
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Cardiovasc. Med. , 22 June 2022
Sec. Cardiovascular Therapeutics
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.936142
This article is part of the Research Topic Women in Cardiovascular Therapeutics View all 16 articles
 Emily E. Bramel1,2
Emily E. Bramel1,2 Rustam Bagirzadeh1
Rustam Bagirzadeh1 Muzna Saqib1
Muzna Saqib1 Tyler J. Creamer1
Tyler J. Creamer1 Wendy A. Espinoza Camejo1,2
Wendy A. Espinoza Camejo1,2 LaToya Ann Roker3Jennifer Pardo Habashi4Harry C. Dietz1,5
LaToya Ann Roker3Jennifer Pardo Habashi4Harry C. Dietz1,5 Elena Gallo MacFarlane1,6*
Elena Gallo MacFarlane1,6*Angiotensin II (Ang II) type 1 receptor (AT1R) signaling controls both physiological and pathogenetic responses in the vasculature. In mouse models of Loeys-Dietz syndrome (LDS), a hereditary disorder characterized by aggressive aortic aneurysms, treatment with angiotensin receptor blockers (ARBs) prevents aortic root dilation and associated histological alterations. In this study we use germline and conditional genetic inactivation of Agtr1a (coding for the AT1a receptor) to assess the effect of systemic and localized AT1R signaling attenuation on aortic disease in a mouse model of LDS (Tgfbr1M318R/+). Aortic diameters and histological features were examined in control and Tgfbr1M318R/+ mice with either germline or Mef2CSHF-Cre mediated genetic inactivation of Agtr1a, the latter resulting in deletion in second heart field (SHF)-derived lineages in the aortic root and proximal aorta. Both systemic and regional AT1R signaling attenuation resulted in reduction of diameters and improvement of tissue morphology in the aortic root of LDS mice; these outcomes were associated with reduced levels of Smad2/3 and ERK phosphorylation, signaling events previously linked to aortic disease in LDS. However, regional AT1a inactivation in SHF-derived lineages resulted in a more modest reduction in aortic diameters relative to the more complete effect of germline Agtr1a deletion, which was also associated with lower blood pressure. Our findings suggest that the therapeutic effects of AT1R antagonisms in preclinical models of aortic disease depend on both regional and systemic factors and suggest that combinatorial approaches targeting both processes may prove beneficial for aneurysm mitigation.
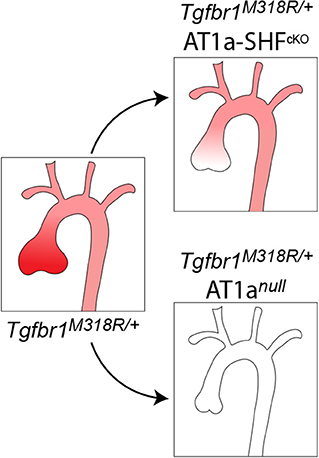
Graphical Abstract. The aortic root, which is composed primarily of SHF-derived cells, is a site of increased susceptibility to dilation in both patients and mouse models of Loeys-Dietz Syndrome (Tgfbr1M318R/+). This increased risk associates with regionally increased expression of AT1a (shown in red). Both SHF-specific (AT1a-SHFcKO) and germline (AT1anull) deletion of AT1a reduce aortic root dilation and associated histopathological alterations in Tgfbr1M318R/+ mice, with the latter, however, having a more pronounced effect.
Aneurysms of the thoracic aorta are characterized by progressive weakening of the aortic wall resulting first in dilation and, ultimately, life-threatening dissection and rupture (1, 2). Aortic pathology is primarily linked to maladaptive changes in vascular smooth muscle cells (VSMC) and defective remodeling of the extracellular matrix; however, systemic factors, such as elevated blood pressure, can further promote disease (3). Angiotensin II (Ang II) signaling via the Ang II type 1 receptor (AT1R) activates several signaling pathways that can influence aneurysm pathogenesis both systemically, through regulation of vasoconstriction and fluid homeostasis, and locally, through regulation of VSMC phenotype, matrix deposition and inflammation (4–6). Although rodents have two types of AT1R, AT1a and AT1b, signaling via the AT1a receptor (encoded by the Agtr1a gene) plays the primary role in promotion of aneurysm pathogenesis (7, 8). Binding of Ang II to AT1R directly activates specific signaling cascades, including those mediated by mitogen-activated protein kinases (MAPK) (6); engagement of AT1R can also lead to transactivation of growth factor receptors and increased expression of components of other signaling pathways, including those activated by Transforming Growth Factor-β (TGF-β), Platelet-derived growth factor (PDGF), and reactive oxygen species (ROS) (5, 9–11).
Loeys-Dietz Syndrome (LDS) is a hereditary aneurysm disorder caused by heterozygous inactivating mutations in positive effectors of the TGF-β signaling pathway; these mutations result in an initial impairment of signaling output, which is followed by compensatory upregulation at sites of disease (12–19). Although these mutations occur in genes expressed ubiquitously in the aorta, the aortic root is a site of increased susceptibility to dilation (18, 19). As observed in other mouse models of aortic aneurysm, aortic root dilation in LDS mouse models is prevented by treatment with angiotensin receptor blockers (ARB), in association with lowered blood pressure and attenuated AT1R-dependent signaling in the aortic wall (5, 18–20).
Second heart field (SHF) progenitors, identified in mice by conditional genetic reporters and the Mef2CSHF-Cre transgene (21), give rise to vascular smooth muscle cells (VSMCs), aortic fibroblasts and endothelial cells, all of which contribute to morphogenesis of the aortic wall (22). Whereas SHF-derived cells predominate in the root and proximal aorta, the contribution of VSMCs derived from the cardiac-neural crest (CNC) increases progressively along the proximal-to-distal axis (23, 24). Our previous work and that of others have shown that SHF-derived VSMCs are intrinsically more sensitive to the effects of LDS-causing mutations (18, 25, 26), express higher levels of Agtr1a, and show increased responsiveness to Ang II in culture (18), suggesting that AT1R signaling in these cells is a contributor to pathogenesis. In this study, we test the systemic and SHF-specific contribution of AT1a receptor signaling to aortic dilation in the Tgfbr1M318R/+ LDS mouse model by examining the aortic phenotype in mice with either germline or Mef2CSHF-Cre mediated Agtr1a deletion.
