 Yuxi Cai1†Yiwen Zhou1†
Yuxi Cai1†Yiwen Zhou1† Zhangwang Li1Panpan Xia1,2,3,4Xinxi ChenFu1,2,3,4Ao Shi5,6
Zhangwang Li1Panpan Xia1,2,3,4Xinxi ChenFu1,2,3,4Ao Shi5,6 Jing Zhang1,7*
Jing Zhang1,7* Peng Yu1,2,3*
Peng Yu1,2,3*- 1The Second Clinical Medical College of Nanchang University, The Second Affiliated Hospital of Nanchang University, Nanchang, China
- 2Department of Metabolism and Endocrinology, the Second Affiliated Hospital of Nanchang University, Nanchang, China
- 3Institute for the Study of Endocrinology and Metabolism in Jiangxi Province, Nanchang, China
- 4Branch of National Clinical Research Center for Metabolic Diseases, Nanchang, China
- 5School of Medicine, University of Nicosia, Nicosia, Cyprus
- 6School of Medicine, St. George University of London, London, United Kingdom
- 7Department of Anesthesiology, The Second Affiliated Hospital of Nanchang University, Nanchang, China
Accumulating evidence has proved that non-coding RNAs (ncRNAs) play a critical role in the genetic programming and gene regulation of cardiovascular diseases (CVDs). Cardiovascular disease morbidity and mortality are rising and have become a primary public health issue that requires immediate resolution through effective intervention. Numerous studies have revealed that new types of cell death, such as pyroptosis, necroptosis, and ferroptosis, play critical cellular roles in CVD progression. It is worth noting that ncRNAs are critical novel regulators of cardiovascular risk factors and cell functions by mediating pyroptosis, necroptosis, and ferroptosis. Thus, ncRNAs can be regarded as promising therapeutic targets for treating and diagnosing cardiovascular diseases. Recently, there has been a surge of interest in the mediation of ncRNAs on three types of cell death in regulating tissue homeostasis and pathophysiological conditions in CVDs. Although our understanding of ncRNAs remains in its infancy, the studies reviewed here may provide important new insights into how ncRNAs interact with CVDs. This review summarizes what is known about the functions of ncRNAs in modulating cell death-associated CVDs and their role in CVDs, as well as their current limitations and future prospects.
Introduction
Despite significant advances in prevention, diagnosis, and early intervention, cardiovascular diseases (CVDs) remain the leading cause of global mortality and a significant contributor to a decline in quality of life for many people (1, 2). According to the Global Burden of Disease Cooperation Organization's official statistics, CVD patients increased from 271 million in 1990 to 523 million in 2019, nearly doubling in < 30 years. Furthermore, CVD-related deaths have risen steadily from 12.1 million in 1990 to 18.6 million in 2019 (1). CVDs include a wide range of disorders, such as atherosclerosis, ischemia-reperfusion injury, myocardial infarction, uremic cardiomyopathy, cardiac hypertrophy, and others, and are also the leading cause of disability and death worldwide (3).
The pathophysiology of CVDs is complicated (4). Cell death plays a vital role in the pathophysiology of CVDs. Programmed cell death (PCD) is a type of death that is both independent and organized. Apoptosis is widely regarded as the most common mode of cell death and the first type of modulated cell death to be discovered. PCD currently involves pyroptosis, necroptosis, ferroptosis, autophagy, and apoptosis. Cells that have been programmed to die have a significant impact on cardiovascular disease (5). Necroptosis, for example, is activated in advanced atherosclerotic plaques in humans and can be used as a target for experimental treatment and diagnostic intervention in atherosclerosis (6).
