 Zhenqi Rao
Zhenqi Rao Yidan Zheng
Yidan Zheng Li Xu1
Li Xu1 Zihao Wang
Zihao Wang Nianguo Dong
Nianguo Dong Zhejun Cai
Zhejun Cai Fei Li
Fei Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 16 June 2022
Sec. Atherosclerosis and Vascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.918056
This article is part of the Research Topic Oxidative Stress, Inflammation and Atherosclerosis-Related Diseases: From basic to clinical research View all 11 articles
Vascular calcification (VC) is characterized by calcium phosphate deposition in blood vessel walls and is associated with many diseases, as well as increased cardiovascular morbidity and mortality. However, the molecular mechanisms underlying of VC development and pathogenesis are not fully understood, thus impeding the design of molecular-targeted therapy for VC. Recently, several studies have shown that endoplasmic reticulum (ER) stress can exacerbate VC. The ER is an intracellular membranous organelle involved in the synthesis, folding, maturation, and post-translational modification of secretory and transmembrane proteins. ER stress (ERS) occurs when unfolded/misfolded proteins accumulate after a disturbance in the ER environment. Therefore, downregulation of pathological ERS may attenuate VC. This review summarizes the relationship between ERS and VC, focusing on how ERS regulates the development of VC by promoting osteogenic transformation, inflammation, autophagy, and apoptosis, with particular interest in the molecular mechanisms occurring in various vascular cells. We also discuss, the therapeutic effects of ERS inhibition on the progress of diseases associated with VC are detailed.
Vascular calcification (VC) commonly occurs with aging, atherosclerosis, diabetes, and chronic kidney disease (CKD) owing to accumulation of apatite calcium salts in the media and/or intima of arteries (1). Based on the location of hydroxyapatite, three main types of VC have been reported, namely, intimal calcification, Mönckeberg medial arterial calcification, and valvular calcification (2). Intimal calcification is closely associated with lipid deposits, inflammatory cell infiltration, atherosclerosis, and atherosclerotic plaque rupture (3). Medial calcification occurs when smooth muscle cells transform into osteoblast-like cells. This transformation is associated with various genes, including bone morphogenetic protein-2 (BMP2), MSH homeobox 2 (MSX2), and alkaline phosphatase (ALP; 4). Valvular calcification occurs in aortic valves as a result of long-term mechanical stress and the effects of proinflammatory cytokines, which can cause aortic stenosis.
The endoplasmic reticulum (ER) is an intracellular organelle with important roles in protein folding and maturation, lipid biosynthesis, and intracellular calcium homeostasis. However, the capacity of the ER protein maturation machinery can become overwhelmed in certain physiological or pathological conditions, leading to the accumulation of defective or superfluous proteins and ER stress (ERS; 5). Over the past two decades, ERS has been widely recognized as an important mechanism implicated in the development of several human diseases. Moreover, numerous studies have shown that ERS contributes to VC through various mechanisms in vascular cells. VC research is currently focused on the intima and tunica media; involvement of the former is observed in atherosclerosis and that of the latter in degenerative vascular diseases, such as CKD and diabetes; however, no effective clinical therapy is available for VC.
In this review, we summarize the roles of ERS in the initiation and progression of VC. Furthermore, we discuss the current understanding of how ERS can promote VC through various mechanisms involving vascular smooth muscle cells (VSMCs), vascular endothelial cells (VECs), and immune cells, leading to an increase in the unfolded protein response (UPR). Finally, we evaluate emerging therapeutic strategies to target VC-associated ERS.
The UPR is a highly conserved mechanism mediated by three ER transmembrane sensor proteins: PKR-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1; Figure 1). In the absence of stress, these proteins are maintained in an inactive state by the binding of their luminal domains with the ER intraluminal glucose-regulated protein 78 (GRP78; 6). ERS conditions activate these proteins and increase the binding of GRP78 to misfolded or unfolded proteins. This results in the dissociation of GRP78 from ATF6, IRE1, and PERK and the subsequent activation of UPR signaling (7).
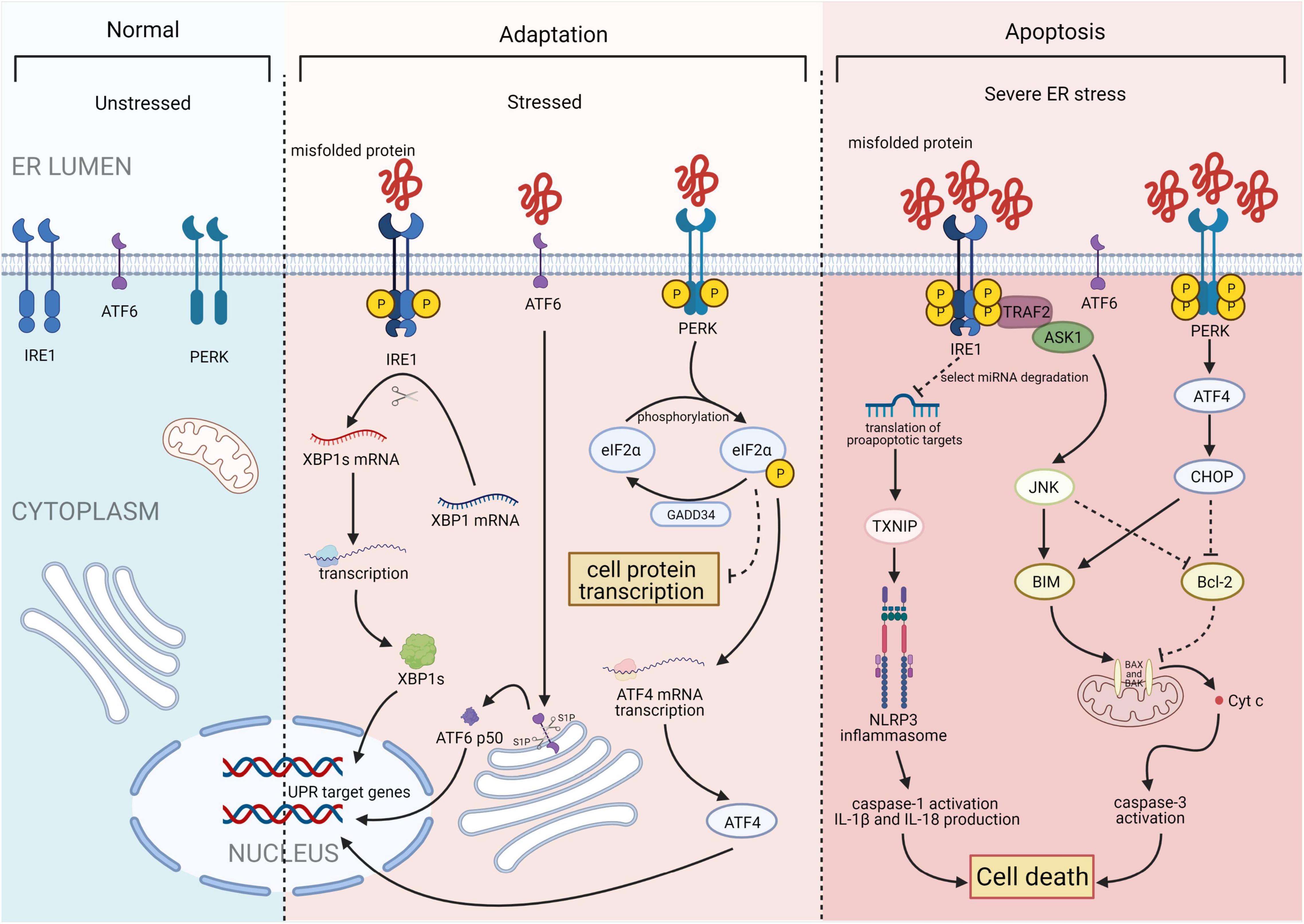
Figure 1. Three major states of endoplasmic reticulum stress (ERS): unstressed, stressed, and severe ERS. In the absence of stress, PKR-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1) are maintained in an inactive state. Upon activation, the IRE1 endoribonuclease domain specifically recognizes and cleaves a 26-base fragment from the X-box binding protein 1 (XBP1) mRNA transcript, resulting in XBP1s. PERK phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF2α), that leads to a downregulation of overall mRNA translation, which reduces the accumulation of unfolded proteins. Phosphorylation of eIF2α also induces the translation of ATF4. ATF6 will be cleaved by two Golgi resident proteases to release its N-terminal transcriptionally active 50-kDa fragment. Thereafter, the 50-kDa N-terminus protein will translocate into the nucleus to activate transcription. Finally, after activation of ATF6, IRE1, and PERK, the unfolded protein response (UPR) is constitutively activated. Under severe ERS, IRE1 forms a complex with tumor necrosis factor receptor-associated factor 2 (TRAF2) and apoptosis signal-regulating kinase 1 (ASK1) to activate c-Jun N-terminal kinase (JNK) and the caspase family. This process activates the NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome. ATF4 and C/EBP homologous protein (CHOP) synergistically induce the expression of multiple genes involved in apoptosis, autophagy, and antioxidant responses, which all lead to cell death. Figures were created using BioRender.com.
