 Xuewen Duan
Xuewen Duan Xingguang Liu
Xingguang Liu Zhenzhen Zhan
Zhenzhen Zhan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 08 July 2022
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.933060
This article is part of the Research Topic Cardiac Regeneration View all 12 articles
The mortality due to heart diseases remains highest in the world every year, with ischemic cardiomyopathy being the prime cause. The irreversible loss of cardiomyocytes following myocardial injury leads to compromised contractility of the remaining myocardium, adverse cardiac remodeling, and ultimately heart failure. The hearts of adult mammals can hardly regenerate after cardiac injury since adult cardiomyocytes exit the cell cycle. Nonetheless, the hearts of early neonatal mammals possess a stronger capacity for regeneration. To improve the prognosis of patients with heart failure and to find the effective therapeutic strategies for it, it is essential to promote endogenous regeneration of adult mammalian cardiomyocytes. Mitochondrial metabolism maintains normal physiological functions of the heart and compensates for heart failure. In recent decades, the focus is on the changes in myocardial energy metabolism, including glucose, fatty acid, and amino acid metabolism, in cardiac physiological and pathological states. In addition to being a source of energy, metabolites are becoming key regulators of gene expression and epigenetic patterns, which may affect heart regeneration. However, the myocardial energy metabolism during heart regeneration is majorly unknown. This review focuses on the role of energy metabolism in cardiac regeneration, intending to shed light on the strategies for manipulating heart regeneration and promoting heart repair after cardiac injury.
Heart failure is a burgeoning public health problem (1). It is a common and complex disease and has a poor prognosis in most patients. Ischemia is one of the main factors that contribute to heart failure. Following myocardial infarction (MI), the function and morphology of the cardiac cells begin to change. In the early phase of ischemia, cardiomyocytes lack oxygen and nutrients, resulting in a large-scale loss. Myofibroblasts in the heart became activated and produce collagen and other extracellular matrix components to compensate for the maintenance of the basic heart structure. In the later phase of ischemia, a large number of myofibroblasts accumulate, which thickens the ventricular wall and changes the mechanics of the heart, thereby impairing the cardiac pump function (2). As the proliferative capacity of adult cardiomyocytes is very low, they fail to repair the damaged area. This results in maladaptive cardiac remodeling and fibrosis, which ultimately progresses to an irreversible late-stage heart failure (3). With the exception of heart transplantation, current treatments, such as ventricular assist devices, fail to replenish the massive loss of cardiomyocytes after injury. Xenotransplantation and cardiac tissue engineering are achieving the encouraging progress for replacing damaged heart tissues (4). However, cardiac regeneration through altering cardiomyocyte fate plasticity is emerging as a promising approach to compensate for the loss of functional cardiomyocytes and repair cardiac functions.
A previous study revealed that in several non-mammalian lower vertebrates, endogenous cardiac regeneration occurs after cardiac damage (5). However, there has been a long-standing assumption that the mammalian heart is a terminally differentiated organ and that the adult mammalian heart cells are incapable of cell division and proliferation. Intriguingly, a study by Engel et al. (6) for the first time showed that adult mammalian ventricular cardiomyocytes could be induced not only to enter the cell cycle but also to undergo cytokinesis resulting in cell division. However, previous research showed that neonatal mice possessed remarkable endogenous cardiomyocyte proliferation and cardiac regeneration capacity before postnatal day 7 (P7) (7). Further evidence revealed that the heart of P3 mice/rat could not regenerate anymore, which might be due to cell cycle withdrawal and changes in energy metabolism occurring around P3 in mice and rats (8). Thus, establishing a neonatal mouse cardiac regeneration model provides a way to elucidate the mechanisms of cardiac regeneration. After birth, there is a shift in the source of nutrition from placenta-dependent to postnatal nutrition. This in turn triggers a dramatic change in the availability of energy substrates and leads to a shift in the metabolism of cardiomyocytes. Moreover, from embryonic to adult life, the oxygen status alters, thereby shifting the energy metabolism of cardiomyocytes. Interestingly, the shift in myocardial energy metabolism coincides with the time point of cardiomyocyte cycle withdrawal and loss of myocardial regenerative capacity in postnatal mice (9). This raises a question whether changes in metabolism are the cause of cell cycle exit or a consequence thereof. To sum up, these researches insinuate that myocardial metabolism plays a key role in the regenerative capacity of the heart.
In this review, we summarize the changes in major energy metabolic pathways, including fatty acid oxidation, glycolysis, amino acid metabolism, and tricarboxylic acid cycle (TCA), occurring during heart regeneration. We also discuss the key metabolic targets that point the way to research on cardiac regeneration.
Lower vertebrates, such as zebrafish (Danio rerio) and axolotl, have the incredible potential for heart regeneration. The Mexican cavefish have been reported to regenerate their hearts after an injury (10). Zebrafish have the ability to regenerate cardiac muscles throughout their lifetime (11). After removing 20% of the ventricular volume, the heart can be completely regenerated within 2 months, with myocardial contractility returning to normal levels (11). The reasons for the high regenerative capacity of lower vertebrates might be divided into three aspects. First, the circulatory system of zebrafish is relatively hypoxic. This is because the zebrafish heart consists of a single atrium and a single ventricle, causing mixing of arterial and venous blood, which leads to low oxygen content in circulating blood (12, 13). Second, previous research found that centrosomes were intact in cardiomyocytes of zebrafish and salamander, while in mammalian cardiomyocytes they were disintegrated, impairing proper cell division (14). Finally, zebrafish cardiomyocytes are small and mononuclear throughout their life cycle and maintain proliferative potential (13, 15, 16). In addition, the oxygen content in the natural aquatic habitat of zebrafish is 1/30 of that in atmospheric air (17, 18). A previous study found that hypoxia promoted myocardial regeneration by altering the metabolic state. In contrast, exposing zebrafish to hyperoxic water at 45 kPa (vs. normoxic water at 21 kPa) inhibited myocardial regeneration (19). In addition, the change in environmental oxygen level from embryonic to adult stage triggers a switch in energy metabolism of the postnatal heart, which generates mitochondrial reactive oxygen species (ROS). The DNA damage response (DDR) is triggered, and the cell cycle of postnatal cardiomyocytes is thus stalled (17). Moreover, a research found that ROS altered the activity of metabolically critical enzymes and hence played a role in cardiac regeneration of zebrafish. Since elevated H2O2 levels are detrimental, investigators identify protein tyrosine phosphatase 1b (Ptp1b) as a downstream target of ROS and propose that inhibition of Ptp1b promotes myocardial regeneration (20). In addition, MSI-1436, a small molecule inhibitor, selectively represses Ptp1b and hence promotes regeneration (21). Dual-specificity phosphatase 6 (Dusp6), a member of the PTP family, is sensitive to the redox effects of ROS. Moreover, the change in Dusp6 expression correlates with the regenerative window of the heart, which suggests Dusp6 might have a key role in the early cardiac regeneration process (22). Furthermore, by single-cell transcriptome analysis of regenerated zebrafish myocardium, researchers found that cardiomyocytes in the proliferative border zone underwent a shift in energy metabolism after heart cryoinjury (23). This suggests that proliferation and regeneration of cardiomyocytes after cardiac injury in zebrafish involve energy metabolism.
Morphology of murine neonatal heart is similar to that of zebrafish heart (24). Hence, researchers turned their attention to find out whether mammalian heart could regenerate after damage. The size of human cardiomyocyte increases approximately 8.6-fold during the first 20 years of life (25). Although adult cardiomyocytes can temporarily self-renew, their self-renewal rate is extremely low (26). In addition, the regeneration ability of adult cardiomyocytes is very limited and is not enough to repair the damaged myocardial tissue. Hence, adult mammalian myocardial tissue cannot regenerate after heart damage. However, recent data suggest that cardiomyocyte renewal occurs in adulthood in mammals including humans (27, 28). Interestingly, in 2011, researchers discovered that neonatal mice had an amazing myocardial regenerative potential (7). This study showed that 1-day-old newborn mice heart had the ability to regenerate resected myocardium after apical resection. Nonetheless, this regenerative ability is lost after 7 days of birth (7).
Further, on investigating the reasons for reduced regenerative capacity of adult mice, researchers found that differences in the type of energy metabolism were influential factors that determined the ability of the myocardium to regenerate. There are two main ways by which cardiac energy in mice is altered. On the one hand, a change in oxygen status from embryonic to postnatal causes a shift in cardiac metabolism. Due to exposure to relatively hypoxic environment, embryonic blood shunt circulation in the mammalian heart results in significant mixing of arteriovenous blood (29). However, after birth, the shift in circulation and the rapid increase in arterial oxygen change the oxygenation status of cardiomyocytes within minutes (17). Therefore, mammalian cardiomyocytes are capable of generating energy through metabolic conversion to adapt to high energy demands after birth (30). The main source of adenosine triphosphate (ATP) is cardiac metabolism, and ATP maintains cardiac homeostasis and function (31, 32). Mitochondria perform energy conversion, and more than 95% of the ATP is produced through energy substrates for the heart (33). After birth of mice, energy metabolism of neonatal cardiomyocytes changes, and the number of cardiomyocyte mitochondria dramatically increases in the adult heart (34). One research found that mitochondrial DNA copy number was lower in the neonatal murine heart compared with the adult murine heart (17). At present, many studies focus on understanding the regulation of mitochondrial metabolism in postnatal cardiomyocyte cycle arrest and influencing future regeneration strategies.
On the other hand, nutrient supply is altered after birth of mice (35), and the shift in metabolic pattern causes cardiomyocyte maturation and cell cycle exit (36). Previous studies on porcine and rabbit hearts indicated that postnatal cardiomyocytes underwent a shift in energy source compared with embryonic cardiomyocytes due to different nutrients (37, 38). Therefore, the increased oxygen content at sharp atmosphere stress and changes in metabolic matrix postnatally together alter the type of energy metabolism in cardiomyocytes and reduce their proliferation capacity (Figure 1). In all, the regenerative capacity of the myocardium is related to the metabolism of cardiomyocytes.
