 Yiwen Zha
Yiwen Zha Wenwen Zhuang1
Wenwen Zhuang1- 1Medical College, Yangzhou University, Yangzhou, China
- 2Institute of Translational Medicine, Medical College, Yangzhou University, Yangzhou, China
- 3Jiangsu Key Laboratory of Integrated Traditional Chinese and Western Medicine for Prevention and Treatment of Senile Diseases, Yangzhou University, Yangzhou, China
Vascular smooth muscle cells (VSMCs) are the primary cell type involved in the atherosclerosis process; senescent VSMCs are observed in both aged vessels and atherosclerotic plaques. Factors associated with the atherosclerotic process, including oxidative stress, inflammation, and calcium-regulating factors, are closely linked to senescence in VSMCs. A number of experimental studies using traditional cellular aging markers have suggested that anti-aging biochemical agents could be used to treat atherosclerosis. However, doubt has recently been cast on such potential due to the increasingly apparent complexity of VSMCs status and an incomplete understanding of the role that these cells play in the atherosclerosis process, as well as a lack of specific or spectrum-limited cellular aging markers. The utility of anti-aging drugs in atherosclerosis treatment should be reevaluated. Promotion of a healthy lifestyle, exploring in depth the characteristics of each cell type associated with atherosclerosis, including VSMCs, and development of targeted drug delivery systems will ensure efficacy whilst evaluation of the safety and tolerability of drug use should be key aims of future anti-atherosclerosis research. This review summarizes the characteristics of VSMC senescence during the atherosclerosis process, the factors regulating this process, as well as an overview of progress toward the development and application of anti-aging drugs.
Introduction
The elderly population is increasing across the world, and vascular aging in particular is a major risk factor for atherosclerotic cardiovascular disease (1). Vascular aging accelerates the progression of atherosclerosis, however, the developmental processes governing atherosclerotic lesions are complex (2). Vascular smooth muscle cells (VSMCs) are located within the vascular architecture and are the predominant cell type involved in all stages of atherosclerotic plaques (3, 4). Cellular senescence has been associated with atherosclerosis development, and can be divided into two discreet categories: replicative senescence (RS) and stress-induced premature senescence (SIPS) (5, 6). Senescent cells cease to proliferate, however, cells remain metabolically active and can promote inflammation, giving rise to the term senescence-associated secretory phenotype (SASP) (7). Senescent VSMCs have been reported in both aged vessels and atherosclerotic plaques (1, 8). The suitability and reliability of traditional markers of senescence have recently been questioned due to a lack of specificity and application across cell types (9–11). Further, existing anti-aging drugs have limitations in their application during atherosclerosis treatment (12). To address these issues in this review, we: (1) attempt to summarize the characteristics of VSMCs senescence during the atherosclerosis process; (2) identify key factors regulating VSMCs senescence; (3) assess potential pathways that may be exploited therapeutically to mitigate VSMC senescence; (4) discuss the progression of atherosclerotic cardiovascular disease, and the current challenges faced in fully characterizing this process.
Accumulation of Senescent VSMCs During Atherosclerosis
Cellular senescence is not a static cellular state, but a dynamic process during which cells undergo quiescence (initial transient senescence), early senescence (stable growth arrest), complete senescence (chromatin changes associated with senescence and SASP), and late/deep senescence (phenotypic diversification) (13). Similar to other cell types, senescent VSMCs have impaired proliferative potential coupled with increased propensity for expression of cellular senescence markers and cell death (1). VSMCs aging is characterized by a shift from a contractile phenotype to a synthetic phenotype, impaired response to contractile or diastolic mediators secreted by endothelial cells, and changes in ion channel expression and abundance in the cell membrane (14). In addition, the occurrence of senescence follows the formation of VSMC polyploidy. A recent study has shown that proprotein convertase subtilisin/kexin type 9 (PCSK9) induced VSMCs senescence, possibly through a cellular mechanism of polyploidization involving downregulation of apolipoprotein E receptor 2 (ApoER2) (15). In atherosclerosis, senescent VSMCs may be present only in the intima rather than the mesenchyme (16), and VSMCs senescence is associated primarily with plaque size rather than plaque formation (17). Advanced atherosclerotic plaques are covered by fibrous caps containing VSMCs and extracellular matrix (ECM) molecules (18). Given that VSMCs can secrete and deposit ECM proteins, they are generally considered to be protective against atherosclerotic plaque instability (19). However, senescent VSMCs promote plaque vulnerability by secreting matrix-degrading proteases. Compared with normal VSMCs, collagen secretion from senescent VSMCs is reduced which further impairs plaque stability (20). Thus, senescent VSMCs not only accumulate in the atherosclerotic setting, but their properties exacerbate the development of atherosclerosis and increase the risk of atherosclerosis-related complications.
The Pro-Atherosclerotic Properties of Senescent VSMCs
Oxidative Stress
In conditions considered to be major risk factors for atherosclerotic cardiovascular disease (including diabetes, hypertension, dyslipidemia and smoking), reactive oxygen species (ROS) are at increased abundance in the vessel wall (21). The free radical theory of aging suggests that aging occurs when cells suffer lasting damage due to sustained attack by free radicals (22). Mitochondria and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) are the two major sources of ROS production in the cell (23). Mitochondria are responsible for the production of adenosine triphosphate (ATP) via oxidative phosphorylation, a process which produces ROS as a by-product (24). The primary function of NOXs is to produce ROS (23). Alterations in generation of ROS are thought to be important contributing factors to VSMCs senescence and atherosclerotic progression (Figure 1). Studies have confirmed that beneficial effects of the active form of vitamin D on vascular health are achieved by inhibiting the production of free radicals and preventing premature aging of VSMCs (25). Aldehyde dehydrogenase 2 (ALDH2) deficiency in mice can reduce atherosclerotic plaque area by accelerating mitochondrial ROS-mediated VSMC senescence, while promoting plaque instability (26). Overexpression of NOX4 in four-month old mice resulted in elevated mitochondrial H2O2 and superoxide production in aortic VSMCs, which, in parallel, induced DNA damage and increased cellular senescence and pro-inflammatory gene expression (23).