ncRNA is a link that cannot be ignored in the occurrence and development of CVDs. ncRNAs account for 98–99% of the human genome and are involved in modulating the expression of genes encoding proteins (7), which can be categorized into long non-coding RNAs (lncRNA), micro RNA (miRNA), circular RNAs (circRNAs), small nuclear RNA (snRNA), piwi-interacting RNA (piRNA), small interfering RNA (siRNA), and small nucleolar RNA (snoRNA) (8). miRNAs are single-stranded RNA molecules about 22 nucleotides in length that can downregulate gene expression at the post-transcriptional level (9). lncRNAs are transcripts of over 200 nucleotides in length that participate in practically every step of gene expression modulation (10). circRNA acts as a novel endogenous closed-chain covalent RNA that is largely expressed in mammalian cells and participate in the transcriptional or post-transcriptional adjustment of gene expression (11). lncRNAs and circRNAs can serve as competing for endogenous RNAs (ceRNAs) to modulate miRNAs, thus interacting with their corresponding mRNA targets. Of note, miRNAs and circRNAs possess the advantage of outstanding stability, resistance to degradation caused by various enzymes, and the ability to circulate in body fluids for an extended duration owing to their short length and circular structure (7). As described in previous studies, small ncRNAs have emerged as critical regulators of gene expression in various cellular pathways and systems (8). One of the most classic action modes of ncRNAs is that miRNAs directly target the partially complementary sites located in the 3′ untranslated region of mRNA and inhibit their expression (12).
Furthermore, ncRNAs play a role in the precise regulation of PCD. In the past few years, an increasing number of studies have identified that ncRNA has functional roles in regulating traditional PCD, such as apoptosis, necrosis, and autophagy (13). These types of cell death can contribute to the progression of certain diseases by regulating the cell cycle through the modulation of their downstream corresponding proteins or genes. In 2022, Xu's study found that overexpression of circ_0007059 can contribute to apoptosis in cardiac tissues of the MI mouse model by regulating its downstream miR-378 and miR-383 (14).
Recently, newly discovered PCDs, such as necroptosis, pyroptosis, and ferroptosis, have been studied, with ample evidence indicating that PCD is a potential key factor in the CVD process (15–17). Lately, ncRNAs have become increasingly crucial in CVDs (coronary heart disease, atherosclerosis, myocardial infarction, and heart failure) (18–21). Through various pathways, ncRNAs regulate gene expression in various cardiovascular diseases. Concurrently, recent research discovered that ncRNAs play an important role in CVDs by modulating PCD.
This review summarizes the progress of the regulatory mechanisms of necroptosis, pyroptosis, and ferroptosis in CVDs and the role of ncRNAs, particularly circRNAs, lncRNAs, and miRNAs, in modulating PCD in CVDs. Identifying these cardiovascular disease-associated ncRNAs will help us understand how CVDs work and provide some new targets and implications for their prevention and treatment. Furthermore, we provide an update on recent developments and perspectives for the clinical and therapeutic use of ncRNAs on pyroptosis, necroptosis, and ferroptosis in CVDs, and their current limitations and prospects for future research.
Necroptosis in CVDs
Necroptosis signaling pathway
Necroptosis is a type of necrosis controlled by death receptors (22). TNF can initiate necroptosis in the first step by binding to its receptor TNFR1, resulting in the formation of complex I, which includes cIAP1/2, RIPK1, TRAF2/5, and TRADD (23, 24). The ubiquitination of RIPK1 in complex I is mediated by clAP1/2 and TRAF2/5, resulting in complex I stability, which in turn motivates the activation of the classical NF-κB pathway and results in cell survival (25). Meanwhile, cylindromatosis (CYLD) and A20 can deubiquitinate RIP1, leading to the formation of complex IIa, which includes pro-caspase-8, FADD, and TRADD, and can activate RIP1-independent apoptosis (23, 26, 27). When the expressions of cIAP, TGF-activated kinase 1 (TAK1), or NF-κB essential modulator (NEMO) are suppressed, the complex IIb, which includes RIPK1, FADD, and pro-caspase-8, is activated, resulting in RIP1-dependent apoptosis. (28, 29). When the level of FLIPL (FLICE inhibitory protein), which interacts with caspase-8, is high, the catalytic activity of caspase-8 is impaired, resulting in necroptosis (30). RIPK1 and RIPK3 communicate with each other through the receptor homology domain (RHD), which results in the formation of necrosomes and the induction of downstream signaling, ultimately resulting in necroptosis (31). After activation, RIPK3 transports MLKL (a mixed-line kinase-like protein) to the plasma membrane for phosphorylation, resulting in necrosis, changes in cell membrane permeability, and, eventually, cell death (32). The specific molecular signaling mechanism of necroptosis is depicted in Figure 1.