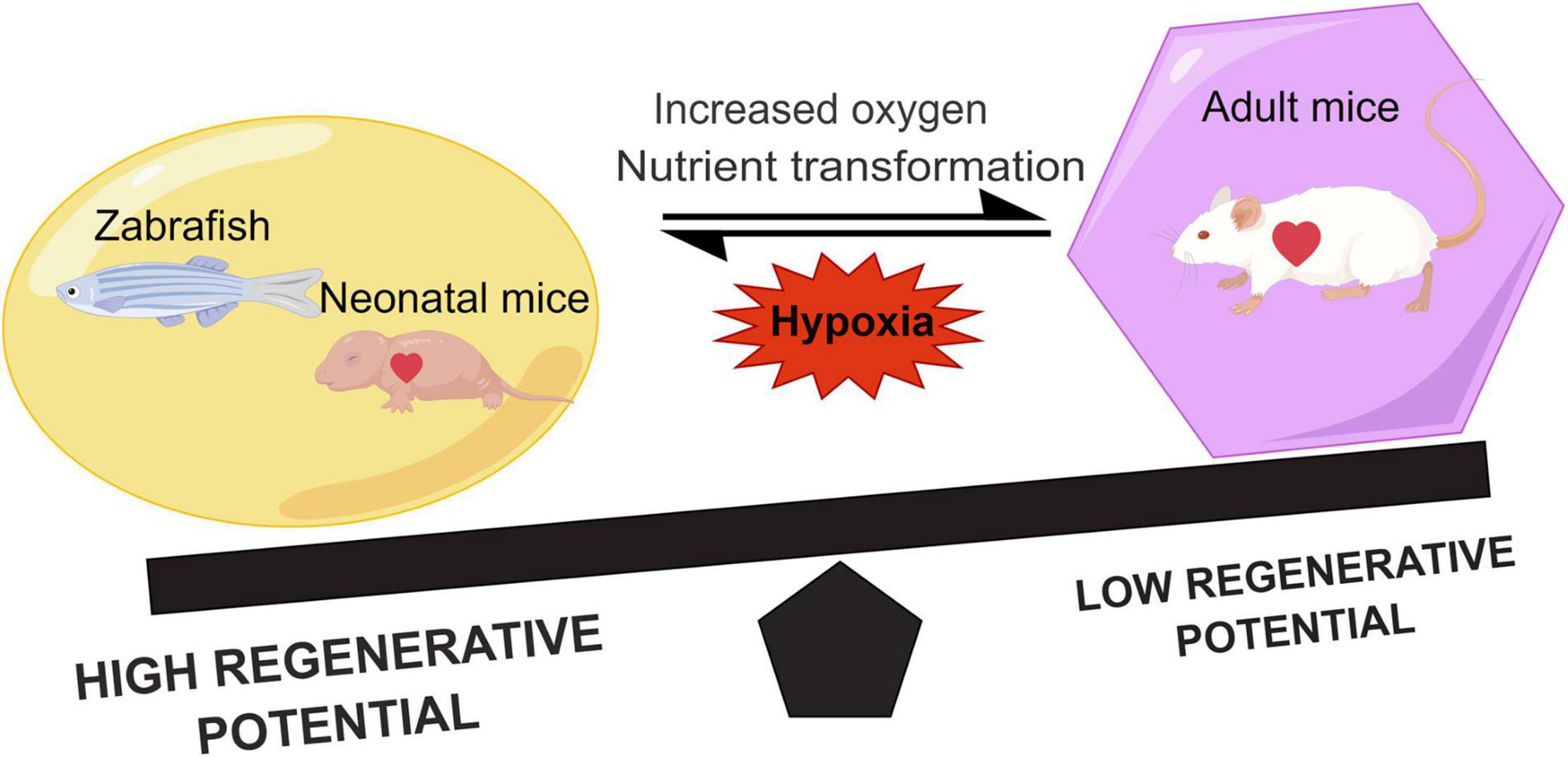
Figure 1. Comparison of the regenerative capacity of representative lower vertebrates, neonatal, and adult mammals. Hypoxia can promote heart regeneration and increase the regenerative capacity. During the growth of neonatal mice, the shift in metabolic state of the heart due to the increased oxygen and nutrients leads to a decrease in the myocardial regenerative capacity.
Hypoxia refers to reduced and insufficient oxygen supply, and it influences myocardial energy metabolism and cardiac contractile function (39, 40). Chronic hypoxia decreases the activity of fatty acid oxidase (40), which in turn reduces fatty acid uptake and oxidation in mitochondria (41). In addition, it increases glycolysis and glycolytic enzyme activity (42). Moreover, chronic hypoxia decreases mitochondrial utilization of fatty acid and pyruvate substrates and reduces the enzymatic activities of electron transport chain (ETC) complexes I, II, and IV in the mitochondria of cardiomyocytes, thereby lowering the production of ROS (43). Hence, the regenerated cardiomyocytes show characteristics of proliferative cardiomyocytes in the embryonic stage, which exhibits hypoxic environment.
Professor Sadek’s team found that moderate hypoxia promoted myocardial regeneration after myocardial infarction (44). They reported that gradually reducing inhaled oxygen by 1% and maintaining oxygen concentration level at 7% for 2 weeks, reduced ROS production and DDR, however, restarted mitosis in cardiomyocytes. Hypoxia-inducible factor (HIF)-1 family adjusts the cells to hypoxic environment. HIF-1 is a class of specialized heterodimers composed of unstable alpha subunits (HIF-α). HIF-1α protein is steady under hypoxic conditions and activates transcription of multiple genes involved in glycolysis, fatty acid metabolism, mitochondrial metabolism, and cell cycle regulation (45). Stem cells or progenitor cells of some organs are relatively hypoxic and maintain their normal physiological functions by stabilizing the HIF-1α subunit (46). In 2012, researchers found that hypoxia promoted cardiomyocyte dedifferentiation and myocardial regeneration in adult zebrafish, and HIF-1α played the important role in this (19). However, another study found that the complete activation of HIF-1α signal led to the dilated cardiomyopathy and heart failure (47, 48). These findings suggest that the moderate activation of HIF-1α hypoxia signal may be a potential strategy for heart regeneration. In addition, the authors found that HIF-1α deficiency impaired glycolysis and inhibited proliferation of hypoxic fetal cardiomyocytes, which in turn led to transient reprograming of amino acid metabolism and activation of HIF2 (19, 49, 50). This shows that the embryonic heart is metabolically flexible.
In conclusion, chronic severe hypoxia enhances the expression of glycolytic and cell cycle genes, whereas it inhibits the expression of fatty acid oxidation genes and cell cycle inhibitory genes (51), and thus, it metabolically reprograms embryonic heart. These genetic and metabolic changes promote myocardial regeneration, which is referred to as the hypoxia-induced cardiomyocyte proliferation and the infarcted zone revascularization.
The heart is the most energy-intensive organ of the body (52). In the physiological state, the heart of adult mouse produces ATP in two ways to maintain its contractile function. Mitochondrial oxidative metabolism produces approximately 95% of the cardiac energy, while glycolysis produces only 5% of it. About 40–60% of the energy in mitochondrial oxidative metabolism is generated through free fatty acids and glucose metabolism, while the remainder of this is produced by the oxidation of ketones, lactate, and amino acids (53, 54). Compared with glucose oxidation, fatty acid oxidation requires more oxygen. Hence, the aerobic oxidation efficiency of glucose is higher than that of fatty acids (53). During the perinatal period, glycolysis and lactate oxidation are the main sources of cardiac ATP (55–57), while fatty acid oxidation produces only a small fraction of ATP (55). In the early postnatal period (from P1 to P7), glycolysis produces almost half of the ATP for the neonatal murine heart (55). While 7 days post-birth, glycolysis gradually decreases, providing only a small amount (10%) of ATP for cardiomyocyte metabolism. However, there is a significant increase in fatty acid oxidation to maintain cardiomyocyte energy metabolism (38, 55). In the neonatal period, the source of cardiac energy is β-oxidation of fatty acids, which produces ATP at levels close to those found in the heart of adult animals (58). The researchers measured metabolite levels, such as fatty acids, in the cardiac arteries of fetal, newborn (1–4 days), and juvenile (7 weeks) lambs (57, 59). They found that fatty acid flux was zero in the embryonic heart. Interestingly, there was an increase in fatty acids in the myocardium of newborn lambs, but there was no net flux of fatty acid (57). And the flux of fatty acids was increased in the juvenile lambs (57). Glucose and lactate were sources of energy metabolism for embryonic and neonatal (3–15 days) lamb hearts (60). In addition, a research found that fatty acids promoted proliferation of P4 cardiomyocytes but inhibited proliferation of P5 cardiomyocytes (61). In all, different developmental stages and oxygen levels result in altered energy sources for the mouse heart. This leads to varying metabolic states of cardiomyocytes that affect their proliferative capacity (Figure 2).
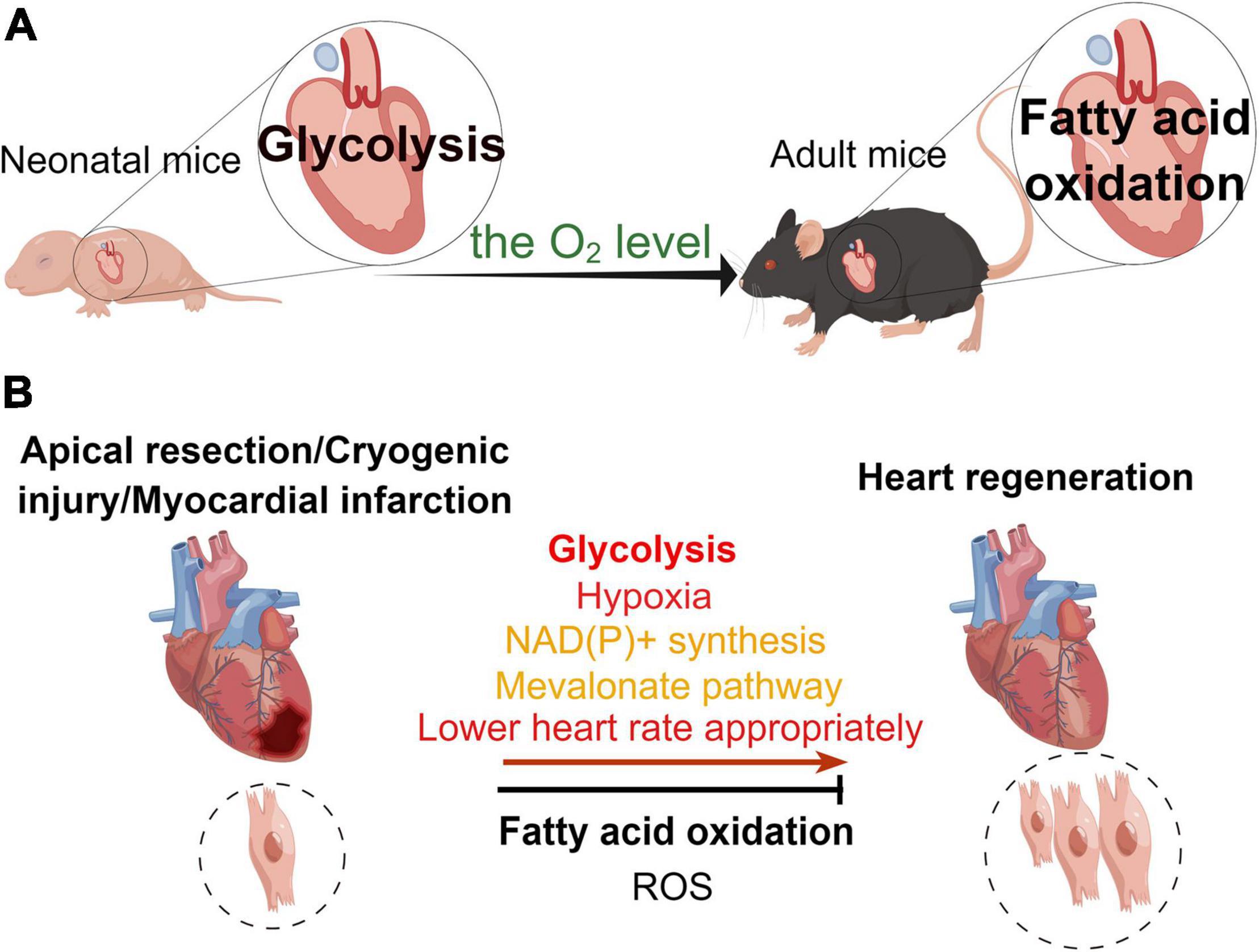
Figure 2. Different metabolic patterns in physiological and pathological states affect the myocardial regenerative capacity. (A) Under physiological conditions, the main mode of metabolism in the murine neonatal heart is glycolysis, whereas that in the hearts of adult mice is fatty acid oxidation. (B) In the pathological state, glycolysis, hypoxia, NAD(P)+ synthesis, mevalonate pathway, and appropriate reduction in heart rate promote heart regeneration, while fatty acid oxidation and reactive oxygen species (ROS) inhibit myocardial regeneration in the neonatal mouse heart regeneration models (apical resection, cryoinjury, or myocardial infarction).
Studies indicate that fatty acid oxidation is the main source of cardiac energy for the adult heart (62). After entering cardiomyocytes, fatty acids generate ATP mainly through oxidation via the TCA in the mitochondria. Among all energy substrates, fatty acids produce the highest ATP content and have the highest oxygen demand. Therefore, fatty acids are myocardial energy substrates with the lowest metabolic efficiency (ATP production/oxygen consumption) (33).