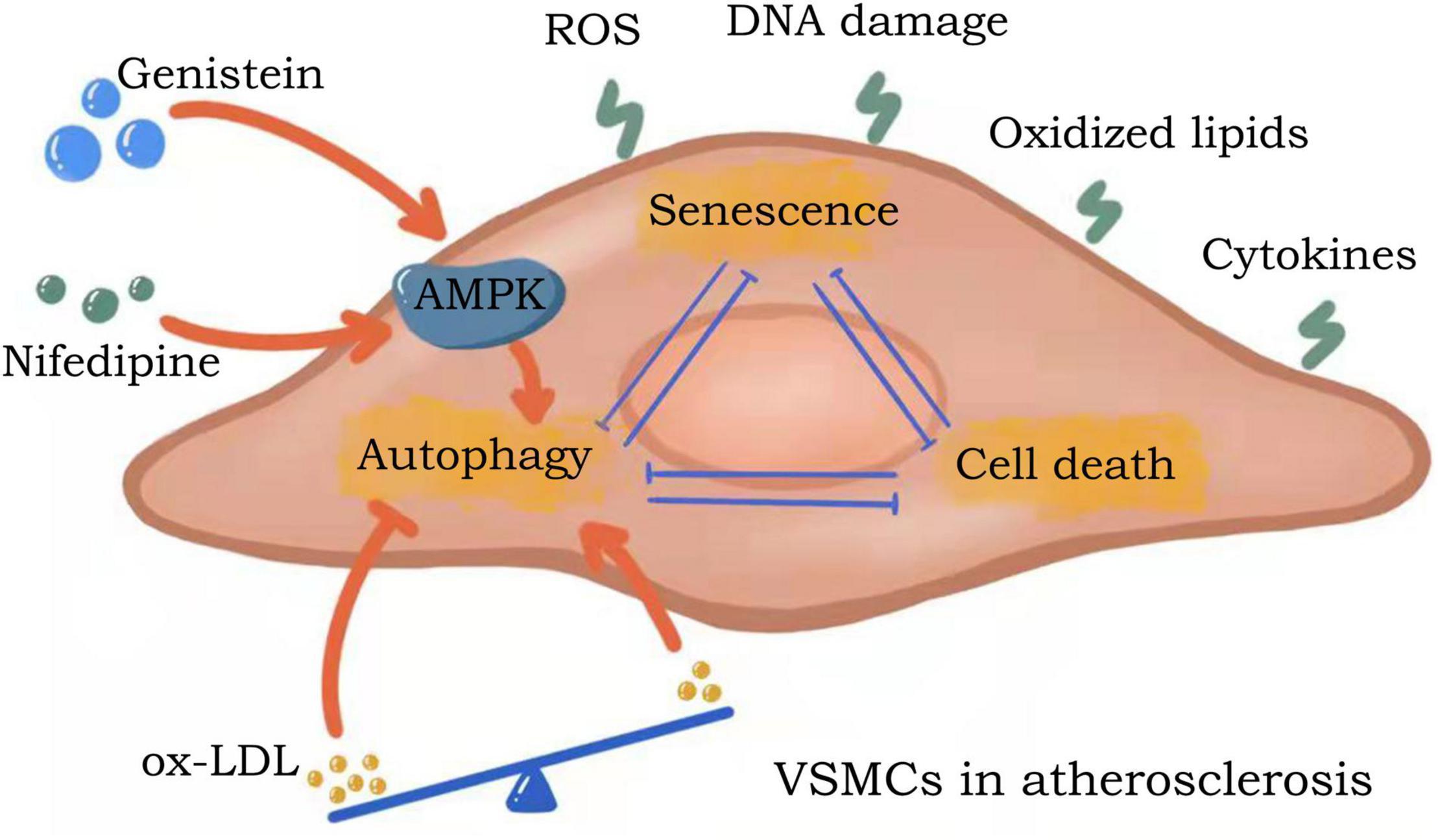
Figure 1. Potential mechanisms of ROS effects on VSMCs senescence and atherosclerosis. Both increased ALHD2 and decreased NOX4 promote ROS production, while vitamin D inhibits ROS production. All of these can regulate the senescence process of VSMCs, thus affecting the formation of atherosclerotic plaques. although oxidative DNA damage occurs in both genomic and mtDNA molecules, the propensity for mtDNA damage due to ROS is disputed. ALDH2, aldehyde dehydrogenase 2; NOX4, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4; ROS, reactive oxygen species; mtDNA, mitochondrial DNA.
DNA damage is primarily mediated by physiological or pathological ROS, and the low redox potential of guanine makes this base particularly vulnerable to oxidation (27). 8-Oxoguanine (8oxoG) is the most abundant damaging DNA modification induced by ROS exposure. Human atherosclerotic plaque VSMCs are proposed to have 8oxoG basal excision repair (BER) defects and are associated with reduced 8oxoG DNA glycosylase (OGG1) expression and acetylation (28). BER is a key process in genome integrity and maintenance, evidenced by the severe premature aging and metabolism-deficient phenotypes of BER-deficient mice (29). Notably, although oxidative DNA damage occurs in both genomic and mitochondrial DNA (mtDNA) molecules, the propensity for mtDNA damage due to ROS is disputed (24, 30). When first described, there was no increase in ROS in mutant mice despite the presence of widespread mtDNA deficiency (31, 32). In subsequent studies, no changes in ROS were observed despite increased mtDNA damage, atherosclerosis, and atherosclerotic plaque vulnerability (33). Nevertheless, long-term mtDNA damage leads to mitochondrial dysfunction and more damaging ROS production, which induces cellular senescence and hastens atherosclerosis development (34).
Oxidative stress has also been shown to cause accelerated telomere shortening as it is rich in guanine and therefore prone to prone to be oxidized to 8oxoG (35). Telomere shortening was strongly associated with increasing severity of atherosclerosis, and telomere length was a putative risk factor for atherosclerotic cardiovascular disease (36, 37). Compared with normal VSMCs, human fibrous cap VSMCs exhibited distinctly shorter telomeres (38). Furthermore, loss of telomeric repeat-binding factor-2 (TRF2), which plays an important role in maintaining telomeres, has been shown to promote plaque VSMCs senescence and exacerbate plaque instability in atherosclerosis (8). In addition, increased ROS and SASP have been shown to linked to immune senescence, which could further contribute to increased inflammation and promotes atherosclerosis. In T-Cells, oxidative stress reduced telomerase activity, enabling T-cell senescence and creating pro-inflammatory phenotypes within the plaques, for example (39). However, whether oxidative stress can modulate VSMCs senescence by affecting telomere length and thus the onset and progression of atherosclerosis has not been elucidated.
Inflammation
A low-grade chronic inflammation which contributes to the development of geriatric disease and persists throughout the geriatric process is known as “inflamm-aging.” Inflamm-aging is caused by the interaction between environmental and genetic factors, and induces a senescent state in VSMCs (40, 41). The aging process in VSMCs is accompanied by SASP, resulting in the production of pro-inflammatory cytokines including, but not limited to, interleukin (IL)-1α, IL-1β, IL-6, IL-8, IL-18 and TNF-α (42). Chronic inflammation is exacerbated by ROS production and reduced antioxidant capacity (43). ROS regulates a variety of cytokines and chemokines at the transcriptional level through modulation of transcription factors, generating a positive feedback loop whereby inflammatory regulators stimulate inflammation and ROS production in the surrounding environment (44). This ultimately leads to an amplification of the aging and atherosclerotic processes.
