 Patrick Müller
Patrick Müller Melvin Khee-Shing Leow
Melvin Khee-Shing Leow Johannes W. Dietrich
Johannes W. Dietrich

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 15 August 2022
Sec. Cardiac Rhythmology
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.942971
This article is part of the Research TopicThyroid Hormones and Cardiac ArrhythmiaView all 12 articles
It is well established that thyroid dysfunction is linked to an increased risk of cardiovascular morbidity and mortality. The pleiotropic action of thyroid hormones strongly impacts the cardiovascular system and affects both the generation of the normal heart rhythm and arrhythmia. A meta-analysis of published evidence suggests a positive association of FT4 concentration with major adverse cardiovascular end points (MACE), but this association only partially extends to TSH. The risk for cardiovascular death is increased in both subclinical hypothyroidism and subclinical thyrotoxicosis. Several published studies found associations of TSH and FT4 concentrations, respectively, with major cardiovascular endpoints. Both reduced and elevated TSH concentrations predict the cardiovascular risk, and this association extends to TSH gradients within the reference range. Likewise, increased FT4 concentrations, but high-normal FT4 within its reference range as well, herald a poor outcome. These observations translate to a monotonic and sensitive effect of FT4 and a U-shaped relationship between TSH and cardiovascular risk. Up to now, the pathophysiological mechanism of this complex pattern of association is poorly understood. Integrating the available evidence suggests a dual etiology of elevated FT4 concentration, comprising both ensuing primary hypothyroidism and a raised set point of thyroid function, e. g. in the context of psychiatric disease, chronic stress and type 2 allostatic load. Addressing the association between thyroid homeostasis and cardiovascular diseases from a systems perspective could pave the way to new directions of research and a more personalized approach to the treatment of patients with cardiovascular risk.
Sudden cardiac death (SCD) is a global health issue that causes more than 600,000 fatalities per annum in the United States and Europe alone (1–3) being responsible for 15–20% of total mortality in industrialized societies (4–7). Therefore, the prevention of SCD continues to be a major task of cardiovascular medicine (8). Several conditions are known to be associated with increased risk of SCD including higher age, male sex, coronary artery disease (previous myocardial infarction), cardiomyopathies, primary electrical disorders, aortopathies or aortic dissection, and decreased left ventricular systolic function (7, 9). The main underlying pathologic substrate that conveys the development of ventricular tachyarrhythmias responsible for SCD involves channelopathies and/or the presence of myocardial fibrosis.
With the advent of implantable cardioverter-defibrillators (ICDs), accurate assessment of risk for SCD becomes crucial in clinical practice, the more as the majority of all first clinical events is fatal (10). Current guidelines on primary prevention for SCD recommend risk stratification solely on heart failure symptoms and reduced left ventricular ejection fraction (LVEF) (11–14). Several studies confirmed LVEF as a strong predictor of arrhythmic death (15, 16). For example, the Autonomic Tone and Reflexes After Myocardial Infarction (ATRAMI) study revealed that LVEF <35% was associated with a relative risk for cardiac mortality of 7.3 in short-term survivors of myocardial infarction (17, 18). Moreover, randomized controlled ICD trials for primary prevention in patients with reduced LVEF demonstrated improved survival within the ICD arms (19). However, using LVEF as a sole risk stratifier for SCD has several limitations. First, a relevant number of SCD continues to occur in subjects with LVEF > 40%. The Maastricht Circulatory Arrest Registry reported that more than 50% of victims of sudden circulatory arrest had LVEF > 40% (20, 21). Therefore, a significant proportion of those at risk for SCD live unnoticed when stratified by LVEF alone. Second, even in patients with LVEF <35% only a small proportion will benefit from ICD. Recent data of the DANISH trial revealed a low incidence of appropriate ICD shocks (11.5%) in patients with non-ischemic cardiomyopathy after a median follow-up of 67.6 months, whereas on the other hand the incidence of device infection (4.9%) and inappropriate shocks (5.9%) was considerable (22). Third, further developments of heart failure medications such as the angiotensin-neprilysin inhibitor combination LCZ696 (sacubitril/valsartan) may improve LVEF and minimize the risk for SCD (23–25).
Therefore, it is highly needed to identify additional markers potentially predicting a higher relative risk for SCD (26). This form of biomarkers would also open avenues to a better understanding, which are the patients that mainly benefit from ICD treatment (27).
Among prevalent, but previously underrecognized conditions, hyperthyroidism and thyrotoxicosis are known to significantly increase the risk for cardiac morbidity and mortality (28–33). Elevated concentration of free thyroxine (FT4) in overt thyrotoxicosis is an established risk factor for major cardiovascular endpoints (including cardiovascular death, hospital admission, ventricular arrhythmia and ICD therapy) (28, 30, 31). Subclinical thyroid disorders and “euthyroid” variations of FT4 within their respective reference range have been linked to cardiovascular outcome measures as well, but their mechanisms are less well understood (34, 35). Likewise, the implications of altered thyrotropin (TSH) concentration continue to be unclear (36–39). Although major cardiovascular endpoints define an important health issue, the available evidence from the literature is ambiguous, leaving important questions unsolved (29, 37).
Despite the established pro-arrhythmic role of thyroid hormones, treatment with levothyroxine may have favorable effects in heart failure (40), and both subclinical and overt hypothyroidism are established risk factors for coronary heart disease and mortality (41). The line between beneficial and harmful effects of substitution therapy, is small, however, since thyrotoxicosis and even subclinical hyperthyroidism may be independent triggers for heart failure and cardiovascular mortality (42–44). In two large population-based studies even the use of antithyroid drugs was found to be a risk factor for SCD (45), but causality remains unclear.
Until recently, the question could not be resolved if screening of subclinical and undiagnosed overt thyroid dysfunction and treatment of subclinical hypothyroidism is beneficial in adults without goiter or thyroid nodules (46). Potential reasons for this persisting vagueness include the complexity of thyroid hormone action in the cardiovascular system, age dependence of the involved effects and misconceptions about the role of thyrotropin in thyroid homeostasis, as well as insufficient sample size and heterogeneity of study designs (37, 38, 47–52).
Our systematic analysis was motivated by the hypothesis that both TSH and T4 concentrations are associated to important end points of cardiac arrhythmia, but that the specific relationships are different from a qualitative perspective. The underlying differences might arise from the dual role of both TSH and thyroid hormones, acting as controlling as well as controlled elements, from the inverse relationship between T4 and TSH in primary thyroid dysfunction and from the fact that TSH (and TRH) secretion is controlled by multiple central afferences reflecting the role of the TRH neuron as an integrator of stress signaling and energy homeostasis (53–59).
This review article is organized in four parts. In the next section we summarize known fundamental concepts including historical notes and insights from basic research. The subsequent section covers the methodology and results of a systematic search and meta-analysis of the association of mild disorders of thyroid homeostasis with major adverse cardiovascular endpoints (MACE). Toward the end of the article, we discuss the implications of the findings and try to integrate the available evidence in a comprehensive model that provides a possible explanation of all observations including previously poorly understood phenomena as well.
Thyroid hormones are key regulators of growth, differentiation and integrative energy homeostasis. They orchestrate the trade-off between–often conflicting–demands of supply with energy and substrates, ontogeny, thermoregulation and fight-and-flight reactions (56). As slow mediators of allostatic load, they represent the fourth level of the stress response after the sensorimotor and autonomic nervous system, the release of catecholamines and the secretion of glucocorticoids (56). It is therefore not surprising that their role as a switch between anabolic and catabolic functions includes the cardiovascular system as well. Cardiac complications of thyrotoxicosis were probably first observed as early as 1,785 by Caleb Hillier Parry, who portrayed a series of cases with exophthalmic goiter that was associated with palpitations and tachycardia (60), shortly later followed by similar observations made in other countries (61–63).
Today it is well recognized that a broad spectrum of heart diseases may ensue from disorders of thyroid homeostasis (31, 32, 64). Established scoring systems for the diagnosis of myxoedema coma and thyroid storm, two life-threatening thyroid conditions, comprise cardiac manifestations including brady- and tachycardia, congestive heart failure and atrial fibrillation (30).
On a molecular and cellular level pleiotropic effects of iodothyronines on gene expression, metabolism and electrophysiological mechanisms have been described (64). The effects of thyroid hormones are mediated by four different signaling types (Figure 1) (48). Type 1 of thyroid hormone action represents classical, genomic effects. Here, iodothyronines dock to thyroid hormone receptors (THR) bound to DNA as monomer, homodimer or heterodimer and modify the expression of regulated genes. THRs may also bind to RNA via a specific binding site, such that their binding and recruitment of RNA can accentuate its transcriptional activity of thyroid hormone responsive genes (70). Five types of THRs are able to both bind thyroid hormones and form active dimers, THRα1, which is the main receptor in myocardial tissue and bone, THRβ1, THRβ2 and THRβ3, which play an important role in extracardiac tissue (e.g., pituitary, liver and adipose tissue) (65) and the mitochondrial p43 receptor (71). In type 2 signaling, hormone-receptor complexes are indirectly bound to DNA via adaptor proteins. In type 3 action, iodothyronines are bound to (cytoplasmic) thyroid hormone receptors, and their effects are mediated via intracellular transmitters (second messengers), e. g. the MAPK and PI3K/AKT/mTOR pathways. The term Type 4 signaling refers to responses mediated via integrin receptors on the cell membrane (48). Of note, genomic effects (type 1 and 2 signaling) occur on a much slower time scale (hours) than non-genomic effects (type 3 and 4 signaling, minutes) (66, 72). The signaling types 1 to 3 require two iodine atoms at the inner ring of the hormone molecule, but a maximum of one iodine atom bound to the outer ring. Therefore, triiodothyronine (T3), triiodothyroacetic acid (TRIAC) and 3,5-diiodothyronine (3,5-T2) exert strong and direct genomic and non-genomic effects, whereas thyroxine (T4) and tetraiodothyroacetic acid (TETRAC) act directly via non-genomic signaling only, and only indirectly (after deiodination to T3, 3,5-T2 or TRIAC) via genomic signaling (67, 73, 74). It could be recently demonstrated that the effects of thyroid hormones on the heart rate involve type 3 signaling in a complex interaction with the autonomic nervous system (66).
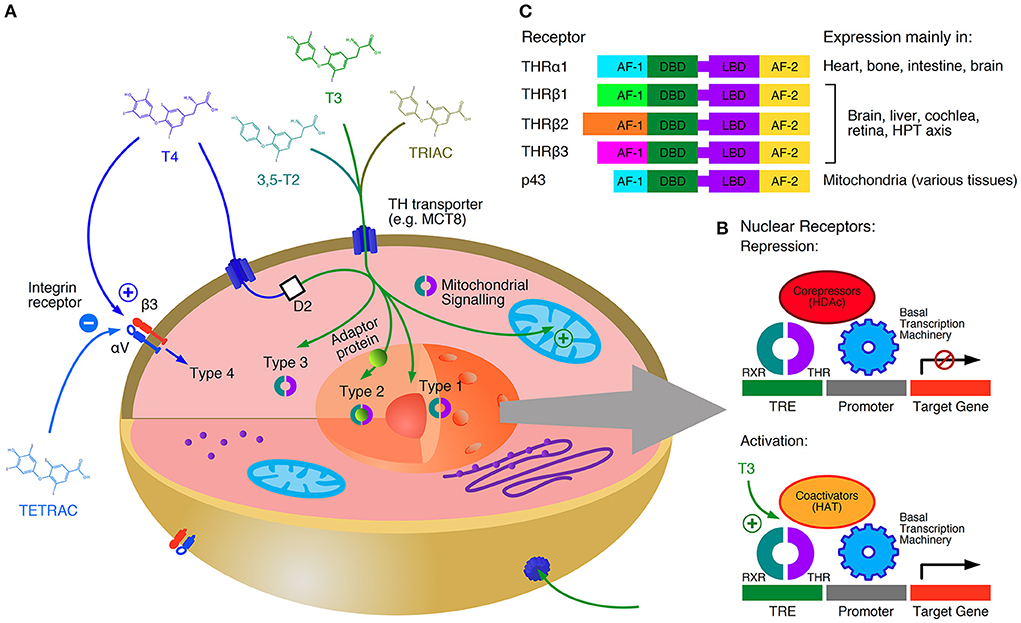
Figure 1. Types and mechanisms of thyroid hormone signaling. T4 is a prohormone with respect to genomic signaling, but a true fast-acting hormone regarding type 4 action (which is inhibited by the iodothyroacetate TETRAC). Genomic action (type 1, type 2 and mitochondrial signaling) occurs on a slow time scale, whereas non-genomic effects (type 3 and type 4 signaling) represent a fast response (A). Thyroid hormone receptors (THR) usually act as heterodimers. Without thyroid hormone bound they block the transcription together with corepressors. Iodothyronines displace the corepressors and stimulate gene expression together with coactivators (B). Tissue specific distributions of THRs further contribute to the diversity of signaling patterns in the organism (C) (48, 65–69). AF-1, activation function 1; AF-2, activation function 2; D2, type 2 deiodinase; DBD, DNA-binding domain; HAT, histone acetyl-transferase; HDAc, histone deacetylase; LBD, ligand-binding domain; RXR, retinoid X receptor; TRE, thyroid-hormone response element.
The transcription of multiple genes is stimulated by classical thyroid hormone signaling. They include alpha-myosin heavy chain, atrial natriuretic hormone, beta1- and beta-2-adrenergic receptors, sarcoplasmic reticulum Ca2+-ATPase (SERCA), Na+/K+-ATPase, and voltage-gated potassium channels (Kv1.5, Kv4.2, Kv4.3). Negatively regulated genes include adenylyl cyclase catalytic subunits, beta-myosin heavy chain, phospholamban, THRα1 and the Na+/Ca2+ exchanger. Animal experiments revealed complex remodeling of cardiac ion channel expression depending on thyroid status (75). Fast responses of ion channels result from direct modulation via non-genomic type 4 signaling (31, 64, 76).
Additional effects on cardiovascular physiology may result from extracardiac mechanisms in the cardiovascular system, e. g., increased pulmonary arterial pressure, stimulation of tissue thermogenesis and a decline of both systemic vascular resistance and diastolic blood pressure, resulting in reduced afterload (31, 49, 64, 76–79).
The mechanisms underlying arrhythmogenesis can be divided into disorders of impulse formation, disorders of impulse conduction or a combination of both (80). All of these scenarios can be critically modulated by thyroid hormones (Table 1).
