 Ronghui Han1
Ronghui Han1 Jing Tang
Jing Tang Zhengyuan Xia
Zhengyuan Xia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 11 August 2022
Sec. Cardiovascular Metabolism
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.951597
Diabetic cardiomyopathy (DCM) is the primary cause of morbidity and mortality in diabetic cardiovascular complications, which initially manifests as cardiac hypertrophy, myocardial fibrosis, dysfunctional remodeling, and diastolic dysfunction, followed by systolic dysfunction, and eventually end with acute heart failure. Molecular mechanisms underlying these pathological changes in diabetic hearts are complicated and multifactorial, including but not limited to insulin resistance, oxidative stress, lipotoxicity, cardiomyocytes apoptosis or autophagy, inflammatory response, and myocardial metabolic dysfunction. With the development of molecular biology technology, accumulating evidence illustrates that members of the class O of Forkhead box (FoxO) transcription factors are vital for maintaining cardiomyocyte metabolism and cell survival, and the functions of the FoxO family proteins can be modulated by a wide variety of post-translational modifications including phosphorylation, acetylation, ubiquitination, arginine methylation, and O-glycosylation. In this review, we highlight and summarize the most recent advances in two members of the FoxO family (predominately FoxO1 and FoxO3a) that are abundantly expressed in cardiac tissue and whose levels of gene and protein expressions change as DCM progresses, with the goal of providing valuable insights into the pathogenesis of diabetic cardiovascular complications and discussing their therapeutic potential and possible effects of salvianolic acids, a natural product.
The improvement of people’s living standards has always been accompanied by a variety of chronic and inconspicuous diseases that exercise a negative influence on our mental and physical health and our longevity. Diabetes Mellitus is increasingly recognized as a serious, worldwide public concern, in particular, patients at a young age and low body max index (1). Though the fleeting high glucose at the very beginning is frequently neglected among patients with type 2 diabetes (T2D), continuous hyperglycemia will damage the vascular system and induce a range of pathological changes in hearts (1, 2). Whatever the origin with nature or nurture, a plethora of clinical and experimental studies have verified that cardiomyocytes impairment provoked directly by type 1 diabetes (T1D) and T2D would contribute to the occurrence and development of diabetic cardiomyopathy (DCM) (3, 4), and eventually, lead to lethal heart failure. Meanwhile, myocardial pathological changes and dysfunction in DCM are extraordinarily tangled and complicated, it involves abnormal insulin sensitivity and signaling, excessive oxidation products, continuous stimulation of inflammation, degeneration of heart tissue, uncontrollable apoptosis, necrosis and autophagy (5–8). As a result, understanding the pathogenic mechanism governing the development and progress of DCM and exploring the effective and controllable cardiac biomarkers for risk prediction is urgently needed.
Recently, mounting evidence from multiple scientific studies shows that Forkhead box O (FoxO) transcription factors, including FoxO1 (FKHR), FoxO3a (FKHRL1), FoxO4 (AFX) and FoxO6, have important functional implications in several signaling pathways refer to human health and diseases (9, 10). So far, two subtypes of FoxO (FoxO1 and FoxO3a) are known to be essential for the maintenance of cardiovascular homeostasis that are nearly expressed in all tissues (11–13). Studies from our laboratory also illustrated a pivotal role for FoxO1 or FoxO3a in cardiac metabolism. Our findings indicated that FoxO1 is a critical initiator of T1D-induced vascular remodeling through activating NOD-like receptor family protein-3 (NLRP3) inflammasome-dependent inflammation (14). Meanwhile, activation of FoxO1 in diabetic rats may promote a change in the preferred substrate from glucose to fatty acid and results in mitochondrial and cardiac dysfunction via stimulating pyruvate dehydrogenase kinase 4 (PDK4) and carnitine palmitoyltransferase 1 (CPT1) (15). Furthermore, we found that up-regulation of FoxO1 or FoxO3a is attributable to the reduction of H9c2 cardiac cells apoptosis and autophagy induced by hypoxia/reoxygenation injury under hyperglycemia in vitro (16). Interestingly, our findings regarding the impact of FoxO1 and FoxO3a on apoptosis and autophagy (16) are in contrast to findings of previous studies conducted in different experimental settings which showed that enhancement of cardiac FoxO1 protein expression is responsible for the increased autophagy and apoptosis in mice with DCM (17), while increase in FoxO3a activation has been shown to be a mechanism by which high glucose induced oxidative stress and apoptosis in cardiac microvascular endothelial cells (18). Available evidence suggest that the FoxO family plays an important role in the development of DCM but its molecular mechanism remains largely unclear.
This review focuses primarily on the regulation and outcomes of FoxO signaling which should be comprehensively investigated to develop novel therapeutics and preventives for DCM.
Human Forkhead-box gene family consists of 19 subfamilies from FoxA to FoxS and there are more than 40 members that have been identified till now (19, 20). It is noticeable that mammalians express four members FoxO1, FoxO3a, FoxO4, and FoxO6 in FoxO transcription factors (21). From the embryo to the adult, FoxO1 and FoxO3a are critical for maintaining cellular homeostasis and genetic integrity of which we will discuss below.
FoxO transcription factors are characterized by four domains, including an extremely conserved DNA-binding Forkhead or wing-helix domain (DBD, consists of W1/W2 wing-like loops, H1/H2/H3 α-helices, and S1/S2/S3 β-strands), a nuclear localization sequence (NLS, the precise amino-acid sequence in the adjacent neighborhood of the DBD for their nuclear translocation), a nuclear export sequence (NES, the reverse function of a nuclear import signaling, which targets proteins out of the cell nucleus), and a C-terminal containing a transactivation domain (TAD) as being demonstrated in previous publications (10, 22).
It is well known that regulation of transactivation domain or activation domain of FoxO transcription factors depends on post-translational modification, containing but not limited to phosphorylation, acetylation/deacetylation, ubiquitination, methylation, and interaction with other transcription protein (23) (Figure 1). Meanwhile, attenuation or enhancement of transcription function in FoxO transcription factors achieved by the above-mentioned post-translational modifications is depend on the magnitude of nuclear/cytoplasmic localization and variation of DNA recognition under some specific physiological and pathological conditions, such as fasting diet, diabetes, sepsis, ischemia and so on, and thereby trigger cellular proliferation, autophagy, apoptosis, self-degradation, etc. (24, 25).
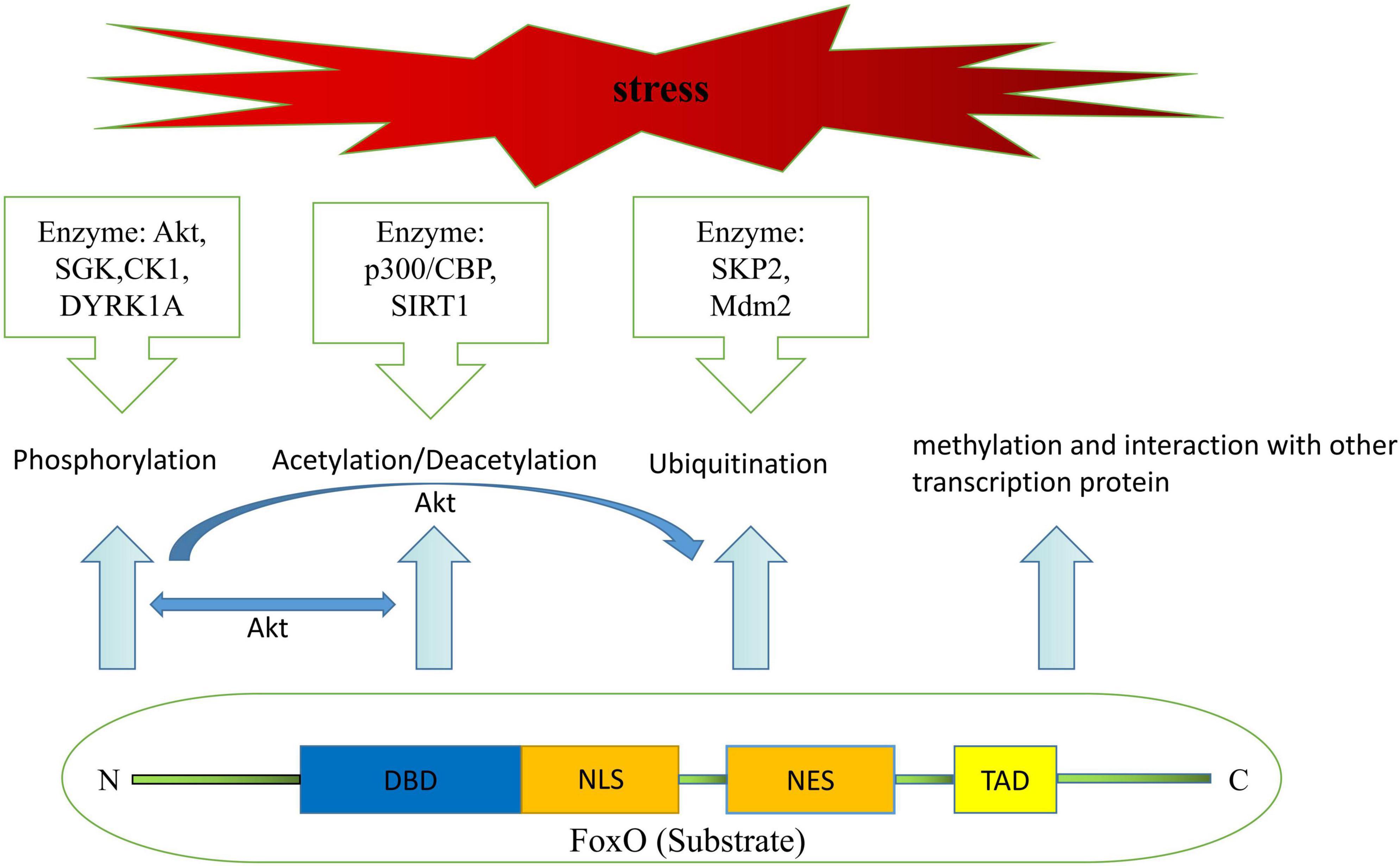
Figure 1. Structural components of FoxO protein and mechanisms leading to post-translational modifications of FoxO.
Akt-dependent phosphorylation has been thought of as a key factor in cellular proliferation and survival by inhibiting the transcriptional functions of FoxO over the past two decades, as this post-translational modification would facilitate FoxO translocation from the nuclear to the cytoplasm and results in the disruption of NLS and DBD function (26–28). Emerging evidence also suggests that FoxO transcription factors could be phosphorylated by other kinases, including serum and glucocorticoid-regulated protein kinase (SGK), casein kinase-1 (CK1), dual-specificity tyrosine-phosphorylated and regulated kinase-1A (29, 30). Interestingly, Chen et al. reported that both SGK and Akt kinase have great effects on lifespan, stress resistance, and FoxO transcription factor activity in Caenorhabditis elegans through completely different mechanisms (31). For instance, whereas Akt-1 and Akt-2 weaken lifespan extension by secluding DAF16/FoxO in the cytoplasm (32), SGK promotes longevity in a DAF16/FoxO-dependent manner as well (30). Despite that there was a broad consensus on the fact that SGK and Akt share similar primary structure and substrate specificity, and both of them form a protein complex regulated by the 3-phosphoinositide-dependent protein kinase-1 (PDK1) or phosphatidylinositide3-kinases (PI3K) are capable of phosphorylating DAF16/FoxO (33–35). However, it is not known whether one could substitute for the other in regulating FoxO transcription factors.