Inositol-requiring enzyme 1, the most conserved UPR transducer, is a transmembrane protein responsible for protein kinase and endoribonuclease activity (8, 9). IRE1 homodimerizes and is transphosphorylated upon dissociation from GRP78, facilitating subsequent allosteric activation of its C-terminal endoribonuclease domain (10, 11). This domain specifically recognizes and cleaves a 26-base fragment from the mRNA transcript of the X-box binding protein 1 (XBP1). The 26-base transcript fragment is subsequently translated into spliced XBP1s (XBP1s), which act as a transcription factors by inducing the expression of genes involved in protein folding, autophagy, and ER-associated degradation to help maintain ER homeostasis (12). In addition, IRE1 forms a complex with tumor necrosis factor receptor-associated factor 2 and apoptosis signal-regulating kinase 1, leading to the activation of c-Jun N-terminal kinases and caspase proteases, which promote apoptosis (13).
PKR-like ER kinase, a type I ER-resident protein kinase, is activated through autophosphorylation and homodimerization after dissociating from GRP78 (14). Once activated, PERK phosphorylates the alpha subunit of eukaryotic translation initiation factor (eIF) 2α, leading to downregulation of overall protein synthesis. This reduces the accumulation of unfolded proteins. However, phosphorylation of eIF2α also induces the translation of ATF4 (15), which stimulates the expression of C/EBP homologous protein (CHOP). Subsequently, ATF4 and CHOP synergistically induce the expression of genes involved in apoptosis, autophagy, and antioxidant response (16).
Activating transcription factor 6 is a type II ER transmembrane protein with a basic leucine zipper transcription activation domain at the N-terminus. The ATF6 C-terminus is localized in the ER cavity. ATF6 has multiple GRP78 binding sites and two Golgi positioning signals. GRP78 dissociates from the luminal domain of ATF6 in response to ERS, exposing two Golgi localization signals and causing ATF6 translocation to the Golgi (17). Following translocation, ATF6 is cleaved by site-1 protease and site-2 protease to release its active N-terminal fragment (18). The N-terminus protein then binds to the ATF/cyclic adenosine monophosphate response element and ERS response element and subsequently migrates into the nucleus to activate transcription. Furthermore, to reduce unfolded protein accumulation, ATF6 regulates the transcription of various genes, such as those encoding ER chaperones and protein folding enzymes. This process is mediated by activating specific UPR-related genes (including XBP1) and three main branches of the UPR (including ATF4, XBP1, and ATF6; 12).
Vascular calcification occurs when calcium phosphate crystals (hydroxyapatite) accumulate in the media and/or intima of vessel walls and is strongly correlated with cardiovascular mortality in patients with CKD, atherosclerosis, and diabetes (19, 20). Hence, delaying and reversing VC can theoretically reduce the case fatality rate of these high-risk groups. However, the specific mechanisms underlying VC development and pathogenesis are not fully understood.
Multiple factors contribute to the occurrence of VC. Physiological calcification is a normal process occurring in bones and teeth; however, its occurrence is closely associated with aging, advanced atherosclerosis, diabetes mellitus, and CKD (21). Moreover, numerous risk factors, including calcium and phosphorus metabolism disorder, inflammation, oxidative stress, apoptosis, autophagy, and aging, can also contribute to VC (22).
In the past decade, VC was defined as a passive, degenerative condition. However, more recent studies have suggested that VC is a highly regulated process resulting from multiple factors acting together over a certain period of time. For example, the transformation of smooth muscle cells from a contractile phenotype to an osteoblast-like phenotype, together with extracellular matrix remodeling, apoptosis, and elevated calcium and phosphorus levels, is involved in the development of VC. Under the action of calcification-stimulating factors, the expression levels of osteoblast cell markers, including Runt-related transcription factor 2 (Runx2), become increased, as does the activity of the transcription factor core binding factor alpha1 and the expression of genes containing the core binding factor alpha1 binding site (i.e., osteopontin, osteocalcin, and ALP; 23). By contrast, the expression levels of smooth muscle cell markers, such as SM22-α and smooth muscle α-actin, are decreased, thereby promoting the transformation of smooth muscle cells from a contractile phenotype to an osteoblast cell phenotype (23). Moreover, disorder of calcium (Ca2+) and phosphorus (Pi) metabolism promotes VC. That is, high Ca2+ and Pi concentrations can promote the expression of BMP2, Runx2, MSX2, and osteocalcin in VSMCs, thus promoting osteogenic-like differentiation of VSMCs. In addition, imbalanced Ca2+ and Pi concentrations can cause accumulation of Ca2+ in VSMCs, thereby promoting the release and mineralization of matrix vesicles as well as the apoptosis of VSMCs (24, 25). However, despite the abundance of research on the various risk factors for VC, including hyperphosphatemia, hypercalcemia, oxidative stress, inflammation, and apoptosis, there is a dearth of information regarding the associated regulatory pathways and molecules. Therefore, as a calcium reservoir, the ER has high value in the field of VC research.
Numerous recent studies have shown that ERS can regulate VC through various mechanisms in vascular cells (Table 1; 26).
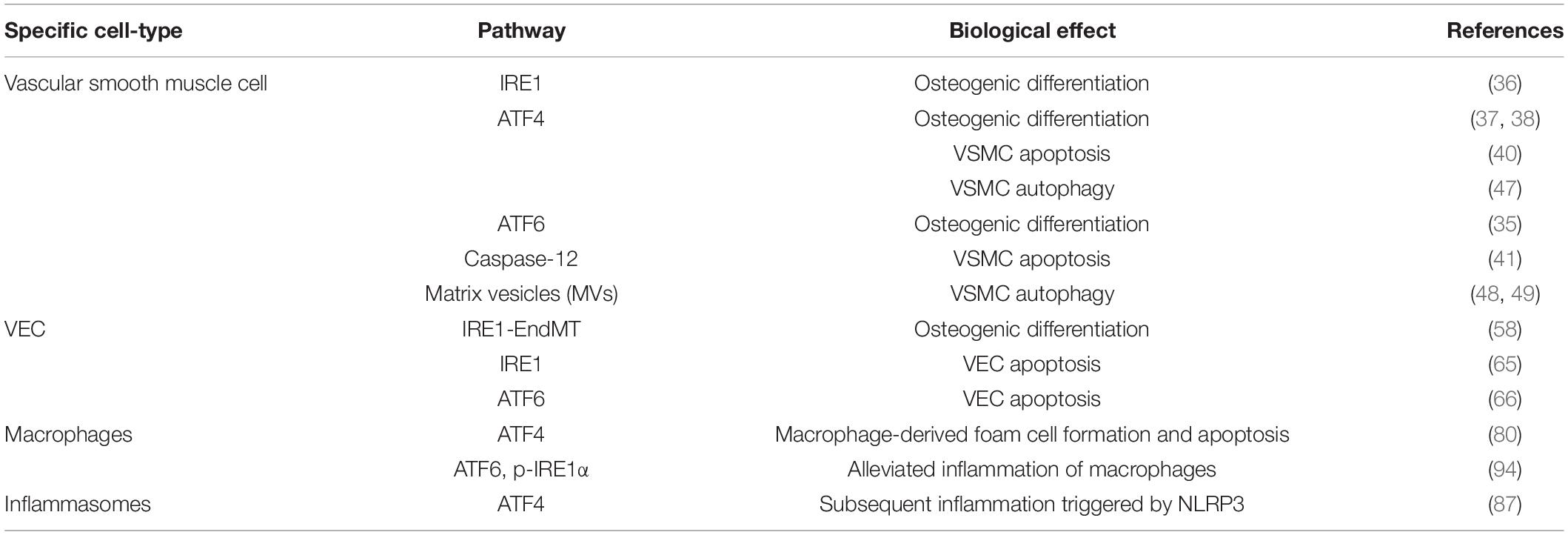
Table 1. Reference table to the specific cell type, signaling pathway leading to ERS, and the mechanism of VCd LDL.
Vascular smooth muscle cells are the most abundant cell type in the arterial vessel wall, and play a key role in regulating atherosclerotic plaque formation and VC (27). In response to ERS, VSMCs differentiate into calcified vascular cells through multiple mechanisms, including osteogenic differentiation (28), apoptosis (29), autophagy induction (30), cellular senescence (31), and oxidative stress (32). Many of these processes also occur with bone formation. Importantly, ERS and the UPR are crucial for bone development. All three branches of the UPR are activated during bone formation to regulate the expression of osteogenic genes. ERS is strongly associated with VC, particularly in VSMCs (Figure 2; 33, 34).