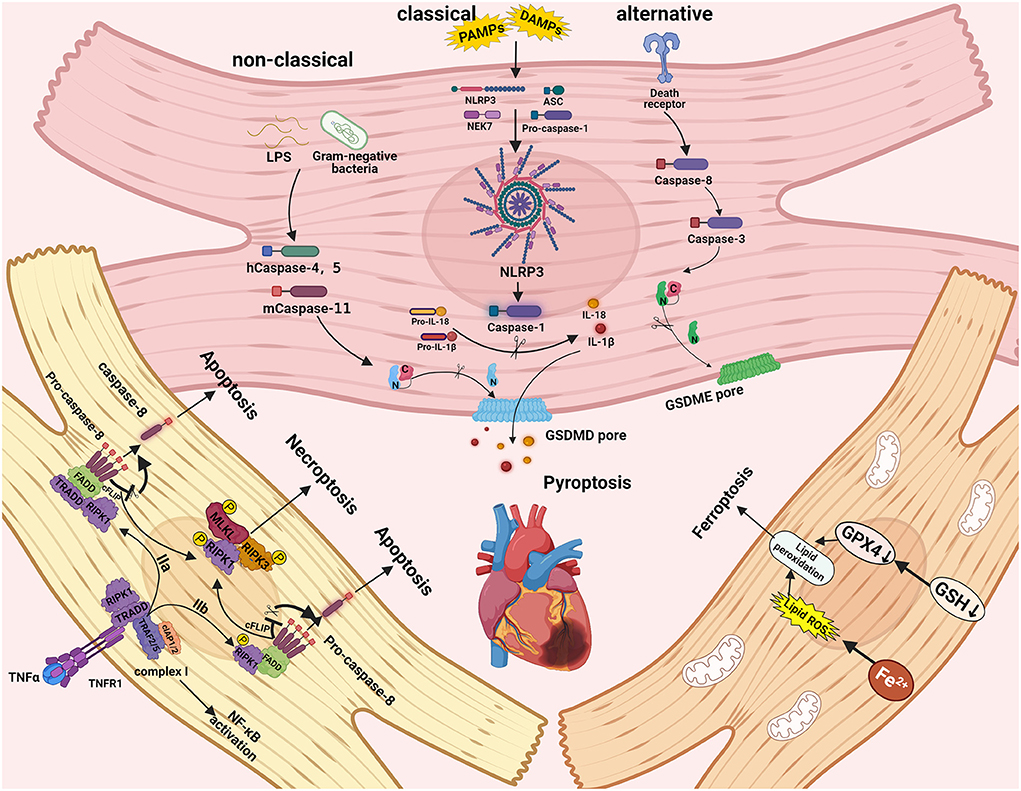
Figure 1. The molecular signaling mechanisms and pathways of pyroptosis, necroptosis, and ferroptosis in cardiac cells. Necroptosis can be triggered by RIPK3 and RIPK1, which recruit MLKL to form the necrosome. The caspase-1 induced classical inflammasome pathway, caspase-4, caspase-5, and caspase-11 induced non-classical pathways, and caspase-3 and caspase-8 induced alternative pathways can trigger pyroptosis. Ferroptosis can be activated by iron accumulation and low levels of GSH-induced lipid peroxidation.
