 Kae-Woei Liang
Kae-Woei Liang Sheng-Kai Chang
Sheng-Kai Chang Yu-Wei Chen
Yu-Wei Chen Wei-Wen Lin
Wei-Wen Lin Wan-Jane Tsai
Wan-Jane Tsai Kuo-Yang Wang
Kuo-Yang Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 22 June 2022
Sec. Cardiovascular Genetics and Systems Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.911649
Background: Genetic variants could be identified in subjects with idiopathic and heritable pulmonary arterial hypertension (PAH). The 6th World Symposium on Pulmonary Hypertension (WSPH) provided a list of genes with evidence of association with PAH. However, reports using whole exome sequencing (WES) from southeastern Asian PAH cohorts were scarce.
Methods: Subjects with idiopathic and heritable PAH (N = 45) from two medical centers in central Taiwan were screened for PAH related gene variants. The genomic DNA was prepared from peripheral blood lymphocytes. We performed WES for all patients enrolled in this study. All identified gene variants were validated by polymerase-chain reaction and Sanger sequencing. The clinical and hemodynamic data were compared between bone morphogenetic protein receptor type-2 (BMPR2) gene variants carriers vs. non-carriers.
Results: Eight patients (8/45 = 17.8%) was identified carrying BMPR2 gene variants and 8 patients (8/45 = 17.8%) had other WSPH-listed PAH-related gene variants (1 with ACVRL1, 1 with ENG, 1 with SMAD9, 1 with SMAD1, 1 with ATP13A3 and 3 with AQP1). In addition, a total of 14 non-WSPH-listed PAH-related genetic variant sites (ABCC8, NOTCH1, NOTCH2, NOTCH3, JAG1, BMP10, GGCX, FBLN2, ABCA3 and PTGIS) were found in this PAH cohort. Subjects carrying BMPR2 gene variant (N = 8) were younger at diagnosis of PAH (30 ± 11 vs 49 ± 13 years, p = 0.001) than the non-carrier group (N = 37). BMPR2 variant carriers had a trend toward having higher mean pulmonary arterial pressure (PAP) (61 ± 19 vs. 51 ± 13 mmHg, p = 0.076) than the non-carriers upon initial diagnosis. Pulmonary vascular resistance, right atrial pressure, cardiac output, as well as functional class were similar between BMPR2 variant carriers and non-carriers at initial diagnosis.
Conclusions: We identified 17.8% of patients with BMPR2 gene variants and 17.8% subjects with other 6th WSPH-listed PAH-related gene variants in a Taiwanese idiopathic and heritable PAH cohort. PAH patients carrying BMPR2 variants presented at a younger age with a trend toward having higher mean PAP at initial diagnosis.
It is now well established that around 70–80% of heritable pulmonary arterial hypertension (PAH) and 10–20% of idiopathic PAH cases are related with genetic variants in bone morphogenetic protein receptor type-2 (BMPR2) (1, 2). BMPR2 deficiency leads to an abnormal over-activation of the transforming growth factor β (TGF-β) signaling pathway, resulting in over-proliferation of vascular smooth muscle cells at pulmonary arteriole (3).
Genetic variants in other components of the TGF-β/BMPR2 signal pathways, including activin receptor-like kinase 1 (ACVRL1) and endoglin (ENG) have been reported in PAH patients (2, 4–6). BMPR2 and ACVRL1 form a signaling complex, utilizing ENG as a co-receptor in endothelial cells of pulmonary arterioles (6). ACVRL1 or ENG genetic variants have been found in patients with hereditary hemorrhagic telangiectasia (HHT) combined with PAH (7). Genetic variants downstream of BMPR2 signaling intermediaries, SMAD1, SMAD4 and SMAD9 are also reported to be associated with PAH pathogenesis (6, 8).
Genes not directly impacting on the TGF-β/BMP pathway, including aquaporin 1 (AQP1), which encodes the plasma membrane water channel aquaporin 1, and ATPase family homolog up-regulated in senescence cells 1 (ATP13A3), an ATPases involved in ion channel transport, were reported to be associated with PAH (2, 6, 8). A distinct form of PAH, pulmonary veno-occlusive disease (PVOD) or pulmonary capillary hemangiomatosis (PCH), was shown recently to be caused by homozygous mutations in eukaryotic translation initiation factor 2 alpha kinase 4 (EIF2AK4), a kinase in the integrated stress response (6, 8–10). The 6th World Symposium on Pulmonary Hypertension (WSPH) provided a list of pulmonary arterial hypertension genes with higher or lower level of evidence (2).
In addition to the genetic variants listed by WSPH (2), there are several new candidate genes with related reports for their associations with PAH. ABCC8 (ATP-binding cassette, subfamily C, member 8) encodes SUR1 (sulfonylurea receptor 1), a regulatory subunit of the ATP-sensitive potassium channel (11). ABCC8 loss-of function mutations in both pediatric and adult-onset PAH had been reported (11, 12). NOTCH signaling is involved in vascular morphogenesis and function (13). A mechanistic link between Notch3 signaling and PAH is supported by the observation that NOTCH3 KO mice are protected from hypoxia-induced PAH and that PAH can be treated by a g-secretase inhibitor that inhibits the NOTCH pathway (14). Chida et al. (15) identified two novel missense mutations (G840E and T900P) of the NOTCH3 gene in two cases of childhood idiopathic PAH.
Patients with PAH carrying BMPR2 genetic variants usually presented at a younger age with higher mean pulmonary arterial pressure (PAP) or pulmonary vascular resistance (PVR) (1, 16, 17). Meta-analysis also showed worse clinical outcomes for death or transplantation in BMPR2 mutants compared with non-mutants with PAH (1). A pediatric idiopathic PAH cohort from Beijing reported 50% primary PAH patients carried PAH-related genetic variants, including BMPR2 (30.5%), ACVRL1 (6.1%), 1 in ENG (1.2%) etc. and the variant group had worse clinical outcomes (18).
