 Jeremy Jong
Jeremy Jong James R. Pinney1,2,3
James R. Pinney1,2,3 René R. Sevag Packard
René R. Sevag Packard- 1Department of Medicine, Division of Cardiology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, United States
- 2Ronald Reagan UCLA Medical Center, Los Angeles, CA, United States
- 3Veterans Affairs West Los Angeles Medical Center, Los Angeles, CA, United States
- 4Department of Physiology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, United States
- 5Jonsson Comprehensive Cancer Center, University of California, Los Angeles, Los Angeles, CA, United States
- 6Molecular Biology Institute, University of California, Los Angeles, Los Angeles, CA, United States
- 7California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA, United States
Anthracyclines are a widely used class of chemotherapy in pediatric and adult cancers, however, their use is hampered by the development of cardiotoxic side-effects and ensuing complications, primarily heart failure. Clinically used imaging modalities to screen for cardiotoxicity are mostly echocardiography and occasionally cardiac magnetic resonance imaging. However, the assessment of diastolic and global or segmental systolic function may not be sensitive to detect subclinical or early stages of cardiotoxicity. Multiple studies have scrutinized molecular nuclear imaging strategies to improve the detection of anthracycline-induced cardiotoxicity. Anthracyclines can activate all forms of cell death in cardiomyocytes. Injury mechanisms associated with anthracycline usage include apoptosis, necrosis, autophagy, ferroptosis, pyroptosis, reactive oxygen species, mitochondrial dysfunction, as well as cardiac fibrosis and perturbation in sympathetic drive and myocardial blood flow; some of which have been targeted using nuclear probes. This review retraces the pathobiology of anthracycline-induced cardiac injury, details the evidence to date supporting a molecular nuclear imaging strategy, explores disease mechanisms which have not yet been targeted, and proposes a clinical strategy incorporating molecular imaging to improve patient management.
Introduction
Anthracyclines are one of the most commonly prescribed chemotherapies and are used to treat a variety of cancers. Although effective agents, their benefits are sometimes compromised by acute and/or late-onset cardiotoxic side effects. A study that compared adult survivors of pediatric cancer with their siblings found that survivors (that had been treated with anthracyclines and/or radiotherapy) had a 15-fold higher risk of developing heart failure (1). The risk of subsequent heart failure in pediatric patients treated with anthracyclines was demonstrated to be highly dose-dependent, particularly in cumulative anthracycline doses ≥300 mg/m2, and to increase over time (3.3% at 2 years, 4.5% at 10 years, and 9.8% at 20 years after the first dose) (2, 3). Similarly, a retrospective analysis of three phase III clinical trials with adult patients indicated that treatment with a cumulative doxorubicin dose of ≥400 mg/m2 led to a 5% incidence of heart failure, rising to up to 26% at a cumulative dose of 550 mg/m2 (4). It must be emphasized, however, that (i) no “safe dose” of anthracyclines truly exists, (ii) late effects leading to heart failure can occur and need to be monitored, and (iii) risk assessment needs to be individualized with a particular focus on pre-existing heart disease and/or cardiovascular risk factors such as hypertension. Thus, given the wide individual variability in patient risk of developing anthracycline-induced cardiac injury, risk stratification must be done on a case-by-case basis.
In clinical practice, side effects of anthracyclines are balanced by limiting the dosage while tightly monitoring for clinical manifestations of cardiotoxicity. Typically, imaging modalities such as echocardiography are used to monitor for cardiotoxicity, commonly defined as a decline in LVEF of ≥10% to a final value <50% (5). A recent International Cardio-Oncology Society (IC-OS) consensus statement (6) proposed a standardized and more nuanced definition of cancer therapy-related cardiac dysfunction (CTRCD), applicable to anthracyclines, as follows: (1) Asymptomatic CTRCD graded as (i) mild (LVEF ≥50% and new decline in GLS >15% from baseline, and/or new rise in troponin I/T, BNP, NT-proBNP), (ii) moderate (new LVEF reduction by ≥10 percentage points to a LVEF 40–49%, or new LVEF reduction by <10 percentage points to a LVEF 40–49% and new decline in GLS >15% from baseline, and/or new rise in troponin I/T, BNP, NT-proBNP), and (iii) severe (new LVEF reduction to <40%). (2) Symptomatic CTRCD with supportive LVEF and diagnostic biomarkers, graded from mild to very severe based on heart failure symptoms, requirement for intensification of heart failure treatment, hospitalization, and/or inotropic or mechanical circulatory support.
However, these prognostic/diagnostic tools have several limitations, including their variability, lack of sensitivity, and inadequate detection of toxicity at a subclinical level. One of the main issues is that these parameters detect cardiotoxicity when the myocardium is already damaged and/or cardiac function impaired, which may hinder treatment options. Indeed, histological biopsy samples of patients that underwent doxorubicin treatment demonstrated that the myocardium could incur significant injury despite patients having a preserved/normal LVEF (7).
Therapeutic treatment for AIC remains limited. One of the most well studied agents, dexrazoxane, has been shown to significantly reduce cardiotoxicity in adults and pediatric patients when concurrently prescribed with anthracyclines (8–10). Recent studies have demonstrated the safety of dexrazoxane as a cardioprotective agent and confirmed its lack of interference with the anti-tumor action of anthracyclines. Indeed, an analysis of three Children’s Oncology Group (COG) trials that had randomized patients to doxorubicin with or without dexrazoxane (dexrazoxane:doxorubicin dose ratio 10:1, cumulative protocol-specified doxorubicin dose 100–360 mg/m2) with a median follow-up of 12.6 years demonstrated that dexrazoxane does not compromise long-term survival, and is not associated with mortality from acute myeloid leukemia or cardiovascular causes (11). This was followed by another COG analysis of four trials with pediatric patients that similarly were treated with doxorubicin with or without dexrazoxane (other than one trial in which all patients were assigned to dexrazoxane upfront), and with a median follow-up of close to 20 years, indicating that dexrazoxane does not negatively affect long-term mortality or second cancer risk (12). Angiotensin-converting enzyme inhibitors and beta-blockers have also been studied. In a study of 201 patients with AIC, Cardinale et al. observed that 64% of patients treated early (i.e., 1–2 months after completion of chemotherapy) with enalapril/carvedilol had complete LVEF recovery, while 0% of the patients treated 6 months post-chemotherapy had complete LVEF recovery (13). Whereas these findings were not corroborated in the prospective CECCY trial which randomized 200 HER2-negative breast cancer patients to carvedilol vs. placebo synchronous with anthracycline initiation (total 240 mg/m2 over 4 cycles), a benefit of betablockade was noted on the development of diastolic dysfunction (14).
These clinical findings underscore the importance of early disease detection and emphasize the need for additional methods to diagnose subclinical AIC. A promising such strategy is nuclear imaging that can map molecular processes perturbed in AIC using radioactively labeled probes (Table 1). Advancements in nuclear imaging have rendered imaging of pathological processes such as mitochondrial dysfunction, sympathetic innervation, and fibrosis, possible. With the ongoing dissection of the pathobiology of AIC at the molecular level, we predict these advances will permit the identification of novel molecular imaging targets and posit a future role for nuclear imaging that will be complementary to that of echocardiography (and/or cardiac MRI). The present review retraces the preclinical and clinical evidence supporting the use of a nuclear molecular imaging strategy in AIC, and offers new avenues for tracer development targeting injury pathways that have not yet been explored.

Table 1. Radiotracer, mechanism of uptake, and application for nuclear imaging of anthracycline-induced cardiotoxicity.
Nuclear imaging targets
I. Metabolic dysfunction
Anthracyclines induce intracellular ROS through several mechanisms. Fe3+ can react with the ketone and hydroxy groups of anthracyclines to form free radicals through the Fenton reaction (15). Anthracyclines also accumulate within the mitochondrial inner membrane, in part due to their high affinity to cardiolipin. In mitochondria, quinone and semiquinone moieties of anthracycline undergo redox cycling, generating large amounts of ROS (16). These events cause oxidative damage to cellular proteins, lipids, and mitochondria, resulting in mitochondrial membrane potential loss, mitochondrial swelling, activation of the mitochondrial-permeability transition pore (mPTP), and the release of cytochrome c (17). Formation of the apoptosome, initiated by cytochrome c release from mitochondria to the cytosol, leads to the cleavage and activation of caspase 3 and cell death (Figure 1). Long-term mitochondrial dysfunction also leads to a compensatory shift in cardiomyocyte metabolism, which may be targeted for AIC imaging (18).
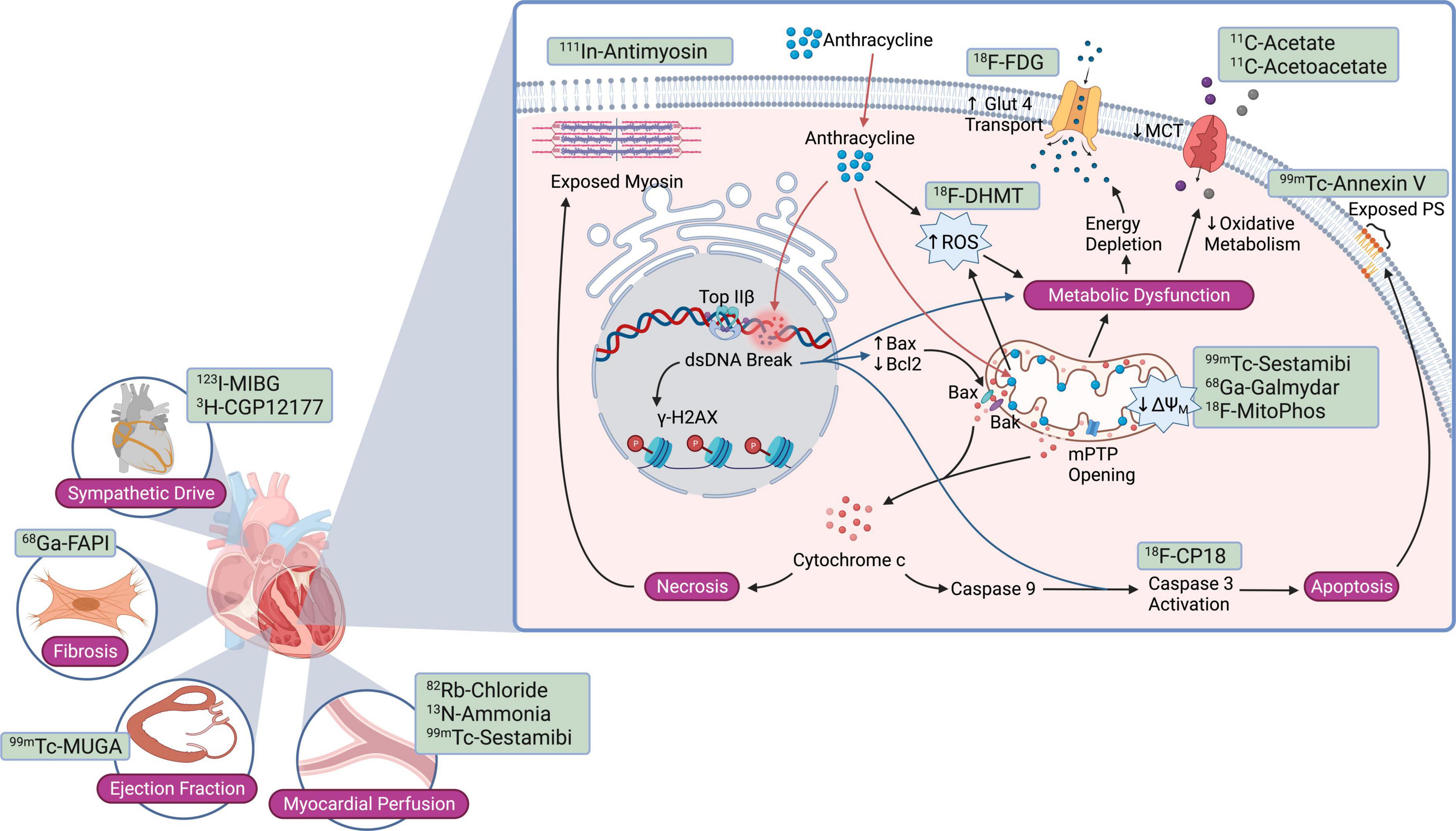
Figure 1. Current nuclear molecular imaging strategies targeting pathways and cell injury mechanisms activated in anthracycline-induced cardiotoxicity. Nuclear probes studied to date target anthracycline-mediated cardiomyocyte injury mechanisms such as apoptosis, necrosis, reactive oxygen species, and mitochondrial dysfunction; downstream consequences such as cardiac fibrosis; and more global changes such as sympathetic drive, myocardial blood flow, and left ventricular ejection fraction. Bax, Bcl2 associated X protein; Bak, Bcl2 antagonist/killer; Bcl2, B-cell CLL/lymphoma 2; dsDNA, double-stranded DNA; GLUT1, glucose transporter 1; γ-H2AX, phosphorylated histone variant H2AX; MCT, monocarboxylate transporter; ΔΨM, mitochondrial membrane potential; mPTP, mitochondrial permeability transition pore; PS, phosphatidylserine; ROS, reactive oxygen species; Top IIβ, topoisomerase IIβ.