Contributions of necroptosis to the pathophysiology of CVDs
Necroptosis is involved in the pathogenesis of various CVDs, including AS, I/R injury, and MI.
In the early stage of AS, circulating monocytes enter the subintima to phagocytose ox-LDL through damaged vascular epithelial cells, turning them into foam cells. Subsequently, the expression of RIP3 and MLKL in foam cells is upregulated so that necroptosis is activated, manifesting as inflammation, ultimately boosting the development of AS (33).
When myocardial I/R injury occurs, necroptosis will also arise soon afterward by triggering the RIP1-RIP3-MLKL complex, which can exacerbate myocardial oxidative stress and aggravate I/R injury, simultaneously accompanied by an inflammatory response (34).
RIP3-mediated inflammation is instrumental to the pathogenesis of MI. This inflammatory response results in adverse ventricular remodeling after MI by promoting ROS production (35). Additionally, chemical inhibition of RIP1, another component of the RIP1-RIP3-MLKL complex, can cut down the size of the necrotic myocardium induced by MI (36).
ncRNAs regulate necroptosis in CVDs
ncRNA manifests as a complete axis that connects the occurrence of PCD with the development of CVDs. In this axis, lncRNA and circRNA compete with target miRNA for the 3′UTR binding site of mRNA to play a negative role in regulating downstream mRNA expression. The activation of this axis can result in the release of tremendous inflammasome that induces the corresponding PCD-like necroptosis and promotes the process of various CVDs. In addition, lncRNA and circRNA can act as “ molecular sponges” to inhibit miRNA expression to participate in the gene expression modulation, thus influencing the development of CVDs.
miRNAs play critical roles in ischemia-reperfusion (I/R) injury (37). I/R injury is defined as a pathological situation characterized by an initial reduction or interruption of blood flow to the organ, followed by restoration of perfusion and reoxygenation, which can result in worsening tissue damage and severe inflammation, as well as severe cell damage and death (38, 39). Qin's team previously demonstrated that miR-223-5p/3p duplex regulated death receptors (DR6 and TNFR1) protein levels and NLRP3 inflammasome signaling pathway, which contributed to the inhibition of myocardial I/R-induced necroptosis (40). Tan et al. discovered that in a mouse model of myocardial I/R injury, RIPK1 was upregulated, worsening myocardial I/R injury via the TNF signaling pathway. At the same time, miR-24-3p, which can protect the heart by suppressing RIPK1, was downregulated (41).
Similarly, miRNAs play a role in myocardial infarction (MI) pathology. MI, also known as a heart attack, is most commonly caused by reduced or interrupted blood flow to a part of the heart, resulting in cardiac cell death. In addition, because the epicardial artery is important in feeding the heart muscle, a blood clot embedded in it is usually the primary cause of ischemic myocardium necrosis. However, it is now recognized that blood clot etiology is not required in all cases. The blood supply must match the oxygen demand of all living tissues, including the heart muscle. This is referred to as a supply–demand relationship. In the absence of blood clots, it is now known that if the heart rate is too high or the blood pressure drops, this imbalance in the ratio will damage the heart muscle (42). According to Zhang et al. (43), miR-325-3p overexpression induced by a specific agomiR significantly reduced MI-associated symptoms by suppressing the RIPK3-based necroptosis pathway, indicating that miR-325-3p upregulation could improve MI. Zaafan et al. found that silencing miR-103 inhibited necroptosis and MI induction by targeting fas-associated protein with a death domain (FADD), which was previously shown to be an essential negative regulator of necroptosis (44, 45). Cardiomyocyte progenitor cells (CMPCs) are thought to be a potential source of cell transplantation therapy for promoting myocardial recovery. Furthermore, a recent study found that miR-155 overexpression could reduce necrotic cell death by 40±2.3% in CMPCs by targeting receptor-interacting serine/threonine-protein kinase 1 (RIPK1) (46).