PAH patients with BMPR2 genetic variants are trended to have earlier onset and worse clinical scenarios (1, 18, 19). Some PAH Asian cohorts had a higher rate for carrying PAH-related genetic variants than the Caucasian cohorts (18–20). However, reports using whole exome sequencing (WES) for identifying genetic variants from Southeasten Asian idiopathic and heritable PAH cohorts are still rare. The aim of the current research is using WES to identify PAH-related genetic variants and investigate their hemodynamic and clinical profiles in a Taiwanese idiopathic and heritable PAH cohort.
We enrolled 45 idiopathic and heritable PAH patients from the Cardiology divisions of Taichung Veterans General Hospital (Taichung, Taiwan) and China Medical University Hospital (Taichung, Taiwan) for PAH genetic analysis between Mar. and Sep. 2021 (Figure 1). The diagnosis of PAH was based on the measurements of a mean PAP>20 mmHg at rest and a pulmonary arterial wedge pressure ≤ 15 mmHg by right heart catheterization (21). Secondary pulmonary hypertension was excluded by history, blood tests, chest computed tomography scan, nuclear ventilation-perfusion scan and echocardiography (21). We retrospectively collected information including age of initial PAH diagnosis, functional class at diagnosis, and medications and clinical worsening events after initial PAH diagnosis. This study was reviewed and approved by the Institutional Review Board of Taichung Veterans General Hospital (Approval Number, SE21052A) and China Medical University Hospital (Approval Number, CMUH109-REC3-198) (Taichung, Taiwan) and all patients gave their written consent to participate.
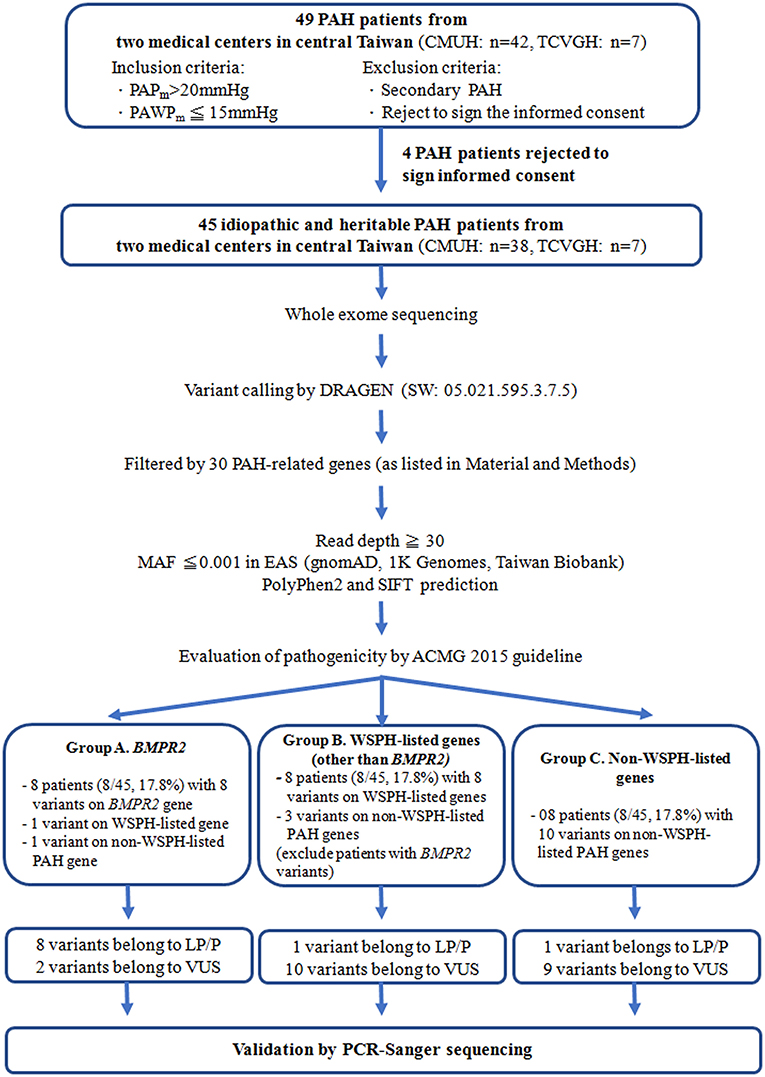
Figure 1. The study flow chart. PAH, pulmonary arterial hypertension; WSPH, The 6th World Symposium on Pulmonary Hypertension; BMPR2, bone morphogenetic protein receptor type-2; TCVGH, Taichung Veterans General Hospital (Taichung, Taiwan); CMUH, China Medical University Hospital (Taichung, Taiwan); ACMG, 2015 The American College of Medical Genetics and Genomics guidelines; P, pathogenic; LP, likely pathogenic; VUS, variant of uncertain significance; MAF, Minor allele frequency of East Asian, PAPm, mean pulmonary arterial pressure, PAWPm, mean pulmonary arterial wedge pressure; PCR, polymerase chain reaction.
All diagnoses were confirmed by right heart catheterization to measure hemodynamic values for all patients at their initial diagnosis (21). The mean PAP, pulmonary arterial wedge pressure, cardiac output, and PVR were recorded by standard techniques (21). The date of 1st right heart catheterization was deemed as the time of initial PAH diagnosis.
The diagnosis is based on the Curacao criteria. These criteria include (1) recurrent and spontaneous epistaxis; (2) visceral localization (gastrointestinal telangiectasia, pulmonary, hepatic, cerebral or spinal arteriovenous malformations) (3) an affected first-degree family member; and (4) the presence of mucocutaneous telangiectases. When an individual shows three or more criteria, they are considered to have HHT. When they meet two criteria the diagnosis is possible and with one or none criteria, HHT is considered unlikely (7, 22).
