 Siyu Jiang
Siyu Jiang Yingjie Ai
Yingjie Ai Liyuan Ni1
Liyuan Ni1- 1Department of Gastroenterology and Hepatology, Zhongshan Hospital, Fudan University, Shanghai, China
- 2Endoscopy Center and Endoscopy Research Institute, Zhongshan Hospital of Fudan University, Shanghai, China
Background: Portal vein thrombosis (PVT) is a serious complication of cirrhosis accompanied by unclear pathogenesis. Transforming growth factor-beta (TGF-β) has been implicated in atherosclerosis and venous thrombosis whereas study regarding its part in PVT is lacking. The aim of this study was to explore the role of cytokine TGF-β1 in PVT and the potential mechanism.
Materials and methods: We included patients with cirrhotic gastroesophageal varices and divided them into two groups according to the presence of PVT. Serum levels of TGF-β1 were detected using Cytometric Bead Array kit and compared between two groups. Coagulation status was assessed using thromboelastography (TEG). Primary liver sinusoidal endothelial cells were treated with TGF-β1 and evaluated for endothelial dysfunction by RT-PCR.
Results: Our results uncovered that TGF-β1 (6,866.55 vs. 3,840.60 pg/ml, P = 0.015) significantly increased in the PVT group. Splenectomy might promote PVT by increasing platelet-derived TGF-β1 levels. Other cytokines showed no difference between PVT and non-PVT groups. Besides, TGF-β1 was correlated with platelet, fibrinogen, TEG-CI, TEG-MA, and TEG-α (coef = 0.733, 0.494, 0.604, 0.608, and 0.511; P < 0.001, 0.027, 0.004, 0.004, and 0.021, respectively), which indicated a hypercoagulable state in PVT patients. RT-PCR of liver sinusoidal endothelial cells showed a markable increment of von Willebrand Factor (vWF), thrombomodulin(TM), intercellular adhesion moleclar-1(ICAM-1), and vascular endothelial growth factor(VEGF) after TGF-β1 treatment, suggesting the involvement of endothelial dysfunction.
Conclusion: Elevated platelet-derived TGF-β1 exhibited association with hypercoagulability and promoting effect on endothelial dysfunction, closely related with PVT in cirrhotic patients.
Introduction
Portal vein thrombosis (PVT) is a serious complication of cirrhosis accompanied with a poor prognosis (1). The pathogenesis of PVT is multifactorial and components of Virchow’s triad (decreased blood flow, endothelial injury, and hypercoagulability) have been thought to play a key role in the onset and development of PVT (2). Although several studies believed that alteration in portal vein flow plays a predominate part in the development of PVT, the function of endothelial damage in the natural course of PVT cannot be overestimated (1, 3, 4). The underlying mechanism of PVT advancement remains unclear and desiderates further investigations.
Inflammation is believed to have crucial effects on thrombi formation as systemic inflammation has been proved to be associated with increased PVT risk (5, 6). Transforming growth factor-beta (TGF-β), a multifunctional cytokine mainly derived from platelets (7, 8), participates in many biological and pathologic functions, including the immune response, angiogenesis, venous thrombosis, and tissue fibrosis (9). As for endothelial damage, TGF-β is associated with apoptosis of epithelial cells induced by continuous stretch (10). Meanwhile, it can also aggravate venous thrombosis by impairing thrombus resolution and promoting endothelial dysfunction without affecting platelet function (11). Despite that TGF-β has been reported to take part in atherosclerosis and venous thrombosis, there is a lack of studies regarding its role in PVT.
The aim of this study was to investigate the role and potential mechanisms of inflammatory cytokine TGF-β1 promoting PVT through clinical and experimental studies.
Materials and methods
Study population
The study was a cross-sectional case-control study to investigate the role of cytokines. Fifty-three patients who had cirrhotic gastroesophageal varices confirmed by CT angiography (CTA) and gastroscopy were enrolled between 1 December 2016, and 31 December 2017. All patients had a history of gastroesophageal variceal hemorrhage. Exclusion criteria were: (1) active bleeding; (2) combination with malignancy; (3) overt infection or sepsis; (4) treatment with systemic antibiotic or anti-inflammatory drugs in the past 2 weeks; (5) severe abdominal pain; (6) coagulation disorders.
All patients were divided into two groups according to the presence of PVT (PVT group, n = 20; non-PVT group, n = 33). None of them received anticoagulation or antiplatelet therapy before. The protocol for conducting the current analysis was approved by the ethic committee of Zhongshan Hospital, Fudan University (B2015-133R), in accordance with the Declaration of Helsinki.
Laboratory methods
Measurements were ascertained while blinded to the sample origin. Routine biochemical tests were carried out using standard procedures. Patients had fasted for the last 12 h before blood sampling and underwent routine biochemical evaluations. Serum TGF-β1 (cat. no. 560429), L-selectin (cat. no. 560420), intercellular adhesion moleclar-1 (ICAM-1, cat. no. 560269), and vascular cell adhesion molecule-1 (VCAM-1, cat. no. 560427) were determined using Cytometric Bead Array kit (CBA; BD Biosciences, San Jose, CA, United States). Serum samples were mixed to capture specific beads for each cytokine. Antibodies were added, conjugated with phycoerythrin, and incubated for 2 h under room temperature, protected from light. Tubes were then centrifuged (200 g for 5 min) and the supernatant was carefully aspired and discarded. The pellets containing beads were resuspended and the samples were analyzed on the Thermo Cytometer. The data obtained were analyzed by the BD™ Cell Quest and FCAP Array software.
