 Hiroyuki Yamamoto
Hiroyuki Yamamoto Katsuya Hashimoto1
Katsuya Hashimoto1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cardiovasc. Med. , 07 July 2022
Sec. General Cardiovascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.913724
This article is part of the Research Topic Case Reports in General Cardiovascular Medicine: 2022 View all 35 articles
Eosinophilic granulomatosis with polyangiitis (EGPA) is a systemic vasculitis involving small-to-medium-sized vessels characterized by asthma, vasculitis, and peripheral eosinophilia. EGPA-associated eosinophilic myocarditis (EM) occurs rarely, yet can be fatal if left untreated. Moreover, the accurate diagnosis of EGPA-associated EM without vasculitis is exceptionally difficult because of the overlapping features with EM of other causes. We report a case of probable EGPA with subclinical neurological involvement that presented with acute EM. The constellation of peripheral eosinophilia, left ventricular dysfunction, and normal epicardial coronary arteries raised suspicion of acute EM, which was confirmed by cardiac magnetic resonance (CMR) investigation and endomyocardial biopsy (EMB). Prompt systemic administration of corticosteroids completely restored and normalized myocardial structure and function. Although the patient's history suggested the presumed hypersensitivity myocarditis, EMB revealed EM without vasculitis, not hypersensitivity, leading to a tentative diagnosis of idiopathic hypereosinophilic syndrome. Interestingly, the characteristic findings of vasculitis on CMR imaging strongly suggested EGPA-associated EM. Although the patient had no clinical neurological manifestations, a nerve conduction study confirmed mononeuritis multiplex, leading to the final diagnosis of probable EGPA. Therefore, this case highlights the diagnostic challenge associated with EGPA and the diagnostic synergy of CMR and EMB for an exploratory diagnosis of EGPA-associated EM.
Eosinophilic granulomatosis with polyangiitis (EGPA), previously known as Churg-Strauss syndrome, is a multisystem disorder characterized by necrotizing vasculitis of small-to-medium-sized vessels, with the coexistence of asthma, rhinosinusitis, and marked peripheral eosinophilia (1). EGPA-associated eosinophilic myocarditis (EM) is rare but can be fatal (1, 2). Because EM has multiple etiologies with overlapping clinical and biological features, a definitive diagnosis of EGPA-associated EM remains challenging in the absence of clinical manifestations of vasculitis. Therefore, a promising strategy to accurately diagnose EGPA-associated EM needs to be formulated.
A 72-year-old woman was admitted to our hospital with worsening chest pain. She had a history of asthma and was treated with a fluticasone furoate/vilanterol (inhaler), theophylline (200 mg/day), ambroxol hydrochloride capsules (45 mg/day), montelukast (10 mg/day), and mequitazine (6 mg/day). Her treatment was aided with short-term use of oral prednisolone as required. The patient's general practitioner switched her from regular branded medications to generic medications 1 month prior to her admission. A week before admission, she experienced chest pain, characterized by chest tightness on exertion that disappeared on rest. Her symptoms worsened and were accompanied by dyspnea on effort (New York Heart Association class II–III). Her vital signs were as follows: blood pressure 118/73 mmHg, heart rate 87 beats/min, temperature 37.3°C, and oxygen saturation 97%. The physical and neurological examination results and chest radiographs were unremarkable. The electrocardiogram (ECG) showed pathologic Q waves in V2 to V3, and negative QRS complexes including rS morphology in the inferior leads, diagnosed as a left anterior fascicular block (Supplementary Figure 1A). The laboratory testing revealed a significant eosinophilia with an eosinophil percentage of 67.6% (normal <6%) and an absolute eosinophil count (AEC) of 10,410/μL (normal <500/μL), elevated brain natriuretic peptide level of 536 pg/mL (normal <18.4 pg/mL), and elevated cardiac biomarkers levels as follows: creatine kinase, 473 U/L (reference: 41–153 U/L); CK-MB, 39 U/L (normal <25 U/L); aspartate aminotransferase, 72 U/L (reference: 13–30 U/L); lactate dehydrogenase, 613 U/L (reference: 124–222 U/L); and high sensitivity cardiac troponin I, 47,875.3 pg/mL (normal <26.2 pg/mL). The levels of the inflammatory markers were also elevated—the C-reactive protein was 0.65 mg/dL (normal <0.3 mg/dL) and the erythrocyte sedimentation rate was >110 mm/h (reference: 3–15 mm/h). The results of renal function and urinalysis were normal. Further laboratory studies revealed elevated levels of serum IgE at 1,840 IU/mL (normal <173 IU/mL) and rheumatoid factor at 204 IU/mL (normal <15 IU/mL). In addition, the levels of the Th2 cytokines-related interleukins (IL) were also raised—IL-4 was 7.7 pg/mL (normal <3.9 pg/mL) and IL-5 was 30 pg/mL (normal <3.9 pg/mL). Anti-neutrophil cytoplasmic antibodies (ANCA) were not detected. Echocardiography revealed a mildly thickened myocardium and significant left ventricular (LV) systolic dysfunction with an ejection fraction of 43%. In