In the course of atherosclerosis, VSMCs senescence is closely linked to the inflammatory response. Studies suggest that senescent VSMCs may drive the transition to SASP in an IL-1α-dependent manner, inducing adjacent cells into a pro-inflammatory state and thus directly contributing to the chronic inflammation associated with atherosclerosis (45). In human atherosclerosis, coagulation factor Xa (FXa) is locally produced and activated. Chronic stimulation of FXa promotes VSMC senescence, upregulates expression of insulin-like growth factor binding protein 5 (IGFBP-5) and p53, and leads to chronic inflammation (46). Conversely, in mice sirtuin protein 6 is reported to delay the senescence process in VSMCs by promoting stability of atherosclerotic plaques via reduced inflammatory cytokine expression and protection of telomere integrity (47). Several recent studies have reported that alogliptin and omarigliptin, inhibitors of dipeptidyl peptidase-4 (DPP-4), have protective effects against IL-1β and TNF-α-induced VSMC senescence, respectively (48, 49). Therefore, therapies targeting the reduction of inflammation to delay VSMCs senescence may be therapeutically beneficial as anti-atherosclerosis agents to promote plaque stability.
Calcium Regulators
Vascular smooth muscle cells are key to the vascular calcification process (50, 51). Two broad and discreet vascular calcification processes have been described: arterial medial calcification (AMC) and arterial intimal calcification (AIC)(52, 53). Although the phenotypic alterations associated with calcification in VSMCs are similar for both AMC and AIC, the factors driving calcification in each are significantly different. AMC is commonly seen in patients with diabetes and chronic renal failure (54–56). By contrast, AIC is more often associated with AS, and patients with AIC tend to express high levels of serum pro-inflammatory cytokines despite calcium phosphate homeostasis (57, 58). Genealogy tracking studies in mouse models of atherosclerosis have shown that osteochondrocyte-like precursor cells and chondrocytes observed in atherosclerotic lesions are typically derived from VSMCs, suggesting that these cells are important mediators of AIC (59). Common mechanisms by which VSMCs contribute to AIC may include transdifferentiation into osteochondrocyte and macrophage lineages, facilitating the release of extracellular vesicles and apoptotic vesicles that promote calcification (60, 61), production of collagen and elastin matrices that facilitate calcification (62, 63), and regulation of the production of pro-calcification molecules and inhibitors of calcification (64–66).
Aging VSMCs exhibit a pro-calcification phenotype and activate multiple osteogenic pathways such as runt-associated transcription factor 2 (Runx-2), bone morphogenetic protein 2 (BMP-2), alkaline phosphatase (ALP), osteopontin (OPN), and osteoprotegerin (OPG) in response to stimulation by inflammation and oxidative stress (42, 67). Downregulation of miR-542-3p in VSMCs plays an important role in the osteogenic transformation of these cells in aging rats and is achieved by targeting BMP-7 (68). Recently, myostatin was reported to reduce the expression of Runx-2 and BMP-2 in rat VSMCs, operating through the mammalian target of rapamycin (mTOR) signaling pathway and thereby attenuating VSMCs calcification (69).
The relationship between VSMCs aging and AMC has been elaborated upon by several studies. For example, circulating miR-34a was positively correlated with IL-6 in a healthy population with an age range between 20 and 90 years; while deletion of miR-34a in mice promoted SASP in VSMCs and exacerbation of AMC (70). Furthermore, in a mouse model of aging, senescence was associated with AMC and the appearance of osteoblast-like VSMCs expressing Runx-2 (67). Notably, although aging increases risk of both AMC and AIC, and aging in VSMCs contributes to the pro-calcification phenotypic shift, it is currently unclear whether VSMCs aging is also involved in atherosclerotic AIC.
Autophagy and Apoptosis
Continuous stimulation of VSMCs in the arterial wall or atherosclerotic lesions by oxidized lipids, ROS, inflammatory cytokines, DNA damage molecules or other stimulants, a response is mounted via three pathways: resistance (autophagy), adaptation (senescence), or death (apoptosis/necrosis) (71–73). VSMCs can promote cell survival by activating autophagy pathways to counteract damage (74). VSMCs adapt to stress conditions by inhibiting proliferation and effectively entering a senescent state. Although VSMCs survive constant stimulation, senescent VSMCs undergo significant metabolic and morphological alterations. When subjected to chronic, excessive stimulation, VSMCs eventually undergo cell death via apoptotic or necrotic pathways.
Vascular smooth muscle cells autophagy, senescence, and apoptosis are all interrelated in the context of atherosclerosis (Figure 2). Autophagy in VSMCs is moderated by a balance of oxidized low-density lipoprotein (ox-LDL), where low to moderate ox-LDL concentrations promote autophagy and elevated concentrations inhibit autophagy (75). Stimulation of autophagy inhibits VSMC senescence, and conversely, inhibition of autophagy promotes VSMC senescence. Studies have shown that genistein, the major isoflavone in soy products, can promote autophagy by inducing LKB1-AMPK activation, thereby inhibiting the aging of VSMCs (76). Similarly, nifedipine-induced AMPK activation can inhibit the aging process in VSMCs by regulating autophagic flux (77). Senescent VSMCs are widely characterized as anti-apoptotic due to their expression of anti-apoptotic proteins (78). During apoptosis, the autophagic pathway is inhibited to ensure the complete execution of cell death. Thus, VSMCs autophagy, senescence and death are interconnected and are negatively correlated in atherosclerosis (20).