Total genomic DNA was isolated from peripheral blood using NucleoSpin® Blood Kit (Macherey-Nagel, Duren, Germany) according to manufacturer protocols. We performed the WES for all patients enrolled in this study. Whole exome libraries were processed using KAPA HyperExome Plus Kit, KAPA Universal Adapter, and KAPA HyperCapture Bead Kit according to the KAPA HyperCap Workflow v3.0 Workflow (Roche Sequencing and Life Science, MA, USA). The paired-end 2 × 150 bp sequencing was performed on the Illumina NovaSeq 6000 Sequencer (Illumina Inc., USA). For single nucleotide variants (SNV), the sequencing raw data was processed by DRAGEN platform (SW: 05.021.595.3.7.5) (Illumina Inc., USA). All obtained variants from Variant Call Format (VCF) file were further filtered by a panel of PAH-related genes including 16 WSPH-listed [BMPR2, KCNK3, EIF2AK4, KLF2, ACVRL1, SMAD1, AQP1, SMAD4, ATP13A3, SMAD9, TBX4, SOX17, CAV1, KCNA5, BMPR1B, GDF2(23) and ENG] (2) and 14 non-WSPH-listed genes [ABCA3(18, 24), NOTCH1(25, 26), NOTCH2(27, 28), NOTCH3(15, 29), ABCC8(11, 12), BMP10(30, 31), FBLN2(32), JAG1(33), PTGIS(34), PDGFD(32), GGCX(35), SMAD5(36, 37), KLK1(35) and TopBP1(26, 38)]. Those variants with read depth <30x were filtered out. The coverage of the specific gene was analyzed using WES Binary Alignment/Map (BAM) files. In this study, the average coverages of all candidate genes have >100×. All variants on those genes were compared with widely used normal subject control databases (gnomAD database, ExAC, 1000 Genomes Project and Taiwan Biobank) to filter those variants allelic frequencies more than 0.1% in East Asian group (EAS). The Polyphen2 and SIFT in silico software were applied to predict the impact of variants on specific genes. For structure variations on BMPR2 gene, we applied ExomeDepth from a free R package (version 1.1.15) to detect CNVs on the BMPR2 gene of all PAH patients (39). The suspected CNVs on BMPR2 from ExomeDepth analysis software were subjected to validation through multiplex ligation-dependent probe amplification technology using SALSA MLPA Probemix P093 HHT/HPAH kit (MRC-Holland, Netherlands). The evaluation of clinical impacts followed the 2015 ACMG guidelines (The American College of Medical Genetics and Genomics) (40).