Within the framework of their normal physiological function thyroid hormones have strong effects on the heart rate. Sinus bradycardia and sinus tachycardia are typical sequelae of hypothyroidism and thyrotoxicosis, respectively.
This association is well explained by modulation of the impulse formation in pacemaker cells of the sinoatrial node and (in pathological conditions) other cell types. The normal automaticity in pacemaker cells is ensured by two redundant, but intertwined loops, as suggested by Maltsev et al. (85) (Figure 2). Both an external membrane loop and an internal calcium loop, buffered by calcium storage in the sarcoplasmic reticulum, are independently able to maintain the generation of a depolarisation rhythm. They are coupled via late diastolic depolarisation and a slow L-type calcium current, thereby providing a better failure tolerance in impulse generation. Thyroid hormones can modulate both to generate a higher frequency of action potentials. The involved mechanisms include an acceleration of diastolic depolarisation (86–88), activation of potassium currents (81, 89) and synchronized loading of the sarcoplasmic reticulum with calcium via SERCA (75, 90, 91).
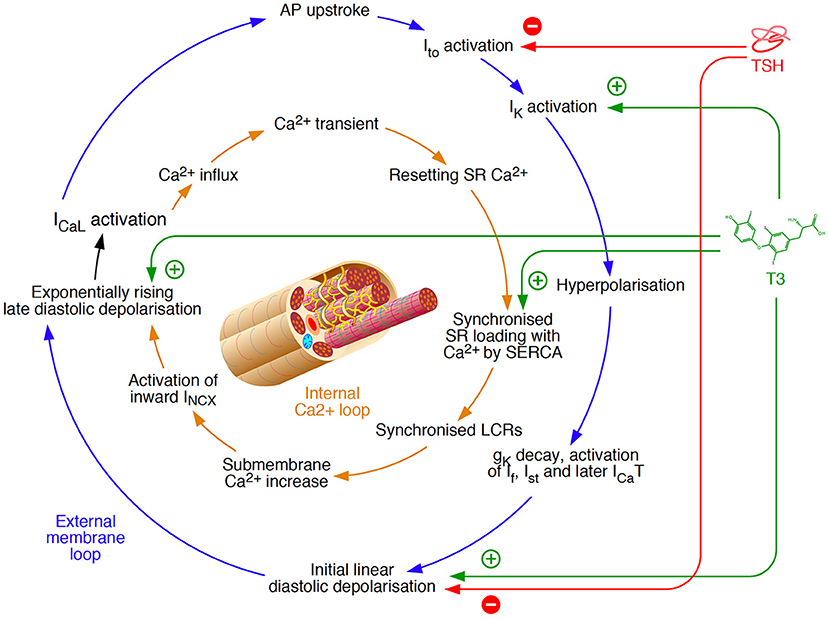
Figure 2. Mechanisms of rhythm generation in cardiomyocytes based on a model by Maltsev et al. (85). Two loops, an external membrane loop and an internal calcium loop independently ensure the generation of impulses. Therefore, they provide some redundancy, but they are also intertwined at the stage of slow L-type Ca2+ current (ICaL) activation. Thyroid hormone signaling is able to modulate both loops simultaneously via interfaces at several sites (75, 81, 86–91), and direct myocardial effects of TSH are largely opposing (92). gK, ionic conductance for K+; INCX, Na+/Ca2+ exchange current; ICaT, T-type Ca2+ current; If, hyperpolarisation-activated “funny” current; IK, voltage-gated K+ current; Ist, sustained non-selective current; Ito, transient outward potassium current; LCR, local Ca2+ release; SERCA, sarcoendoplasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum.
In addition to thyroid hormones, thyroid-stimulating hormone (TSH) is able to modulate the function of the sinoatrial node as well. Among its extrathyroidal actions are myocardial effects that are mediated via G protein-coupled TSH receptors (TSHR), which are expressed in various tissues. The resulting impact of TSH on the physiological rhythm generator is mainly of decelerating nature by inhibiting several ion currents (Figure 2) (92). Since the effects of TSH action are largely antagonistic to those of beta-adrenergic stimulation they are probably mediated by pathways other than the cAMP signaling, e.g., via inositol trisphosphate (IP3) (93, 94).
Although we write about the “normal heart rhythm” and “physiological function” here, the mentioned mechanisms may lead to substantial pathology, e. g., in cases of severe bradycardia or tachycardia in thyroid dysfunction or activation of ectopic rhythm generators. “Normality” refers to the fact that it is physiological mechanisms that are controlled by thyroid hormone signaling. This is different in the situations mentioned in the subsequent section, which ensue from a qualitative difference and represent pathological processes per se.
In addition to the modulation of physiological mechanisms, high concentrations of thyroid hormones can ignite triggers for additional scenarios of arrhythmogenesis.
Afterdepolarisations, i.e., depolarising oscillations of membrane potential after one or more preceding action potentials, are able to initiate myocardial activity. Both early (occurring before full repolarisation) and delayed afterdepolarisations (arising after completion of repolarisation) can be mediated by elevated concentrations of thyroid hormones, giving rise to triggered activity (81).
Additionally, iodothyronines shorten the effective refractory period (ERP) of cardiomyocytes in a concentration-dependent manner (95–97), and they reduce the conduction velocity (θ), e.g. by downregulating the expression of connexin 43, a key component of gap junctions (Figure 3) (75, 96–98). As a result, the wavelength of excitation (λ), which is defined by the product of ERP and θ, is considerably diminished, so that it may become shorter than the dimensions of potential re-entry circuits, e.g., in the vicinity of scars (99) (Figure 4). Consequently, an electrical impulse traversing a cardiac fiber bundle is slowed down and may, provided electrical irregularities are present in the tissue, meet excitable fibers after the refractory period has passed. Therefore, the combination of both mechanisms permits the evolution of re-entry or spiral wave processes (100).
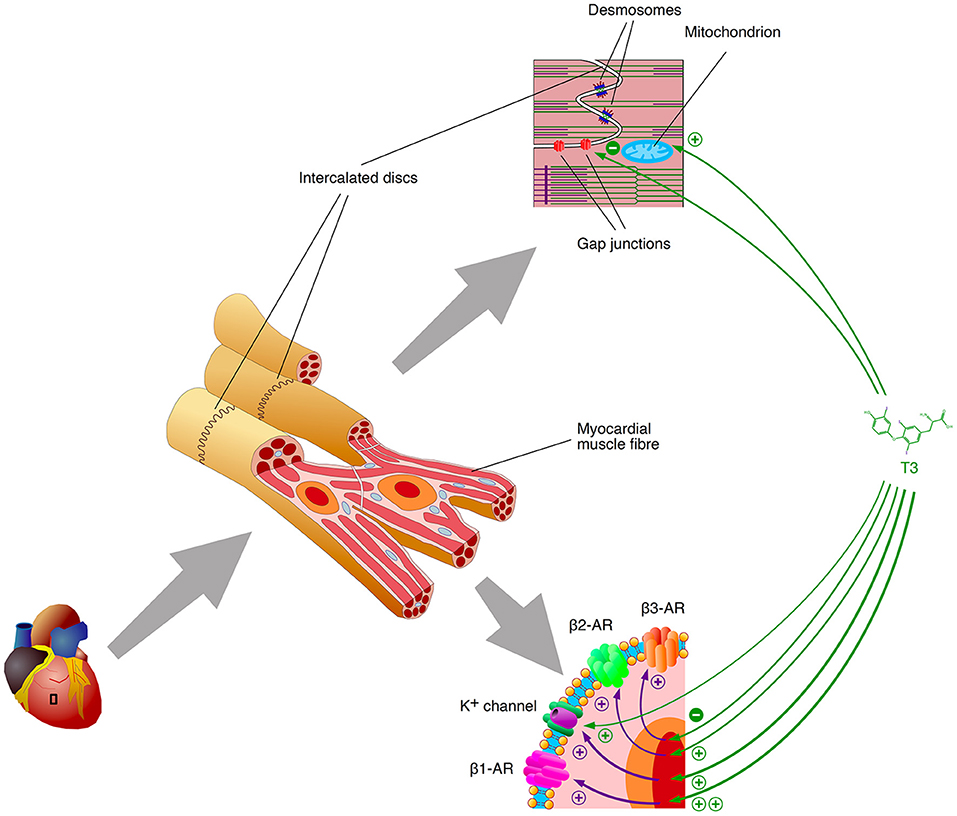
Figure 3. Selected mechanisms of arrhythmogenesis by thyroid hormones. T3 (and other active thyroid hormones) upregulate the gene expression of beta1 and beta2 adrenoceptors via classical genomic signaling, but downregulate protective beta3 adrenoceptor expression. The transcription of critical genes for the formation of gap junctions is downregulated as well. Potassium channels are regulated via classical type 1 signaling and via non-genomic effects (type 4 action) as well. Purple arrows indicate the effects of gene expression (transcription, translation and associated processing steps), green arrows visualize the impact of T3-agonistic thyroid hormones.
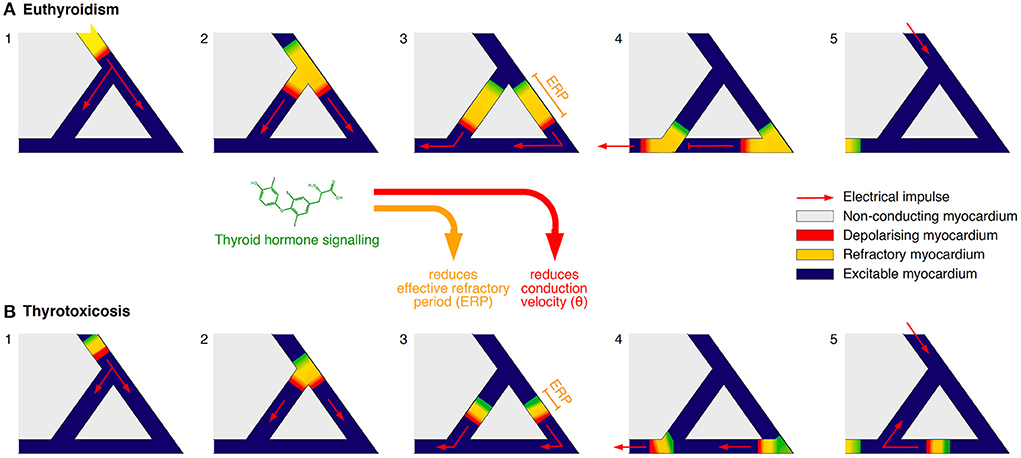
Figure 4. (A,B) Simplified model of the effects of thyroid hormone signaling facilitating disorders of impulse conduction. Iodothyronine action reduces both the effective refractory period (ERP) and the conduction velocity (θ). As a consequence, the wavelength of excitation λ = ERP x θ may get shorter than the dimensions of a potential re-entry circuit, thus giving rise to re-entrant tachycardia (99).
The probability for successful action potential propagation depends on the safety factor for conduction, i.e., the ratio of the energy for normal conduction to the minimum propagating energy (80). Impaired function of gap junctions may lead to decremental conduction, where an impulse with a low safety factor loses activation effectiveness along with its propagation. This mechanism may cause rare paradoxical bradycardia or heart blocks in thyrotoxicosis (82, 83).
The term of “subclinical” thyroid diseases continues to be debated. It is unclear if this class of diseases is a proper representation of physiological processes (101–104), and the implications with respect to meaningful endpoints are unclear (105, 106). Therefore, useful cut-off values serving as a threshold for initiating therapy, could not be established (35). Recently, a more detailed grading system has been suggested, including FT3 and different zones of TSH concentration (107). It may be beneficial for a better classification of primary thyroid dysfunction.
This new classification cannot, however, address the dual role of TSH as both a regulator and indicator of thyroid function (53, 102, 103, 108, 109). Deviations of thyrotropin concentration may indicate ensuing or manifest primary thyroid disorders, but pituitary disease and variations in the central hypothalamic set point as well (37, 47). This physiological heterogeneity may explain why thyroid hormones are better predictors of clinical outcome measures than TSH concentration (38).
Furthermore, universal reference ranges for thyrotropin and thyroid hormones have recently been more and more questioned, since the intraindividual variation of TSH and thyroid hormone concentration is smaller than the interindividual variation (110, 111). This non-ergodicity of laboratory results reflects an individual set point of the homeostatic systems, and deviations from this setpoint may from a personalized perspective be of higher importance than the position of laboratory results with respect to population-derived reference ranges (37, 112–116).
To compile the most comprehensive list of pertinent publications electronic literature searches in English, German and Mandarin were performed in the PubMed and Web of Science databases for relevant publications up to December 2021, evaluating a potential connection between minimal disorders of thyroid function and sudden cardiac death or malignant arrhythmia. The following query formula was used: “[(thyroid function) OR (free t4)] AND [(sudden cardiac death) OR (ventricular fibrillation) OR (ventricular tachycardia)]”. Our search was limited to studies that were performed in humans and investigated the thyroid function in relation to sudden cardiac death, a combination of major endpoints including cardiovascular death (CVD) or the result of continuous recording via an implanted device (pacemaker, ICD or event recorder).
Among the larger set of identified studies, the meta-analysis subproject was restricted to longitudinal investigations that reported quantitative information on thyroid homeostasis and a hazard or odds ratio for one of the mentioned cardiovascular endpoints. If adjusted and unadjusted analyses with the same outcome measures and thyroid-related predictors were reported in one paper a fully adjusted multivariable analysis was selected for further evaluation.
Exclusion criteria were case reports, animal or cell culture experiments, therapeutic trials, studies on non-thyroidal illness (TACITUS) syndrome, studies on amiodarone effects, surveys, review articles or correspondence without original data. After removing duplicates, three authors (P.M, M.K.L and J.W.D.) screened all found studies for eligibility by abstract screening and full-text reviewing. From the included publications quantitative data, including sample size, mean and standard error (or 95% confidence interval) of hormone concentrations, hazard ratio or odds ratio for cardiovascular endpoints and type of study (prospective or retrospective) were extracted.
In order to address a potentially low number of studies and to reduce the effects of bias arising from heterogeneities in MACE definitions (117–119), we used a hierarchical approach, where we analyzed the association of thyroid function separately with SCD, CVD, study-specific MACE (as defined by the authors of included publications) and inclusive MACE (the union set of MACE and cardiovascular death).