FoxO transcriptional activity is also modulated by acetylation/deacetylation. As more acetylation sites have been discovered, mostly in the W2 region of the DNA-binding domain, mounting evidence has described that the effects of FoxO acetylation/deacetylation are associated with two types of proteins namely histone deacetylases and histone actyltransferase which initiate fundamentally distinct feedback signals during various biological processes (36, 37). Early studies found that acetylation of FoxO factors by calcium response element-binding (CREB)-binding protein (CBP) and/or p300 increased nuclear export, which was made feasible by phosphorylation, and that this effect could be reversed by over-expression of the NAD-dependent protein deacetylase silent information regulator (SIRT1) (38–40). Deacetylation, however, has also been shown to influence FoxO activity in a dual manner (41). On the one hand, in response to stress and nutritional restriction, SIRT1 stimulates catabolic gene expression under fasting condition via deacetylating the Forkhead factor FoxO (42, 43). Meanwhile, SIRT1 deacetylates FoxO, thus increasing FoxO’s ability to prevent oxidative stress and inflammation (44, 45). But on the other, deacetylation promotes FoxO-induced cell-cycle arrest (41, 46). These findings pave the way to a better understanding of the processing of FoxO acetylation/deacetylation. From metabolism to longevity and open new perspectives for therapeutic targets to treat maladaptive processing of pathophysiology.
Excessive activation of FoxO transactivation activity disturbs the metabolic equilibrium in vivo. Scientists have revealed that there exists a way to ameliorate abrupt FoxO-toxicity, namely ubiquitination (29, 47). Until now, researchers believed that this post-translational modification, poly-ubiquitination-dependent proteasomal degradation, is regulated by PI3K/Akt-mediated phosphorylation and activated by S-phase kinase-associated protein-2 (SKP2) binding to FoxO at Ser256 site in the cytoplasm, but not in the nuclear (23, 48). Besides the canonical pathway of poly-ubiquitination, the mono-ubiquitination, Murine double minute-2 (Mdm2) has been identified as a novel ubiquitin E3 ligase which induces mono-ubiquitination with increased FoxO transcriptional activity via promoting its nuclear localization, and, it suggested an analogical model of regulation between FoxO transcription factors and p53 as well (49). It also has been validated that Mdm2 is required for extracellular signal-regulated kinase (ERK)-induced phosphorylation of FoxO3a and may function as an ubiquitin ligase for the further degradation of FoxO3a (50). Interestingly, FoxO3a protein also exhibits a poly-ubiquitination pattern in degradation that is associated with Mdm2 as mentioned in the research by Wang et al. (42). That is to say, both poly and mono ubiquitination could be switched or interconverted by Mdm2. Furthermore, neither type of ubiquitination is far from controlled, and USP7 is reported to remove ubiquitin from FoxO, and serves as a deubiquitinating enzyme (51).
In 1972, Rubler and his co-workers initially described the existence of a DCM based on postmortem pathological findings of four adult diabetic patients who suffered from heart failure without apparent evidence of classic clinical manifestations, such as hypertension, coronary artery disease, congenital heart defect or excessive drinking (52). Until now, multiple researches have uncovered a far-ranging potential mechanisms that attribute to the process of deterioration of DCM to heart failure, which are related to changes of metabolism (hyperglycemia, insulin resistance/hyperinsulinaemia, and dyslipidemia), various forms of programmed active cell death (apoptosis, pyroptosis, autophagic cell death, necroptosis, and ferroptosis) and mitochondrial dysfunction (53) (Figure 2). In other words, fully understanding of those possible pathogenesis is beneficial to develop pharmacological strategies combating DCM. The following sections provide an update on the current understanding of the pathophysiology of DCM.
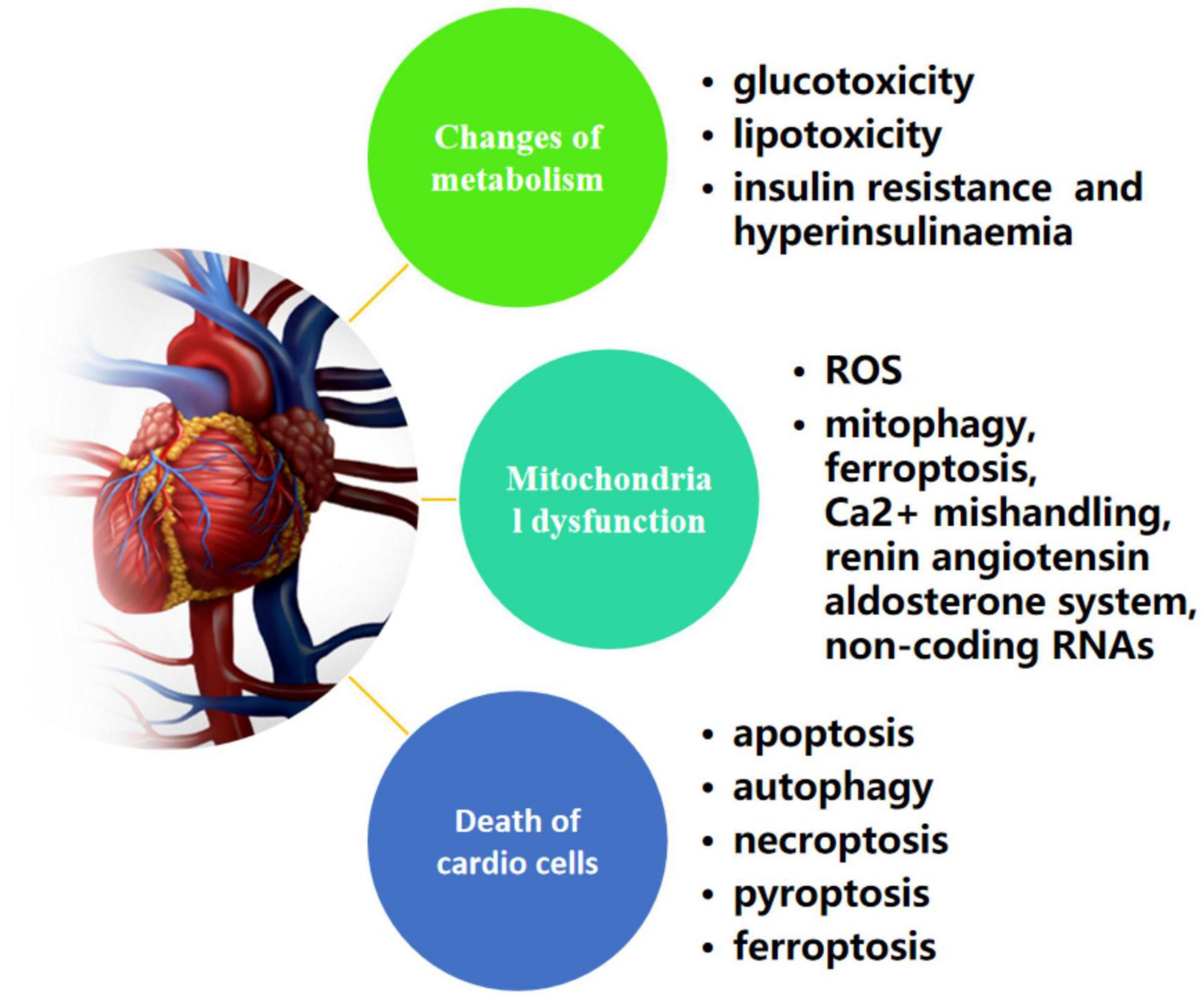
Figure 2. Possible mechanisms of metabolism, cell death, and mitochondrial dysfunction in diabetic cardiomyopathy.
Metabolic disturbance including high glucose, insulin resistance and activation of fatty acid which all are attributable to adverse myocardial structural remodeling in diabetes and often lead to cellular malfunction, and early interventions such as drugs that low blood sugar and/or reduce triglyceride levels in diabetic dyslipidemia would be beneficial in preventing or attenuating cardiac remodeling in diabetes (54).
The healthy heart can always remain delicately equilibrium to utilize myocardial energy substrates, however, this common balance would be destroyed in patients who endured unremitting high glucose levels, a situation termed as “glucotoxicity.” Both animal and cellular studies have demonstrated that glucose overload in cardiomyocytes promotes the malformation of morphology and cardiac dysfunction, for instance, hypertrophy, fibrosis, systolic or diastolic dysfunction and other pathophysiological changes (55), which can be achieved through direct or indirect activation of oxidative stress, accumulation of advanced glycation end-products (AGEs), maladaptive hexosamine biosynthesis and polyol pathway (56). Interestingly, even though epidemiological evidence has shown that glycemic control is an essential part to reduce diabetic complications (57), a phenomenon called “hyperglycemic memory” suggested that hyperglycemic stress persists in the cardiovascular system of patients with diabetes regardless of the level of blood sugar is strictly controlled (58).
In the uncontrolled diabetic state, free fatty acids oxidation nearly become the major energy resource to produce enough adenosine triphosphate (ATP) that satisfies the high demand of the heart following the distinct limitation of glucose oxidation (59). It is also noticeable that excess free fatty acids and other lipids (triglycerides, etc.) could result in lipotoxicity when metabolic disorder occurs in cardiomyocytes (60). While cardiomyocytes use fatty acids almost exclusively to support ATP synthesis, the process will consume more oxygen than using glucose for energy generation, and thus it is less efficient to sustain cardiac work as a consequence of increased production of reactive oxygen species (ROS) and accumulation of lipid intermediates such as diacylglycerols and ceramide (61–63).
The metabolic shift toward fatty acids oxidation rather than glucose oxidation is partially due to the insulin resistance or hyperinsulinaemia (64, 65). In the normal heart, 60–80% of cardiac energy provided for myocardial metabolism is derived from fatty acid oxidation, and the remaining 20–40% is derived from glucose and lactic acid metabolism (66). The utilization of glucose is further cut down when cardiac energy relies more on fatty acids oxidation under insulin resistance condition or hyperinsulinaemic state (67). And beyond that, numerous studies have reported that impaired myocardial insulin signaling under insulin resistance or hyperinsulinaemia are independently linked with the development of DCM and cardiac diastolic dysfunction through a variety of mechanisms (6, 68). For instance, Mellor et al. showed that cardiomyocytes from Glut-4-knockout mice demonstrated extreme glucose uptake deficiency and exhibited cardiac hypertrophy and marked excitation-contraction coupling abnormalities (69). On the other hand, cardiac energy deprivation due to reduction in fatty acid uptake and inhibition of ATP generation resulted from impaired FoxO1/CD36 signaling may accelerate the occurrence of concentric hypertrophy and myocardial fibrosis, exacerbating the progression of DCM (70).