All filtered variants identified from candidate genes were validated by polymerase-chain reaction (PCR) and Sanger sequencing. The specific PCR primers were designed manually to cover entire exons of genes. The PCR was conducted using PCRBIO Ultra Polymerase (PCR Biosystems, London, UK) and SensoQuest Labcycler (SensoQuest, Göttingen, Germany). The purified PCR products were directly sequenced with an ABI PRISM terminator cycle sequencing kit v3.1 on the ABI 3730 DNA sequencer (Applied Biosystems, CA, USA).
Continuous variables are expressed as mean ± SD and categorical data as percentages. The study subjects were grouped as BMPR2 genetic variant carriers and non-carriers. Non-normally distributed continuous variables were compared with the Mann-Whitney U test, while normally distributed variables were compared with Student's t test. Categorical variables were compared by the Chi-square test with Yate's correction or Fisher's exact test. A two-tailed p-value < 0.05 was considered statistically significant. The SPSS 12.1 statistical software package (SPSS, Inc., Chicago, IL, USA) was used for all calculations.
A total of 45 idiopathic and heritable PAH patients were enrolled for analysis. We found BMPR2 gene variants in 17.8 % (8/45) of PAH patients in central Taiwan (Table 1 and Figure 1). Among the 8 patients with BMPR2 gene variants, 3 had frameshifts, 2 had non-sense, 2 had missense variants and 1 had BMPR2 exon 8-9 deletions (Table 1 and Supplementary Figure 1). Among the 8 BPMPR2 variants, 4 variant genetic types were previously reported in the literature (4, 5, 41–45). We identified three novel BMPR2 gene variants (c.1512 deletion, c.479 duplicate, c.1165 G>A) in the current study and one patient with exon 8-9 deletions (Table 1). The patient (A365) with “c.1165 G>A” novel variant (resulting in missense amino acid change of Glu389Lys) presented with heritable PAH and a large pulmonary arteriovenous malformation (Table 2). However, she did not fit the diagnosis criterion for HHT (22).
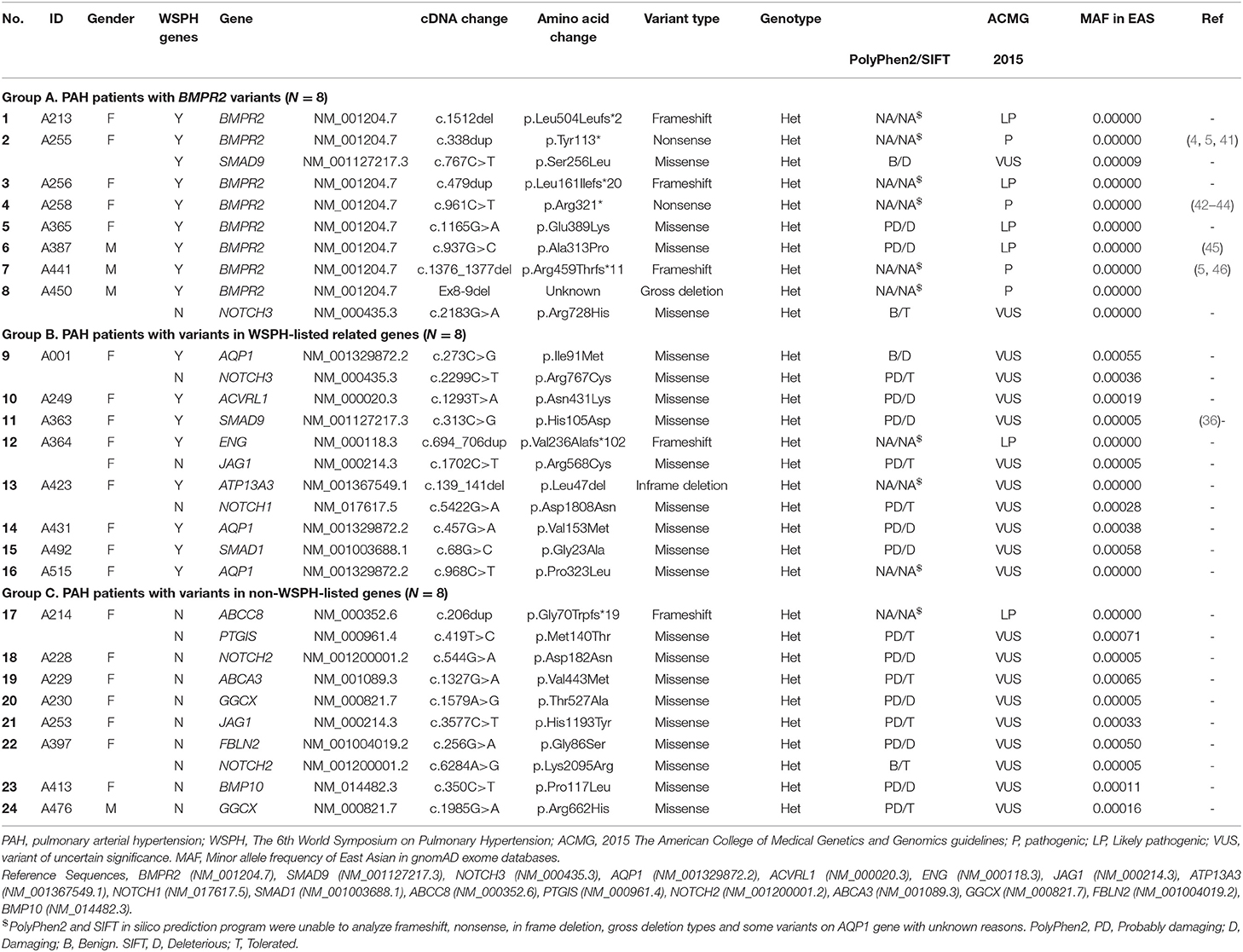
Table 1. Details of genetic variants in PAH-related genes.
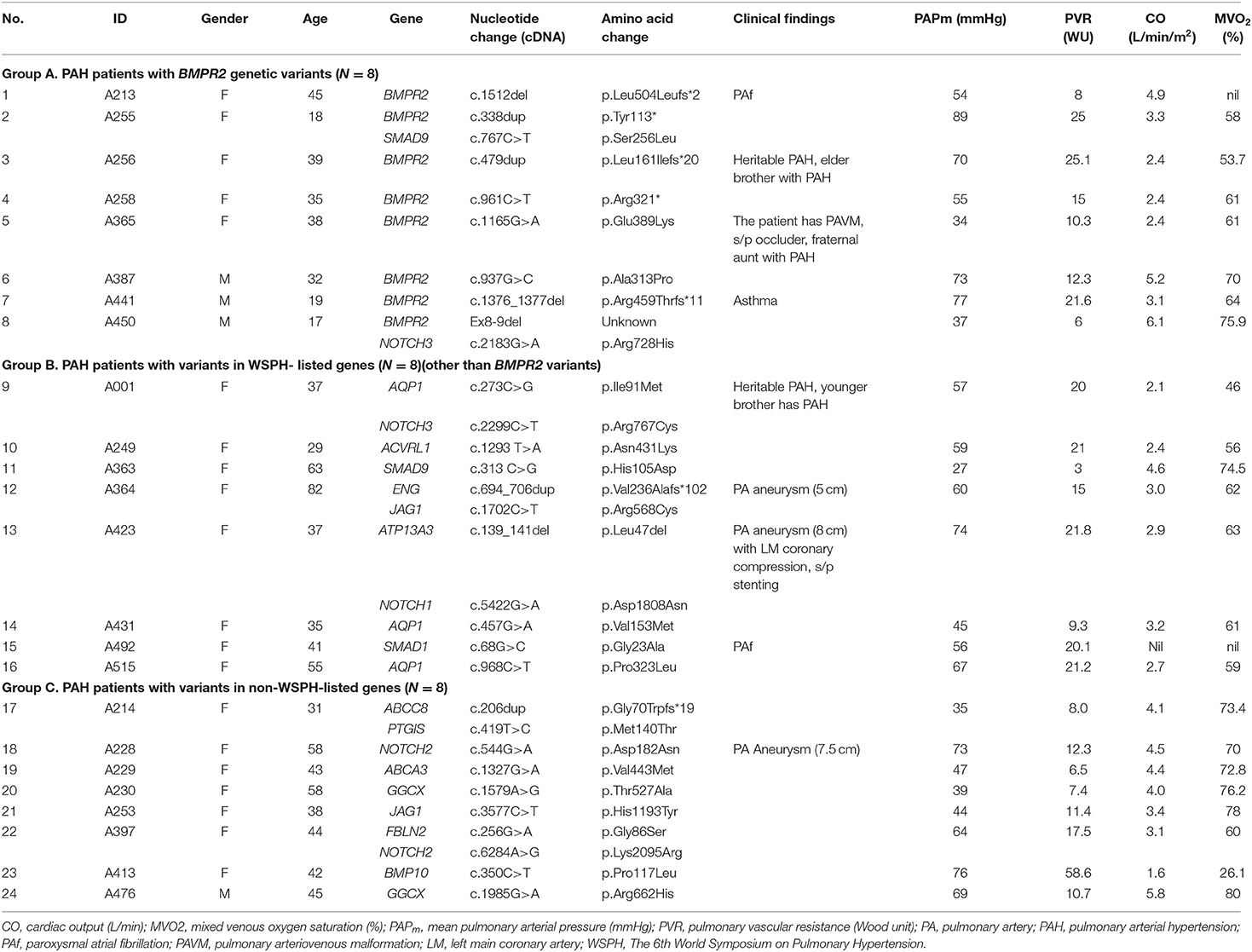
Table 2. Pulmonary arterial hypertension patients with genetic variants and their clinical findings.