Cell culture and treatment
Liver sinusoidal endothelial cells (LSECs, Sciencell, San Diego, California, USA), were cultured in the endothelial cell medium (ECM, 1001, Sciencell, San Diego, California, USA) supplemented with 5% fetal bovine serum and 1% penicillin–streptomycin. Cells were incubated in humidified atmosphere of 5% CO2 at 37°C.
After seeding, LSEC cells were stimulated with different concentrations of TGF-β1 (0, 1, 5, 10, 50, 100 ng/mL, 100-21, PeproTech, Cranbury, New Jersey, USA) for 48 h. The cells were collected for real-time PCR to measure the effects of TGF-β1 on endothelial cells.
Reverse transcription-polymerase chain reaction
The cellular RNA was extracted by TRIzol (15596018, Thermo Fisher Scientific, Carlsbad, California, USA) and reverse-transcribed into cDNA using a commercial reverse transcription kit (A5000, Promega, Beijing, China). The real-time PCR was performed using a SYBR Green PCR Master Mix (FP215, Tiangen, Beijing, China) in accordance with the manufacturer’s protocol. Target genes were quantified using the 2–ΔΔCt method and normalization with the expression of GAPDH.
The primers used were as follows:
| Gene | Species | Forward 5′ →3′ | Reverse 5′→3′ |
| thrombomodulin (TM) | Homo | CGACCTTCCT CAATGCCAGT CAG |
CGTCGCCGTT CAGTAGCAAGG |
| vWF | Homo | GAAGCAGACG ATGGTGGATTC CTC |
AGCAATGGT GTCGCAGAA GCAG |
| ICAM-1 | Homo | GTCACCTATGGC AACGACTCCTTC |
AGTGTCTCCT GGCTCTGG TTCC |
| VEGF | Homo | AGAAGGAGGAG GGCAGAATCAT CAC |
GGGCACACAGG ATGGCTTGAAG |
Thromboelastography
Thromboelastography (TEG) (TEG 5000, Thrombelastograph Hemostasis Analyzer System, Haemonetics Corporation, Braintree, MA, USA) was performed to assess coagulation profile. Clot formation was triggered using Kaolin in vitro. The variables of TEG include rate of clot formation (α angle), maximum amplitude (MA), clot time (R), clot formation time (K), and coagulation index (CI).
Statistics
Continuous variable data were reported as mean ± standard error (when the values were normally distributed) or median and interquartile range (IQR) (when the values had a distribution that departed significantly from normal). All data regarding categorical variables are shown as n (proportion). We tested the independence of categorical variables by x2 test. We used Student’ unpaired t-test and Pearson correlation analysis to evaluate normally distributed continuous variables. Mann–Whitney U test was employed for all the other variables between the two groups. Levene’s test was used for testing the equality of variances. Pearson correlation analysis were performed to explore the relationship between TGF-β1 and factors associated with coagulation and endothelial injury. Only two-tailed probabilities were used for testing statistical significance. Probability values < 0.05 were regarded as statistically significant. All calculations were made with the SPSS 24.0.
Results
General characteristics
A total of 53 patients with cirrhotic varices were included and the characteristics of all individuals were summarized in Table 1. The baseline characteristics indicated that males accounted for 43.40% and the mean age of included individuals was 54 ± 11 years old. Of 53 patients, there were 6 (13.04%) participants who had hypertension and 23 (43.40%) diabetes. A total of 17 (36.17%) patients had experienced splenectomy. Most patients were Child-Pugh A (n = 32, 60.38%) while 21 (39.62%) patients were Child-Pugh B. About half of the included patients suffered from hepatitis B cirrhosis (54.72%).
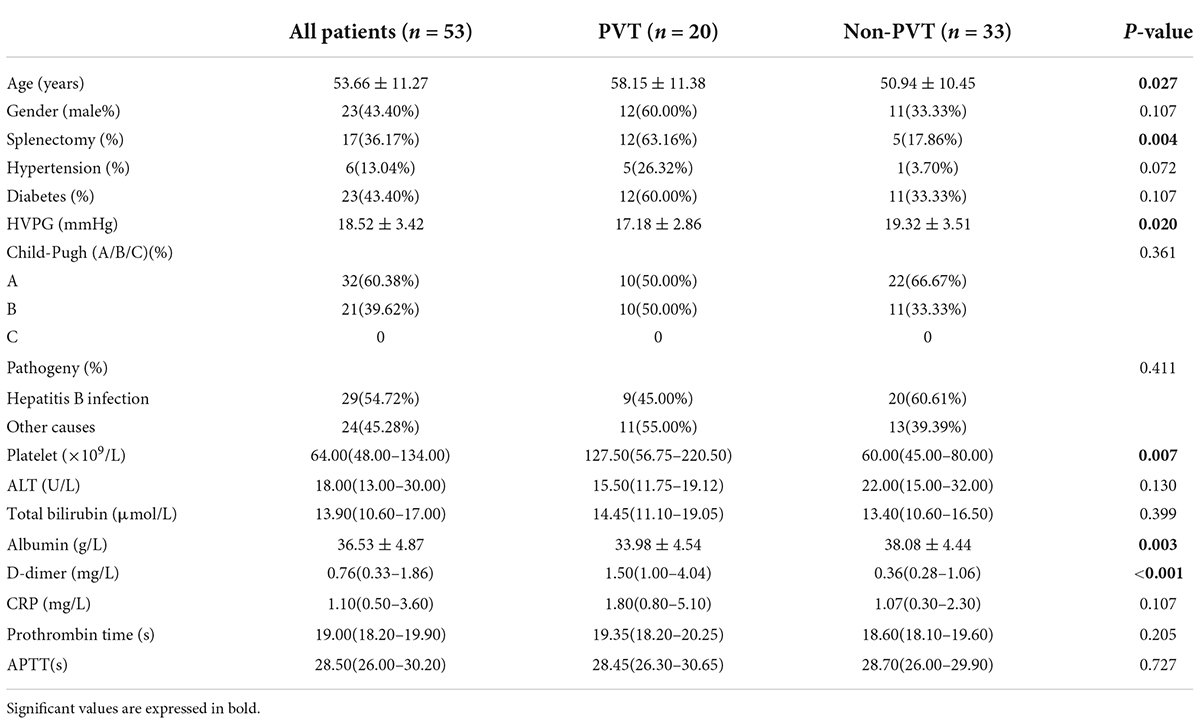
Table 1. Demographic characteristics and laboratory exanimations of cirrhotic patients.