Studies have verified the existence of defects of mitochondrial function in animal models of diabetes, and the potential regulations in mitochondrial biology in diabetes have been consistently unmasked (71–73). In particular, distinct pathways and mechanisms governing the development of mitochondrial dysfunction in DCM have been proposed in recent years (74, 75). Under normal circumstance, the energy needed to support heart function is mainly derived from free fatty acids oxidation but glucose oxidation also contribute significantly to heart energy generation. Given that fatty acid oxidation yields fewer ATP production per mole of oxygen than that from glucose oxidation, excessive fatty acid oxidant in the circumstance of impaired glucose oxidation in diabetes causes mitochondrial swelling and uncoupling due in part to the excessive consumption of molecule oxygen and the concomitantly increased production of ROS (76, 77). The damage caused by accumulation of ROS-mediated mitochondrial malfunction is not limited to intensive lipotoxicity induced by fatty acid oxidation or impaired adiponectin receptor 1 signaling, but can also be related to mitophagy, ferroptosis, Ca2+ mishandling, renin angiotensin aldosterone system, non-coding RNAs, and so forth (78–80). Initially, researchers observed the significant mitochondrial damage mediated by ROS-induced alteration of topoisomerase activity under chronically elevated glucose concentration in rat cardiomyocytes (81). Afterward, further studies revealed that mitophagy resulted from burst of ROS might be a compensation strategy for inconsistent autophagy flux in T2D models while the diminished autophagy appears to be an adaptive response that protects against cardiac injury in type 1 diabetic animal models (62, 82, 83). Indeed, numerous studies have demonstrated that various ROS scavengers or antioxidants are able to reduce cardiomyocyte death in vitro and cardiac injury in vivo via eliminating impaired mitochondria and protecting of the remaining intact mitochondria, however, these studies were still restricted to diabetic experiments in animals (84), and effects of treatment with various antioxidants remain largely disappointing in clinical trials (85) despite marginal treatment effects has been reported showing that alpha-lipoic acid may have a role in preventing the development of DCM in type 1 diabetes with sub-clinical left ventricular dysfunction (86).
In both T1D and T2D, it is widely accepted that there are 3 pivotal forms of cell death in cardiomyocytes including apoptosis, autophagy and necrosis, while necrosis can be further classified as necroptosis, pyroptosis, and ferroptosis (87, 88). From the mass of evidence available, the effective interference linked with a certain degree of suppression or activation in cardiomyocyte death of any forms yields tremendous protective consequences (89, 90).
There are two important apoptosis pathways that have been extensively studied in animal experiments including intrinsic apoptotic pathway (also known as death receptor pathway) and extrinsic pathway (called mitochondrial apoptotic program). For the intrinsic pathway, researchers confirmed that the cytochrome c-activated caspase-3 pathway caused myocardial apoptosis triggered by ROS derived from high levels of glucose in H9c2 cardiac myoblast cells (91). Meanwhile, it has been shown that galangin (Gal) attenuated cardiac apoptosis through inhibiting caspase activity in diabetic rats (92). Angiotensin II (Ang II)-dependent process is another mechanism implicated in the induction of cardiomyocyte apoptosis in diabetic rat hearts (93), and the subsequent evidence that myocardial cell apoptosis induced by Ang II could be suppressed by the application of metallothionein confirmed the involvement of the above mechanism (94). During the past few years, various non-coding RNAs (such as microRNAs and long non-coding RNAs) and exosomes are emerging as potential key regulators of apoptosis pathway in diabetes (95–98), but the specific mechanism remains incompletely elucidated. However, it is worth noting that all these complicated signaling pathways in diabetes-induced cell death seem to point to the formation of excessive ROS in the diabetic hearts.
In the heart, autophagy is important to maintain homeostasis via eliminating impaired or useless organelles, and thus, disruption in this pathway leads to uncontrolled and untargeted cellular suicide. However, the real role of autophagy in DCM is quite controversial. Qiao et al. found that activating the RIPK1-RIPK3 pathway by impairing the autophagic flux which increased the expression of autophagic related proteins such as LC3-II, p62, and active-cathepsin D eventually leads to myocardial fibrosis and cardiac dysfunction in diabetic rats (99). On the other hand, selective autophagic removal of mitochondria (mitophagy) appears to be an adaptive response through clearing away defective mitochondria that protects against cardiac injury in type 1 diabetes (100, 101). By contrast, other studies suggested that cardiomyocyte apoptosis may suppress autophagy in diabetic hearts (102, 103), that is to say that mutual effects between the autophagy and apoptotic cell death pathways are crucial in the pathological progress of DCM. Therefore, the impact of diabetes on cardiac autophagy and its consequences still need to be further investigated.
Necroptosis is involved in the pathogenesis of many diseases, but its role in DCM is still not clear as the relevant investigation is quite rare in both animal models or in in vitro studies. Some studies confirmed that myocardial cell necroptosis is observed in the hearts of diabetic patients and animal models (90, 104). Recent studies described that activation of Ca2+/calmodulin-dependent protein kinase (CaMKII) via a RIPK3-dependent manner or deficiency of sirtuin-3 (SIRT3) can exacerbate DCM through necroptosis enhancement in diabetic rats (105, 106). Even so, direct evidence for the exact mechanism of necroptosis in DCM is not very clear and more in depth investigations are merited.
There is also limited knowledge about the role of pyroptosis in DCM. As a unique and programmed form of cell death, pyroptosis has been revealed to be associated with inflammatory response in DCM via releasing of pro-inflammatory intracellular contents including interleukin (IL)-1β, IL-18, and other inflammatory substances that are induced subsequent to the activation of the NLRP3 inflammasome which is linked to key cardiovascular risk factors (107, 108). Meanwhile, emerging evidence has verified the role of non-coding RNAs in the pathogenesis of pyroptosis in DCM (109), including micro RNA (miRNA), circular RNA (circRNA), and long non-coding RNA (lncRNA) (110). Xu et al. proved that suppression of NLRP3 inflammasome activation-mediated pyroptosis by targeted miR-34b-3p/AHR axis from long non-coding RNA GAS5 is valuable for the attenuation of DCM (111). ROS scavengers, p38 and FoxO1 inhibitors have been shown to inhibit NLRP3 inflammasome assembly and activation in diabetic conditions and attenuate diabetic atherosclerosis through downregulating the p38-FoxO1-TXNIP (thioredoxin-interacting protein) pathway (112), and TXNIP regulates myocardial fatty acid oxidation via miR-33a signaling (113). Unlike previous study, another report suggested that methyltransferase-like 14 (METTL14) inhibits pyroptosis by down-regulating of lncRNA TINCR (114). Regardless of these discrepancies regarding the mechanisms of pyroptosis regulation in the context of diabetes and DCM, there is no doubt that pyroptosis may be a new therapeutic target in the prevention or treatment of DCM.
Ferroptosis is a newly described form of iron-dependent regulated necrosis that distinctly varied from apoptosis, autophagy and other types of cell death (115). Until now, numerous emerging evidences presented that ferroptosis plays an important role in the pathophysiology of cardiovascular disease including ischemia/reperfusion injury, myocardial infarction, heart failure, DCM, and cardiac arrhythmia controlled by several mechanisms, including mitochondrial activity and metabolism of iron, lipid, and amino acids (116, 117). Zang et al. substantiated that cardiac iron deposition was significantly increased in T1D rats which was promoted by activated nuclear factor erythroid2-related factor 2 (Nrf2) signaling subsequent to autophagy inhibition via cardiomyocyte-restricted knockout of autophagy-related 5 gene (107). Consistently, suppression of autophagy in H9C2 cardiomyocytes enhanced Nrf2-coordinated ferroptosis (118). Meanwhile, recent study described that ferroptosis was also evidenced in the heart of type 2 diabetic mice with DCM and could be inhibited by sulforaphane via AMPK-mediated Nrf2 activation (119). Taken together, credible and further evidences for developmental mechanism in ferroptosis of clinical patients remain blurred and the dual role of Nrf2 in the development of DCM needs to be investigated deeply.
It is well known that FoxO transcription factor plays a vital role in cardiovascular homeostasis (120), at the same time, the available evidence indicates that FoxO may impact DCM through vascular remodeling, cellular apoptosis and autophagy, oxidative stress, inflammation, and other pathophysiological processes (11, 121, 122) (Figure 3).
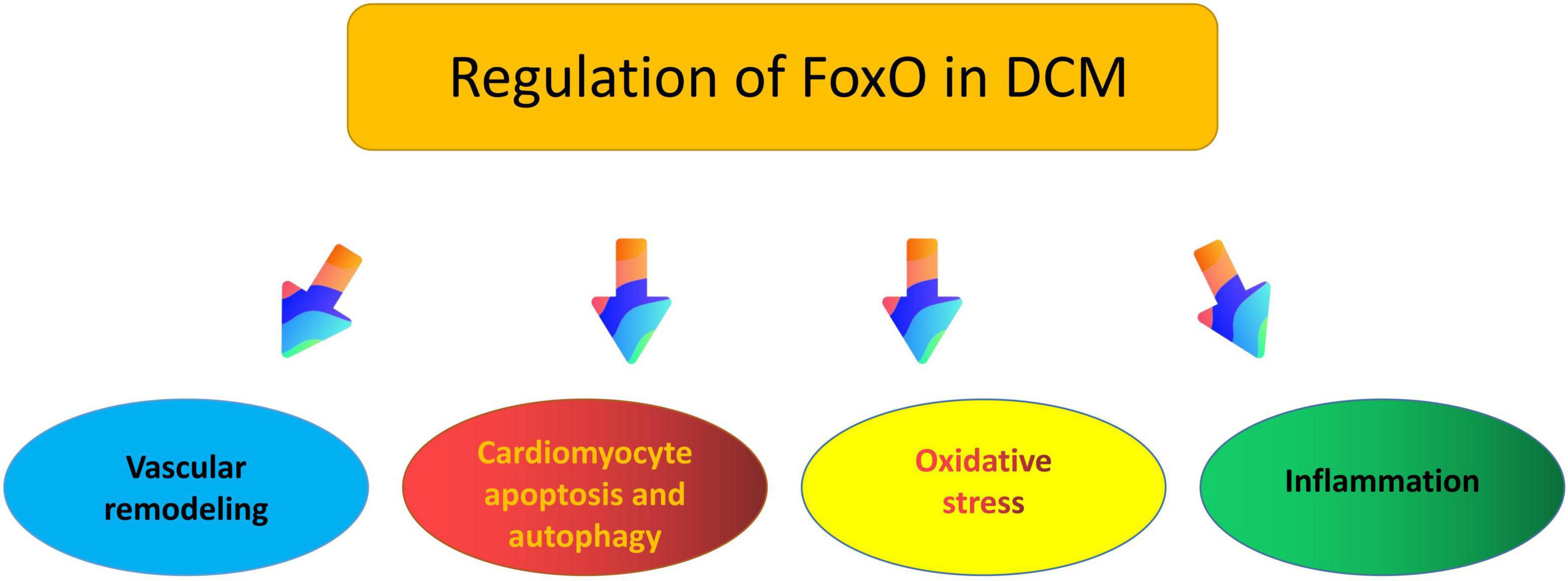
Figure 3. Putative roles of FoxO in the pathogenesis of diabetic cardiomyopathy.