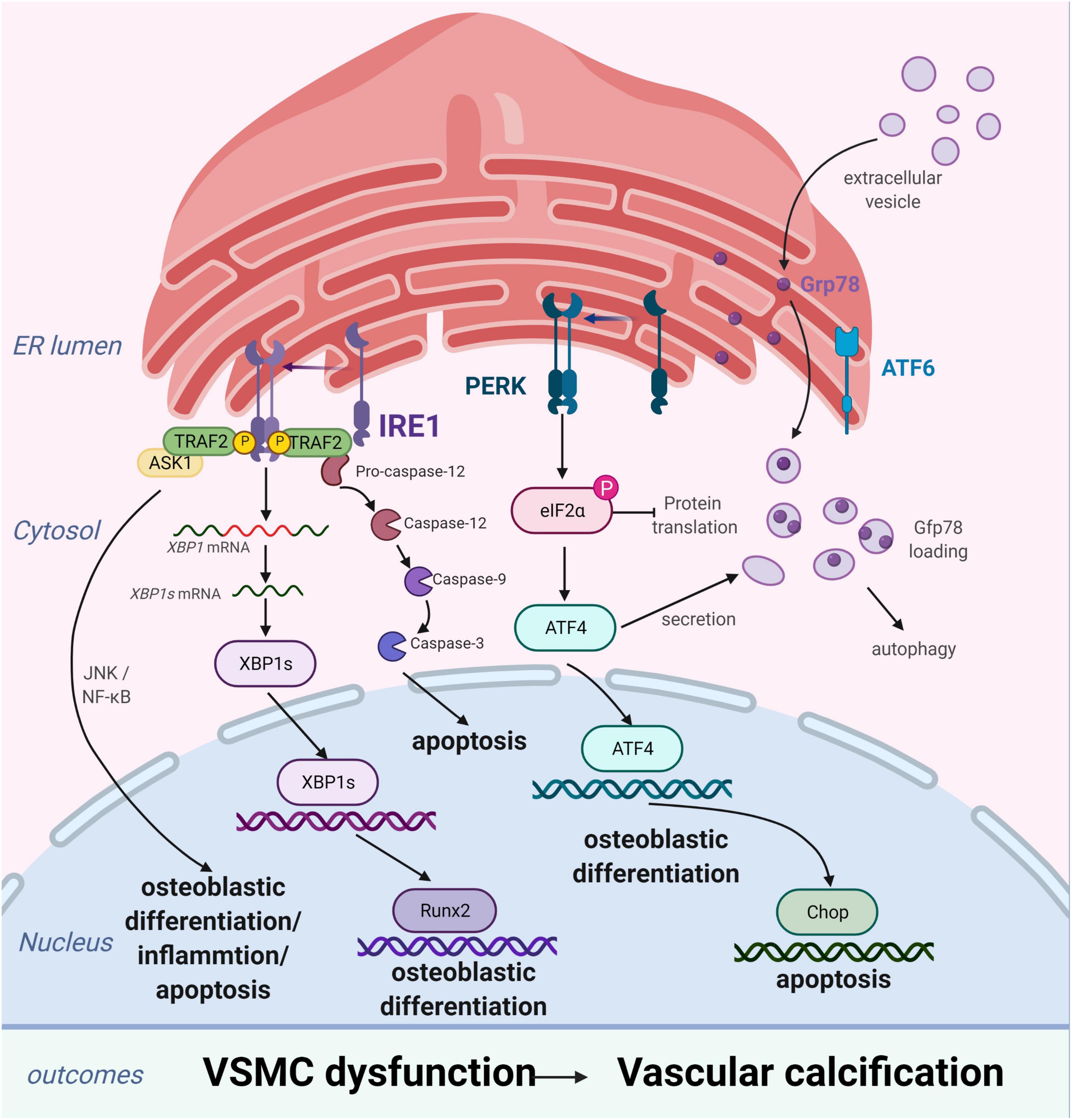
Figure 2. Endoplasmic reticulum stress (ERS) promotes vascular calcification (VC) by inducing the osteogenic differentiation, autophagy, and apoptosis of vascular smooth muscle cells (VSMCs). In response to ERS, VSMCs differentiate into calcifying VSMCs via multiple mechanisms, including osteogenic differentiation, apoptosis, and autophagy. Inositol-requiring enzyme 1 (IRE1) can promote osteoblastic differentiation via nuclear factor kappa B (NF-κB), the IRE1α-XBP1 axis, and RUNX2 signaling pathways and aggravate apoptosis by the IRE1α-ASK-JNK and caspase-12 pathways. PKR-like ER kinase (PERK) induces apoptosis via the PERK-eIF2α-CHOP signaling pathway and osteogenic differentiation via ATF4 activation. Extracellular vesicles, induced by increased expression of Grp78 and ATF4, attract inflammatory cells and induce VC. Figures were created using BioRender.com.
The process of VC involves vascular cells, mainly VSMCs, undergoing osteogenic processes that resemble osteoblast formation, such as osteogenic differentiation, matrix maturation, and matrix mineralization stages. The osteogenic differentiation of VSMCs induced by ERS plays an important role in VC development. This is supported by studies demonstrating that ERS can promote osteogenic differentiation of VSMCs through three pathways, namely IRE1-XBP1, PERK-eIF2α-ATF4, and ATF6. Downregulation of the expression of genes involved in the UPR, including HSPA5, XBP1, ATF4, DDIT3, and ATF6, has been shown to suppress osteogenic gene expression and mineralization of VSMCs (35). Runx2 is a key transcription factor involved in osteoblast differentiation and its expression can be directly regulated by XBP1 (36). ATF4 is a key transcription factor involved in osteoblastogenesis and ERS-induced apoptosis. Notably, ATF4 deficiency has been reported to inhibits osteogenic differentiation and calcification of VSMCs, both in vitro and in vivo (37, 38).
Apoptosis is closely related to calcification. Calcified VSMCs are prone to apoptosis, and apoptosis can in turn promote calcification. VSMC apoptosis provides a suitable microenvironment for the nucleation of hydroxyapatite crystals, which play a key role in the initiation of VC. Moreover, VSMC apoptosis directly affects the morphology and structure of advanced atherosclerosis and plaque stability (39). VSMC apoptosis process can be activated by ERS through three apoptotic pathways: the IRE1α-ASK-JNK, PERK-eIF2α-CHOP signaling, and caspase-12 pathways. CHOP, an ERS-specific transcription factor, is activated by the PERK-eIF2α-ATF4 pathway and induces apoptosis by decreasing the expression of the anti-apoptotic protein, B-cell lymphoma 2. Shiozaki et al. found that transgenic mice with SMC-specific CHOP expression develop severe vascular apoptosis and medial calcification with CKD (40). They further demonstrated that the cyclin-dependent kinase 9 (CDK9)-cyclin T1 complex mediates VC through CHOP induction and phosphorylation-mediated ATF4 activation. Caspase-12, activated exclusively by ERS, activates caspase-9 directly, which subsequently activates caspase-3, resulting in apoptosis (41). Shi et al. found that the metabolic hormone fibroblast growth factor 21 (FGF21) inhibits VC progression by alleviating ERS-mediated apoptosis in rats. The caspase-12 pathway, but not the phospho-JNK-JNK pathway, is involved in FGF21 expression (34). Moreover, expression levels of JNK, which plays a key role in cell differentiation, inflammation, and apoptosis, did not change with ERS-induced apoptosis in a rat model of periodontitis and VC (42). Therefore, the role of the IRE1α-ASK-JNK pathway in VC development needs further investigation.
Autophagy is a catabolic and tightly regulated subcellular process in which long-lived proteins and damaged organelles are degraded by lysosomes. Autophagy can regulate endothelial cell homeostasis, VSMC phenotype transition, and Ca2+ homeostasis in VSMCs (2). Emerging evidence has demonstrated that autophagy directly protects against VC. Morciano et al. found that the autophagy and mitochondrial phagocytosis levels of caveolins increased and that rapamycin can enhance the calcification phenotype by promoting autophagy in vitro (43). In addition, crosstalk among endosomes, dysfunctional mitochondria, autophagic vesicles, and Ca2+- and Pi-enriched matrix vesicles (MVs) may underlie the pathogenesis of VC. Autophagy is an adaptive response that protects against phosphate-induced VSMC calcification by regulating apoptosis and releasing mineralizing MVs from VSMCs (44–46). Moreover, autophagosomes are formed by shedding the double-layer membrane of the ribosome free attachment area of the rough ER and wrapping some cytoplasmic and intracellular organelles, proteins, and other components that need to be degraded. Therefore, the ER plays an indispensable role in autophagy regulation. ERS activates autophagy through the UPR and calcium-mediated signaling cascade pathway. However, the associations of ERS, autophagy, and VC remain poorly understood. Furmanik et al. reported that ERS mediates VSMC calcification via increased release of extracellular vesicles, induced by increased expression of GRP78 and ATF4 (47). However, another study on VC revealed that autophagy results in the release of MVs, which attract inflammatory cells and induce VC (48, 49). Therefore, further studies of the mechanisms linking ERS and autophagy in VSMCs are warranted.