Patients were divided into the PVT and non-PVT groups (n = 20 vs. 33). There was no significant difference observed in gender, hypertension, diabetes, or cirrhosis etiology between the two groups. More patients in the PVT group had experienced splenectomy (12 vs. 5, P = 0.004). Platelet counts significantly increased in the PVT group (127.50 vs. 60.00 × 109/L, P = 0.007) as albumin(33.98 ± 4.54 vs. 38.08 ± 4.44 g/L, P = 0.003) remarkably decreased. Correspondingly, ALT, total bilirubin, prothrombin time, and APTT showed no significant difference.
Transforming growth factor-beta1 was related to the occurrence of portal vein thrombosis in cirrhosis
The comparison of inflammatory mediators was performed in cirrhotic patients with or without PVT. As shown in Table 2, TGF-β1 [8.78 ± 0.71 vs. 8.25 ± 0.64 log (pg/ml), P = 0.009] interleukin 6 (IL-6, 4.80 vs. 2.40, P = 0.045) significantly increased in the PVT group, whereas other inflammatory mediators [procalcitonin (PCT), C-reactive protein (CRP), lipopolysaccharides (LPS), erythrocyte sedimentation rate (ESR), tumor necrosis factorα (TNFα), interleukin 2 receptor (IL-2R), interleukin 6 (IL-6), and interleukin 8 (IL-8)] showed no difference between two groups. After being stratified by splenectomy, no significant difference in TGF-β1 was observed between the two groups [8.26 ± 0.68 vs. 8.12 ± 0.59, P = 0.611 log (pg/ml); 9.06 ± 0.60 vs. 8.77 ± 0.84 log (pg/ml), P = 0.515 in non-splenectomized and splenectomized patients, respectively] (Supplementary Tables 1, 2). Besides, we found that patients with splenectomy exhibited significantly increased platelets (166.82 ± 94.67 vs. 73.95 ± 55.27 × 109, P = 0.001) and TGF-β1 levels [8.98 ± 0.66 vs. 8.15 ± 0.60 log (pg/ml), P < 0.001] compared with those without splenectomy.
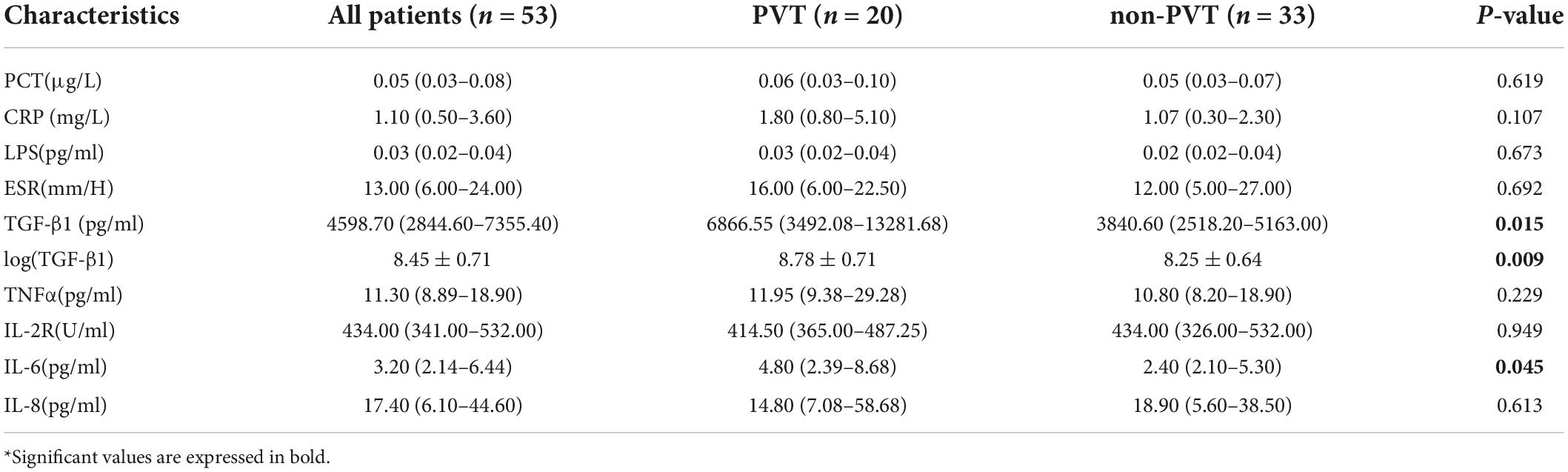
Table 2. The comparison of levels of inflammatory cytokines between portal vein thrombosis (PVT) and non-PVT groups.