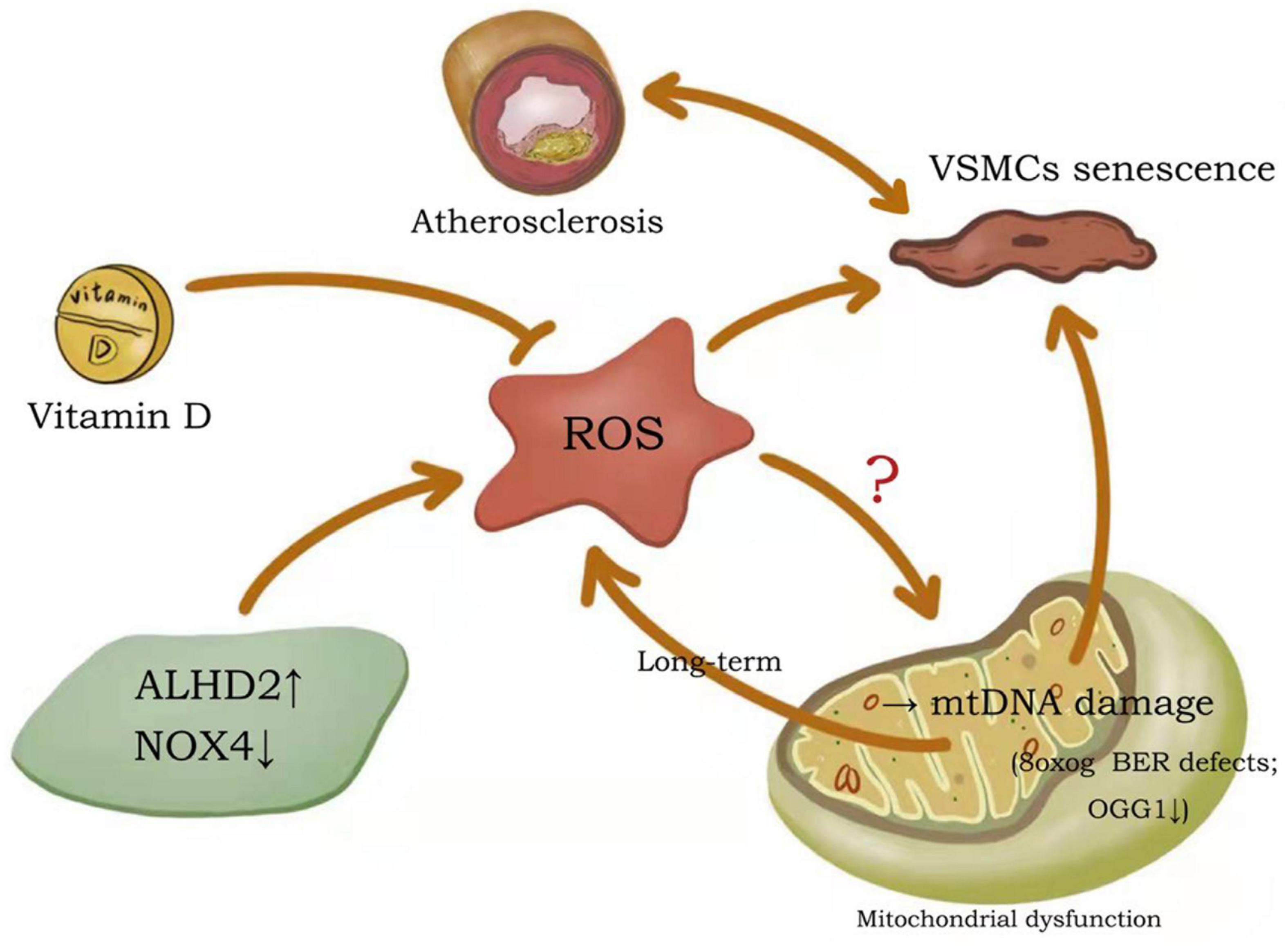
Figure 2. Crosstalk between autophagy, senescence and apoptosis in VSMCs. Continuous stimulation of VSMCs in the arterial wall or atherosclerotic lesions by oxidized lipids, ROS, inflammatory cytokines, DNA damage molecules or other stimulants, a response is mounted via three pathways: resistance (autophagy), adaptation (senescence), or death (apoptosis/necrosis). Autophagy in VSMCs is moderated by a balance of ox-LDL. VSMCs autophagy, senescence and death are interconnected and are negatively correlated in atherosclerosis.
Lifestyle Influences VSMCs Senescence
Living a healthy lifestyle may be crucial for retarding aging and preventing age-related diseases. Poor lifestyle practices include smoking and disturbed circadian rhythms, among others. Factors such as the ones listed above may increase cardiovascular risk by negatively affecting VSMCs. More than one billion people worldwide smoke, and smoking accounts for one-third of cardiovascular disease deaths among lifestyle risk factors, and the additional harmful effects of smoking put older adults at higher risk for cardiovascular disease (79). For VSMCs, the source of smoking-induced oxidative stress is NOX-1 (80) and smoking increases the expression of MMP-9. Cotinine, a metabolite of nicotine, has been shown to increase telomerase activity in VSMCs. Thus, even following nicotine metabolism, there can still be long-term adverse effects on vascular function (81). However, whether NOX-1 mediates the effects of nicotine and its metabolites on aging remains to be elucidated. Additionally, circadian rhythms are strongly correlated with atherothrombotic events (82). Researchers have found that circadian misalignment promotes VSMCs apoptosis through defective autophagy (83). However, it is not yet known if circadian rhythms influence VSMCs senescence. To conclude, a healthy lifestyle is almost certainly beneficial to reduce the risk of atherosclerosis, and delaying VSMCs senescence may be one important link of that.
Senotherapeutics
Pharmacological interventions targeting senescent cells, known as senotherapeutics, fall into two primary categories: senolytics and senomorphics. These agents aim to prevent or delay cellular senescence, thereby delaying age-related pathological processes (84). Senomorphics indirectly prevent cellular senescence by inhibiting SASP, and do not promote cell death (85). Therapies based on senomorphics mainly neutralize SASP by targeting pathways which influence SASP expression (including mTOR, JAK/STAT and PI3k/Akt) and related transcription factors (such as NF-κB and STAT3), or by using targeted antibodies (antibodies targeting IL-1α, IL-6 and IL-8 for example) (84). Previous studies have shown that blackberry, raspberry and black raspberry polyphenol extracts attenuate angiotensin II-induced senescence in VSMCs (86). Recent findings suggest that the atherogenic protective effect of blackberries is mediated through a Nox-1-dependent mechanism, but this is limited to ApoE–/– male mice, not females, and the anti-atherogenic effect of blackberries is independent of circulating total cholesterol, low-density lipoprotein and triglyceride levels (87). Calorie restriction as well as exercise has been shown to be senomorphic. Research has confirmed that calorie restriction without causing malnutrition appears to be the most effective lifestyle-based anti-aging strategy. Calorie restriction reduces risk factors for vascular disease that accompany aging (for example hypertension, glucose/insulin sensitivity and circulating lipid levels) and has direct beneficial effects on vascular oxidative stress resistance and suppression of inflammation (88). Systemic metabolism was altered in calorie-restricted ApoE knockout (ApoE–/–) mice, and sirtuin 1 (SIRT1) was significantly upregulated in VSMCs, although the metabolism of VSMCs was not altered. VSMC-SIRT1 is a major sensor of aortic calorie restriction and is associated with matrix metalloproteinase-2 (MMP-2) activation (89). There is growing evidence that sensible and effective exercise regimes improve vascular function and reduce the risk of vascular aging and disease. Telomerase reverse transcriptase (TERT) is the major protein component of telomerase, which extends telomere length and delays cellular aging. Exercise training, an inexpensive lifestyle factor, can increase TERT expression and telomerase activity, thereby reducing telomere wear and tear and effectively extending lifespan (90). In addition, studies have shown that the preventive effect of long-term moderate-intensity continuous training (MICT) on cardiovascular disease is achieved through VSMCs sarc-KATP channels (91).
However, senolytics promote apoptosis in senescent cells (85). Natural compounds quercetin, fisetin and curcumin all belong to the class of senolytics. Natural compounds are mainly found in food and are therefore referred to as “nutraceuticals” (92). Quercetin is a plant flavonoid with powerful antioxidant activity. Fisetin is a bioactive flavonol found in fruits and vegetables, such as apples, strawberries, grapes, onions and cucumbers, with antioxidant activity and anti-inflammatory properties (93). Curcumin, derived from turmeric, has anti-inflammatory, antioxidant, antibacterial and anticancer properties (94). In addition to natural compounds, dasatinib, which is a protein tyrosine kinase inhibitor (95, 96), and ABT-263 and ABT-737, which are BH3-fitted proteins, also belong to the senolytic class of agents (97, 98). There is still uncertainty as to whether senolytics can reduce atherosclerotic plaques by eliminating senescent cells.