Previous study shown that FoxO1 is required for embryonic vascular development and FoxO1-null embryo died in the early stage as a consequence of incomplete vascular growth (123), while the deletion of FoxO3a may results in cardiac hypertrophy and eventual cardiac failure for the offspring (124). To elucidate the character of FoxO transcription factor, Potente et al. showed that overexpression of FoxO1 or FoxO3a led to profound inhibition of endothelial cell migration and tube formation, however, such effect is counterproductive while silencing either of FoxO1 or FoxO3a via using small interfering RNA (siRNA). And beyond that, researchers also further suggested that several genes with vital vascular functions are regulated by FoxO1 or FoxO3a, and gene silencing study revealed that eNOS is integral for postnatal neovascularization as a novel FoxO target gene (125). Consistent with these findings, our group first demonstrated that suppression of FoxO1 might attenuate vascular remodeling induced by type 1 diabetes in rats through decreasing the expression of NLRP3 inflammasome activation and activating related underlying mechanism including PDK1/FoxO1 pathway (14). As a result, inhibition of FoxO may provide a valuable tool to alleviate the adverse vascular remodeling in DCM but the specific and unique role of each FoxO isoform remains unclear.
Appropriate programmed cell death such as apoptosis and autophagy seems to be an essential part in the elimination of cytotoxic proteins and dysfunctional organelles which would otherwise accelerate the development of DCM and the deterioration of DCM to cardiac failure. And, more and more evidences revealed that suppression of excessive cardiomyocyte death of aforementioned death modes occurred in the diabetic heart may yield tremendous advantageous effects on DCM (126–128). It is well known that FoxO1 and/or FoxO3a participate in the regulation of various cell death seen during insulin resistance and diabetes. With apoptosis, FoxO transcription factors are downstream targets of the Akt kinase which regulates processes of cellular proliferation and survival and also may indirectly stimulate expression of death receptor ligands such as Fas ligand through the unstable mitochondrial channels or directly induce the expression of multiple pro-apoptotic proteins like Bcl-2 family (18, 25, 129). As for autophagy, researchers have shown that plenty of the regulation and control mechanisms include but are not limited to the mTOR/AMPK, FoxOs, SIRTs, and others signalings, and further studies to unmask the detailed roles and in particular the potentially complicated interplays of the pathways should help to elucidate the inconsistent outcomes observed following the intervention of the individual pathways in the treatment of DCM (127, 130). Convincing evidences showed that certain drugs protected pancreatic β-cells by enhancing autophagy such as liraglutide (a long-acting human glucagon-like peptide-1 analog), which protected insulinoma cells via alleviating apoptosis accompanied by a significant increase of autophagy under hyperglycemia in vitro (131). And, Guo et al. proved that the improvement of islet function by liraglutide involves FoxO signaling pathway through modulating miRNA expression profiling (132). However, a recent study suggests that inadequate autophagy with impaired autophagosome-lysosomal fusion exists in the aortic intima and endothelial cells from diabetic patients and that FoxO1 may inhibit autophagosome-lysosome fusion and lead to endothelial autophagic-apoptosis in diabetes (133). In a previous study, we revealed that propofol postconditioning could attenuate hypoxia/reoxygenation-induced apoptosis and autophagy by regulating the expression of FoxO1 and FoxO3a in H9c2 cell under hyperglycemia (16). Of note, FoxO emerges as a potential participant in regulating cell death, but its role under conditions of insulin resistance and diabetes still need to be further explored.
It is indisputable that oxidative stress plays a vital role in cardiac remodeling and malfunction, and reverse of excessive burst production of ROS by antioxidants contributes to the improvement of cardiac metabolism and function in the heart of experimental diabetic models (134, 135). Our recent study demonstrated that activation of FoxO1 via stimulating the expression of PDK4 and CPT led to imbalanced oxidative metabolism which may exacerbate the progress of DCM in streptozotocin-induced type 1 diabetes in rats and such disadvantageous effects like mitochondrial and cardiac dysfunction is reversible by administration of AS1842856 (AS), a selective FoxO1-selective inhibitor (15). Interestingly, we also found that up-regulation of FoxO1 or FoxO3a decreased the ROS level in H9c2 cells, a cardiac cell strain derived from the S-D Rat left ventricle, under the condition of hyperglycemia (16). In addition, researchers demonstrated that propofol could up-regulate FoxO1 to attenuate myocardial cell injury after oxygen and glucose deprivation and reperfusion (136), and Peng et al. showed that FoxO3a silencing strengthened ROS accumulation (18), these data are in agreement with our experimental findings in vitro but contradictory to other finding in vivo (137). These inconsistent results implied that oxidative stress regulates FoxO activity through various post-translational modifications including phosphorylation, acetylation, ubiquitination and so on, which may be restricted to the experimental environment as well (138). For example, inhibition of SIRT1 by Exendin-4 down-regulated the phosphorylation of FoxO1 and reduced post-ischemic infarct size and preserved the function and structure of the left ventricles in rats through scavenging free radicals directly (139), and enhancement of H4 acetylation of the FoxO3a promoter region has been shown to confer myocardium protection from oxidative stress-mediated ischemia/reperfusion injury (140). Therefore, understanding the complicated and changeable mechanisms in redox regulation of FoxO signaling is essential for the initiation of interventions that may help to maintain cardiac function in diabetes.
Inflammation is significantly involved in the progression of DCM, and chronic inflammation eventually leads to decreased insulin sensitivity and diabetic complications. Hyperglycemia-induced FoxO over-activation is central to the production of pro-inflammatory cytokines.
Lundell et al. showed that several inflammatory signaling pathways are enriched by FoxO1 transfection which including Cd68, Cd48, Itgax, Cd3g, Ncr1, Itgam, and Ly6c, while only three markers of immune cells involving Cd68, Itgam, and Ly6c can be enriched by FoxO3a transfection (141). A recent study also indicated that triggering of FoxO localization by 9-hydroxy-octadecadienoic acid mediated JNK signaling pathway is accompanied with antagonism of insulin signaling, providing a novel linking between dietary fatty acid balance and meta-inflammation (142). In this regard, we assume that inhibition of inflammatory pathways via FoxO transcription factors may increasingly be recognized as exciting prospects for the prevention of multiple diabetic disorders.
Despite multiple studies have provided a variety of potential mechanisms acting on DCM and support a dual role of FoxO transcript factors in DCM presumably dependent on the level of oxidative stress, it is noticeable that the participation of oxidative stress possibly proceeds the development of certain signaling transduction and pathophysiological changes including most types of cell death, especially in modulating the relationship between autophagy and ferroptosis that are both targets for FoxO regulation. Ferritinophagy, an unique form of selective autophagy of ferritin has been proved to contribute significantly in the development of cardiovascular diseases (143). Li and colleagues reported that enhanced ferroptosis and activated ubiquitin specific peptidase-19/beclin1-mediated autophagy were observed in oxygen–glucose deprivation/reoxygenation-induced H9c2 cell death, which could be reversed by resveratrol treatment via attenuating the oxidative stress injury (144). Given that ROS are also associated with other cell death pathways, and that FoxO is recognized as playing an dual role in transcriptional regulation of proteins in autophagy as we described previously, we assume that FoxO may impact on ferroptosis in both autophagy dependent and independent manners via ROS as the trigger during the development of DCM. On the other hand, FoxO1 and FoxO3a exerted the same protective effects in H9C2 cells under hyperglycemia as we investigated before (16) and the deficiency of either of them leads to the identical consequences that aggravates meniscus lesions in experimental models through abolishing autophagy and antioxidant defense genes (145), whereas FoxO1 or FoxO3a may have radically different effects in diabetic hearts and they even interfere with each other. For instance, FoxO3a may increase autophagy that is mediated and associated with the promotion of the translocation of FoxO1 from the nucleus to the cytoplasm, which suggested that the process dominated by FoxO3a cannot be separated from FoxO1 (146). However, it is yet hard to know whether FoxO1 or FoxO3a may play a leading role in the development of DCM (Figure 4).
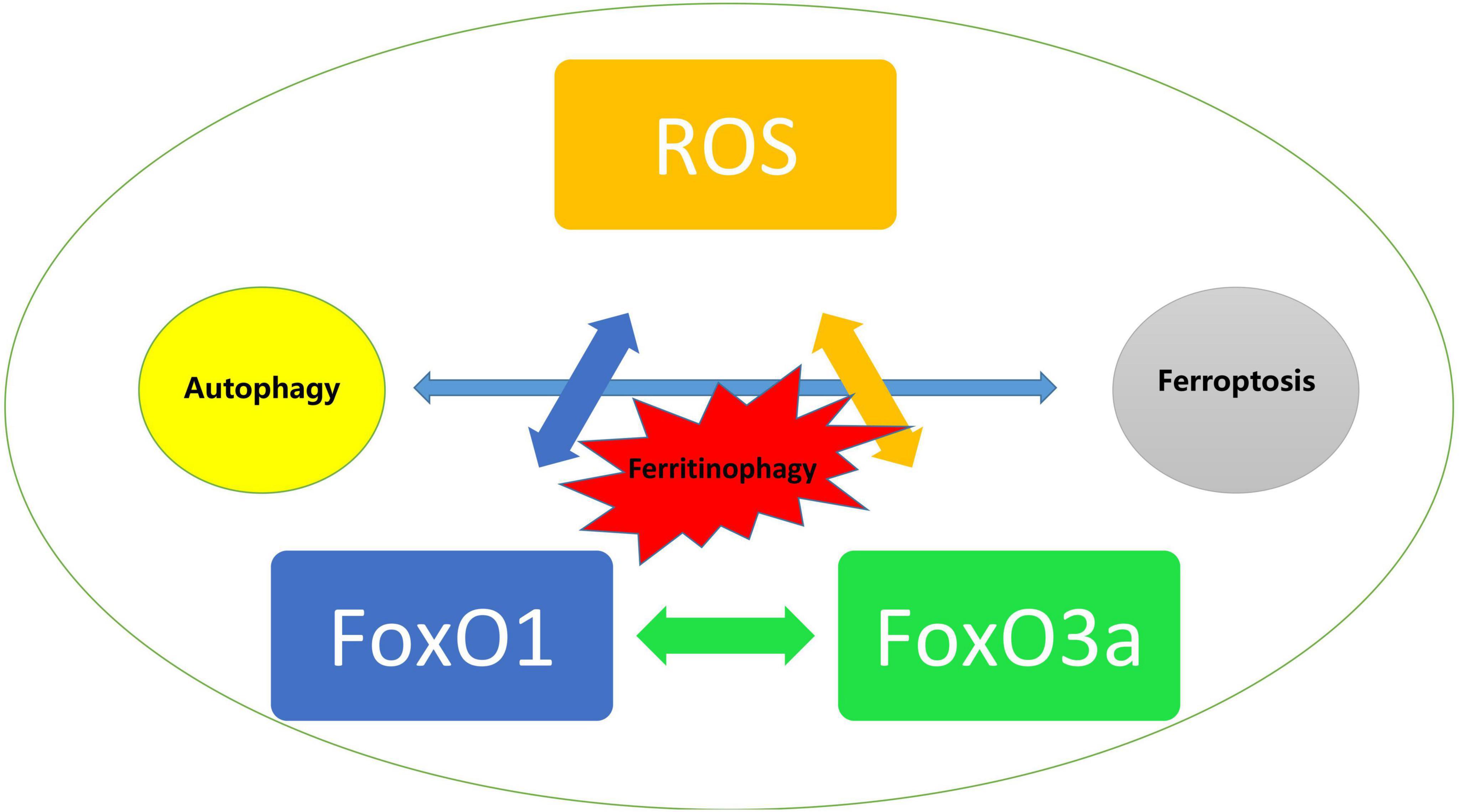
Figure 4. Trigger of ROS mediated activation of FoxO and the impact on autophagy and ferroptosis in diabetic cardiomyopathy.