To investigate the relationship between cirrhosis and TGF-β1, the levels of inflammatory mediators were compared between healthy individuals and cirrhotic patients. A slight increase tendency of TGF-b1 levels (8.45 ± 0.71 vs. 8.56 ± 0.17 log(pg/ml), P = 0.303) was observed in cirrhosis (Supplementary Table 3).
To ascertain whether a change of TGF-β1 influences the pathogenesis of PVT, the correlation between TGF-β1 and parameters associated with coagulation and endothelial dysfunction were explored (Figure 1). The Pearson’s correlation coefficient between TGF-β1 and platelet as well as fibrinogen was 0.733 (P < 0.001) and 0.494 (P = 0.027), respectively, which is correspondent with TGF-β1’s derivation from platelets. TEG showed that TEG-CI (P = 0.004), TEG-MA (P = 0.004), and TEG-α (P = 0.021) were closely related to TGF-β1 as Pearson’s correlation coefficients were 0.604, 0.608, and 0.511, respectively. TEG-MA represents the interaction between platelet and fibrinogen, and TEG-α represents fibrinogen levels, reflecting the strength and rate of clot formation. Taken together, the relationships between TGF-β1 and fibrinogen and parameters of TEG indicate that TGF-β1 may partake in the hypercoagulable status in PVT.
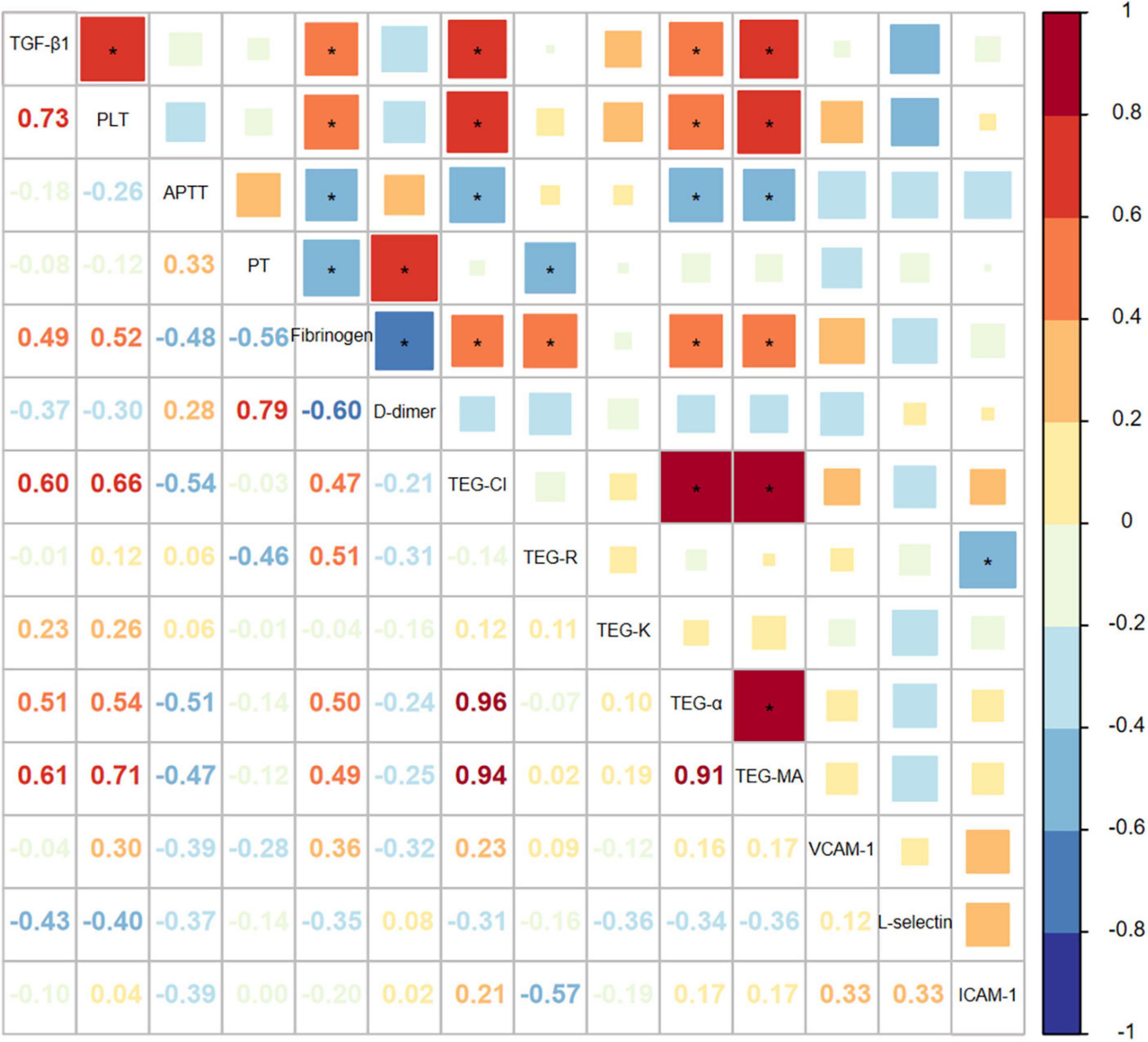
Figure 1. The correlation between TGF-β1 and factors associated with coagulation and endothelial dysfunction in portal vein thrombosis (PVT) patients.