miRNAs play an important role in atherosclerosis (AS). AS is an inflammatory disease of the arterial intima caused by lipids. The final clinical outcome is determined by balancing its pro-inflammatory and anti-inflammatory mechanisms. Macrophages are primarily responsible for the infiltration, modification, and uptake of intimal plasma-derived lipoproteins, resulting in the formation of lipid-filled foam cells and atherosclerotic lesions (47). Recent research found that deleting HIF-1α from myeloid cells could reduce AS and necrotic core formation in apolipoprotein E-deficient mice by inhibiting macrophage necroptosis. By acting on 2, 4-dienoyl-CoA reductase, HIF-1α can mechanically upregulate miR-210, resulting in necroptosis and a reduction in mitochondrial respiration. Simultaneously, HIF-1-mediated downregulation of miR-383 in inflammatory macrophages increased ATP depletion by suppressing poly (ADP-ribose) glycohydrolase, which exacerbated atherosclerosis in lesional macrophages and macrophages derived from inflammatory bone marrow (48).
In other CVDs like myocardial necroptosis prompted by Se deficiency, Yang's study found that overexpression of miR-200a-5p rendered receptor-interacting serine/threonine kinase 3 (RIP3)-dependent necroptosis in cardiomyocytes and animal models by targeting gene ring finger protein 11 (RNF11) (49). Jiang et al. discovered that by targeting MLKL in human HK2 cells, hsa-miR-500a-3P could attenuate toxic and ischemic insults caused by cell necroptosis and the inflammatory response in acute kidney injury (AKI) (50). Furthermore, Huang's research discovered that miR-223-3p was upregulated in AKI caused