Glucose uptake
The most studied PET tracer for AIC imaging is 18F-FDG. Anthracyclines impair mitochondrial phosphorylation and the oxidation of all substrates – including fatty acids, carbohydrates, and ketones – thus driving cardiac myocytes to shift toward utilization of substrates with a more favorable ATP production phosphate/oxygen ratio, such as glucose (18, 19). Cardiomyocyte uptake of the glucose analog 18F-FDG is mediated by glucose transporter (GLUT) −4 and −1 (20). Whereas GLUT-1 is considered responsible for basal intracellular glucose transport, GLUT-4 and to a lesser extent GLUT-1 translocate to the plasma membrane and increase intracellular glucose uptake in response to stimuli such as insulin (21), ischemia (22, 23), anoxia (24), and catecholamines (20). Upon cardiomyocyte uptake, 18F-FDG is phosphorylated by hexokinase and not metabolized further (25). Additionally, the reverse reaction (dephosphorylation by glucose-6-phosphatase) is minimal (25). Thus, phosphorylated 18F-FDG remains trapped within cardiomyocytes. To infer glucose metabolic rate from 18F-FDG metabolic rate, a “lumped constant” – initially formulated by Sokoloff et al. (26) – or correction factor, is used. The lumped constant is based on competitive substrate kinetics between glucose and 18F-FDG, and accounts for differences in transport and phosphorylation rates (27). Importantly, the lumped constant for 18F-FDG in the myocardium is dependent on fasting state and serum insulin levels (28–31). The lumped constant has not been evaluated in the setting of anthracycline chemotherapy-induced cardiac injury.
Preclinical studies have assessed the potential utility of 18F-FDG in AIC. Bulten et al. observed a progressive increase in 18F-FDG uptake in mice treated with doxorubicin (15 mg/kg, once every 3 weeks for up to four cycles) (32). They further noted a significant correlation between myocardial 18F-FDG uptake and hypoxia-inducible factor (HIF)-1α, a hypoxia-driven transcription factor that activates GLUTs and glycolytic enzymes. In another study of doxorubicin treated mice, increased 18F-FDG myocardial uptake had a direct correlation with histologically determined myocardial redox stress (33).
Several retrospective clinical studies have documented higher 18F-FDG uptake in patients treated with anthracyclines. Borde et al. observed in lymphoma patients treated with doxorubicin-based chemotherapy a higher post-therapy 18F-FDG uptake than before treatment (34). In a retrospective study of 43 Hodgkin lymphoma patients that developed AIC, Sarocchi et al. observed that a decrease in LVEF several months to years following treatment was inversely correlated with LV uptake of 18F-FDG during doxorubicin containing chemotherapeutic treatment (R2 = 0.30, P < 0.01) (35). Similarly, in a recent study of 121 consecutive breast cancer patients undergoing treatment with anthracycline or trastuzumab, Kim et al. found that patients who developed cardiotoxicity had a higher 18F-FDG right ventricular uptake than patients who did not (2.4 ± 1.1 vs. 1.6 ± 0.7, P = 0.012) (36). Though it remains unclear whether the observed right ventricular uptake preceded or was a result of LV dysfunction, other have reported that left ventricular and right ventricular global longitudinal strain are both similarly impaired during trastuzumab treatment (37). Additional studies have suggested that baseline LV 18F-FDG SUV may also be an indicator of patient susceptibility to AIC. Bauckneht et al. observed that among a cohort of 36 Hodgkin lymphoma patients that had previously undergone doxorubicin treatment, the 11 patients that developed significant post treatment reduction in ejection fraction had lower pre-treatment LV 18F-FDG uptake compared to the remaining 25 patients that didn’t develop cardiac abnormalities (mean SUV = 1.53 ± 0.9 vs. 3.34 ± 2.54, P < 0.01) (38). Heckmann et al. (39) demonstrated in a retrospective study (n = 337 consecutive patients) that Hodgkin’s lymphoma (n = 52) was associated with a higher cardiac 18F-FDG uptake (mean SUV = 3.5 ± 3.6, odds ratio = 2.4, P < 0.01) whereas non-Hodgkin’s lymphoma (n = 57) and non-lymphatic cancer (n = 228) were not. Interestingly, the authors observed that the increase in cardiac 18F-FDG uptake in Hodgkin’s lymphoma was not determined by prior chemotherapy and/or serum glucose levels, however, with the caveat that patient preparations were not optimized or standardized for cardiac 18F-FDG PET imaging, in addition to the retrospective nature of the study (39). A significant limitation of these retrospective studies is that protocols were designed for cancer staging and not to measure cardiomyocyte 18F-FDG uptake per se, which were done post hoc in a retrospective manner. In this setting, patients were only required to fast a minimum of 6 h during which myocardial metabolic patterns still have a high degree of variability (40). Furthermore, the pattern of myocardial 18F-FDG uptake may also need to be taken into consideration when determining physiologic vs. pathologic signals (41). Whereas standardized protocols incorporating adequate nutritional preparation are required (42), these preliminary studies are promising and set the stage for the prospective evaluation of cardiomyocyte 18F-FDG uptake in AIC.
Oxidative metabolism
Acetate is utilized by cardiomyocytes in the tricarboxylic acid (TCA) cycle and can thus serve as a metric to quantify myocardial oxygen consumption. A preclinical model of chronic doxorubicin treatment in rats (2 mg/kg IV weekly for 6 weeks) observed that doxorubicin decreased myocardial oxygen consumption reserve (2.3 ± 0.3 vs. 1.8 ± 0.4, P = 0.02) (43). 11C- acetoacetate, a ketone body that utilizes the same monocarboxylic acid transporter as acetate, exhibited similar changes in an analogous rat model treated with doxorubicin (44).
Nony et al. investigated 11C-acetate uptake to assess myocardial oxidative metabolism and myocardial blood flow in patients treated with anthracyclines (45). The resting myocardial blood flow of 6 patients were serially measured during a doxorubicin treatment course of 50 mg/m2 every 3 weeks for 15 weeks (cumulative dose of 300 mg/m2). The investigators observed that compared to baseline, there was no significant change in resting myocardial blood flow during or after completion of doxorubicin treatment. Similarly, no significant changes were noted in Kmono, an index of myocardial oxygen consumption (45, 46).
Fatty acid metabolism
Whereas 70 to 90% of cardiac ATP production is derived from fatty acid β-oxidation under physiologic conditions, fatty acid usage decreases significantly in heart failure and cardiomyopathy models (47), thus making it a possible target for AIC imaging. There are no preclinical or clinical studies to date that have applied radiolabeled fatty acids to monitor AIC. Potential candidates include 11C-palmitate, although its clinical utility is hindered by a lack of kinetic data that models and accounts for the redistribution of 11C metabolites within various lipid pools (48). 18F-FTHA (14[R,S]-18F-fluoro-6-thia-heptadecanoic acid) is another PET tracer that could circumvent this limitation by utilizing a sulfur atom in its backbone that prevents it from undergoing further β-oxidation (49).
Mitochondrial membrane potential
99mTc-sestamibi is a SPECT tracer clinically used to image myocardial perfusion, though its utility as a lipophilic cation has proven useful to detect disruptions in mitochondrial membrane potential. The ability of these cations to accumulate inside the mitochondria has been used as a proxy index for mitochondrial membrane potential (ΔΨM) (50). Animal studies conducted by Safee et al. indicated that rats treated with a single dose of doxorubicin had lower levels of 99mTc-sestamibi uptake in the myocardium, corresponding to a loss in ΔΨM. A significant 2.5-fold decrease in 99mTc-sestamibi 2 weeks post-treatment was detected only in rats treated with the highest doxorubicin dose (10 mg/kg), which was associated with a 7% and 9.5% drop in ejection fraction and fractional shortening, respectively, that was also only significant 2 weeks post-treatment (51). While promising, usage of 99mTc-sestamibi is hindered by its pharmacokinetics and the limited sensitivity inherent to most SPECT tracers. In comparison, PET tracers such as the metalloprobe 68Ga-galmydar have been developed for this same application (52). In live-cell fluorescent imaging of H9c2 cells, Sivapackiam et al. observed a dose- and time-dependent decrease in 68Ga-galmydar that correlated with an increase of caspase-3 activation (52). These findings were subsequently confirmed in in vivo models, where the authors reported a nearly 2-fold decrease in myocardial 68Ga-galmydar uptake 5 days following a single doxorubicin dose of 15 mg/kg in rats, verified by post-imaging quantitative biodistribution. Another PET lipophilic cation that bears promise is [1-(2-18F-fluoroethyl),1H[1,2,3]triazole-4-ethylene]triphenylphosphonium bromide (18F-MitoPhos) (53). In a Langendorff perfusion heart model, 18F-MitoPhos exhibited more than double cardiac retention compared to 99mTc-sestamibi. Moreover, in vivo studies in an acute doxorubicin rat model indicated a close to 50% decrease in the left ventricular retention of 18F-MitoPhos compared to controls 48 h after treatment. While 68Ga-galmydar and 18F-MitoPhos represent encouraging alternatives to 99mTc-sestamibi for measuring ΔΨM, further studies are needed to corroborate these findings with reference parameters such as ejection fraction and fractional shortening.
An additional promising strategy for the scrutiny of ΔΨM involves the radiolabeled lipophilic cation 18F-tetraphenylphosphonium (18F-TPP+) (54). Using a pig model, Alpert et al. demonstrated its in vivo myocardial applicability via a novel method accounting for extracellular space and employing kinetic analysis to estimate tracer volume of distribution (55). Additionally, Pelletier-Galarneau et al. demonstrated excellent agreement of in vivo measures of myocardial 18F-TPP+ in healthy humans subjects with previous in vitro assessments (56), paving the way for human studies quantifying temporal changes in mitochondrial membrane potential using this radiopharmaceutical in the context of anthracycline-induced cardiotoxicity (57). These tracers require prospective clinical trial evaluation to determine their clinical utility.