A research found that feeding fatty acid-deficient milk to postnatal mice prolonged the proliferation window of their hearts (63). Carnitine palmitoyltransferase 1 (CPT1) transfers fatty acids from the cytoplasm to the mitochondria and is the key enzyme regulating fatty acid oxidation. CPT1 activity in the neonatal murine heart is very low, while it increases significantly in the heart of a 7-day-old mouse, which coincides with the time when mammalian cardiomyocytes lose their ability to proliferate (64). One research found that etomoxir (ETO) could inhibit the activity of CPT1, thereby reducing fatty acid oxidation and promoting proliferation of neonatal mouse cardiomyocytes and heart regeneration in neonatal mice (61). Previous study showed that the low expression of carnitine palmitoyltransferase 2 (CPT2), which is an essential enzyme for fatty acid oxidation (65), could promote cardiomyocyte proliferation in P14 mice (66). Acyl-CoA synthetase long-chain family member 1 (ACSL1) mediates the uptake of cellular lipids and is a key enzyme regulating lipid metabolism. On examining the expression of glycolipid-related enzymes in heart tissue of mice at different ages, it was found that ASCL1 expression increases with the age of mice (67). Inhibition of ACSL1 expression with a corresponding decrease in cardiomyocyte lipid uptake in cardiomyocytes of adult mice post-MI and primary neonatal mouse cardiomyocytes upregulated the expression of cell cycle-activating genes, but downregulated the expression of cell cycle-inhibiting genes (67).
However, the view that fatty acid oxidation inhibits myocardial regeneration is still debatable. A research found that peroxisome proliferator-activated receptor alpha (PPARα)-mediated β-oxidation of fatty acid promoted primary neonatal mouse cardiomyocyte hypertrophy and maturation and enhanced the presence of binucleated cardiomyocytes at postnatal day 5. Moreover, it caused the withdrawal of cardiomyocytes from cell cycle (61). Nevertheless, it is interesting to note that PPARα-mediated β-oxidation of fatty acid promoted G0/G1 cell cycle entry rate and proliferation of cardiomyocytes in mice at postnatal day 4 (61). What makes fatty acid oxidation has opposite regulatory effects on myocardial regeneration? In this context, the authors speculate that the reduced proliferation rate of cardiomyocytes at postnatal day 5 is due to the presence of binucleated cardiomyocytes and cardiomyocytes leaving the cell cycle (61). These results suggest that the effect of fatty acids on myocardial regeneration is inconclusive and needs to be studied in the future.
Glycolysis is the primary mode of energy metabolism in the neonatal murine heart. A previous study reported that glycolysis promoted myocardial regeneration in adult zebrafish (68). Glucose is an important fuel and produces ATP through cytoplasmic glycolysis and mitochondrial oxidation in the heart. In the heart, glucose transporter 1 (GLUT1) and GLUT4 transport glucose into cardiomyocytes. Under physiological conditions, GLUT1, whose the expression increases by HIF-1α (69), is the main glucose transporter in embryonic and neonatal hearts (70, 71). A research found that overexpression of GLUT1 promoted glycolysis in the neonatal murine heart (72, 73) and increased nucleotide synthesis, thereby promoting myocardial regeneration by enhancing glucose metabolism after cryogenic injury in neonatal mice at P21 and P40 (9).
The key glycolytic enzymes play important roles in myocardial regeneration. Phosphofructokinase 2 (PFK2) regulates the level of fructose-2,6-bisphosphate (Fru-2,6-P2) by phosphorylating fructose-6-phosphate (F6P) during glycolysis. A research revealed that PFK2 could increase the contractility of hypoxic cardiomyocytes in mice, suggesting that it affects the metabolism of cardiomyocytes while maintaining their function (74). Pyruvate dehydrogenase kinase (PDK) inhibits the activity of pyruvate dehydrogenase (PDH). Previous studies found a significantly high expression of PDK in the infarct area of the adult heart (71) and zebrafish (68) after cryogenic injury of the heart. Moreover, PDK4 deletion promoted cardiac regeneration in adult mice after MI and proliferation of primary adult mouse cardiomyocytes by increasing glycolysis (63). In contrast, a report implied that PDK3 overexpression promoted cardiomyocyte proliferation by enhancing glycolysis during the early stages of cardiac regeneration in zebrafish, while there was no scar repair in the later stages (68). What is the reason for the difference between early and late stages? Since PDK3 overexpression can promote cardiomyocyte proliferation, can it also promote maturation of new cardiomyocytes to replace injured cardiomyocytes, and thus promote scar repair? Of note, PDK3 and PDK4 are both isozymes of PDK, so why do they exhibit opposite effects on cardiomyocyte proliferation? Their distributions are different, but the mechanisms involved require further study. Notably, studies have shown that M2-pyruvate kinase (PKM2) can promote the phosphopeptide pathway and inhibit oxidative stress (75, 76). A research found that knockout of PKM2 in mice post-MI promoted catenin beta 1 (Ctnnb1) translocation to the nucleus and trans-activation of key genes that mediate cell cycle progression (77). In zebrafish, the Pkma2 gene encodes M2-pyruvate kinase. It has been found that pkma2 knockdown in zebrafish decreases glycolysis and thereby inhibits cardiomyocyte proliferation (68). Magadan’s team found that overexpression of PKM2 promoted cardiomyocyte proliferation and regeneration after MI in adult mice, by increasing the expression of glucose-6-phosphate dehydrogenase (G6PD) (78). Therefore, PDK and PKM2 serve as promising treatment strategies for promoting cardiac repair and regeneration.
Interestingly, some key regulatory genes can regulate cardiac regeneration by promoting glycolysis. Myeloid ecotropic viral integration site 1 (MEIS1) is an important transcription factor that regulates the cardiomyocyte cycle. A research found that myocardium-specific knockout of MEIS1 in neonatal mice could extend the proliferation window of cardiomyocytes and that these cardiomyocytes could re-enter the cell cycle (79). Conversely, another finding indicated that inhibiting MEIS1 expression in primary fetal and neonatal sheep cardiomyocytes resulted in the increased mitochondrial activity and decreased glycolytic genes, leading to the enhanced cardiomyocyte maturation (80). The discrepancy in MEIS1 function between mice and sheep may be related to the differences in cardiomyocytes themselves from these two species. Further studies are needed to understand the mechanism of MEIS1 regulation on the metabolic state as well as the cell cycle of cardiomyocytes.
It is worth noting that cardiomyocyte proliferation-related pathways can also affect glycolysis. Furthermore, Yes-associated protein (YAP), a component of the Hippo pathway, is involved in glycolytic metabolism during heart development (81). A previous study found that the defect in Hippo pathway could activate Yap signaling to promote cardiomyocyte proliferation in adult mice (82–84). Studies on adult cardiomyocytes showed that direct and indirect target genes of activated Yap were associated with glycolysis and mitochondrial metabolism (84–86). In addition, the interaction between neuregulin 1 (NRG1) and its receptors, ErbB2 or ErbB4, facilitated glycolysis and inhibited mitochondrial oxidative phosphorylation (87), which was vital for cardiomyocyte proliferation in zebrafish (88) and newborn mice (89). Researchers showed that ErbB2 affected cardiac regeneration via downstream activation of YAP (81). Moreover, Wnt/β-catenin and ERK/MYC signaling pathways promoted glycolysis and cell proliferation by upregulating cyclin–CDK complex in neonatal mouse cardiomyocytes and immature human pluripotent stem cell-derived cardiomyocyte (hPSC-CM) (90, 91). Overexpression of c-Myc could promote glycolysis and cardiomyocyte proliferation in mice during both gestation and adult life (92). In contrast, Wnt/β-catenin signaling in adult mice was cardioprotective but did not induce cardiomyocyte proliferation (91). Thus, the role of Wnt/β-catenin signaling in mice at different stages depends on the metabolic state of cardiomyocytes (91). Taken together, these pathways and genes can regulate cardiomyocyte proliferation by influencing glycolysis. However, which gene or component of the glycolytic pathway is affected is not yet elucidated. Moreover, whether these pathways only affect glycolysis or also affect lipid and other metabolic pathways remains to be determined.
In all, these studies indicate that glycolysis plays an essential role in modulating cardiomyocyte proliferation and cardiac regeneration after injury. As a result, targeting glucose metabolism is an interesting strategy to promote adult heart regeneration.
Cardiomyocytes require protein synthesis during growth, after birth, and during maturation. The increased protein synthesis results in the increased amino acid metabolism. Amino acid oxidation is also a source of cardiac ATP, with branched-chain amino acids (BCAAs) playing a major role. BCAA includes three amino acids, namely, leucine, isoleucine, and valine. Although BCAA oxidation produces only 2% of the total ATP generated by the heart (93), BCAAs play an important role in regulating cardiac signaling pathways, including insulin and mTOR signaling pathways (94).
Metabolomic analysis of mouse heart tissues at different developmental stages revealed that BCAAs showed differential levels in the heart at different developmental stages (95). Valine, leucine, and isoleucine concentrations gradually increased postnatally reaching a peak at day 9 and then decreased until they reached to the postnatal day 1 (P1) level on P23 (95). Most of the differentially expressed genes (DEGs) directly correlated with BCAA concentrations and were upregulated at the mRNA level between P9 and P23 (95). Therefore, we speculate whether inhibiting enzymes of the BCAA breakdown pathway could promote cardiomyocyte proliferation.
Glutamine is an amino acid transporter. Elevated glutamine expression in zebrafish and neonatal mouse cardiomyocytes activated the amino acid-driven mTOR signaling pathway, which promoted mitochondrial maturation and regulated cardiomyocyte proliferation (96). In addition, high concentrations of leucine and glutamate were found to activate the mTOR pathway and promote regeneration (96). Since leucine concentration rises in the postnatal week, does the concentration of BCAA also affect the ability of cardiomyocytes to proliferate? Researchers found that cardiomyocyte injury initiated Wnt/β-catenin signaling during zebrafish cardiomyocyte regeneration, which in turn initiated mTOR activation and metabolic remodeling (97). As previously described, Wnt/β-catenin signaling pathway promoted glycolysis and activated mTOR signaling pathway together with BCAAs (91, 98). This suggests that metabolic pathways are not independent but interact with each other. Thus, alterations in amino acid metabolism in cardiomyocytes affect their proliferation. However, studies in this research area are limited and need to be investigated in the future.
Tricarboxylic acid cycle occurs in the mitochondrial matrix. It is the metabolic center of the carbohydrate–lipid–amino acid linkage and the last metabolic pathway of the three nutrients. The transformation of energy metabolism and an increase in mitochondrial DNA after birth of mice lead to a significant increase in ROS produced by mitochondria (17). This increased ROS production damages proteins, lipids, and DNA and leads to cell cycle arrest (99–102). Therefore, clarifying the role of mitochondrial metabolites in regulating metabolic switch is essential for identifying metabolic targets that will promote adult heart regeneration.