Transforming growth factor-beta1 contributes to endothelial dysfunction
To further investigate the influence of TGF-β1 on endothelial cells, in vitro experiments were subsequently performed. LSECs were cultured and treated with different concentrations of TGF-β1. The mRNA expression of endothelial damage [von Willebrand Factor (vWF), thrombomodulin (TM)], adhesion molecule (ICAM-1), and angiogenesis vascular endothelial growth factor (VEGF) was significantly increased after the treatment of TGF-β1 (Figure 2). The alterations of ICAM-1, and VEGF mRNA expression were in parallel with the concentration of TGF-β1. These results provided mechanistic evidence for pro-thrombotic effects of TGF-β1 in PVT.
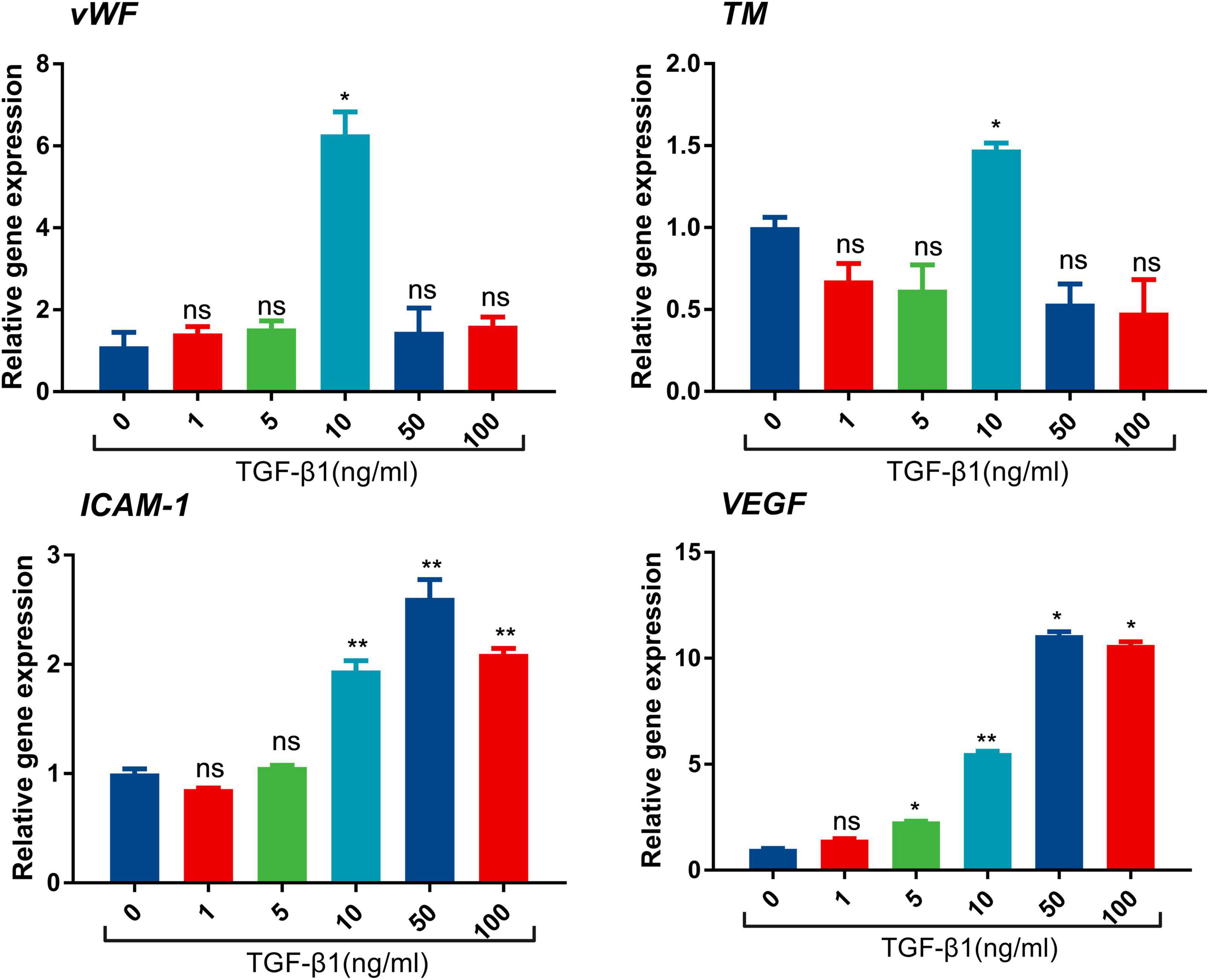
Figure 2. mRNA expression of vWF, intercellular adhesion moleclar-1 (ICAM-1), TM, and VEGF after the treatment of TGF-β1 in endothelial cells. (*p < 0.05, **p < 0.01). Data are expressed as mean ± SEM. Three replicates per group.