The 6th WSPH provided a list of pulmonary arterial hypertension genes with higher or lower level of evidence (2). In this study, 8 patients (8/45 = 17.8%) had WSPH-listed PAH-related gene (other than BMPR2) variants (1 with ACVRL1, 1 with ENG, 1 with SMAD9, 1 with SMAD1, 1 with ATP13A3 and 3 with AQP1) (Table 1 and Figure 1). All the mis-sense variant effects were checked by software PolyPhen2 and SIFT and were listed in Table 1. The patient (A249) with ACVRL1 variant and the patient (A364) with ENG variant did not show clinical evidence of HHT (Table 2) (22).
Among the study cohort, we totally identified 14 genetic variants sites, which are not listed in WSPH PAH-related genes, from 12 patients (Figure 1 and Table 1). Novel genetic variants related with Notch pathway, such as NOTCH3, NOTCH2, NOTCH1, and JAG1 were identified in this cohort, too (Table 1). One patient carried an ABCC8 genetic variant, which is deemed likely pathogenic (Table 1).
Among the 45 study subjects, 3 of them had family aggregate of heritable PAH (Table 2). Patient (A256) with BMPR2 variant of c.479dup had a family history heritable PAH with her younger brother (not included in this study, data not shown) diagnosed with PAH, too. Patient (A365) with BMPR2 variant of c.1165 G>A had a family history heritable PAH with her fraternal aunt (not included in this study, data not shown) diagnosed with PAH and carried the same BMPR2 variant site. Though she had a large pulmonary arteriovenous malformation, she or her aunt did not fit the diagnosis of HHT. Her two siblings did not have PAH and did not receive genetic tests. Patient (A1) with AQP1 variant of c.273C>G had a family history heritable PAH with her younger brother having PAH and carried the same AQP1 variant site (not included in this study, data not shown) (Table 2).
A total of 45 idiopathic and heritable PAH patients were enrolled for analysis (Table 3). Subjects carrying BMPR2 gene variant (N = 8) were younger at diagnosis of PAH (30 ± 11 vs. 49 ± 13 years, p = 0.001) than the non-BMPR2 variant group (N = 37). BMPR2 variant carriers had a trend toward having higher mean PAP (61 ± 19 vs. 51 ± 13 mmHg, p = 0.076) than the non-carriers upon initial diagnosis. PVR, right atrial pressure, cardiac output, as well as functional class were similar between BMPR2 variant carriers and non-carriers at initial diagnosis.
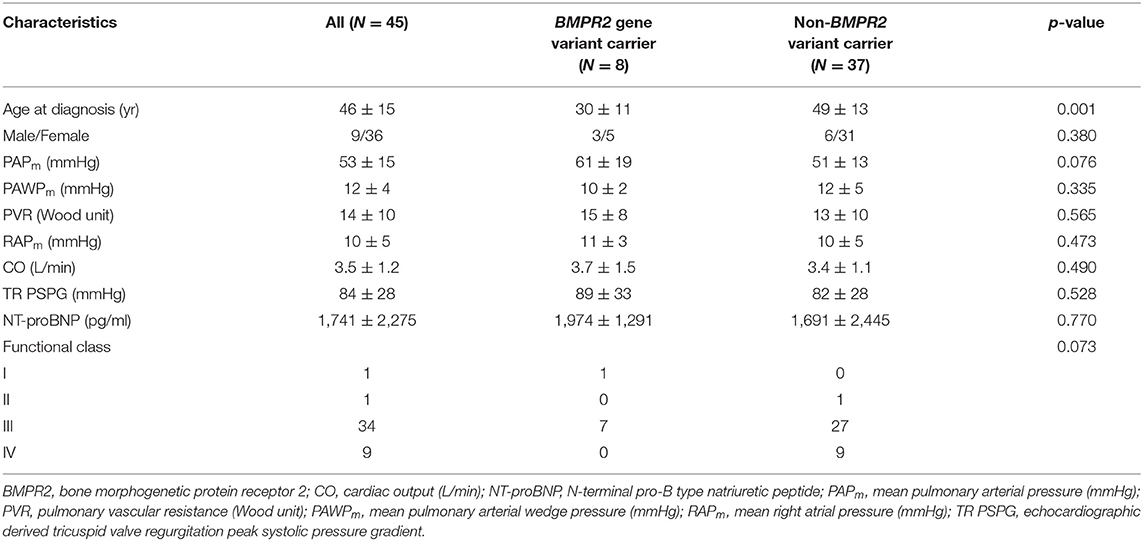
Table 3. Clinical and hemodynamic presentations at initial diagnosis for pulmonary arterial hypertension patients with or without BMPR2 genetic variant.
Idiopathic and heritable PAH patients with genetic variants are usually younger with worse hemodynamic or clinical profiles in Caucasians and Northeastern Asian cohorts (1, 18). However, reports from southeastern Asian cohorts are scarce. In this study, we identified 17.8% of patients with BMPR2 gene variants and 17.8% subjects with WSPH-listed PAH-related gene (other than BMPR2) variants in a Taiwanese PAH cohort. Idiopathic and heritable PAH patients carrying BMPR2 variant presented at a younger age with a trend toward having higher mean PAP at initial diagnosis.
BMPR2 genetic variants will lead to receptor protein deficiency and result in abnormal overactivation of the TGF-β1 signaling pathway (3). The TGF-β pathway over-activation leads to proliferation of vascular smooth muscle cells and increase of PVR in pulmonary circulation in primary PAH (3, 46). Asian cohort from Korea reported that the prevalence of BMPR2 variants in Korean primary PAH patients was 22% (19). A Japanese group reported BMPR2 variants in all 4 familial (4/4 = 100%) and 12 (12/30 = 40%) of the sporadic PAH cases, which was higher than the proportions reported in Caucasian patients (20). Another Japanese group demonstrated that with state-of-the-art therapy, the long-term survival of patients with PAH was high, regardless of the BMPR2 mutation status (47). Evans et al. conducted a meta-analysis of 1,550 patients with idiopathic, heritable, and anorexigen-associated PAH from eight cohorts and showed that 29% had BMPR2 gene mutations (1). In our study from central Taiwan, we identified BMPR2 variants in 17.8 % (8/45) of primary PAH patients, which is somewhat lower than previously reported prevalence from other cohorts. However, we identified three novel BMPR2 gene variants (c.1512 deletion, c.479 duplicate, c.1165 G>A) in the current study (Table 1), which were not reported in literature before.