addition, speckle-tracking echocardiography showed a reduced baseline global longitudinal strain of −9.9% (Figures 1A–C and Supplementary Video 1). Accordingly, we made a tentative diagnosis of acute coronary syndrome (ACS). An emergency coronary angiography was performed after pre-treatment with methylprednisolone (250 mg) that was administered to prevent allergic contrast reactions for the patient with asthma. The angiogram revealed normal epicardial coronary arteries. Therefore, acute EM was suspected, and cardiac magnetic resonance (CMR) was performed to assess the myocardial tissue (Figures 2A–C and Supplementary Video 2). Myocardial first-pass perfusion (FPP) imaging showed patchy and circumferential subendocardial perfusion defects (arrowheads), suggesting microvascular disorders. CMR also showed subendocardial late gadolinium enhancement (LGE) as multiple and lobulated high-signal intensity spots (arrows), which suggested vasculitis. The T2-weighted image showed a transmural high-intensity signal throughout the myocardium, corresponding to myocardial edema. Acute myocarditis was diagnosed based on the Lake–Louise criteria. These findings were consistent with acute EM. Simultaneously, an exhaustive diagnostic workup for hypereosinophilia was performed. Its differential diagnoses include hypersensitivity myocarditis (HSM), EGPA, parasitic infections, hematologic malignancies, and lymphocytic/idiopathic hypereosinophilic syndrome (HES). Considering the patient's recent medical history, HSM was initially suspected as the cause of EM. The generic drugs being administered to the patient were discontinued after admission. A subsequent endomyocardial biopsy (EMB) was performed, which demonstrated marked extravascular eosinophilic infiltrates without granulomatous and fibrinoid necrotizing vasculitis (Figure 3A). Numerous eosinophilic infiltrations, with degranulated eosinophils admixed with lymphocytes and myocyte necrosis, were observed in the myocardial interstitium that extended to the endocardium (Figures 3B,C). Moderate endocardial thickening and perivascular interstitial fibrosis were observed (data not shown). Immunostaining was performed to identify the major basic proteins revealed extensive staining in the endocardium and myocardial interstitium (Figure 3D). These findings led to the final diagnosis of acute EM. Subsequently, the patient was treated with intravenous methylprednisolone (1 g/day for 3 days), followed by oral prednisolone (1 mg/kg/day). The clinical response to steroid treatment was remarkable, with significant recovery of LV dysfunction, hypereosinophilia, and elevated cardiac enzyme levels within 21 days of steroid treatment (Figures 1D–F and Supplementary Video 3). Follow-up ECG showed resolution of all abnormal findings recognized during the initial ECG (Supplementary Figure 1B). On day 33, the patient was discharged after administration of prednisolone (15 mg/day), with a gradual tapering of the doses. On day 56, the patient remained asymptomatic, with fully recovered cardiac function observed on echocardiography (Supplementary Figure 2 and Supplementary Video 4). Moreover, the abnormal findings of the CMR resolved completely (Figures 2D–F and Supplementary Video 5). On day 75, prednisolone was tapered and finally discontinued in the outpatient clinic. However, her asthma precipitated again 2 weeks later, which was concurrent with an eosinophilia count of 1,512/μL. Since a thorough diagnostic workup for hypereosinophilia was negative, idiopathic HES was also considered. Although the patient had no clinical neurological manifestations, her CMR findings were suggestive of vasculitis, which encouraged us to perform a nerve conduction study that revealed mononeuritis multiplex. Eventually, as per the diagnostic criteria of the American College of Rheumatology (ACR) for EGPA, the patient met four of the six items (asthma, eosinophilia >10%, mononeuritis multiplex, and extravascular eosinophilia). However, a histological diagnosis of vasculitis could not be performed because the patient refused nerve biopsies. Therefore, the final diagnosis of probable EGPA was made. The patient was restarted on prednisolone treatment (15 mg/day). At the 1-year follow-up, the patient remained clinically stable, with prednisolone tapered to 4 mg/day. As a supplement, we have presented a timeline for the case presentation (Supplementary Figure 3).
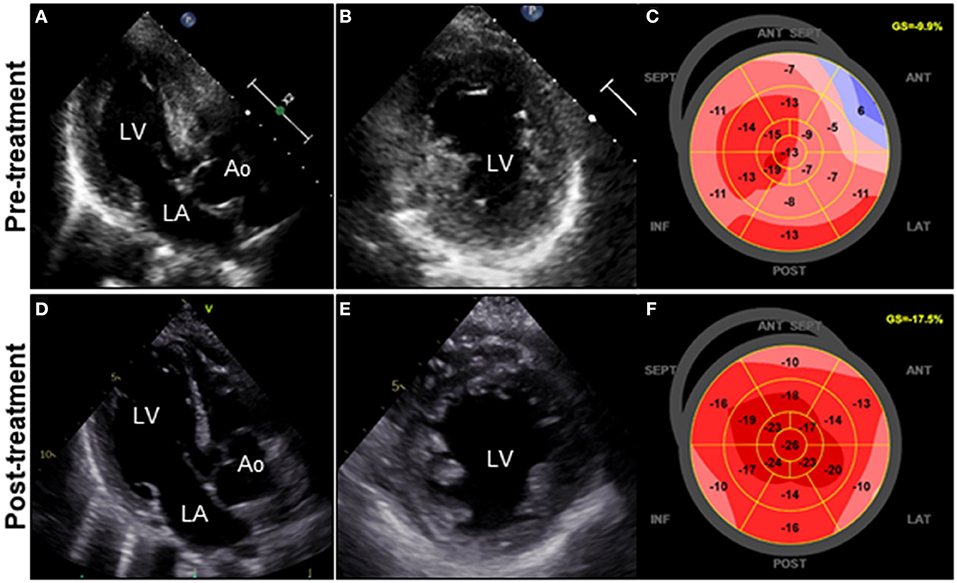
Figure 1. Clinical effects of corticosteroid treatment on TTE. TTE on admission reveals diffuse and symmetrical LV wall thickening (11 mm), decreased cavity size, reduced ventricular function, and GLS values of the LV (LVDd, 44 mm; LVEF, 43%; and GLS, −9.9%; respectively) (A–C). Follow-up TTE on day 21 after corticosteroid therapy reveals a significant decrease in LV wall thickness (8 mm) with concomitant improvement in cavity size, ventricular function, and GLS values of the LV (LVDd, 48 mm; LVEF, 50%; and GLS, −17.5%; respectively) (D–F). Ao, aorta; GLS, global longitudinal strain; LA, left atrium; LV, left ventricle; LVDd, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; TTE, transthoracic echocardiography.