Discussion
In this study, we discovered that TGF-β1 was significantly elevated in cirrhotic patients with PVT. The results suggested that TGF-β1 was positively correlated with platelet and fibrinogen, TEG-CI, TEG-MA, as well as TEG-α, which uncovered a hypercoagulable state. In vitro experiments verified the pro-thrombotic effects of TGF-β1 through up-regulating vWF, TM, ICAM-1, and VEGF. Our study showed that TGF-β1 plays a pivotal role in PVT by partaking in hypercoagulability and promoting endothelial dysfunction in cirrhosis.
As a common and serious complication of cirrhosis, the clinical impact of PVT varies from asymptomatic to life-threatening conditions, which is related to accumulating risk of variceal bleeding and mortality (12–15). The mechanism of cirrhosis-induced PVT is multifactorial. Previous studies mainly focused on Virchow’s triad, but further investigations regarding the role of the inflammatory mediator, TGF-β1, in PVT development are lacking. Anticoagulation therapy is a well-accepted therapeutic method for PVT. However, the efficacy of anticoagulation treatment is far from ideal, as only 42–53% of cases could achieve complete portal vein recanalization (16). Besides, some patients exhibited poor responses to anticoagulation treatment in clinical practice (16, 17). The unsatisfactory performance of anticoagulation treatment suggested that PVT might have other underlying mechanisms, which deserves further investigation.
Accumulating evidence indicated that inflammatory response could simultaneously act as a cause and consequence of venous thrombosis (18). Systemic and local inflammation contributes to the occurrence of PVT (5, 19–21). TGF-β1 family is composed of many multifunctional cytokines, including TGF-β1, bone morphogenetic proteins and anti-Müllerian hormone (22).
Previous studies have explored the role of TGF-β1 in bleeding events and coagulation. TGF-β1 polymorphisms could result in vascular malformations, which are associated with an increased risk of bleeding complications. Kim (23) proposed that TGF-β1 polymorphisms were associated with bleeding complications in patients with warfarin therapy after cardiac valve replacement. For patients with cirrhotic portal hypertension, TGF-β1 plays a prominent role in angiogenesis and vascular development, resulting in the formation of collateral vessels. As a result of that, elevated TGF-β1 in cirrhosis may not increase the risk of bleeding. The relationships between TGF-β1 and hemorrhage in cirrhosis need further investigations. Numerous researches have been carried out on the effects of TGF-β1 on atherosclerosis and venous thrombosis, but little attention has been paid to the role of TGF-β1 in PVT development. The effects of TGF-β1 are controversial in atherosclerosis. On the one hand, TGF-β1 was believed to exert antiatherogenic effects (24) and on the other hand, some studies proposed that endothelial-derived TGF-β1 lead to vessel wall inflammation and atherosclerotic plaque growth (25–27). In venous thrombosis, the knockout of TGF-β1 resulted in smaller thrombi in mice without affecting platelet function. TGF β1 signaling via TGF-β1RI impairs thrombus resolution by favoring fibrosis and endothelial dysfunction (11). Our previous study has confirmed that continuous stretch of hepatic endothelial cells leads to upregulation of TGF-β1, caused by increased ROS levels (10). Lee (28) found a significant increase in plasma TGF-β1 levels in hepatocellular carcinoma (HCC) patients with PVT compared with those without tumoral PVT. It is difficult to distinguish blood clot formation in the portal venous system from tumoral PVT but their mechanisms are completely different (1). In order to investigate the effects of TGF-β1 on blood clot formation in cirrhotic PVT, patients with hepatic carcinoma and other malignancy were excluded. Our results elucidate that elevated TGF-β1 is closely associated with non-tumoral PVT. As it is widely believed that 40–45% of plasma TGF-β1 was secreted by platelets, the relationship between TGF-β1 and platelets was consistent with our results, suggesting the potent potential value of TGF-β1 in PVT development.
Splenectomy is the independent risk factor of PVT and significantly contributes to the increase of platelets. At the same time, platelets are the leading carrier of TGFβ in the body, responsible for about 40–45% of all TGF-β1 in peripheral blood. According to our results, a relatively higher prevalence of splenectomy was observed in the PVT group and patients with splenectomy had higher platelet levels and almost the same TGF-β1 levels in the PVT group. The absence of association between TGF-β1 and PVT after stratifying splenectomy indicated that splenectomy had a pivotal effect on PVT onset. Previous studies have elucidated increased TGF-β1 production after splenectomy, inconsistent with our results (29, 30). Besides, platelets and TGF-β1 exhibited a strong positive correlation (coef: 0.733, P < 0.001) based on our findings. Therefore, we supposed that elevated TGF-β1 levels derived from platelets might be owing to splenectomy. Splenectomy may promote PVT development by upregulating TGF-β1 levels in cirrhosis.