To begin with, the primary approach to promote cardiomyocyte proliferation by inhibiting mitochondrial metabolism is to suppress ROS production by mitochondrial oxidation. Studies have reported that overexpression of mitochondrial catalase (mCAT) and the ROS scavenger N-acetylcysteine (NAC) system reduces DDR and promotes cardiomyocyte proliferation (17). Martin’s team found that the activation of the antioxidant response after cardiac injury could reduce ROS and thereby promote cardiac repair (84). In cardiomyocytes, nuclear factor-erythroid-2-related factor 2 (Nrf2) directly regulates the expression and subcellular location of paired-like homeodomain 2 (Pitx2). Genome analysis indicated that Pitx2 activates genes in response to oxidative response by recruiting YAP (103). The knockdown of Pitx2 in the neonatal mouse heart leads to the failure of regeneration after cardiac injury. This suggests that antioxidant stress response pathways are critical for cardiomyocyte proliferation and cardiac regeneration (84). These include genes encoding ETC components and ROS eliminator that protect cells from ROS damage and promote cardiomyocyte regeneration (84, 103). Pei et al. (104) confirmed that hydrogen sulfide (H2S) could scavenge ROS, and hence, cardiomyocytes could re-enter the proliferation cycle. Researchers used propargylglycine (PAG) to inhibit H2S synthesis and found that it promoted the deposition of ROS in the murine neonatal heart and inhibited proliferation of cardiomyocytes (104). In contrast, sodium hydrosulfide hydrate (NaHS), an H2S donor, could reduce ROS accumulation and promote cardiomyocyte proliferation and regeneration by alleviating H2S-mediated cardiomyocyte injury (104). These findings reveal that antioxidant pathways play an important role in the regeneration of the heart. In addition, cardiac sarcomere contraction after birth promoted mitochondrial metabolism and activated p53 to produce ROS and DNA damage responses (105). It is known that p53 causes cardiomyocytes to exit the cell cycle by repressing Cyclin B1 gene (106). We speculate that cardiac sarcomere contraction affects the proliferative capacity of cardiomyocytes by regulating their metabolism. However, this speculation remains to be validated. Overall, sarcomere disassembly in cardiomyocytes can preclude ROS production and promote proliferation of cardiomyocytes (105).
Succinate dehydrogenase (SDH) converts succinate to fumarate in the TCA cycle. During ischemia, succinate concentration increases and inhibits SDH activity. A previous study revealed that injecting succinic acid in neonatal mice inhibited the proliferation of cardiomyocytes. In contrast, malonate, an SDH inhibitor, prolonged the regeneration window of the heart after birth and promoted the proliferation of cardiomyocytes and heart regeneration in adult mice after MI (107). Similarly, malonate injection in neonatal mice promoted myocardial regeneration after an injury (108). In addition, malonic acid induced the formation of a new coronary vascular system within the infarcted tissue (108), given that glycolysis promoted angiogenesis through apical cell formation (109). In short, malonate promotes proliferation of cardiomyocytes and vascularization would be a candidate therapeutic target. In terms of clinical translation, delivery of SDH inhibitors directly to the heart may be more effective because SDH deficiency can lead to tumorigenesis (108). Nonetheless, the role of other critical enzymes of the TCA cycle is not yet clear. In short, these studies suggest that in addition to glycolipid metabolism, changes in the activity of critical enzymes of the TCA cycle also have different impacts on cardiomyocyte proliferation.
Interestingly, researchers found that in addition to classical metabolic pathways, several biosynthetic pathways could affect cardiomyocyte proliferation. A study revealed that simvastatin inhibited cardiomyocyte proliferation by inhibiting the mevalonate pathway of isoprene synthesis in cardiomyocytes of human-induced pluripotent stem cell (hiPSC)-cardiac organoids (110). Previous studies found that the expression of genes that regulated ancillary biosynthetic pathway enzymes increased in cardiomyocytes in response to cell cycle stimulation by four cell cycle factors, namely, cyclin B1, cyclin D1, CDK1, and CDK4. These enzymes were related to the hexosamine biosynthesis pathway (HBP), serine biosynthesis pathway, protein O-GlcNAcylation, and those involved in NAD(P)+ (111). Researchers found that overexpression of O-GlcNAcase (OGA) was associated with HBP and prevented cardiomyocyte cycle entry and progression, while knockdown of nicotinamide phosphoribosyltransferase (NAMPT) inhibited cycle entry of human iPSC cardiomyocytes (hiPSC-CM) (111). In addition, investigators found that overexpression of phosphoenolpyruvate carboxykinase 2 (PCK2), a gluconeogenic pathway enzyme that catalyzed the transformation of oxaloacetate to phosphoenolpyruvate, promoted proliferation of hiPSC-CM (111). In general, the crossover point metabolites in these synthetic pathways are provided by the glycolytic pathway, which again highlights the importance of the glycolytic pathway in the process of myocardial regeneration.
Previous research found that cardiomyocyte-specific overexpression of PPARδ induced cell cycle progression in cardiomyocytes after MI in mice and improved cardiac function (112). In addition, carbacyclin, a PPARδ activator, induced proliferation of neonatal and adult rat mononuclear cardiomyocytes via (PPARδ)/PDK1/Akt/GSK3β/β-catenin pathway (112). Glycogen synthase kinase-3β (GSK-3β) is a serine/threonine kinase originally identified as a key enzyme in glycogen synthesis. The authors do not indicate whether myocardial metabolism is altered during PPARδ-mediated myocardial regeneration. In addition, we cannot speculate whether PPARδ ultimately promotes myocardial regeneration by enhancing glycogen synthesis during this process. In contrast, previous studies have shown that PPARβ/δ overexpression in the heart increases glucose utilization by enhancing the expression of glucose transporter protein type 4 (Glut4) (113, 114). Can this be considered that PPARδ promotes cardiomyocytes to enter the cell cycle by enhancing glycolysis? This should be further delved into future studies.
In addition to its ability to regulate glycolysis, PKM2 has been reported to redirect glucose carbon flow to oxidative pentose phosphate pathway (PPP), thereby reducing oxidative stress and causing increased expression of cell cycle genes in postnatal cardiomyocytes (78). It has been shown that overexpression of the glucose transporter protein Glut1 leads to an increase in glucose metabolites and thus nucleotide supply in heart regeneration of neonatal mice (9). Recently, researchers have found that metabolic pathways can combine with biosynthetic pathways to affect the cardiomyocyte proliferation. Moderate heart rate reduction (HRR) can induce cardiomyocyte proliferation by altering the metabolic pattern of cardiomyocytes (115). On the one hand, glucose metabolism enzymes exert non-enzymatic activity to promote cell cycle progression. On the other hand, the PPP is activated to supply the biosynthetic metabolic demand required for promoting cardiomyocyte proliferation (115). In addition, researchers found that lowering the heart rate upregulated the expression of cyclin D1. Moreover, they also observed an increase in the nuclear transport of PKM2, which promoted cyclin D1 and thus restarted cardiomyocyte proliferation (115). This suggests that the three biological properties of cardiomyocytes, namely continuous rhythmic beating, unique energy metabolic pattern, and limited proliferative capacity, are intrinsically linked. In addition, PPP may be a prospective regenerative strategy for cardiac injury. Overall, reducing the heart rate of cardiomyocytes can affect their energy metabolic patterns and thus promote cardiac regeneration. However, some questions have not been explored yet. For example, what specific molecules and signaling pathways of PPP are affected by HRR regulation? Are there any other biosynthetic pathways that are also affected? Furthermore, this suggests that future studies combine metabolism and synthesis, rather than a single aspect, to explain the phenomenon of cardiac regeneration.
Heart failure is the end stage of many heart diseases, including the ischemic cardiomyopathy and non-ischemic cardiomyopathy. In this regard, MI is a common ischemic cardiomyopathy, while cardiac hypertrophy due to pressure-loaded cardiomyopathy is a common non-ischemic cardiomyopathy. These processes include a wide range of remodeling in metabolism, cardiac structure, and cardiac electrophysiology (116). There is growing evidence that metabolic remodeling precedes most pathological processes and may play an important role in myocardial hypertrophy and heart failure (33, 117–119). In the failing heart, mitochondrial ROS production increases and also mitochondrial autophagy decreases. This results in the impaired mitochondrial function and reduced mitochondrial oxidative metabolism (120, 121). Hence, to increase the ATP supply, there is a compensatory increase in glycolysis (122). In addition, the energy metabolism shifts to a “fetal”-like pattern of energy substrate metabolism after cardiac hypertrophy (123, 124). That is, glycolysis increases and fatty acid oxidation decreases in pressure-overload cardiac hypertrophy (122, 125).
Although major changes in glycolysis and fatty acid oxidation in both failing and hypertrophic hearts resemble the major metabolic pattern of the embryonic heart, the cardiomyocyte proliferation capacity varies. For this reason, the specific metabolic patterns in the embryonic and failing hearts are intrinsically different. The primary difference is the substrate of energy metabolism and the way of metabolism. In the ischemic heart, anaerobic glycolysis is the main source of cardiac ATP and leads to the production of lactate (126). In the absence of blood perfusion, glucose is not available to cardiomyocytes, and hence, previously stored glycogen is used to produce ATP (127). Since glycolysis produces less ATP than oxidative phosphorylation, ATP is consumed more than it is produced. The intracellular lactic acid accumulates, leading to cellular acidosis, and inhibits enzymes of the glycolytic pathway (116). Hence, the efficiency of glycolysis decreases during prolonged ischemia (127). Glycolysis ceases despite the availability of glycogen stores in cardiomyocytes (127). In addition, the lack of blood perfusion leads to the accumulation of intracellular protons that inhibits glycolysis in a feedback manner (128, 129). However, during the embryonic period, the heart obtains energy mainly through glucose. Due to low oxygen concentration during the embryonic period, the embryonic heart produces energy mainly through anaerobic glycolysis, which promotes heart regeneration (68). This suggests that the substrates of energy metabolism are different in the ischemic and regenerative heart.
On the one hand, researchers found that aerobic glycolysis was the main source of cardiac ATP in pressure-overload-induced heart failure (130–132), whereas, anaerobic glycolysis was the main source of cardiac ATP during myocardial regeneration in neonatal mice (133). On the other hand, glucose oxidation was lower in hypertrophied hearts than that in non-hypertrophied hearts (134, 135). When the heart rates were appropriately reduced, P1 cardiomyocytes were more dependent on glucose oxidation (115). This suggests that glucose oxidation promotes myocardial proliferation. Thus, regenerative and hypertrophic hearts are metabolized in different ways. Besides, researchers tested 13 key genes for energy metabolic substrates, including “adult” isoform genes and “fetal” isoform genes. They found that the expression of “adult” isoform of metabolic genes was reduced in the failing adult heart (124). This suggests that the reversion of the metabolic pattern in the failing heart is similar to the metabolic pattern of the embryonic heart (124). However, some studies had different conclusions. It was found that fatty acid oxidation (FAO) was either unchanged or increased, whereas glycolysis was either unchanged or decreased in the hypertrophied heart (132). Another research showed that myocardial FAO was unchanged, glycolysis was slightly reduced, and glucose and lactate oxidation was decreased in angiotensin II-induced myocardial hypertrophy (136). These discrepancies suggest that the metabolism of failing and regenerating hearts is different. Hence, the failing cardiac metabolism evolving from infarction and myocardial hypertrophy is different from the cardiac metabolism of neonatal mice.
Heart failure is by far the leading cause of mortality and disability throughout the world. Neonatal mice have an ability of myocardial regeneration and thus serve as a model for the treatment of heart failure. In this review, we summarize the current status of research on the metabolic regulation of myocardial regeneration, including recent findings and controversies (Table 1 and Figure 3). Glycolipid–amino acid metabolism and biosynthesis are critical for the regulation of heart regeneration. Based on this, changing metabolism of related components, making cardiomyocytes to re-enter the cell cycle, promoting proliferation of cardiomyocytes, and enhancing cardiac regeneration serve as effectives therapies for heart diseases that eventually evolve into heart failure (Figure 4 and Table 1). However, further studies are required to find solutions for the following problems: For example, several metabolic pathways are closely related to each other, so which molecule or mechanism controls the direction of metabolic changes? What are the mechanisms by which the metabolic alterations eventually affect the cardiomyocytes proliferation and heart regeneration? Many metabolisms end up affecting the intra-nuclear epigenetic mechanisms. So, whether the combination of metabolism and epigenesis influences the process of heart regeneration? In addition to the four major metabolisms discussed in this review, are other metabolic pathways involved in the regulation of cardiac regeneration? For example, is the current and popularly studied novel metabolism of metal ions (iron and copper metabolism) related to other heart diseases?