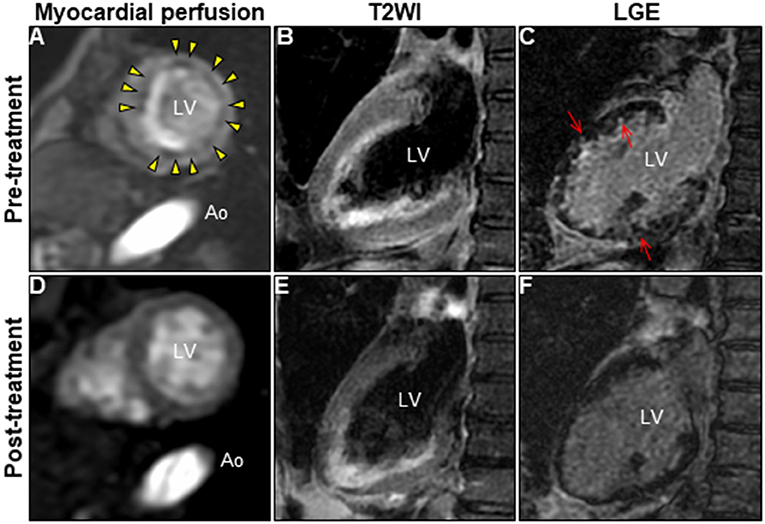
Figure 2. Changes in CMR findings in patients following corticosteroid treatment. CMR findings before (A–C) and after corticosteroid treatment (D–F). Myocardial first-pass perfusion imaging at rest (A,D), T2WI of the 2-chamber view (B,E), and LGE image (C,F). Ao, aorta; CMR, cardiac magnetic resonance; LGE, late gadolinium enhancement; LV, left ventricle; T2WI, T2-weighted image.
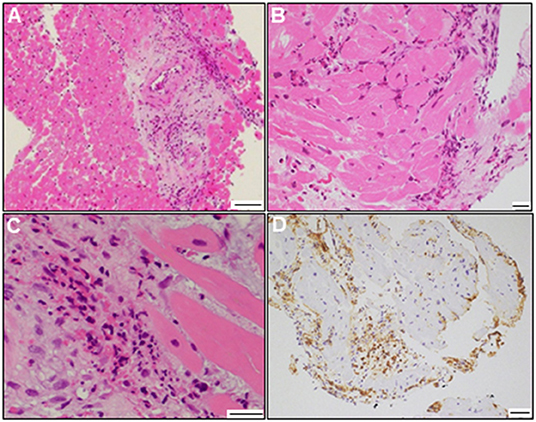
Figure 3. Endomyocardial biopsy findings. Photomicrograph with hematoxylin and eosin staining (A–C) (bars: A, 100 μm; B and C, 20 μm). Photomicrograph showing immunostaining against the major basic protein (D) (bar: 50 μm).
EGPA is classified among ANCA-positive vasculitides, and its underlying pathological mechanism remains poorly understood. The widely accepted ACR criteria for EGPA include asthma, eosinophilia (>10% in the differential count), neuropathy, non-fixed pulmonary infiltrates, paranasal sinus abnormalities, and extravascular eosinophils. According to a study, the ACR criteria for the classification of vasculitis as EGPA yielded a sensitivity of 85.0% and specificity of 99.7%, when four or more of the aforementioned conditions were applicable (3). EGPA is commonly diagnosed at ~40 years of age and exhibits no gender predominance. Its reported prevalence is 10.7–18 per million in Europe and the United States (4, 5). EGPA progresses through three chronological phases: the prodromal phase with the occurrence of asthma and allergic manifestations; the eosinophilic phase, which is characterized by eosinophilic infiltration of the organs involving the lungs and the myocardium; and the vasculitic phase, which is characterized by organ damage due to vasculitis in small-to-medium-sized vessels in the skin, peripheral nerves, and kidneys. These three phases may overlap. Generally, EGPA has an excellent prognosis, with a 5-year survival rate of 97% (6). Cardiovascular involvement rarely occurs (about 16% of organ involvements), but is the leading cause of death in about 50% of cases (7). Although data on cardiac involvement in patients with EGPA are available in the literature, the number of reported cases of EGPA-associated EM is small (2, 8). Therefore, we conducted an updated systematic review of case reports to investigate the characteristics of EGPA-associated EM (Table 1). The literature search was performed using PubMed databases documented from 1983 to 2021, with the search restricted to studies published in English. We used the following MeSH terms: “eosinophilic granulomatosis with polyangiitis” or “Churg–Strauss syndrome” and “case report” and “myocarditis.” Moreover, we selected cases of EGPA-associated EM confirmed by EMB and/or CMR findings. Finally, 38 cases were included (9–46). The mean age of the participants included was 47 years with no gender predominance. The main extracardiac involvement was asthma, followed by peripheral neuropathy and constitutional symptoms (94.7, 52.6, and 23.7%, respectively). At presentation, the patients were commonly diagnosed with cardiac insufficiency (51.6%), ACS (21.1%), and cardiogenic shock (13.2%). As in our case, patients with EGPA-associated EM had a high rate of ANCA-negativity and presented with marked peripheral eosinophilia. Presence of LV dysfunction (66%) and pericardial effusion (47%) were frequently observed. A higher five-factor score was indicative of poor prognosis in 39% of the patients. Considering the number of cases with a poor clinical course, including sudden cardiac death (21%) and incomplete normalized cardiac function despite intensive treatment (26%), early recognition and treatment of EGPA-associated EM is required.