by 3-chloro-1, 2-propanediol (3-MCPD)-dipalmitate, and regulated RIPK3 expression by targeting the 3′UTR of RIPK3 (51). Similarly, Gu et al. reported in sepsis that miR-425-5p could target the 3′UTR of RIPK1 mRNA to suppress RIPK1 expression and activate RIPK1, reversing the suppression of necroptosis and inflammasome induced by miR-425-5p in septic hepatocytes (52).
There have only been a few reports that lncRNA regulates necroptosis in CVDs. Previous research in I/R injury found that necrosis-related factor (NRF), a long non-coding RNA, regulated cardiomyocyte necrosis by targeting miR-873, which inhibited RIPK1/RIPK3 translation and suppressed RIPK1/RIPK3-mediated necroptosis in cardiomyocytes induced by I/R injury (53).
In general, what has been discussed thus far indicates that ncRNA plays an important role in CVDs by regulating necroptosis, as shown in Table 1 and Figure 2.
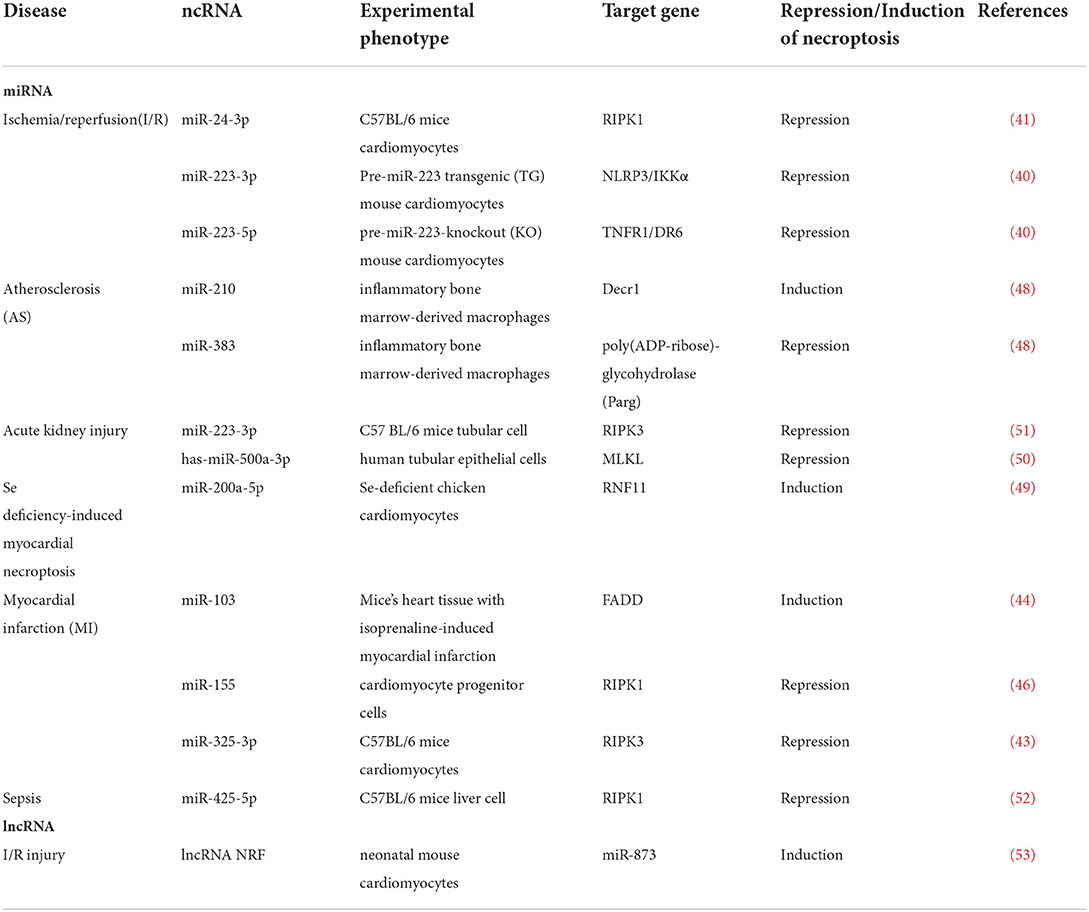
Table 1. Non-coding RNAs regulate necroptosis in various type of CVDs.
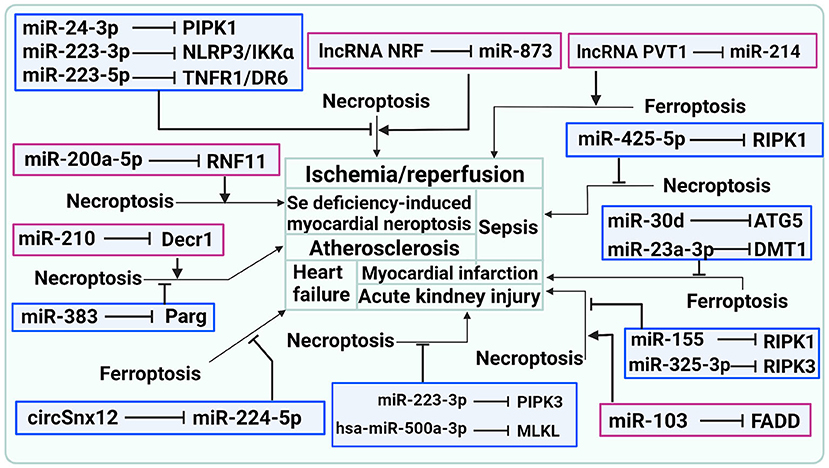
Figure 2. ncRNAs are involved in CVDs by influencing the occurrence of necroptosis and ferroptosis.
How to detect necroptosis
The activation of RIPK1, RIPK3, and MLKL is core links in the necroptosis pathway, which are associated with their phosphorylation. Accordingly, phosphorylation of RIP1, RIP3, and MLKL can be used as the hallmarks of the induction of necroptosis in vivo and vitro (54). It is of note that two newly developed anti-phosphorylated RIP1 antibodies have been explored as biomarkers for the sensitization of RIP1, which respectively recognize the phospho-Ser166 and phospho-Ser14/15 of hRIP1 (55).