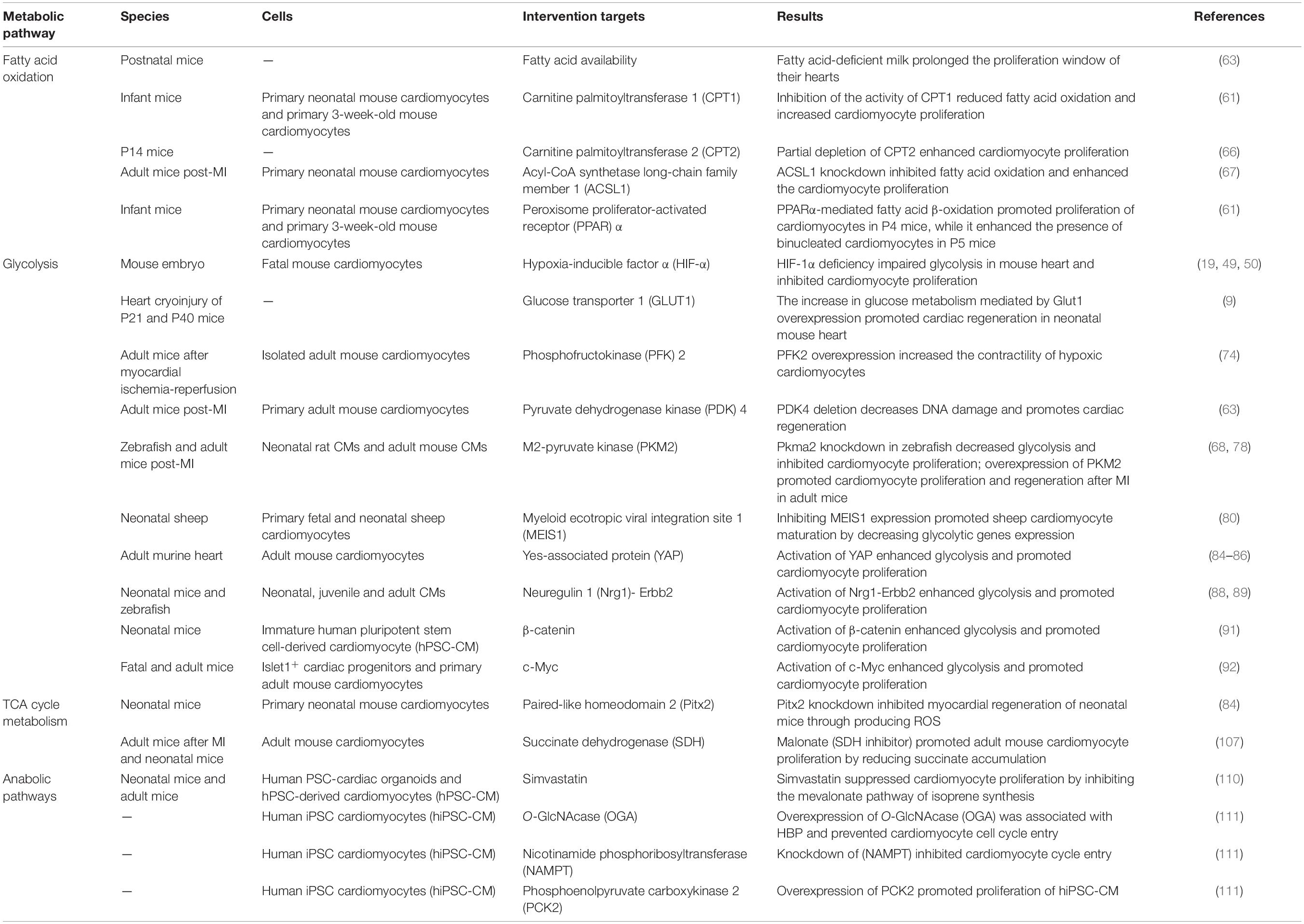
Table 1. Summary of recent studies demonstrating the role of metabolism in heart regeneration.
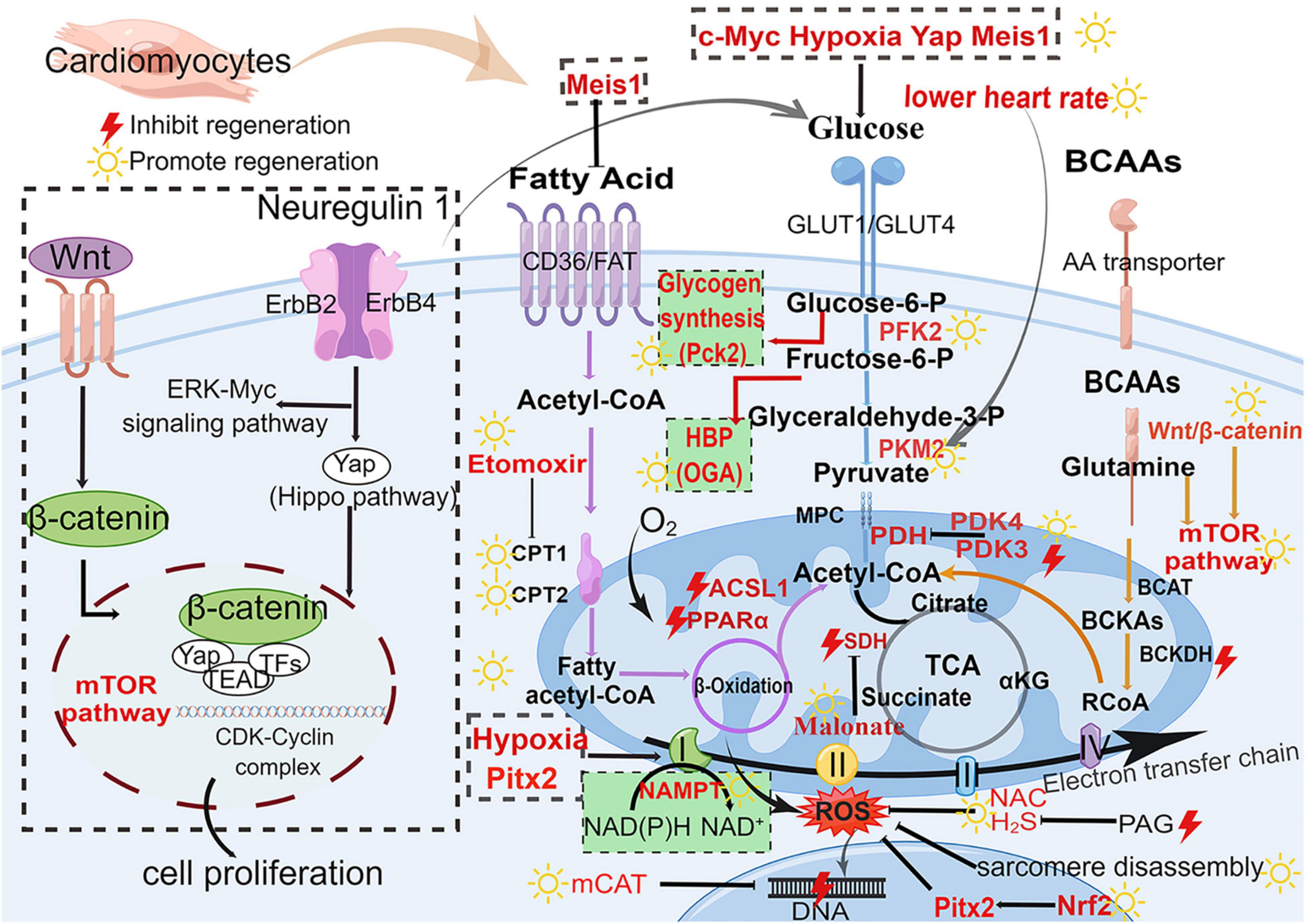
Figure 3. Major metabolic pathways and signaling pathways in heart regeneration after cardiac injury. Glucose metabolism (blue), fatty acid metabolism (purple), BCAA metabolism (orange), and biosynthetic pathways (green background). The boxed section shows the factors affecting the metabolic pathways. Wnt/β-catenin pathway and Nrg1-ErbB pathway affect the metabolism of heart regeneration. Acetyl-CoA is the final effector of these three metabolism pathways (fatty acid oxidation, glycolysis, and amino acid metabolism) and regulates the initiation of the TCA cycle. GLUT, glucose transporter type; Glucose-6-P, glucose-6-phosphate; Fructose-6-P, fructose-6-phosphate; Glyceraldehyde-3-P, glyceraldehyde-3-phosphate; PFK, phosphofructokinase; PKM2, M2-pyruvate kinase; PDK, pyruvate dehydrogenase kinase; PDH, pyruvate dehydrogenase; CD36, cluster of differentiation; FAT, fatty acid transport carrier; CPT, carnitine palmitoyltransferase; BCAAs, branched-chain amino acids; BCAT, branched-chain amino acid transferase; BCKA, branched-chain alpha-keto acids; BCKDH, branched-chain alpha-keto acid dehydrogenase; mTOR, mammalian target of rapamycin; TCA, tricarboxylic acid cycle; SDH, succinate dehydrogenase; αKG, α-ketoglutarate; ROS, reactive oxygen species; H2S, hydrogen sulfide; NAC, N-acetylcysteine; NAMPT, nicotinamide phosphoribosyltransferase; PAG, propargylglycine; mCAT, mitochondrial catalase; Pitx2, paired-liked homeodomain transcription factor 2; Nrf2, nuclear factor-erythroid-2-related factor 2; CDK, cyclin-dependent kinases; HBP, hexosamine biosynthesis pathway; Pck2, phosphoenolpyruvate carboxykinase 2; OGA, O-GlcNAcase. The yellow sun represents the promotion of heart regeneration, while the red lightning represents the inhibition of heart regeneration.
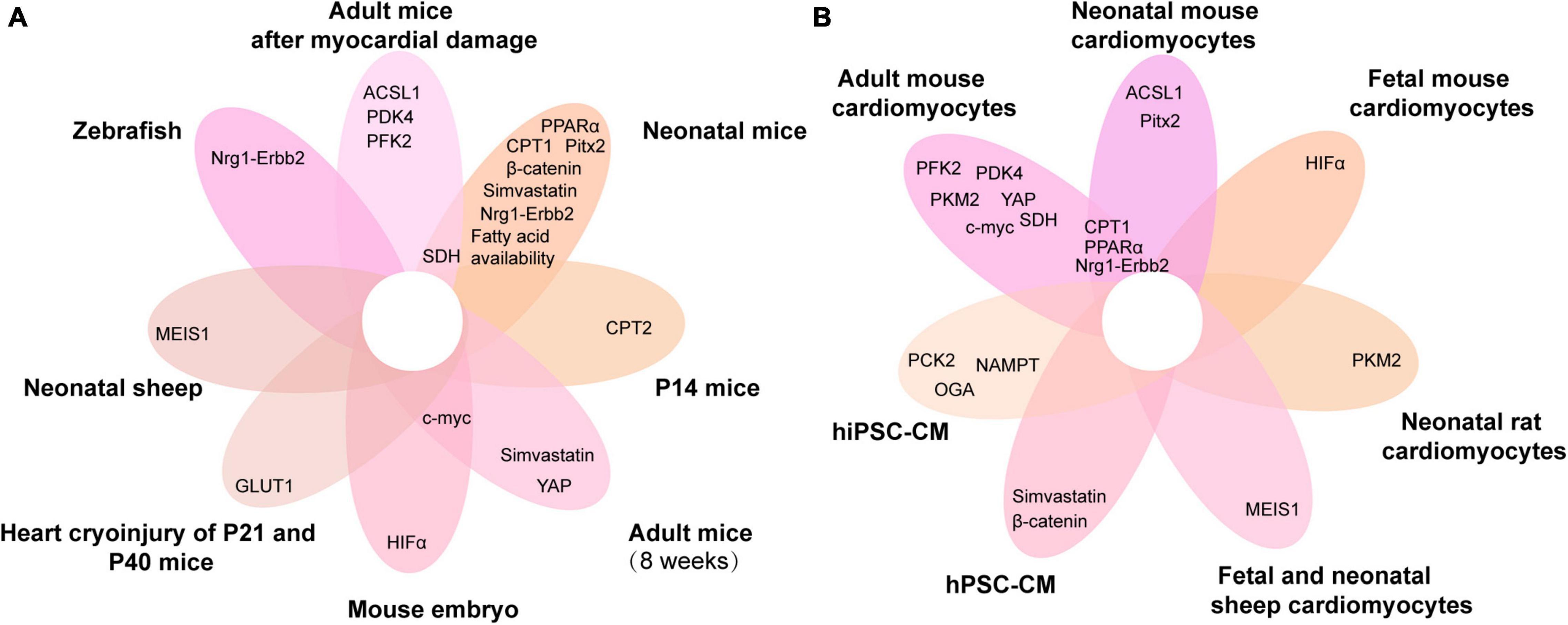
Figure 4. Venn diagram of metabolic genes associated with cardiomyocyte proliferation overlapping in different animals and cardiomyocytes. (A,B) The metabolic genes associated with cardiomyocyte proliferation in different animals (A) and cardiomyocytes (B). ACSL1, acyl-CoA synthetase long-chain family member 1; PFK, phosphofructokinase 2; PPARα, peroxisome proliferator-activated receptor; HIF-α, hypoxia-inducible factor α; PDK4, pyruvate dehydrogenase kinase 4; MEIS1, myeloid ecotropic viral integration site 1; PCK2, phosphoenolpyruvate carboxykinase 2; CPT, carnitine palmitoyltransferase; NAMPT, nicotinamide phosphoribosyltransferase; YAP, yes-associated protein; Nrg1, neuregulin 1; SDH, succinate dehydrogenase; GLUT1, glucose transporter 1; Pitx2, paired-like homeodomain 2; hiPSC-CM, human-induced pluripotent stem cell cardiomyocytes; hPSC-CM, human pluripotent stem cell-derived cardiomyocytes; P14, postnatal day 14; P21, postnatal day 21; P40, postnatal day 40.