All animal experiments were conducted following protocols approved by the Animal Care and Use Committee at Johns Hopkins University School of Medicine. Mice were housed in the animal facility with unlimited access to standard chow and water with a light/dark cycle of 10/14 h. All mice were backcrossed to 129S6/SvEv mice (Taconic, 129SVE) for at least five generations; all experiments used littermates and cohort-mates as controls. Tgfbr1+/+and Tgfbr1M318R/+(19) were bred to Agtr1aflox/flox (The Jackson Laboratory, strain #016211) (27) mice, some bearing the Mef2cSHF-Cre transgene (gifted by the K.R. Chien lab at the Cardiovascular Research Center, Massachusetts General Hospital, Boston, Massachusetts, USA), to generate mice with second heart field-specific deletion of Agtr1a. These mice are referred to as AT1aSHFcKO. Tgfbr1+/+and Tgfbr1M318R/+ were also bred to mice with a global deletion of Agtr1aD/D, which were generated by deletion of the Agtr1aflox allele via a germline recombination event. These mice are referred to as AT1anull. Mice were genotyped twice, once at the beginning and then at the end of the study, using protocols described in Chen et al. (8) for the Agtr1a locus and Gallo et al. (19) for the Tgfbr1 locus.
Aortic dimensions were monitored by serial echocardiography using the Visual Sonics Vivo 2100 machine and a 30 mHz probe using a parasternal long-axis view, as previously described (18). Three independent measurements of the maximal internal diameters at the sinus of Valsalva were averaged for aortic root measurements; for ascending aorta measurements, measurements were taken at the maximal diameter. All measurements were taken during systole, with on open aortic valve. Operators blinded to genotype were responsible for imaging and measurements. Tail cuff blood pressure measurements were taken for mice using the Visitech BP-2000 Non-Invasive tail cuff device, also as previously described (19).
RNA was extracted according to previously described protocols (28). In brief, dissected aortic root tissue was placed in TRIzol (ThermoFisher, 15596018) and lysed using an MP Biomedicals FastPrep-24 5G automatic bead homogenizer. A Direct-zol RNA MiniPrep kit (Zymo Research, R2052) was used to extract and purify RNA according to the manufacturer's instructions. The High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, 4368813) was used according to the manufacturer's protocol and qPCR was performed using TaqMan reagents and probes (Applied Biosystems, 4369016; Agtr1a Mm01166161_m1, Hprt Mm00446968_m1) and run on the QuantStuido7 Flex.
After euthanasia by halothane inhalation (Millipore Sigma, H0150000) at a standard concentration (4%, 0.2 ml/L of container volume), the heart and thoracic aorta were dissected en bloc. Samples were then fixed overnight at 4oC in 4% paraformaldehyde in PBS (Electron Microscopy Sciences, 15710). The following day, samples were transferred to six well plates containing 70% ethanol and left overnight at 4oC. The entire sample was embedded in 2% agarose prior to paraffin embedding. Paraffin blocks were then cut into 5-μm radial sections (resulting in a longitudinal view of the vessel) that were either stained with Verhoeff-van Gieson (VVG; StatLab, STVGI) or used for immunofluorescence as described below. Slides stained with VVG were imaged using an Eclipse E400 microscope (Nikon Inc.) at 40 × magnification.
Elastic fiber content per area unit was quantified by a staff member of the Johns Hopkins School of Medicine Microscope Facility, who was blinded to the genotype of the VVG-stained sections. Color-decon2 (29) was used for unbiased automated selection of the two regions of interest (ROIs), elastic fibers and the cellular area in-between, and separation of corresponding vectors to individual channels. High intensity results, which identified blood cells, and very low intensity results, which identified non-vascular areas, were excluded from further analysis. Individual channels for each ROI were then converted to “binary” to measure the corresponding gray value (30), and this value was then converted to area to obtain the relative ratio of “elastic fiber” to “cells” content.
The following protocol was adapted from Cell Signaling Technology's Immunofluorescence Protocol with Formaldehyde Fixation. Paraffin-embedded sections were baked at 60°C for 15 min. Slides were deparaffinized in xylene and rehydrated by immersing in a graded alcohol series: 100% ethanol, 95% ethanol, 70% ethanol and 1 × PBS for 3 min each. Slides were then incubated in an antigen retrieval solution (10 mM sodium citrate buffer, 0.05% tween, pH 6.0) for 15 min at 90°C in a water bath. After cooling to room temperature, slides were incubated in fresh sodium borohydride solution (10 mg/ml PBS; Sigma-Aldrich, 452882) for 20 min. Slides were permeabilized with 1 × TBS (Quality Biological, 351086101) + 0.1% Triton X-100 (Sigma-Aldrich, T9284) + 0.1 M glycine (Sigma-Aldrich, G8898) for 20 min, then incubated with Fc Receptor Blocker (Innovex, NB309) for 20 min at room temperature, and then Background Buster (Innovex, NB306) for another 20 min. Slides were rinsed with 1 × TBS + Triton X-100 (TBT) and then incubated with either P-Smad3 (Abcam, ab52903) at 1:50 or P-ERK (Cell Signaling Technology, 4370) at 1:200 overnight at 4°C in a humid chamber. Slides were rinsed twice with TBT for 5 min in Donkey Anti-Rabbit Alexa Fluor 555 (ThermoFisher, A32794) at 1:100 for 45 min. Slides were again washed two times with TBT and once with TBS prior to mounting with Hard Set Mounting Media with DAPI (VECTASHIELD, H-1500). Images were acquired on a Zeiss LSM880 Airyscan FAST confocal microscope at 20× magnification and are presented as maximal intensity projection. Image adjustments to enhance visualization of information present in the original were applied equally across samples.
