 Abrahim Mahmood
Abrahim Mahmood Kinza Ahmed
Kinza Ahmed Youhua Zhang
Youhua Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Cardiovasc. Med., 01 July 2022
Sec. Heart Failure and Transplantation
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.925692
This article is part of the Research TopicFunctional Modifications of Ion Channels in ArrhythmogenesisView all 6 articles
Cardiac sympathetic activation, mediated by β-adrenergic receptors (β-ARs), normally increases cardiac contraction and relaxation. Accomplishing this task requires a physiological, concerted Ca2+ signaling, being able to increase Ca2+ release from sarcoplasmic reticulum (SR) in systole and speed up Ca2+ re-uptake in diastole. In heart failure (HF) myocardial β-ARs undergo desensitization/down-regulation due to sustained sympathetic adrenergic activation. β-AR desensitization/down-regulation diminishes adrenergic signaling and cardiac contractile reserve, and is conventionally considered to be detrimental in HF progression. Abnormal Ca2+ handling, manifested as cardiac ryanodine receptor (RyR2) dysfunction and diastolic Ca2+ leak (due to sustained adrenergic activation) also occur in HF. RyR2 dysfunction and Ca2+ leak deplete SR Ca2+ store, diminish Ca2+ release in systole and elevate Ca2+ levels in diastole, impairing both systolic and diastolic ventricular function. Moreover, elevated Ca2+ levels in diastole promote triggered activity and arrhythmogenesis. In the presence of RyR2 dysfunction and Ca2+ leak, further activation of the β-AR signaling in HF would worsen the existing abnormal Ca2+ handling, exacerbating not only cardiac dysfunction, but also ventricular arrhythmogenesis and sudden cardiac death. Thus, we conclude that β-AR desensitization/down-regulation may be a self-preserving, adaptive process (acting like an intrinsic β-AR blocker) protecting the failing heart from developing lethal ventricular arrhythmias under conditions of elevated sympathetic drive and catecholamine levels in HF, rather than a conventionally considered detrimental process. This also implies that medications simply enhancing β-AR signaling (like β-AR agonists) may not be so beneficial unless they can also correct dysfunctional Ca2+ handling in HF.
Beta-adrenergic receptors (β-ARs) are members of a large family of cell surface receptors known as G-protein-coupled receptors (GPCRs) (1). β-ARs mediate the sympathetic adrenergic effect (the fight or flight response) on the heart. There are 3 β-AR subtypes: β1-AR, β2-AR and β3-AR in the heart (2). These receptors are predominantly found in the myocardium, vascular smooth muscle, and adipose tissue, respectively. Cardiomyocytes express all 3 subtypes, with β1-ARs being the predominant subtype, representing ~80%, whereas β2-ARs and β3-ARs comprising the remaining 17 and 3%, respectively (3, 4). In cardiomyocytes, β1-ARs are widely distributed across the cell membrane and can be activated by both norepinephrine (released from sympathetic nerve fibers) and epinephrine (from the chromaffin cells of the adrenal medulla) (5). Activation of β1-ARs and the subsequent Gs-protein coupled signaling cascade (adenylyl cyclase-cAMP-protein kinase A) leads to increased myocardial contraction (inotropy), relaxation (lusitropy), heart rate (chronotropy) and AV conduction (dromotropy), respectively (6). β2-ARs are more localized within the transverse (T)-tubules (7), have a higher affinity for epinephrine and are coupled to the Gs and Gi subunits, allowing for both stimulatory and inhibitory effects on cardiac contractile function (8). β3-ARs are expressed at low levels on cardiomyocytes, and their physiological role is less clear (9).
Heart failure (HF) is a global epidemic problem, affecting more than 37.7 million individuals globally and imposes substantial economic burden to the health care system (10). HF occurs when various assaults result in myocardial injury/loss, impairing cardiac function. In response, the neurohumoral system, particularly the sympathetic nervous system is activated. The sympathetic activation, mediated by the β-AR (mainly β1-AR) signaling cascade (as mentioned before), increases cardiac contractility, relaxation, and cardiac output. Thus, sympathetic activation acutely (or in short term) is a beneficial compensatory process. However, sustained long-term sympathetic activation in HF can lead to pathological remodeling in β-ARs, manifested as uncoupling of the β-AR signaling (desensitization) and reduced number of β-ARs (mainly β1-ARs) on cell membrane (β-AR down-regulation) (6). β-AR desensitization/down-regulation diminishes adrenergic signaling and contractile reserve, and is conventionally considered to be detrimental in HF deterioration/progression (3, 11, 12). This notion is further supported by the fact that effective treatments (e.g., beta-blockers) are associated with re-sensitization/up-regulation of the β-ARs (6). As a result, re-sensitizing/up-regulating β-ARs is currently believed to be beneficial and efforts are directed to develop medications to up-regulate/re-sensitize the β-ARs as therapeutic strategies in HF (6, 12, 13).