Although previous experimental studies have suggested that many anti-aging biochemicals have the potential to treat atherosclerosis, the potential toxic side effects of these drugs should be fully considered before being put into therapeutic application. Senomorphics inhibit SASP but do not permanently eliminate the source of SASP activation, thus requiring chronic or repeated treatment which may increase the incidence of toxic side effects (84). ABT-263 causes transient thrombocytopenia and neutropenia (99). ABT-737 activates a process called minority mitochondrial outer membrane permeabilization (miMOMP), which not only fails to trigger cell death, but also causes DNA damage that promotes tumorigenesis. The above conclusions, however, were drawn only based on in vitro experiments, and There is no in vivo evidence to support this (100). The possibility of long-term use of these drugs should be considered for patients with atherosclerosis, as a chronic disease. Intriguingly, however, studies have found that the toxicity of senolytics may not represent a clinical problem as short-term treatments may be sufficient for treatment. A treatment regime of 3-days of D&Q once per week over 3 was found to be safe in idiopathic pulmonary fibrosis patients (101). Similar observations were reported in a trial on patients with diabetic kidney disease (102). In view of these facts, such a short-term hit and run approach to senolytic therapy may have the potential to improve tissue function in the long-term while reducing detrimental side effects clinically.
In order to minimize the side effects of such drugs, and to further explore the properties of various senescent cells (including VSMCs) in the pathological process of atherosclerosis, it is necessary to develop delivery systems that can specifically target drugs to the cells of interest. First, it is not currently known at which stage in the disease process do VSMCs express specific senescence markers. VSMCs are not in a static state during the complex atherosclerosis process, and the aging of VSMCs is a multi-step process of changes. Recently, data suggested that traditional cellular senescence markers used to identify senescent VSMCs are no longer expressed during the atherosclerosis process. Therefore, specific, lineage-restricted markers are needed (9). Second, it is unclear whether senolytic drugs prevent atherosclerosis through multiple mechanisms or whether they do so only by clearing senescent cells (9). Not all anti-aging drugs are effective against atherosclerosis; long-term oral administration of dasatinib + quercetin (D + Q) significantly reduced aortic medial senescent cell markers in chronic hypercholesterolemic mice and naturally aging mice, as well as improving vasomotor function, but the mice still developed atherosclerosis. Further, the size of atherosclerotic plaques did not decrease following these treatments (103). Third, before entering the clinical stage of application, optimal conditions for the use of anti-aging drugs should be fully clarified, and the efficacy, specificity and safety of the drugs should be rigourously evaluated.
Conclusion
Factors associated with the atherosclerotic process, including oxidative stress, inflammation, and calcium-regulating factors, are closely linked to senescence in VSMCs. Despite similarities in characteristic features associated with senescence between VSMCs and other cell types, senescent VSMCs exhibit unique properties during the atherosclerosis process. Promotion of a healthy lifestyle, exploring in depth the characteristics of each cell type associated with atherosclerosis, including VSMCs, and development of targeted drug delivery systems will ensure efficacy whilst evaluation of the safety and tolerability of drug use should be key aims of future anti-atherosclerosis research.
Author Contributions
YiZ performed the systematic literature search and preparation of the manuscript. JL and HL oversaw the literature review and wrote the manuscript with YiZ. WZ, YY, and YuZ helped with the literature search. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (82100413), China Postdoctoral Science Foundation (2020-M671633), National Key Research Program of China (2016YFE0126000), “Six Talent Peaks” in Jiangsu Province (WSN-082), and Postgraduate Research & Practice Innovation Program of Jiangsu Province (Yangzhou University) (XKYCX20_036).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. (2012) 111:245–59. doi: 10.1161/CIRCRESAHA.111.261388
2. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. (2015) 21:1424–35. doi: 10.1038/nm.4000
3. Aherrahrou R, Guo L, Nagraj VP, Aguhob A, Hinkle J, Chen L, et al. Genetic regulation of atherosclerosis-relevant phenotypes in human vascular smooth muscle cells. Circ Res. (2020) 127:1552–65. doi: 10.1161/CIRCRESAHA.120.317415
4. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702.
5. Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. (2021) 22:75–95. doi: 10.1038/s41580-020-00314-w
6. Villot R, Poirier A, Bakan I, Boulay K, Fernandez E, Devillers R, et al. ZNF768 links oncogenic RAS to cellular senescence. Nat Commun. (2021) 12:4841. doi: 10.1038/s41467-021-24932-w
7. Colpani O, Spinetti G. MicroRNAs orchestrating senescence of endothelial and vascular smooth muscle cells. Vasc Biol. (2019) 1:H75–81. doi: 10.1530/VB-19-0017
8. Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. (2015) 132:1909–19. doi: 10.1161/CIRCULATIONAHA.115.016457
9. Garrido AM, Kaistha A, Uryga AK, Oc S, Foote K, Shah A, et al. Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc Res. (2021) cvab208. doi: 10.1093/cvr/cvab208
10. Sikora E, Bielak-Zmijewska A, Mosieniak G. A common signature of cellular senescence; does it exist? Ageing Res Rev. (2021) 71:101458. doi: 10.1016/j.arr.2021.101458
11. Kohli J, Wang B, Brandenburg SM, Basisty N, Evangelou K, Varela-Eirin M, et al. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nat Protoc. (2021) 16:2471–98. doi: 10.1038/s41596-021-00505-5
12. Balistreri CR, Madonna R, Ferdinandy P. Is it the time of seno-therapeutics application in cardiovascular pathological conditions related to ageing? Curr Res Pharmacol Drug Discov. (2021) 2:100027. doi: 10.1016/j.crphar.2021.100027
14. Rubio-Ruiz ME, Perez-Torres I, Soto ME, Pastelin G, Guarner-Lans V. Aging in blood vessels. Medicinal agents FOR systemic arterial hypertension in the elderly. Ageing Res Rev. (2014) 18:132–47. doi: 10.1016/j.arr.2014.10.001
15. Guo Y, Tang Z, Yan B, Yin H, Tai S, Peng J, et al. PCSK9 (proprotein convertase subtilisin/Kexin Type 9) triggers vascular smooth muscle cell senescence and apoptosis: implication of its direct role in degenerative vascular disease. Arterioscler Thromb Vasc Biol. (2022) 42:67–86. doi: 10.1161/ATVBAHA.121.316902
16. Minamino T. Role of cellular senescence in lifestyle-related disease. Circ J. (2010) 74:2527–33. doi: 10.1253/circj.cj-10-0916
17. Chi C, Li DJ, Jiang YJ, Tong J, Fu H, Wu YH, et al. Vascular smooth muscle cell senescence and age-related diseases: state of the art. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:1810–21. doi: 10.1016/j.bbadis.2018.08.015
18. Holm Nielsen S, Jonasson L, Kalogeropoulos K, Karsdal MA, Reese-Petersen AL, Auf dem Keller U, et al. Exploring the role of extracellular matrix proteins to develop biomarkers of plaque vulnerability and outcome. J Intern Med. (2020) 287:493–513. doi: 10.1111/joim.13034
19. Johnson JL. Matrix metalloproteinases: influence on smooth muscle cells and atherosclerotic plaque stability. Expert Rev Cardiovasc Ther. (2007) 5:265–82. doi: 10.1586/14779072.5.2.265
20. Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. (2018) 114:622–34. doi: 10.1093/cvr/cvy007
21. Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. (2017) 19:42.