The 6th WSPH listed BMPR2, EIF2AK4, TBX4, ATP13A3, GDF2, SOX17, AQP1, ACVRL1, SMAD9, ENG, KCNK3 and CAV1 mutations as higher level of evidence and SMAD4, SMAD1, KLF2, BMPR1B and KCNA5 with lower level of evidence of association with primary PAH (2). Mutations of ACVRL1, ENG, SMAD1, SMAD4, and SMAD9 are closely related with alterations of the TGF-β/BMPR2 signal pathways in pathogenesis of primary PAH (2, 4–6). In this study, we used WES and identified 17.8% subjects with 6th WSPH listed PAH-related gene variants (other than BMPR2) in a Taiwanese idiopathic and heritable PAH cohort. The protein product of EIF2AK4 belongs to a family of kinases that regulates angiogenesis. Specifically, these kinases phosphorylate the α subunit of eukaryotic translation initiation factor 2, a protein that downregulates protein synthesis in response to varied cellular stresses (9). In this study, none of our study subjects had homozygous EIF2AK4 variants and nor did anyone present with evidence of PVOD or PCH (9, 10, 48). Three of study patients had EIF2AK4 heterozygous variants (Supplementary Table 1) (48). The mutations of ACVRL1, a TGF-β/BMPR2 signal pathway member, have been found to be associated with PAH. In addition, ACVRL1 mutation has been found in patients with HHT combined with PAH (7). One study subject (A249) carried ACVRL1 variant but did not show clinical evidence of HHT. Previous report from Zhang et al. (49) reported primary PAH patients with ACVRL1 mutation have rapid disease progression, high overall mortality rate and no response to the acute pulmonary vasodilation test.
In terms of non-WSPH-listed PAH related genes, we identified 14 novel sites in 12 patients (Table 1). ABCC8 encodes SUR1, a regulatory subunit of the ATP-sensitive potassium channel (11). ABCC8 loss-of function mutations in both pediatric and adult-onset PAH had been reported (11, 12). We identified a genetic variant of c.206dup, which is deemed likely pathogenic. Notch receptors, several ligands, and components of the downstream signaling machinery are expressed in the vessel and have been reported to be related with PAH (13, 14, 50). We identified several novel genetic variant sites in NOTCH1, NOTCH 2, NOTCH 3 and JAG1 in this PAH study cohort (Table 1). However, we did not conduct further in vitro studies to prove their genetic impacts on the downstream cell signals.
This study had several limitations. First, the PAH patients were recruited from two medical centers in central Taiwan. Thus, the patient numbers were limited. Second, the PAH patients were enrolled for genetic analysis in a specific time frame between Mar. and Sep. 2021. Their clinical and hemodynamic data were then retrospectively traced back to their initial diagnosis date. Those who had the worst clinical scenario with rapid disease progression to death had no chance to be retrospectively enrolled in this genetic study. Third, we identified several novel variant sites for BMPR2 as well as other WSPH-listed or non-listed PAH-related genes. We only used software PolyPhen2 and SIFT to exam all the mis-sense mutations but did not have further in vivo studies to confirm that the genetic variants did have functional changes to the downstream proteins.
In conclusion, we identified 17.8% of patients with BMPR2 gene variants and 17.8% subjects with other WSPH-listed PAH-related gene variants in a Taiwanese idiopathic and heritable PAH cohort. In addition, we also identified 14 none WSPH-listed PAH-related genetic variants sites from 12 patient. PAH patients carrying BMPR2 variants presented at a younger age with a trend toward having higher mean PAP at initial diagnosis.
The datasets presented in this study can be found in online repositories (NCBI BioProject Accession: PRJNA824211).
The studies involving human participants were reviewed and approved by Taichung Veterans General Hospital, China Medical University Hospital (Taichung, Taiwan). The patients/participants provided their written informed consent to participate in this study.
K-WL wrote the manuscript and analyzed/interpreted the data. S-KC performed the laboratory work. K-YW, K-WL, W-WL, W-JT, and Y-WC recruited the patients and critically reviewed the manuscript for important intellectual content. K-YW designed the study. All authors read and approved the final manuscript.
The whole exome sequencing, polymerase-chain reaction and Sanger sequencing of the genomic DNA were sponsored by Excelsior Biopharma Inc. and conducted at its Genetic Laboratory. The author S-KC is employed by Excelsior Biopharma Inc. The Excelsior Biopharma Inc. was not involved in the study design, collection, analysis, interpretation of data, and the writing of this article or the decision to submit it for publication.
S-KC was employed by Excelsior Biopharma Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.911649/full#supplementary-material
Supplementary Figure 1. The result of CNVs on BMPR2 gene of pulmonary arterial hypertension (PAH) patient A450 by multiplex ligation-dependent probe amplification (MLPA) technology. (A) The probe signals indicated that there was a heterozygous deletion from BMPR2 exon 8 to exon 9 in PAH patient A450. (B) The quantification plot of copy number variation of A450 on BMPR2, ENG, and ACVRL1 genes using the SALSA MLPA Probemix P093 HHT/HPAH kit (MRC-Holland, Netherlands). BMPR2 5: the sample analytical number of A450. WT: the sample derived from normal subjects.
Supplementary Table 1. Details of genetic variants in EIF2AK4 (NM_001013703.4).