Reactive oxygen species
[18F]6-{4-[(1-(2-fluoroethyl)-1H-1,2,3-triazol-4-yl) methoxy]phenyl}-5-methyl-5,6-dihydrophenanthridine-3,8- diamine (18F-DHMT), an analog of the superoxide indicator dihydroethidium, has previously been identified as a promising PET radiotracer of ROS generation (58). In a chronic doxorubicin-induced cardiotoxicity rodent model, Boutagy et al. described the significant increase in myocardial 18F-DHMT uptake 4 weeks post treatment which preceded degradation in LVEF that was significant only 6 weeks post treatment (P = 0.0012) (59). Correlation analysis suggested an inverse correlation (r2 = 0.6; P = 0.01) between LV 18F-DHMT uptake and LVEF as well as a direct correlation (r2 = 0.72; P = 0.007) between LV 18F-DMHT and LV ESV. These results suggest that 18F-DHMT may be a viable radiotracer for early assessment of cardiotoxicity that precedes left ventricular systolic dysfunction.
II. Cell death
Dox intercalates in the DNA and induces single- and double-strand DNA breaks in target cells in a topoisomerase (Top)-2-dependent manner (60). By producing temporary single- or double-stranded DNA breaks, Top regulates topological changes during DNA replication, transcription, or recombination (61). Top-2α is overexpressed in tumors and is the molecular basis of Dox anticancer activity (62, 63).
Adult cardiomyocytes express Top-2β but not Top-2α (62), and Top-2β is also a Dox target, forming a Top-2β-Dox-DNA ternary cleavage complex that induces DNA strand breaks and ensuing cell death (64, 65). These DNA breaks rapidly result in the phosphorylation of histone variant γ-H2AX, a sensitive marker of the DNA damage response (66, 67) (Figure 1). Subsequently, mediator of DNA damage checkpoint protein (MDC)-1 binds to γ-H2AX (68) and facilitates DNA damage repair protein recruitment (69, 70). Furthermore, Dox/Top-2β bind to selective promoters, significantly affecting the cardiomyocyte transcriptome (65, 71). Ensuingly, key antioxidative enzymes are reduced, providing a mechanism linking Dox-induced reactive oxygen species (ROS) production in a Top-2β-dependent manner. For example, peroxisome proliferator activated receptor-γ (PPAR-γ) coactivator –1-α and -β, pivotal transcription factors implicated in mitochondrial biogenesis, are decreased in the setting of Dox cardiotoxicity (65, 71).
Anthracyclines triggers various cell death mechanisms (Figures 1, 2), though the two most well characterized pathways in AIC are apoptosis and necrosis, mediated in part by Bax-induced mitochondrial damage (Figure 1). Bax (Bcl-2 associated X protein) is a member of the Bcl-2 family. Under homeostatic conditions, Bax resides in an inactive conformation in the cytosol (72). Upon anthracycline treatment, Bax undergoes a conformational change that results in its translocation to the mitochondrial membrane (Figure 1). There, Bax mediates opening of the mitochondrial permeability transition pore (mPTP) located in the inner mitochondrial membrane (73). In turn, mPTP opening leads to swelling of the mitochondrial intermembrane space followed by rupture of the outer mitochondrial membrane, release of intermembrane space proteins – including the small soluble electron carrier cytochrome c – into the cytosol, and cardiomyocyte necrosis (74) (Figure 1). Another proposed mechanism of anthracycline-induced, Bax-mediated cytochrome c release is the oligomerization of Bak and Bax within the outer mitochondrial membrane, leading to its permeabilization and activation of apoptotic pathways (72, 75).

Figure 2. Additional anthracycline cardiotoxicity mechanisms for the development of novel molecular nuclear probes. Highlighted mechanisms have not yet been targeted using a nuclear imaging strategy, and certain key molecular or biochemical targets are presented for the potential development of new SPECT/PET tracers. (A) Anthracyclines impair autophagy by compromising lysosomal acidification, increasing beclin-1 expression, inhibiting mTOR, and inhibiting transcription factor EB, thereby blocking autophagic flux and causing an accumulation of autolysosomes that leads to increased ROS production. New probes may be developed to target molecules implicated in the accumulation of undegraded autolysosomes. (B) Anthracycline-induced ferroptosis is caused by iron overload through upregulation of TfR, inactivation of ferritin, inhibition of ABC-B8, and downregulation of GPX4, which may be targets for probe development. (C) Anthracycline induces pyroptosis by upregulating TINCR, which increases the expression of NLRP3 and caspase 1 activation. Future probes may for example target NLRP3 or caspase1. ABC-B8, ATP binding cassette subfamily B member 8; Dox, doxorubicin; GPX4, glutathione peroxidase 4; GSDMD, gasdermin D; IGF2BP1, insulin-like growth factor 2 mRNA binding protein 1; IL, interleukin; Lipid-OOH, lipid hydroperoxides; mTOR, mammalian target of rapamycin; NLRP3, nucleotide-binding oligomerization domain (NOD-), leucine-rich repeat (LRR-), and pyrin domain (PYD)-containing protein 3; ROS, reactive oxygen species; TFEB, transcription factor EB; TfR, transferrin receptor; TINCR, terminal differentiation-induced non-coding RNA.
Annexin V
Annexin V is a well-established method of detecting externalized phosphatidylserine (PS), a phospholipid that is translocated from the inner to the outer leaflet of the plasma membrane early in apoptosis. 99mTc-annexin-V was scrutinized by Bennink et al. in an acute doxorubicin-induced cardiotoxicity rat model. Doxorubicin treated rats displayed a significant increase in myocardial 99mTc-annexin-V uptake, with longer doxorubicin treatment regimens corresponding to an even higher uptake (76). Furthermore, heart-to-body weight ratio decreased in response to doxorubicin treatment, which may have been an indication of cardiomyocyte death. These findings correlated well with cardiotoxicity measured through immunohistochemistry and the TUNEL assay. Clinical studies utilizing annexin V probes have been limited; 99mTc-HYNIC-annexin-V, was tested in early clinical trials but its performance in detecting apoptosis in head and neck carcinoma was limited by moderate non-specific binding and slow clearance times (77).
Caspase
A more direct way of measuring apoptosis is by targeting caspase activation. Caspases are intracellular enzymes that are essential for executing apoptosis. Different initiator caspases can be activated extrinsically or intrinsically, though both pathways ultimately converge with caspase-3 and –7 activation. Several analogs of 5-Dialkylaminosulfonylisatins, a potent non-peptide inhibitor of caspase-3 and –7, have been adapted as PET tracers. 18F-ICMT-11, 11C-WC-98, and 18F-WC-IV-3 have demonstrated high caspase-3 affinity in vitro but all exhibited poor specificity in vivo models, likely due to the dicarbonyl moiety caspase binding region of isatin being recognized by other proteases such as cathepsins (78–80). A substrate-based probe, 18F-CP18, was designed to improve specificity by taking advantage of caspase-3′s unique substrate recognition motif for aspartic acid residues in the P1 and P4 positions (75 - 81). In this probe, the caspase-3 substrate sequence Asp-Glu-Val-Asp connects a radioactively labeled metabolite to a short polyethylene glycol (PEG) chain. The hydrophilic PEG chain facilitates the probe’s transport across the cell membrane and upon encountering activated caspase-3 it is cleaved away, leaving the radioactively labeled metabolite inside the cell. While initially developed for visualization of apoptosis in tumors, this radiotracer has also been adapted for AIC imaging (82). Su et al. (83) detected increased accumulation of 18F-CP18 in the myocardium starting at 3 weeks after doxorubicin treatment in mice, which was histologically validated using a TUNEL assay. The authors also observed that 18F-CP18 detects myocardial apoptosis at a stage prior to significant changes in LVEF. Clinical studies are required to further expand on these promising results.
Necrosis
111In-antimyosin is a tracer that binds to the exposed myosin of damaged cells, an indicator used to quantify regions of myocardial necrosis. Early clinical studies evaluated 111In-antimyosin in AIC (84–86). Carrió et al. studied 30 sarcoma patients who underwent serial 111In-antimyosin imaging prior to chemotherapy and at intermediate (240–300 mg/m2) and maximal (420–600 mg/m2) cumulative doxorubicin doses. Whereas an abnormal heart-to-lung 111In-antimyosin uptake ratio was observed with both doxorubicin dosages, maximal exposure to doxorubicin led to a more pronounced 111In-antimyosin uptake with a ratio of 2.02 ± 0.3 (P < 0.01) and was associated with a significant ≥10% decrease in LVEF (85). Furthermore, in a follow up study of patients that had discontinued anthracycline treatment due to a decrease in LV function leading to a LVEF <50%, Olmos et al. demonstrated that patients with an 111In-antimyosin heart-to-lung uptake ratio ≥1.87 experienced a persistent decline in LVEF at 2–26 weeks follow-up, with 4 out of 11 of these patients subsequently developing congestive heart failure (86). In contrast, patients with a transient change in LVEF following the discontinuation of anthracycline treatment had a mean 111In-antimyosin heart-to-lung uptake ratio of 1.52. Despite these promising results, interest in 111In-antimyosin cardiac imaging has waned and this tracer is not commonly used in contemporary practice due to its detection of necrotic cell death which occurs at more at more advanced stages of the disease process and thereby limits options for clinical intervention.
III. Sympathetic innervation
The clinical signs of early myocardial cell injury are often masked by a compensatory rise in sympathetic drive. Indeed, an increase in chronotropy and inotropy preserve LVEF during the early stages of AIC (87, 88). While initially beneficial, long term cardiac sympathetic activation is detrimental, with cardiac sympathetic dysinnervation occurring in cardiomyopathy and heart failure (89). The main catecholamine released by sympathetic postganglionic fibers is norepinephrine which accounts for 70% of circulating levels, with the remainder mainly released by the adrenal gland (90, 91). The majority of clinical tracers that monitor sympathetic innervation are radiolabeled analogs of norepinephrine.
123I-metaiodobenzylguanidine
123I-metaiodobenzylguanidine (123I-MIBG), a norepinephrine analog that shares with it similar release and uptake mechanisms, can be used to identify areas of abnormal adrenergic innervation in the myocardium. Approximately 80–90% of norepinephrine released at sympathetic nerve terminals is taken up again via norepinephrine transporter uptake-1 (89). Reduction of norepinephrine uptake at these sites has been documented in various cardiovascular diseases (92, 93). Like norepinephrine, reuptake of 123I-MIBG is mediated by norepinephrine transporters along myocardial sympathetic nerve terminals. Given 123I-MIBG is not metabolized, its retention can be used as an indicator for neuronal integrity.
123I-metaiodobenzylguanidine uptake is reduced in preclinical models of AIC. In a chronic doxorubicin rat model (2 mg/kg IV weekly for 1, 2, 3, 4, 5 and 8 weeks), a significant decrease in cardiac 123I-MIBG uptake was detected at week 4, which correlated with histologically examined myocardial tissue damage (94). A comparative study of 123I-MIBG with 18F-FDG in a chronic doxorubicin-induced cardiotoxicity rat model (15 mg/kg cumulative dose) indicated that 18F-FDG uptake decreased significantly in doxorubicin treated groups at weeks 4 and 6 (4.2 ± 0.5%ID/g vs. 9.2 ± 0.8%ID/g at week 6), which correlated with LVEF (r = 0.49, P = 0.002) (95). In contrast, a significant decrease in 123I-MIBG heart-to-mediastinum (H/M) ratio between groups was detected earlier at week 2 (∼1.9% vs. ∼1.4%, P < 0.05), maintained at weeks 4 and 6, but was not correlated with LVEF decrease at week 6 (r = 0.24, P = 0.15). While promising, a limiting point of these preclinical studies is the large variability in anthracycline doses and temporal administration patterns which may lead to systemic toxicity in animals.