In the above sections, we reviewed the recent progress on FoxO multiple post-translational modifications and mechanisms, which involve various signal transduction pathways in DCM. In the meanwhile, it should be noted that currently pharmacological therapies that can specifically and effectively target FoxOs in the clinical settings are lacking. Interestingly, we noticed that salvianolic acids (mainly refer to hydrophilic constituents of salvianolic A and B) extracted from the roots of a traditional Chinese medicinal herb, Salvia miltiorrhiza, have been emerged as potent antioxidants that confer protection against cardiovascular disease (147, 148). For instance, several studies demonstrated salvianolic acid A conferred cardio-protective effects as a free radical scavenger through targeting the Akt/GSK-3β, JNK, PI3K/Akt, and ERK1/2 signaling pathways, and reduced myocardial ischemia/reperfusion injury in diabetic rats and cellular hypoxia/reoxygenation injury in in vitro models that involved the reduction cardiomyocytes apoptosis (149–151). Similar to salvianolic acid A, salvianolic acid B can also act on the PI3K/Akt signaling pathway and the ERK1/2 signaling pathway, both of which are closely associated with myocardial damage (152, 153). Furthermore, cardiac dysfunction and fibrosis in the heart of diabetic rats could be ameliorated by salvianolic acid B through down-regulating of insulin-like growth factor-binding protein 3 (IGFBP3) activity by enhancing myocardial angiogenesis (154). The above-mentioned cardio protective effects of salvianolic acids might stem from their unique polyphenolic structure (135, 155, 156). Additionally, salvianolic acid A treatment yield distinct protective effects against cerebral ischemia/reperfusion injury via inhibition of the FoxO3a/BIM pathway both in vivo and in vitro (157), whilst salvianolic acid B could mitigate tunicamycin-mediated cell pyroptosis via modulating of AMPK/FoxO4 and Syndecan-4/Rac1 signaling pathways (158). Salvianolic Acid B has also been shown to decrease oxidative stress reaction by regulating SIRT3/FoxO1 signaling pathway and play a therapeutic role in the treatment of non-alcoholic steatohepatitis in rats (159). The above findings collectively suggest that salvianolic acids may serve as a potential drug candidates in cardiovascular disease therapy in general and in DCM in particular through targeting FoxOs directly or indirectly via their strong ROS scavenging properties (160, 161), given that FoxO represent viable target of DCM, although relevant and existing investigations are rare. Further in depth studies are merited to figure out explicit mechanisms between FoxO and salvianolic acids in the prevention and/or treatment of DCM in view that these natural products are safe and do not have major side effects at the doses used to treat cardiovascular diseases (162).
In the current review, we discussed mechanisms and pathways that are disturbed in the diabetic heart and structure as well as regulation of FoxO in DCM. However, above discussions mainly based on animal and cell experiments instead of real clinical applications while the integrated and precise signaling of diabetic cardiac injury or the relate contribution of FoxO transcription factors in DCM remain largely unknown. In order to seize the target spot of FoxO in DCM, it is necessary to determinate whether the observed changes is available and adaptive both in TID and T2D, while oxidative stress is widely accepted as a promising pointcut under high glucose condition. Meanwhile, given the interplay between different mechanisms, it is also important to achieve a comprehensive confirmation of many of these molecular underpinnings that regulate and coordinate these processes to attenuate injury in the human diabetic heart to gain more insight if potential treatment targets identified in animal models may also be a valid target in humans.
RH, HH, WX, JL, HL, JT, and ZX: conceptualization, literature review, tables, writing – review and editing, revisions, and final editing. All authors have read and agreed to the published version of the manuscript.
This project was funded by the National Natural Science Foundation of China (Nos. 81970247 and 81670770).
We acknowledge VanScholar Editors Co., Ltd., Vancouver, Canada, for the English editing assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
DCM, diabetic cardiomyopathy; FoxO, the class O of Forkhead box; T2D, type 2 diabetes; T1D, type 1 diabetes; PDK4, pyruvate dehydrogenase kinase 4; Akt, protein kinase B; NLRP3, NOD-like receptor family protein-3; SGK, serum and glucocorticoid-regulated protein kinase; CK1, casein kinase-1; PI3K, phosphatidylinositide3-kinases; PDK1, 3-phosphoinositide-dependent protein kinase-1; SIRT1, silent information regulator; Mdm2, murine double minute-2; ERK, extracellular signal-regulated kinase; ATP, adenosine triphosphate; ROS, reactive oxygen species; Ang II, Angiotensin II; SIRT3, sirtuin-3; IL, Interleukin; TXNIP, thioredoxin-interacting protein; Nrf2, nuclear factor erythroid2-related factor 2.
1. Hu C, Jia W. Diabetes in China: epidemiology and genetic risk factors and their clinical utility in personalized medication. Diabetes. (2018) 67:3–11. doi: 10.2337/dbi17-0013
2. Tchobroutsky G. Relation of diabetic control to development of microvascular complications. Diabetologia. (1978) 15:143–52. doi: 10.1007/BF00421230
4. Hardin NJ. The myocardial and vascular pathology of diabetic cardiomyopathy. Coron Artery Dis. (1996) 7:99–108. doi: 10.1097/00019501-199602000-00002
5. Ponugoti B, Dong G, Graves DT. Role of forkhead transcription factors in diabetes-induced oxidative stress. Exp Diabetes Res. (2012) 2012:939751. doi: 10.1155/2012/939751
6. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. (2016) 12:144–53. doi: 10.1038/nrendo.2015.216
7. Eid S, Sas KM, Abcouwer SF, Feldman EL, Gardner TW, Pennathur S, et al. New insights into the mechanisms of diabetic complications: role of lipids and lipid metabolism. Diabetologia. (2019) 62:1539–49. doi: 10.1007/s00125-019-4959-1
8. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. (2020) 126:1501–25. doi: 10.1161/CIRCRESAHA.120.315913
9. Du S, Zheng H. Role of FoxO transcription factors in aging and age-related metabolic and neurodegenerative diseases. Cell Biosci. (2021) 11:188. doi: 10.1186/s13578-021-00700-7
10. van der Vos KE, Coffer PJ. The extending network of FOXO transcriptional target genes. Antioxid Redox Signal. (2011) 14:579–92. doi: 10.1089/ars.2010.3419
11. Wang Y, Zhou Y, Graves DT. FOXO transcription factors: their clinical significance and regulation. Biomed Res Int. (2014) 2014:925350. doi: 10.1155/2014/925350
12. Tsuchiya K, Ogawa Y. Forkhead box class O family member proteins: the biology and pathophysiological roles in diabetes. J Diabetes Investig. (2017) 8:726–34. doi: 10.1111/jdi.12651
13. Ronnebaum SM, Patterson C. The FoxO family in cardiac function and dysfunction. Annu Rev Physiol. (2010) 72:81–94. doi: 10.1146/annurev-physiol-021909-135931
14. Liu J, Xie X, Yan D, Wang Y, Yuan H, Cai Y, et al. Up-regulation of FoxO1 contributes to adverse vascular remodelling in type 1 diabetic rats. J Cell Mol Med. (2020) 24:13727–38. doi: 10.1111/jcmm.15935
15. Yan D, Cai Y, Luo J, Liu J, Li X, Ying F, et al. FOXO1 contributes to diabetic cardiomyopathy via inducing imbalanced oxidative metabolism in type 1 diabetes. J Cell Mol Med. (2020) 24:7850–61. doi: 10.1111/jcmm.15418
16. Han RH, Huang HM, Han H, Chen H, Zeng F, Xie X, et al. Propofol postconditioning ameliorates hypoxia/reoxygenation induced H9c2 cell apoptosis and autophagy via upregulating forkhead transcription factors under hyperglycemia. Mil Med Res. (2021) 8:58. doi: 10.1186/s40779-021-00353-0
17. Zhang M, Sui W, Xing Y, Cheng J, Cheng C, Xue F, et al. Angiotensin IV attenuates diabetic cardiomyopathy via suppressing FoxO1-induced excessive autophagy, apoptosis and fibrosis. Theranostics. (2021) 11:8624–39. doi: 10.7150/thno.48561
18. Peng C, Ma J, Gao X, Tian P, Li W, Zhang L. High glucose induced oxidative stress and apoptosis in cardiac microvascular endothelial cells are regulated by FoxO3a. PLoS One. (2013) 8:e79739. doi: 10.1371/journal.pone.0079739
20. Gui T, Burgering BMT. FOXOs: masters of the equilibrium. FEBS J. (2021) [Epub ahead of print]. doi: 10.1111/febs.16221
21. Link W. Introduction to FOXO Biology. Methods Mol Biol. (2019) 1890:1–9. doi: 10.1007/978-1-4939-8900-3_1
22. Obsil T, Obsilova V. Structure/function relationships underlying regulation of FOXO transcription factors. Oncogene. (2008) 27:2263–75. doi: 10.1038/onc.2008.20
23. Jiramongkol Y, Lam EW. FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev. (2020) 39:681–709. doi: 10.1007/s10555-020-09883-w
24. Furukawa-Hibi Y, Kobayashi Y, Chen C, Motoyama N. FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antioxid Redox Signal. (2005) 7:752–60. doi: 10.1089/ars.2005.7.752
25. Puthanveetil P, Wan A, Rodrigues B. FoxO1 is crucial for sustaining cardiomyocyte metabolism and cell survival. Cardiovasc Res. (2013) 97:393–403. doi: 10.1093/cvr/cvs426
