2013) 5:1128–45. doi: 10.1002/emmm.201202168
99. Moretti A, Fonteyne L, Giesert F, Hoppmann P, Meier AB, Bozoglu T, et al. Kupatt: Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nature Med. (2020) 26:207–14. doi: 10.1038/s41591-019-0738-2
100. Amoasii L, Hildyard JCW, Li H, Sanchez-Ortiz E, Mireault A, Caballero D, et al. Olson: Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science (New York, NY.). (2018) 362:86–91. doi: 10.1126/science.aau1549
101. Li J, Wang K, Zhang Y, Qi T, Yuan J, Zhang L, et al. Therapeutic exon skipping through a crispr-guided cytidine deaminase rescues dystrophic cardiomyopathy in vivo. Circulation. (2021) 144:1760–76. doi: 10.1161/CIRCULATIONAHA.121.054628
102. Yue Y, Li Z, Harper SQ, Davisson RL, Chamberlain JS, Duan D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. (2003) 108:1626–32. doi: 10.1161/01.CIR.0000089371.11664.27
103. Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, et al. Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure. Circulation Res. (2014) 114:101–8. doi: 10.1161/CIRCRESAHA.113.302421
104. Butter C, Rastogi S, Minden H-H, Meyhöfer J, Burkhoff D, Sabbah NH. Cardiac contractility modulation electrical signals improve myocardial gene expression in patients with heart failure. J Am College Cardiol. (2008) 51:1784–9. doi: 10.1016/j.jacc.2008.01.036
105. Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. The Lancet. (2016) 387:1178–86. doi: 10.1016/S0140-6736(16)00082-9
106. Sitsel A, De Raeymaecker J, Drachmann ND, Derua R, Smaardijk S, Andersen JL, et al. Nissen: Structures of the heart specific SERCA2a Ca(2+)-ATPase. EMBO Journal. (2019) 38:e100020. doi: 10.15252/embj.2018100020
107. Zhang Y, Jiao L, Sun L, Li Y, Gao Y, Xu C, et al. LncRNA ZFAS1 as a SERCA2a inhibitor to cause intracellular Ca(2+) overload and contractile dysfunction in a mouse model of myocardial Infarction. Circulat Res. (2018) 122:1354–368. doi: 10.1161/CIRCRESAHA.117.312117
108. Mariani JA, Smolic A, Preovolos A, Byrne MJ, Power JM, Kaye MD. Augmentation of left ventricular mechanics by recirculation-mediated AAV2/1–SERCA2a gene delivery in experimental heart failure. Eur J Heart Failure. (2011) 13:247–53. doi: 10.1093/eurjhf/hfq234
109. Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, et al. Calcium up-regulation by percutaneous administration of gene therapy in cardiac disease trial: Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Cardiac Failure. (2009) 15:171–81. doi: 10.1016/j.cardfail.2009.01.013
110. Wang J, Shi Q, Wang Y, Dawson LW, Ciampa G, Zhao W, et al. Gene therapy with the N-terminus of junctophilin-2 improves heart failure in mice. Circulat Res. (2022) 130:1306–17. doi: 10.1161/CIRCRESAHA.121.320680
111. Burke MA, Cook SA, Seidman JG, Seidman EC. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J Am College Cardiol. (2016) 68:2871–86. doi: 10.1016/j.jacc.2016.08.079
112. Semsarian C, Semsarian RC. Variable penetrance in hypertrophic cardiomyopathy: in search of the holy grail*. J Am College Cardiol. (2020) 76:560–2. doi: 10.1016/j.jacc.2020.06.023
113. McKenna WJ, Maron BJ, Thiene G. Classification, epidemiology, and global burden of cardiomyopathies. Circulat Res. (2017) 121:722–30. doi: 10.1161/CIRCRESAHA.117.309711
114. Martinez EC, Li J, Ataam JA, Tokarski K, Thakur H, Karakikes I, et al. Kapiloff: Targeting mAKAPβ expression as a therapeutic approach for ischemic cardiomyopathy. Gene Therapy. (2022). doi: 10.1038/s41434-022-00321-w
115. Gaul S, Shahzad K, Medert R, Gadi I, Mäder C, Schumacher D, et al. Novel Nongenetic Murine Model of Hyperglycemia and Hyperlipidemia-Associated Aggravated Atherosclerosis. Front Cardiovascular Med. (2022) 9:813215. doi: 10.3389/fcvm.2022.813215
116. Jarrett KE, Lee C, De Giorgi M, Hurley A, Gillard BK, Doerfler AM, et al. Somatic editing of ldlr with adeno-associated viral-crispr is an efficient tool for atherosclerosis research. Arterioscler Thromb Vasc Biol. (2018) 38:1997–2006. doi: 10.1161/ATVBAHA.118.311221
Keywords: AAV, gene therapy, CVD, cardiomyocyte, heart failure
Citation: Zhang H, Zhan Q, Huang B, Wang Y and Wang X (2022) AAV-mediated gene therapy: Advancing cardiovascular disease treatment. Front. Cardiovasc. Med. 9:952755. doi: 10.3389/fcvm.2022.952755
Received: 25 May 2022; Accepted: 08 August 2022;
Published: 19 August 2022.
Edited by:
Kiyotake Ishikawa, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Julie R. McMullen, Baker Heart and Diabetes Institute, AustraliaCopyright © 2022 Zhang, Zhan, Huang, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyan Wang, d3h5OTgwMTExMEBzaW5hLmNvbQ==; Yigang Wang, d2FuZ3lpZ2FuZzQzQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.