Early and late H/M ratios of 123I-MIBG uptake were proposed as an index for stratifying prognosis and risk in patients with chronic heart failure (96). Carrió et al. (96) observed a significant 1.5-fold decrease in cardiac 123I-MIBG uptake in sarcoma patients undergoing maximal cumulative doxorubicin treatment (420–600 mg/m2), which also corresponded with a significant (≥10%) reduction in LVEF. A similar reduction in ejection fraction was not detected in patients at intermediate cumulative doxorubicin doses (240–300 mg/m2), though 25% of patients exhibited slight decreases in 123I-MIBG uptake that were not significant compared to baseline. A study by Laursen et al. (97) further reinforced that 123I-MIBG imaging is only applicable in patients undergoing high cumulative doxorubicin doses. Moreover, the authors observed that patients undergoing intermediate cumulative doses of doxorubicin had a non-significant increase post therapy on both background-corrected WOR – which reflects norepinephrine retention in adrenergic neurons, closely connected to sympathetic tone (18.6% vs. 23.4%, P = 0.09), and H/Mearly – a measure of the anatomical distribution of functioning myocardial adrenergic neurons (2.7% vs. 2.9%, P = 0.4). No association was observed between follow-up decreases in LVEF and WOR (P = 0.5). Moreover, in a study of asymptomatic patients who had completed anthracycline treatment ≥2 years prior, 123I-MIBG uptake did not differ between anthracycline treated patients (cumulative anthracycline dose 257.6 ± 117.1 g/m2) and control patients, as assessed either by mean H/Mlate – which reflects overall neuronal functioning, i.e., the product of norepinephrine uptake, storage, and release (2.24% vs. 2.26%, P = 0.5) – or WOR (10.32% vs. 9.64%, P = 0.8) (98). These results suggest that the myocardial adrenergic activation initially detected by 123I-MIBG during treatment is reversible upon discontinuation of anthracycline treatment. Overall, 123I-MIBG adrenergic imaging is not presently a clinically applicable strategy for the early detection of AIC, though further clinical studies may be warranted to investigate its sensitivity at lower cumulative anthracycline doses.
Additional tracers
Other PET radiotracers for cardiac sympathetic innervation have been studied in animal models. Ex vivo biodistribution studies 3 weeks after chronic doxorubicin treatment in rats found no change in cardiac uptake of the norepinephrine analog 11C-meta-hydroxyephedrine (11C-HED) or the phosphodiesterase-4 inhibitor (R)-11C-rolipram (which provides an index of myocardial cyclic AMP activity, downstream of norepinephrine) (99). On the other hand, the authors noted a decreased uptake of the β-adrenergic antagonist 3H-CGP12177 3 weeks post treatment while there was no change in ejection fraction or heart-to-weight ratio. Similarly, Kizaki et al. also reported that β-adrenergic receptor gene expression decreases following doxorubicin treatment in rats (100).
IV. Myocardial function, perfusion, and blood flow
99mTc-MUGA imaging is an accurate method to assess ventricular contraction. While generally avoided in pediatric patients due to radiation concerns, 99mTc-MUGA may be used to monitor cardiotoxicity in adult patients due to its high accuracy and low inter-observer variability (101–103). In a prospective study of 28 non-Hodgkin lymphoma patients undergoing doxorubicin treatment, Nousiainen et al. observed that a decrease of ≥4% in LVEF quantified by 99mTc-MUGA following a cumulative doxorubicin dose of 200 mg/m2 predicted AIC with 90% sensitivity and 72% specificity (104). However, these findings were not supported by a large-scale retrospective study of 630 patients grouped according to increasing doxorubicin doses or placebo (4). Overall, assessment of LVEF – even if done accurately by 99mTc-MUGA – is not a favored approach to assess AIC as it only provides a global and often delayed assessment of cardiac mechanical abnormalities following injury.
Microvascular dysfunction is a possible complication of AIC (105). The SPECT myocardial perfusion imaging tracer 99mTc-sestamibi was assessed in AIC in a prospective study of breast cancer patients undergoing radiation therapy and doxorubicin treatment (106). Hardenbergh et al. observed that 7 out of 10 patients developed new visible perfusion defects 6 months post-radiation. More recently, PET tracers such as 13N-ammonia and 82Rb-chloride have become more common for myocardial perfusion assessment due to their superior diagnostic accuracy compared to SPECT (107–109). In a small prospective study (n = 10) using 13N-ammonia, Nehmeh et al. reported decreased MFR 1-year post-radiation in 50% of breast cancer patients (n = 4) receiving radiotherapy and anthracycline (110). However, these findings are suggested by a small dataset only, and it is unknown if the observed cardiotoxic effects were caused by the anthracycline treatment or the radiation. PET myocardial perfusion tracers are amenable to myocardial blood flow quantitation, an integrated measure of epicardial and microvascular coronary artery disease (109). 82Rb-chloride assessment of myocardial blood flow in lymphoma patients by Laursen et al. revealed a mild reduction in myocardial flow reserve (MFR: 2.69 vs. 2.51, P = 0.03) – calculated as the stress/rest ratio of myocardial blood flow – 3 days after initial doxorubicin treatment, with 13 out of 54 patients exhibiting low cardiotoxicity threshold (>20% decline in MFR) (105). Importantly, the MFR decline was independent of perfusion defects determined using the summed stress score and summed difference score. Stratifying patient risk of developing AIC in this manner is relevantly new, and additional studies are needed to determine whether an acute reduction in MFR shortly after anthracycline administration may identify patients at higher cardiotoxicity risk. Furthermore, the PET tracers 13N-ammonia (110), 15O-water (111) and 18F-flurpiridaz (112, 113) should also be considered to assess myocardial blood flow changes in AIC given their higher myocardial extraction fractions (108).
V. Cardiac fibrosis
Myocardial fibrosis (114) may occur as a result of AIC. While the exact role cardiac fibroblasts play in AIC remains underexplored, doxorubicin was recently reported to induce fibroblast differentiation (115). 68Ga-fibroblast activation protein inhibitor (FAPI) is a robust fibroblast PET tracer that was initially developed to detect high FAP-expressing, cancer-associated fibroblasts. In a retrospective study of n = 32 patients, Siebermair et al. evaluated the cardiac uptake of 68Ga-FAPI in a heterogenous population of cancer patients treated with various anticancer therapies (116). While only 3 patients with FAPI uptake had been treated with anthracyclines, the authors observed a significant association of myocardial FAPI uptake with CAD and LVEF. Using modeling (n = 185) and confirmatory (n = 44) consecutive cohorts of patients with cancer metastasis who had FAPI-positive PET scans, Heckmann et al. (117) did not observe an association of focal myocardial FAPI uptake with anthracycline treatment, however, noted a correlation of high signal intensities with cardiovascular risk factors and metabolic disease. Additional studies are warranted to investigate cardiac fibroblast FAP expression patterns in the context of AIC (116).
Additional anthracycline cardiotoxicity mechanisms for future development of candidate molecular targets
Several AIC mechanisms have not been targeted to date using a nuclear imaging strategy. Anthracycline-induced cardiac injury is multi-factorial and -genic, with no single mechanism fully explaining all aspects of the injury process. Importantly, anthracyclines induce all forms of cell death (118, 119). We highlight below three additional injury pathways that may lead to the development of new radiotracers for nuclear molecular imaging in AIC.
Autophagy
Autophagy is a homeostatic process in which cells utilize lysosomes to remove unnecessary or damaged cellular components (75). Anthracyclines block cardiomyocyte autophagic flux by impairing lysosomal acidification – critical for lysosomal hydrolytic enzyme activity and lysosomal maturation (120) – leading to the accumulation of undegraded autolysosomes (121) (Figure 2A). Another mechanism of anthracycline interference with autophagic flux is the inhibition of transcription factor EB expression which is a regulator of lysosomal proteolysis mediated primarily by cathepsin B activity (122). Furthermore, phosphoinositide 3-kinase γ (PI3Kγ) is induced downstream of Toll-like receptor 9 by cardiomyocytes following anthracycline treatment (123). PI3Kγ leads to Akt phosphorylation and inactivation of mTOR (mammalian target of rapamycin) targets, thus causing autophagy inhibition and a reduced ability to remove damaged organelles such as mitochondria (123). Expression of beclin-1, a mediator of autophagy initiation, increases following doxorubicin treatment in mice (121). Furthermore, Li et al. indicated that haploinsufficiency of beclin-1 diminishes autophagy initiation, leading to fewer unprocessed autolysosomes and decreased ROS production. Conversely, doxorubicin-induced cardiac injury is accentuated in mice with beclin-1 overexpression. Taken together, these mechanisms indicate the contribution of autophagy perturbation to AIC-induced cardiomyocyte death, cardiac remodeling, and failure (124).
Ferroptosis
Ferroptosis is a type of cell death characterized by the iron-related accumulation of lipid peroxides (75). Iron plays a significant role in AIC injury (125) (Figure 2B). Heart biopsies of patients who experienced anthracycline-related heart failure demonstrated excessive mitochondrial iron accumulation (126). This excessive iron load in the mitochondria can be explained by doxorubicin downregulation of ATP-binding cassette protein-B8 (ABC-B8), which mitigates iron transport out of the mitochondria (126). Doxorubicin also downregulates the key anti-ferroptosis protein glutathione peroxidase-4, resulting in lipid peroxidation. Additionally, doxorubicin can interact with the iron response elements of ferritin, reducing cytosolic ferritin and increasing labile iron (127).
Pyroptosis
Pyroptosis is a cell death mechanism characterized by increased proinflammatory signaling and activation of caspase-1, –4, –5, and –11, leading to plasma membrane rupture mediated by gasdermin D (GSDMD) (75). Previous research has determined that doxorubicin induces pyroptosis via induction of terminal differentiation-induced non-coding RNA (TINCR) and activation of the NLRP-3-caspase-1 pathway (128–130). Specifically, doxorubicin upregulation of TINCR leads to recruitment of the adapter protein IGF2BP1 (insulin-like growth factor 2 mRNA-binding protein 1) and stabilization of NLRP3 mRNA (128). This increases NLRP3 expression and activates caspase-1, thereby leading to GSDMD cleavage, plasma membrane rupture, and interleukin (IL)-1β and IL-18 release (Figure 2C).
Need for improved cardiac imaging approaches in the context of contemporary anthracycline use
While cancer affects more than one in three people over their lifetime, improved long-term cancer survival has led to an increase in the incidence of adverse cardiac side-effects of cancer treatments (131). The U.S. National Cancer Institute estimates that in 2022 there will be ∼ 18 million cancer survivors which mounts to >5% of the U.S. population (132). Anthracyclines are a cornerstone of chemotherapy in various cancers (133), however, their use is complicated by anthracycline-induced cardiotoxicity (134, 135) which has been appreciated for decades (4, 136–138).
Despite the continued discovery of alternative chemotherapeutic strategies, and their known cardiotoxic side-effects, anthracyclines remain a mainstay of many cancer treatments (139, 140). Indeed, anthracyclines are used in 30–35% of breast cancer patients (141–143) and 60–70% of elderly lymphoma patients (144, 145). Moreover, 50–60% of childhood cancer survivors were treated with a chemotherapy regimen containing anthracyclines (146, 147). In parallel, continued advances in cancer therapy have increased the survival rate of childhood cancer to ∼ 80% (132). Furthermore, long-term follow-up of childhood cancer survivors indicate that up to 30% of patients treated with anthracyclines have signs of cardiac dysfunction in adulthood that are unmasked when more sensitive detection techniques are used (148), indicating significant under-estimation of long-term complications. These observations highlight the need for additional and improved imaging strategies in the context of AIC.