All statistical analyses were performed using GraphPad Prism 9. A Q = 5% in ROUT test was selected a priori as an exclusion criterion for outliers. If present, outliers are shown in figures as gray circles, but not included in tests for assessment of normality, which were performed using the Shapiro-Wilk test. Data that passed normality test upon exclusion of outliers was considered normally distributed and analyzed using Brown-Forsythe and Welch ANOVA test, with no assumptions as to equal variance among groups. The two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli was used for multiple comparison correction. Dataset that failed the normality test were analyzed using Kruskal–Wallis test, also followed by the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli for multiple comparison correction.
For individual time points, data are presented as a box-and-whiskers plot, with whiskers indicating minimum to maximum points, with all points shown. Growth curves over time were compared using a linear regression model, with least-square regression and no weighting; comparison between slopes was performed using the extra-sum-of-squares F-test, with P = 0.05. Error bars in growth curve plots refer to the 95% confidence interval (CI). Survival tables were analyzed using Fisher's exact test.
Tgfbr1M318R/+ mice (also referred to as LDS mice in this text), recapitulate many of the features observed in LDS patients, including dilation of the aortic root (18, 19). To determine if global or SHF-specific attenuation of AT1R signaling mitigated aortic dilation in LDS mice, we crossed these and control mice to either mice homozygous for the Agtr1aD allele (Agtr1aD/D, referred to as AT1anull) or to Agtr1aflox/flox mice also expressing the Mef2cSHF-Cre (21) transgene. Serial echocardiography was performed from 8 to 24 weeks of age, and aortic tissue collected and processed for histological analysis at the 24-week timepoint as previously described (18, 19). Blood pressure was also measured prior to sacrifice. Analysis of an initial experimental cohort showed that there were no significant differences in aortic measurements or rate of aortic enlargement between male and female Tgfbr1M318R/+ mice (Supplementary Figures 1A–C) however, control male mice were significantly larger than their female counterparts, and the difference in aortic diameters between female and male Tgfbr1M318R/+ mice at the 24-week time point approached significance (P = 0.06; Supplementary Figure 1B). For these reasons, male and female mice were analyzed separately according to current guidelines.
We found that the presence of the Mef2cSHF-Cre transgene in mice also carrying a Agtr1afloxallele resulted in relatively frequent recombination in the germline, leading to generation of both Agtr1aflox/D; Mef2cSHF-Cre and Agtr1aflox/flox; Mef2cSHF-Cre litters when breeding Agtr1aflox/+; Mef2cSHF-Cre or Agtr1aflox/flox Mef2cSHF-Cre mice. Germline recombination occurred in both male and female breeders whenever the Agtr1afloxallele was present in mice also carrying the Mef2cSHF-Cre transgene. However, the presence of the Agtr1aD null allele in heterozygosity did not significantly affect blood pressure, Agtr1a expression in the aorta, nor aortic size in either Tgfbr1+/+or Tgfbr1M318R/+ mice (Supplementary Figures 2, 3). Therefore, Agtr1aflox/D and Agtr1aflox/floxare collectively referred to as controls (AT1aCtrl) in the absence of the Cre recombinase, and as AT1a SHF-deficient mice (AT1aSHFcKO) if also expressing the Mef2cSHF-Cre recombinase. All ultrasound measurements and genotypes are provided in Supplementary Table 1.
Homozygous deletion of Agtr1a in SHF-derived lineages resulted in reduced aortic root dimeters in both female and male Tgfbr1M318R/+ mice relative to AT1aCtrl controls at the 16-week time-point; however, this effect remained significant only in female mice by 24 weeks of age (Figures 1A–G). Female AT1aSHFcKO Tgfbr1M318R/+ mice but not male mice also showed a reduced rate of growth from 8 to 24 weeks relative to AT1aCtrl Tgfbr1M318R/+ mice (Figures 1D,G). Blood pressure was not significantly affected by deletion of Agtr1a in SHF-derived lineages (Figures 1H,I).