The primary step of heart regeneration is to promote the cardiomyocytes to enter the cell cycle for division and proliferation. The interphase of the cell cycle is metabolically active and is the period during which the cells synthesize various enzymes, RNA, DNA, and proteins. During the S-phase, the cell cycle can be promoted by metabolic enzymes and proliferation pathways that target nucleic acid and protein synthesis, thereby promoting entry of cardiomyocytes into the cell cycle. However, specific biosynthetic pathways and metabolic enzymes need to be further explored, in order to increase the proliferative capacity of cardiomyocytes.
Certain questions are still unanswered. For example, are there other cardiac component cells (such as immune cells, endothelial cells, and fibroblasts) that interact with cardiomyocytes to influence their metabolism and thus alter their proliferative potential? Macrophages are required for myocardial regeneration in neonatal mice (137). However, it is unclear whether macrophages reprogram cardiomyocyte metabolism through intercellular interaction or paracrine effects. The angiogenesis of vascular endothelium supports regenerating cardiomyocytes after MI (138). The vascular endothelium supplies paracrine factors to cardiomyocytes so that they participate in regeneration. However, whether it affects the metabolism of cardiomyocytes is unclear. Besides, the investigators suggest that in situ modulation of cardiac cells in infarct foci can promote cardiomyocyte proliferation to repair scars and thus avoids the need of transplantation (139). Cardiac fibroblasts constitute a large proportion of the cardiac cells, and previous studies suggest that the cocktail therapy could potentially reprogram the fibroblasts in infarct foci directly into cardiomyocytes by modulating the transcription factors (139). Nonetheless, it is unclear whether cardiomyocyte metabolism is altered in this case? In addition, is it possible to enhance the proliferation of cardiomyocytes directly with small molecule drug-targeted metabolites?
Currently, hypoxia-induced glycolysis is the most studied among the metabolic pathways and has the greatest impact on myocardial regeneration. In future, translational studies can be conducted in depth from glycolysis to provide new targets for future clinical translation. It is important to further explore the relationship between myocardial regeneration mechanisms and changes in myocardial energy metabolism. However, we should note that almost none of the published studies have proven that their manipulations improve cardiac function by directly increasing the number of cardiomyocytes. The improved heart function is mainly correlated with the enhanced cell cycle activity and the improved functioning of the remaining cardiomyocytes, which might be due to a metabolic shift. Thus, it is essential to uncover the signaling pathways and key regulatory factors that affect energy metabolism accompanied by myocardial regeneration to develop effective therapeutic approaches. In all, we are optimistic that it will be possible to achieve great progress in human heart regeneration.
ZZ and XL conceived and designed the study and revised the manuscript. XD analyzed the data and wrote the manuscript. All authors listed and approved the manuscript for publication.
This work was supported by the National Key R&D Program of China (2019YFA0801502), the National Natural Science Foundation of China (82070415, 82071790, and 81771695), the Shuguang Program sponsored by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (19SG17 and 18SG33), and the Shanghai Science & Technology Development Foundation (AX-2105).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Figdraw (www.figdraw.com) for the help in drawing the figures.
1. Eriksson H. Heart failure: a growing public health problem. J Intern Med. (1995) 237:135–41. doi: 10.1111/j.1365-2796.1995.tb01153.x
2. Laflamme MA, Zbinden S, Epstein SE, Murry CE. Cell-based therapy for myocardial ischemia and infarction: pathophysiological mechanisms. Annu Rev Pathol. (2007) 2:307–39. doi: 10.1146/annurev.pathol.2.010506.092038
4. Reardon S. First pig-to-human heart transplant: what can scientists learn? Nature. (2022) 601:305–6. doi: 10.1038/d41586-022-00111-9
5. Becker RO, Chapin S, Sherry R. Regeneration of the ventricular myocardium in amphibians. Nature. (1974) 248:145–7. doi: 10.1038/248145a0
6. Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, et al. P38 map kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. (2005) 19:1175–87. doi: 10.1101/gad.1306705
7. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. (2011) 331:1078–80. doi: 10.1126/science.1200708
8. Notari M, Ventura-Rubio A, Bedford-Guaus SJ, Jorba I, Mulero L, Navajas D, et al. The local microenvironment limits the regenerative potential of the mouse neonatal heart. Sci Adv. (2018) 4:eaao5553. doi: 10.1126/sciadv.aao5553
9. Fajardo VM, Feng I, Chen BY, Perez-Ramirez CA, Shi B, Clark P, et al. Glut1 overexpression enhances glucose metabolism and promotes neonatal heart regeneration. Sci Rep. (2021) 11:8669. doi: 10.1038/s41598-021-88159-x
10. Stockdale WT, Lemieux ME, Killen AC, Zhao J, Hu Z, Riepsaame J, et al. Heart regeneration in the Mexican cavefish. Cell Rep. (2018) 25:1997–2007.e7. doi: 10.1016/j.celrep.2018.10.072
11. Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. (2002) 298:2188–90. doi: 10.1126/science.1077857
12. Staudt D, Stainier D. Uncovering the molecular and cellular mechanisms of heart development using the zebrafish. Annu Rev Genet. (2012) 46:397–418. doi: 10.1146/annurev-genet-110711-155646
13. Leone M, Magadum A, Engel FB. Cardiomyocyte proliferation in cardiac development and regeneration: a guide to methodologies and interpretations. Am J Physiol Heart Circ Physiol. (2015) 309:H1237–50. doi: 10.1152/ajpheart.00559.2015
14. Zebrowski DC, Vergarajauregui S, Wu CC, Piatkowski T, Becker R, Leone M, et al. Developmental alterations in centrosome integrity contribute to the post-mitotic state of mammalian cardiomyocytes. Elife. (2015) 4:e05563. doi: 10.7554/eLife.05563
15. Poss KD. Getting to the heart of regeneration in zebrafish. Semin Cell Dev Biol. (2007) 18:36–45. doi: 10.1016/j.semcdb.2006.11.009
16. Wills AA, Holdway JE, Major RJ, Poss KD. Regulated addition of new myocardial and epicardial cells fosters homeostatic cardiac growth and maintenance in adult zebrafish. Development. (2008) 135:183–92. doi: 10.1242/dev.010363
17. Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. (2014) 157:565–79. doi: 10.1016/j.cell.2014.03.032
18. Rees BB, Sudradjat FA, Love JW. Acclimation to hypoxia increases survival time of zebrafish, danio rerio, during lethal hypoxia. J Exp Zool. (2001) 289:266–72. doi: 10.1002/1097-010x(20010401/30)289:43.0.co;2-5
19. Jopling C, Suñé G, Faucherre A, Fabregat C, Izpisua Belmonte JC. Hypoxia induces myocardial regeneration in zebrafish. Circulation. (2012) 126:3017–27. doi: 10.1161/circulationaha.112.107888
20. Helston O, Amaya E. Reactive oxygen species during heart regeneration in zebrafish: lessons for future clinical therapies. Wound Repair Regen. (2021) 29:211–24. doi: 10.1111/wrr.12892
21. Smith AM, Maguire-Nguyen KK, Rando TA, Zasloff MA, Strange KB, Yin VP. The protein tyrosine phosphatase 1b inhibitor Msi-1436 stimulates regeneration of heart and multiple other tissues. NPJ Regen Med. (2017) 2:4. doi: 10.1038/s41536-017-0008-1
22. Missinato MA, Saydmohammed M, Zuppo DA, Rao KS, Opie GW, Kühn B, et al. Dusp6 attenuates Ras/Mapk signaling to limit zebrafish heart regeneration. Development. (2018) 145:dev157206. doi: 10.1242/dev.157206
23. Honkoop H, de Bakker DE, Aharonov A, Kruse F, Shakked A, Nguyen PD, et al. Single-cell analysis uncovers that metabolic reprogramming by Erbb2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife. (2019) 8:e50163. doi: 10.7554/eLife.50163
24. Rubin N, Harrison MR, Krainock M, Kim R, Lien CL. Recent advancements in understanding endogenous heart regeneration-insights from adult zebrafish and neonatal mice. Semin Cell Dev Biol. (2016) 58:34–40. doi: 10.1016/j.semcdb.2016.04.011
25. Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. (1996) 28:1737–46. doi: 10.1006/jmcc.1996.0163
26. Ali SR, Hippenmeyer S, Saadat LV, Luo L, Weissman IL, Ardehali R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci U S A. (2014) 111:8850–5. doi: 10.1073/pnas.1408233111
27. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, et al. Evidence for cardiomyocyte renewal in humans. Science. (2009) 324:98–102. doi: 10.1126/science.1164680
28. Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, et al. Dynamics of cell generation and turnover in the human heart. Cell. (2015) 161:1566–75. doi: 10.1016/j.cell.2015.05.026
29. Dawes GS, Mott JC, Widdicombe JG. The foetal circulation in the lamb. J Physiol. (1954) 126:563–87. doi: 10.1113/jphysiol.1954.sp005227
30. Vivien CJ, Hudson JE, Porrello ER. Evolution, comparative biology and ontogeny of vertebrate heart regeneration. NPJ Regen Med. (2016) 1:16012. doi: 10.1038/npjregenmed.2016.12
31. Neely JR, Morgan HE. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu Rev Physiol. (1974) 36:413–59. doi: 10.1146/annurev.ph.36.030174.002213
32. Kolwicz SC Jr., Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. (2013) 113:603–16. doi: 10.1161/circresaha.113.302095
33. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. (2013) 113:709–24. doi: 10.1161/circresaha.113.300376
34. Ritterhoff J, Tian R. Metabolism in cardiomyopathy: every substrate matters. Cardiovasc Res. (2017) 113:411–21. doi: 10.1093/cvr/cvx017
35. Maroli G, Braun T. The long and winding road of cardiomyocyte maturation. Cardiovasc Res. (2021) 117:712–26. doi: 10.1093/cvr/cvaa159
36. Mills RJ, Titmarsh DM, Koenig X, Parker BL, Ryall JG, Quaife-Ryan GA, et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc Natl Acad Sci U S A. (2017) 114:E8372–81. doi: 10.1073/pnas.1707316114
37. Ascuitto RJ, Ross-Ascuitto NT, Chen V, Downing SE. Ventricular function and fatty acid metabolism in neonatal piglet heart. Am J Physiol. (1989) 256:H9–15. doi: 10.1152/ajpheart.1989.256.1.H9
38. Lopaschuk GD, Spafford MA. Energy substrate utilization by isolated working hearts from newborn rabbits. Am J Physiol. (1990) 258:H1274–80. doi: 10.1152/ajpheart.1990.258.5.H1274
39. Essop MF. Cardiac metabolic adaptations in response to chronic hypoxia. J Physiol. (2007) 584:715–26. doi: 10.1113/jphysiol.2007.143511
40. Cole MA, Abd Jamil AH, Heather LC, Murray AJ, Sutton ER, Slingo M, et al. On the pivotal role of PPARα in adaptation of the heart to hypoxia and why fat in the diet increases hypoxic injury. Faseb J. (2016) 30:2684–97. doi: 10.1096/fj.201500094R
41. Daneshrad Z, Garcia-Riera MP, Verdys M, Rossi A. Differential responses to chronic hypoxia and dietary restriction of aerobic capacity and enzyme levels in the rat myocardium. Mol Cell Biochem. (2000) 210:159–66. doi: 10.1023/a:1007137909171
42. Dang YM, Huang YS, Zhou JL, Zhang JP, Yan H, Zhang M. [An experimental study on the influence of hypoxia induction Factor-1alpha on the glycolysis of the rat myocardial cell under hypoxic condition]. Zhonghua Shao Shang Za Zhi. (2005) 21:339–42.