While echocardiography continues to be the most widely used tool for AIC monitoring, it is important to consider the limitations and advantages of each modality when selecting the proper screening exam for an individual patient. Echocardiography enjoys attractive features such as wide availability, rapid interpretation, lack of ionizing radiation exposure and relatively low cost which have firmly established it as the staple of AIC monitoring (6). However, the quality of the study is highly dependent on patient anatomy, acoustic windows, technician skill, and interobserver variability to a greater degree than other available methods (149–151).
In contrast, automated segmentation and ventricular volume algorithms offer precise and accurate evaluations of chamber function in cardiac MRI and multi-detector gated cardiac CT. However, cardiac MRI is more expensive and less widely available at many clinical centers. It is also a time-consuming exam that requires significant patient cooperation and can be undermined by rapid heart rates, arrhythmias, or by the presence of intrathoracic hardware such as implantable pacemakers or defibrillators that can introduce excessive signal artifact (152, 153).
Cardiac CT is also challenged by susceptibility to gating artifacts with rapid or irregular heart rhythms, need for iodinated contrast which is limiting in patients with kidney disease, as well as less robust data on the assessment of muscle strain or mechanics that can indicate early toxicity as compared to echocardiography or cardiac MRI (154).
Lastly, although largely fallen in clinical desuetude, patients with poor echocardiographic acoustic windows and contraindications to MRI or CT imaging may be referred for a radionuclide MUGA scan. This modality has proven to be a viable alternative to cardiac MRI or CT in patients where the precision or accuracy of echocardiographic measurements are in question, and thus remains a clinical option when choosing a strategy to monitor for AIC (155, 156). However, the advent of unique and specific markers in the growing field of nuclear molecular imaging may offer an additional toolset to detect and treat early manifestations of AIC.
Whereas the utility of perfusion-based imaging and myocardial blood flow quantitation for the detection of sub-clinical or early AIC has been studied in several small trials, additional research is needed to bring the evidence to the level of routine clinical utility. In particular, PET-derived myocardial blood flow has been examined as a potential marker of patients who may be at increased risk for AIC (157, 158). Whether changes in myocardial blood flow metrics are a sign of early cardiac stress or indicative of irreversible toxicity in response to anthracyclines require further scrutiny (157). If the value of myocardial flow reserve assessment in AIC can be further demonstrated in large-scale clinical studies, it could be added as an early evaluation strategy in company of echo-derived left ventricular global longitudinal strain or biomarkers such as plasma troponin levels.
Similarly, although no societal guidelines exist for routine myocardial perfusion imaging of patients undergoing anthracycline based chemotherapy regimens, the high correlation of CAD and subsequent acute coronary syndromes in cancer patients often prompts clinicians to screen patients with intermediate or high risk of CAD prior to initiation of therapy. In one retrospective analysis of 6.5 million cases of acute coronary syndromes, 9% of the patients had a diagnosis of cancer, either active or in remission, suggesting that pre-chemotherapy evaluation and revascularization, if indicated, may be appropriate in this patient group (159).
Several of the probes discussed in this review such as 18F-FDG and 68Ga-FAPI are used for cancer staging or progression evaluation. Retrospective analyses of these clinical studies suggest abnormal myocardial uptake in certain patients, paving the way for dedicated cardiac studies to detect and monitor AIC. There is also an opportunity to investigate these probes for oncologic and cardiac assessment in tandem. Doing so, however, will require patients to have a standardized preparation and clinicians to follow a more rigid imaging protocol that adheres to both oncologic and cardiac quality control requirements.
Echocardiography, including the segmental assessment of myocardial function by strain or displacement vectors, remains the first imaging modality of choice for AIC screening. Therefore, we posit a complementary role – dependent on the imaging targets – for the clinical utilization of nuclear molecular imaging applications in cardio-oncology, that we separate into two aims: 1) improving the prediction of AIC development in the setting of a normal echocardiogram, and 2) improving AIC reclassification in the setting of an abnormal echocardiogram (Figure 3). The clinical adoption of nuclear molecular imaging approaches remains limited at present, however, is poised to significantly affect current imaging strategies that have limitations in both sensitivity and specificity to screen and monitor AIC.

Figure 3. Potential clinical applications of nuclear molecular imaging in predicting and reclassifying cardiotoxicity in cancer patients undergoing anthracycline treatment. Different clinical algorithms may be followed according to the mechanistic pathway targeted for imaging – whether expression of cardiac injury related proteins, or alternatively activation of cardioprotective pathways – and the presence or absence of abnormal echocardiographic features suggestive of cardiotoxicity. (A) Molecular imaging may be used to predict the risk of developing cardiotoxicity during and/or following anthracycline treatment in patients with a normal echocardiogram. (B) In patients with an abnormal echocardiogram suggesting a new impairment in cardiac function, molecular imaging may be able to finetune the cardiotoxicity diagnosis with implications on the duration of anthracycline-induced cardiac dysfunction, or to reclassify the echocardiographic diagnosis, thereby providing oncologists and cardiologists greater diagnostic certainty with ensuing clinical implications.
Conclusion
Anthracycline-induced cardiotoxicity involves a broad range of pathophysiological pathways that lead to cardiomyocyte injury and that may be further complicated by cardiomyopathy and heart failure. Mechanisms implicated in the disease process, as well as molecular responses thereto, can be probed for nuclear imaging, e.g., metabolic dysfunction, cardiomyocyte death, sympathetic innervation, and changes in myocardial blood flow. Detecting these AIC-induced processes at a subclinical level, prior to the onset of irreversible cardiac impairment, may provide clinicians with valuable information permitting changes in chemotherapeutic strategies and/or timely initiation of cardioprotective strategies.
Author contributions
JJ, JP, and RP wrote the manuscript. JJ drew the figures and wrote the table. All authors read and approved the final manuscript.
Funding
This study was supported by VA Merit BX004558, UCLA Cardiovascular Discovery Fund/Lauren B. Leichtman and Arthur E. Levine Investigator Award, and NIH NCATS UCLA CTSI UL1TR00188.
Acknowledgments
We thank the Cardiovascular Research Foundation of Southern California who covered the publication costs of this article. The figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AIC, anthracycline-induced cardiotoxicity; BNP, B-type natriuretic peptide; CAD, coronary artery disease; CT, computerized tomography; Dox, doxorubicin; 18F-FDG, 18F-fluorodeoxyglucose; GLS, global longitudinal strain; H/M, heart-to-mediastinum; ID/g, injected dose/g; LV, left ventricle; LVEF, left ventricular ejection fraction; MRI, magnetic resonance imaging; MUGA, multigated acquisition; NLRP3, nucleotide-binding oligomerization domain (NOD-), leucine-rich repeat (LRR-), and pyrin domain (PYD)-containing protein 3; NT-proBNP, N-terminal proBNP; PET, positron emission tomography; PS, phosphatidylserine; ROS, reactive oxygen species; SPECT, single-photon emission computed tomography; TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling; WOR, washout rate.
References
1. Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. (2006) 355:1572–82.
2. van Dalen EC, van der Pal HJ, Kok WE, Caron HN, Kremer LC. Clinical heart failure in a cohort of children treated with anthracyclines: a long-term follow-up study. Eur J Cancer. (2006) 42:3191–8. doi: 10.1016/j.ejca.2006.08.005
4. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. (2003) 97:2869–79.
5. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. Circulation. (2022) 145:e895–1032.
6. Herrmann J, Lenihan D, Armenian S, Barac A, Blaes A, Cardinale D, et al. Defining cardiovascular toxicities of cancer therapies: an International cardio-oncology society (IC-OS) consensus statement. Eur Heart J. (2022) 43:280–99. doi: 10.1093/eurheartj/ehab674
7. Ewer MS, Ali MK, Mackay B, Wallace S, Valdivieso M, Legha SS, et al. A comparison of cardiac biopsy grades and ejection fraction estimations in patients receiving Adriamycin. J Clin Oncol. (1984) 2:112–7. doi: 10.1200/JCO.1984.2.2.112
8. Lipshultz SE, Rifai N, Dalton VM, Levy DE, Silverman LB, Lipsitz SR, et al. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N Engl J Med. (2004) 351:145–53. doi: 10.1056/NEJMoa035153
9. Lipshultz SE, Scully RE, Lipsitz SR, Sallan SE, Silverman LB, Miller TL, et al. Assessment of dexrazoxane as a cardioprotectant in doxorubicin-treated children with high-risk acute lymphoblastic leukaemia: long-term follow-up of a prospective, randomised, multicentre trial. Lancet Oncol. (2010) 11:950–61. doi: 10.1016/S1470-2045(10)70204-7
10. Armenian SH, Lacchetti C, Barac A, Carver J, Constine LS, Denduluri N, et al. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: American society of clinical oncology clinical practice guideline. J Clin Oncol. (2017) 35:893–911.
11. Chow EJ, Asselin BL, Schwartz CL, Doody DR, Leisenring WM, Aggarwal S, et al. Late mortality after dexrazoxane treatment: a report from the children’s oncology group. J Clin Oncol. (2015) 33:2639–45.
12. Chow EJ, Aplenc R, Vrooman LM, Doody DR, Huang YV, Aggarwal S, et al. Late health outcomes after dexrazoxane treatment: a report from the children’s oncology group. Cancer. (2022) 128:788–96. doi: 10.1002/cncr.33974
13. Cardinale D, Colombo A, Lamantia G, Colombo N, Civelli M, De Giacomi G, et al. Anthracycline-induced cardiomyopathy: clinical relevance and response to pharmacologic therapy. J Am Coll Cardiol. (2010) 55:213–20.
14. Avila MS, Ayub-Ferreira SM, de Barros Wanderley MR Jr., das Dores Cruz F, Gonçalves Brandão SM, Rigaud VOC, et al. Carvedilol for prevention of chemotherapy-related cardiotoxicity: the CECCY trial. J Am Coll Cardiol. (2018) 71:2281–90. doi: 10.1016/j.jacc.2018.02.049
15. Horenstein MS, Vander Heide RS, L’Ecuyer TJ. Molecular basis of anthracycline-induced cardiotoxicity and its prevention. Mol Genet Metab. (2000) 71:436–44.
16. Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem. (1986) 261:3068–74.
17. Tsang WP, Chau SP, Kong SK, Fung KP, Kwok TT. Reactive oxygen species mediate doxorubicin induced p53-independent apoptosis. Life Sci. (2003) 73:2047–58.
18. Carvalho RA, Sousa RP, Cadete VJ, Lopaschuk GD, Palmeira CM, Bjork JA, et al. Metabolic remodeling associated with subchronic doxorubicin cardiomyopathy. Toxicology. (2010) 270:92–8. doi: 10.1016/j.tox.2010.01.019
19. Peng X, Chen B, Lim CC, Sawyer DB. The cardiotoxicology of anthracycline chemotherapeutics: translating molecular mechanism into preventative medicine. Mol Interv. (2005) 5:163–71. doi: 10.1124/mi.5.3.6
20. Egert S, Nguyen N, Schwaiger M. Contribution of alpha-adrenergic and beta-adrenergic stimulation to ischemia-induced glucose transporter (GLUT) 4 and GLUT1 translocation in the isolated perfused rat heart. Circ Res. (1999) 84:1407–15. doi: 10.1161/01.res.84.12.1407
21. Zaninetti D, Greco-Perotto R, Assimacopoulos-Jeannet F, Jeanrenaud B. Effects of insulin on glucose transport and glucose transporters in rat heart. Biochem J. (1988) 250:277–83.