For studies reporting FT4 concentration in other units of measurement than pmol/L, HRs were converted with
Likewise, if TSH concentration was reported in logarithmic presentation, the HR was adjusted with
In studies that reported HRs for increases of TSH and FT4 in standard deviations adjustments were performed with
The quality of included studies was assessed with the Newcastle-Ottawa score (NOS). In order to control for small-study effects leading to potential bias, funnel plots were drawn for analyses with five or more studies. Quantitative papers were pooled and summary measures were included in random-effects meta-analysis. The between-study variance was assessed with the DerSimonian-Laird estimator, Cochran's Q and tau squared, and heterogeneity with Higgins' and Thompson's I2. Calculations were performed with custom S scripts for the statistical environment R (version 4.1.1 for macOS) and supported by the R packages meta and metaphor.
The protocol for the meta-analysis has been registered in the PROSPERO International prospective register of systematic reviews with the ID CRD42022311340 and is available from https://www.crd.york.ac.uk/prospero/display_record.php?ID=CRD42022311340.
Via the described search strategy and other sources, we identified 32 eligible publications that were included in the systematic review (Figure 5). From these, 18 studies reported an association of thyroid homeostasis to the hazard ratio of SCD, CVD, ICD therapy or major cardiovascular events comprising one of these outcome measures. These studies could be included in the meta-analysis. See Supplementary figure 1 for the numbers of included studies with respect to different outcome measures.
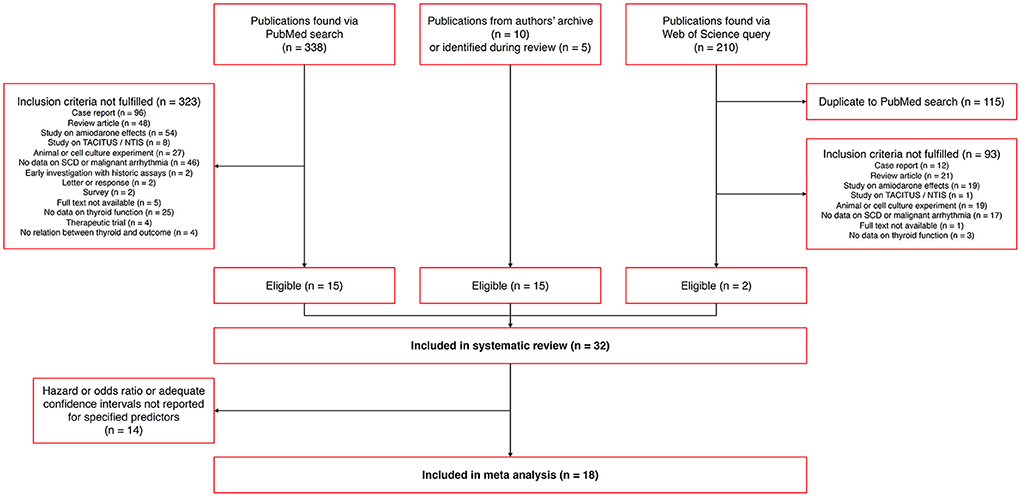
Figure 5. Flowchart of identified, eligible and included publications.
Seven studies, including more than 1.2 million subjects, observed an association of reduced TSH concentration with major cardiovascular endpoints (Table 2). A very large register-based study including more than half a million citizens of Copenhagen found reduced TSH concentration to be associated with MACE, heart failure and all-cause mortality (42). In another large cohort study with elderly community-dwelling individuals diminished TSH levels were associated with a higher incidence of all-cause death (121). In a similar study with subjects aged 85 years or older reduced TSH concentration predicted both all-cause and cardiovascular mortality (120). A prospective multicentre cohort study observed a suppressed TSH concentration to be associated with higher cardiovascular mortality and risk of stroke. However, after multivariable association it was associated with stroke only (123). Another multicentre cohort study observed increased all-cause mortality in subjects with systolic heart failure and reduced TSH concentration, but the association vanished in an adjusted model (124). A study with 1 000 diabetic patients on haemodialysis found a reduced TSH level to be associated with a doubled risk of SCD, and this effect was preserved in an adjusted model (126).
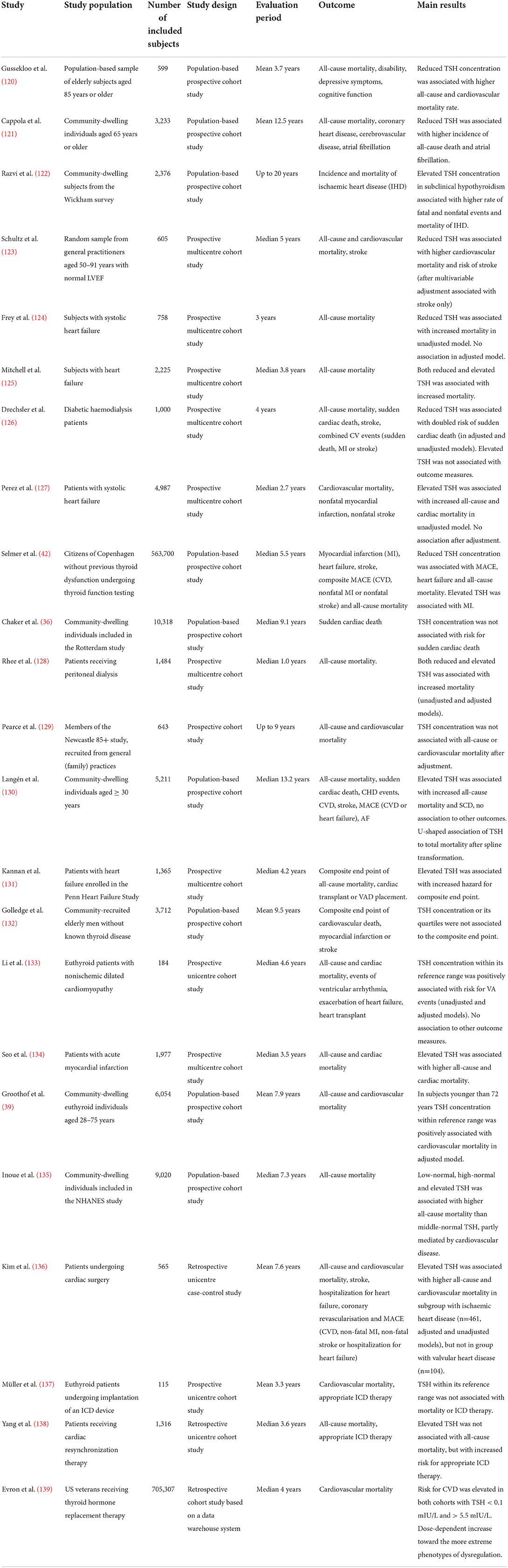
Table 2. Association between TSH concentration and major cardiovascular outcome measures.
Elevated TSH levels were identified in seven studies as risk factors for MACE as well. In sum, these studies comprised about 720 000 subjects. In the community-based Wickham survey elevated TSH concentration predicted a higher mortality of ischaemic heart disease and an elevated rate of fatal and nonfatal events (122). A study including subjects with systolic heart failure found TSH elevations to be associated with increased all-cause and cardiovascular mortality, but this association was lost after adjustment (127). Similar associations were observed in subjects with myocardial infarction and community-dwelling adults aged 30 years and older (130, 134). In subjects with heart failure elevated TSH concentration was associated with increased hazard ratio for a composite end point (131). Patients undergoing cardiac surgery with ischaemic heart disease demonstrated elevated TSH concentration to be associated with higher all-cause and cardiovascular mortality (136).
A small study in subjects with cardiomyopathy found TSH concentration within its reference range to be positively associated with risk for ventricular arrhythmia events in both adjusted and unadjusted models (133). The cardiovascular mortality was also associated to within-reference TSH in the PREVEND study covering community dwelling subjects younger than 72 years (39).
Additionally, several studies did not identify a relation between TSH concentration and major cardiovascular end points. These studies included in sum about 15 000 subjects. In the large community-based Rotterdam study, the TSH concentration did not predict the risk for SCD (36). Likewise, the Health in Men Study did not observe an association of TSH concentration or its quartiles to a composite end point in more than 3 000 elderly men (132). TSH concentration was not associated with all-cause or cardiovascular mortality in the Newcastle 85+ study as well. This study included more than 600 elderly subjects (129). Two studies including subjects undergoing device implantation did not find an association of TSH levels with all-cause mortality (137, 138), but one of these studies observed elevated TSH to predict appropriate ICD therapy (138).
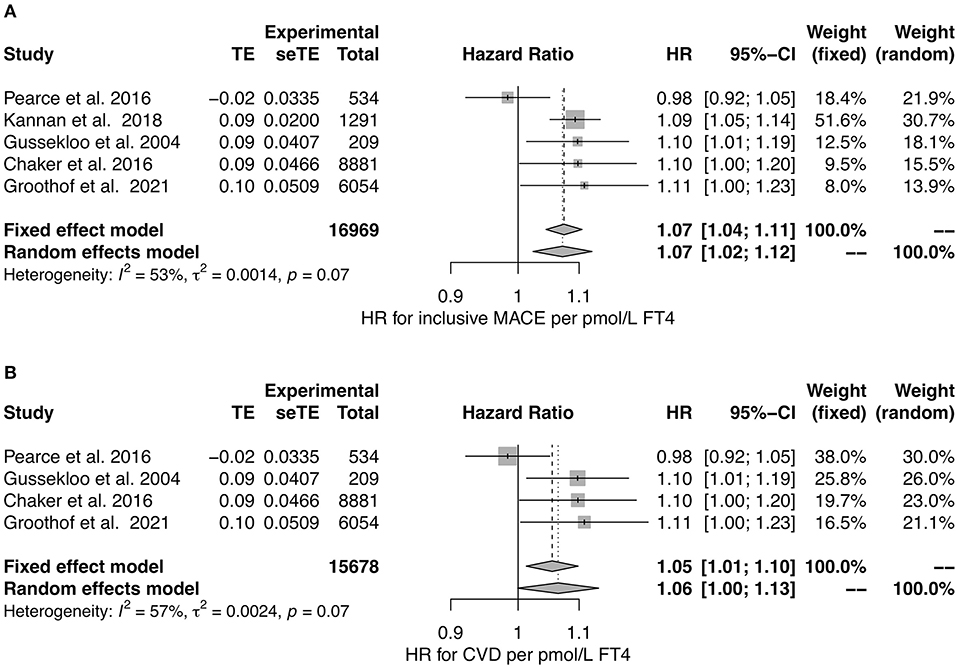
In a meta-analysis of studies reporting hazard ratios (36, 39, 120, 129, 131), we observed no association of TSH concentration to MACE in random effect models, but a significant negative association in both fixed and random effects models with CVD as end point (Figure 6). However, the number of eligible studies was small for CVD and the unexplained heterogeneity was substantial for MACE, as demonstrated by an I2 value of 75%.
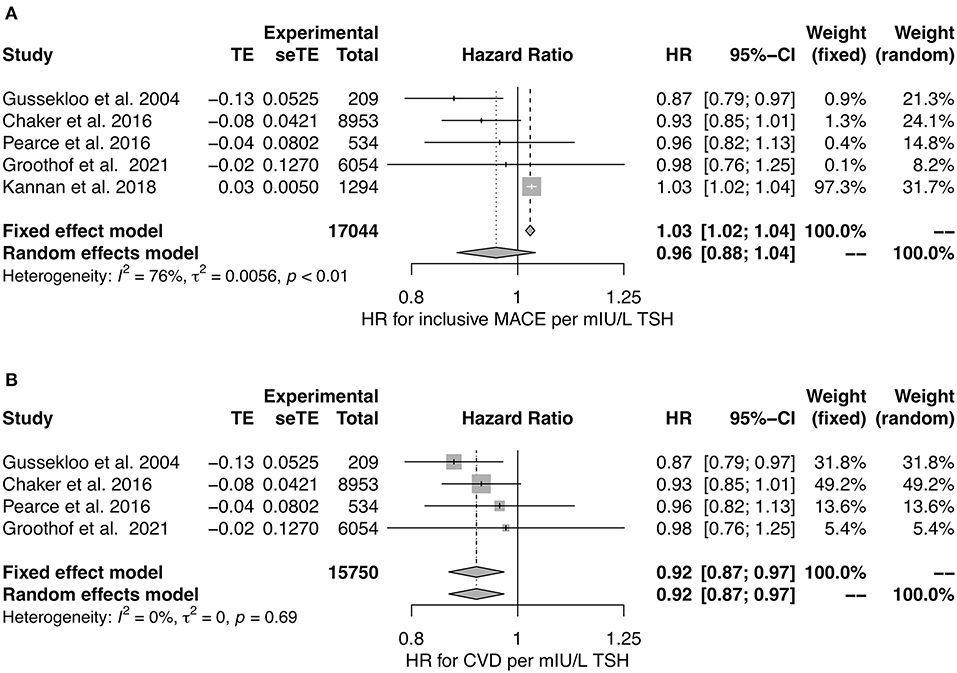
Figure 6. Hazard ratio for major cardiovascular events (A) and cardiovascular death (B) in relation to TSH concentration (36, 39, 120, 129, 131).
U-shaped relationships between TSH and end points were described in 5 studies covering more than 720 000 subjects. In patients with heart failure both reduced and elevated TSH predicted increased mortality (125). Another study with patients receiving peritoneal dialysis observed both reduced and elevated TSH to be associated with increased mortality (128). This applied to unadjusted and adjusted models. After spline transformation, a study with community-dwelling individuals found a U-shaped association of TSH to total mortality (130). Similar observations were made in another population-based study that found low-normal, high-normal and elevated TSH to be associated with higher all-cause mortality than middle-normal TSH (135). This effect was partly mediated by cardiovascular disease. In the large population-based study by Selmer et al. both reduced and elevated TSH concentration was associated to cardiovascular endpoints (42). However, high TSH concentration predicted myocardial infarction only, whereas low TSH concentration was associated to MACE (CVD, non-fatal MI and non-fatal stroke), heart failure and all-cause mortality. An even larger data warehouse-based study by Evron et al. found a strong association of both reduced and elevated TSH concentration with cardiovascular mortality (139) in adults receiving thyroid hormone replacement therapy. In this study the increase in the adjusted hazard ratio was concentration-dependent, with a positive gradient toward both lower and higher TSH concentrations.
Elevated FT4 concentrations were associated with major endpoints in seven studies comprising in sum more than 720 000 subjects (Table 3). A positive association to the risk of SCD was observed in large community-based studies (36, 39, 120, 132). Two of these studies also observed FT4 concentration to be related to all-cause mortality (39, 120). The FT4 concentration predicted the hazard for a composite endpoint in subjects with heart failure (131). In a small study the FT4 concentration predicted the risk for appropriate ICD therapy and the hazard for ICD therapy-free survival (137). Two of the described studies also saw a positive association of within-reference FT4 concentration to major endpoints (36, 137). In a large data warehouse-based study including US veterans aged 18 or older on levothyroxine replacement therapy, both reduced and elevated FT4 concentrations were associated with the risk of cardiovascular mortality (139).