During blood clotting, various agents, such as thrombin, could activate platelet and thus lead to the secretion of TGF-β1 (31). The close association between TGF-β1 and fibrinogen as well as parameters of TEG in our results corroborated that TGF-β1 was related to activated coagulation process in the course of blood clotting. The augment in markers of endothelial injury, adhesion molecules, and angiogenesis indicated endothelial dysfunction and reflected a thrombotic tendency. RT-PCR confirmed the prothrombotic effects of TGF-β1 by promoting endothelial dysfunction. Our investigation of the mechanisms of TGF-β1 in PVT helps gain greater insight into molecular pathogenesis. The pathogenetic effects of TGF-β1 in promoting thrombosis deserve further investigation.
This study has a few limitations. First of all, this is a cross-sectional study so only the association between TGF-β1 and PVT could be explored. Secondly, only reverse transcription-polymerase chain reaction (RT-PCR) was performed to evaluate the function of TGF-β1 on endothelial cells. Protein assessment and other molecular biology experiments are needed. Moreover, larger prospective validation studies, as well as mechanistic studies, should be conducted in the future.
Conclusion
Transforming growth factor-1 was related to PVT in cirrhotic patients, which was closely associated with hypercoagulable status and promoting hepatic endothelial dysfunction. Further clinical and mechanistic studies are needed to investigate the role of TGF-β1 as a potential pathogenetic factor of PVT in the cirrhotic population.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethic Committee of Zhongshan Hospital, Fudan University. The patients/participants provided their written informed consent to participate in this study.
Author contributions
SJ and YA performed the experiments, analyzed, interpreted the data, and drafted the manuscript. LN and LW helped in data collection and data analysis. XH conceived the study, collected, and performed the critical revision of the manuscript. SC contributed significantly to critical revision of the manuscript. All authors contributed to the study and involved in critical revision of the manuscript for important intellectual content.
Funding
This study was supported by the Innovation Fund of Science and Technology Commission of Shanghai Municipality (No. 19411970200), Shanghai Sailing Program (No. 19YF1406500), and Zhongshan Hospital Foundation (No. 2021ZSYQ03). The funders played no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer YW declared a past co-authorship with several of the authors XH and SC to the handling editor.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.938397/full#supplementary-material
Abbreviations
PVT, portal vein thrombosis; TGF- β, transforming growth factor-beta; LSECs, liver sinusoidal endothelial cells; ICAM-1, intercellular adhesion moleclar-1; VCAM-1, vascular cell adhesion molecule-1; TEG, thromboelastography; PCT, procalcitonin; CRP, C-reactive protein; LPS, lipopolysaccharides; ESR, erythrocyte sedimentation rate; TNF α, tumor necrosis factor α; IL-2R, interleukin 2 receptor; IL-6, interleukin 6; IL-8, interleukin 8.
References
1. Senzolo M, Garcia-Tsao G, Garcia-Pagan JC. Current knowledge and management of portal vein thrombosis in cirrhosis. J Hepatol. (2021) 75:442–53. doi: 10.1016/j.jhep.2021.04.029
2. Praktiknjo M, Trebicka J, Carnevale R, Pastori D, Queck A, Ettorre E, et al. Von Willebrand and Factor VIII portosystemic circulation gradient in cirrhosis: implications for portal vein thrombosis. Clin Transl Gastroenterol. (2020) 11:e00123. doi: 10.14309/ctg.0000000000000123
3. Turon F, Driever EG, Baiges A, Cerda E, García-Criado A, Gilabert R, et al. Predicting portal thrombosis in cirrhosis: a prospective study of clinical, ultrasonographic and hemostatic factors. J Hepatol. (2021) 75:1367–76. doi: 10.1016/j.jhep.2021.07.020
4. Intagliata NM, Caldwell SH, Tripodi A. Diagnosis, development, and treatment of portal vein thrombosis in patients with and without cirrhosis. Gastroenterology. (2019) 156:1582–99.e1. doi: 10.1053/j.gastro.2019.01.265
5. Huang X, Fan X, Zhang R, Jiang S, Yang K, Chen S, et al. Systemic inflammation and portal vein thrombosis in cirrhotic patients with gastroesophageal varices. Eur J Gastroenterol Hepatol. (2020) 32:401–5. doi: 10.1097/MEG.0000000000001526
6. Nery F, Carneiro P, Correia S, Macedo C, Gandara J, Lopes V, et al. Systemic inflammation as a risk factor for portal vein thrombosis in cirrhosis: a prospective longitudinal study. Eur J Gastroenterol Hepatol. (2021) 33(Suppl. 1):e108–13. doi: 10.1097/MEG.0000000000001982
7. Ghafoory S, Varshney R, Robison T, Kouzbari K, Woolington S, Murphy B, et al. Platelet TGF-β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. (2018) 2:470–80. doi: 10.1182/bloodadvances.2017010868
8. Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. (1983) 258:7155–60. doi: 10.1016/S0021-9258(18)32345-7
9. Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. (2000) 342:1350–8. doi: 10.1056/NEJM200005043421807
10. Dong G, Huang X, Jiang S, Ni L, Chen S. Simvastatin mitigates apoptosis and transforming growth factor-beta upregulation in stretch-induced endothelial cells. Oxid Med Cell Longev. (2019) 2019:6026051. doi: 10.1155/2019/6026051
11. Bochenek ML, Leidinger C, Rosinus NS, Gogiraju R, Guth S, Hobohm L, et al. Activated endothelial TGFbeta1 signaling promotes venous thrombus nonresolution in mice via endothelin-1: potential role for chronic thromboembolic pulmonary hypertension. Circ Res. (2020) 126:162–81. doi: 10.1161/CIRCRESAHA.119.315259
12. Zhang Y, Xu BY, Wang XB, Zheng X, Huang Y, Chen J, et al. Prevalence and clinical significance of portal vein thrombosis in patients with cirrhosis and acute decompensation. Clin Gastroenterol Hepatol. (2020) 18:2564–72.e1. doi: 10.1016/j.cgh.2020.02.037
13. Faccia M, Ainora ME, Ponziani FR, Riccardi L, Garcovich M, Gasbarrini A, et al. Portal vein thrombosis in cirrhosis: why a well-known complication is still matter of debate. World J Gastroenterol. (2019) 25:4437–51. doi: 10.3748/wjg.v25.i31.4437
14. Cool J, Rosenblatt R, Kumar S, Lucero C, Fortune B, Crawford C, et al. Portal vein thrombosis prevalence and associated mortality in cirrhosis in a nationally representative inpatient cohort. J Gastroenterol Hepatol. (2019) 34:1088–92. doi: 10.1111/jgh.14501
15. Williams S, Chan AK. Neonatal portal vein thrombosis: diagnosis and management. Semin Fetal Neonatal Med. (2011) 16:329–39.