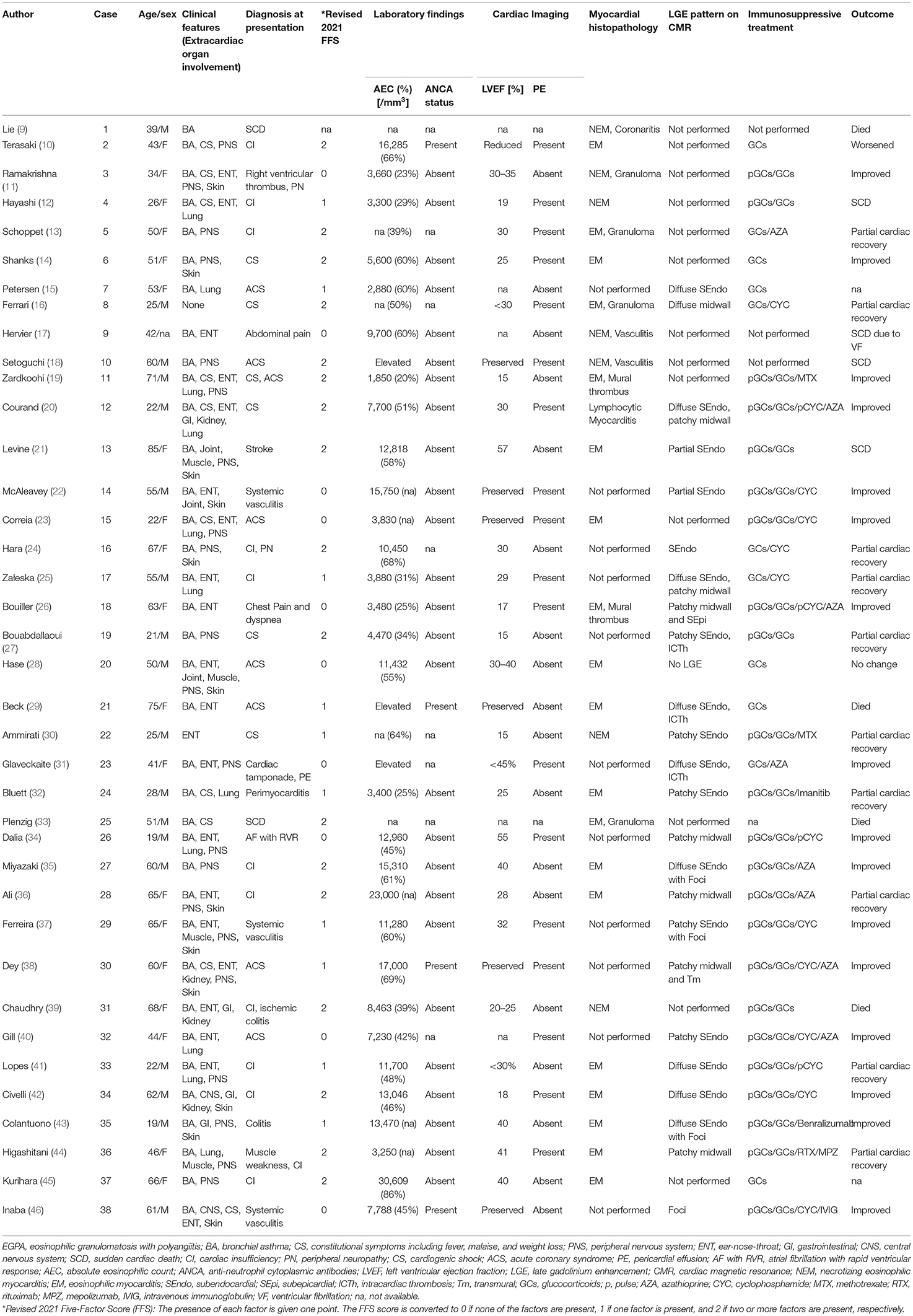
Table 1. Cases of EGPA-associated EM.
Herein, we describe a previously undiagnosed case of probable EGPA with the development of acute EM, which was successfully treated with corticosteroid treatment. This case provides three clinical suggestions.
First, the clinical course, in this case, was acute EM without obvious vasculitis, which led to a delayed diagnosis of probable EGPA.
The typical symptoms of systemic vasculitis in EGPA include constitutional symptoms (fever, malaise, and weight loss), myalgia, mono/polyneuropathy (numbness, tingling, muscle weakness, and pain), skin symptoms (purpura and non-pruritic nodules), and gastrointestinal ischemic symptoms such as abdominal pain. However, none of the above symptoms, except for low-grade fever, were observed in our case. The present case exemplifies the following three diagnostic challenges of EGPA.
First, HSM was suspected based on the medication history. External factors such as exposure to allergens, infection, or vaccination can induce the development of EGPA. However, the symptoms precipitated even after discontinuation of the suspected drugs, and the histological findings on EMB confirmed that HSM was less likely. Anti-asthmatic drugs, such as leukotriene receptor antagonists and anti-IgE antibodies can trigger the development of EGPA; however, a direct causal relationship in our case was unclear. The anti-asthmatic effects of the drugs might have delayed the systemic administration of corticosteroids, resulting in the manifestation of EGPA (47).
Second, the main histopathological findings of EGPA, other than eosinophilic infiltration, were not observed in our case. EMBs in patients with EGPA-associated EM often show EM without vasculitis (2), which is probably attributed to the possibility of heterogeneous distribution of vasculitis, the small number of EMB samples, and the limited biopsy sites including the left ventricular apex and interventricular septum (Table 1). Besides, in the autopsy cases, it was very likely to detect evidence of vasculitis because the whole heart was able to be analyzed. Therefore, EGPA-associated EM cannot be ruled out based on the lack of histologic evidence of vasculitis in EMBs. In addition, because the histopathologic findings may vary by the phase of the disease, our patient might have been during the early vasculitic phase of the disease. Furthermore, the cases of EGPA-associated EM have a high frequency of coexisting asthma (94.7%) (Table 1). Therefore, inhaled corticosteroids for asthma, and corticosteroid prophylaxis for allergic reactions to the contrast media, might have contributed to the absence of typical histopathologic findings in our patient. Subsequently, this patient presented with a mononeuritis multiplex which was significantly associated with systemic vasculitis (48), leading us to believe that this patient was diagnosed with probable EGPA.
A third diagnostic challenge is the potential clinical and biological overlap between EGPA and idiopathic HES. HES is a sporadic disorder that is diagnosed based on the following criteria: elevated AEC (>1,500 cells/μL on at least two occasions), and/or pathologic confirmation of tissue hypereosinophilia. With great advances in the knowledge of eosinophil biology and molecular diagnostics, the classification of HES subgroups is evolving rapidly. The following six variants of HES have been proposed (49): (i) myeloid HES, (ii) lymphocytic HES, (iii) overlap HES—eosinophilic disorders overlapping in presentation with idiopathic HES (e.g., EGPA), (iv) associated HES (eg, parasite infections, drug hypersensitivities, or immunodeficiency), (v) familial HES, and (vi) idiopathic HES—eosinophilic disorders of unknown etiology. Because AECs are high (average: 6,716; range: 1,850–30,609) and ANCAs are often undetectable (87%) in EGPA-associated EM, the clinical characteristics of EGPA-associated EM are similar to those of idiopathic HES (50). Thus, it is crucial to distinguish between the two because they differ in treatment and prognosis. EGPA and idiopathic HES can be differentiated based on vasculitis (clinical or histological). Our case review showed that patients with established EGPA-associated EM had a high prevalence of neurological involvement (52.6%, Table 1). Considering ANCA-negativity and the absence of significant vasculitis, our case was difficult to distinguish from idiopathic HES. Generally, asthma is not mandatory for the diagnosis of EGPA (3) and can also be found in patients with any variants of HES. Nevertheless, given that the overwhelming majority of patients with EGPA-associated EM have concomitant asthma of 94.7% as shown in Table 1, our case underscores the critical importance of preferentially considering EGPA as the causative etiology in cases of EM with asthma. A study analyzing 179 cases with histologically proven EM reported the prevalence of a history of asthma to be 68% in the EGPA group, 21% in the idiopathic/undefined group, and 23% in the HES group (51), which suggested that EGPA might not have been diagnosed due to the absence of obvious vasculitis in the latter two groups.