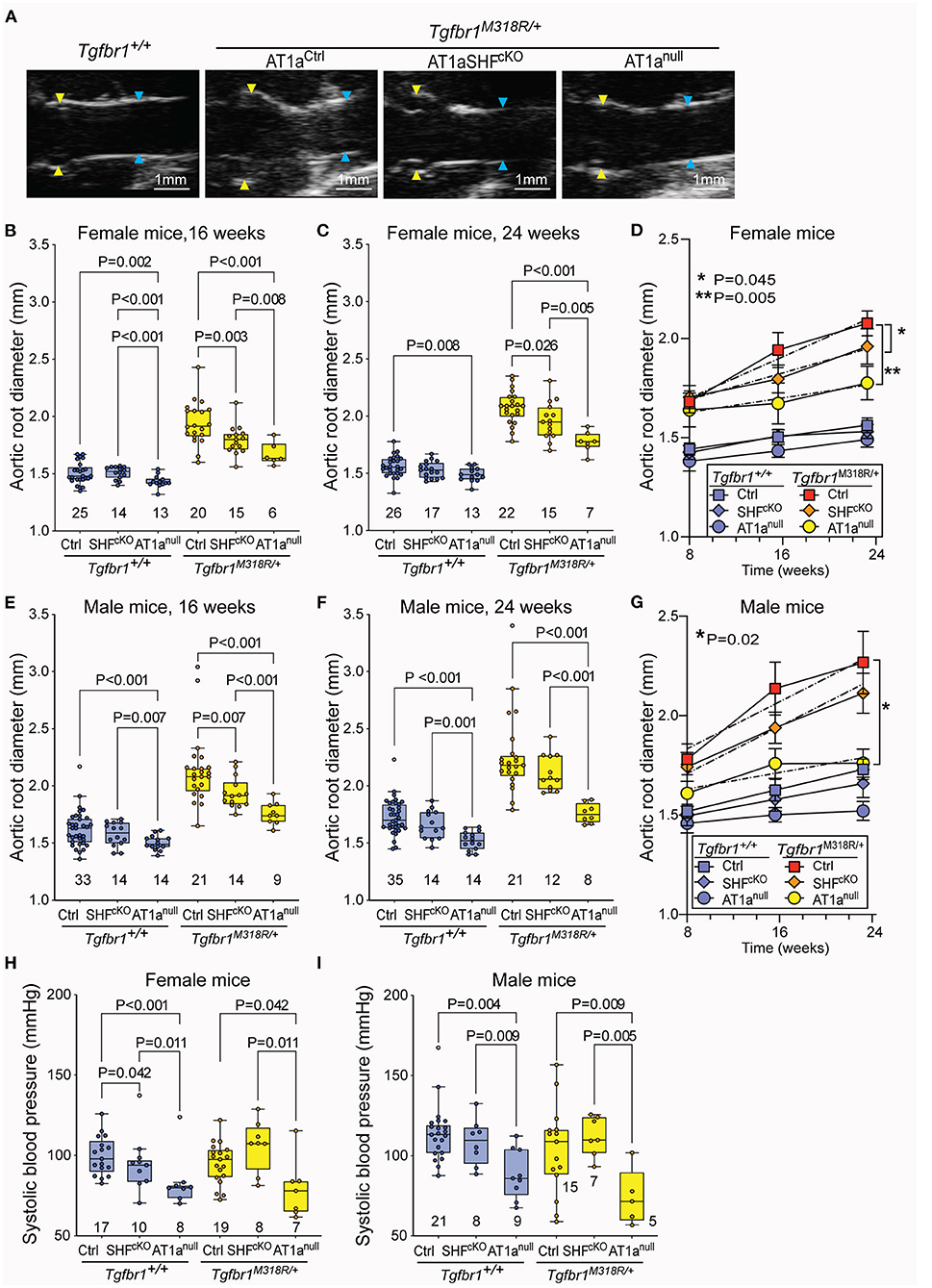
Figure 1. Global and SHF-specific deletion of Agtr1a result in reduction of aortic root dilation in LDS mice. (A) Representative echocardiograms of Tgfbr1+/+ and Tgfbr1M318R/+mice with and without conditional (AT1aSHFcKO) or global (AT1anull) deletion of Agtr1a, the aortic root is indicated by yellow arrows while the ascending aorta is indicated by blue arrows. Aortic root diameter as measured by echocardiography in both females (B–D) and males (E–G) at indicated time points. Tgfbr1+/+ mice are shown in blue and Tgfbr1M318R/+are shown in yellow. The number of animals per group is indicated. P-values refer to Brown-Forsythe ANOVA, followed by post-hoc test with multiple comparison FDR correction. In panel (C,E), the error bars represent the 95% Confidence Interval (CI), the dashed line indicates a simple linear-regression of serial echocardiographic measurements of aortic root diameter from 8 to 24 weeks of age; P-value refers to comparison between slopes using the extra-sum-of-squares F-test in GraphPad. (H,I) Systolic blood pressure as measured at 24 weeks of age. P-values refer to Brown-Forsythe ANOVA, followed by post-hoc test with multiple comparison FDR correction.
Homozygous germline Agtr1a deletion resulted in reduction of aortic root diameters and rate of growth in both male and female LDS mice up to 24 weeks of age (Figures 1A–G), and also associated with a reduction in blood pressure, consistent with previous analyses of AT1anull mice (31) (Figures 1H,I). No significant differences were observed in the diameter of the ascending aorta (Supplementary Figure 4A). Although our study was not designed nor powered to detect differences in survival, a significant decrease in survival between male Tgfbr1M318R/+ relative to control Tgfbr1+/+ mice was noted (Supplementary Figure 4B).
Histological sections of aortas were stained to visualize tissue architecture and elastic fibers, and the relative content of elastic fiber to cellular area was quantified using an ImageJ macro. Both AT1anull and AT1aSHFcKO Tgfbr1M318R/+ mice, of either sex, showed improved elastic fiber content relative to AT1aCtrl Tgfbr1M318R/+ mice (Figure 2 and Supplementary Figure 5). This improvement correlated with reduced levels of phosphorylation of both Smad2 and Smad3 (p-Smad2/3) and extracellular signal-regulated kinase 1 and 2 (p-ERK1/2), two signaling events previously shown to correlate with severity of aortic disease in mouse models of LDS and related conditions (5) (Figures 3A,B and Supplementary Figure 6). No specific localization relative to inner or outer media was noted for either p-Smad2/3 or p-ERK1/2 signal, possibly in consequence of the advanced stage of aortic disease and media disruption at the time point examined. However, whereas p-ERK1/2 signal was detectable in the endothelial and adventitial layer regardless of genotype or disease status, both systemic and SHF-specific AT1a deletion resulted in reduced p-ERK1/2 levels across the media compared Tgfbr1M318R/+ mice, which is consistent with previous observations (19).