22. Sun D, Nguyen N, DeGrado TR, Schwaiger M, Brosius FCIII. Ischemia induces translocation of the insulin-responsive glucose transporter GLUT4 to the plasma membrane of cardiac myocytes. Circulation. (1994) 89:793–8. doi: 10.1161/01.cir.89.2.793
23. Young LH, Renfu Y, Russell R, Hu X, Caplan M, Ren J, et al. Low-flow ischemia leads to translocation of canine heart GLUT-4 and GLUT-1 glucose transporters to the sarcolemma in vivo. Circulation. (1997) 95:415–22. doi: 10.1161/01.cir.95.2.415
24. Wheeler TJ. Translocation of glucose transporters in response to anoxia in heart. J Biol Chem. (1988) 263:19447–54.
26. Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, et al. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. (1977) 28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x
27. Barrio JR, Huang SC, Satyamurthy N, Scafoglio CS, Yu AS, Alavi A, et al. Does 2-FDG PET accurately reflect quantitative in vivo glucose utilization? J Nucl Med. (2020) 61:931–7. doi: 10.2967/jnumed.119.237446
28. Hariharan R, Bray M, Ganim R, Doenst T, Goodwin GW, Taegtmeyer H. Fundamental limitations of [18F]2-deoxy-2-fluoro-D-glucose for assessing myocardial glucose uptake. Circulation. (1995) 91:2435–44. doi: 10.1161/01.cir.91.9.2435
29. Bøtker HE, Böttcher M, Schmitz O, Gee A, Hansen SB, Cold GE, et al. Glucose uptake and lumped constant variability in normal human hearts determined with [18F]fluorodeoxyglucose. J Nucl Cardiol. (1997) 4:125–32.
30. Ng CK, Soufer R, McNulty PH. Effect of hyperinsulinemia on myocardial fluorine-18-FDG uptake. J Nucl Med. (1998) 39:379–83.
31. Botker HE, Goodwin GW, Holden JE, Doenst T, Gjedde A, Taegtmeyer H. Myocardial glucose uptake measured with fluorodeoxyglucose: a proposed method to account for variable lumped constants. J Nucl Med. (1999) 40:1186–96.
32. Bulten BF, Sollini M, Boni R, Massri K, de Geus-Oei LF, van Laarhoven HWM, et al. Cardiac molecular pathways influenced by doxorubicin treatment in mice. Sci Rep. (2019) 9:2514.
33. Bauckneht M, Pastorino F, Castellani P, Cossu V, Orengo AM, Piccioli P, et al. Increased myocardial (18)F-FDG uptake as a marker of Doxorubicin-induced oxidative stress. J Nucl Cardiol. (2020) 27:2183–94. doi: 10.1007/s12350-019-01618-x
34. Borde C, Kand P, Basu S. Enhanced myocardial fluorodeoxyglucose uptake following Adriamycin-based therapy: evidence of early chemotherapeutic cardiotoxicity? World J Radiol. (2012) 4:220–3. doi: 10.4329/wjr.v4.i5.220
35. Sarocchi M, Bauckneht M, Arboscello E, Capitanio S, Marini C, Morbelli S, et al. An increase in myocardial 18-fluorodeoxyglucose uptake is associated with left ventricular ejection fraction decline in Hodgkin lymphoma patients treated with anthracycline. J Transl Med. (2018) 16:295. doi: 10.1186/s12967-018-1670-9
36. Kim J, Cho SG, Kang SR, Yoo SW, Kwon SY, Min JJ, et al. Association between FDG uptake in the right ventricular myocardium and cancer therapy-induced cardiotoxicity. J Nucl Cardiol. (2020) 27:2154–63. doi: 10.1007/s12350-019-01617-y
37. Keramida K, Farmakis D, Bingcang J, Sulemane S, Sutherland S, Bingcang RA, et al. Longitudinal changes of right ventricular deformation mechanics during trastuzumab therapy in breast cancer patients. Eur J Heart Fail. (2019) 21:529–35. doi: 10.1002/ejhf.1385
38. Bauckneht M, Ferrarazzo G, Fiz F, Morbelli S, Sarocchi M, Pastorino F, et al. Doxorubicin effect on myocardial metabolism as a prerequisite for subsequent development of cardiac toxicity: a translational (18)F-FDG PET/CT observation. J Nucl Med. (2017) 58:1638–45. doi: 10.2967/jnumed.117.191122
39. Heckmann MB, Totakhel B, Finke D, Anker MS, Müller-Tidow C, Haberkorn U, et al. Evidence for a cardiac metabolic switch in patients with Hodgkin’s lymphoma. ESC Heart Fail. (2019) 6:824–9. doi: 10.1002/ehf2.12475
40. Dilsizian V, Bacharach SL, Beanlands RS, Bergmann SR, Delbeke D, Dorbala S, et al. ASNC imaging guidelines/SNMMI procedure standard for positron emission tomography (PET) nuclear cardiology procedures. J Nucl Cardiol. (2016) 23:1187–226. doi: 10.1007/s12350-016-0522-3
41. Nose H, Otsuka H, Otomi Y, Terazawa K, Takao S, Iwamoto S, et al. The physiological uptake pattern of (18)F-FDG in the left ventricular myocardium of patients without heart disease. J Med Invest. (2014) 61:53–8.
42. Jong J, Packard RRS. (18)F-FDG PET imaging of myocardial inflammation and viability following experimental infarction and anti-inflammatory treatment with compound MCC950. J Nucl Cardiol. (2021) 28:2358–60. doi: 10.1007/s12350-020-02104-5
43. Croteau E, Gascon S, Bentourkia M, Langlois R, Rousseau JA, Lecomte R, et al. [11C]Acetate rest-stress protocol to assess myocardial perfusion and oxygen consumption reserve in a model of congestive heart failure in rats. Nucl Med Biol. (2012) 39:287–94.
44. Croteau E, Tremblay S, Gascon S, Dumulon-Perreault V, Labbé SM, Rousseau JA, et al. [(11)C]-Acetoacetate PET imaging: a potential early marker for cardiac heart failure. Nucl Med Biol. (2014) 41:863–70. doi: 10.1016/j.nucmedbio.2014.08.006
45. Nony P, Guastalla JP, Rebattu P, Landais P, Lievre M, Bontemps L, et al. In vivo measurement of myocardial oxidative metabolism and blood flow does not show changes in cancer patients undergoing doxorubicin therapy. Cancer Chemother Pharmacol. (2000) 45:375–80.
46. Armbrecht JJ, Buxton DB, Schelbert HR. Validation of [1-11C]acetate as a tracer for noninvasive assessment of oxidative metabolism with positron emission tomography in normal, ischemic, postischemic, and hyperemic canine myocardium. Circulation. (1990) 81:1594–605. doi: 10.1161/01.cir.81.5.1594
47. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. (2013) 113:709–24.
48. Christensen NL, Jakobsen S, Schacht AC, Munk OL, Alstrup AKO, Tolbod LP, et al. Whole-body biodistribution, dosimetry, and metabolite correction of [(11)C]palmitate: a PET tracer for imaging of fatty acid metabolism. Mol Imaging. (2017) 16:1536012117734485. doi: 10.1177/1536012117734485
49. DeGrado TR, Coenen HH, Stocklin G. 14(R,S)-[18F]fluoro-6-thia-heptadecanoic acid (FTHA): evaluation in mouse of a new probe of myocardial utilization of long chain fatty acids. J Nucl Med. (1991) 32:1888–96.
50. Murphy MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta. (2008) 1777:1028–31.
51. Safee ZM, Baark F, Waters ECT, Veronese M, Pell VR, Clark JE, et al. Detection of anthracycline-induced cardiotoxicity using perfusion-corrected (99m)Tc sestamibi SPECT. Sci Rep. (2019) 9:216. doi: 10.1038/s41598-018-36721-5
52. Sivapackiam J, Kabra S, Speidel S, Sharma M, Laforest R, Salter A, et al. 68Ga-Galmydar: a PET imaging tracer for noninvasive detection of Doxorubicin-induced cardiotoxicity. PLoS One. (2019) 14:e0215579. doi: 10.1371/journal.pone.0215579
53. McCluskey SP, Haslop A, Coello C, Gunn RN, Tate EW, Southworth R, et al. Imaging of chemotherapy-induced acute cardiotoxicity with (18)F-labeled lipophilic cations. J Nucl Med. (2019) 60:1750–6. doi: 10.2967/jnumed.119.226787
54. Min JJ, Biswal S, Deroose C, Gambhir SS. Tetraphenylphosphonium as a novel molecular probe for Imaging tumors. J Nucl Med. (2004) 45:636–43.
55. Alpert NM, Guehl N, Ptaszek L, Pelletier-Galarneau M, Ruskin J, Mansour MC, et al. Quantitative in vivo mapping of myocardial mitochondrial membrane potential. PLoS One. (2018) 13:e0190968. doi: 10.1371/journal.pone.0190968
56. Pelletier-Galarneau M, Petibon Y, Ma C, Han P, Kim SJW, Detmer FJ, et al. In vivo quantitative mapping of human mitochondrial cardiac membrane potential: a feasibility study. Eur J Nucl Med Mol Imaging. (2021) 48:414–20. doi: 10.1007/s00259-020-04878-9
57. Pelletier-Galarneau M, Detmer FJ, Petibon Y, Normandin M, Ma C, Alpert NM, et al. Quantification of myocardial mitochondrial membrane potential using PET. Curr Cardiol Rep. (2021) 23:70.
58. Chu W, Chepetan A, Zhou D, Shoghi KI, Xu J, Dugan LL, et al. Development of a PET radiotracer for non-invasive imaging of the reactive oxygen species, superoxide, in vivo. Org Biomol Chem. (2014) 12:4421–31. doi: 10.1039/c3ob42379d
59. Boutagy NE, Wu J, Cai Z, Zhang W, Booth CJ, Kyriakides TC, et al. In vivo reactive oxygen species detection with a novel positron emission tomography tracer, (18)F-DHMT, allows for early detection of anthracycline-induced cardiotoxicity in rodents. JACC Basic Transl Sci. (2018) 3:378–90. doi: 10.1016/j.jacbts.2018.02.003
60. Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. (1984) 226:466–8.
61. Stingele J, Bellelli R, Boulton SJ. Mechanisms of DNA-protein crosslink repair. Nat Rev Mol Cell Biol. (2017) 18:563–73.
62. Capranico G, Tinelli S, Austin CA, Fisher ML, Zunino F. Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim Biophys Acta. (1992) 1132:43–8. doi: 10.1016/0167-4781(92)90050-a
63. Damiani RM, Moura DJ, Viau CM, Caceres RA, Henriques JAP, Saffi J. Pathways of cardiac toxicity: comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch Toxicol. (2016) 90:2063–76. doi: 10.1007/s00204-016-1759-y
64. Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, et al. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. (2007) 67:8839–46. doi: 10.1158/0008-5472.CAN-07-1649
65. Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. (2012) 18:1639–42. doi: 10.1038/nm.2919
66. Plesca D, Mazumder S, Almasan A. DNA damage response and apoptosis. Methods Enzymol. (2008) 446:107–22.
67. Harada A, Matsuzaki K, Takeiri A, Mishima M. The predominant role of apoptosis in gamma H2AX formation induced by aneugens is useful for distinguishing aneugens from clastogens. Mutat Res Genet Toxicol Environ Mutagen. (2014) 771:23–9. doi: 10.1016/j.mrgentox.2014.05.010
68. Scully R, Xie A. Double strand break repair functions of histone H2AX. Mutat Res. (2013) 750:5–14.
69. Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. (2006) 21:201–14.