As a second clinical suggestion, both CMR and EMB were valuable for the final diagnosis of probable EGPA in our case.
CMR enables the characterization of myocardial tissue properties (inflammation, thrombus, and fibrosis) and yields high diagnostic performance in identifying acute myocarditis (81% sensitivity, 71% specificity, and 79% accuracy) (52). Typical CMR findings of EGPA-associated EM include subendocardial LGE in the apical and mid-left ventricles. In our case, the following unique CMR findings led to a strong suspicion of EGPA-associated EM. CMR revealed diffuse patchy subendocardial defects on FPP at rest, which were superimposed on abnormal lesions on LGE and T2-weighted images. Taken together, these observations were suggestive of microvascular dysfunction caused by acute inflammation, which was consistent with previous reports (53). Similar findings were observed in a patient with cardiac syndrome X, which is characterized by unexplained chest pain (54). Although coronary luminal stenosis, thrombus, or spasms have been proposed as possible causes of angina symptoms in patients with EGPA, the ACS-like symptoms reported in our case can be explained by microvascular dysfunction. Notably, in our case, multiple foci on the initial LGE were resolved following the corticosteroid treatment. Similar foci have been reported in some cases of EGPA-associated EM, and are presumed to be specific to vasculitis, which can be an indicator for EGPA-associated EM (35, 43, 55). Further studies on the relationship between foci on LGE and their respective pathologies are warranted. In addition, EMB provides useful information on the nature and distribution of inflammatory infiltrates. EGPA and HSM can be differentiated based on the histopathological findings. The histological features of EGPA include tissue eosinophilia, extravascular eosinophilic granuloma, and necrotizing vasculitis. However, the latter two are rarely seen in cardiac pathology (56) (Table 1), whereas HSM is characterized by interstitial prominent eosinophilic infiltrates without myocardial necrosis or fibrosis (57). The distinct pattern of endo/peri-myocardial eosinophilic infiltration and degranulation, accompanied by the presence of myocardial necrosis and fibrosis in EMB, was a crucial finding for the definitive diagnosis of acute EM and differentiating it from HSM. EMB is the golden standard for histological diagnosis of myocarditis, but it is an invasive method with limitations such as serious procedure-related complications or sampling errors. With an excellent diagnostic accuracy of CMR in acute myocarditis due to great advances in imaging technology, the usefulness of non-invasive CMR alone in diagnosing acute myocarditis has been widely reported (52, 58, 59). Therefore, a single approach of either CMR or EMB is now considered sufficient for the diagnosis of acute myocarditis. However, each approach has its advantages and disadvantages in cases of acute EM that develops as the first manifestation of EGPA without clinical vasculitis as per our case. Therefore, our case underscores that the combination of CMR and EMB provides diagnostic synergy in the exploratory diagnosis of EGPA in patients with suspected acute EM.
Our final clinical suggestion is that timely corticosteroid treatment allowed significant recovery and normalization of the cardiac structure and function in our case.
The primary treatment for EGPA is systemic glucocorticoids. Additional immunosuppressive agents should be considered in patients with progressive, refractory, or relapsing diseases. Despite administering multiple immunomodulators, many cases of EGPA-associated EM were directly linked to fatal cardiac complications and incomplete recovery of cardiac function (Table 1). In our case, systemic and oral corticosteroids resulted in complete recovery and normalization of the cardiac structure and function within about 2 months. Considering the malignant features of EGPA-associated EM, our case underscores the significance of early recognition and treatment of this disease.
We present a case of probable EGPA with subclinical mononeuritis multiplex that developed acute EM and was successfully treated with systemic corticosteroid therapy. The EGPA-associated EM is a rare yet potentially life-threatening disorder that can be cured if treated appropriately and timely. However, without obvious vasculitis, accurate diagnosis of EGPA-associated EM is challenging. Therefore, clinicians should be aware of this rare disease and consider using both CMR and EMB diagnostic approaches for patients with suspected EM.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
Authorization for the use of case information and materials was obtained from the Institutional Review Board of Narita-Tomisato Tokushukai Hospital. The authors confirm that written consent for the submission and publication of this case report, including the images and associated movie, was obtained from the patient.
HY contributed in creating the clinical design and concept, interpreted the data, drafted, and revised the manuscript. HY, KH, and TH acquired the clinical data. JI performed the CMR analyses. YI performed the pathological analyses. All authors discussed and approved the manuscript and authorized its submission for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.913724/full#supplementary-material
Supplementary Figure 1. ECG at admission (A) and on day 21 (B). An initial ECG reveals evidence of pathologic Q waves in V2 to V3, and a significant left axis deviation at −57°, which is consistent with a left anterior fascicular block. Notably, the follow-up ECG after treatment of corticosteroid shows the resolution of all abnormal findings recognized during the initial ECG. ECG, electrocardiogram.
Supplementary Figure 2. Clinical effects of the corticosteroid treatment on TTE. Follow-up TTE on day 56 after the corticosteroid therapy reveals a full recovery of the ventricular function and GLS values of the LV (LVDd, 49 mm; LVEF, 62%; GLS −16.1%, respectively) (A–C). Ao, aorta; GLS, global longitudinal strain; LA, left atrium; LV, left ventricle; LVDd, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; TTE, transthoracic echocardiography.
Supplementary Figure 3. A timeline for the case presentation. EGPA, eosinophilic granulomatosis with polyangiitis; EM, eosinophilic myocarditis; HES, hypereosinophilic syndrome; LGE, late gadolinium enhancement; LV, left ventricular.
Supplementary Video 1. Pre-treatment transthoracic echocardiography. Transthoracic echocardiography at the initial presentation in a parasternal short-axis view reveals mild diffuse thickening of the left ventricular wall and decreased cavity size with a reduced ejection fraction of 43%.