Vascular endothelial cells, which form a biological barrier that controls the passage of immune cells and biomolecules between the vascular walls and mediates physiological functions, are the main components of the vascular intima. During homeostatic conditions, endothelial cells maintain microvascular integrity and exert vasodilatory, anti-inflammatory, and antithrombotic activities (50). However, under various pathological conditions, these biological functions become compromised (51) and may lead to VC through osteogenic differentiation, endothelial microparticles, or cytokines. ERS plays an important role in these changes primarily by inducing osteogenic differentiation and apoptosis of VECs (Figure 3).
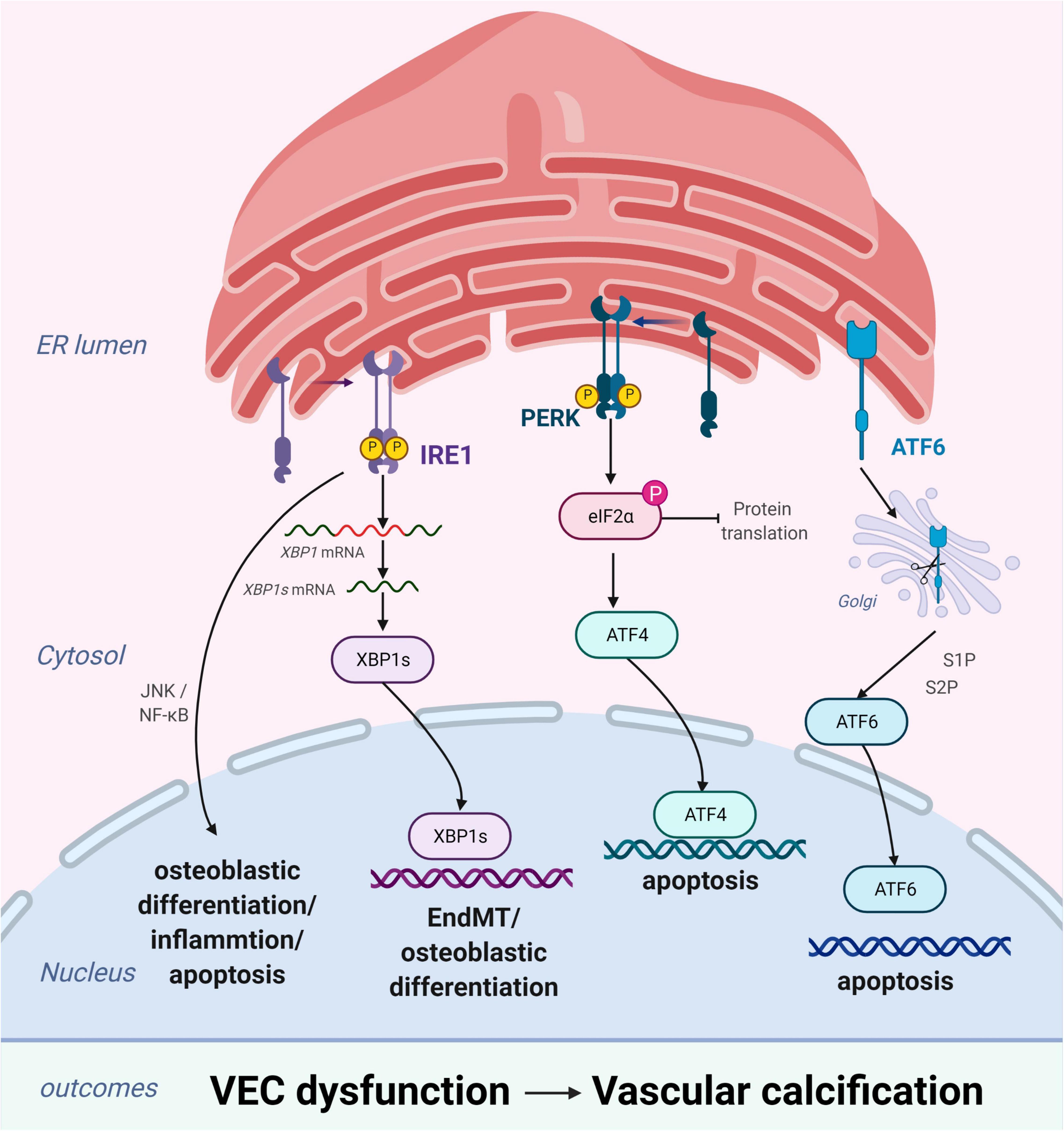
Figure 3. Endoplasmic reticulum stress (ERS) promotes vascular calcification (VC) by inducing the osteogenic differentiation and apoptosis of vascular endothelial cells (VECs). In response to ERS, inositol-requiring enzyme 1 (IRE1) can aggravate EndMT through the IRE1α-XBP1 axis, promote osteoblastic differentiation via the nuclear factor kappa B (NF-κB) signaling pathway and the IRE1α-XBP1 axis, and induce apoptosis via the IRE1α-ASK-JNK pathway. PKR-like ER kinase (PERK) promotes apoptosis via the PERK-eIF2α-ATF4 signaling pathway. High expression of active ATF6 may exacerbate VEC apoptosis through the mitochondrial apoptotic pathway. Figures were created using BioRender.com.
Similar to VSMCs, VECs promote VC by inducing osteogenic differentiation (52). Changes in VEC physiological homeostasis promotes osteoblastic differentiation via the extracellular signal-regulated protein kinase 1/2 and nuclear factor kappa B (NF-κB) signaling pathways, leading to VC (53). Moreover, studies have substantiated that endothelial-to-mesenchymal transition (EndMT) plays a critical role in the osteogenic differentiation of VECs (54–56). EndMT is a process through which endothelial lineage cells lose cell polarity, acquire migratory and aggressive characteristics, and differentiate into mesenchymal stem cells. Moreover, the EndMT gives VECs the potential for osteogenesis differentiation in different physiological states. Yao et al. found that reducing the EndMT by knocking out serine protease or Sox2 in in vitro experiments improves VC (57). EndMT occurs when endothelial cells acquire mesenchymal and stem-cell-like characteristics and is closely related to ERS (58, 59). ERS can aggravate EndMT via the IRE1α-XBP1 axis and thereby contribute to VC (58).
Apoptosis of endothelial cells plays a pivotal role in the development of VC. Increased endothelial cell apoptosis has been observed in the atherosclerosis-prone regions of the vasculature (60) and in the endothelium of human atherosclerotic plaques (61). Under homeostatic conditions, clearance of apoptotic cells occurs in the absence of immune activation and is mediated by various phagocytic cells (7, 62). However, dysregulation of apoptotic cell clearance can lead to secondary necrosis and release of proinflammatory intracellular contents (62). Moreover, this can promote the development of chronic inflammatory diseases, such as atherosclerosis (63). ERS-induced VEC apoptosis plays an important role in the pathogenesis and development of several vascular diseases (64, 65). Recent studies have demonstrated that high expression of active ATF6 may exacerbate VEC apoptosis through the mitochondrial apoptotic pathway (66). Similarly, the activation of IRE1α may enhance VEC apoptosis via the pro-apoptotic molecule JNK and the p38-mitogen-activated protein kinase pathway (65). Furthermore, VEC apoptosis changes the balance between pro-apoptotic and anti-apoptotic proteins of the Bcl-2 family, leading to VC (67).
Vascular calcification is associated with the development of chronic inflammation, as inflammatory cell infiltration has been detected in all stages of VC (68–71). Therefore, in addition to VSMCs and VECs, VC therapeutic strategies targeting inflammation warrant further investigation. ERS is associated with various pathological conditions linked to chronic inflammation (72, 73). Studies indicate that ERS can trigger inflammatory pathways and proinflammatory stimuli, such as Toll-like receptor ligands, reactive oxygen species, and cytokines. These proinflammatory signals can then initiate ERS and result in UPR activation, which further amplifies inflammatory responses (74). Collectively, these processes increase the chances of developing VC (75).
Macrophages are central effectors of innate immunity and play a crucial role in VC development. Studies have shown that activated macrophages contribute to VC by differentiating into osteoclasts (76) and secreting inflammatory factors (77). ERS induces an inflammatory response in macrophages via IRE1α-XBP1 (78) and PERK-ATF4 (79). Yao et al. found that D4F, an apoA-I mimetic peptide, can alleviate macrophage-derived foam cell formation and apoptosis by inhibiting CD36-mediated ox-LDL uptake and subsequent activation of the ERS-CHOP pathway (80). Ren et al. found that intermedin1-53, a cardiovascular protective peptide, protects against homocysteine-promoted atherosclerotic calcification in ApoE–/– mice by inhibiting ERS markers in rat VSMCs and mouse peritoneal macrophages (81). Further investigation is needed to better understand the role of ERS in macrophage-induced VC.
The NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome is an essential component of the innate immune system and can induce the secretion of the proinflammatory cytokine interleukin 1 beta (IL-1β) in a caspase-1-dependent manner (82, 83). Moreover, IL-1β can activate the secretion of the receptor activator of NF-κB ligand, which promotes the formation of osteoclasts, leading to VC (84, 85). ERS stimulates the NLRP3 inflammasome activation through oxidative stress, NF-κB activation, and calcium homeostasis (86). Ren et al. found that intermedin may attenuate the progression of atherosclerotic lesions and plaque susceptibility by inhibiting ERS-CHOP-mediated macrophage apoptosis and subsequent inflammation triggered by NLRP3 both in vivo and in vitro (87). Therefore, further studies are needed to elucidate the mechanisms of ERS-mediated and NLRP3 inflammasome-mediated VC.
It is important to identify effective treatments for the globally prevalent metabolic disorders, such as aging, CKD, diabetes, and atherosclerosis-related VC (88). However, no effective clinical therapy is currently available. ERS is a key feature of metabolic disorders. Hence, herein we aim to identify new therapeutic targets for the treatment of VC by discussing the effects of ERS in different diseases.
Although there have been recent improvements in the systemic management of CKD, cardiovascular disease remains a leading cause of death in patients with CKD (89, 90). VC is a common complication in patients with CKD and is associated with increased cardiovascular disease-related mortality. Recent evidence suggests that ERS is the primary cause of VC in CKD via several pathways. Tumor necrosis factor α induces the PERK-eIF2α-ATF4-CHOP axis of the ERS response, leading to CKD-associated VC (91). Other positive regulators of the PERK-eIF2α-ATF4-CHOP axis of the ERS response in VSMCs include high phosphate levels, oxidized lipids, BMP2, and basic fibroblast growth factor (92, 93). Furthermore, oxysterol accumulation in the ER induces ERS and activates CKD-dependent VC via the PERK-eIF2α-ATF4-CHOP pathway. Oxysterol-mediated ERS can be reduced by ezetimibe-simvastatin combination therapy, thereby attenuating CKD-dependent vascular diseases (94). In addition, ATF4 activity, which is activated by the CDK9-cyclin T1 complex during ERS, can lead to VC in CKD. Moreover, inhibition of the cyclin T1-CDK9-CHOP pathway may decrease ERS-induced CHOP expression and CKD-dependent VC (40).
Atherosclerosis is a chronic inflammatory disease characterized by the progressive accumulation of lipids and plaques in arteries (95). Pathological conditions, such as inflammation, oxidized lipids, and metabolic stress, can activate ERS (96, 97). The UPR is chronically activated in atherosclerotic lesion cells, particularly advanced lesional macrophages, and endothelial cells. Tabas found that ERS is a significant cause of apoptosis of endothelial cells and macrophages in advanced lesions (60). In addition, Oh et al. reported that suppression of macrophage ERS can lead to polarization of differentiated M2 macrophages toward an M1 phenotype and can subsequently suppressed foam cell formation (98). Furthermore, intermedin1-53 protects against homocysteine-related atherosclerotic calcification in Apoe–/– mice by inhibiting ERS (81). These studies indicate that ERS plays a significant role in atherosclerosis and that inhibiting ERS can alleviate the pathological damage associated with atherosclerotic calcification.
Despite improvements in CVD treatment over the past few decades, diabetes remains a significantly independent cardiovascular risk factor (61, 99). Therefore, reducing adverse events caused by diabetes-induced CVD is a clinical challenge (100). Diabetes-related VC presents with disturbed vessel wall homeostasis, endothelial dysfunction, and phenotypic switching of VSMCs (101–103). High glucose levels trigger apoptosis and phenotypic transformation of VSMCs (in the presence of ERS). In addition, compared with continued high glucose conditions, increased glycemic variability is more strongly associated with VC (104). Chronic exposure of VSMCs to high glucose conditions can exacerbate inflammation and calcification through the induction of CD36 scavenger receptors (71). Moreover, CD36 signaling may contribute to diabetic atherosclerosis via ERS induction. Collectively, these findings suggest that ERS crucially contributes to diabetes; however, further investigation is warranted.
Endoplasmic reticulum stress plays a key pathological role in promoting the occurrence and development of CVDs. Over the past few decades, research has focused on the signaling proteins involved in ERS, resulting in the development of an increasing array of drugs (Table 2). This section aims to explain the fundamentals, value, and limitations of existing drugs targeting ERS.
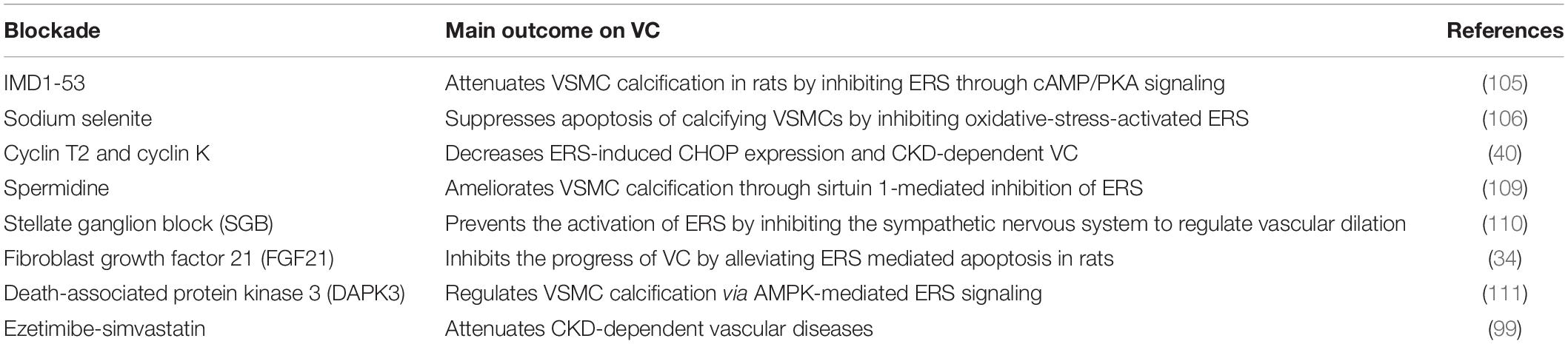
Table 2. Table summarizing ERS blockade attempt in diseases and their main outcome on VC.
Intermedin1-53, a paracrine/autocrine peptide in the vasculature, attenuates VSMC calcification in rats by inhibiting ERS through cyclic adenosine monophosphate-protein kinase A signaling (105). Sodium selenite may suppress apoptosis of calcifying VSMCs, in part, by inhibiting oxidative-stress-activated ERS (106). The CDK9-cyclin T1 complex, an essential component in ERS, mediates pro-apoptotic CHOP expression and VC by activating ATF4. Cyclin T2 and cyclin K inhibit CHOP induction by competitively binding CDK9. Hence, inhibition of the cyclin T1/CDK9-CHOP pathway may be a potential therapeutic strategy for VC treatment (107). Spermidine, an endogenously synthesized polyamine, has been shown to protect against CVD and extend lifespan (108). VSMC calcification has recently been shown to be ameliorated through sirtuin 1-mediated inhibition of ERS (109). Stellate ganglion block, which regulates vascular dilation through sympathetic blockade, is used to treat several CVDs. Recently, stellate ganglion block has been shown to prevent the activation of ERS by inhibiting sympathetic nervous activity (110). For the first time, FGF21 has been shown to reduce ERS-mediated VC progression in rats. Some studies suggest that FGF21 has the potential to regulate many metabolic diseases and CVDs because of its pleiotropic biological effects. Thus, FGF21 is a promising new therapeutic target for preventing and treating VC (34). Death-associated protein kinase 3 is involved in hypertension-related vascular remodeling and has been shown to regulate VSMC calcification via AMPK-mediated ERS signaling (111). Although these studies provide important translational insights into ERS-targeted prevention of VC, they present only basic experiments and therefore, clinical studies are needed. Notably, inappropriate alteration of ERS may also cause harm, as it is a key mechanism in body maintenance. Hence, the safety of ERS inhibitors requires further evaluation prior to their clinical application.
Despite various clinical prevention strategies, CVD remains a common complication of aging, atherosclerosis, hypertension, diabetes, and CKD. Accumulating evidence indicates that ERS can regulate the development of VC by promoting osteogenic transformation, inflammation, autophagy, and apoptosis, and by increasing the UPR. Although these studies comprehensively demonstrate that ERS inhibitors can ameliorate VC, they all have certain limitations. That is, most of these studies conducted basic experiments that did not fully nor accurately reflect the pathological changes in human diseases. Additionally, considering that the ERS-UPR pathway is ubiquitous in humans and sensitive to external stimuli, the repression of excessive ERS can promote cellular damage and lead to increased disease progression. Therefore, clinical trials are needed to validate the results of these studies; this may aid in the development of new therapies for VC.
ZR, LX, YDZ, ZW, YZ, MC, ND, ZC, and FL: conceptualization. All authors writing-original draft preparation, editing, and revising. FL and ZC: supervision.