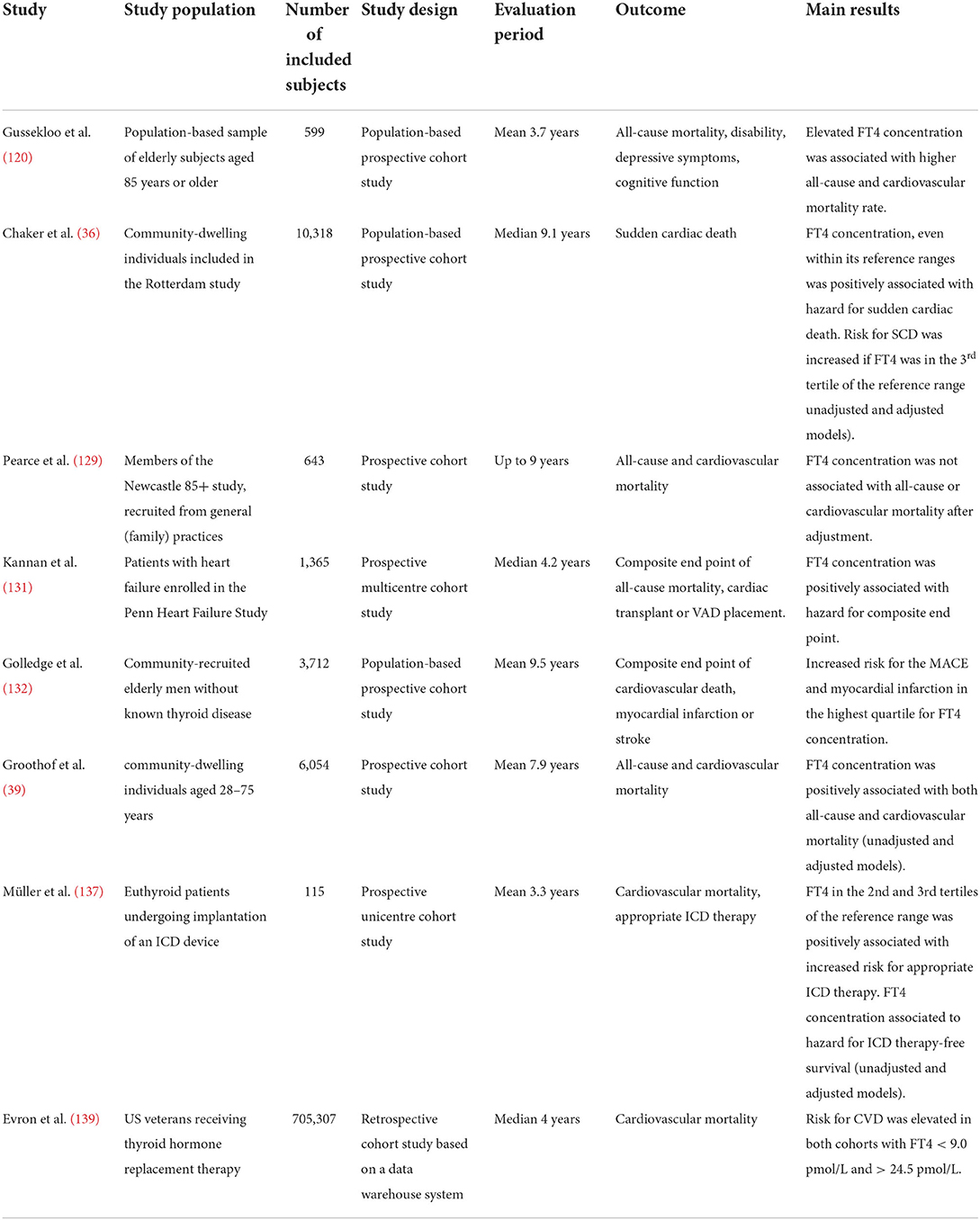
Table 3. Association between FT4 concentration and major cardiovascular outcome measures.
After adjustment, the Newcastle 85+ study did not find an association of FT4 concentration with all-cause or cardiovascular mortality. This study had a comparably small sample size of 643 elderly subjects (129).
Including studies reporting hazard ratios in a meta-analysis (36, 39, 120, 129, 131), we observed a significant association of FT4 concentration to MACE and a potential association to CVD (Figure 7). The heterogeneity between studies was moderate for both MACE and CVD with I2 values being 53 and 57%, respectively. Therefore, random effects models may be preferable over fixed-effects models to draw conclusions.
The heterogeneity between studies was moderate to substantial in the meta-analysis for subclinical hypothyroidism (42, 121, 123, 126, 130, 131, 136, 140–143). Random effects models reveal an association to inclusive MACE, but not to study-specific MACE (Figures 8A,B). Both fixed effects and random effects models confirm subclinical hypothyroidism to predict CVD (Figure 8C).
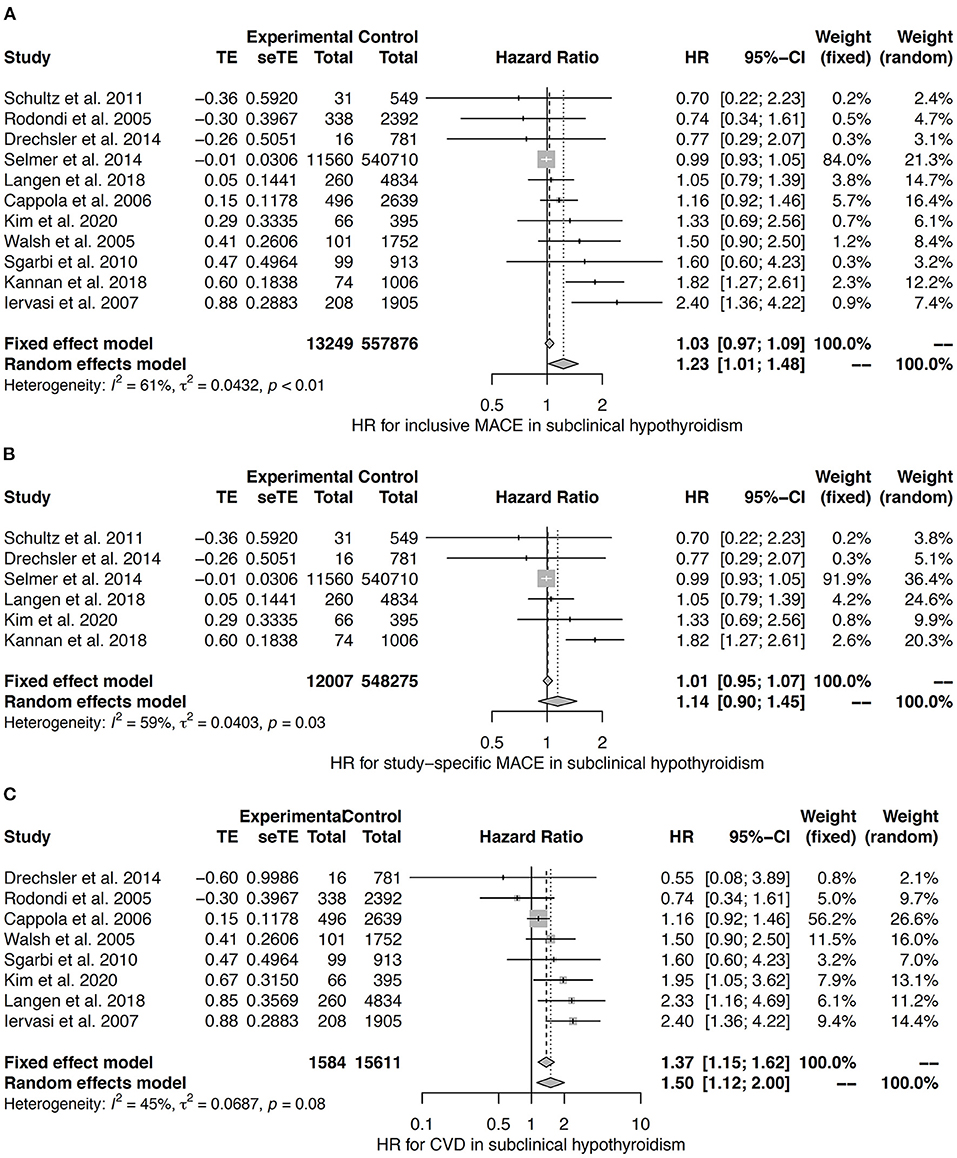
Figure 8. HR for inclusive MACE (A), study-specific MACE (B), and CVD (C) in subclinical hypothyroidism (42, 121, 123, 126, 130, 131, 136, 140–143).
In the meta-analyses for subclinical hyperthyroidism the heterogeneity was very low (study-specific MACE) or moderate (MACE and CVD) (42, 121, 123, 126, 140, 142–144). The fixed effects model suggests a small effect with a hazard ratio of 1.10 with respect to both inclusive MACE and study-specific MACE (Figures 9A,B). An association to CVD was confirmed by both fixed effects and random effects models (Figure 9C).
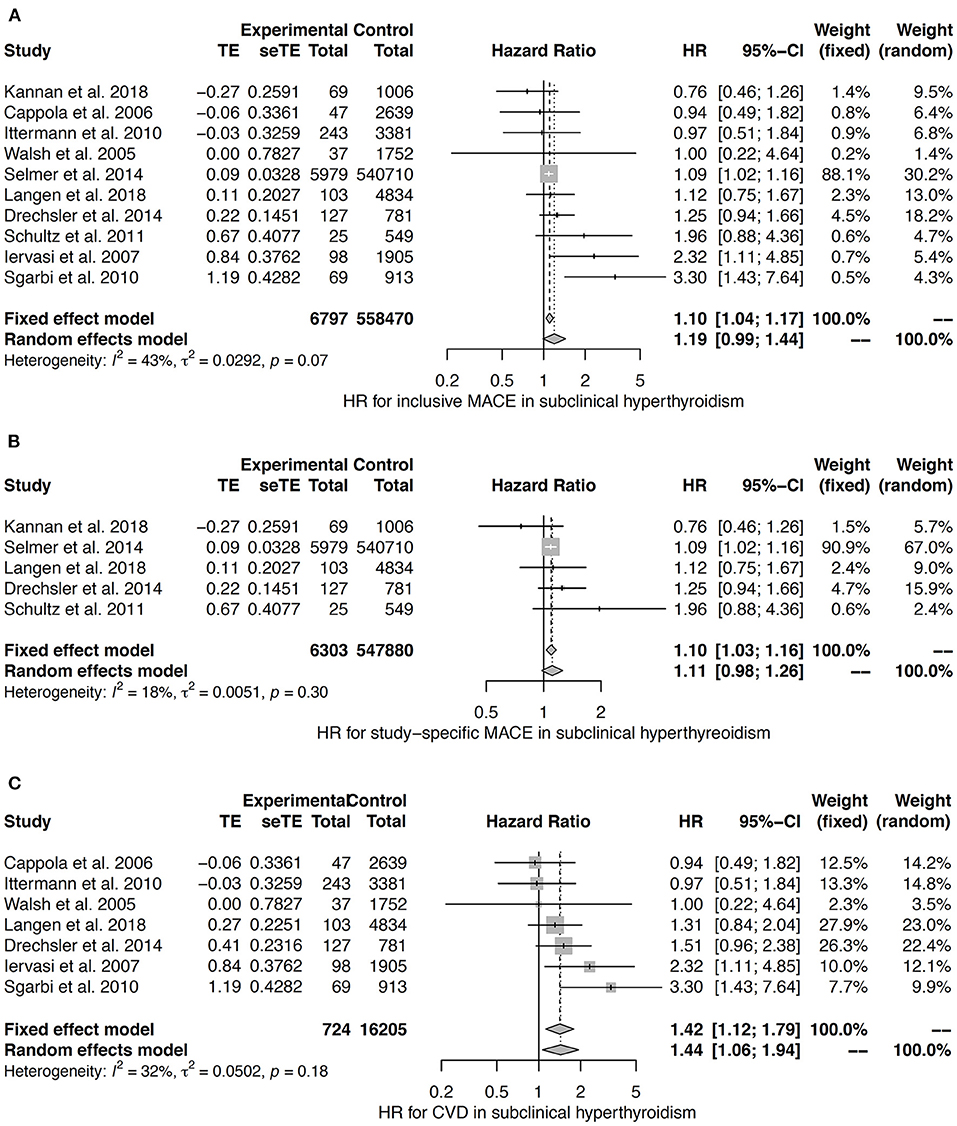
Figure 9. HR for MACE (A,B) and CVD (C) in subclinical hyperthyroidism (42, 121, 123, 126, 140, 142–144).
The studies included in meta-analyses had a mean Newcastle-Ottawa score of 7.7, suggesting a sufficient quality of the available evidence. Funnel plots did not signify considerable bias, but support the assumption of heterogeneity among included studies. Detailed information is provided in the Supplementary material.
Both the generation of normal cardiac automaticity and the pathogenesis of arrhythmia are critically dependent on thyroid hormone signaling. Basic research identified multiple mechanisms linking thyroid hormone action to the formation of the normal and pathological heart rhythm. On a molecular level, classical thyroid hormone signaling (type 1 action) modulates the expression of multiple genes involved in the generation and conduction of excitation. On the level of electrophysiological processes, critical components of the rhythm-generating loops are sensitive to active iodothyronines. Interestingly, TSH has fundamentally opposing effects on generator loops, probably mediated via IP3 signaling.
Non-canonical thyroid hormone signaling has been linked to heart rate as well. The detailed mechanisms are not well understood up to now.
Although thyroid hormones increase the heart rate, they slow down the velocity of impulse conduction (θ), mainly due to down-regulation of critical components of gap junctions. Together with a shortened effective refractory period (ERP), this may lead to re-entrant tachycardia, if the wavelength of excitation
gets shorter than the length of potential anatomical re-entry circles. Necessary for this mechanism to come into effect is some heterogeneity in the myocardial tissue, e.g., due to the existence of scars forming the center of latent re-entry circles and to some electrophysiological variety of conducting tissue. This may explain why the probability of thyrogenic arrhythmia increases with age and in subjects with pre-existing cardiac disease.
From a clinical perspective, a relation of thyroid function to mortality is well established. Both the described physiological mechanisms and several prospective studies suggest this association to be mediated by arrhythmia.