Supplementary Video 2. Pre-treatment myocardial first-pass perfusion deficits on CMR. Short-axis CMR imaging demonstrates that myocardial first-pass perfusion at rest shows patchy and circumferential subendocardial perfusion deficits. CMR: cardiac magnetic resonance.
Supplementary Video 3. Post-treatment transthoracic echocardiography on day 21. Post-treatment follow-up transthoracic echocardiography in a parasternal short-axis view reveals significant resolution of the left ventricular wall thickening with concomitant improvement in cavity size and ventricular function with an ejection fraction of 50%.
Supplementary Video 4. Post-treatment transthoracic echocardiography on day 56. Note the fully recovered cardiac function observed on echocardiography.
Supplementary Video 5. Changes in myocardial first-pass perfusion deficits on CMR following corticosteroid treatment. Note the significant improvement in the abnormal first-pass perfusion deficits at admission on post-treatment follow-up CMR. CMR, cardiac magnetic resonance.
EGPA, eosinophilic granulomatosis with polyangiitis; EM, eosinophilic myocarditis; ECG, electrocardiogram; AEC, absolute eosinophilic count; ANCA, anti-neutrophil cytoplasmic antibodies; LV, left ventricular; ACS, acute coronary syndrome; CMR, cardiac magnetic resonance; FPP, first-pass perfusion; LGE, late gadolinium enhancement; HES, hypereosinophilic syndrome; HSM, hypersensitivity myocarditis; EMB, endomyocardial biopsy; ACR, American College of Rheumatology.
1. Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine. (1999) 78:26–37. doi: 10.1097/00005792-199901000-00003
2. Neumann T, Manger B, Schmid M, Kroegel C, Hansch A, Kaiser WA, et al. Cardiac involvement in Churg-Strauss syndrome: impact of endomyocarditis. Medicine. (2009) 88:236–43. doi: 10.1097/MD.0b013e3181af35a5
3. Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. (1990) 33:1094–100. doi: 10.1002/art.1780330806
4. Mohammad AJ. An update on the epidemiology of ANCA-associated vasculitis. Rheumatology. (2020) 59(Suppl. 3):iii42–50. doi: 10.1093/rheumatology/keaa089
5. Conron M, Beynon HL. Churg-Strauss syndrome. Thorax. (2000) 55:870–7. doi: 10.1136/thorax.55.10.870
6. Ribi C, Cohen P, Pagnoux C, Mahr A, Arène JP, Lauque D, et al. French Vasculitis Study Group. Treatment of Churg-Strauss syndrome without poor-prognosis factors: a multicenter, prospective, randomized, open-label study of seventy-two patients. Arthritis Rheum. (2008) 58:586–94. doi: 10.1002/art.23198
7. Sada KE, Amano K, Uehara R, Yamamura M, Arimura Y, Nakamura Y, et al. A nationwide survey on the epidemiology and clinical features of eosinophilic granulomatosis with polyangiitis (Churg-Strauss) in Japan. Mod Rheumatol. (2014) 24:640–4. doi: 10.3109/14397595.2013.857582
8. Qiao L, Gao D. A case report and literature review of Churg-Strauss syndrome presenting with myocarditis. Medicine. (2016) 95:e5080. doi: 10.1097/MD.0000000000005080
9. Lie JT, Bayardo RJ. Isolated eosinophilic coronary arteritis and eosinophilic myocarditis. A limited form of Churg-Strauss syndrome. Arch Pathol Lab Med. (1989) 113:199–201.
10. Terasaki F, Hayashi T, Hirota Y, Okabe M, Suwa M, Deguchi H, et al. Evolution to dilated cardiomyopathy from acute eosinophilic pancarditis in Churg-Strauss syndrome. Heart Vessels. (1997) 12:43–8. doi: 10.1007/BF01747501
11. Ramakrishna G, Connolly HM, Tazelaar HD, Mullany CJ, Midthun DE. Churg-Strauss syndrome complicated by eosinophilic endomyocarditis. Mayo Clin Proc. (2000) 75:631–5. doi: 10.4065/75.6.631
12. Hayashi S, Furuya S, Imamura H. Fulminant eosinophilic endomyocarditis in an asthmatic patient treated with pranlukast after corticosteroid withdrawal. Heart. (2001) 86:E7. doi: 10.1136/heart.86.3.e7
13. Schoppet M, Pankuweit S, Maisch B. CD83+ dendritic cells in inflammatory infiltrates of Churg-Strauss myocarditis. Arch Pathol Lab Med. (2003) 127:98–101. doi: 10.5858/2003-127-98-CDCIII
14. Shanks M, Ignaszewski AP, Chan SY, Allard MF. Churg-Strauss syndrome with myocarditis manifesting as acute myocardial infarction with cardiogenic shock: case report and review of the literature. Can J Cardiol. (2003) 19:1184–8.