26. Bouchard C, Marquardt J, Brás A, Medema RH, Eilers M. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J. (2004) 23:2830–40. doi: 10.1038/sj.emboj.7600279
27. Birkenkamp KU, Coffer PJ. Regulation of cell survival and proliferation by the FOXO (Forkhead box, class O) subfamily of Forkhead transcription factors. Biochem Soc Trans. (2003) 31(Pt 1):292–7. doi: 10.1042/bst0310292
28. Van Der Heide LP, Hoekman MF, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J. (2004) 380(Pt 2):297–309. doi: 10.1042/BJ20040167
29. Vogt PK, Jiang H, Aoki M. Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle. (2005) 4:908–13. doi: 10.4161/cc.4.7.1796
30. Shin MG, Lee JW, Han JS, Lee B, Jeong JH, Park SH, et al. Bacteria-derived metabolite, methylglyoxal, modulates the longevity of C. elegans through TORC2/SGK-1/DAF-16 signaling. Proc Natl Acad Sci USA. (2020) 117:17142–50. doi: 10.1073/pnas.1915719117
31. Chen AT, Guo C, Dumas KJ, Ashrafi K, Hu PJ. Effects of Caenorhabditis elegans sgk-1 mutations on lifespan, stress resistance, and DAF-16/FoxO regulation. Aging Cell. (2013) 12:932–40. doi: 10.1111/acel.12120
32. Murphy CT, Hu PJ. Insulin/insulin-like growth factor signaling in C. elegans. WormBook. (2013) 26:1–43. doi: 10.1895/wormbook.1.164.1
33. Hertweck M, Göbel C, Baumeister R. C. elegans SGK-1 is the critical component in the Akt/PKB kinase complex to control stress response and life span. Dev Cell. (2004) 6:577–88. doi: 10.1016/s1534-5807(04)00095-4
34. Wang M, Qiao X, Cooper T, Pan W, Liu L, Hayball J, et al. HPV E7-mediated NCAPH ectopic expression regulates the carcinogenesis of cervical carcinoma via PI3K/AKT/SGK pathway. Cell Death Dis. (2020) 11:1049. doi: 10.1038/s41419-020-03244-9
35. Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. (2011) 13:224. doi: 10.1186/bcr3039
36. Daitoku H, Sakamaki J, Fukamizu A. Regulation of FoxO transcription factors by acetylation and protein-protein interactions. Biochim Biophys Acta. (2011) 1813:1954–60. doi: 10.1016/j.bbamcr.2011.03.001
37. Zhao Y, Wang Y, Zhu WG. Applications of post-translational modifications of FoxO family proteins in biological functions. J Mol Cell Biol. (2011) 3276–82. doi: 10.1093/jmcb/mjr013
38. Hatta M, Liu F, Cirillo LA. Acetylation curtails nucleosome binding, not stable nucleosome remodeling, by FoxO1. Biochem Biophys Res Commun. (2009) 379:1005–8. doi: 10.1016/j.bbrc.2009.01.014
39. Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci USA. (2005) 102:11278–83. doi: 10.1073/pnas.0502738102
40. van der Heide LP, Smidt MP. Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem Sci. (2005) 30:81–6. doi: 10.1016/j.tibs.2004.12.002
41. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. (2004) 303:2011–5. doi: 10.1126/science.1094637
42. Wang B, Moya N, Niessen S, Hoover H, Mihaylova MM, Shaw RJ, et al. A hormone-dependent module regulating energy balance. Cell. (2011) 145:596–606. doi: 10.1016/j.cell.2011.04.013
43. Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. (2010) 107:1470–82. doi: 10.1161/CIRCRESAHA.110.227371
44. Singh V, Ubaid S. Role of Silent Information Regulator 1 (SIRT1) in regulating oxidative stress and inflammation. Inflammation. (2020) 43:1589–98. doi: 10.1007/s10753-020-01242-9
45. Yun JM, Chien A, Jialal I, Devaraj S. Resveratrol up-regulates SIRT1 and inhibits cellular oxidative stress in the diabetic milieu: mechanistic insights. J Nutr Biochem. (2012) 23:699–705. doi: 10.1016/j.jnutbio.2011.03.012
46. Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. (2004) 14:408–12. doi: 10.1016/j.tcb.2004.07.006
47. Manola MS, Gumeni S, Trougakos IP. Differential dose- and tissue-dependent effects of foxo on aging, metabolic and proteostatic pathways. Cells. (2021) 10:3577. doi: 10.3390/cells10123577
48. Huang H, Regan KM, Wang F, Wang D, Smith DI, van Deursen JM, et al. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci USA. (2005) 102:1649–54. doi: 10.1073/pnas.0406789102
49. Brenkman AB, de Keizer PL, van den Broek NJ, Jochemsen AG, Burgering BM. Mdm2 induces mono-ubiquitination of FOXO4. PLoS One. (2008) 3:e2819. doi: 10.1371/journal.pone.0002819
50. Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. (2008) 10:138–48. doi: 10.1038/ncb1676
51. van der Horst A, de Vries-Smits AM, Brenkman AB, van Triest MH, van den Broek N, Colland F, et al. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol. (2006) 8:1064–73. doi: 10.1038/ncb1469
53. Avagimyan A. The pathophysiological basis of diabetic cardiomyopathy development. Curr Probl Cardiol. (2022) 19:101156. doi: 10.1016/j.cpcardiol.2022.101156
54. Nakamura M, Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. J Physiol. (2020) 598:2977–93. doi: 10.1113/JP276747
55. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. (2020) 17:585–607. doi: 10.1038/s41569-020-0339-2
56. Salvatore T, Pafundi PC, Galiero R, Albanese G, Di Martino A, Caturano A, et al. The diabetic cardiomyopathy: the contributing pathophysiological mechanisms. Front Med. (2021) 8:695792. doi: 10.3389/fmed.2021.695792
57. Lachin JM, Orchard TJ, Nathan DM. DCCT/EDIC Research Group. Update on cardiovascular outcomes at 30 years of the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care. (2014) 37:39–43. doi: 10.2337/dc13-2116
58. Zhan J, Chen C, Wang DW, Li H. Hyperglycemic memory in diabetic cardiomyopathy. Front Med. (2021) 16:25–38. doi: 10.1007/s11684-021-0881-2
59. Karwi QG, Sun Q, Lopaschuk GD. The contribution of cardiac fatty acid oxidation to diabetic cardiomyopathy severity. Cells. (2021) 10:3259. doi: 10.3390/cells10113259
60. Borghetti G, von Lewinski D, Eaton DM, Sourij H, Houser SR, Wallner M. Diabetic cardiomyopathy: current and future therapies. beyond glycemic control. Front Physiol. (2018) 9:1514. doi: 10.3389/fphys.2018.01514
61. Lorenzo O, Ramírez E, Picatoste B, Egido J, Tuñón J. Alteration of energy substrates and ROS production in diabetic cardiomyopathy. Mediators Inflamm. (2013) 2013:461967. doi: 10.1155/2013/461967
62. Dirkx E, Schwenk RW, Glatz JF, Luiken JJ, van Eys GJ. High fat diet induced diabetic cardiomyopathy. Prostaglandins Leukot Essent Fatty Acids. (2011) 85:219–25. doi: 10.1016/j.plefa.2011.04.018
63. Sowton AP, Griffin JL, Murray AJ. Metabolic profiling of the diabetic heart: toward a richer picture. Front Physiol. (2019) 10:639. doi: 10.3389/fphys.2019.00639
64. Sangaleti CT, Katayama KY, De Angelis K, Lemos de Moraes T, Araújo AA, Lopes HF, et al. Consolim Colombo FM. The cholinergic drug galantamine alleviates oxidative stress alongside anti-inflammatory and cardio-metabolic effects in subjects with the metabolic syndrome in a randomized trial. Front Immunol. (2021) 12:613979. doi: 10.3389/fimmu.2021.613979
65. Ren Z, Okyere SK, Xie L, Wen J, Wang J, Chen Z, et al. Oral Administration of Bacillus toyonensis Strain SAU-20 improves insulin resistance and ameliorates hepatic steatosis in type 2 diabetic mice. Front Immunol. (2022) 13:837237. doi: 10.3389/fimmu.2022.837237
66. Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate and lipids. Prog Cardiovasc Dis. (1972) 15:289–329. doi: 10.1016/0033-0620(72)90029-1
67. Kok BP, Brindley DN. Myocardial fatty acid metabolism and lipotoxicity in the setting of insulin resistance. Heart Fail Clin. (2012) 8:643–61. doi: 10.1016/j.hfc.2012.06.008
68. Letonja M, Petroviè D. Is diabetic cardiomyopathy a specific entity? World J Cardiol. (2014) 6:8–13. doi: 10.4330/wjc.v6.i1.8
69. Mellor KM, Bell JR, Ritchie RH, Delbridge LM. Myocardial insulin resistance, metabolic stress and autophagy in diabetes. Clin Exp Pharmacol Physiol. (2013) 40:56–61. doi: 10.1111/j.1440-1681.2012.05738.x
70. Ying F, Liu H, Ching Tang EH, Lakhani I, Liu N, Xia Z, et al. Prostaglandin E receptor subtype 4 protects against diabetic cardiomyopathy by modulating cardiac fatty acid metabolism via FOXO1/CD36 signalling. Biochem Biophys Res Commun. (2021) 548:196–203. doi: 10.1016/j.bbrc.2021.01.038
71. Tanaka Y, Konno N, Kako KJ. Mitochondrial dysfunction observed in situ in cardiomyocytes of rats in experimental diabetes. Cardiovasc Res. (1992) 26:409–14. doi: 10.1093/cvr/26.4.409
72. Montgomery MK, Turner N. Mitochondrial dysfunction and insulin resistance: an update. Endocr Connect. (2015) 4:R1–15. doi: 10.1530/EC-14-0092
73. Hathaway QA, Pinti MV, Durr AJ, Waris S, Shepherd DL, Hollander JM. Regulating microRNA expression: at the heart of diabetes mellitus and the mitochondrion. Am J Physiol Heart Circ Physiol. (2018) 314:H293–310. doi: 10.1152/ajpheart.00520.2017
74. Liang Q, Kobayashi S. Mitochondrial quality control in the diabetic heart. J Mol Cell Cardiol. (2016) 95:57–69. doi: 10.1016/j.yjmcc.2015.12.025
75. Gollmer J, Zirlik A, Bugger H. Mitochondrial mechanisms in diabetic cardiomyopathy. Diabetes Metab J. (2020) 44:33–53. doi: 10.4093/dmj.2019.0185
76. Seager MJ, Singal PK, Orchard R, Pierce GN, Dhalla NS. Cardiac cell damage: a primary myocardial disease in streptozotocin-induced chronic diabetes. Br J Exp Pathol. (1984) 65:613–23.