Abnormal Ca2+ signaling has been identified as another form of pathological remodeling in HF (14). Several critical Ca2+ handling components are dysfunctional in HF, including sarcoplasmic reticulum (SR) cardiac ryanodine receptor (RyR2, the calcium release channel) dysfunction and diastolic Ca2+ leak, reduced Ca2+ pump (the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase, SERCA) activity, etc. (14). RyR2 dysfunction and Ca2+ leak could lead to depleted SR Ca2+ stores and elevated cytoplasmic Ca2+ levels in diastole, resulting in both systolic and diastolic dysfunction. In addition, by activating the inward Na+/Ca2+ exchanger (NCX) current (15), diastolic Ca2+ leak and elevated cytoplasmic Ca2+ levels could depolarize membrane potential, contributing to electrical instability, delayed afterdepolarizations, triggered arrhythmias, and sudden cardiac death (16–19). RyR2 dysfunction and Ca2+ leak in HF is caused by sustained sympathetic activation and elevated β-AR signaling (14). Thus, it is foreseeable that further activation of β-AR signaling would worsen the existing RyR2 dysfunction and Ca2+ leak, not only affecting cardiac function, but also enhancing arrhythmogenesis. Accordingly, we present our viewpoint here that in the presence of RyR2 dysfunction and Ca2+ leak, further enhancing β-AR signaling is detrimental rather than beneficial, and β-AR desensitization/down-regulation in HF may be a self-preserving, adaptive process rather than a conventionally considered detrimental process. This viewpoint will be detailed in the following discussion.
In HF cardiomyocytes develop 2 distinct yet related abnormalities: β-AR desensitization/down-regulation and RyR2 dysfunction/Ca2+ leak (Figure 1). Both abnormalities are allegedly caused by the sustained sympathetic activation and β-AR signaling in HF (6, 14). β-AR desensitization/down-regulation (Figure 1, abnormality 1) in HF is well-characterized, which diminishes sympathetic signaling, cardiac functional capacity and contractile reserve, and is conventionally considered to be detrimental in HF progression (3, 11, 12). On the other hand, when RyR2 dysfunction and Ca2+ leak occur (abnormality 2), further enhancing β-AR signaling would worsen the existing RyR2 dysfunction and Ca2+ leak, leading to exacerbation of cardiac dysfunction and arrhythmogenesis (Figure 1). Accordingly, we postulate that β-AR desensitization/down-regulation, acting like an intrinsic β-AR blocker and by diminishing adrenergic signaling, can limit RyR2 dysfunction and Ca2+ leak from further deteriorating under sustained sympathetic adrenergic drive with persistent high-level of catecholamines in HF. In other words, β-AR desensitization/down-regulation in HF may be a self-preserving, adaptive process (a friend) rather than a conventionally considered detrimental/pathological process (a foe). This viewpoint is supported by the following several lines of evidence.
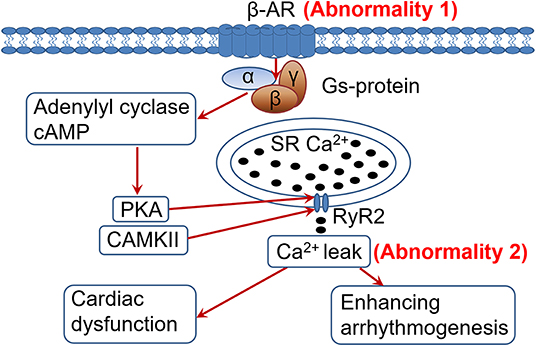
Figure 1. Heightened adrenergic signaling in the presence of cardiac ryanodine receptor (RyR2) dysfunction and Ca2+ leak is detrimental. In heart failure (HF), sustained adrenergic activation leads to hyperphosphorylation (among other posttranslational modifications) of the RyR2 by PKA and CaMKII (through PKA-independent signaling mediated by β1-AR activation), resulting in RyR2 dysfunction and pathological Ca2+ leak. Desensitization/down-regulationof the β-AR (abnormality 1) would diminish adrenergic signaling, alleviating RyR2 dysfunction and Ca2+ leak (abnormality 2) than they would otherwise. In contrast, enhancing adrenergic signaling in HF would worsen the RyR2 dysfunction and Ca2+ leak (abnormality 2), exacerbating cardiac dysfunction and arrhythmogenesis. PKA, protein kinase A; CaMKII, Ca2+/calmodulin-dependent protein kinase II; SR, sarcoplasmic reticulum; RyR2, cardiac ryanodine receptor.