15. Petersen SE, Kardos A, Neubauer S. Subendocardial and papillary muscle involvement in a patient with Churg-Strauss syndrome, detected by contrast enhanced cardiovascular magnetic resonance. Heart. (2005) 91:e9. doi: 10.1136/hrt.2004.050070
16. Ferrari M, Pfeifer R, Poerner TC, Figulla HR. Bridge to recovery in a patient with Churg-Strauss myocarditis by long-term percutaneous support with microaxial blood pump. Heart. (2007) 93:1419. doi: 10.1136/hrt.2006.101881
17. Hervier B, Masseau A, Bossard C, Agard C, Hamidou M. Vasa-vasoritis of the aorta and fatal myocarditis in fulminant Churg-Strauss syndrome. Rheumatology. (2008) 47:1728–9. doi: 10.1093/rheumatology/ken329
18. Setoguchi M, Okishige K, Sugiyama K, Shimura T, Maeda M, Aoyagi H, et al. Sudden cardiac death associated with Churg-Strauss syndrome. Circ J. (2009) 73:2355–9. doi: 10.1253/circj.CJ-08-0926
19. Zardkoohi O, Hobbs R, Tan CD. A rare shock. Am J Med. (2011) 124:1019–22. doi: 10.1016/j.amjmed.2011.06.008
20. Courand PY, Croisille P, Khouatra C, Cottin V, Kirkorian G, Bonnefoy E. Churg-Strauss syndrome presenting with acute myocarditis and cardiogenic shock. Heart Lung Circ. (2012) 21:178–81. doi: 10.1016/j.hlc.2011.09.002
21. Levine AB, Kalliolias G, Heaney M, Endo Y, Gersten A, Weinsaft JW, et al. Churg-strauss syndrome with eosinophilic myocarditis: a clinical pathology conference held by the division of rheumatology at hospital for special surgery. HSS J. (2012) 8:313–9. doi: 10.1007/s11420-012-9276-x
22. McAleavey N, Millar A, Pendleton A. Cardiac involvement as the main presenting feature in eosinophilic granulomatosis with polyangiitis. BMJ Case Rep. (2013) 2013:bcr2013009394. doi: 10.1136/bcr-2013-009394
23. Correia AS, Gonçalves A, Araújo V, Almeida e Silva J, Pereira JM, Rodrigues Pereira P, et al. Churg-Strauss syndrome presenting with eosinophilic myocarditis: a diagnostic challenge. Rev Port Cardiol. (2013) 32:707–11. doi: 10.1016/j.repc.2012.10.017
24. Hara T, Yamaguchi K, Iwase T, Kadota M, Bando M, Ogasawara K, et al. Eosinophilic myocarditis due to Churg-Strauss syndrome with markedly elevated eosinophil cationic protein. Int Heart J. (2013) 54:51–3. doi: 10.1536/ihj.54.51
25. Załeska J, Wiatr E, Zych J, Szopiński J, Oniszh K, Kober J, et al. Severe congestive heart failure as the main symptom of eosinophilic granulomatosis and polyangiitis (Churg-Strauss syndrome). Pneumonol Alergol Pol. (2014) 82:582–9. doi: 10.5603/PiAP.2014.0077
26. Bouiller K, Samson M, Eicher JC, Audia S, Berthier S, Leguy V, et al. Severe cardiomyopathy revealing antineutrophil cytoplasmic antibodies-negative eosinophilic granulomatosis with polyangiitis. Intern Med J. (2014) 44:928–31. doi: 10.1111/imj.12525
27. Bouabdallaoui N, Arlet JB, Hagege AA. Cardiogenic shock, asthma, and hypereosinophilia. Am J Emerg Med. (2015) 33:309.e1–2. doi: 10.1016/j.ajem.2014.08.019
28. Hase H, Yamamoto T, Saito T, Yamazaki H, Kuno T, Tabei R, et al. Successful treatment using corticosteroids in early phase of eosinophilic myocarditis with eosinophilic granulomatosis with polyangiitis. J Cardiol Cases. (2016) 14:177–80. doi: 10.1016/j.jccase.2016.08.006
29. Beck KS, Jeong SY, Lee KY, Chang K, Jung JI. Native T1 Mapping Demonstrating Apical Thrombi in Eosinophilic Myocarditis Associated with Churg-Strauss Syndrome. Korean Circ J. (2016) 46:882–5. doi: 10.4070/kcj.2016.46.6.882
30. Ammirati E, Cipriani M, Musca F, Bonacina E, Pedrotti P, Roghi A, et al. A life-threatening presentation of eosinophilic granulomatosis with polyangiitis. J Cardiovasc Med. (2016) 17(Suppl. 2):e109–11. doi: 10.2459/JCM.0000000000000330
31. Glaveckaite S, Valeviciene N, Palionis D, Kontrimaviciute E, Lesinskas E. Heart involvement in Churg-Strauss syndrome. Kardiol Pol. (2017) 75:184. doi: 10.5603/KP.2017.0028
32. Bluett R, McDonnell D, O'Dowling C, Vaughan C. Eosinophilic myocarditis as a first presentation of eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). BMJ Case Rep. (2017) 2017:bcr2017221227. doi: 10.1136/bcr-2017-221227
33. Plenzig S, Heinbuch S, Held H, Verhoff MA, Lux C. A case of fatal perimyocarditis due to a rare disease. Forensic Sci Med Pathol. (2017) 13:454–458. doi: 10.1007/s12024-017-9920-3
34. Dalia T, Parashar S, Patel NV, Gautam A, Dai H, Bormann S. Eosinophilic myocarditis demonstrated using cardiac magnetic resonance imaging in a patient with eosinophilic granulomatosis with polyangiitis (Churg-Strauss Disease). Cureus. (2018) 10:e2792. doi: 10.7759/cureus.2792
35. Miyazaki M, Hattori H, Suzuki A, Serizawa N, Uto K, Fukushima K, et al. Successfully treated eosinophilic granulomatosis with polyangiitis relapse presenting as myocarditis and followed by multimodality imaging. J Cardiol Cases. (2018) 18:145–8. doi: 10.1016/j.jccase.2018.06.008
36. Ali D, Snead D, Dhakshinamurthy VA, Banerjee P. Rise and fall of the eosinophils in heart failure: a rare but important phenomenon seen with cardiomyopathy. BMJ Case Rep. (2018) 2018:bcr2017221081. doi: 10.1136/bcr-2017-221081
37. Ferreira RM, Madureira P, Pinho T, Martins E, Pimenta S, Costa L. Silent acute myocarditis in eosinophilic granulomatosis with polyangiitis. Acta Reumatol Port. (2018) 43:309–13.