22. Mehdi MM, Solanki P, Singh P. Oxidative stress, antioxidants, hormesis and calorie restriction: the current perspective in the biology of aging. Arch Gerontol Geriatr. (2021) 95:104413. doi: 10.1016/j.archger.2021.104413
23. Canugovi C, Stevenson MD, Vendrov AE, Hayami T, Robidoux J, Xiao H, et al. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. (2019) 26:101288. doi: 10.1016/j.redox.2019.101288
24. Yu EP, Bennett MR. The role of mitochondrial DNA damage in the development of atherosclerosis. Free Radic Biol Med. (2016) 100:223–30. doi: 10.1016/j.freeradbiomed.2016.06.011
25. Valcheva P, Cardus A, Panizo S, Parisi E, Bozic M, Lopez Novoa JM, et al. Lack of vitamin D receptor causes stress-induced premature senescence in vascular smooth muscle cells through enhanced local angiotensin-II signals. Atherosclerosis. (2014) 235:247–55. doi: 10.1016/j.atherosclerosis.2014.05.911
26. Zhu H, Wang Z, Dong Z, Wang C, Cao Q, Fan F, et al. Aldehyde dehydrogenase 2 deficiency promotes atherosclerotic plaque instability through accelerating mitochondrial ROS-mediated vascular smooth muscle cell senescence. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:1782–92. doi: 10.1016/j.bbadis.2018.09.033
27. Uryga A, Gray K, Bennett M Damage DNA and repair in vascular disease. Annu Rev Physiol. (2016) 78:45–66.
28. Shah A, Gray K, Figg N, Finigan A, Starks L, Bennett M. Defective base excision repair of oxidative DNA damage in vascular smooth muscle cells promotes atherosclerosis. Circulation. (2018) 138:1446–62. doi: 10.1161/CIRCULATIONAHA.117.033249
29. Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S, et al. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc Natl Acad Sci U.S.A. (2006) 103:1864–9. doi: 10.1073/pnas.0507444103
30. Davidson SM, Yellon DM. Mitochondrial DNA damage, oxidative stress, and atherosclerosis: where there is smoke there is not always fire. Circulation. (2013) 128:681–3. doi: 10.1161/CIRCULATIONAHA.113.004531
31. Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. (2005) 309:481–4. doi: 10.1126/science.1112125
32. Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U.S.A. (2005) 102:17993–8. doi: 10.1073/pnas.0508886102
33. Yu E, Calvert PA, Mercer JR, Harrison J, Baker L, Figg NL, et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation. (2013) 128:702–12.
34. Yu EP, Bennett MR. Mitochondrial DNA damage and atherosclerosis. Trends Endocrinol Metab. (2014) 25:481–7.
35. De Rosa M, Johnson SA, Opresko PL. Roles for the 8-oxoguanine DNA repair system in protecting telomeres from oxidative stress. Front Cell Dev Biol. (2021) 9:758402. doi: 10.3389/fcell.2021.758402
36. Samani NJ, Boultby R, Butler R, Thompson JR, Goodall AH. Telomere shortening in atherosclerosis. Lancet. (2001) 358:472–3.
37. De Meyer T, Nawrot T, Bekaert S, De Buyzere ML, Rietzschel ER, Andres V. Telomere length as cardiovascular aging biomarker: JACC review topic of the week. J Am Coll Cardiol. (2018) 72:805–13. doi: 10.1016/j.jacc.2018.06.014
38. Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. (2006) 99:156–64. doi: 10.1161/01.RES.0000233315.38086.bc
39. Richardson GD, Sage A, Bennaceur K, Al Zhrany N, Coelho-Lima J, Dookun E, et al. Telomerase mediates lymphocyte proliferation but not the atherosclerosis-suppressive potential of regulatory T-cells. Arterioscler Thromb Vasc Biol. (2018) 38:1283–96.
40. Liberale L, Montecucco F, Tardif JC, Libby P, Camici GG. Inflamm-ageing: the role of inflammation in age-dependent cardiovascular disease. Eur Heart J. (2020) 41:2974–82. doi: 10.1093/eurheartj/ehz961
41. Geng S, Chen K, Yuan R, Peng L, Maitra U, Diao N, et al. The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat Commun. (2016) 7:13436. doi: 10.1038/ncomms13436
42. Stojanovic SD, Fiedler J, Bauersachs J, Thum T, Sedding DG. Senescence-induced inflammation: an important player and key therapeutic target in atherosclerosis. Eur Heart J. (2020) 41:2983–96. doi: 10.1093/eurheartj/ehz919
43. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. (2011) 21:103–15. doi: 10.1038/cr.2010.178
44. Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. (2010) 40:616–9. doi: 10.1002/eji.200940168
45. Gardner SE, Humphry M, Bennett MR, Clarke MC. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1alpha-dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol. (2015) 35:1963–74. doi: 10.1161/ATVBAHA.115.305896
46. Sanada F, Muratsu J, Otsu R, Shimizu H, Koibuchi N, Uchida K, et al. Local production of activated factor X in atherosclerotic plaque induced vascular smooth muscle cell senescence. Sci Rep. (2017) 7:17172. doi: 10.1038/s41598-017-17508-6
47. Grootaert MOJ, Finigan A, Figg NL, Uryga AK, Bennett MR. SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ Res. (2021) 128:474–91.