77. Ansari M, Gopalakrishnan S, Kurian GA. Streptozotocin-induced type II diabetic rat administered with non-obesogenic high-fat diet is highly susceptible to myocardial ischemia-reperfusion injury: an insight into the function of mitochondria. J Cell Physiol. (2019) 234:4104–14. doi: 10.1002/jcp.27217
78. Makrecka-Kuka M, Liepinsh E, Murray AJ, Lemieux H, Dambrova M, Tepp K, et al. Altered mitochondrial metabolism in the insulin-resistant heart. Acta Physiol. (2020) 228:e13430. doi: 10.1111/apha.13430
79. Jaquenod De Giusti C, Palomeque J, Mattiazzi A. Ca2+ mishandling and mitochondrial dysfunction: a converging road to prediabetic and diabetic cardiomyopathy. Pflugers Arch. (2022) 474:33–61. doi: 10.1007/s00424-021-02650-y
80. Sukumaran V, Gurusamy N, Yalcin HC, Venkatesh S. Understanding diabetes-induced cardiomyopathy from the perspective of renin angiotensin aldosterone system. Pflugers Arch. (2022) 474:63–81. doi: 10.1007/s00424-021-02651-x
81. Medikayala S, Piteo B, Zhao X, Edwards JG. Chronically elevated glucose compromises myocardial mitochondrial DNA integrity by alteration of mitochondrial topoisomerase function. Am J Physiol Cell Physiol. (2011) 300:C338–48. doi: 10.1152/ajpcell.00248.2010
82. Zheng H, Zhu H, Liu X, Huang X, Huang A, Huang Y. Mitophagy in diabetic cardiomyopathy: roles and mechanisms. Front Cell Dev Biol. (2021) 9:750382. doi: 10.3389/fcell.2021.750382
83. Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta. (2015) 1852:252–61. doi: 10.1016/j.bbadis.2014.05.020
84. Verma SK, Garikipati VNS, Kishore R. Mitochondrial dysfunction and its impact on diabetic heart. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1098–105. doi: 10.1016/j.bbadis.2016.08.021
85. Celik T, Yuksel C, Iyisoy A. Alpha tocopherol use in the management of diabetic cardiomyopathy: lessons learned from randomized clinical trials. J Diabetes Complicat. (2010) 24:286–8. doi: 10.1016/j.jdiacomp.2009.02.005
86. Hegazy SK, Tolba OA, Mostafa TM, Eid MA, El-Afify DR. Alpha-lipoic acid improves subclinical left ventricular dysfunction in asymptomatic patients with type 1 diabetes. Rev Diabet Stud. (2013) 10:58–67. doi: 10.1900/RDS.2013.10.58
87. Chen Y, Hua Y, Li X, Arslan IM, Zhang W, Meng G. Distinct types of cell death and the implication in diabetic cardiomyopathy. Front Pharmacol. (2020) 11:42. doi: 10.3389/fphar.2020.00042
88. Lu LQ, Tian J, Luo XJ, Peng J. Targeting the pathways of regulated necrosis: a potential strategy for alleviation of cardio-cerebrovascular injury. Cell Mol Life Sci. (2021) 78:63–78. doi: 10.1007/s00018-020-03587-8
89. Muñoz-Córdova F, Hernández-Fuentes C, Lopez-Crisosto C, Troncoso MF, Calle X, Guerrero-Moncayo A, et al. Novel insights into the pathogenesis of diabetic cardiomyopathy and pharmacological strategies. Front Cardiovasc Med. (2021) 8:707336. doi: 10.3389/fcvm.2021.707336
90. Cai L, Kang YJ. Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol. (2003) 3:219–28. doi: 10.1385/ct:3:3:219
91. Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes. (2002) 51:1938–48. doi: 10.2337/diabetes.51.6.1938
92. Abukhalil MH, Althunibat OY, Aladaileh SH, Al-Amarat W, Obeidat HM, Al-Khawalde AAA, et al. Galangin attenuates diabetic cardiomyopathy through modulating oxidative stress, inflammation and apoptosis in rats. Biomed Pharmacother. (2021) 138:111410. doi: 10.1016/j.biopha.2021.111410
93. Fiordaliso F, Li B, Latini R, Sonnenblick EH, Anversa P, Leri A, et al. Myocyte death in streptozotocin-induced diabetes in rats in angiotensin II- dependent. Lab Invest. (2000) 80:513–27. doi: 10.1038/labinvest.3780057
94. Xu J, Wang G, Wang Y, Liu Q, Xu W, Tan Y, et al. Diabetes- and angiotensin II-induced cardiac endoplasmic reticulum stress and cell death: metallothionein protection. J Cell Mol Med. (2009) 13:1499–512. doi: 10.1111/j.1582-4934.2009.00833.x
95. Jakubik D, Fitas A, Eyileten C, Jarosz-Popek J, Nowak A, Czajka P, et al. MicroRNAs and long non-coding RNAs in the pathophysiological processes of diabetic cardiomyopathy: emerging biomarkers and potential therapeutics. Cardiovasc Diabetol. (2021) 20:55. doi: 10.1186/s12933-021-01245-2
96. Singla R, Garner KH, Samsam M, Cheng Z, Singla DK. Exosomes derived from cardiac parasympathetic ganglionic neurons inhibit apoptosis in hyperglycemic cardiomyoblasts. Mol Cell Biochem. (2019) 462:1–10. doi: 10.1007/s11010-019-03604-w
97. Zhao SF, Ye YX, Xu JD, He Y, Zhang DW, Xia ZY, et al. Long non-coding RNA KCNQ1OT1 increases the expression of PDCD4 by targeting miR-181a-5p, contributing to cardiomyocyte apoptosis in diabetic cardiomyopathy. Acta Diabetol. (2021) 58:1251–67. doi: 10.1007/s00592-021-01713-x
98. Ge L, Cai Y, Ying F, Liu H, Zhang D, He Y, et al. miR-181c-5p exacerbates hypoxia/reoxygenation-induced cardiomyocyte apoptosis via targeting PTPN4. Oxid Med Cell Longev. (2019) 2019:1957920. doi: 10.1155/2019/1957920
99. Qiao S, Hong L, Zhu Y, Zha J, Wang A, Qiu J, et al. RIPK1-RIPK3 mediates myocardial fibrosis in type 2 diabetes mellitus by impairing autophagic flux of cardiac fibroblasts. Cell Death Dis. (2022) 13:147. doi: 10.1038/s41419-022-04587-1
100. Morales PE, Arias-Durán C, Ávalos-Guajardo Y, Aedo G, Verdejo HE, Parra V, et al. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol Aspects Med. (2020) 71:100822. doi: 10.1016/j.mam.2019.09.006
101. Bharath LP, Rockhold JD, Conway R. Selective autophagy in hyperglycemia-induced microvascular and macrovascular diseases. Cells. (2021) 10:2114. doi: 10.3390/cells10082114
102. Shen GY, Shin JH, Song YS, Joo HW, Park IH, Seong JH, et al. Role of autophagy in granulocyte-colony stimulating factor induced anti-apoptotic effects in diabetic cardiomyopathy. Diabetes Metab J. (2021) 45:594–605. doi: 10.4093/dmj.2020.0049
103. Ouyang C, You J, Xie Z. The interplay between autophagy and apoptosis in the diabetic heart. J Mol Cell Cardiol. (2014) 71:71–80. doi: 10.1016/j.yjmcc.2013.10.014
104. Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, et al. Myocardial cell death in human diabetes. Circ Res. (2000) 87:1123–32. doi: 10.1161/01.res.87.12.1123
105. Song S, Ding Y, Dai GL, Zhang Y, Xu MT, Shen JR, et al. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol Sin. (2021) 42:230–41. doi: 10.1038/s41401-020-0490-7
106. Chen Y, Li X, Hua Y, Ding Y, Meng G, Zhang W. RIPK3-mediated necroptosis in diabetic cardiomyopathy requires CaMKII activation. Oxid Med Cell Longev. (2021) 2021:6617816. doi: 10.1155/2021/6617816
107. Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One. (2014) 9:e104771. doi: 10.1371/journal.pone.0104771
108. Luo B, Huang F, Liu Y, Liang Y, Wei Z, Ke H, et al. NLRP3 inflammasome as a molecular marker in diabetic cardiomyopathy. Front Physiol. (2017) 8:519. doi: 10.3389/fphys.2017.00519
109. Wang C, Zhu L, Yuan W, Sun L, Xia Z, Zhang Z, et al. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J Cell Mol Med. (2020) 24:6670–9. doi: 10.1111/jcmm.15318
110. Lu Y, Lu Y, Meng J, Wang Z. Pyroptosis and its regulation in diabetic cardiomyopathy. Front Physiol. (2022) 12:791848. doi: 10.3389/fphys.2021.791848
111. Xu Y, Fang H, Xu Q, Xu C, Yang L, Huang C. LncRNA GAS5 inhibits NLRP3 inflammasome activation-mediated pyroptosis in diabetic cardiomyopathy by targeting miR-34b-3p/AHR. Cell Cycle. (2020) 19:3054–65. doi: 10.1080/15384101.2020.1831245
112. Nyandwi JB, Ko YS, Jin H, Yun SP, Park SW, Kim HJ. Rosmarinic acid inhibits oxLDL-induced inflammasome activation under high-glucose conditions through downregulating the p38-FOXO1-TXNIP pathway. Biochem Pharmacol. (2020) 182:114246. doi: 10.1016/j.bcp.2020.114246
113. Chen J, Young ME, Chatham JC, Crossman DK, Dell’Italia LJ, Shalev A. TXNIP regulates myocardial fatty acid oxidation via miR-33a signaling. Am J Physiol Heart Circ Physiol. (2016) 311:H64–75. doi: 10.1152/ajpheart.00151.2016
114. Meng L, Lin H, Huang X, Weng J, Peng F, Wu S. METTL14 suppresses pyroptosis and diabetic cardiomyopathy by downregulating TINCR lncRNA. Cell Death Dis. (2022) 13:38. doi: 10.1038/s41419-021-04484-z
115. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. (2016) 73:2195–209. doi: 10.1007/s00018-016-2194-1
116. Hu H, Chen Y, Jing L, Zhai C, Shen L. The link between ferroptosis and cardiovascular diseases: a novel target for treatment. Front Cardiovasc Med. (2021) 8:710963. doi: 10.3389/fcvm.2021.710963
117. Duan JY, Lin X, Xu F, Shan SK, Guo B, Li FX, et al. Ferroptosis and its potential role in metabolic diseases: a curse or revitalization? Front Cell Dev Biol. (2021) 9:701788. doi: 10.3389/fcell.2021.701788
118. Zang H, Wu W, Qi L, Tan W, Nagarkatti P, Nagarkatti M, et al. Autophagy inhibition enables Nrf2 to exaggerate the progression of diabetic cardiomyopathy in mice. Diabetes. (2020) 69:2720–34. doi: 10.2337/db19-1176
119. Wang X, Chen X, Zhou W, Men H, Bao T, Sun Y, et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. (2022) 12:708–22. doi: 10.1016/j.apsb.2021.10.005
120. Menghini R, Casagrande V, Iuliani G, Rizza S, Mavilio M, Cardellini M, et al. Metabolic aspects of cardiovascular diseases: Is FoxO1 a player or a target? Int J Biochem Cell Biol. (2020) 118:105659. doi: 10.1016/j.biocel.2019.105659
121. Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest. (2012) 122:1109–18. doi: 10.1172/JCI60329
122. Tsuchiya K, Tanaka J, Shuiqing Y, Welch CL, DePinho RA, Tabas I, et al. FoxOs integrate pleiotropic actions of insulin in vascular endothelium to protect mice from atherosclerosis. Cell Metab. (2012) 15:372–81. doi: 10.1016/j.cmet.2012.01.018
123. Hosaka T, Biggs WH III, Tieu D, Boyer AD, Varki NM, Cavenee WK, et al. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci USA. (2004) 101:2975–80. doi: 10.1073/pnas.0400093101
124. Evans-Anderson HJ, Alfieri CM, Yutzey KE. Regulation of cardiomyocyte proliferation and myocardial growth during development by FOXO transcription factors. Circ Res. (2008) 102:686–94. doi: 10.1161/CIRCRESAHA.107.163428
125. Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, et al. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. (2005) 115:2382–92. doi: 10.1172/JCI23126
126. El Hayek MS, Ernande L, Benitah JP, Gomez AM, Pereira L. The role of hyperglycaemia in the development of diabetic cardiomyopathy. Arch Cardiovasc Dis. (2021) 114:748–60. doi: 10.1016/j.acvd.2021.08.004
127. Dewanjee S, Vallamkondu J, Kalra RS, John A, Reddy PH, Kandimalla R. Autophagy in the diabetic heart: a potential pharmacotherapeutic target in diabetic cardiomyopathy. Ageing Res Rev. (2021) 68:101338. doi: 10.1016/j.arr.2021.101338
128. Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. (2018) 14:207–15. doi: 10.1080/15548627.2017.1378838
129. Duan P, Wang J, Li Y, Wei S, Su F, Zhang S, et al. Opening of mitoKATP improves cardiac function and inhibits apoptosis via the AKT-Foxo1 signaling pathway in diabetic cardiomyopathy. Int J Mol Med. (2018) 42:2709–19. doi: 10.3892/ijmm.2018.3832
130. Yao F, Zhang M, Chen L. 5’-Monophosphate-activated protein kinase (AMPK) improves autophagic activity in diabetes and diabetic complications. Acta Pharm Sin B. (2016) 6:20–5. doi: 10.1016/j.apsb.2015.07.009
131. Chen ZF, Li YB, Han JY, Yin JJ, Wang Y, Zhu LB, et al. Liraglutide prevents high glucose level induced insulinoma cells apoptosis by targeting autophagy. Chin Med J. (2013) 126:937–41.