Pyroptosis in CVDs
Pyroptosis signaling pathway
Pyroptosis, also known as a form of distinct programmed cell death; compared to other forms (e.g., apoptosis and autophagic cell death), its morphology is unique that is influenced by inflammatory caspases (56, 57), as first demonstrated in the death of macrophages caused by Salmonella (58). Classical inflammatory pathways mediated by caspase-1 and non-classical inflammatory pathways mediated by caspase-4/5/11 can both induce pyrolysis (59). Activated caspases can also cleave and multimerize members of the gasdermin family, including GSDMD. During the process of pyroptosis, active GSDMD-NT is transferred to the plasma membrane and forms pores, resulting in water inflow, cellular edema, and dissolution during pyroptosis (60, 61). Pathogen-associated molecular patterns (PAMPs) or endogenous damage-associated molecular patterns (DAMPs) can recognize and combine with PRRs in the non-classical pathway, including NLPR3, the best-studied inflammasomes receptor that plays an important role in immune defense (62). NLPR3 interacts with apoptosis-associated speck-like protein containing a CARD (ASC) via the PYD domain after recognizing a signal. ASC is a speck-like protein with an N-terminal PYD domain and a C-terminal CARD domain. The inflammasome is formed by recruiting the CARD domain of ASC, recognizing, and interacting with pro-caspase-1. Caspase-1 activation can result in the cleavage of pro-IL-18 and pro-IL-1β and the release of mature IL-18 and IL-1β (63). While in a non-classical way, gram-negative bacterium's LPS can trigger caspase-11 in mice by binding with their CARD domains, caspase-4 and caspase-5 can also bind with LPS, leading to caspase-4/5/11 oligomerization and excitation in humans (64). Following activation, caspase-4, caspase-5, and caspase-11 cleave GSDMD to generate GSDMD-NT, which can self-assemble in the plasma membrane, causing pores to form and motivating the NLRP3 inflammasome to activate maturation and release of cytokines IL-1β and IL-18, ultimately promoting pyroptosis (65). It is worth noting that alternative pathways mediated by caspase-8 and caspase-3 can also result in pyroptosis. Caspase-8 acts as an apoptotic initiator, while caspase-3 acts as an executioner caspase, cleaving gasdermin E (GSDME) to form an N-terminal fragment of GSDME (GSDME-NT), promoting ATP-induced pyroptosis in macrophages (66). Caspase-8 also causes gasdermin D cleavage, which exacerbates pyroptosis in Yersinia infections (67). The molecular signaling mechanism of pyroptosis is depicted in Figure 1.
Contributions of pyroptosis to the pathophysiology of CVDs
Pyroptosis plays a critical role in the pathophysiology of different CVDs.
In I/R injury, although ischemia itself can damage the heart muscle, most of the myocardial infarctions stem from injury during reperfusion after a transient period of coronary artery occlusion. The occurrence of pyroptosis results from the activation of caspase and the cleavage of GSDMD induced by inflammasomes. This active fragment creates large pores in the cell membrane to destroy the myocardiocyte, thus aggravating myocardial injury (68).
Vascular endothelial cells (VEC) damage is indispensable to As lesions. The caspase-1 inflammasome pathway can upregulate pyroptosis-related proteins and ultimately trigger VEC pyroptosis, which brings about the loss of endothelium integrity and the increase of vascular permeability, thus promoting As development (69).
The inflammatory response is involved in the development of cardiac hypertrophy (CH) and heart failure (HF). NLRP3 inflammasome components have been discovered in cardiomyocytes, resulting in pyroptosis. This process eventually causes cardiomyocyte proliferation and cytoskeletal remodeling, which can exacerbate the impairment of cardiac function and promote the development of CH and HF (70, 71).