16. Rodrigues SG, Sixt S, Abraldes JG, De Gottardi A, Klinger C, Bosch J, et al. Systematic review with meta-analysis: portal vein recanalisation and transjugular intrahepatic portosystemic shunt for portal vein thrombosis. Aliment Pharmacol Ther. (2019) 49:20–30. doi: 10.1111/apt.15044
17. Francoz C, Belghiti J, Vilgrain V, Sommacale D, Paradis V, Condat B, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut. (2005) 54:691–7. doi: 10.1136/gut.2004.042796
18. Branchford BR, Carpenter SL. The role of inflammation in venous thromboembolism. Front Pediatrics. (2018) 6:142. doi: 10.3389/fped.2018.00142
19. Ijaz S, Yang W, Winslet MC, Seifalian AM. Impairment of hepatic microcirculation in fatty liver. Microcirculation. (2003) 10:447–56.
20. Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med. (2011) 365:147–56. doi: 10.1056/NEJMra1011170
21. Queck A, Carnevale R, Uschner FE, Schierwagen R, Klein S, Jansen C, et al. Role of portal venous platelet activation in patients with decompensated cirrhosis and TIPS. Gut. (2020) 69:1535–6. doi: 10.1136/gutjnl-2019-319044
22. Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. (2006) 86:515–81. doi: 10.1152/physrev.00024.2005
23. Kim W, Yee J, Chang BC, Chung JE, Lee KE, Gwak HS, et al. TGF-β1 polymorphism increases the risk of bleeding complications in patients on oral anticoagulant after cardiac valve replacement. Heart Vessels. (2021) 36:1885–91. doi: 10.1007/s00380-021-01867-2
24. Mallat Z, Gojova A, Marchiol-Fournigault C, Esposito B, Kamaté C, Merval R, et al. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ Res. (2001) 89:930–4. doi: 10.1161/hh2201.099415
25. Chen PY, Qin L, Li G, Wang Z, Dahlman JE, Malagon-Lopez J, et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat Metab. (2019) 1:912–26. doi: 10.1038/s42255-019-0102-3
26. Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. (2015) 125:4514–28. doi: 10.1172/JCI82719
27. Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. (2016) 7:11853. doi: 10.1038/ncomms11853
28. Lee D, Chung YH, Kim JA, Lee YS, Lee D, Jang MK. Transforming growth factor beta 1 overexpression is closely related to invasiveness of hepatocellular carcinoma. Oncology. (2012) 82:11–8. doi: 10.1159/000335605
29. Haraguchi K, Shimura H, Ogata R, Inoue H, Saito T, Kondo T, et al. Focal segmental glomerulosclerosis associated with essential thrombocythemia. Clin Exp Nephrol. (2006) 10:74–7. doi: 10.1007/s10157-005-0391-6
30. Karakantza M, Mouzaki A, Theodoropoulou M, Bussel JB, Maniatis A. Th1 and Th2 cytokines in a patient with Evans’ syndrome and profound lymphopenia. Br J Haematol. (2000) 110:968–70. doi: 10.1046/j.1365-2141.2000.02296.x
Keywords: portal vein thrombosis, transforming growth factor-beta, cirrhosis, endothelial damage, hypercoagulability
Citation: Jiang S, Ai Y, Ni L, Wu L, Huang X and Chen S (2022) Platelet-derived TGF-β1 is related to portal vein thrombosis in cirrhosis by promoting hypercoagulability and endothelial dysfunction. Front. Cardiovasc. Med. 9:938397. doi: 10.3389/fcvm.2022.938397
Received: 10 May 2022; Accepted: 08 September 2022;
Published: 26 September 2022.
Edited by:
Jingyan Han, Boston University, United StatesReviewed by:
Hui-Chun Huang, Taipei Veterans General Hospital, TaiwanYichen Wang, Mercy Medical Center, United States
Copyright © 2022 Jiang, Ai, Ni, Wu, Huang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoquan Huang, aHVhbmcueGlhb3F1YW5AenMtaG9zcGl0YWwuc2guY24=
†These authors have contributed equally to this work