Cardiac excitation-contraction coupling (ECC) refers to the conversion from electrical excitation (in the form of an action potential) into mechanical contraction. Briefly, cardiac action potential propagates along the cell membrane, including the specialized invaginations (T-tubules) (20). Depolarization activates voltage-gated L-type Ca2+ channels (LTCCs) on the T-tubules and allows Ca2+ to enter the cell. The entering Ca2+ then binds to and activates Ca2+ release channels (RyR2) on SR, releasing Ca2+ to cytoplasm from the SR (where Ca2+ is stored at high concentrations). This process by which Ca2+ entering cardiomyocyte through LTCCs activates RyR2 is known as Ca2+ induced Ca2+ release (21). The spike of Ca2+ concentration in cytoplasm initiates actin–myosin cross-bridging, causing muscle contraction (systole). Cardiac relaxation (diastole) is initiated when RyR2 close (terminating SR Ca2+ release) and Ca2+ are pumped back into the SR by the SERCA or extruded from the cell by the Na+/Ca2+ exchanger (NCX). This coordinated Ca2+ release and reuptake in cardiomyocytes are essential for ECC.
Normal Ca2+ signaling is required in mediating physiological sympathetic response. Sympathetic activation, mediated by the β-ARs-Gs-adenylyl cyclase-cAMP-protein kinase A (PKA) signaling pathway, activates and phosphorylates LTCCs, increasing inward Ca2+ current (22). The same cascade also phosphorylates RyR2 (23), increasing Ca2+ release from SR, which in turn increases cardiac contractility (14). Adrenergic signaling also activates SERCA, speeding up Ca2+ removal from cytosol, thus improving cardiac relaxation. This normal Ca2+ signaling is essential in realizing sympathetic adrenergic effects on the heart.
It is known that ventricular arrhythmias are common in HF patients and about 50% of HF patients die due to sudden cardiac death (24). Accumulating evidence indicates that a pathogenic process of defective cardiac Ca2+ signaling plays a critical role in HF (16, 25, 26). As discussed, RyR2 open and close in a concerted manner during myocyte contraction and relaxation (27). In HF, chronic sympathetic adrenergic activation leads to unstable channels that leak Ca2+ during diastole because the channel cannot close properly (28). The RyR2 dysfunction in HF is reportedly caused by enhanced adrenergic signaling resulting in hyperphosphorylation, oxidation or S-nitrosylation of RyR2, etc., which results in Ca2+ leak by destabilizing the closed state of the channel (14). RyR2 dysfunction and Ca2+ leak can be observed in isolated cardiomyocytes as short, unsynchronized SR Ca2+ release events during diastole (29).
RyR2 dysfunction and Ca2+ leak could impair cardiac function and enhance arrhythmogenesis. Chronic Ca2+ leak via dysfunctional RyR2 depletes Ca2+ store in the SR, lowers Ca2+ transient amplitude in systole and reduces cardiac contractility (30). Ca2+ leak can lead to elevated Ca2+ levels in diastole, impairing myocyte relaxation. Thus, RyR2 dysfunction and Ca2+ leak could lead to both systolic and diastolic dysfunction. Moreover, Ca2+ leak and elevated diastolic Ca2+ levels increase Ca2+ extrusion via NCX, promoting an inward depolarizing current which is electrogenic and promotes triggered activity and arrhythmia formation (15). The detrimental effect of β-AR activation in HF is supported by evidence that treatment with β-AR agonists is associated with increased mortality in HF (31, 32), despite their short-term hemodynamic benefits. We have demonstrated recently that failing hearts are more vulnerable to develop both atrial and ventricular arrhythmias under sympathetic stimulation, despite less positive inotropy in HF (33, 34). Finally, this is also supported by the fact that β-AR blocker treatment can reduce cardiac arrhythmias and sudden cardiac death in HF (35).