43. Heather LC, Cole MA, Tan JJ, Ambrose LJ, Pope S, Abd-Jamil AH, et al. Metabolic adaptation to chronic hypoxia in cardiac mitochondria. Basic Res Cardiol. (2012) 107:268. doi: 10.1007/s00395-012-0268-2
44. Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, et al. Hypoxia induces heart regeneration in adult mice. Nature. (2017) 541:222–7. doi: 10.1038/nature20173
45. Kaelin WG Jr. The Von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem Biophys Res Commun. (2005) 338:627–38. doi: 10.1016/j.bbrc.2005.08.165
46. Wu D, Liu L, Fu S, Zhang J. Osteostatin improves the osteogenic differentiation of mesenchymal stem cells and enhances angiogenesis through Hif-1α under hypoxia conditions in vitro. Biochem Biophys Res Commun. (2022) 606:100–7. doi: 10.1016/j.bbrc.2022.02.085
47. Moslehi J, Minamishima YA, Shi J, Neuberg D, Charytan DM, Padera RF, et al. Loss of hypoxia-inducible factor Prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation. (2010) 122:1004–16. doi: 10.1161/circulationaha.109.922427
48. Lei L, Mason S, Liu D, Huang Y, Marks C, Hickey R, et al. Hypoxia-inducible factor-dependent degeneration, failure, and malignant transformation of the heart in the absence of the Von Hippel-Lindau protein. Mol Cell Biol. (2008) 28:3790–803. doi: 10.1128/mcb.01580-07
49. Menendez-Montes I, Escobar B, Gomez MJ, Albendea-Gomez T, Palacios B, Bonzon-Kulichenko E, et al. Activation of amino acid metabolic program in cardiac HIF1-alpha-deficient mice. iScience. (2021) 24:102124. doi: 10.1016/j.isci.2021.102124
50. Guimarães-Camboa N, Stowe J, Aneas I, Sakabe N, Cattaneo P, Henderson L, et al. HIF1α represses cell stress pathways to allow proliferation of hypoxic fetal cardiomyocytes. Dev Cell. (2015) 33:507–21. doi: 10.1016/j.devcel.2015.04.021
51. Cardoso AC, Pereira AHM, Sadek HA. Mechanisms of neonatal heart regeneration. Curr Cardiol Rep. (2020) 22:33. doi: 10.1007/s11886-020-01282-5
52. Wang Z, Ying Z, Bosy-Westphal A, Zhang J, Schautz B, Later W, et al. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr. (2010) 92:1369–77. doi: 10.3945/ajcn.2010.29885
53. Karwi QG, Uddin GM, Ho KL, Lopaschuk GD. Loss of metabolic flexibility in the failing heart. Front Cardiovasc Med. (2018) 5:68. doi: 10.3389/fcvm.2018.00068
54. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. (2007) 356:1140–51. doi: 10.1056/NEJMra063052
55. Lopaschuk GD, Spafford MA, Marsh DR. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am J Physiol. (1991) 261:H1698–705. doi: 10.1152/ajpheart.1991.261.6.H1698
56. Werner JC, Sicard RE. Lactate metabolism of isolated, perfused fetal, and newborn pig hearts. Pediatr Res. (1987) 22:552–6. doi: 10.1203/00006450-198711000-00016
57. Bartelds B, Gratama JW, Knoester H, Takens J, Smid GB, Aarnoudse JG, et al. Perinatal changes in myocardial supply and flux of fatty acids, carbohydrates, and ketone bodies in lambs. Am J Physiol. (1998) 274:H1962–9. doi: 10.1152/ajpheart.1998.274.6.H1962
58. Itoi T, Lopaschuk GD. The contribution of glycolysis, glucose oxidation, lactate oxidation, and fatty acid oxidation to ATP production in isolated biventricular working hearts from 2-week-old rabbits. Pediatr Res. (1993) 34:735–41. doi: 10.1203/00006450-199312000-00008
59. Onay-Besikci A. Regulation of cardiac energy metabolism in newborn. Mol Cell Biochem. (2006) 287:1–11. doi: 10.1007/s11010-006-9123-9
60. Bartelds B, Knoester H, Beaufort-Krol GC, Smid GB, Takens J, Zijlstra WG, et al. Myocardial lactate metabolism in fetal and newborn lambs. Circulation. (1999) 99:1892–7. doi: 10.1161/01.cir.99.14.1892
61. Cao T, Liccardo D, LaCanna R, Zhang X, Lu R, Finck BN, et al. Fatty acid oxidation promotes cardiomyocyte proliferation rate but does not change cardiomyocyte number in infant mice. Front Cell Dev Biol. (2019) 7:42. doi: 10.3389/fcell.2019.00042
62. Isu G, Robles Diaz D, Grussenmeyer T, Gaudiello E, Eckstein F, Brink M, et al. Fatty acid-based monolayer culture to promote in vitro neonatal rat cardiomyocyte maturation. Biochim Biophys Acta Mol Cell Res. (2020) 1867:118561. doi: 10.1016/j.bbamcr.2019.118561
63. Cardoso AC, Lam NT, Savla JJ, Nakada Y, Pereira AHM, Elnwasany A, et al. Mitochondrial substrate utilization regulates cardiomyocyte cell cycle progression. Nat Metab. (2020) 2:167–78.
64. Brown NF, Weis BC, Husti JE, Foster DW, McGarry JD. Mitochondrial carnitine palmitoyltransferase I isoform switching in the developing rat heart. J Biol Chem. (1995) 270:8952–7. doi: 10.1074/jbc.270.15.8952
65. Ceccarelli SM, Chomienne O, Gubler M, Arduini A. Carnitine Palmitoyltransferase (CPT) modulators: a medicinal chemistry perspective on 35 years of research. J Med Chem. (2011) 54:3109–52. doi: 10.1021/jm100809g
66. Hirose K, Payumo AY, Cutie S, Hoang A, Zhang H, Guyot R, et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science. (2019) 364:184–8. doi: 10.1126/science.aar2038
67. Li Y, Yang M, Tan J, Shen C, Deng S, Fu X, et al. Targeting ACSL1 promotes cardiomyocyte proliferation and cardiac regeneration. Life Sci. (2022) 294:120371. doi: 10.1016/j.lfs.2022.120371
68. Fukuda R, Marín-Juez R, El-Sammak H, Beisaw A, Ramadass R, Kuenne C, et al. Stimulation of glycolysis promotes cardiomyocyte proliferation after injury in adult zebrafish. EMBO Rep. (2020) 21:e49752. doi: 10.15252/embr.201949752
69. Huang Y, Lei L, Liu D, Jovin I, Russell R, Johnson RS, et al. Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1α-dependent function. Proc Natl Acad Sci U S A. (2012) 109:17478–83. doi: 10.1073/pnas.1209281109
70. Shao D, Tian R. Glucose transporters in cardiac metabolism and hypertrophy. Compr Physiol. (2015) 6:331–51. doi: 10.1002/cphy.c150016
71. Sugden MC, Langdown ML, Harris RA, Holness MJ. Expression and regulation of pyruvate dehydrogenase kinase isoforms in the developing rat heart and in adulthood: role of thyroid hormone status and lipid supply. Biochem J. (2000) 352(Pt 3):731–8.
72. Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, et al. Inducible overexpression of Glut1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J Am Heart Assoc. (2013) 2:e000301. doi: 10.1161/jaha.113.000301
73. Liao R, Jain M, Cui L, D’Agostino J, Aiello F, Luptak I, et al. Cardiac-specific overexpression of Glut1 prevents the development of heart failure attributable to pressure overload in mice. Circulation. (2002) 106:2125–31. doi: 10.1161/01.cir.0000034049.61181.f3
74. Wang Q, Donthi RV, Wang J, Lange AJ, Watson LJ, Jones SP, et al. Cardiac phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase increases glycolysis, hypertrophy, and myocyte resistance to hypoxia. Am J Physiol Heart Circ Physiol. (2008) 294:H2889–97. doi: 10.1152/ajpheart.91501.2007
75. Mazurek S. Pyruvate kinase Type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. (2011) 43:969–80. doi: 10.1016/j.biocel.2010.02.005
76. Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget. (2011) 2:551–6. doi: 10.18632/oncotarget.299
77. Hauck L, Dadson K, Chauhan S, Grothe D, Billia F. Inhibiting the Pkm2/B-catenin axis drives in vivo replication of adult cardiomyocytes following experimental MI. Cell Death Differ. (2021) 28:1398–417. doi: 10.1038/s41418-020-00669-9
78. Magadum A, Singh N, Kurian AA, Munir I, Mehmood T, Brown K, et al. Pkm2 regulates cardiomyocyte cell cycle and promotes cardiac regeneration. Circulation. (2020) 141:1249–65. doi: 10.1161/circulationaha.119.043067
79. Muralidhar SA, Sadek HA. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. In: T Nakanishi, RR Markwald, HS Baldwin, BB Keller, D Srivastava, H Yamagishi editors. Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology. Tokyo: Springer Copyright 2016, The Author(s) (2016). p. 93–101.