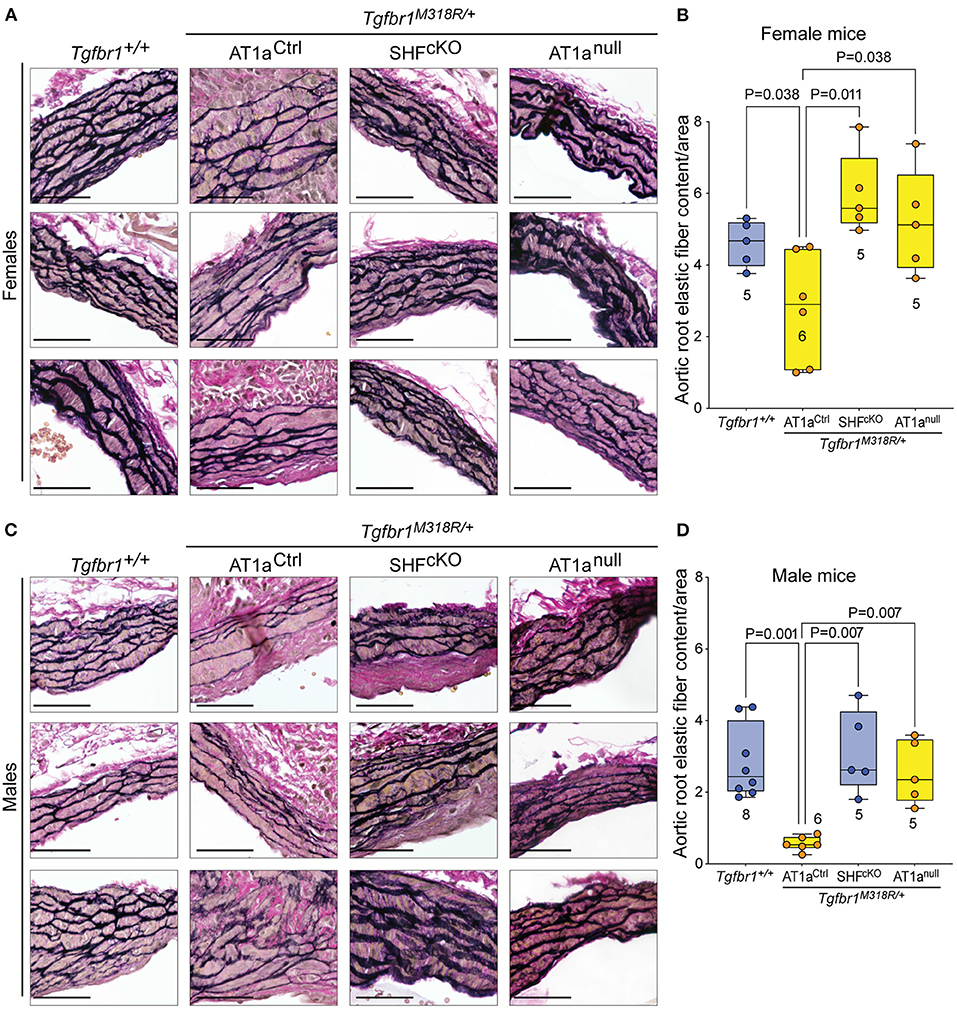
Figure 2. Global and SHF-specific deletion of Agtr1a result in improved histopathology. Representative sections of VVG-stained aortic roots and quantification of elastic fiber content relative to cellular area for female (A,B) and male (C,D) samples of indicated genotypes. Three representative images are shown per genotype for each sex. Images are shown at 20× magnification. Scale bar is 50 μm. Quantification of elastic fiber content relative to cellular area is shown in (C) for female samples, and in (D) for male samples, with higher value indicating a greater content of elastic fiber per area unit. The number of mice scored per group is indicated. P-values refer to Brown-Forsythe ANOVA, followed by post-hoc test with multiple comparison FDR correction.
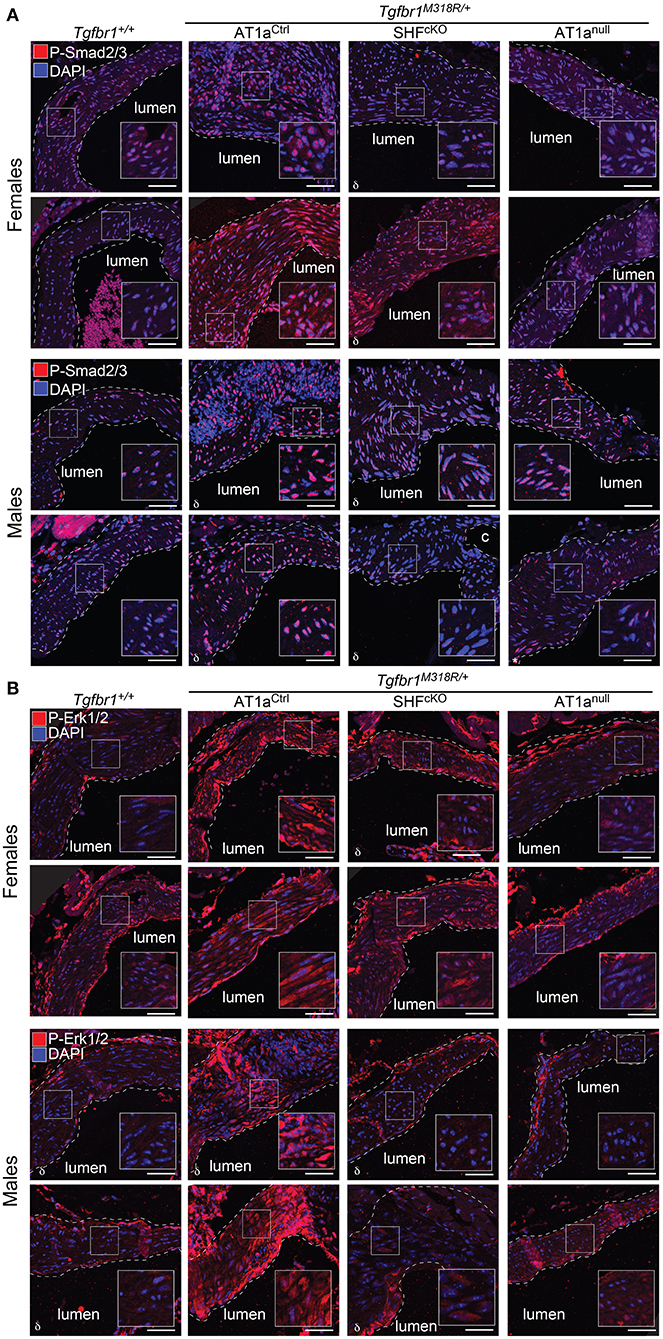
Figure 3. Global and SHF-specific deletion of Agtr1a result in reduced p-Smad2/3 and p-ERK1/2 signaling. Representative images of immunofluorescence for (A) p-Smad2/3 and (B) p-ERK1/2 at 24 weeks of age. Four independent biological replicates are shown per genotype, two from male animals and two from female animals. Insets identify locations shown at higher magnification. Images were acquired at 20× magnification. Scale bar is 50 μm. δ symbol in the left-hand corner of a given image indicates the section is from an animal heterozygous for the Agtr1aD null allele.