This work was supported by funding from the National Natural Science Foundation of China (82170377 and 81974034).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. (2011) 109:697–711. doi: 10.1161/CIRCRESAHA.110.234914
2. Phadwal K, Feng D, Zhu D, MacRae VE. Autophagy as a novel therapeutic target in vascular calcification. Pharmacol Ther. (2020) 206:107430. doi: 10.1016/j.pharmthera.2019.107430
3. Amann K. Media calcification and intima calcification are distinct entities in chronic kidney disease. Clin J Am Soc Nephrol. (2008) 3:1599–605. doi: 10.2215/CJN.02120508
4. Chen Y, Zhao X, Wu H. Arterial stiffness: a focus on vascular calcification and its link to bone mineralization. Arterioscler Thromb Vasc Biol. (2020) 40:1078–93. doi: 10.1161/ATVBAHA.120.313131
5. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. (2016) 529:7586. doi: 10.1038/nature17041
6. Braakman I, Hebert DN. Protein folding in the endoplasmic reticulum. Cold Spring Harb Perspect Biol. (2013) 5:a013201. doi: 10.1101/cshperspect.a013201
7. Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. (2007) 18:716–31. doi: 10.1016/j.semcdb.2007.09.003
8. Hassler J, Cao SS, Kaufman RJ. IRE1, a double-edged sword in pre-miRNA slicing and cell death. Dev Cell. (2012) 23:921–3. doi: 10.1016/j.devcel.2012.10.025
9. Shamu CE, Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. (1996) 15:3028–39.
10. Tringali G, Mancuso C, Mirtella A, Pozzoli G, Parente L, Preziosi P, et al. Evidence for the neuronal origin of immunoreactive interleukin-1 beta released by rat hypothalamic explants. Neurosci Lett. (1996) 219:143–6. doi: 10.1016/s0304-3940(96)13195-5
11. Korennykh AV, Korostelev AA, Egea PF, Finer-Moore J, Stroud RM, Zhang C, et al. Structural and functional basis for RNA cleavage by Ire1. BMC Biol. (2011) 9:47. doi: 10.1186/1741-7007-9-47
12. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. (2011) 334:1081–6. doi: 10.1126/science.1209038
13. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. (2013) 1833:3460–70. doi: 10.1016/j.bbamcr.2013.06.028
14. Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. (2002) 3:99–111. doi: 10.1016/s1534-5807(02)00203-4
15. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. (2012) 13:89–102. doi: 10.1038/nrm3270
16. Brewer JW. Regulatory crosstalk within the mammalian unfolded protein response. Cell Mol Life Sci. (2014) 71:1067–79. doi: 10.1007/s00018-013-1490-2
17. Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J Biol Chem. (2002) 277:13045–52. doi: 10.1074/jbc.M110636200
18. Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. (2000) 6:1355–64. doi: 10.1016/s1097-2765(00)00133-7
19. Duer MJ, Friscić T, Proudfoot D, Reid DG, Schoppet M, Shanahan CM, et al. Mineral surface in calcified plaque is like that of bone: further evidence for regulated mineralization. Arterioscler Thromb Vasc Biol. (2008) 28:2030–4. doi: 10.1161/atvbaha.108.172387
20. Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manage. (2009) 5:185–97. doi: 10.2147/vhrm.s4822
21. Jing L, Li L, Sun Z, Bao Z, Shao C, Yan J, et al. Role of matrix vesicles in bone-vascular cross-talk. J Cardiovasc Pharmacol. (2019) 74:372–8. doi: 10.1097/FJC.0000000000000720
22. Pescatore LA, Gamarra LF, Liberman M. Multifaceted mechanisms of vascular calcification in aging. Arterioscler Thromb Vasc Biol. (2019) 39:1307–16. doi: 10.1161/ATVBAHA.118.311576
23. Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. (2001) 89:1147–54. doi: 10.1161/hh2401.101070
24. Abbasian N. Vascular calcification mechanisms: updates and renewed insight into signaling pathways involved in high phosphate-mediated vascular smooth muscle cell calcification. Biomedicines. (2021) 9:804. doi: 10.3390/biomedicines9070804
25. Li T, Yu H, Zhang D, Feng T, Miao M, Li J, et al. Matrix vesicles as a therapeutic target for vascular calcification. Front Cell Dev Biol. (2022) 10:825622. doi: 10.3389/fcell.2022.825622
26. Lee SJ, Lee IK, Jeon JH. Vascular calcification-new insights into its mechanism. Int J Mol Sci. (2020) 21:2685. doi: 10.3390/ijms21082685
27. Ma WQ, Sun XJ, Zhu Y, Liu NF. PDK4 promotes vascular calcification by interfering with autophagic activity and metabolic reprogramming. Cell Death Dis. (2020) 11:991. doi: 10.1038/s41419-020-03162-w
28. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
29. Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. (2018) 114:622–34. doi: 10.1093/cvr/cvy007
30. Dong Y, Chen H, Gao J, Liu Y, Li J, Wang J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J Mol Cell Cardiol. (2019) 136:27–41. doi: 10.1016/j.yjmcc.2019.09.001
31. Shi J, Yang Y, Cheng A, Xu G, He F. Metabolism of vascular smooth muscle cells in vascular diseases. Am J Physiol Heart Circ Physiol. (2020) 319:H613–31. doi: 10.1152/ajpheart.00220.2020
32. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:727–44. doi: 10.1038/s41569-019-0227-9
33. Dong Q, Chen Y, Liu W, Liu X, Chen A, Yang X, et al. 25-Hydroxycholesterol promotes vascular calcification via activation of endoplasmic reticulum stress. Eur J Pharmacol. (2020) 880:173165. doi: 10.1016/j.ejphar.2020.173165
34. Shi Y, Wang S, Peng H, Lv Y, Li W, Cheng S, et al. Fibroblast growth factor 21 attenuates vascular calcification by alleviating endoplasmic reticulum stress mediated apoptosis in rats. Int J Biol Sci. (2019) 15:138–47. doi: 10.7150/ijbs.28873
35. Duangchan T, Tawonsawatruk T, Angsanuntsukh C, Trachoo O, Hongeng S, Kitiyanant N, et al. Amelioration of osteogenesis in iPSC-derived mesenchymal stem cells from osteogenesis imperfecta patients by endoplasmic reticulum stress inhibitor. Life Sci. (2021) 278:119628. doi: 10.1016/j.lfs.2021.119628
36. Liberman M, Johnson RC, Handy DE, Loscalzo J, Leopold JA. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem Biophys Res Commun. (2011) 413:436–41. doi: 10.1016/j.bbrc.2011.08.114
37. Masuda M, Miyazaki-Anzai S, Keenan AL, Shiozaki Y, Okamura K, Chick WS, et al. Activating transcription factor-4 promotes mineralization in vascular smooth muscle cells. JCI Insight. (2016) 1:e88646. doi: 10.1172/jci.insight.88646
38. Duan XH, Chang JR, Zhang J, Zhang BH, Li YL, Teng X, et al. Activating transcription factor 4 is involved in endoplasmic reticulum stress-mediated apoptosis contributing to vascular calcification. Apoptosis. (2013) 18:1132–44. doi: 10.1007/s10495-013-0861-3
39. Zeini M, Lopez-Fontal R, Traves PG, Benito G, Hortelano S. Differential sensitivity to apoptosis among the cells that contribute to the atherosclerotic disease. Biochem Biophys Res Commun. (2007) 363:444–50. doi: 10.1016/j.bbrc.2007.09.004
40. Shiozaki Y, Okamura K, Kohno S, Keenan AL, Williams K, Zhao X, et al. The CDK9-cyclin T1 complex mediates saturated fatty acid-induced vascular calcification by inducing expression of the transcription factor CHOP. J Biol Chem. (2018) 293:17008–20. doi: 10.1074/jbc.RA118.004706
41. Garcia de la Cadena S, Massieu L. Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase-12. Apoptosis. (2016) 21:763–77. doi: 10.1007/s10495-016-1247-0
42. Song X, Li J, Jiao M, Chen Y, Pan K. Effect of endoplasmic reticulum stress-induced apoptosis in the role of periodontitis on vascular calcification in a rat model. J Mol Histol. (2021) 52:1097–104. doi: 10.1007/s10735-021-10015-z
43. Morciano G, Patergnani S, Pedriali G, Cimaglia P, Mikus E, Calvi S, et al. Impairment of mitophagy and autophagy accompanies calcific aortic valve stenosis favoring cell death and the severity of disease. Cardiovasc Res. (2021). [Epub ahead of print]. doi: 10.1093/cvr/cvab267
44. Pi S, Mao L, Chen J, Shi H, Liu Y, Guo X, et al. The P2RY12 receptor promotes VSMC-derived foam cell formation by inhibiting autophagy in advanced atherosclerosis. Autophagy. (2021) 17:980–1000. doi: 10.1080/15548627.2020.1741202