38. Dey M, Nair J, Sankaranarayanan R, Kanagala P. Myopericarditis as a presentation of eosinophilic granulomatosus with polyangiitis (EGPA). BMJ Case Rep. (2019) 12:e230593. doi: 10.1136/bcr-2019-230593
39. Chaudhry MA, Grazette L, Yoon A, Correa A, Fong MW. Churg-Strauss syndrome presenting as acute necrotizing eosinophilic myocarditis: concise review of the literature. Curr Hypertens Rev. (2019) 15:8–12. doi: 10.2174/1573402114666180903164900
40. Gill JS, Fontana M, Knight D, Kalra SS. A case report of eosinophilic granulomatosis and polyangiitis myocarditis presenting as ST elevation myocardial infarction and showing positive response to immunotherapy. Eur Heart J Case Rep. (2019) 3:1–6. doi: 10.1093/ehjcr/ytz161
41. Lopes PM, Rocha BML, Cunha GJL, Ranchordas S, Albuquerque C, Ferreira AM, et al. Fulminant eosinophilic myocarditis: a rare and life-threatening presentation of eosinophilic granulomatosis with polyangiitis. JACC Case Rep. (2020) 2:802–8. doi: 10.1016/j.jaccas.2020.01.031
42. Civelli VF, Narang VK, Sharma R, Sharma R, Kim J, Bhandohal J, et al. A progressive case of eosinophilic myocarditis due to eosinophilic granulomatosis with polyangiitis in a caucasian male. J Investig Med High Impact Case Rep. (2020) 8:2324709620966855. doi: 10.1177/2324709620966855
43. Colantuono S, Pellicano C, Leodori G, Cilia F, Francone M, Visentini M. Early benralizumab for eosinophilic myocarditis in eosinophilic granulomatosis with polyangiitis. Allergol Int. (2020) 69:483–4. doi: 10.1016/j.alit.2020.03.001
44. Higashitani K, Yoshimi R, Sato Y, Watanabe T, Ihata A. Rituximab and mepolizumab combination therapy for glucocorticoid-resistant myocarditis related to eosinophilic granulomatosis with polyangiitis. Mod Rheumatol Case Rep. (2021) 6:87–92. doi: 10.1093/mrcr/rxab022
45. Kurihara K, Tsugawa J, Ouma S, Ogata T, Aoki M, Omoto M, et al. Eosinophilic granulomatosis with polyangiitis presenting with myocarditis as an initial symptom: a case report and review of the literature. Case Rep Neurol. (2021) 13:329–33. doi: 10.1159/000516255
46. Inaba R, Fuse Y, Kurimoto F, Suzuki S, Watanabe K. A rare case of eosinophilic granulomatosis with polyangiitis presenting as ischemic stroke and splenic infarction. J Stroke Cerebrovasc Dis. (2021) 30:105539. doi: 10.1016/j.jstrokecerebrovasdis.2020.105539
47. Gioffredi A, Maritati F, Oliva E, Buzio C. Eosinophilic granulomatosis with polyangiitis: an overview. Front Immunol. (2014) 5:549. doi: 10.3389/fimmu.2014.00549
48. Cottin V, Bel E, Bottero P, Dalhoff K, Humbert M, Lazor R, et al. Groupe d'Etudes et de Recherche sur les Maladies Orphelines Pulmonaires (GERM“O”P). Revisiting the systemic vasculitis in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): a study of 157 patients by the Groupe d'Etudes et de Recherche sur les Maladies Orphelines Pulmonaires and the European Respiratory Society Taskforce on eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Autoimmun Rev. (2017) 16:1–9. doi: 10.1016/j.autrev.2016.09.018
49. Klion A. Hypereosinophilic syndrome: approach to treatment in the era of precision medicine. Hematology Am Soc Hematol Educ Program. (2018) 2018:326–31. doi: 10.1182/asheducation-2018.1.326
50. Khoury P, Zagallo P, Talar-Williams C, Santos CS, Dinerman E, Holland NC, et al. Serum biomarkers are similar in Churg-Strauss syndrome and hypereosinophilic syndrome. Allergy. (2012) 67:1149–56. doi: 10.1111/j.1398-9995.2012.02873.x
51. Brambatti M, Matassini MV, Adler ED, Klingel K, Camici PG, Ammirati E. Eosinophilic myocarditis: characteristics, treatment, and outcomes. J Am Coll Cardiol. (2017) 70:2363–75. doi: 10.1016/j.jacc.2017.09.023
52. Lurz P, Eitel I, Adam J, Steiner J, Grothoff M, Desch S, et al. Diagnostic performance of CMR imaging compared with EMB in patients with suspected myocarditis. JACC Cardiovasc Imaging. (2012) 5:513–24. doi: 10.1016/j.jcmg.2011.11.022
53. Hansch A, Pfeil A, Rzanny R, Neumann T, Kaiser WA. First-pass myocardial perfusion abnormalities in Churg-Strauss syndrome with cardiac involvement. Int J Cardiovasc Imaging. (2009) 25:501–10. doi: 10.1007/s10554-009-9457-y
54. Panting JR, Gatehouse PD, Yang GZ, Grothues F, Firmin DN, Collins P, et al. Abnormal subendocardial perfusion in cardiac syndrome X detected by cardiovascular magnetic resonance imaging. N Engl J Med. (2002) 346:1948–53. doi: 10.1056/NEJMoa012369
55. Cui X, Peng Y, Liu J, Dong Y, Wu Z, Chen Y. Case report: area of focus of myocardial infarction with non-obstructive coronary arteries in eosinophilic granulomatosis with polyangiitis. Front Cardiovasc Med. (2021) 8:731897. doi: 10.3389/fcvm.2021.731897
56. Vaglio A, Buzio C, Zwerina J. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): state of the art. Allergy. (2013) 68:261–73. doi: 10.1111/all.12088
57. Fenoglio JJ Jr, McAllister HA Jr, Mullick FG. Drug related myocarditis. I. Hypersensitivity myocarditis. Hum Pathol. (1981) 12:900–7. doi: 10.1016/S0046-8177(81)80195-5
58. Ferreira VM, Schulz-Menger J, Holmvang G, Kramer CM, Carbone I, Sechtem U, et al. cardiovascular magnetic resonance in nonischemic myocardial inflammation: expert recommendations. J Am Coll Cardiol. (2018) 72:3158–76. doi: 10.1016/j.jacc.2018.09.072
Keywords: EGPA, acute EM, hypereosinophilia, CMR, EMB, corticosteroid treatment
Citation: Yamamoto H, Hashimoto K, Ikeda Y, Isogai J and Hashimoto T (2022) The Diagnostic Challenge of Eosinophilic Granulomatosis With Polyangiitis Presenting as Acute Eosinophilic Myocarditis: Case Report and Literature Review. Front. Cardiovasc. Med. 9:913724. doi: 10.3389/fcvm.2022.913724
Received: 06 April 2022; Accepted: 13 June 2022;
Published: 07 July 2022.
Edited by:
Jinwei Tian, The Second Affiliated Hospital of Harbin Medical University, ChinaReviewed by:
Fady Gerges, Mediclinic Al Jowhara Hospital, United Arab EmiratesCopyright © 2022 Yamamoto, Hashimoto, Ikeda, Isogai and Hashimoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroyuki Yamamoto, aHlhbWFtb3RvMTk3MDA5MDhAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.