With our search strategy we identified 32 publications investigating a potential relation between minimal deviations of thyroid function and major cardiovascular end points. Several studies with large sample sizes found reduced and/or elevated TSH concentration to predict cardiovascular or all-cause mortality, or composite MACE endpoints. Two studies described even a positive correlation of the TSH concentration within its reference range with cardiovascular events. However, several studies, even with large sample size, did not find an association of TSH concentration to major outcome measures. Our meta-analyses identified a negative relation of TSH to hazard ratios of CVD, but no clear association to MACE.
The situation was clearer for FT4 concentration. Six studies with a large cumulative sample size found a significant positive association between FT4 and cardiovascular events, including composite endpoints, mortality and appropriate ICD therapy. In two studies this association was also present if FT4 concentration was restricted to its reference interval. Only one study described both reduced and elevated FT4 concentrations to be associated to CVD (139), but this study included subjects on levothyroxine replacement therapy and may, therefore, represent slightly different pathophysiological mechanisms. Via meta-analysis we found a positive association of FT4 to MACE and a tendency for CVD.
The dissimilarity between these results, with a strong monotone prediction model for FT4, but a much less clear association of TSH to major endpoints, may be traced back to several causes. One may be the considerable heterogeneity of the results in the included studies, so that less powerful random effects models had to be preferred to fixed effects models, especially for the association of TSH concentration to MACE. Another explanation may be a U-shaped relation between TSH concentration and the risk for cardiovascular events (145). This form of nonlinear interaction was revealed by four studies that found both reduced and elevated TSH to predict mortality. Studies with negative results may have missed this complex association due to the use of over-simplified statistical models. It is corroborated, however, by our meta-analyses revealing both subclinical hypothyroidism and hyperthyroidism to predict cardiovascular mortality (Figures 8C, 9C).
Our results are consistent with the outcomes of previous studies and meta-analyses (146). However, a U-shaped link between TSH and cardiovascular risk is not straightforward to be explained from a physiological perspective. A recent study described very similar results for the link between thyroid function to Takotsubo syndrome, and the pathogenesis was explained by nosological heterogeneity, where two different scenarios overlapped (147). These considerations may be extended to cardiovascular endpoints in general (Figure 10). A dyshomeostatic type of thyrogenic arrhythmia may result from primary hyperthyroidism and other forms of non-central thyrotoxicosis. Here, the TSH secretion responds inversely to rising FT4 concentrations, so that FT4 is positively, but TSH negatively associated to the event rate (53, 148, 149). In an allostatic type of thyro-cardiac linkage, however, TSH concentrations rise with acute stress levels and long-term type 2 allostatic load, with consecutive, centrally-mediated, increasing T4 secretion (47, 56, 147). This case reflects a raised set-point of thyroid homeostasis. Therefore, both TSH and FT4 are positively associated to mortality and other major endpoints. Taken together, the blending of both types in population-based studies yields a U-shaped association between TSH concentration and the hazard of cardiovascular events, but a simpler monotonic prediction model for FT4 (Figure 10).
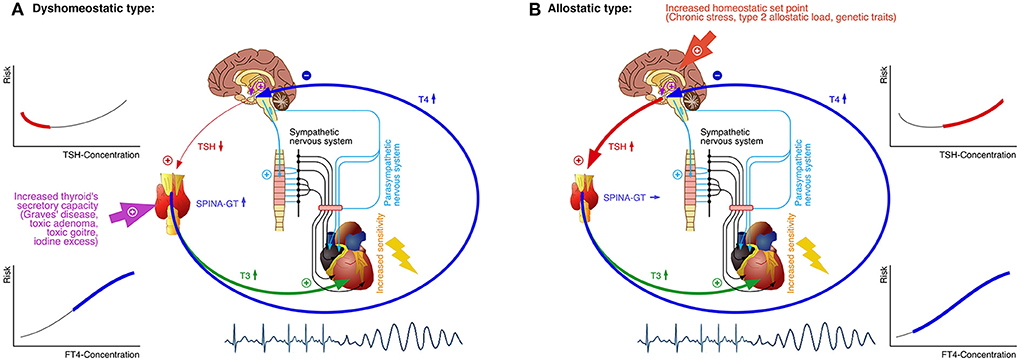
Figure 10. An integrated model for the association between thyroid homeostasis and major cardiovascular events. In the dyshomeostatic type of thyrogenic arrhythmia elevated FT4 concentration, caused by primary thyrotoxicosis, increases the risk for severe arrhythmia as a major cause for cardiovascular mortality. The TSH concentration is reduced in this case, represented by the left, declining, branch of the U-shaped relation between TSH level and risk (A). In the allostatic type, mainly caused by type 2 allostatic load and genetic traits, the set point of thyroid homeostasis is raised, resulting in increased TSH and FT4 concentration and subsequently elevated risk for arrhythmia. This situation is mirrored in the right, rising, branch of the relation between TSH concentration and cardiovascular risk (B). In any case, elevated concentrations of T3, T4 and other T3-agonistic thyroid hormones increase the sensitivity to catecholamines and sympathetic signaling (via upregulated expression of beta1 and beta2 adrenoceptors), thereby contributing to reduced stress tolerance. SPINA-GT, “gain of thyroid,” i.e., thyroid's secretory capacity (150).
Multiple studies, reviews and meta-analyses described reduced T3 concentrations and other phenotypes of non-thyroidal illness syndrome (representing a type 1 allostatic response being also known as euthyroid sick syndrome or TACITUS—thyroid allostasis in critical illness, tumors, uraemia and starvation) to be linked to cardiovascular and general mortality and arrhythmia as well (56, 78, 151–153). Up to now, it is still not established whether low-T3 syndrome represents an adaptive response, a maladaptive mechanism or a combination of both (79). We did not address this topic in detail here and even excluded studies on TACITUS from the systematic review part, since this association may reflect common causes of mortality rather than a direct effect of low-T3 syndrome on cardiac electrophysiology. However, some non-classical thyroid hormones, especially 3,5-T2, which is thyromimetic and upregulated in critical illness, may increase both heart rate and risk for arrhythmia (152, 154).
Type 1 allostatic response in heart failure may to a certain extent limit our conclusions since FT4 concentrations could rise if the pathway via deiodinases to T3 formation is down-regulated (56). Decreased deiodinase activity may result from critical illness, but also from therapy with certain drugs, including amiodarone, beta-blockers and glucocorticoids (155). Additionally, the free fraction of thyroid hormones may increase due to drug interference with plasma protein binding, which may originate, e.g., from the use of furosemide, aspirin, heparin or other substances (156–158). Therefore, elevated FT4 concentration might as well partly reflect therapeutic decisions in subjects with cardiac failure. It is, however, unlikely that major bias arises from this fact since most of the described effects are rapidly settled by the hypothalamus-pituitary-thyroid feedback loop and, therefore, of temporary nature, whereas the follow-up time in most of the included studies extended to several years (55, 159).
The selection criteria for the systematic review may have led to the under-representation of studies reporting the risk in alternative form, e.g., in form of quantiles. This potential bias may have been mitigated by the fact that we included publications from other sources, e. g., own archives. Unfortunately, it was impossible to sensibly combine quantile-based studies in a meta-analysis, since the numbers of quantiles (tertiles, quartiles and quintiles were used) differed among the publications. Qualitative analysis suggests, however, an association of FT4 in the highest tertiles or quartiles to cardiovascular risk.
In summary, basic research and physiological studies suggest a strong link of thyroid hormones to cardiac rhythm generation and the pathogenesis of arrhythmia. The clinical evidence suggests a monotonic and unambiguous relation of FT4 concentration to arrhythmia, which seems to be one of the most important mediators of major cardiovascular endpoints. The relationship of TSH levels to arrhythmia, mortality and other outcome measures is less clear and best explained by a U-shaped link, which may reflect the overlap of two different scenarios, a dyshomeostatic type of thyrogenic arrhythmia resulting from ensuing primary thyrotoxicosis, and an allostatic response with an increased set point of the feedback loop. This hypothesis may serve as a starting point for future targeted research projects.
Variations of thyroid function, even the slightest forms within the respective reference ranges of TSH and thyroid hormones, are strong predictors of major cardiovascular endpoints. However, the nature of this relation is more complex than previously assumed.
In addition to the well-established effects of primary thyrotoxicosis, central mechanisms of the hypothalamus-pituitary-thyroid (HPT) axis may play an important, but previously under recognized, role as well. This class of diseases includes secondary and tertiary hyperthyroidism in pituitary-related and hypothalamic disorders and in central resistance to thyroid hormone (RTH). These conditions are rare diseases, however, with respective prevalence of about 1 in 100 000 subjects. Much more common are adaptive responses representing type 2 allostatic load (160). Examples include post-traumatic stress disorder (PTSD), certain psychiatric diseases and long-term effects of social disparity. It could be demonstrated that conditions associated to type 2 allostatic load involve an elevated set point of the HPT axis and that they convey an increased risk for major cardiovascular events (5, 47, 56, 147, 161–163). The heterogeneity of pathophysiological mechanisms provides a plausible explanation for the U-shaped relation between TSH concentration and cardiovascular risk.
The new understanding of the dual etiology of the thyro-cardiac link has important therapeutic implications. First of all, the threshold for the treatment of thyrotoxicosis may be in future adjusted to include subclinical hyperthyroidism (SH) as well. This decision may be supported by results of a meta-analysis revealing a 24% increased risk of overall mortality in SH (164). However, the potentially beneficial effects of a more intensive correction of low-grade thyrotoxicosis have to be balanced against the risks resulting from treatment with thyrostatic agents and from definitive therapy (surgery or radioiodine treatment). The European Thyroid Association recommended treatment of SH in subjects of 65 years or older and in younger persons with concomitant cardiovascular disease (165).
The situation is more straightforward regarding the treatment of hypothyroidism. Here, it may prove advantageous if dosage titration algorithms for levothyroxine approach the target from below and prevent the free T4 concentration from entering the zone of the highest quartile of its reference range (166). This consideration also applies to the secondary prophylaxis of differentiated thyroid cancer (DTC). Accordingly, recent guidelines have also addressed cardiovascular concerns when overturning the previous recommendation of universal TSH suppression in low- and intermediate-risk DTC (167).
Treatment of type 2 allostatic load with a subsequently elevated set point of the HPT axis should address its origins. Sensible starting points include psychosocial support and lifestyle interventions (168–172). Early addressing of socioeconomic disparity and environmental factors may provide the basis for primary prevention of cardiovascular mortality (173, 174), and this may in part be due to a readjustment of thyroid homeostasis toward a healthier phenotype.
Where possible, future studies on the association of thyroid function with major cardiovascular endpoints should be based on validated criteria, including standardized MACE sets and uniform quantile definitions, in order to support replication, comparison and aggregation of results (118).
PM, JD, and ML defined the selection criteria and screened all found studies for eligibility by abstract screening and full-text reviewing. JD maintained the study table and performed the meta-analyses. JD and PM drafted a first version of the manuscript. ML edited the text and contributed additional ideas, material, and text passages. All authors contributed to the article and approved the submitted version.
Publication fees were defrayed via the Open Access Publication Funds of the Ruhr-Universität Bochum.
We acknowledge support by the Open Access Publication Funds of the Ruhr-Universität Bochum. The authors also want to thank Dr. Dion Groothof, University of Groningen, for providing important details on the results of the PREVEND study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.942971/full#supplementary-material
1. Jean-Philippe E, Lerner I, Valentin E, Folke F, Bottiger B, Gislason G, et al. Incidence of sudden cardiac death in the European Union. J Am Coll Cardiol. (2022) 79:1818–27. doi: 10.1016/j.jacc.2022.02.041
2. Kumar A, Avishay DM, Jones CR, Shaikh JD, Kaur R, Aljadah M, et al. Sudden cardiac death: epidemiology, pathogenesis and management. Rev Cardiovasc Med. (2021) 22:147–58. doi: 10.31083/j.rcm.2021.01.207
3. Rogers AJ, Selvalingam A, Alhusseini MI, Krummen DE, Corrado C, Abuzaid F, et al. Machine learned cellular phenotypes in cardiomyopathy predict sudden death. Circ Res. (2021) 128:172–84. doi: 10.1161/CIRCRESAHA.120.317345
4. Albert CM, Chae CU, Grodstein F, Rose LM, Rexrode KM, Ruskin JN, et al. Prospective study of sudden cardiac death among women in the United States. Circulation. (2003) 107:2096–101. doi: 10.1161/01.CIR.0000065223.21530.11
5. Batelaan NM, Seldenrijk A, van den Heuvel OA, van Balkom A, Kaiser A, Reneman L, et al. Anxiety, mental stress, and sudden cardiac arrest: epidemiology, possible mechanisms and future research. Front Psychiatry. (2021) 12:813518. doi: 10.3389/fpsyt.2021.813518
6. Gräsner JT, Lefering R, Koster RW, Masterson S, Bottiger BW, Herlitz J, et al. EuReCa ONE-27 Nations, ONE Europe, ONE Registry: A prospective one month analysis of out-of-hospital cardiac arrest outcomes in 27 countries in Europe. Resuscitation. (2016) 105:188–95. doi: 10.1016/j.resuscitation.2016.10.001
7. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. (2015) 116:1887–906. doi: 10.1161/CIRCRESAHA.116.304521
8. Trayanova NA. Learning for prevention of sudden cardiac death. Circ Res. (2021) 128:185–7. doi: 10.1161/CIRCRESAHA.120.318576
9. Wong CX, Brown A, Lau DH, Chugh SS, Albert CM, Kalman JM, et al. Epidemiology of sudden cardiac death: global and regional perspectives. Heart Lung Circ. (2019) 28:6–14. doi: 10.1016/j.hlc.2018.08.026
10. Breitenstein A, Steffel J. Devices in heart failure patients-who benefits from ICD and CRT? Front Cardiovasc Med. (2019) 6:111. doi: 10.3389/fcvm.2019.00111
11. Kusumoto FM, Calkins H, Boehmer J, Buxton AE, Chung MK, Gold MR, et al. HRS/ACC/AHA expert consensus statement on the use of implantable cardioverter-defibrillator therapy in patients who are not included or not well represented in clinical trials. Circulation. (2014) 130:94–125. doi: 10.1161/CIR.0000000000000056
12. NICE. Technology appraisal guidance: Implantable cardioverter defibrillators and cardiac resynchronisation therapy for arrhythmias and heart failure. London: National Institute for Health and Care Excellence (2014).
13. Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. (2015) 36:2793–867. doi: 10.1093/eurheartj/ehv316
14. Writing Committee M, Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. (2013) 128:e240–327. doi: 10.1161/CIR.0b013e3182742cf6
15. Huikuri HV, Makikallio TH, Raatikainen MJ, Perkiomaki J, Castellanos A, Myerburg RJ. Prediction of sudden cardiac death: appraisal of the studies and methods assessing the risk of sudden arrhythmic death. Circulation. (2003) 108:110–5. doi: 10.1161/01.CIR.0000077519.18416.43
16. Lopera G, Curtis AB. Risk stratification for sudden cardiac death: current approaches and predictive value. Curr Cardiol Rev. (2009) 5:56–64. doi: 10.2174/157340309787048130
17. La Rovere MT, Bigger JT. Jr., Marcus FI, Mortara A, Schwartz PJ. Baroreflex sensitivity and heart-rate variability in prediction of total cardiac mortality after myocardial infarction ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigators. Lancet. (1998) 351:478–84. doi: 10.1016/S0140-6736(97)11144-8
18. La Rovere MT, Pinna GD, Hohnloser SH, Marcus FI, Mortara A, Nohara R, et al. Baroreflex sensitivity and heart rate variability in the identification of patients at risk for life-threatening arrhythmias: implications for clinical trials. Circulation. (2001) 103:2072–7. doi: 10.1161/01.CIR.103.16.2072
19. Moss AJ, Zareba W, Hall WJ, Klein H, Wilber DJ, Cannom DS, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. (2002) 346:877–83. doi: 10.1056/NEJMoa013474
20. de Vreede-Swagemakers JJ, Gorgels AP, Dubois-Arbouw WI, van Ree JW, Daemen MJ, Houben LG, et al. Out-of-hospital cardiac arrest in the 1990's: a population-based study in the Maastricht area on incidence, characteristics and survival. J Am Coll Cardiol. (1997) 30:1500–5. doi: 10.1016/S0735-1097(97)00355-0
21. Gorgels AP, Gijsbers C, de Vreede-Swagemakers J, Lousberg A, Wellens HJ. Out-of-hospital cardiac arrest–the relevance of heart failure. The Maastricht Circulatory arrest registry. Eur Heart J. (2003) 24:1204–9. doi: 10.1016/S0195-668X(03)00191-X
22. Køber L, Thune JJ, Nielsen JC, Haarbo J, Videbæk L, Korup E, et al. Defibrillator implantation in patients with nonischemic systolic heart failure. N Engl J Med. (2016) 375:1221–30. doi: 10.1056/NEJMoa1608029
23. Desai AS, McMurray JJ, Packer M, Swedberg K, Rouleau JL, Chen F, et al. Effect of the angiotensin-receptor-neprilysin inhibitor LCZ696 compared with enalapril on mode of death in heart failure patients. Eur Heart J. (2015) 36:1990–7. doi: 10.1093/eurheartj/ehv186
24. Suematsu Y, Miura S, Goto M, Matsuo Y, Arimura T, Kuwano T, et al. LCZ696, an angiotensin receptor-neprilysin inhibitor, improves cardiac function with the attenuation of fibrosis in heart failure with reduced ejection fraction in streptozotocin-induced diabetic mice. Eur J Heart Fail. (2016) 18:386–93. doi: 10.1002/ejhf.474
25. von Lueder TG, Wang BH, Kompa AR, Huang L, Webb R, Jordaan P, et al. Angiotensin receptor neprilysin inhibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ Heart Fail. (2015) 8:71–8. doi: 10.1161/CIRCHEARTFAILURE.114.001785
26. Wu KC, Calkins H. Powerlessness of a number: why left ventricular ejection fraction matters less for sudden cardiac death risk assessment. Circ Cardiovasc Imaging. (2016) 9:e005519. doi: 10.1161/CIRCIMAGING.116.005519
27. Zaman S, Goldberger JJ, Kovoor P. Sudden Death Risk-Stratification in 2018-2019: the old and the new. Heart Lung Circ. (2019) 28:57–64. doi: 10.1016/j.hlc.2018.08.027
28. Dahl P, Danzi S, Klein I. Thyrotoxic cardiac disease. Curr Heart Fail Rep. (2008) 5:170–6. doi: 10.1007/s11897-008-0026-9
29. Dalan R, Leow MK. Cardiovascular collapse associated with beta blockade in thyroid storm. Exp Clin Endocrinol Diabetes. (2007) 115:392–6. doi: 10.1055/s-2007-971065
30. Dietrich JW. Thyroid storm. Med Klin Intensivmed Notfmed. (2012) 107:448–53. doi: 10.1007/s00063-012-0113-2
31. Klein I, Danzi S. Thyroid disease and the heart. Circulation. (2007) 116:1725–35. doi: 10.1161/CIRCULATIONAHA.106.678326
32. Fadel BM, Ellahham S, Ringel MD, Lindsay J. Jr., Wartofsky L, Burman KD. Hyperthyroid heart disease. Clin Cardiol. (2000) 23:402–8. doi: 10.1002/clc.4960230605
33. Yamakawa H, Kato TS, Noh JY, Yuasa S, Kawamura A, Fukuda K, et al. Thyroid hormone plays an important role in cardiac function: from bench to bedside. Front Physiol. (2021) 12:606931. doi: 10.3389/fphys.2021.606931
34. Biondi B, Palmieri EA, Lombardi G, Fazio S. Effects of subclinical thyroid dysfunction on the heart. Ann Intern Med. (2002) 137:904–14. doi: 10.7326/0003-4819-137-11-200212030-00011
35. Floriani C, Gencer B, Collet TH, Rodondi N. Subclinical thyroid dysfunction and cardiovascular diseases: 2016 update. Eur Heart J. (2018) 39:503–7. doi: 10.1093/eurheartj/ehx050
36. Chaker L, van den Berg ME, Niemeijer MN, Franco OH, Dehghan A, Hofman A, et al. Thyroid function and sudden cardiac death: a prospective population-based cohort study. Circulation. (2016) 134:713–22. doi: 10.1161/CIRCULATIONAHA.115.020789
37. Dietrich JW, Makimoto H, Muller P. Letter by dietrich et al. regarding article, “thyroid dysfunction in heart failure and cardiovascular outcomes”. Circ Heart Fail. (2019) 12:e005854. doi: 10.1161/CIRCHEARTFAILURE.118.005854
38. Fitzgerald SP, Bean NG, Falhammar H, Tuke J. Clinical parameters are more likely to be associated with thyroid hormone levels than with thyrotropin levels: a systematic review and meta-analysis. Thyroid. (2020) 30:1695–709. doi: 10.1089/thy.2019.0535
39. Groothof D, Flores-Guerrero JL, Nolte IM, Bouma HR, Gruppen EG, Bano A, et al. Thyroid function and risk of all-cause and cardiovascular mortality: a prospective population-based cohort study. Endocrine. (2021) 71:385–96. doi: 10.1007/s12020-020-02397-z
40. Abdel-Moneim A, Gaber AM, Gouda S, Osama A, Othman SI, Allam G. Relationship of thyroid dysfunction with cardiovascular diseases: updated review on heart failure progression. Hormones (Athens). (2020) 19:301–9. doi: 10.1007/s42000-020-00208-8
41. Rodondi N, den Elzen WP, Bauer DC, Cappola AR, Razvi S, Walsh JP, et al. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA. (2010) 304:1365–74. doi: 10.1001/jama.2010.1786
42. Selmer C, Olesen JB, Hansen ML, von Kappelgaard LM, Madsen JC, Hansen PR, et al. Subclinical and overt thyroid dysfunction and risk of all-cause mortality and cardiovascular events: a large population study. J Clin Endocrinol Metab. (2014) 99:2372–82. doi: 10.1210/jc.2013-4184
43. Sohn SY, Lee E, Lee MK, Lee JH. The association of overt and subclinical hyperthyroidism with the risk of cardiovascular events and cardiovascular mortality: meta-analysis and systematic review of cohort studies. Endocrinol Metab (Seoul). (2020) 35:786–800. doi: 10.3803/EnM.2020.728
44. Faber J, Selmer C. Cardiovascular disease and thyroid function. Front Horm Res. (2014) 43:45–56. doi: 10.1159/000360558
45. van Noord C, Sturkenboom MC, Straus SM, Hofman A, Witteman JC, Stricker BH. Population-based studies of antithyroid drugs and sudden cardiac death. Br J Clin Pharmacol. (2009) 68:447–54. doi: 10.1111/j.1365-2125.2009.03474.x
46. Rugge JB, Bougatsos C, Chou R. Screening and treatment of thyroid dysfunction: an evidence review for the US Preventive Services Task Force. Ann Intern Med. (2015) 162:35–45. doi: 10.7326/M14-1456
47. Dietrich JW, Hoermann R, Midgley JEM, Bergen F, Muller P. The two faces of janus: why thyrotropin as a cardiovascular risk factor may be an ambiguous target. Front Endocrinol (Lausanne). (2020) 11:542710. doi: 10.3389/fendo.2020.542710
48. Flamant F, Cheng SY, Hollenberg AN, Moeller LC, Samarut J, Wondisford FE, et al. Thyroid hormone signaling pathways: time for a more precise nomenclature. Endocrinology. (2017) 158:2052–7. doi: 10.1210/en.2017-00250
49. Stamatouli A, Bedoya P, Yavuz S. Hypothyroidism: cardiovascular endpoints of thyroid hormone replacement. Front Endocrinol (Lausanne). (2019) 10:888. doi: 10.3389/fendo.2019.00888
50. Recker S, Voigtlander R, Viehmann A, Dunschen K, Kerp H, Frank-Raue K, et al. Thyroid related quality of life in elderly with subclinical hypothyroidism and improvement on levothyroxine is distinct from that in young patients (TSAGE). Horm Metab Res. (2019) 51:568–74. doi: 10.1055/a-0897-8785
51. Dietrich JW, Midgley JEM, Hoermann R. Editorial: “homeostasis and allostasis of thyroid function”. Front Endocrinol (Lausanne). (2018) 9:287. doi: 10.3389/fendo.2018.00287
52. Midgley JE, Larisch R, Dietrich JW. Hoermann R. Letter to the editor: comment on “Reconciling the Log-Linear and Non-Linear Aspects of the TSH-Free T4 Relationship: Intra-Individual Analysis of a Large Population” by Rothacker KM, et al. J Clin Endocrinol Metab. (2016) 101:L40–1. doi: 10.1210/jc.2016-1115
53. Hoermann R, Midgley JE, Larisch R, Dietrich JW. Homeostatic control of the thyroid-pituitary axis: perspectives for diagnosis and treatment. Front Endocrinol (Lausanne). (2015) 6:177. doi: 10.3389/fendo.2015.00177
54. Hoermann R, Midgley JE, Larisch R, Dietrich JW. Relational stability in the expression of normality, variation, and control of thyroid function. Front Endocrinol (Lausanne). (2016) 7:142. doi: 10.3389/fendo.2016.00142
55. Hoermann R, Pekker MJ, Midgley JE, Larisch R, Dietrich JW. Principles of endocrine regulation: reconciling tensions between robustness in performance and adaptation to change. Front Endocrinol. (Lausanne). (2022) 13:825107. doi: 10.3389/fendo.2022.825107
56. Chatzitomaris A, Hoermann R, Midgley JE, Hering S, Urban A, Dietrich B, et al. Thyroid allostasis-adaptive responses of thyrotropic feedback control to conditions of strain, stress, and developmental programming. Front Endocrinol (Lausanne). (2017) 8:163. doi: 10.3389/fendo.2017.00163
57. Joseph-Bravo P. Hypophysiotropic thyrotropin-releasing hormone neurons as transducers of energy homeostasis. Endocrinology. (2004) 145:4813–5. doi: 10.1210/en.2004-0979
58. Joseph-Bravo P, Jaimes-Hoy L, Charli JL. Advances in TRH signaling. Rev Endocr Metab Disord. (2016) 17:545–58. doi: 10.1007/s11154-016-9375-y
59. Lechan RM, Fekete C. The TRH neuron: a hypothalamic integrator of energy metabolism. Prog Brain Res. (2006) 153:209–35. doi: 10.1016/S0079-6123(06)53012-2
60. Parry CH. Enlargement of the thyroid gland in connection with enlargement or palpitation of the heart. Collections from the unpublished medical writings of the late Caleb Hillier Parry. London: Underwoods. (1825). p. 111–25.
61. Flajani G. Collezione d'osservazioni e riflessioni di chirurgia. Roma: stamp. di S. Michele a Ripa (1802).
62. Graves RJ. Newly observed affection of the thyroid gland in females. London Med Surg J. (1835) 7:516–7.
63. von Basedow K. Exophtalmos durch Hypertrophie des Zellgewebes in der Augenhöhle. Wschr ges Heilk. (1840) 13:197–228.
64. Kahaly GJ, Dillmann WH. Thyroid hormone action in the heart. Endocr Rev. (2005) 26:704–28. doi: 10.1210/er.2003-0033
65. Ortiga-Carvalho TM, Sidhaye AR, Wondisford FE. Thyroid hormone receptors and resistance to thyroid hormone disorders. Nat Rev Endocrinol. (2014) 10:582–91. doi: 10.1038/nrendo.2014.143
66. Hönes GS, Rakov H, Logan J, Liao XH, Werbenko E, Pollard AS, et al. Noncanonical thyroid hormone signaling mediates cardiometabolic effects in vivo. Proc Natl Acad Sci U S A. (2017) 114:E11323–E32. doi: 10.1073/pnas.1706801115
67. Saponaro F, Sestito S, Runfola M, Rapposelli S, Chiellini G. Selective thyroid hormone receptor-beta (TRbeta) agonists: new perspectives for the treatment of metabolic and neurodegenerative disorders. Front Med (Lausanne). (2020) 7:331. doi: 10.3389/fmed.2020.00331
68. Davis PJ, Davis FB, Lin HY, Mousa SA, Zhou M, Luidens MK. Translational implications of nongenomic actions of thyroid hormone initiated at its integrin receptor. Am J Physiol Endocrinol Metab. (2009) 297:E1238–46. doi: 10.1152/ajpendo.00480.2009
69. Köhrle J. The colorful diversity of thyroid hormone metabolites. Eur Thyroid J. (2019) 8:115–29. doi: 10.1159/000497141
70. Xu B, Koenig RJ. An RNA-binding domain in the thyroid hormone receptor enhances transcriptional activation. J Biol Chem. (2004) 279:33051–6. doi: 10.1074/jbc.M404930200
71. Lanni A, Moreno M, Goglia F. Mitochondrial actions of thyroid hormone. Compr Physiol. (2016) 6:1591–607. doi: 10.1002/cphy.c150019
72. Milo R, Phillips R. Cell Biology by the Numbers. New York, Abingdon: Garland Science. (2016). doi: 10.1201/9780429258770
73. Giammanco M, Liegro CMD, Schiera G, Liegro ID. Genomic and non-genomic mechanisms of action of thyroid hormones and their catabolite 3,5-Diiodo-L-thyronine in mammals. Int J Mol Sci. (2020) 21:4140. doi: 10.3390/ijms21114140
74. Köhrle J. Thyroid hormones and derivatives: endogenous thyroid hormones and their targets. Methods Mol Biol. (2018) 1801:85–104. doi: 10.1007/978-1-4939-7902-8_9
75. Le Bouter S, Demolombe S, Chambellan A, Bellocq C, Aimond F, Toumaniantz G, et al. Microarray analysis reveals complex remodeling of cardiac ion channel expression with altered thyroid status: relation to cellular and integrated electrophysiology. Circ Res. (2003) 92:234–42. doi: 10.1161/01.RES.0000053185.75505.8E
76. Chatzitomaris A, Scheeler M, Gotzmann M, Koditz R, Schildroth J, Knyhala KM, et al. Second degree AV block and severely impaired contractility in cardiac myxedema: a case report. Thyroid Res. (2015) 8:6. doi: 10.1186/s13044-015-0018-2
77. Panagoulis C, Halapas A, Chariatis E, Driva P, Matsakas E. Hyperthyroidism and the heart. Hellenic J Cardiol. (2008) 49:169–75.