48. Zhao J, He X, Zuo M, Li X, Sun Z. Anagliptin prevented interleukin 1beta (IL-1beta)-induced cellular senescence in vascular smooth muscle cells through increasing the expression of sirtuin1 (SIRT1). Bioengineered. (2021) 12:3968–77. doi: 10.1080/21655979.2021.1948289
49. Zhang X, Yuan J, Zhou N, Shen K, Wang Y, Wang K, et al. Omarigliptin prevents TNF-alpha-induced cellular senescence in rat aorta vascular smooth muscle cells. Chem Res Toxicol. (2021) 34:2024–31. doi: 10.1021/acs.chemrestox.1c00076
50. Furmanik M, Chatrou M, van Gorp R, Akbulut A, Willems B, Schmidt H, et al. Reactive oxygen-forming Nox5 links vascular smooth muscle cell phenotypic switching and extracellular vesicle-mediated vascular calcification. Circ Res. (2020) 127:911–27. doi: 10.1161/CIRCRESAHA.119.316159
51. Liu Y, Drozdov I, Shroff R, Beltran LE, Shanahan CM. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res. (2013) 112:e99–109. doi: 10.1161/CIRCRESAHA.111.300543
52. Neven E, D’Haese PC. Vascular calcification in chronic renal failure: what have we learned from animal studies? Circ Res. (2011) 108:249–64. doi: 10.1161/CIRCRESAHA.110.225904
53. Zwakenberg SR, de Jong PA, Hendriks EJ, Westerink J, Spiering W, de Borst GJ, et al. Intimal and medial calcification in relation to cardiovascular risk factors. PLoS One. (2020) 15:e0235228. doi: 10.1371/journal.pone.0235228
54. Krishnan P, Moreno PR, Turnbull IC, Purushothaman M, Zafar U, Tarricone A, et al. Incremental effects of diabetes mellitus and chronic kidney disease in medial arterial calcification: synergistic pathways for peripheral artery disease progression. Vasc Med. (2019) 24:383–94. doi: 10.1177/1358863X19842276
55. Lanzer P, Hannan FM, Lanzer JD, Janzen J, Raggi P, Furniss D, et al. Medial arterial calcification: JACC state-of-the-art review. J Am Coll Cardiol. (2021) 78:1145–65. doi: 10.1016/j.jacc.2021.06.049
56. Ho CY, Shanahan CM. Medial arterial calcification: an overlooked player in peripheral arterial disease. Arterioscler Thromb Vasc Biol. (2016) 36:1475–82. doi: 10.1161/ATVBAHA.116.306717
57. Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. (2016) 15:335–43. doi: 10.1038/nmat4519
58. Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol. (2014) 34:724–36. doi: 10.1161/ATVBAHA.113.302642
59. Naik V, Leaf EM, Hu JH, Yang HY, Nguyen NB, Giachelli CM, et al. Sources of cells that contribute to atherosclerotic intimal calcification: an in vivo genetic fate mapping study. Cardiovasc Res. (2012) 94:545–54. doi: 10.1093/cvr/cvs126
60. Nguyen N, Naik V, Speer MY. Diabetes mellitus accelerates cartilaginous metaplasia and calcification in atherosclerotic vessels of LDLr mutant mice. Cardiovasc Pathol. (2013) 22:167–75. doi: 10.1016/j.carpath.2012.06.007
61. New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, et al. Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res. (2013) 113:72–7. doi: 10.1161/CIRCRESAHA.113.301036
62. McRobb LS, McGrath KCY, Tsatralis T, Liong EC, Tan JTM, Hughes G, et al. Estrogen receptor control of atherosclerotic calcification and smooth muscle cell osteogenic differentiation. Arterioscler Thromb Vasc Biol. (2017) 37:1127–37. doi: 10.1161/ATVBAHA.117.309054
63. Jover E, Silvente A, Marin F, Martinez-Gonzalez J, Orriols M, Martinez CM, et al. Inhibition of enzymes involved in collagen cross-linking reduces vascular smooth muscle cell calcification. FASEB J. (2018) 32:4459–69. doi: 10.1096/fj.201700653R
64. Qian S, Regan JN, Shelton MT, Hoggatt A, Mohammad KS, Herring PB, et al. The P2Y2 nucleotide receptor is an inhibitor of vascular calcification. Atherosclerosis. (2017) 257:38–46.
65. Yang Y, Sun Y, Chen J, Bradley WE, Dell’Italia LJ, Wu H, et al. AKT-independent activation of p38 MAP kinase promotes vascular calcification. Redox Biol. (2018) 16:97–103. doi: 10.1016/j.redox.2018.02.009
66. Willems BA, Furmanik M, Caron MMJ, Chatrou MLL, Kusters DHM, Welting TJM, et al. Ucma/GRP inhibits phosphate-induced vascular smooth muscle cell calcification via SMAD-dependent BMP signalling. Sci Rep. (2018) 8:4961. doi: 10.1038/s41598-018-23353-y
67. Nakano-Kurimoto R, Ikeda K, Uraoka M, Nakagawa Y, Yutaka K, Koide M, et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol. (2009) 297:H1673–84. doi: 10.1152/ajpheart.00455.2009
68. Liu H, Wang H, Yang S, Qian D. Downregulation of miR-542-3p promotes osteogenic transition of vascular smooth muscle cells in the aging rat by targeting BMP7. Hum Genomics. (2019) 13:67. doi: 10.1186/s40246-019-0245-z
69. Huang Y, Wang J, Luo M, Yan D, Zhang C. Carnosine attenuates vascular smooth muscle cells calcification through mTOR signaling pathway. Aging Med (Milton). (2020) 3:153–8. doi: 10.1002/agm2.12125
70. Zuccolo E, Badi I, Scavello F, Gambuzza I, Mancinelli L, Macri F, et al. The microRNA-34a-induced senescence-associated secretory phenotype (SASP) favors vascular smooth muscle cells calcification. Int J Mol Sci. (2020) 21:4454. doi: 10.3390/ijms21124454
71. Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. (2011) 36:30–8. doi: 10.1016/j.tibs.2010.07.007
72. Wu X, Zheng X, Cheng J, Zhang K, Ma C. LncRNA TUG1 regulates proliferation and apoptosis by regulating miR-148b/IGF2 axis in ox-LDL-stimulated VSMC and HUVEC. Life Sci. (2020) 243:117287. doi: 10.1016/j.lfs.2020.117287
73. Yu H, Fellows A, Foote K, Yang Z, Figg N, Littlewood T, et al. FOXO3a (forkhead transcription factor O subfamily member 3a) links vascular smooth muscle cell apoptosis, matrix breakdown, atherosclerosis, and vascular remodeling through a novel pathway involving MMP13 (matrix metalloproteinase 13). Arterioscler Thromb Vasc Biol. (2018) 38:555–65. doi: 10.1161/ATVBAHA.117.310502
74. Luo Z, Xu W, Ma S, Qiao H, Gao L, Zhang R, et al. Moderate autophagy inhibits vascular smooth muscle cell senescence to stabilize progressed atherosclerotic plaque via the mTORC1/ULK1/ATG13 signal pathway. Oxid Med Cell Longev. (2017) 2017:3018190. doi: 10.1155/2017/3018190
75. Larroque-Cardoso P, Swiader A, Ingueneau C, Negre-Salvayre A, Elbaz M, Reyland ME, et al. Role of protein kinase C delta in ER stress and apoptosis induced by oxidized LDL in human vascular smooth muscle cells. Cell Death Dis. (2013) 4:e520. doi: 10.1038/cddis.2013.47
76. Lee KY, Kim JR, Choi HC. Genistein-induced LKB1-AMPK activation inhibits senescence of VSMC through autophagy induction. Vascul Pharmacol. (2016) 81:75–82. doi: 10.1016/j.vph.2016.02.007
77. Kim SG, Sung JY, Kim JR, Choi HC. Nifedipine-induced AMPK activation alleviates senescence by increasing autophagy and suppressing of Ca(2+) levels in vascular smooth muscle cells. Mech Ageing Dev. (2020) 190:111314. doi: 10.1016/j.mad.2020.111314
78. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. (2005) 122:927–39. doi: 10.1016/j.cell.2005.07.002