HF is characterized by sympathetic activation and elevated blood catecholamine levels (3). As discussed, sympathetic adrenergic activation, mediated through β-AR signaling, increases cardiac contraction and relaxation. These responses are initially beneficial in supporting the failing heart by increasing cardiac output and sustaining circulation. However, long-term overstimulation of β-ARs eventually leads to loss of responsiveness to sympathetic signaling (β-ARs desensitization) and reduced density (number) of β-ARs on cell membrane (down-regulation), resulting in diminished β-AR signaling, and loss of contractile reserve. It has been reported that β-AR desensitization/down-regulation correlates with HF severity (11). These effects underlie the pathogenesis of HF (6).
β-AR desensitization in HF is caused by phosphorylation of β-ARs themselves by PKA and G-protein coupled receptor kinases (GRKs) (6). GRK activity has been found to be augmented by prolonged β-AR activation under catecholamine exposure. GRK levels are elevated in the setting of HF and contribute to its pathogenesis (36). In addition, GRK activation and β-AR uncoupling can lead to β-ARs (mainly β1-ARs) internalization (β1-ARs shifted from the sarcolemma membrane into cytosolic compartments), contributing to β-ARs down-regulation (6). It has been reported that prolonged stimulation of β-AR signaling can induce β-AR down-regulation even without HF (37).
RyR2 dysfunction and Ca2+ leak in HF are caused by sustained adrenergic signaling (14). It would be obvious that further activation of the adrenergic signaling can further worsen RyR2 dysfunction and Ca2+ leak (Figure 1). In HF the Ca2+ leak is usually less at rest. With enhanced adrenergic signaling during sympathetic stress, the RyR2 dysfunction and Ca2+ leak would deteriorate (14). This would exacerbate cardiac dysfunction and promote arrhythmogenesis (15). As shown in the figure, the stronger the β-AR signaling, the worse the RyR2 dysfunction and Ca2+ leak, and thus more arrhythmogenesis. It could be speculated that if there were no β-AR desensitization/down-regulation in HF, there would be an even greater risk of developing arrhythmias and sudden cardiac deaths in HF patients due to persistent sympathetic activation and elevated catecholamine levels. Our recent findings that failing hearts are more vulnerable to develop arrhythmias under sympathetic stimulation (33, 34) support this concept. In line with this, β-AR agonist treatment is associated with increased mortality in HF (31, 32) and β-AR blocker treatment can reduce cardiac arrhythmias and sudden cardiac death in HF (35).
Thus, in the presence of RyR2 dysfunction and Ca2+ leak in HF, β-ARs desensitization/down-regulation, by diminishing adrenergic signaling, helps to attenuate RyR2 dysfunction and Ca2+leak, protecting the heart from developing serious cardiac arrhythmias and sudden cardiac death. In other words, β-ARs desensitization/down-regulation in HF may act in a way similar to a β-AR blocker, attenuating adrenergic signaling, which is beneficial especially under heightened sympathetic activation with persistent high-level of catecholamines seen in HF. From this point of view, β-AR desensitization/down-regulation in HF is a self-preserving, adaptive process (a friend) rather than a detrimental, pathological process (a foe), as conventionally considered.
β-AR desensitization/down-regulation is one of the many characteristic abnormalities seen in HF that is conventionally considered to be detrimental in HF progression (3, 11, 12). However, emerging evidence indicates that this concept may be too simplistic and may not be correct. Despite the fact that β-AR desensitization/down-regulation in HF diminishes adrenergic signaling and cardiac contractile reserve, this process may act as an intrinsic β-AR blocker, protecting the heart from developing more severe ventricular arrhythmias and sudden cardiac death. In line with this, new medications designed to re-sensitize/up-regulate β-ARs have to also normalize Ca2+ handling to be beneficial. Otherwise, simply enhancing adrenergic signaling without correcting RyR2 dysfunction (like β-AR agonists) may exacerbate RyR2 dysfunction and Ca2+ leak, increasing the arrhythmia risk and sudden cardiac death in HF. It should be noted that β-AR blocker treatment in HF is associated with normalization of RyR2 function and Ca2+ signaling (38), besides re-sensitizing/up-regulating the β-ARs.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
YZ contributed to conception of the manuscript. AM and KA wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hakak Y, Shrestha D, Goegel MC, Behan DP, Chalmers DT. Global analysis of G-protein-coupled receptor signaling in human tissues. FEBS Lett. (2003) 550:11–7. doi: 10.1016/S0014-5793(03)00762-2
2. Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. (2002) 415:206–12. doi: 10.1038/415206a
3. Bristow MR, Hershberger RE, Port JD, Minobe W, Rasmussen R. Beta 1- and beta 2-adrenergic receptor-mediated adenylate cyclase stimulation in nonfailing and failing human ventricular myocardium. Mol Pharmacol. (1989) 35:295–303.