78. Razvi S, Jabbar A, Pingitore A, Danzi S, Biondi B, Klein I, et al. Thyroid hormones and cardiovascular function and diseases. J Am Coll Cardiol. (2018) 71:1781–96. doi: 10.1016/j.jacc.2018.02.045
79. Galli E, Pingitore A, Iervasi G. The role of thyroid hormone in the pathophysiology of heart failure: clinical evidence. Heart Fail Rev. (2010) 15:155–69. doi: 10.1007/s10741-008-9126-6
80. Nattel S, Tomaselli GF. Mechanisms of Cardiac Arrhythmia. In: Libby P, Bonow RO, Mann DL, Tomaselli GF, Bhatt D, Solomon SD, . editors. Braunwald's Heart Disease. 2. Philadelphia, PA: Elsevier (2021). p. 1163-90.
81. Chen YC, Chen SA, Chen YJ, Chang MS, Chan P, Lin CI. Effects of thyroid hormone on the arrhythmogenic activity of pulmonary vein cardiomyocytes. J Am Coll Cardiol. (2002) 39:366–72. doi: 10.1016/S0735-1097(01)01731-4
82. Kausel A, Korniyenko A, Sandhu G. Bradyarrhythmia as a presenting feature of subclinical hyperthyroidism. QJM. (2012) 105:461–2. doi: 10.1093/qjmed/hcr049
83. Krishnamoorthy S, Narain R, Creamer J. Unusual presentation of thyrotoxicosis as complete heart block and renal failure: a case report. J Med Case Rep. (2009) 3:9303. doi: 10.1186/1752-1947-3-9303
84. Gavrilescu S, Luca C, Streian C, Lungu G, Deutsch G. Monophasic action potentials of right atrium and electrophysiological properties of AV conducting system in patients with hypothyroidism. Br Heart J. (1976) 38:1350–4. doi: 10.1136/hrt.38.12.1350
85. Maltsev VA, Vinogradova TM, Lakatta EG. The emergence of a general theory of the initiation and strength of the heartbeat. J Pharmacol Sci. (2006) 100:338–69. doi: 10.1254/jphs.CR0060018
86. Freedberg AS, Papp JG, Williams EM. The effect of altered thyroid state on atrial intracellular potentials. J Physiol. (1970) 207:357–69. doi: 10.1113/jphysiol.1970.sp009066
87. Johnson PN, Freedberg AS, Marshall JM. Action of thyroid hormone on the transmembrane potentials from sinoatrial node cells and atrial muscle cells in isolated atria of rabbits. Cardiology. (1973) 58:273–89. doi: 10.1159/000169643
88. Sun ZQ, Ojamaa K, Nakamura TY, Artman M, Klein I, Coetzee WA. Thyroid hormone increases pacemaker activity in rat neonatal atrial myocytes. J Mol Cell Cardiol. (2001) 33:811–24. doi: 10.1006/jmcc.2001.1353
89. Sun ZQ, Ojamaa K, Coetzee WA, Artman M, Klein I. Effects of thyroid hormone on action potential and repolarizing currents in rat ventricular myocytes. Am J Physiol Endocrinol Metab. (2000) 278:E302–7. doi: 10.1152/ajpendo.2000.278.2.E302
90. Chang KC, Figueredo VM, Schreur JH, Kariya K, Weiner MW, Simpson PC, et al. Thyroid hormone improves function and Ca2+ handling in pressure overload hypertrophy. Association with increased sarcoplasmic reticulum Ca2+-ATPase and alpha-myosin heavy chain in rat hearts. J Clin Invest. (1997) 100:1742–9. doi: 10.1172/JCI119699
91. Kaasik A, Minajeva A, Paju K, Eimre M, Seppet EK. Thyroid hormones differentially affect sarcoplasmic reticulum function in rat atria and ventricles. Mol Cell Biochem. (1997) 176:119–26. doi: 10.1023/A:1006887231150
92. Alonso H, Fernandez-Ruocco J, Gallego M, Malagueta-Vieira LL, Rodriguez-de-Yurre A, Medei E, et al. Thyroid stimulating hormone directly modulates cardiac electrical activity. J Mol Cell Cardiol. (2015) 89:280–6. doi: 10.1016/j.yjmcc.2015.10.019
93. Thyroid stimulating hormone (TSH) signaling pathway (Homo sapiens) [Internet]. WikiPathways. (2011-2022). Available online at: https://www.wikipathways.org/instance/WP2032_r122770.
94. Holzapfel HP, Bergner B, Wonerow P, Paschke R. Expression of G(alpha)(s) proteins and TSH receptor signalling in hyperfunctioning thyroid nodules with TSH receptor mutations. Eur J Endocrinol. (2002) 147:109–16. doi: 10.1530/eje.0.1470109
95. Bosch RF, Wang Z, Li GR, Nattel S. Electrophysiological mechanisms by which hypothyroidism delays repolarization in guinea pig hearts. Am J Physiol. (1999) 277:H211–20. doi: 10.1152/ajpheart.1999.277.1.H211
96. Goel BG, Hanson CS, Han J. A-V conduction in hyper- and hypothyroid dogs. Am Heart J. (1972) 83:504–11. doi: 10.1016/0002-8703(72)90041-5
97. Zhang Y, Dedkov EI, Teplitsky D, Weltman NY, Pol CJ, Rajagopalan V, et al. Both hypothyroidism and hyperthyroidism increase atrial fibrillation inducibility in rats. Circ Arrhythm Electrophysiol. (2013) 6:952–9. doi: 10.1161/CIRCEP.113.000502
98. Tribulova N, Kurahara LH, Hlivak P, Hirano K, Szeiffova Bacova B. Pro-arrhythmic signaling of thyroid hormones and its relevance in subclinical hyperthyroidism. Int J Mol Sci. (2020) 21:2844. doi: 10.3390/ijms21082844
99. King JH, Huang CL, Fraser JA. Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front Physiol. (2013) 4:154. doi: 10.3389/fphys.2013.00154
100. Richter Y, Lind PG, Seemann G, Maass P. Anatomical and spiral wave reentry in a simplified model for atrial electrophysiology. J Theor Biol. (2017) 419:100–7. doi: 10.1016/j.jtbi.2017.02.008
101. Hoermann R, Midgley JE, Larisch R, Dietrich JW. Is pituitary TSH an adequate measure of thyroid hormone-controlled homoeostasis during thyroxine treatment? Eur J Endocrinol. (2013) 168:271–80. doi: 10.1530/EJE-12-0819
102. Hoermann R, Midgley JEM, Larisch R, Dietrich JW. Heterogenous biochemical expression of hormone activity in subclinical/overt hyperthyroidism and exogenous thyrotoxicosis. J Clin Transl Endocrinol. (2020) 19:100219. doi: 10.1016/j.jcte.2020.100219
103. Midgley JE, Hoermann R, Larisch R, Dietrich JW. Physiological states and functional relation between thyrotropin and free thyroxine in thyroid health and disease: in vivo and in silico data suggest a hierarchical model. J Clin Pathol. (2013) 66:335–42. doi: 10.1136/jclinpath-2012-201213
104. Midgley JEM, Toft AD, Larisch R, Dietrich JW, Hoermann R. Time for a reassessment of the treatment of hypothyroidism. BMC Endocr Disord. (2019) 19:37. doi: 10.1186/s12902-019-0365-4
105. Pasqualetti G, Tognini S, Polini A, Caraccio N, Monzani F. Is subclinical hypothyroidism a cardiovascular risk factor in the elderly? J Clin Endocrinol Metab. (2013) 98:2256–66. doi: 10.1210/jc.2012-3818
106. Hoermann R, Midgley JEM, Larisch R, Dietrich JW. Lessons from randomised clinical trials for triiodothyronine treatment of hypothyroidism: have they achieved their objectives? J Thyroid Res. (2018) 2018:3239197. doi: 10.1155/2018/3239197
107. Wiersinga WM. Guidance in subclinical hyperthyroidism and subclinical hypothyroidism: are we making progress? Eur Thyroid J. (2015) 4:143–8. doi: 10.1159/000438909
108. Hoermann R, Midgley JEM, Dietrich JW, Larisch R. Dual control of pituitary thyroid stimulating hormone secretion by thyroxine and triiodothyronine in athyreotic patients. Ther Adv Endocrinol Metab. (2017) 8:83–95. doi: 10.1177/2042018817716401
109. Hoermann R, Midgley JEM, Larisch R, Dietrich JW. The role of functional thyroid capacity in pituitary thyroid feedback regulation. Eur J Clin Invest. (2018) 48:e13003. doi: 10.1111/eci.13003
110. Andersen S, Pedersen KM, Bruun NH, Laurberg P. Narrow individual variations in serum T(4) and T(3) in normal subjects: a clue to the understanding of subclinical thyroid disease. J Clin Endocrinol Metab. (2002) 87:1068–72. doi: 10.1210/jcem.87.3.8165
111. Larisch R, Giacobino A, Eckl W, Wahl HG, Midgley JE, Hoermann R. Reference range for thyrotropin. Post hoc assessment. Nuklearmedizin. (2015) 54:112–7. doi: 10.3413/Nukmed-0671-14-06
112. Goede SL, Latham KR, Leow MK, Jonklaas J. High resolution free triiodothyronine-thyrotropin (FT3-TSH) responses to a single oral dose of liothyronine in humans: evidence of distinct inter-individual differences unraveled using an electrical network model. J Biol Syst. (2017) 25:119–43. doi: 10.1142/S0218339017500073
113. Hoermann R, Midgley JEM, Larisch R, Dietrich JW. Functional and symptomatic individuality in the response to levothyroxine treatment. Front Endocrinol (Lausanne). (2019) 10:664. doi: 10.3389/fendo.2019.00664
114. Hoermann R, Midgley JEM, Larisch R, Dietrich JW. Individualised requirements for optimum treatment of hypothyroidism: complex needs, limited options. Drugs Context. (2019) 8:212597. doi: 10.7573/dic.212597
115. Leow MK, Goede SL. The homeostatic set point of the hypothalamus-pituitary-thyroid axis–maximum curvature theory for personalized euthyroid targets. Theor Biol Med Model. (2014) 11:35. doi: 10.1186/1742-4682-11-35
116. Meng F, Li E, Yen PM, Leow MKS. Hyperthyroidism in the personalized medicine era: the rise of mathematical optimization. J R Soc Interface. (2019) 16:20190083. doi: 10.1098/rsif.2019.0083
117. Bonsu JM, Guha A, Charles L, Yildiz VO, Wei L, Baker B, et al. Reporting of cardiovascular events in clinical trials supporting FDA approval of contemporary cancer therapies. J Am Coll Cardiol. (2020) 75:620–8. doi: 10.1016/j.jacc.2019.11.059
118. Bosco E, Hsueh L, McConeghy KW, Gravenstein S, Saade E. Major adverse cardiovascular event definitions used in observational analysis of administrative databases: a systematic review. BMC Med Res Methodol. (2021) 21:241. doi: 10.1186/s12874-021-01440-5
119. Poudel I, Tejpal C, Rashid H, Jahan N. Major Adverse Cardiovascular Events: An Inevitable Outcome of ST-elevation myocardial infarction? A Literature Review. Cureus. (2019) 11:e5280. doi: 10.7759/cureus.5280
120. Gussekloo J, van Exel E, de Craen AJ, Meinders AE, Frolich M, Westendorp RG. Thyroid status, disability and cognitive function, and survival in old age. JAMA. (2004) 292:2591–9. doi: 10.1001/jama.292.21.2591
121. Cappola AR, Fried LP, Arnold AM, Danese MD, Kuller LH, Burke GL, et al. Thyroid status, cardiovascular risk, and mortality in older adults. JAMA. (2006) 295:1033–41. doi: 10.1001/jama.295.9.1033
122. Razvi S, Weaver JU, Vanderpump MP, Pearce SH. The incidence of ischemic heart disease and mortality in people with subclinical hypothyroidism: reanalysis of the Whickham Survey cohort. J Clin Endocrinol Metab. (2010) 95:1734–40. doi: 10.1210/jc.2009-1749
123. Schultz M, Kistorp C, Raymond I, Dimsits J, Tuxen C, Hildebrandt P, et al. Cardiovascular events in thyroid disease: a population based, prospective study. Horm Metab Res. (2011) 43:653–9. doi: 10.1055/s-0031-1283162
124. Frey A, Kroiss M, Berliner D, Seifert M, Allolio B, Guder G, et al. Prognostic impact of subclinical thyroid dysfunction in heart failure. Int J Cardiol. (2013) 168:300–5. doi: 10.1016/j.ijcard.2012.09.064
125. Mitchell JE, Hellkamp AS, Mark DB, Anderson J, Johnson GW, Poole JE, et al. Thyroid function in heart failure and impact on mortality. JACC Heart Fail. (2013) 1:48–55. doi: 10.1016/j.jchf.2012.10.004
126. Drechsler C, Schneider A, Gutjahr-Lengsfeld L, Kroiss M, Carrero JJ, Krane V, et al. Thyroid function, cardiovascular events, and mortality in diabetic hemodialysis patients. Am J Kidney Dis. (2014) 63:988–96. doi: 10.1053/j.ajkd.2013.10.009
127. Perez AC, Jhund PS, Stott DJ, Gullestad L, Cleland JG, van Veldhuisen DJ, et al. Thyroid-stimulating hormone and clinical outcomes: the CORONA trial (controlled rosuvastatin multinational study in heart failure). JACC Heart Fail. (2014) 2:35–40. doi: 10.1016/j.jchf.2013.07.008
128. Rhee CM, Ravel VA, Streja E, Mehrotra R, Kim S, Wang J, et al. Thyroid functional disease and mortality in a national peritoneal dialysis cohort. J Clin Endocrinol Metab. (2016) 101:4054–61. doi: 10.1210/jc.2016-1691
129. Pearce SH, Razvi S, Yadegarfar ME, Martin-Ruiz C, Kingston A, Collerton J, et al. Serum thyroid function, mortality and disability in advanced old age: the newcastle 85+ study. J Clin Endocrinol Metab. (2016) 101:4385–94. doi: 10.1210/jc.2016-1935