Administration of ARBs such as losartan has been shown to ameliorate aortic pathology in several mouse models of aneurysm, including LDS (5, 8, 18–20, 32). However, experiments based on pharmacological antagonism cannot disentangle the potential benefits of local antagonism from those accrued thanks to systemic effects. In addition, the existence of potential off-targets for drugs such as losartan has led to the hypothesis that some benefits of ARB administration may result from AT1R-independent effects (33).
In this study, we show that genetic deletion of AT1a in SHF-derived cells, which include VSMC residing primarily in the aortic root but also fibroblasts and subsets of endothelial cells (22), mitigates aortic dilation and improves aortic tissue architecture in LDS mice. However, this intervention fails to recapitulate the more robust reduction in aortic diameters observed after germline AT1a inactivation, particularly at later time points in male mice. We hypothesize that the additional benefit of systemic AT1a inactivation on aortic diameters may be due to both ablation in AT1a receptor signaling in non-SHF-derived cells and, possibly, lessening of mechanical stresses on the weakened wall secondary to reduction in blood pressure. Although our previous work showed that only SHF-derived VSMCs had higher expression of Agtr1a and increased responsiveness to Ang II (18), it is possible that AT1a receptor signaling in non-SHF-derived cells, including endothelial cells, may also play a role in LDS aortic dilation, similarly to what has been observed in other mouse models (27, 34).
Although we did not observe significant sexual dimorphism in the absence of additional genetic perturbations, there was a trend for larger aortic root diameters in male Tgfbr1M318R/+ mice relative to their female counterparts, especially at later time points. This was accompanied by a significant reduction of aortic root diameters in female but not male Tgfbr1M318R/+ mice with homozygous Agtr1a deletion in SHF-derived cells relative to controls. The presence of sexual dimorphism in LDS mouse models under specific circumstances is consistent with reports showing similar patterns in other hereditary connective tissue disorders, including both patients and mouse models of Marfan and Ehlers-Danlos syndrome (35–38). Although the mechanisms remain unclear, angiotensin II-driven aortic pathogenesis has been consistently shown to be accentuated in males relatively to females (39). We hypothesize that the severity of aortic disease in LDS mouse models generally masks the subtle effect of sexually dimorphic hormonal or chromosomal contributions, and that these effects may become more apparent in specific contexts that result in moderate amelioration of pathogenesis.
Despite its well-documented efficacy in preclinical animal models, randomized trials using the ARB losartan at doses sufficient to successfully reduced blood pressure have shown more modest and sometimes mixed results in the treatment of aneurysm in Marfan syndrome (MFS) (5, 40–42). Our study suggests that regional AT1R inhibition may be important for amelioration of aortic tissue architecture, and that better outcomes may be possible using strategies able to suppress both local and systemic AT1R signaling. Although this study explored the effects of AT1R inhibition only in the proximal thoracic aorta, the multiplicity of roles played by this signaling pathway in both physiological and pathological processes suggests that its inhibition may influence other LDS-associated phenotypes (12–19). An exploration of the effects of AT1 inhibition on both vascular and non-vascular LDS connective tissue anomalies would provide stronger evidence of therapeutic benefit.
Notably, a recent study by Chen et al. (8) has shown that administration of antisense oligonucleotides results in stable and durable reduction in levels of angiotensinogen, the precursor to Ang II, resulting in protection from aortic disease in the Fbn1C1041G/+ MFS mouse model. We speculate strategies that better mimic the early, robust, and continuous suppression of AT1R signaling achieved in animal models by germline deletion of Agtr1a (or by early ARB administration in drinking water and/or osmotic pump) may prove more efficacious for medical therapy in LDS and related conditions.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by the Johns Hopkins University Animal Care and Use Committee.