4. Gauthier C, Leblais V, Kobzik L, Trochu JN, Khandoudi N, Bril A, et al. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Invest. (1998) 102:1377–84. doi: 10.1172/JCI2191
5. Perlman RL, Chalfie M. Catecholamine release from the adrenal medulla. Clin Endocrinol Metab. (1977) 6:551–76. doi: 10.1016/S0300-595X(77)80071-6
6. Pfleger J, Gresham K, Koch WJ. G protein-coupled receptor kinases as therapeutic targets in the heart. Nat Rev Cardiol. (2019) 16:612–22. doi: 10.1038/s41569-019-0220-3
7. Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, et al. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. (2010) 327:1653–7. doi: 10.1126/science.1185988
8. Molinoff PB. Alpha- and beta-adrenergic receptor subtypes properties, distribution and regulation. Drugs. (1984) 28(Suppl. 2):1–15. doi: 10.2165/00003495-198400282-00002
9. Dessy C, Balligand JL. Beta3-adrenergic receptors in cardiac and vascular tissues emerging concepts and therapeutic perspectives. Adv Pharmacol. (2010) 59:135–63. doi: 10.1016/S1054-3589(10)59005-7
10. Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. (2016) 13:368–78. doi: 10.1038/nrcardio.2016.25
11. Brodde OE, Zerkowski HR, Doetsch N, Motomura S, Khamssi M, Michel MC. Myocardial beta-adrenoceptor changes in heart failure: concomitant reduction in beta 1- and beta 2-adrenoceptor function related to the degree of heart failure in patients with mitral valve disease. J Am Coll Cardiol. (1989) 14:323–31. doi: 10.1016/0735-1097(89)90181-2
12. de Lucia C, Eguchi A, Koch WJ. New insights in cardiac beta-adrenergic signaling during heart failure and aging. Front Pharmacol. (2018) 9:904. doi: 10.3389/fphar.2018.00904
13. Ali DC, Naveed M, Gordon A, Majeed F, Saeed M, Ogbuke MI, et al. beta-Adrenergic receptor, an essential target in cardiovascular diseases. Heart Fail Rev. (2020) 25:343–54. doi: 10.1007/s10741-019-09825-x
14. Dridi H, Kushnir A, Zalk R, Yuan Q, Melville Z, Marks AR. Intracellular calcium leak in heart failure and atrial fibrillation: a unifying mechanism and therapeutic target. Nat Rev Cardiol. (2020) 17:732–47. doi: 10.1038/s41569-020-0394-8
15. Sipido KR, Bito V, Antoons G, Volders PG, Vos MA. Na/Ca exchange and cardiac ventricular arrhythmias. Ann N Y Acad Sci. (2007) 1099:339–48. doi: 10.1196/annals.1387.066
16. Shannon TR, Lew WY. Diastolic release of calcium from the sarcoplasmic reticulum: a potential target for treating triggered arrhythmias and heart failure. J Am Coll Cardiol. (2009) 53:2006–8. doi: 10.1016/j.jacc.2009.02.032
17. Hartmann N, Pabel S, Herting J, Schatter F, Renner A, Gummert J, et al. Antiarrhythmic effects of dantrolene in human diseased cardiomyocytes. Heart Rhythm. (2017) 14:412–9. doi: 10.1016/j.hrthm.2016.09.014
18. Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. (2007) 100:391–8. doi: 10.1161/01.RES.0000258172.74570.e6
19. Zamiri N, Masse S, Ramadeen A, Kusha M, Hu X, Azam MA, et al. Dantrolene improves survival after ventricular fibrillation by mitigating impaired calcium handling in animal models. Circulation. (2014) 129:875–85. doi: 10.1161/CIRCULATIONAHA.113.005443
20. Franzini-Armstrong C, Protasi F, Tijskens P. The assembly of calcium release units in cardiac muscle. Ann N Y Acad Sci. (2005) 1047:76–85. doi: 10.1196/annals.1341.007
21. Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. (1983) 245:C1–14. doi: 10.1152/ajpcell.1983.245.1.C1
22. Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. (2000) 87:1095–102. doi: 10.1161/01.RES.87.12.1095
23. Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, et al. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. (2010) 120:4388–98. doi: 10.1172/JCI32726
24. Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. (2004) 95:754–63. doi: 10.1161/01.RES.0000145047.14691.db
25. Oda T, Yano M, Yamamoto T, Tokuhisa T, Okuda S, Doi M, et al. Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation. (2005) 111:3400–10. doi: 10.1161/CIRCULATIONAHA.104.507921
26. Kobayashi S, Yano M, Suetomi T, Ono M, Tateishi H, Mochizuki M, et al. Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. J Am Coll Cardiol. (2009) 53:1993–2005. doi: 10.1016/j.jacc.2009.01.065
27. Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR. Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ Res. (2001) 88:1151–8. doi: 10.1161/hh1101.091268
28. Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. (2000) 101:365–76. doi: 10.1016/S0092-8674(00)80847-8
29. Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. (2008) 88:1491–545. doi: 10.1152/physrev.00030.2007
30. Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest. (2013) 123:46–52. doi: 10.1172/JCI62834
31. O'Connor CM, Gattis WA, Uretsky BF, Adams KF Jr., McNulty SE, et al. Continuous intravenous dobutamine is associated with an increased risk of death in patients with advanced heart failure: insights from the Flolan international randomized survival trial (FIRST). Am Heart J. (1999) 138:78–86. doi: 10.1016/S0002-8703(99)70250-4
32. El-Armouche A, Eschenhagen T. Beta-adrenergic stimulation and myocardial function in the failing heart. Heart Fail Rev. (2009) 14:225–41. doi: 10.1007/s10741-008-9132-8
33. Delfiner MS, Nofi C, Li Y, Gerdes AM, Zhang Y. Failing hearts are more vulnerable to sympathetic, but not vagal stimulation-induced, atrial fibrillation-ameliorated with dantrolene treatment. J Card Fail. (2018) 24:460–9. doi: 10.1016/j.cardfail.2018.05.008
34. Greco LV, Li Y, Zhang Y. Sympathetic β-Adrenergic stimulation evokes less positive inotropic response but more ventricular arrhythmias in heart failure. FASEB J. (2020) 34(Suppl. 1). doi: 10.1096/fasebj.2020.34.s1.02439
35. Al-Gobari M, El Khatib C, Pillon F, Gueyffier F. beta-Blockers for the prevention of sudden cardiac death in heart failure patients: a meta-analysis of randomized controlled trials. BMC Cardiovasc Disord. (2013) 13:52. doi: 10.1186/1471-2261-13-52
36. Vinge LE, Oie E, Andersson Y, Grogaard HK, Andersen G, Attramadal H. Myocardial distribution and regulation of GRK and beta-arrestin isoforms in congestive heart failure in rats. Am J Physiol Heart Circ Physiol. (2001) 281:H2490–9. doi: 10.1152/ajpheart.2001.281.6.H2490
37. Soltysinska E, Thiele S, Olesen SP, Osadchii OE. Chronic sympathetic activation promotes downregulation of beta-adrenoceptor-mediated effects in the guinea pig heart independently of structural remodeling and systolic dysfunction. Eur J Physiol. (2011) 462:529–43. doi: 10.1007/s00424-011-1005-7
Keywords: β-adrenergic receptor, β-adrenergic receptor desensitization/down-regulation, cardiac ryanodine receptor, calcium leak, arrhythmia, heart failure
Citation: Mahmood A, Ahmed K and Zhang Y (2022) β-Adrenergic Receptor Desensitization/Down-Regulation in Heart Failure: A Friend or Foe? Front. Cardiovasc. Med. 9:925692. doi: 10.3389/fcvm.2022.925692
Received: 21 April 2022; Accepted: 06 June 2022;
Published: 01 July 2022.
Edited by:
Susumu Minamisawa, Jikei University School of Medicine, JapanReviewed by:
Paramjit S. Tappia, I.H. Asper Clinical Research Institute, CanadaCopyright © 2022 Mahmood, Ahmed and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youhua Zhang, eXpoYW5nNDlAbnlpdC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.