45. Smith M, Wilkinson S. ER homeostasis and autophagy. Essays Biochem. (2017) 61:625–35. doi: 10.1042/EBC20170092
46. Zhao Y, Zhao MM, Cai Y, Zheng MF, Sun WL, Zhang SY, et al. Mammalian target of rapamycin signaling inhibition ameliorates vascular calcification via Klotho upregulation. Kidney Int. (2015) 88:711–21. doi: 10.1038/ki.2015.160
47. Furmanik M, van Gorp R, Whitehead M, Ahmad S, Bordoloi J, Kapustin A, et al. Endoplasmic reticulum stress mediates vascular smooth muscle cell calcification via increased release of Grp78 (Glucose-regulated protein, 78 kDa)-loaded extracellular vesicles. Arterioscler Thromb Vasc Biol. (2021) 41:898–914. doi: 10.1161/atvbaha.120.315506
48. Zazzeroni L, Faggioli G, Pasquinelli G. Mechanisms of arterial calcification: the role of matrix vesicles. Eur J Vasc Endovasc Surg. (2018) 55:425–32. doi: 10.1016/j.ejvs.2017.12.009
49. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. (2011) 147:728–41. doi: 10.1016/j.cell.2011.10.026
50. Guglielmetti G, Quaglia M, Sainaghi PP, Castello LM, Vaschetto R, Pirisi M, et al. “War to the knife” against thromboinflammation to protect endothelial function of COVID-19 patients. Crit Care. (2020) 24:365. doi: 10.1186/s13054-020-03060-9
51. Werner N, Wassmann S, Ahlers P, Kosiol S, Nickenig G. Circulating CD31+/annexin V+ apoptotic microparticles correlate with coronary endothelial function in patients with coronary artery disease. Arterioscler Thromb Vasc Biol. (2006) 26:112–6. doi: 10.1161/01.ATV.0000191634.13057.15
52. Cianciolo G, Capelli I, Cappuccilli M, Scrivo A, Donadei C, Marchetti A, et al. Is chronic kidney disease-mineral and bone disorder associated with the presence of endothelial progenitor cells with a calcifying phenotype? Clin Kidney J. (2017) 10:389–96. doi: 10.1093/ckj/sfw145
53. Cheng ZY, Ye T, Ling QY, Wu T, Wu GY, Zong GJ. Parathyroid hormone promotes osteoblastic differentiation of endothelial cells via the extracellular signal-regulated protein kinase 1/2 and nuclear factor-kappaB signaling pathways. Exp Ther Med. (2018) 15:1754–60. doi: 10.3892/etm.2017.5545
54. Cheng SL, Shao JS, Behrmann A, Krchma K, Towler DA. Dkk1 and MSX2-Wnt7b signaling reciprocally regulate the endothelial-mesenchymal transition in aortic endothelial cells. Arterioscler Thromb Vasc Biol. (2013) 33:1679–89. doi: 10.1161/ATVBAHA.113.300647
55. Bostrom KI, Yao J, Guihard PJ, Blazquez-Medela AM, Yao Y. Endothelial-mesenchymal transition in atherosclerotic lesion calcification. Atherosclerosis. (2016) 253:124–7. doi: 10.1016/j.atherosclerosis.2016.08.046
56. Sanchez-Duffhues G, Garcia de Vinuesa A, van de Pol V, Geerts ME, de Vries MR, Janson SG, et al. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J Pathol. (2019) 247:333–46. doi: 10.1002/path.5193
57. Yao J, Guihard PJ, Blazquez-Medela AM, Guo Y, Moon JH, Jumabay M, et al. Serine protease activation essential for endothelial-mesenchymal transition in vascular calcification. Circ Res. (2015) 117:758–69. doi: 10.1161/CIRCRESAHA.115.306751
58. Luo R, Li L, Liu X, Yuan Y, Zhu W, Li L, et al. Mesenchymal stem cells alleviate palmitic acid-induced endothelial-to-mesenchymal transition by suppressing endoplasmic reticulum stress. Am J Physiol Endocrinol Metab. (2020) 319:E961–80. doi: 10.1152/ajpendo.00155.2020
59. Ying R, Wang XQ, Yang Y, Gu ZJ, Mai JT, Qiu Q, et al. Hydrogen sulfide suppresses endoplasmic reticulum stress-induced endothelial-to-mesenchymal transition through Src pathway. Life Sci. (2016) 144:208–17. doi: 10.1016/j.lfs.2015.11.025
60. Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. (2010) 107:839–50. doi: 10.1161/circresaha.110.224766
61. Malmberg K, Yusuf S, Gerstein HC, Brown J, Zhao F, Hunt D, et al. Impact of diabetes on long-term prognosis in patients with unstable angina and non-Q-wave myocardial infarction: results of the OASIS (organization to assess strategies for ischemic syndromes) registry. Circulation. (2000) 102:1014–9. doi: 10.1161/01.cir.102.9.1014
62. Poon IK, Hulett MD, Parish CR. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ. (2010) 17:381–97. doi: 10.1038/cdd.2009.195
63. Szondy Z, Garabuczi E, Joos G, Tsay GJ, Sarang Z. Impaired clearance of apoptotic cells in chronic inflammatory diseases: therapeutic implications. Front Immunol. (2014) 5:354. doi: 10.3389/fimmu.2014.00354
64. Toya SP, Malik AB. Role of endothelial injury in disease mechanisms and contribution of progenitor cells in mediating endothelial repair. Immunobiology. (2012) 217:569–80. doi: 10.1016/j.imbio.2011.03.006
65. Legeay S, Fautrat P, Norman JB, Antonova G, Kennard S, Bruder-Nascimento T, et al. Selective deficiency in endothelial PTP1B protects from diabetes and endoplasmic reticulum stress-associated endothelial dysfunction via preventing endothelial cell apoptosis. Biomed Pharmacother. (2020) 127:110200. doi: 10.1016/j.biopha.2020.110200
66. Huang J, Wan L, Lu H, Li X. High expression of active ATF6 aggravates endoplasmic reticulum stress-induced vascular endothelial cell apoptosis through the mitochondrial apoptotic pathway. Mol Med Rep. (2018) 17:6483–9. doi: 10.3892/mmr.2018.8658
67. Lu CL, Liao MT, Hou YC, Fang YW, Zheng CM, Liu WC, et al. Sirtuin-1 and its relevance in vascular calcification. Int J Mol Sci. (2020) 21:1593. doi: 10.3390/ijms21051593
68. Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. (2014) 34:715–23. doi: 10.1161/ATVBAHA.113.302070
69. Goody PR, Hosen MR, Christmann D, Niepmann ST, Zietzer A, Adam M, et al. Aortic valve stenosis: from basic mechanisms to novel therapeutic targets. Arterioscler Thromb Vasc Biol. (2020) 40:885–900. doi: 10.1161/ATVBAHA.119.313067
70. Chernomordik F, Cercek B, Lio WM, Mihailovic PM, Yano J, Herscovici R, et al. The role of T cells reactive to the cathelicidin antimicrobial peptide LL-37 in acute coronary syndrome and plaque calcification. Front Immunol. (2020) 11:575577. doi: 10.3389/fimmu.2020.575577
71. Navas-Madronal M, Castelblanco E, Camacho M, Consegal M, Ramirez-Morros A, Sarrias MR, et al. Role of the scavenger receptor CD36 in accelerated diabetic atherosclerosis. Int J Mol Sci. (2020) 21:7360. doi: 10.3390/ijms21197360
72. Bettigole SE, Glimcher LH. Endoplasmic reticulum stress in immunity. Annu Rev Immunol. (2015) 33:107–38. doi: 10.1146/annurev-immunol-032414-112116
73. Di Conza G, Ho PC. ER stress responses: an emerging modulator for innate immunity. Cells. (2020) 9:695. doi: 10.3390/cells9030695
74. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. (2016) 16:469–84. doi: 10.1038/nri.2016.62
75. Bailey KA, Haj FG, Simon SI, Passerini AG. Atherosusceptible shear stress activates endoplasmic reticulum stress to promote endothelial inflammation. Sci Rep. (2017) 7:8196. doi: 10.1038/s41598-017-08417-9
76. Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, et al. Runx2-upregulated receptor activator of nuclear factor kappaB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol. (2011) 31:1387–96. doi: 10.1161/ATVBAHA.110.222547
77. Buendia P, Montes de Oca A, Madueno JA, Merino A, Martin-Malo A, Aljama P, et al. Endothelial microparticles mediate inflammation-induced vascular calcification. FASEB J. (2015) 29:173–81. doi: 10.1096/fj.14-249706
78. Kim S, Joe Y, Kim HJ, Kim YS, Jeong SO, Pae HO, et al. Endoplasmic reticulum stress-induced IRE1alpha activation mediates cross-talk of GSK-3beta and XBP-1 to regulate inflammatory cytokine production. J Immunol. (2015) 194:4498–506. doi: 10.4049/jimmunol.1401399
79. Iwasaki Y, Suganami T, Hachiya R, Shirakawa I, Kim-Saijo M, Tanaka M, et al. Activating transcription factor 4 links metabolic stress to interleukin-6 expression in macrophages. Diabetes. (2014) 63:152–61. doi: 10.2337/db13-0757