1. Evans JD, Girerd B, Montani D, Wang XJ, Galie N, Austin ED, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med. (2016) 4:129–37. doi: 10.1016/S2213-2600(15)00544-5
2. Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F, et al. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J. (2019) 53:1801899. doi: 10.1183/13993003.01899-2018
3. Sharmin N, Nganwuchu CC, Nasim MT. Targeting the TGF-beta signaling pathway for resolution of pulmonary arterial hypertension. Trends Pharmacol Sci. (2021) 42:510–3. doi: 10.1016/j.tips.2021.04.002
4. Fujiwara M, Yagi H, Matsuoka R, Akimoto K, Furutani M, Imamura S, et al. Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ J. (2008) 72:127–33. doi: 10.1253/circj.72.127
5. Chida A, Shintani M, Yagi H, Fujiwara M, Kojima Y, Sato H, et al. Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am J Cardiol. (2012) 110:586–93. doi: 10.1016/j.amjcard.2012.04.035
6. Graf S, Haimel M, Bleda M, Hadinnapola C, Southgate L, Li W, et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun. (2018) 9:1416. doi: 10.1038/s41467-018-03672-4
7. Vorselaars VMM, Hosman AE, Westermann CJJ, Snijder RJ, Mager JJ, Goumans MJ, et al. Pulmonary arterial hypertension and hereditary haemorrhagic telangiectasia. Int J Mol Sci. (2018) 19:3203. doi: 10.3390/ijms19103203
8. Southgate L, Machado RD, Graf S, Morrell NW. Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol. (2020) 17:85–95. doi: 10.1038/s41569-019-0242-x
9. Eyries M, Montani D, Girerd B, Perret C, Leroy A, Lonjou C, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet. (2014) 46:65–9. doi: 10.1038/ng.2844
10. Best DH, Sumner KL, Austin ED, Chung WK, Brown LM, Borczuk AC, et al. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest. (2014) 145:231–6. doi: 10.1378/chest.13-2366
11. Lago-Docampo M, Tenorio J, Hernandez-Gonzalez I, Perez-Olivares C, Escribano-Subias P, Pousada G, et al. Characterization of rare ABCC8 variants identified in Spanish pulmonary arterial hypertension patients. Sci Rep. (2020) 10:15135. doi: 10.1038/s41598-020-72089-1
12. Bohnen MS, Ma L, Zhu N, Qi H, McClenaghan C, Gonzaga-Jauregui C, et al. Loss-of-function ABCC8 mutations in pulmonary arterial hypertension. Circ Genom Precis Med. (2018) 11:e002087. doi: 10.1161/CIRCGEN.118.002087
13. Morris HE, Neves KB, Montezano AC, MacLean MR, Touyz RM. Notch3 signalling and vascular remodelling in pulmonary arterial hypertension. Clin Sci. (2019) 133:2481–98. doi: 10.1042/CS20190835
14. Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, et al. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med. (2009) 15:1289–97. doi: 10.1038/nm.2021
15. Chida A, Shintani M, Matsushita Y, Sato H, Eitoku T, Nakayama T, et al. Mutations of NOTCH3 in childhood pulmonary arterial hypertension. Mol Genet Genomic Med. (2014) 2:229–39. doi: 10.1002/mgg3.58
16. Pfarr N, Szamalek-Hoegel J, Fischer C, Hinderhofer K, Nagel C, Ehlken N, et al. Hemodynamic and clinical onset in patients with hereditary pulmonary arterial hypertension and BMPR2 mutations. Respir Res. (2011) 12:99. doi: 10.1186/1465-9921-12-99
17. Sztrymf B, Coulet F, Girerd B, Yaici A, Jais X, Sitbon O, et al. Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am J Respir Crit Care Med. (2008) 177:1377–83. doi: 10.1164/rccm.200712-1807OC
18. Zhang H-S, Liu Q, Piao C-M, Zhu Y, Li Q-Q, Du J, et al. Genotypes and phenotypes of Chinese pediatric patients with idiopathic and heritable pulmonary arterial hypertension-a single-center study. Can J Cardiol. (2019) 35:1851–6. doi: 10.1016/j.cjca.2019.07.628
19. Jang AY, Kim BG, Kwon S, Seo J, Kim HK, Chang HJ, et al. Prevalence and clinical features of bone morphogenetic protein receptor type 2 mutation in Korean idiopathic pulmonary arterial hypertension patients: the PILGRIM explorative cohort. PLoS One. (2020) 15:e0238698. doi: 10.1371/journal.pone.0238698
20. Morisaki H, Nakanishi N, Kyotani S, Takashima A, Tomoike H, Morisaki T. BMPR2 mutations found in Japanese patients with familial and sporadic primary pulmonary hypertension. Hum Mutat. (2004) 23:632. doi: 10.1002/humu.9251
21. Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. (2016) 37:67–119. doi: 10.1093/eurheartj/ehv317
22. Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. (2011) 48:73–87. doi: 10.1136/jmg.2009.069013
23. Wang X-J, Lian T-Y, Jiang X, Liu S-F, Li S-Q, Jiang R, et al. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur Respir J. (2019) 53:1801609. doi: 10.1183/13993003.01609-2018
24. Kunig AM, Parker TA, Nogee LM, Abman SH, Kinsella JP. ABCA3 deficiency presenting as persistent pulmonary hypertension of the newborn. J Pediatr. (2007) 151:322–4. doi: 10.1016/j.jpeds.2007.05.054
25. Wang S, Zhu G, Jiang D, Rhen J, Li X, Liu H, et al. Reduced Notch1 cleavage promotes the development of pulmonary hypertension. Hypertension. (2022) 79:79–92. doi: 10.1161/HYPERTENSIONAHA.120.16065
26. Hernandez-Gonzalez I, Tenorio-Castano J, Ochoa-Parra N, Gallego N, Perez-Olivares C, Lago-Docampo M, et al. Novel genetic and molecular pathways in pulmonary arterial hypertension associated with connective tissue disease. Cells. (2021) 10:1488. doi: 10.3390/cells10061488
27. Hurst LA, Dunmore BJ, Long L, Crosby A, Al-Lamki R, Deighton J, et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat Commun. (2017) 8:14079. doi: 10.1038/ncomms14079
28. Sahoo S, Li Y, de Jesus D, Sembrat J, Rojas MM, Goncharova E, et al. Notch2 suppression mimicking changes in human pulmonary hypertension modulates Notch1 and promotes endothelial cell proliferation. Am J Physiol Heart Circ Physiol. (2021) 321:H542–57. doi: 10.1152/ajpheart.00125.2021