EG and HD are responsible for the conception and design of this study. RB initiated much of the experimental work, including animal breeding and echocardiography. JP acquired and analyzed echocardiographic images. EB and TC assisted with echocardiography and animal breeding. MS assisted in genotyping and performed the histological staining. EB and WE performed the immunofluorescence. LR developed a macro in Image J to quantify the elastic fiber content relative to cellular area in aortic sections. EB assisted EG in writing and preparing figures for the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R01HL147947 and by a generous gift from the Loeys-Dietz Foundation. EM was also supported by funding provided to Johns Hopkins by the Broccoli family. Image acquisition was also supported by NIH award number S10OD023548 to the School of Medicine Microscope Facility.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Djahida Bedja for assistance in the acquisition of ultrasound images, and the Dietz laboratory for sharing resources, protocols, and helpful advice for this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.936142/full#supplementary-material
1. Shen YH, LeMaire SA, Webb NR, Cassis LA, Daugherty A, Lu HS. Aortic aneurysms and dissections series. Arterioscler Thromb Vasc Biol. (2020) 40:e37–46. doi: 10.1161/ATVBAHA.120.313991
2. Shen YH, LeMaire SA, Webb NR, Cassis LA, Daugherty A, Lu HS. Aortic aneurysms and dissections series: part II: dynamic signaling responses in aortic aneurysms and dissections. Arterioscler Thromb Vasc Biol. (2020) 40:e78–86. doi: 10.1161/ATVBAHA.120.313804
3. Boczar KE, Boodhwani M, Beauchesne L, Dennie C, Chan KL, Wells GA, et al. Aortic stiffness, central blood pressure, and pulsatile arterial load predict future thoracic aortic aneurysm expansion. Hypertension. (2021) 77:126–34. doi: 10.1161/HYPERTENSIONAHA.120.16249
4. Kawai T, Forrester SJ, O'Brien S, Baggett A, Rizzo V, Eguchi S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol Res. (2017) 125(Pt A):4–13. doi: 10.1016/j.phrs.2017.05.008
5. van Dorst DCH, de Wagenaar NP, van der Pluijm I, Roos-Hesselink JW, Essers J, Danser AHJ. Transforming growth factor-beta and the renin-angiotensin system in syndromic thoracic aortic aneurysms: implications for treatment. Cardiovasc Drugs Ther. (2020) 41:1233–52. doi: 10.1007/s10557-020-07116-4
6. Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. (2018) 98:1627–738. doi: 10.1152/physrev.00038.2017
7. Poduri A, Owens AP. 3rd, Howatt DA, Moorleghen JJ, Balakrishnan A, Cassis LA, et al. Regional variation in aortic AT1b receptor mRNA abundance is associated with contractility but unrelated to atherosclerosis and aortic aneurysms. PLoS ONE. (2012) 7:e48462. doi: 10.1371/journal.pone.0048462
8. Chen JZ, Sawada H, Ye D, Katsumata Y, Kukida M, Ohno-Urabe S, et al. Deletion of AT1a (Angiotensin II Type 1a) receptor or inhibition of angiotensinogen synthesis attenuates thoracic aortopathies in Fibrillin1c1041g/+ mice. Arterioscler Thromb Vasc Biol. (2021) 41:2538–50. doi: 10.1161/ATVBAHA.121.315715
9. Gibbons GH, Pratt RE, Dzau VJ. Vascular smooth muscle cell hypertrophy vs. hyperplasia autocrine transforming growth factor-beta 1 expression determines growth response to angiotensin II. J Clin Invest. (1992) 90:456–61. doi: 10.1172/JCI115881
10. Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci. (2008) 29:367–74. doi: 10.1016/j.tips.2008.05.003
11. Sanchez-Guerrero E, Midgley VC, Khachigian LM. Angiotensin II induction of PDGF-C expression is mediated by AT1 receptor-dependent Egr-1 transactivation. Nucleic Acids Res. (2008) 36:1941–51. doi: 10.1093/nar/gkm923
12. Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. (2005) 37:275–81. doi: 10.1038/ng1511
13. van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. (2011) 43:121–6. doi: 10.1038/ng.744
14. Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. (2012) 44:922–7. doi: 10.1038/ng.2349
15. Wischmeijer A, Van Laer L, Tortora G, Bolar NA, Van Camp G, Fransen E, et al. Thoracic aortic aneurysm in infancy in aneurysms-osteoarthritis syndrome due to a novel SMAD3 mutation: further delineation of the phenotype. Am J Med Genet A. (2013) 161A:1028–35. doi: 10.1002/ajmg.a.35852
16. MacCarrick G, Black JH 3rd, Bowdin S, El-Hamamsy I, Frischmeyer-Guerrerio PA, Guerrerio AL, et al. Loeys-Dietz Syndrome: a primer for diagnosis and management. Genet Med. (2014) 16:576–87. doi: 10.1038/gim.2014.11
17. Bertoli-Avella AM, Gillis E, Morisaki H, Verhagen JM, de Graaf BM, van de Beek G, et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. (2015) 65:1324–36.
18. MacFarlane EG, Parker SJ, Shin JY, Kang BE, Ziegler SG, Creamer TJ, et al. Lineage-specific events underlie aortic root aneurysm pathogenesis in Loeys-Dietz syndrome. J Clin Invest. (2019) 129:659–75. doi: 10.1172/JCI123547
19. Gallo EM, Loch DC, Habashi JP, Calderon JF, Chen Y, Bedja D, et al. Angiotensin II-dependent TGF-beta signaling contributes to Loeys-Dietz Syndrome vascular pathogenesis. J Clin Invest. (2014) 124:448–60. doi: 10.1172/JCI69666
20. Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, et al. Dimorphic effects of transforming growth factor-beta signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol. (2015) 35:911–7. doi: 10.1161/ATVBAHA.114.305150
21. Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. (2005) 287:134–45. doi: 10.1016/j.ydbio.2005.08.041
22. Sawada H, Katsumata Y, Higashi H, Zhang C, Li Y, Morgan S, et al. Second heart field-derived cells contribute to angiotensin II-mediated ascending aortopathies. Circulation. (2022) 145:987–1001. doi: 10.1161/CIRCULATIONAHA.121.058173
23. Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. (2005) 6:826–35. doi: 10.1038/nrg1710
24. Sawada H, Rateri DL, Moorleghen JJ, Majesky MW, Daugherty A. Smooth muscle cells derived from second heart field and cardiac neural crest reside in spatially distinct domains in the media of the ascending aorta-brief report. Arterioscler Thromb Vasc Biol. (2017) 37:1722–6. doi: 10.1161/ATVBAHA.117.309599
25. Gong J, Zhou D, Jiang L, Qiu P, Milewicz DM, Chen YE, et al. In vitro lineage-specific differentiation of vascular smooth muscle cells in response to Smad3 deficiency: implications for Smad3-related thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol. (2020) 40:1651–63. doi: 10.1161/ATVBAHA.120.313033
26. Zhou D, Feng H, Yang Y, Huang T, Qiu P, Zhang C, et al. hiPSC modeling of lineage-specific smooth muscle cell defects caused by Tgfbr1(A230T) variant, and its therapeutic implications for Loeys-Dietz Syndrome. Circulation. (2021) 144:1145–59. doi: 10.1161/CIRCULATIONAHA.121.054744
27. Rateri DL, Moorleghen JJ, Balakrishnan A, Owens AP 3rd, Howatt DA, Subramanian V, et al. Endothelial cell-specific deficiency of Ang II type 1a receptors attenuates Ang II-induced ascending aortic aneurysms in Ldl receptor-/- mice. Circ Res. (2011) 108:574–81. doi: 10.1161/CIRCRESAHA.110.222844
28. Bramel EE, Creamer TJ, Saqib M, Camejo Nunez WA, Bagirzadeh R, Roker LA, et al. Postnatal Smad3 inactivation in murine smooth muscle cells elicits a temporally and regionally distinct transcriptional response. Front Cardiovasc Med. (2022) 9:826495. doi: 10.3389/fcvm.2022.826495
29. Landini G, Martinelli G, Piccinini F. Colour deconvolution: stain unmixing in histological imaging. Bioinformatics. (2021) 37:1485–7. doi: 10.1093/bioinformatics/btaa847
30. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. (2012) 9:676–82. doi: 10.1038/nmeth.2019
31. Chen D, La Greca L, Head GA, Walther T, Mayorov DN. Blood pressure reactivity to emotional stress is reduced in AT1a-receptor knockout mice on normal, but not high salt intake. Hypertens Res. (2009) 32:559–64. doi: 10.1038/hr.2009.59
32. Lu H, Rateri DL, Bruemmer D, Cassis LA, Daugherty A. Involvement of the renin-angiotensin system in abdominal and thoracic aortic aneurysms. Clin Sci. (2012) 123:531–43. doi: 10.1042/CS20120097
33. Sellers SL, Milad N, Chan R, Mielnik M, Jermilova U, Huang PL, et al. Inhibition of Marfan syndrome aortic root dilation by Losartan: role of angiotensin II receptor type 1-independent activation of endothelial function. Am J Pathol. (2018) 188:574–85. doi: 10.1016/j.ajpath.2017.11.006
34. Galatioto J, Caescu CI, Hansen J, Cook JR, Miramontes I, Iyengar R, et al. Cell type-specific contributions of the angiotensin II type 1a receptor to aorta homeostasis and aneurysmal disease-brief report. Arterioscler Thromb Vasc Biol. (2018) 38:588–91. doi: 10.1161/ATVBAHA.117.310609
35. Roman MJ, Devereux RB, Preiss LR, Asch FM, Eagle KA, Holmes KW, et al. Associations of age and sex with Marfan phenotype: the National Heart, Lung, and Blood Institute Gentac (Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions) registry. Circ Cardiovasc Genet. (2017) 10:e001647. doi: 10.1161/CIRCGENETICS.116.001647
36. Tashima Y, He H, Cui JZ, Pedroza AJ, Nakamura K, Yokoyama N, et al. Androgens accentuate TGF-beta dependent ERK/Smad activation during thoracic aortic aneurysm formation in Marfan syndrome male mice. J Am Heart Assoc. (2020) 9:e015773. doi: 10.1161/JAHA.119.015773
37. Bowen CJ, Calderon Giadrosic JF, Burger Z, Rykiel G, Davis EC, Helmers MR, et al. Targetable cellular signaling events mediate vascular pathology in vascular Ehlers-Danlos syndrome. J Clin Invest. (2020) 130:686–98. doi: 10.1172/JCI130730
38. Renard M, Muino-Mosquera L, Manalo EC, Tufa S, Carlson EJ, Keene DR, et al. Sex, pregnancy and aortic disease in Marfan syndrome. PLoS ONE. (2017) 12:e0181166. doi: 10.1371/journal.pone.0181166
39. Sawada H, Lu HS, Cassis LA, Daugherty A. Twenty years of studying Ang II (Angiotensin II)-induced abdominal aortic pathologies in mice: continuing questions and challenges to provide insight into the human disease. Arterioscler Thromb Vasc Biol. (2022) 42:277–88. doi: 10.1161/ATVBAHA.121.317058
40. Al-Abcha A, Saleh Y, Mujer M, Boumegouas M, Herzallah K, Charles L, et al. Meta-analysis examining the usefulness of angiotensin receptor blockers for the prevention of aortic root dilation in patients with the Marfan syndrome. Am J Cardiol. (2020) 128:101–6. doi: 10.1016/j.amjcard.2020.04.034
41. van Andel MM, Indrakusuma R, Jalalzadeh H, Balm R, Timmermans J, Scholte AJ, et al. Long-term clinical outcomes of losartan in patients with Marfan syndrome: follow-up of the multicentre randomized controlled compare trial. Eur Heart J. (2020) 35:4181–7. doi: 10.1093/eurheartj/ehaa377
Keywords: Loeys-Dietz Syndrome, aortic aneurysm, ARBs, angiotensin II type 1 receptor, VSMC
Citation: Bramel EE, Bagirzadeh R, Saqib M, Creamer TJ, Espinoza Camejo WA, Roker LA, Pardo Habashi J, Dietz HC and Gallo MacFarlane E (2022) Distinct Contribution of Global and Regional Angiotensin II Type 1a Receptor Inactivation to Amelioration of Aortopathy in Tgfbr1M318R/+ Mice. Front. Cardiovasc. Med. 9:936142. doi: 10.3389/fcvm.2022.936142
Received: 04 May 2022; Accepted: 30 May 2022;
Published: 22 June 2022.
Edited by:
Hong S. Lu, University of Kentucky, United StatesReviewed by:
Jeff Zheying Chen, University of Texas Southwestern Medical Center, United StatesCopyright © 2022 Bramel, Bagirzadeh, Saqib, Creamer, Espinoza Camejo, Roker, Pardo Habashi, Dietz and Gallo MacFarlane. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Gallo MacFarlane, ZWdhbGxvMUBqaG1pLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.