80. Lindgren IM, Drake RR, Chattergoon NN, Thornburg KL. Down-regulation of Meis1 promotes the maturation of oxidative phosphorylation in perinatal cardiomyocytes. Faseb J. (2019) 33:7417–26. doi: 10.1096/fj.201801330RR
81. Aharonov A, Shakked A, Umansky KB, Savidor A, Genzelinakh A, Kain D, et al. Erbb2 drives yap activation and EMT-like processes during cardiac regeneration. Nat Cell Biol. (2020) 22:1346–56. doi: 10.1038/s41556-020-00588-4
82. Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, et al. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature. (2017) 550:260–4. doi: 10.1038/nature24045
83. Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, et al. Hippo pathway effector yap promotes cardiac regeneration. Proc Natl Acad Sci U S A. (2013) 110:13839–44. doi: 10.1073/pnas.1313192110
84. Tao G, Kahr PC, Morikawa Y, Zhang M, Rahmani M, Heallen TR, et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. (2016) 534:119–23. doi: 10.1038/nature17959
85. Monroe TO, Hill MC, Morikawa Y, Leach JP, Heallen T, Cao S, et al. Yap partially reprograms chromatin accessibility to directly induce adult cardiogenesis in vivo. Dev Cell. (2019) 48:765–79.e7. doi: 10.1016/j.devcel.2019.01.017
86. Li L, Tao G, Hill MC, Zhang M, Morikawa Y, Martin JF. Pitx2 maintains mitochondrial function during regeneration to prevent myocardial fat deposition. Development. (2018) 145:dev168609. doi: 10.1242/dev.168609
87. Ding Y, Liu Z, Desai S, Zhao Y, Liu H, Pannell LK, et al. Receptor tyrosine kinase Erbb2 translocates into mitochondria and regulates cellular metabolism. Nat Commun. (2012) 3:1271. doi: 10.1038/ncomms2236
88. Gemberling M, Karra R, Dickson AL, Poss KD. Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. Elife. (2015) 4:e05871. doi: 10.7554/eLife.05871
89. D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. (2015) 17:627–38. doi: 10.1038/ncb3149
90. Ahuja P, Zhao P, Angelis E, Ruan H, Korge P, Olson A, et al. Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J Clin Invest. (2010) 120:1494–505. doi: 10.1172/jci38331
91. Quaife-Ryan GA, Mills RJ, Lavers G, Voges HK, Vivien CJ, Elliott DA, et al. B -Catenin drives distinct transcriptional networks in proliferative and nonproliferative cardiomyocytes. Development. (2020) 147:dev193417. doi: 10.1242/dev.193417
92. Villa Del Campo C, Clavería C, Sierra R, Torres M. Cell competition promotes phenotypically silent cardiomyocyte replacement in the mammalian heart. Cell Rep. (2014) 8:1741–51. doi: 10.1016/j.celrep.2014.08.005
93. Murashige D, Jang C, Neinast M, Edwards JJ, Cowan A, Hyman MC, et al. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science. (2020) 370:364–8. doi: 10.1126/science.abc8861
94. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res. (2021) 128:1487–513. doi: 10.1161/circresaha.121.318241
95. Talman V, Teppo J, Pöhö P, Movahedi P, Vaikkinen A, Karhu ST, et al. Molecular atlas of postnatal mouse heart development. J Am Heart Assoc. (2018) 7:e010378. doi: 10.1161/jaha.118.010378
96. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
97. Miklas JW, Levy S, Hofsteen P, Mex DI, Clark E, Muster J, et al. Amino acid primed mTOR activity is essential for heart regeneration. iScience. (2022) 25:103574. doi: 10.1016/j.isci.2021.103574
98. Zhang P, Shan T, Liang X, Deng C, Kuang S. Mammalian target of rapamycin is essential for cardiomyocyte survival and heart development in mice. Biochem Biophys Res Commun. (2014) 452:53–9. doi: 10.1016/j.bbrc.2014.08.046
99. Moos PJ, Edes K, Fitzpatrick FA. Inactivation of wild-type P53 tumor suppressor by electrophilic prostaglandins. Proc Natl Acad Sci U S A. (2000) 97:9215–20. doi: 10.1073/pnas.160241897
100. Marnett LJ, Riggins JN, West JD. Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J Clin Invest. (2003) 111:583–93. doi: 10.1172/jci18022
101. Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. (2009) 361:1475–85. doi: 10.1056/NEJMra0804615
102. Elhelaly WM, Lam NT, Hamza M, Xia S, Sadek HA. Redox regulation of heart regeneration: an evolutionary tradeoff. Front Cell Dev Biol. (2016) 4:137. doi: 10.3389/fcell.2016.00137
103. Liu S, Martin JF. The regulation and function of the hippo pathway in heart regeneration. Wiley Interdiscip Rev Dev Biol. (2019) 8:e335. doi: 10.1002/wdev.335
104. Pei J, Wang F, Pei S, Bai R, Cong X, Nie Y, et al. Hydrogen sulfide promotes cardiomyocyte proliferation and heart regeneration via ros scavenging. Oxid Med Cell Longev. (2020) 2020:1412696. doi: 10.1155/2020/1412696
105. Pettinato AM, Yoo D, VanOudenhove J, Chen YS, Cohn R, Ladha FA, et al. Sarcomere function activates a P53-Dependent DNA damage response that promotes polyploidization and limits in vivo cell engraftment. Cell Rep. (2021) 35:109088. doi: 10.1016/j.celrep.2021.109088
106. Taylor WR, Stark GR. Regulation of the G2/M transition by P53. Oncogene. (2001) 20:1803–15. doi: 10.1038/sj.onc.1204252
107. Bae J, Salamon RJ, Brandt EB, Paltzer WG, Zhang Z, Britt EC, et al. Malonate promotes adult cardiomyocyte proliferation and heart regeneration. Circulation. (2021) 143:1973–86. doi: 10.1161/circulationaha.120.049952
108. McNamara JW, Porrello ER. From fragrances to heart regeneration: malonate repairs broken hearts. Circulation. (2021) 143:1987–90. doi: 10.1161/circulationaha.121.054313
109. Yetkin-Arik B, Vogels IMC, Nowak-Sliwinska P, Weiss A, Houtkooper RH, Van Noorden CJF, et al. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci Rep. (2019) 9:12608. doi: 10.1038/s41598-019-48676-2
110. Mills RJ, Parker BL, Quaife-Ryan GA, Voges HK, Needham EJ, Bornot A, et al. Drug screening in human PSC-cardiac organoids identifies pro-proliferative compounds acting via the mevalonate pathway. Cell Stem Cell. (2019) 24:895–907.e6. doi: 10.1016/j.stem.2019.03.009
111. Abouleisa RRE, McNally L, Salama ABM, Hammad SK, Ou Q, Wells C, et al. Cell cycle induction in human cardiomyocytes is dependent on biosynthetic pathway activation. Redox Biol. (2021) 46:102094. doi: 10.1016/j.redox.2021.102094
112. Magadum A, Ding Y, He L, Kim T, Vasudevarao MD, Long Q, et al. Live cell screening platform identifies PPARδ as a regulator of cardiomyocyte proliferation and cardiac repair. Cell Res. (2017) 27:1002–19. doi: 10.1038/cr.2017.84
113. Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM, et al. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. (2007) 117:3930–9. doi: 10.1172/jci32578
114. Magadum A, Engel FB. PPARβ/Δ: linking metabolism to regeneration. Int J Mol Sci. (2018) 19:2013. doi: 10.3390/ijms19072013
115. Tan J, Yang M, Wang H, Shen C, Wu M, Xu H, et al. Moderate heart rate reduction promotes cardiac regeneration through stimulation of the metabolic pattern switch. Cell Rep. (2022) 38:110468. doi: 10.1016/j.celrep.2022.110468
116. Tran DH, Wang ZV. Glucose metabolism in cardiac hypertrophy and heart failure. J Am Heart Assoc. (2019) 8:e012673. doi: 10.1161/jaha.119.012673
117. Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation. (2007) 116:434–48. doi: 10.1161/circulationaha.107.702795
118. Kundu BK, Zhong M, Sen S, Davogustto G, Keller SR, Taegtmeyer H. Remodeling of glucose metabolism precedes pressure overload-induced left ventricular hypertrophy: review of a hypothesis. Cardiology. (2015) 130:211–20. doi: 10.1159/000369782
119. Gibb AA, Hill BG. Metabolic coordination of physiological and pathological cardiac remodeling. Circ Res. (2018) 123:107–28. doi: 10.1161/circresaha.118.312017
120. Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. (2018) 128:3716–26. doi: 10.1172/jci120849
121. Knowlton AA, Chen L, Malik ZA. Heart failure and mitochondrial dysfunction: the role of mitochondrial fission/fusion abnormalities and new therapeutic strategies. J Cardiovasc Pharmacol. (2014) 63:196–206. doi: 10.1097/01.fjc.0000432861.55968.a6
122. Allard MF, Schönekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. (1994) 267:H742–50. doi: 10.1152/ajpheart.1994.267.2.H742
123. Lorell BH, Grossman W. Cardiac hypertrophy: the consequences for diastole. J Am Coll Cardiol. (1987) 9:1189–93. doi: 10.1016/s0735-1097(87)80326-1
124. Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. (2001) 104:2923–31. doi: 10.1161/hc4901.100526
125. Wambolt RB, Henning SL, English DR, Dyachkova Y, Lopaschuk GD, Allard MF. Glucose utilization and glycogen turnover are accelerated in hypertrophied rat hearts during severe low-flow ischemia. J Mol Cell Cardiol. (1999) 31:493–502. doi: 10.1006/jmcc.1998.0804
126. Kübler W, Spieckermann PG. Regulation of glycolysis in the ischemic and the anoxic myocardium. J Mol Cell Cardiol. (1970) 1:351–77. doi: 10.1016/0022-2828(70)90034-9
127. Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol. (2015) 5:1841–75. doi: 10.1002/cphy.c150006
128. Opie LH. Myocardial ischemia–metabolic pathways and implications of increased glycolysis. Cardiovasc Drugs Ther. (1990) 4(Suppl. 4):777–90. doi: 10.1007/bf00051275
129. Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD. Targeting fatty acid and carbohydrate oxidation–a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta. (2011) 1813:1333–50. doi: 10.1016/j.bbamcr.2011.01.015
130. Ritterhoff J, Young S, Villet O, Shao D, Neto FC, Bettcher LF, et al. Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ Res. (2020) 126:182–96. doi: 10.1161/circresaha.119.315483
131. Kolwicz SC Jr., Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of Acetyl Coa Carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. (2012) 111:728–38. doi: 10.1161/circresaha.112.268128
132. Zhang L, Jaswal JS, Ussher JR, Sankaralingam S, Wagg C, Zaugg M, et al. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ Heart Fail. (2013) 6:1039–48. doi: 10.1161/circheartfailure.112.000228
133. Graham N, Huang GN. Endocrine influence on cardiac metabolism in development and regeneration. Endocrinology. (2021) 162:bqab081. doi: 10.1210/endocr/bqab081
134. Allard MF, Henning SL, Wambolt RB, Granleese SR, English DR, Lopaschuk GD. Glycogen metabolism in the aerobic hypertrophied rat heart. Circulation. (1997) 96:676–82. doi: 10.1161/01.cir.96.2.676
135. Wambolt RB, Lopaschuk GD, Brownsey RW, Allard MF. Dichloroacetate improves postischemic function of hypertrophied rat hearts. J Am Coll Cardiol. (2000) 36:1378–85. doi: 10.1016/s0735-1097(00)00856-1
136. Mori J, Basu R, McLean BA, Das SK, Zhang L, Patel VB, et al. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: a metabolic contribution to heart failure with normal ejection fraction. Circ Heart Fail. (2012) 5:493–503. doi: 10.1161/circheartfailure.112.966705
137. Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, et al. Macrophages are required for neonatal heart regeneration. J Clin Invest. (2014) 124:1382–92. doi: 10.1172/jci72181
138. Li M, Wu Y, Ye L. The role of amino acids in endothelial biology and function. Cells. (2022) 11:1372. doi: 10.3390/cells11081372
Keywords: heart regeneration, cardiomyocyte proliferation, fatty acid metabolism, glucose metabolism, amino acid metabolism, metabolism regulation
Citation: Duan X, Liu X and Zhan Z (2022) Metabolic Regulation of Cardiac Regeneration. Front. Cardiovasc. Med. 9:933060. doi: 10.3389/fcvm.2022.933060
Received: 30 April 2022; Accepted: 13 June 2022;
Published: 08 July 2022.
Edited by:
Claudio de Lucia, Scientific Clinical Institute Maugeri (ICS Maugeri), ItalyReviewed by:
Felix B. Engel, University Hospital Erlangen, GermanyCopyright © 2022 Duan, Liu and Zhan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenzhen Zhan, emhhbnp6QHRvbmdqaS5lZHUuY24=; Xingguang Liu, bGl1eGdAaW1tdW5vbC5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.