70. Marteijn JA, Bekker-Jensen S, Mailand N, Lans H, Schwertman P, Gourdin AM, et al. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J Cell Biol. (2009) 186:835–47. doi: 10.1083/jcb.200902150
71. Yeh ET, Chang HM. Oncocardiology-past, present, and future: a review. JAMA Cardiol. (2016) 1:1066–72. doi: 10.1001/jamacardio.2016.2132
72. Amgalan D, Garner TP, Pekson R, Jia XF, Yanamandala M, Paulino V, et al. A small-molecule allosteric inhibitor of BAX protects against doxorubicin-induced cardiomyopathy. Nat Cancer. (2020) 1:315–28. doi: 10.1038/s43018-020-0039-1
73. Kuhlbrandt W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. (2015) 13:89. doi: 10.1186/s12915-015-0201-x
74. Kinnally KW, Peixoto PM, Ryu SY, Dejean LM. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta. (2011) 1813:616–22. doi: 10.1016/j.bbamcr.2010.09.013
75. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. (2019) 99:1765–817.
76. Bennink RJ, van den Hoff MJ, van Hemert FJ, de Bruin KM, Spijkerboer AL, Vanderheyden JL, et al. Annexin V imaging of acute doxorubicin cardiotoxicity (apoptosis) in rats. J Nucl Med. (2004) 45:842–8.
77. Kemerink GJ, Liu X, Kieffer D, Ceyssens S, Mortelmans L, Verbruggen AM, et al. Safety, biodistribution, and dosimetry of 99mTc-HYNIC-annexin V, a novel human recombinant annexin V for human application. J Nucl Med. (2003) 44:947–52.
78. Nguyen QD, Smith G, Glaser M, Perumal M, Arstad E, Aboagye EO. Positron emission tomography imaging of drug-induced tumor apoptosis with a caspase-3/7 specific [18F]-labeled isatin sulfonamide. Proc Natl Acad Sci USA. (2009) 106:16375–80. doi: 10.1073/pnas.0901310106
79. Zhou D, Chu W, Chen DL, Wang Q, Reichert DE, Rothfuss J, et al. [18F]- and [11C]-labeled N-benzyl-isatin sulfonamide analogues as PET tracers for apoptosis: synthesis, radiolabeling mechanism, and in vivo imaging study of apoptosis in Fas-treated mice using [11C]WC-98. Org Biomol Chem. (2009) 7:1337–48.
80. Lee D, Long SA, Murray JH, Adams JL, Nuttall ME, Nadeau DP, et al. Potent and selective nonpeptide inhibitors of caspases 3 and 7. J Med Chem. (2001) 44:2015–26.
81. Su H, Chen G, Gangadharmath U, Gomez LF, Liang Q, Mu F, et al. Evaluation of [(18)F]-CP18 as a PET imaging tracer for apoptosis. Mol Imaging Biol. (2013) 15:739–47.
82. Xia CF, Chen G, Gangadharmath U, Gomez LF, Liang Q, Mu F, et al. In vitro and in vivo evaluation of the caspase-3 substrate-based radiotracer [(18)F]-CP18 for PET imaging of apoptosis in tumors. Mol Imaging Biol. (2013) 15:748–57. doi: 10.1007/s11307-013-0646-7
83. Su H, Gorodny N, Gomez LF, Gangadharmath U, Mu F, Chen G, et al. Noninvasive molecular imaging of apoptosis in a mouse model of anthracycline-induced cardiotoxicity. Circ Cardiovasc Imaging. (2015) 8:e001952. doi: 10.1161/CIRCIMAGING.114.001952
84. Estorch M, Carrió I, Martínez-Duncker D, Berná L, Torres G, Alonso C, et al. Myocyte cell damage after administration of doxorubicin or mitoxantrone in breast cancer patients assessed by indium 111 antimyosin monoclonal antibody studies. J Clin Oncol. (1993) 11:1264–8.
85. Carrió I, Lopez-Pousa A, Estorch M, Duncker D, Berná L, Torres G, et al. Detection of doxorubicin cardiotoxicity in patients with sarcomas by indium-111-antimyosin monoclonal antibody studies. J Nucl Med. (1993) 34:1503–7.
86. Valdés Olmos RA, ten Bokkel Huinink WW, ten Hoeve RF, van Tinteren H, Bruning PF, van Vlies B, et al. Usefulness of indium-111 antimyosin scintigraphy in confirming myocardial injury in patients with anthracycline-associated left ventricular dysfunction. Ann Oncol. (1994) 5:617–22. doi: 10.1093/oxfordjournals.annonc.a058933
87. Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. (2009) 54:1747–62.
88. Eschenhagen T, Force T, Ewer MS, de Keulenaer GW, Suter TM, Anker SD, et al. Cardiovascular side effects of cancer therapies: a position statement from the heart failure association of the European society of cardiology. Eur J Heart Fail. (2011) 13:1–10. doi: 10.1093/eurjhf/hfq213
89. Schroeder C, Jordan J. Norepinephrine transporter function and human cardiovascular disease. Am J Physiol Heart Circ Physiol. (2012) 303:H1273–82.
90. Lymperopoulos A, Rengo G, Koch WJ. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res. (2013) 113:739–53.
91. Eaton MJ, Duplan H. Useful cell lines derived from the adrenal medulla. Mol Cell Endocrinol. (2004) 228:39–52.
92. Bohm M, La Rosee K, Schwinger RH, Erdmann E. Evidence for reduction of norepinephrine uptake sites in the failing human heart. J Am Coll Cardiol. (1995) 25:146–53. doi: 10.1016/0735-1097(94)00353-r
93. Zelt JGE, deKemp RA, Rotstein BH, Nair GM, Narula J, Ahmadi A, et al. Nuclear imaging of the cardiac sympathetic nervous system: a disease-specific interpretation in heart failure. JACC Cardiovasc Imaging. (2020) 13:1036–54. doi: 10.1016/j.jcmg.2019.01.042
94. Wakasugi S, Wada A, Hasegawa Y, Nakano S, Shibata N. Detection of abnormal cardiac adrenergic neuron activity in adriamycin-induced cardiomyopathy with iodine-125-metaiodobenzylguanidine. J Nucl Med. (1992) 33:208–14.
95. Oudot A, Courteau A, Guillemin M, Vrigneaud JM, Walker PM, Brunotte F, et al. [(123)I]MIBG is a better early marker of anthracycline cardiotoxicity than [(18)F]FDG: a preclinical SPECT/CT and simultaneous PET/MR study. EJNMMI Res. (2021) 11:92.
96. Carrió I, Estorch M, Berná L, López-Pousa J, Tabernero J, Torres G. Indium-111-antimyosin and iodine-123-MIBG studies in early assessment of doxorubicin cardiotoxicity. J Nucl Med. (1995) 36:2044–9.
97. Laursen AH, Ripa RS, Hasbak P, Kjær A, Elming MB, Køber L, et al. (123)I-MIBG for detection of subacute doxorubicin-induced cardiotoxicity in patients with malignant lymphoma. J Nucl Cardiol. (2020) 27:931–9. doi: 10.1007/s12350-018-01566-y
98. Dos Santos MJ, da Rocha ET, Verberne HJ, da Silva ET, Aragon DC, Junior JS. Assessment of late anthracycline-induced cardiotoxicity by (123)I-mIBG cardiac scintigraphy in patients treated during childhood and adolescence. J Nucl Cardiol. (2017) 24:256–64. doi: 10.1007/s12350-015-0309-y
99. Kenk M, Thackeray JT, Thorn SL, Dhami K, Chow BJ, Ascah KJ, et al. Alterations of pre- and postsynaptic noradrenergic signaling in a rat model of adriamycin-induced cardiotoxicity. J Nucl Cardiol. (2010) 17:254–63. doi: 10.1007/s12350-009-9190-x
100. Kizaki K, Akatsuka K, Momozaki M, Fujimori Y, Uchide T, Temma K, et al. Changes in myocardial beta1-adrenergic receptor and stimulatory G-protein gene expression after chronic treatment with doxorubicin in rat. J Vet Med Sci. (2004) 66:989–92. doi: 10.1292/jvms.66.989
101. Steinherz LJ, Graham T, Hurwitz R, Sondheimer HM, Schwartz RG, Shaffer EM, et al. Guidelines for cardiac monitoring of children during and after anthracycline therapy: report of the Cardiology Committee of the Childrens Cancer Study Group. Pediatrics. (1992) 89:942–9.
102. Ganz WI, Sridhar KS, Forness TJ. Detection of early anthracycline cardiotoxicity by monitoring the peak filling rate. Am J Clin Oncol. (1993) 16:109–12.
103. Schwartz RG, McKenzie WB, Alexander J, Sager P, D’Souza A, Manatunga A, et al. Congestive heart failure and left ventricular dysfunction complicating doxorubicin therapy. Seven-year experience using serial radionuclide angiocardiography. Am J Med. (1987) 82:1109–18. doi: 10.1016/0002-9343(87)90212-9
104. Nousiainen T, Jantunen E, Vanninen E, Hartikainen J. Early decline in left ventricular ejection fraction predicts doxorubicin cardiotoxicity in lymphoma patients. Br J Cancer. (2002) 86:1697–700.
105. Laursen AH, Elming MB, Ripa RS, Hasbak P, Kjær A, Køber L, et al. Rubidium-82 positron emission tomography for detection of acute doxorubicin-induced cardiac effects in lymphoma patients. J Nucl Cardiol. (2020) 27:1698–707. doi: 10.1007/s12350-018-1458-6
106. Hardenbergh PH, Munley MT, Bentel GC, Kedem R, Borges-Neto S, Hollis D, et al. Cardiac perfusion changes in patients treated for breast cancer with radiation therapy and doxorubicin: preliminary results. Int J Radiat Oncol Biol Phys. (2001) 49:1023–8. doi: 10.1016/s0360-3016(00)01531-5
107. Dreyfuss AD, Bravo PE, Koumenis C, Ky B. Precision Cardio-Oncology. J Nucl Med. (2019) 60:443–50.
108. Maddahi J, Packard RRS. Cardiac PET perfusion tracers: current status and future directions. Semin Nucl Med. (2014) 44:333–43.
109. Maddahi J, Packard RRS. PET should replace SPECT in cardiac imaging for diagnosis and risk assessment of patients with known or suspected CAD: pro. J Nucl Cardiol. (2017) 24:1955–9.
110. Nehmeh SA, Fox JJ, Schwartz J, Ballangrud ÅM, Schöder H, Zhao Y. A pilot study of (13)N-ammonia cardiac PET imaging to assess subacute cardiotoxicity following adjuvant intensity-modulated radiotherapy for locally advanced breast cancer. Clin Imaging. (2020) 68:283–90. doi: 10.1016/j.clinimag.2020.07.026
111. Maaniitty T, Knuuti J, Saraste A. 15O-water PET MPI: current status and future perspectives. Semin Nucl Med. (2020) 50:238–47. doi: 10.1053/j.semnuclmed.2020.02.011
112. Packard RRS, Huang SC, Dahlbom M, Czernin J, Maddahi J. Absolute quantitation of myocardial blood flow in human subjects with or without myocardial ischemia using dynamic flurpiridaz F 18 PET. J Nucl Med. (2014) 55:1438–44.