130. Langen VL, Niiranen TJ, Puukka P, Lehtonen AO, Hernesniemi JA, Sundvall J, et al. Thyroid-stimulating hormone and risk of sudden cardiac death, total mortality and cardiovascular morbidity. Clin Endocrinol (Oxf). (2018) 88:105–13. doi: 10.1111/cen.13472
131. Kannan L, Shaw PA, Morley MP, Brandimarto J, Fang JC, Sweitzer NK, et al. Thyroid dysfunction in heart failure and cardiovascular outcomes. Circ Heart Fail. (2018) 11:e005266. doi: 10.1161/CIRCHEARTFAILURE.118.005266
132. Golledge J, Hankey GJ, Almeida OP, Flicker L, Norman PE, Yeap BB. Plasma free thyroxine in the upper quartile is associated with an increased incidence of major cardiovascular events in older men that do not have thyroid dysfunction according to conventional criteria. Int J Cardiol. (2018) 254:316–21. doi: 10.1016/j.ijcard.2017.10.100
133. Li X, Yao Y, Chen Z, Fan S, Hua W, Zhang S, et al. Thyroid-stimulating hormone within the normal range and risk of major adverse cardiovascular events in nonischemic dilated cardiomyopathy patients with severe left ventricular dysfunction. Clin Cardiol. (2019) 42:120–8. doi: 10.1002/clc.23117
134. Seo SM, Koh YS, Park HJ, Kim DB, Her SH, Lee JM, et al. Thyroid stimulating hormone elevation as a predictor of long-term mortality in patients with acute myocardial infarction. Clin Cardiol. (2018) 41:1367–73. doi: 10.1002/clc.23062
135. Inoue K, Ritz B, Brent GA, Ebrahimi R, Rhee CM, Leung AM. Association of subclinical hypothyroidism and cardiovascular disease with mortality. JAMA Netw Open. (2020) 3:e1920745. doi: 10.1001/jamanetworkopen.2019.20745
136. Kim H, Kong SH, Moon JH, Kim SY, Park KH, Kim JS, et al. Subclinical hypothyroidism affects the long-term outcomes of patients who undergo coronary artery bypass grafting surgery but not heart valve surgery. Endocrinol Metab (Seoul). (2020) 35:308–18. doi: 10.3803/EnM.2020.35.2.308
137. Müller P, Dietrich JW, Lin T, Bejinariu A, Binnebössel S, Bergen F, et al. Usefulness of serum free thyroxine concentration to predict ventricular arrhythmia risk in euthyroid patients with structural heart disease. Am J Cardiol. (2020) 125:1162–9. doi: 10.1016/j.amjcard.2020.01.019
138. Yang M, Li X, Morris JC. 3rd, Liang J, Deshmukh AJ, Hodge D, et al. Outcomes of cardiac resynchronization therapy in patients with hypothyroidism and heart failure. BMC Cardiovasc Disord. (2020) 20:424. doi: 10.1186/s12872-020-01693-w
139. Evron JM, Hummel SL, Reyes-Gastelum D, Haymart MR, Banerjee M, Papaleontiou M. Association of thyroid hormone treatment intensity with cardiovascular mortality among US veterans. JAMA Netw Open. (2022) 5:e2211863. doi: 10.1001/jamanetworkopen.2022.11863
140. Iervasi G, Molinaro S, Landi P, Taddei MC, Galli E, Mariani F, et al. Association between increased mortality and mild thyroid dysfunction in cardiac patients. Arch Intern Med. (2007) 167:1526–32. doi: 10.1001/archinte.167.14.1526
141. Rodondi N, Newman AB, Vittinghoff E, de Rekeneire N, Satterfield S, Harris TB, et al. Subclinical hypothyroidism and the risk of heart failure, other cardiovascular events, and death. Arch Intern Med. (2005) 165:2460–6. doi: 10.1001/archinte.165.21.2460
142. Sgarbi JA, Matsumura LK, Kasamatsu TS, Ferreira SR, Maciel RM. Subclinical thyroid dysfunctions are independent risk factors for mortality in a 75-year follow-up: the Japanese-Brazilian thyroid study. Eur J Endocrinol. (2010) 162:569–77. doi: 10.1530/EJE-09-0845
143. Walsh JP, Bremner AP, Bulsara MK, O'Leary P, Leedman PJ, Feddema P, et al. Subclinical thyroid dysfunction as a risk factor for cardiovascular disease. Arch Intern Med. (2005) 165:2467–72. doi: 10.1001/archinte.165.21.2467
144. Ittermann T, Haring R, Sauer S, Wallaschofski H, Dorr M, Nauck M, et al. Decreased serum TSH levels are not associated with mortality in the adult northeast German population. Eur J Endocrinol. (2010) 162:579–85. doi: 10.1530/EJE-09-0566
145. Osman F, Gammage MD, Sheppard MC, Franklyn JA. Clinical review 142: cardiac dysrhythmias and thyroid dysfunction: the hidden menace? J Clin Endocrinol Metab. (2002) 87:963–7. doi: 10.1210/jcem.87.3.8217
146. Sun J, Yao L, Fang Y, Yang R, Chen Y, Yang K, et al. Relationship between subclinical thyroid dysfunction and the risk of cardiovascular outcomes: a systematic review and meta-analysis of prospective cohort studies. Int J Endocrinol. (2017) 2017:8130796. doi: 10.1155/2017/8130796
147. Aweimer A, El-Battrawy I, Akin I, Borggrefe M, Mugge A, Patsalis PC, et al. Abnormal thyroid function is common in takotsubo syndrome and depends on two distinct mechanisms: results of a multicentre observational study. J Intern Med. (2021) 289:675–87. doi: 10.1111/joim.13189
148. Goede SL, Leow MK, Smit JW, Dietrich JW. A novel minimal mathematical model of the hypothalamus-pituitary-thyroid axis validated for individualized clinical applications. Math Biosci. (2014) 249:1–7. doi: 10.1016/j.mbs.2014.01.001
149. Goede SL, Leow MK, Smit JW, Klein HH, Dietrich JW. Hypothalamus-pituitary-thyroid feedback control: implications of mathematical modeling and consequences for thyrotropin (TSH) and free thyroxine (FT4) reference ranges. Bull Math Biol. (2014) 76:1270–87. doi: 10.1007/s11538-014-9955-5
150. Dietrich JW, Landgrafe-Mende G, Wiora E, Chatzitomaris A, Klein HH, Midgley JE, et al. Calculated parameters of thyroid homeostasis: emerging tools for differential diagnosis and clinical research. Front Endocrinol (Lausanne). (2016) 7:57. doi: 10.3389/fendo.2016.00057
151. Chang CY, Chien YJ, Lin PC, Chen CS, Wu MY. Nonthyroidal illness syndrome and hypothyroidism in ischemic heart disease population: a systematic review and meta-analysis. J Clin Endocrinol Metab. (2020) 105:e3414–e3425. doi: 10.1210/clinem/dgaa310
152. Dietrich JW, Muller P, Schiedat F, Schlomicher M, Strauch J, Chatzitomaris A, et al. Nonthyroidal illness syndrome in cardiac illness involves elevated concentrations of 3,5-diiodothyronine and correlates with atrial remodeling. Eur Thyroid J. (2015) 4:129–37. doi: 10.1159/000381543
153. Özturk S, Kayacioglu I, Sensoz Y, Ozdemir K, Özturk I. Efficiency of Thyroid Function Test in the Prediction of Atrial Fibrillation Following Open Heart Surgery. J Coll Physicians Surg Pak. (2020) 30:740–4. doi: 10.29271/jcpsp.2020.07.740
154. Langouche L, Lehmphul I, Perre SV, Kohrle J, Van den Berghe G. Circulating 3-T1AM and 3,5-T2 in critically ill patients: a cross-sectional observational study. Thyroid. (2016) 26:1674–80. doi: 10.1089/thy.2016.0214
155. Surks MI, Sievert R. Drugs and thyroid function. N Engl J Med. (1995) 333:1688–94. doi: 10.1056/NEJM199512213332507
156. Carvalho GA, Perez CL, Ward LS. The clinical use of thyroid function tests. Arq Bras Endocrinol Metabol. (2013) 57:193–204. doi: 10.1590/S0004-27302013000300005
157. Dietrich JW, Ackermann A, Kasippillai A, Kanthasamy Y, Tharmalingam T, Urban A, et al. Adaptive Veränderungen des Schilddrüsenstoffwechsels als Risikoindikatoren bei Traumata. Trauma und Berufskrankheit. (2019) 21:260–7. doi: 10.1007/s10039-019-00438-z
158. Stockigt JR, Lim CF. Medications that distort in vitro tests of thyroid function, with particular reference to estimates of serum free thyroxine. Best Pract Res Clin Endocrinol Metab. (2009) 23:753–67. doi: 10.1016/j.beem.2009.06.004
159. Dietrich JW, Landgrafe G, Fotiadou EH. TSH and thyrotropic agonists: key actors in thyroid homeostasis. J Thyroid Res. (2012) 2012:351864. doi: 10.1155/2012/351864
160. Peters A, McEwen BS. Stress habituation, body shape and cardiovascular mortality. Neurosci Biobehav Rev. (2015) 56:139–50. doi: 10.1016/j.neubiorev.2015.07.001
161. Guidi J, Lucente M, Sonino N, Fava GA. Allostatic load and its impact on health: a systematic review. Psychother Psychosom. (2021) 90:11–27. doi: 10.1159/000510696
162. Steptoe A, Hackett RA, Lazzarino AI, Bostock S, La Marca R, Carvalho LA, et al. Disruption of multisystem responses to stress in type 2 diabetes: investigating the dynamics of allostatic load. Proc Natl Acad Sci U S A. (2014) 111:15693–8. doi: 10.1073/pnas.1410401111
163. Logan JG, Barksdale DJ. Allostasis and allostatic load: expanding the discourse on stress and cardiovascular disease. J Clin Nurs. (2008) 17:201–8. doi: 10.1111/j.1365-2702.2008.02347.x
164. Collet TH, Gussekloo J, Bauer DC, den Elzen WP, Cappola AR, Balmer P, et al. Subclinical hyperthyroidism and the risk of coronary heart disease and mortality. Arch Intern Med. (2012) 172:799–809. doi: 10.1001/archinternmed.2012.402
165. Biondi B, Bartalena L, Cooper DS, Hegedus L, Laurberg P, Kahaly GJ. The 2015 European Thyroid Association guidelines on diagnosis and treatment of endogenous subclinical hyperthyroidism. Eur Thyroid J. (2015) 4:149–63. doi: 10.1159/000438750
166. Wolff TM, Dietrich JW, Müller MA. Optimal hormone replacement therapy in hypothyroidism - a model predictive control approach. Front Endocrinol. (2022) 13:884018. doi: 10.3389/fendo.2022.884018
167. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American thyroid association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: the american thyroid association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid. (2016) 26:1–133. doi: 10.1089/thy.2015.0020
168. D'Alessio L, Korman GP, Sarudiansky M, Guelman LR, Scevola L, Pastore A, et al. Reducing allostatic load in depression and anxiety disorders: physical activity and yoga practice as add-on therapies. Front Psychiatry. (2020) 11:501. doi: 10.3389/fpsyt.2020.00501
169. Ferrara I, Egan CG, Foletti A. Pilot study on the effect of biophysical therapy on salivary alpha-amylase as a surrogate measure of anxiety/stress: in search of a novel noninvasive molecular approach for the management of stress. Int J Mol Sci. (2020) 21:415. doi: 10.3390/ijms21020415
170. Macit MS, Acar-Tek N. Current perspectives for diabetes and allostatic load: the role of nutrition. Curr Nutr Food Sci. (2018) 14:1–7. doi: 10.2174/1573401314666180620164859
171. Tan M, Mamun A, Kitzman H, Dodgen L. Longitudinal changes in allostatic load during a randomized church-based, lifestyle intervention in African American women. Ethn Dis. (2019) 29:297–308. doi: 10.18865/ed.29.2.297
172. Lipowicz A, Szklarska A, Malina RM. Allostatic load and socioeconomic status in Polish adult men. J Biosoc Sci. (2014) 46:155–67. doi: 10.1017/S0021932013000345
Keywords: thyroid function, sudden cardiac death, ventricular arrhythmia, cardiac electrophysiology, MACE, hypothyroidism, thyrotoxicosis, type 2 allostatic load
Citation: Müller P, Leow MK-S and Dietrich JW (2022) Minor perturbations of thyroid homeostasis and major cardiovascular endpoints—Physiological mechanisms and clinical evidence. Front. Cardiovasc. Med. 9:942971. doi: 10.3389/fcvm.2022.942971
Received: 13 May 2022; Accepted: 22 July 2022;
Published: 15 August 2022.
Edited by:
Chayakrit Krittanawong, New York University, United StatesReviewed by:
Antoine Avignon, University of Montpellier 1, FranceCopyright © 2022 Müller, Leow and Dietrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johannes W. Dietrich, am9oYW5uZXMuZGlldHJpY2hAcnVoci11bmktYm9jaHVtLmRl
†ORCID: Patrick Müller orcid.org/0000-0003-1641-5449
Melvin Khee-Shing Leow orcid.org/0000-0002-2837-5889
Johannes W. Dietrich orcid.org/0000-0002-1185-3549
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.