79. Centner AM, Bhide PG, Salazar G. Nicotine in senescence and atherosclerosis. Cells (2020) 9:1035.
80. Chang KH, Park JM, Lee CH, Kim B, Choi KC, Choi SJ, et al. NADPH oxidase (NOX) 1 mediates cigarette smoke-induced superoxide generation in rat vascular smooth muscle cells. Toxicol Vitro. (2017) 38:49–58. doi: 10.1016/j.tiv.2016.10.013
81. Shinohara M, Adachi Y, Mitsushita J, Kuwabara M, Nagasawa A, Harada S, et al. Reactive oxygen generated by NADPH oxidase 1 (Nox1) contributes to cell invasion by regulating matrix metalloprotease-9 production and cell migration. J Biol Chem. (2010) 285:4481–8. doi: 10.1074/jbc.M109.071779
82. Penaloza-Martinez E, Moreno G, Aroca-Crevillen A, Huertas S, Vicent L, Rosillo N, et al. Circadian rhythms in thrombosis and atherothrombotic events. Front Biosci (Landmark Ed). (2022) 27:51. doi: 10.31083/j.fbl2702051
83. Guo Z, Yu B, Li X, Yang X, Wang C, Fan L. Circadian misalignment promotes vascular smooth muscle cell apoptosis via defective autophagy. J Mol Histol. (2021) 52:799–808. doi: 10.1007/s10735-021-10000-6
84. Lagoumtzi SM, Chondrogianni N. Senolytics and senomorphics: natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free Radic Biol Med. (2021) 171:169–90. doi: 10.1016/j.freeradbiomed.2021.05.003
85. Boccardi V, Mecocci P. Senotherapeutics: targeting senescent cells for the main age-related diseases. Mech Ageing Dev. (2021) 197:111526. doi: 10.1016/j.mad.2021.111526
86. Feresin RG, Huang J, Klarich DS, Zhao Y, Pourafshar S, Arjmandi BH, et al. Blackberry, raspberry and black raspberry polyphenol extracts attenuate angiotensin II-induced senescence in vascular smooth muscle cells. Food Funct. (2016) 7:4175–87. doi: 10.1039/c6fo00743k
87. Serino A, Zhao Y, Hwang J, Cullen A, Deeb C, Akhavan N, et al. Gender differences in the effect of blackberry supplementation in vascular senescence and atherosclerosis in ApoE(-/-) mice. J Nutr Biochem. (2020) 80:108375. doi: 10.1016/j.jnutbio.2020.108375
88. Prata L, Ovsyannikova IG, Tchkonia T, Kirkland JL. Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin Immunol. (2018) 40:101275. doi: 10.1016/j.smim.2019.04.003
89. Liu Y, Wang TT, Zhang R, Fu WY, Wang X, Wang F, et al. Calorie restriction protects against experimental abdominal aortic aneurysms in mice. J Exp Med. (2016) 213:2473–88. doi: 10.1084/jem.20151794
90. Denham J, Sellami M. Exercise training increases telomerase reverse transcriptase gene expression and telomerase activity: a systematic review and meta-analysis. Ageing Res Rev. (2021) 70:101411. doi: 10.1016/j.arr.2021.101411
91. Feng R, Wang L, Li Z, Yang R, Liang Y, Sun Y, et al. A systematic comparison of exercise training protocols on animal models of cardiovascular capacity. Life Sci. (2019) 217:128–40.
92. Alizadeh SR, Ebrahimzadeh MA. Quercetin derivatives: drug design, development, and biological activities, a review. Eur J Med Chem. (2022) 229:114068. doi: 10.1016/j.ejmech.2021.114068
93. Garg S, Khan SI, Malhotra RK, Sharma MK, Kumar M, Kaur P, et al. The molecular mechanism involved in cardioprotection by the dietary flavonoid fisetin as an agonist of PPAR-gamma in a murine model of myocardial infarction. Arch Biochem Biophys. (2020) 694:108572. doi: 10.1016/j.abb.2020.108572
94. Tohamy HG, El Okle OS, Goma AA, Abdel-Daim MM, Shukry M. Hepatorenal protective effect of nano-curcumin against nanocopper oxide-mediated toxicity in rats: behavioral performance, antioxidant, anti-inflammatory, apoptosis, and histopathology. Life Sci. (2022) 292:120296. doi: 10.1016/j.lfs.2021.120296
95. Ghamar Talepoor A, Khosropanah S, Doroudchi M. Partial recovery of senescence in circulating follicular helper T cells after Dasatinib treatment. Int Immunopharmacol. (2021) 94:107465. doi: 10.1016/j.intimp.2021.107465
96. Chen Z, Lee FY, Bhalla KN, Wu J. Potent inhibition of platelet-derived growth factor-induced responses in vascular smooth muscle cells by BMS-354825 (dasatinib). Mol Pharmacol. (2006) 69:1527–33. doi: 10.1124/mol.105.020172
97. Lopez-Dominguez JA, Rodriguez-Lopez S, Ahumada-Castro U, Desprez PY, Konovalenko M, Laberge RM, et al. Cdkn1a transcript variant 2 is a marker of aging and cellular senescence. Aging (Albany NY). (2021) 13:13380–92. doi: 10.18632/aging.203110
98. Song JH, Kandasamy K, Zemskova M, Lin YW, Kraft AS. The BH3 mimetic ABT-737 induces cancer cell senescence. Cancer Res. (2011) 71:506–15. doi: 10.1158/0008-5472.CAN-10-1977
99. Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. (2012) 18:3163–9. doi: 10.1158/1078-0432.CCR-11-3090
100. Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell. (2015) 57:860–72. doi: 10.1016/j.molcel.2015.01.018
101. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
102. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine. (2019) 47:446–56.
Keywords: atheroscelorsis, senescence, VSMCs, SASP, sentherapeutic
Citation: Zha Y, Zhuang W, Yang Y, Zhou Y, Li H and Liang J (2022) Senescence in Vascular Smooth Muscle Cells and Atherosclerosis. Front. Cardiovasc. Med. 9:910580. doi: 10.3389/fcvm.2022.910580
Received: 05 April 2022; Accepted: 04 May 2022;
Published: 01 June 2022.
Edited by:
Emiel Van Der Vorst, Institute for Molecular Cardiovascular Research (IMCAR), GermanyReviewed by:
Andrew Newby, University of Bristol, United KingdomGavin Richardson, Newcastle University, United Kingdom
Copyright © 2022 Zha, Zhuang, Yang, Zhou, Li and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongliang Li, bGlob25nbGlhbmcwODE4QHl6dS5lZHUuY24=; Jingyan Liang, anlsaWFuZ0B5enUuZWR1LmNu