80. Yao S, Tian H, Miao C, Zhang DW, Zhao L, Li Y, et al. D4F alleviates macrophage-derived foam cell apoptosis by inhibiting CD36 expression and ER stress-CHOP pathway. J Lipid Res. (2015) 56:836–47. doi: 10.1194/jlr.M055400
81. Ren JL, Hou YL, Ni XQ, Zhu Q, Chen Y, Zhang LS, et al. Intermedin1-53 ameliorates homocysteine-promoted atherosclerotic calcification by inhibiting endoplasmic reticulum stress. J Cardiovasc Pharmacol Ther. (2020) 25:251–64. doi: 10.1177/1074248419885633
82. Chen TC, Yen CK, Lu YC, Shi CS, Hsieh RZ, Chang SF, et al. The antagonism of 6-shogaol in high-glucose-activated NLRP3 inflammasome and consequent calcification of human artery smooth muscle cells. Cell Biosci. (2020) 10:5. doi: 10.1186/s13578-019-0372-1
83. Schuchardt M, Herrmann J, Henkel C, Babic M, van der Giet M, Tolle M. Long-term treatment of azathioprine in rats induces vessel mineralization. Biomedicines. (2021) 9:327. doi: 10.3390/biomedicines9030327
84. Yu C, Zhang C, Kuang Z, Zheng Q. The role of NLRP3 inflammasome activities in bone diseases and vascular calcification. Inflammation. (2021) 44:434–49. doi: 10.1007/s10753-020-01357-z
85. Zhang X, Li Y, Yang P, Liu X, Lu L, Chen Y, et al. Trimethylamine-N-Oxide promotes vascular calcification through activation of NLRP3 (Nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome and NF-kappaB (Nuclear Factor kappaB) signals. Arterioscler Thromb Vasc Biol. (2020) 40:751–65. doi: 10.1161/ATVBAHA.119.313414
86. Li W, Cao T, Luo C, Cai J, Zhou X, Xiao X, et al. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl Microbiol Biotechnol. (2020) 104:6129–40. doi: 10.1007/s00253-020-10614-y
87. Ren JL, Chen Y, Zhang LS, Zhang YR, Liu SM, Yu YR, et al. Intermedin1-53 attenuates atherosclerotic plaque vulnerability by inhibiting CHOP-mediated apoptosis and inflammasome in macrophages. Cell Death Dis. (2021) 12:436. doi: 10.1038/s41419-021-03712-w
88. Poterucha TJ, Goldhaber SZ. Warfarin and vascular calcification. Am J Med. (2016) 129:635.e1–4. doi: 10.1016/j.amjmed.2015.11.032
89. Parikh NI, Hwang SJ, Larson MG, Levy D, Fox CS. Chronic kidney disease as a predictor of cardiovascular disease (from the Framingham heart study). Am J Cardiol. (2008) 102:47–53. doi: 10.1016/j.amjcard.2008.02.095
90. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. (2004) 351:1296–305. doi: 10.1056/NEJMoa041031
91. Masuda M, Miyazaki-Anzai S, Levi M, Ting TC, Miyazaki M. PERK-eIF2α-ATF4-CHOP signaling contributes to TNFα-induced vascular calcification. J Am Heart Assoc. (2013) 2:e000238. doi: 10.1161/jaha.113.000238
92. Masuda M, Ting TC, Levi M, Saunders SJ, Miyazaki-Anzai S, Miyazaki M. Activating transcription factor 4 regulates stearate-induced vascular calcification. J Lipid Res. (2012) 53:1543–52. doi: 10.1194/jlr.M025981
93. Ting TC, Miyazaki-Anzai S, Masuda M, Levi M, Demer LL, Tintut Y, et al. Increased lipogenesis and stearate accelerate vascular calcification in calcifying vascular cells. J Biol Chem. (2011) 286:23938–49. doi: 10.1074/jbc.M111.237065
94. Miyazaki-Anzai S, Masuda M, Demos-Davies KM, Keenan AL, Saunders SJ, Masuda R, et al. Endoplasmic reticulum stress effector CCAAT/enhancer-binding protein homologous protein (CHOP) regulates chronic kidney disease-induced vascular calcification. J Am Heart Assoc. (2014) 3:e000949. doi: 10.1161/jaha.114.000949
95. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. (2011) 17:1410–22. doi: 10.1038/nm.2538
96. Ivanova EA, Orekhov AN. The role of endoplasmic reticulum stress and unfolded protein response in atherosclerosis. Int J Mol Sci. (2016) 17:193. doi: 10.3390/ijms17020193
97. Zhang C, Syed TW, Liu R, Yu J. Role of endoplasmic reticulum stress, autophagy, and inflammation in cardiovascular disease. Front Cardiovasc Med. (2017) 4:29. doi: 10.3389/fcvm.2017.00029
98. Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, et al. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J Biol Chem. (2012) 287:11629–41. doi: 10.1074/jbc.M111.338673
99. Mukamal KJ, Nesto RW, Cohen MC, Muller JE, Maclure M, Sherwood JB, et al. Impact of diabetes on long-term survival after acute myocardial infarction: comparability of risk with prior myocardial infarction. Diabetes Care. (2001) 24:1422–7. doi: 10.2337/diacare.24.8.1422
100. Barrett H, O’Keeffe M, Kavanagh E, Walsh M, O’Connor EM. Is matrix Gla protein associated with vascular calcification? a systematic review. Nutrients. (2018) 10:415. doi: 10.3390/nu10040415
101. Zhu L, Liu J, Gao C, Zhao W, Que J, Wang X, et al. Comparison of coronary plaque, coronary artery calcification and major adverse cardiac events in Chinese outpatients with and without type 2 diabetes. Springerplus. (2016) 5:1678. doi: 10.1186/s40064-016-3373-0
102. Casella S, Bielli A, Mauriello A, Orlandi A. Molecular pathways regulating macrovascular pathology and vascular smooth muscle cells phenotype in type 2 diabetes. Int J Mol Sci. (2015) 16:24353–68. doi: 10.3390/ijms161024353
103. Dhananjayan R, Koundinya KS, Malati T, Kutala VK. Endothelial dysfunction in type 2 diabetes mellitus. Indian J Clin Biochem. (2016) 31:372–9. doi: 10.1007/s12291-015-0516-y
104. Zhang L, Sun H, Liu S, Gao J, Xia J. Glycemic variability is associated with vascular calcification by the markers of endoplasmic reticulum stress-related apoptosis, Wnt1, galectin-3 and BMP-2. Diabetol Metab Syndr. (2019) 11:67. doi: 10.1186/s13098-019-0464-4
105. Chang JR, Duan XH, Zhang BH, Teng X, Zhou YB, Liu Y, et al. Intermedin1-53 attenuates vascular smooth muscle cell calcification by inhibiting endoplasmic reticulum stress via cyclic adenosine monophosphate/protein kinase A pathway. Exp Biol Med (Maywood). (2013) 238:1136–46. doi: 10.1177/1535370213502619
106. Liu H, Li X, Qin F, Huang K. Selenium suppresses oxidative-stress-enhanced vascular smooth muscle cell calcification by inhibiting the activation of the PI3K/AKT and ERK signaling pathways and endoplasmic reticulum stress. J Biol Inorg Chem. (2014) 19:375–88. doi: 10.1007/s00775-013-1078-1
107. Yao S, Yang N, Song G, Sang H, Tian H, Zhang Y, et al. Induction of mildly oxidized low-density lipoprotein on endoplasmic reticulum stress in macrophages and its signaling pathway. Chin J Arterioscl. (2011) 19:242. doi: 10.1016/j.freeradbiomed.2017.02.006
108. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. (2016) 22:1428–38. doi: 10.1038/nm.4222
109. Liu X, Chen A, Liang Q, Yang X, Dong Q, Fu M, et al. Spermidine inhibits vascular calcification in chronic kidney disease through modulation of SIRT1 signaling pathway. Aging Cell. (2021) 20:e13377. doi: 10.1111/acel.13377
110. Hao W, Yang R, Yang Y, Jin S, Li Y, Yuan F, et al. Stellate ganglion block ameliorates vascular calcification by inhibiting endoplasmic reticulum stress. Life Sci. (2018) 193:1–8. doi: 10.1016/j.lfs.2017.12.002
Keywords: vascular calcification, endoplasmic reticulum stress, unfolded protein response, chronic kidney disease, atherosclerosis, diabetes
Citation: Rao Z, Zheng Y, Xu L, Wang Z, Zhou Y, Chen M, Dong N, Cai Z and Li F (2022) Endoplasmic Reticulum Stress and Pathogenesis of Vascular Calcification. Front. Cardiovasc. Med. 9:918056. doi: 10.3389/fcvm.2022.918056
Received: 12 April 2022; Accepted: 30 May 2022;
Published: 16 June 2022.
Edited by:
Wen-Jun Tu, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Florent Allagnat, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandCopyright © 2022 Rao, Zheng, Xu, Wang, Zhou, Chen, Dong, Cai and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhejun Cai, Y2FpemhlanVuQHpqdS5lZHUuY24=; Fei Li, bGlmZWlfdW5pb25AaHVzdC5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.