132. Guo Q, Xu Y, Li J, An W, Luo D, Huang C, et al. Explore the effect and target of liraglutide on islet function in type 2 diabetic rats by miRNA Omics technology. Diabetes Metab Syndr Obes. (2021) 14:3795–807. doi: 10.2147/DMSO.S325030
133. Zhang H, Ge S, He K, Zhao X, Wu Y, Shao Y, et al. FoxO1 inhibits autophagosome-lysosome fusion leading to endothelial autophagic-apoptosis in diabetes. Cardiovasc Res. (2019) 115:2008–20. doi: 10.1093/cvr/cvz014
134. Jubaidi FF, Zainalabidin S, Taib IS, Hamid ZA, Budin SB. The potential role of flavonoids in ameliorating diabetic cardiomyopathy via alleviation of cardiac oxidative stress, inflammation and apoptosis. Int J Mol Sci. (2021) 22:5094. doi: 10.3390/ijms221050940
135. Ren BC, Zhang YF, Liu SS, Cheng XJ, Yang X, Cui XG, et al. Curcumin alleviates oxidative stress and inhibits apoptosis in diabetic cardiomyopathy via Sirt1-Foxo1 and PI3K-Akt signalling pathways. J Cell Mol Med. (2020) 24:12355–67. doi: 10.1111/jcmm.15725
136. Zhao D, Li Q, Huang Q, Li X, Yin M, Wang Z, et al. Cardioprotective effect of propofol against oxygen glucose deprivation and reperfusion injury in H9c2 cells. Oxid Med Cell Longev. (2015) 2015:184938. doi: 10.1155/2015/184938
137. Alhoshani A, Alanazi FE, Alotaibi MR, Attwa MW, Kadi AA, Aldhfyan A, et al. Inhibitor Gefitinib Induces Cardiotoxicity through the Modulation of Cardiac PTEN/Akt/FoxO3a Pathway and Reactive Metabolites Formation: In Vivo and in Vitro Rat Studies. Chem Res Toxicol. (2020) 33:1719–28. doi: 10.1021/acs.chemrestox.0c00005
138. Klotz LO, Sánchez-Ramos C, Prieto-Arroyo I, Urbánek P, Steinbrenner H, Monsalve M. Redox regulation of FoxO transcription factors. Redox Biol. (2015) 6:51–72. doi: 10.1016/j.redox.2015.06.019
139. Eid RA, Bin-Meferij MM, El-Kott AF, Eleawa SM, Zaki MSA, Al-Shraim M, et al. Exendin-4 protects against myocardial ischemia-reperfusion injury by upregulation of SIRT1 and SIRT3 and activation of AMPK. J Cardiovasc Transl Res. (2021) 14:619–35. doi: 10.1007/s12265-020-09984-5
140. Guo Y, Li Z, Shi C, Li J, Yao M, Chen X. Trichostatin A attenuates oxidative stress-mediated myocardial injury through the FoxO3a signaling pathway. Int J Mol Med. (2017) 40:999–1008. doi: 10.3892/ijmm.2017.3101
141. Lundell LS, Massart J, Altıntaş A, Krook A, Zierath JR. Regulation of glucose uptake and inflammation markers by FOXO1 and FOXO3 in skeletal muscle. Mol Metab. (2019) 20:79–88. doi: 10.1016/j.molmet.2018.09.011
142. Kwon SY, Massey K, Watson MA, Hussain T, Volpe G, Buckley CD, et al. Oxidised metabolites of the omega-6 fatty acid linoleic acid activate dFOXO. Life Sci Alliance. (2020) 3:e201900356. doi: 10.26508/lsa.201900356
143. Qin Y, Qiao Y, Wang D, Tang C, Yan G. Ferritinophagy and ferroptosis in cardiovascular disease: mechanisms and potential applications. Biomed Pharmacother. (2021) 141:111872. doi: 10.1016/j.biopha.2021.111872
144. Li T, Tan Y, Ouyang S, He J, Liu L. Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene. (2022) 808:145968. doi: 10.1016/j.gene.2021.145968
145. Lee KI, Choi S, Matsuzaki T, Alvarez-Garcia O, Olmer M, Grogan SP, et al. FOXO1 and FOXO3 transcription factors have unique functions in meniscus development and homeostasis during aging and osteoarthritis. Proc Natl Acad Sci USA. (2020) 117:3135–43. doi: 10.1073/pnas.1918673117
146. Zhou J, Liao W, Yang J, Ma K, Li X, Wang Y, et al. FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy. (2012) 8:1712–23. doi: 10.4161/auto.21830
147. Li ZM, Xu SW, Liu PQ. Salvia miltiorrhizaBurge (Danshen): a golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol Sin. (2018) 39:802–24. doi: 10.1038/aps.2017.193
148. Wu Y, Xu S, Tian XY. The effect of salvianolic acid on vascular protection and possible mechanisms. Oxid Med Cell Longev. (2020) 2020:5472096. doi: 10.1155/2020/5472096
149. Qian W, Wang Z, Xu T, Li D. Anti-apoptotic effects and mechanisms of salvianolic acid A on cardiomyocytes in ischemia-reperfusion injury. Histol Histopathol. (2019) 34:223–31. doi: 10.14670/HH-18-048
150. Li XL, Fan JP, Liu JX, Liang LN. Salvianolic acid a protects neonatal cardiomyocytes against hypoxia/reoxygenation-induced injury by preserving mitochondrial function and activating Akt/GSK-3β Signals. Chin J Integr Med. (2019) 25:23–30. doi: 10.1007/s11655-016-2747-z
151. Pan H, Li D, Fang F, Chen D, Qi L, Zhang R, et al. Salvianolic acid A demonstrates cardioprotective effects in rat hearts and cardiomyocytes after ischemia/reperfusion injury. J Cardiovasc Pharmacol. (2011) 58:535–42. doi: 10.1097/FJC.0b013e31822de355
152. Liu H, Liu W, Qiu H, Zou D, Cai H, Chen Q, et al. Salvianolic acid B protects against myocardial ischaemia-reperfusion injury in rats via inhibiting high mobility group box 1 protein expression through the PI3K/Akt signalling pathway. Naunyn Schmiedebergs Arch Pharmacol. (2020) 393:1527–39. doi: 10.1007/s00210-019-01755-7
153. Yu J, Chen R, Tan Y, Wu J, Qi J, Zhang M, et al. Salvianolic Acid B alleviates heart failure by inactivating ERK1/2/GATA4 signaling pathway after pressure overload in mice. PLoS One. (2016) 11:e0166560. doi: 10.1371/journal.pone.0166560
154. Li CL, Liu B, Wang ZY, Xie F, Qiao W, Cheng J, et al. Salvianolic acid B improves myocardial function in diabetic cardiomyopathy by suppressing IGFBP3. J Mol Cell Cardiol. (2020) 139:98–112. doi: 10.1016/j.yjmcc.2020.01.009
155. Cao W, Guo XW, Zheng HZ, Li DP, Jia GB, Wang J. Current progress of research on pharmacologic actions of salvianolic acid B. Chin J Integr Med. (2012) 18:316–20. doi: 10.1007/s11655-012-1052-8
156. Zhang L, Zhang W, Zhao Y, Yang X, Fang L, Wang S, et al. [Research progress of salvianolic acid A]. Zhongguo Zhong Yao Za Zhi. (2011) 36:2603–9.
157. Song J, Zhang W, Wang J, Yang H, Zhou Q, Wang H, et al. Inhibition of FOXO3a/BIM signaling pathway contributes to the protective effect of salvianolic acid A against cerebral ischemia/reperfusion injury. Acta Pharm Sin B. (2019) 9:505–15. doi: 10.1016/j.apsb.2019.01.010
158. Tang Y, Wa Q, Peng L, Zheng Y, Chen J, Chen X, et al. Salvianolic acid B suppresses ER stress-induced NLRP3 inflammasome and pyroptosis via the AMPK/FoxO4 and Syndecan-4/Rac1 signaling pathways in human endothelial progenitor cells. Oxid Med Cell Longev. (2022) 2022:8332825. doi: 10.1155/2022/8332825
159. Wang Y, Chen J, Kong W, Zhu R, Liang K, Kan Q, et al. Regulation of SIRT3/FOXO1 signaling pathway in rats with non-alcoholic steatohepatitis by salvianolic acid B. Arch Med Res. (2017) 48:506–12. doi: 10.1016/j.arcmed.2017.11.016
160. Nie R, Xia R, Zhong X, Xia Z. Salvia miltiorrhiza treatment during early reperfusion reduced postischemic myocardial injury in the rat. Can J Physiol Pharmacol. (2007) 85:1012–9. doi: 10.1139/y07-092
161. Xia Z, Gu J, Ansley DM, Xia F, Yu J. Antioxidant therapy with Salvia miltiorrhiza decreases plasma endothelin-1 and thromboxane B2 after cardiopulmonary bypass in patients with congenital heart disease. J Thorac Cardiovasc Surg. (2003) 126:1404–10. doi: 10.1016/s0022-5223(03)00970-x
Keywords: diabetic cardiomyopathy (DCM), the class O of Forkhead box (FoxO) transcription factors, FoxO1, FoxO3a, salvianolic acid
Citation: Han R, Huang H, Xia W, Liu J, Luo H, Tang J and Xia Z (2022) Perspectives for Forkhead box transcription factors in diabetic cardiomyopathy: Their therapeutic potential and possible effects of salvianolic acids. Front. Cardiovasc. Med. 9:951597. doi: 10.3389/fcvm.2022.951597
Received: 25 May 2022; Accepted: 27 July 2022;
Published: 11 August 2022.
Edited by:
Shinichi Oka, Rutgers, The State University of New Jersey, United StatesReviewed by:
Jianzhong An, Independent Researcher, Suzhou, ChinaCopyright © 2022 Han, Huang, Xia, Liu, Luo, Tang and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiyi Xia, d2VpeWlwaWdAZ21haWwuY29t; Zhengyuan Xia, enl4aWFAaGt1Lmhr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.