29. Gómez J, Reguero JR, Junquera MR, Alvarez C, Morís C, Alonso B, et al. Next generation sequencing of the NOTCH3 gene in a cohort of pulmonary hypertension patients. Int J Cardiol. (2016) 209:149–50. doi: 10.1016/j.ijcard.2016.02.024
30. Eyries M, Montani D, Nadaud S, Girerd B, Levy M, Bourdin A, et al. Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur Respir J. (2019) 53:1801371. doi: 10.1183/13993003.01371-2018
31. Hodgson J, Swietlik EM, Salmon RM, Hadinnapola C, Nikolic I, Wharton J, et al. Characterization of GDF2 mutations and levels of BMP9 and BMP10 in pulmonary arterial hypertension. Am J Respir Crit Care Med. (2020) 201:575–85. doi: 10.1164/rccm.201906-1141OC
32. Zhu N, Swietlik EM, Welch CL, Pauciulo MW, Hagen JJ, Zhou X, et al. Rare variant analysis of 4241 pulmonary arterial hypertension cases from an international consortium implicates FBLN2, PDGFD, and rare de novo variants in PAH. Genome Med. (2021) 13:80. doi: 10.1186/s13073-021-00915-w
33. Xiao Y, Gong D, Wang W. Soluble JAGGED1 inhibits pulmonary hypertension by attenuating notch signaling. Arterioscler Thromb Vasc Biol. (2013) 33:2733–9. doi: 10.1161/ATVBAHA.113.302062
34. Wang X-J, Xu X-Q, Sun K, Liu K-Q, Li S-Q, Jiang X, et al. Association of rare PTGIS variants with susceptibility and pulmonary vascular response in patients with idiopathic pulmonary arterial hypertension. JAMA Cardiol. (2020) 5:677–84. doi: 10.1001/jamacardio.2020.0479
35. Zhu N, Pauciulo MW, Welch CL, Lutz KA, Coleman AW, Gonzaga-Jauregui C, et al. Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med. (2019) 11:69. doi: 10.1101/550327
36. Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM, et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat. (2011) 32:1385–9. doi: 10.1002/humu.21605
37. Maron BA. Clarifying the Pulmonary Arterial Hypertension Molecular Landscape Using Functional Genetics. Am J Respir Crit Care Med. (2020) 202:488–90. doi: 10.1164/rccm.202004-1411ED
38. de Jesus Perez VA, Yuan K, Lyuksyutova MA, Dewey F, Orcholski ME, Shuffle EM, et al. Whole-exome sequencing reveals TopBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. (2014) 189:1260–72. doi: 10.1164/rccm.201310-1749OC
39. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. (2012) 28:2747–54. doi: 10.1093/bioinformatics/bts526
40. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
41. Gamou S, Kataoka M, Aimi Y, Chiba T, Momose Y, Isobe S, et al. Genetics in pulmonary arterial hypertension in a large homogeneous Japanese population. Clin Genet. (2018) 94:70–80. doi: 10.1111/cge.13154
42. Long L, Yang X, Southwood M, Moore S, Crosby A, Upton PD, et al. Targeting translational read-through of premature termination mutations in BMPR2 with PTC124 for pulmonary arterial hypertension. Pulm Circ. (2020) 10:2045894020935783. doi: 10.1177/2045894020935783
43. Drake KM, Dunmore BJ, McNelly LN, Morrell NW, Aldred MA. Correction of nonsense BMPR2 and SMAD9 mutations by ataluren in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. (2013) 49:403–9. doi: 10.1165/rcmb.2013-0100OC
44. Wang H, Cui QQ, Sun K, Song L, Zou YB, Wang XJ, et al. Identities and frequencies of BMPR2 mutations in Chinese patients with idiopathic pulmonary arterial hypertension. Clin Genet. (2010) 77:189–92. doi: 10.1111/j.1399-0004.2009.01335.x
45. Machado RD, Aldred MA, James V, Harrison RE, Patel B, Schwalbe EC, et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat. (2006) 27:121–32. doi: 10.1002/humu.20285
46. Sankelo M, Flanagan JA, Machado R, Harrison R, Rudarakanchana N, Morrell N, et al. BMPR2 mutations have short lifetime expectancy in primary pulmonary hypertension. Hum Mutat. (2005) 26:119–24. doi: 10.1002/humu.20200
47. Kabata H, Satoh T, Kataoka M, Tamura Y, Ono T, Yamamoto M, et al. Bone morphogenetic protein receptor type 2 mutations, clinical phenotypes and outcomes of Japanese patients with sporadic or familial pulmonary hypertension. Respirology. (2013) 18:1076–82. doi: 10.1111/resp.12117
48. Hadinnapola C, Bleda M, Haimel M, Screaton N, Swift A, Dorfmuller P, et al. Phenotypic characterization of EIF2AK4 mutation carriers in a large cohort of patients diagnosed clinically with pulmonary arterial hypertension. Circulation. (2017) 136:2022–33. doi: 10.1161/CIRCULATIONAHA.118.033970
49. Zhang X, Zhang C, Li Q, Piao C, Zhang H, Gu H. Clinical characteristics and prognosis analysis of idiopathic and hereditary pulmonary hypertension patients with ACVRL1 gene mutations. Pulm Circ. (2021) 11:20458940211044577. doi: 10.1177/20458940211044577
Keywords: activin receptor-like kinase 1 (ACVRL1), aquaporin 1 (AQP1), bone morphogenetic protein receptor type-2 (BMPR2), pulmonary arterial hypertension (PAH), whole exome sequencing (WES), gene variants
Citation: Liang K-W, Chang S-K, Chen Y-W, Lin W-W, Tsai W-J and Wang K-Y (2022) Whole Exome Sequencing of Patients With Heritable and Idiopathic Pulmonary Arterial Hypertension in Central Taiwan. Front. Cardiovasc. Med. 9:911649. doi: 10.3389/fcvm.2022.911649
Received: 04 April 2022; Accepted: 25 May 2022;
Published: 22 June 2022.
Edited by:
Christoph Sinning, University Medical Center Hamburg-Eppendorf, GermanyReviewed by:
Ayako Chida-Nagai, Hokkaido University Hospital, JapanCopyright © 2022 Liang, Chang, Chen, Lin, Tsai and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kuo-Yang Wang, d2t5ODMwQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.