113. Packard RRS, Votaw JR, Cooke CD, Van Train KF, Garcia EV, Maddahi J. 18F-flurpiridaz positron emission tomography segmental and territory myocardial blood flow metrics: incremental value beyond perfusion for coronary artery disease categorization. Eur Heart J Cardiovasc Imaging. (2021). [Epub ahead of print]. doi: 10.1093/ehjci/jeab267
115. Nebigil CG, Desaubry L. Updates in anthracycline-mediated cardiotoxicity. Front Pharmacol. (2018) 9:1262. doi: 10.3389/fphar.2018.01262
116. Siebermair J, Köhler MI, Kupusovic J, Nekolla SG, Kessler L, Ferdinandus J, et al. Cardiac fibroblast activation detected by Ga-68 FAPI PET imaging as a potential novel biomarker of cardiac injury/remodeling. J Nucl Cardiol. (2021) 28:812–21. doi: 10.1007/s12350-020-02307-w
117. Heckmann MB, Reinhardt F, Finke D, Katus HA, Haberkorn U, Leuschner F, et al. Relationship between cardiac fibroblast activation protein activity by positron emission tomography and cardiovascular disease. Circ Cardiovasc Imaging. (2020) 13:e010628.
118. Carvalho FS, Burgeiro A, Garcia R, Moreno AJ, Carvalho RA, Oliveira PJ. Doxorubicin-induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy. Med Res Rev. (2014) 34:106–35.
119. Christidi E, Brunham LR. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. (2021) 12:339.
120. Pillay CS, Elliott E, Dennison C. Endolysosomal proteolysis and its regulation. Biochem J. (2002) 363:417–29.
121. Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. (2016) 133:1668–87. doi: 10.1161/CIRCULATIONAHA.115.017443
122. Bartlett JJ, Trivedi PC, Yeung P, Kienesberger PC, Pulinilkunnil T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem J. (2016) 473:3769–89.
123. Li M, Sala V, De Santis MC, Cimino J, Cappello P, Pianca N, et al. Phosphoinositide 3-kinase gamma inhibition protects from anthracycline cardiotoxicity and reduces tumor growth. Circulation. (2018) 138:696–711. doi: 10.1161/CIRCULATIONAHA.117.030352
124. Russo M, Bono E, Ghigo A. The interplay between autophagy and senescence in anthracycline cardiotoxicity. Curr Heart Fail Rep. (2021) 18:180–90. doi: 10.1007/s11897-021-00519-w
125. Xu X, Persson HL, Richardson DR. Molecular pharmacology of the interaction of anthracyclines with iron. Mol Pharmacol. (2005) 68:261–71.
126. Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. (2014) 124:617–30.
127. Canzoneri JC, Oyelere AK. Interaction of anthracyclines with iron responsive element mRNAs. Nucleic Acids Res. (2008) 36:6825–34. doi: 10.1093/nar/gkn774
128. Meng L, Lin H, Zhang J, Lin N, Sun Z, Gao F, et al. Doxorubicin induces cardiomyocyte pyroptosis via the TINCR-mediated posttranscriptional stabilization of NLR family pyrin domain containing 3. J Mol Cell Cardiol. (2019) 136:15–26. doi: 10.1016/j.yjmcc.2019.08.009
129. Tavakoli Dargani Z, Singla R, Johnson T, Kukreja R, Singla DK. Exosomes derived from embryonic stem cells inhibit doxorubicin and inflammation-induced pyroptosis in muscle cells. Can J Physiol Pharmacol. (2018) 96:304–7. doi: 10.1139/cjpp-2017-0340
130. Kobayashi M, Usui F, Karasawa T, Kawashima A, Kimura H, Mizushina Y, et al. NLRP3 deficiency reduces macrophage interleukin-10 production and enhances the susceptibility to doxorubicin-induced cardiotoxicity. Sci Rep. (2016) 6:26489. doi: 10.1038/srep26489
131. McGowan JV, Chung R, Maulik A, Piotrowska I, Walker JM, Yellon DM. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc Drugs Ther. (2017) 31:63–75.
133. Vejpongsa P, Yeh ET. Prevention of anthracycline-induced cardiotoxicity: challenges and opportunities. J Am Coll Cardiol. (2014) 64:938–45. doi: 10.1016/j.jacc.2014.06.1167
134. Chang HM, Moudgil R, Scarabelli T, Okwuosa TM, Yeh ETH. Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 1. J Am Coll Cardiol. (2017) 70:2536–51.
135. Chang HM, Okwuosa TM, Scarabelli T, Moudgil R, Yeh ETH. Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 2. J Am Coll Cardiol. (2017) 70:2552–65.
136. Von Hoff DD, Layard MW, Basa P, Davis HL Jr., Von Hoff AL, Rozencweig M, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. (1979) 91:710–7.
137. Lipshultz SE, Adams MJ, Colan SD, Constine LS, Herman EH, Hsu DT, et al. Long-term cardiovascular toxicity in children, adolescents, and young adults who receive cancer therapy: pathophysiology, course, monitoring, management, prevention, and research directions: a scientific statement from the American Heart Association. Circulation. (2013) 128:1927–95. doi: 10.1161/CIR.0b013e3182a88099
138. Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. (2015) 131:1981–8.
139. Bloom MW, Hamo CE, Cardinale D, Ky B, Nohria A, Baer L, et al. Cancer therapy-related cardiac dysfunction and heart failure: part 1: definitions, pathophysiology, risk factors, and imaging. Circ Heart Fail. (2016) 9:e002661. doi: 10.1161/CIRCHEARTFAILURE.115.002661
140. Hamo CE, Bloom MW, Cardinale D, Ky B, Nohria A, Baer L, et al. Cancer therapy-related cardiac dysfunction and heart failure: part 2: prevention, treatment, guidelines, and future directions. Circ Heart Fail. (2016) 9:e002843. doi: 10.1161/CIRCHEARTFAILURE.115.002843
141. Giordano SH, Lin YL, Kuo YF, Hortobagyi GN, Goodwin JS. Decline in the use of anthracyclines for breast cancer. J Clin Oncol. (2012) 30:2232–9.
142. Jasra S, Anampa J. Anthracycline use for early stage breast cancer in the modern era: a review. Curr Treat Options Oncol. (2018) 19:30. doi: 10.1007/s11864-018-0547-8
143. Guo F, Yi Z, Wang W, Han Y, Yu P, Zhang S, et al. Profile, treatment patterns, and influencing factors of anthracycline use in breast cancer patients in China: a nation-wide multicenter study. Cancer Med. (2021) 10:6744–61. doi: 10.1002/cam4.4215
144. Nabhan C, Byrtek M, Rai A, Dawson K, Zhou X, Link BK, et al. Disease characteristics, treatment patterns, prognosis, outcomes and lymphoma-related mortality in elderly follicular lymphoma in the United States. Br J Haematol. (2015) 170:85–95.
145. Chihara D, Westin JR, Oki Y, Ahmed MA, Do B, Fayad LE, et al. Management strategies and outcomes for very elderly patients with diffuse large B-cell lymphoma. Cancer. (2016) 122:3145–51.
146. Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, et al. Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer. (2010) 10:337. doi: 10.1186/1471-2407-10-337
147. Bansal N, Joshi C, Adams MJ, Hutchins K, Ray A, Lipshultz SE. Cardiotoxicity in pediatric lymphoma survivors. Expert Rev Cardiovasc Ther. (2021) 19:957–74.
148. Armstrong GT, Joshi VM, Ness KK, Marwick TH, Zhang N, Srivastava D, et al. Comprehensive echocardiographic detection of treatment-related cardiac dysfunction in adult survivors of childhood cancer: results from the St. Jude lifetime cohort study. J Am Coll Cardiol. (2015) 65:2511–22. doi: 10.1016/j.jacc.2015.04.013
149. Thavendiranathan P, Grant AD, Negishi T, Plana JC, Popovic ZB, Marwick TH. Reproducibility of echocardiographic techniques for sequential assessment of left ventricular ejection fraction and volumes: application to patients undergoing cancer chemotherapy. J Am Coll Cardiol. (2013) 61:77–84. doi: 10.1016/j.jacc.2012.09.035
150. Piveta RB, Rodrigues ACT, Vieira MLC, Fischer CH, Afonso TR, Daminello E, et al. Early change in area strain detected by 3D speckle tracking is associated with subsequent cardiotoxicity in patients treated with low doses of anthracyclines. Front Cardiovasc Med. (2022) 9:842532. doi: 10.3389/fcvm.2022.842532
151. Mitchell C, Rahko PS, Blauwet LA, Canaday B, Finstuen JA, Foster MC, et al. Guidelines for performing a comprehensive transthoracic echocardiographic examination in adults: recommendations from the American society of echocardiography. J Am Soc Echocardiogr. (2019) 32:1–64.
152. Loffler AI, Salerno M. Cardiac MRI for the evaluation of oncologic cardiotoxicity. J Nucl Cardiol. (2018) 25:2148–58.
153. O’Quinn R, Ferrari VA, Daly R, Hundley G, Baldassarre LA, Han Y, et al. Cardiac magnetic resonance in cardio-oncology: advantages, importance of expediency, and considerations to navigate pre-authorization. JACC CardioOncol. (2021) 3:191–200. doi: 10.1016/j.jaccao.2021.04.011
154. Feher A, Baldassarre LA, Sinusas AJ. Novel cardiac computed tomography methods for the assessment of anthracycline induced cardiotoxicity. Front Cardiovasc Med. (2022) 9:875150. doi: 10.3389/fcvm.2022.875150
155. Alexandre J, Cautela J, Ederhy S, Damaj GL, Salem JE, Barlesi F, et al. Cardiovascular toxicity related to cancer treatment: a pragmatic approach to the American and European cardio-oncology guidelines. J Am Heart Assoc. (2020) 9:e018403. doi: 10.1161/JAHA.120.018403
156. Hyafil F, Mirabel M, Tavitian B. Molecular imaging of free radicals for anthracycline-induced cardiotoxicity: see the burn? J Nucl Cardiol. (2022) 29:226–9. doi: 10.1007/s12350-020-02254-6
157. Ziadi MC, de Kemp R, Beanlands RSB, Small GR. Looking for trouble: reduced myocardial flow reserve following anthracyclines. J Nucl Cardiol. (2020) 27:1708–13. doi: 10.1007/s12350-018-01564-0
158. Lin Y, Wang J, Xu M, Qiu C, Xu P, Shang L, et al. Summed rest score in gated myocardial perfusion imaging is a good predicator for treatment-related cardiotoxicity after anthracycline chemotherapy in patients with diffuse large B-cell lymphoma. Oncol Lett. (2020) 20:330. doi: 10.3892/ol.2020.12194
Keywords: anthracycline, cardiotoxicity, molecular imaging, nuclear medicine, clinical application
Citation: Jong J, Pinney JR and Packard RRS (2022) Anthracycline-induced cardiotoxicity: From pathobiology to identification of molecular targets for nuclear imaging. Front. Cardiovasc. Med. 9:919719. doi: 10.3389/fcvm.2022.919719
Received: 13 April 2022; Accepted: 28 June 2022;
Published: 03 August 2022.
Edited by:
Alessandra Ghigo, University of Turin, ItalyReviewed by:
Edoardo Bertero, University Hospital Würzburg, GermanyVlad Gabriel Zaha, University of Texas Southwestern Medical Center, United States
Copyright © 2022 Jong, Pinney and Packard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: René R. Sevag Packard, cnBhY2thcmRAbWVkbmV0LnVjbGEuZWR1