Cardiac fibrosis, one of the primary pathogenesis of diabetic cardiomyopathy (DC), plays a significant role in the process of DC induced by cardiac fibroblasts (CFs) (70). NLRP3 inflammasome components have also been identified in CFs. Hyperglycemia (HG) -induced ROS overproduction contributes to the activation of the NLRP3 inflammasome, of which components have been identified in CFs. In this process, pyroptosis will occur and eventually lead to dysfunction in CFs, thus promoting cardiac fibrosis in DC (72).
ncRNAs regulate pyroptosis in CVDs
miRNAs
MicroRNAs (miRNAs) carry out a variety of cellular functions (73). Recent research has shown that miRNA plays an important role in regulating pyroptosis in CVDs (16, 74).
NLRP3 plays an important role in the development of I/R injury, acting as a molecular platform and activating caspase-1 and cleaving pro-IL-1β, pro-IL-18, and GSDMD (75). A recent study discovered that miR-703 targets NLRP3 and inhibits its expression. Overexpression of miR-703 inhibited the upregulation of NLRP3, caspase-1 mRNA, and protein expression, reducing cardiomyocyte pyroptosis, implying that pyroptosis can be induced via the NLRP3/caspase-1 signaling pathway during the I/R-induced cardiomyocyte injury (76). Similarly, miR-135b may inhibit pyroptosis via NLRP3/caspase-1/IL-1β pathway, thereby reducing myocardial damage and protecting cardiac function (77). Furthermore, exosomal miR-320b protects the myocardium from I/R damage by inhibiting the production of pyroptosis-related proteins such as NLRP3 and caspase-1 (78).
SIRT1 is another important pyroptosis molecule in I/R injury. SIRT1/PGC-1α/Nrf2 signaling has been shown to mediate oxidative stress, which is important in myocardial I/R injury (79, 80). SIRT1 deficiency was associated with I/R injury. Furthermore, inhibiting miR-29a may improve myocardial I/R injury by downregulating SIRT1, as it can reduce oxidative stress and NLRP3-mediated pyroptosis, thereby protecting I/R injury (81). Similarly, miR 132 can directly target SIRT1 and suppress its expression to prevent oxidative stress and pyroptosis in myocardial cells (82).
Furthermore, FOXO3 is a key player in I/R injury, which increases oxidative damage and decreases myocardial function (83). A previous study found that a high level of miR-100-5p in hucMSC-exo could inhibit FOXO3 expression while suppressing the activation of NLRP3 inflammasomes, limiting cytokine release and protecting against I/R-induced pyroptosis (84). Yao et al. discovered that miRNA-1 could mediate the downregulation of PIK3R1 expression while upregulating FOXO3 and caspase-1 expression as well as the production of IL-1β and IL-18, which induced pyroptosis in cardiomyocytes injured by I/R (85). Then, by targeting FOXO3, miR-149 inhibition could also downregulate inflammatory factors like IL-18 and IL-1β in both mRNA and protein levels, preventing cardiomyocyte pyroptosis (86). FOXO3a, on the other hand, is a positive regulator of pyroptosis. miR-29b acted as a negative regulator, inhibiting the protective effect of FOXO3a and its downstream protein ARC, causing pyroptosis in I/R-injured myocardial cells via FOXO3a/ARC axis (87).
Other molecules, such as CRISPLD2, RP105, and MCL-1, also play protective roles in cardiomyocyte pyroptosis. By directly targeting CRISPLD2 and regulating cardiomyocyte pyroptosis, miR-424 promoted cardiac I/R injury (88). Piperine protected against pyroptosis in myocardial I/R injury by mediating the miR-383/RP105/AKT pathway, and RP105 worked as a miR-383 target (89). Exosomal miR-29a from macrophages treated with I/R plays a role in mediating cardiomyocyte pyroptosis by targeting MCL-1 (90). TXNIP, on the other hand, acts as a positive regulator in pyroptosis. M2-exos increased miR-148a expression, which could mediate TXNIP downregulation and inactivation of the TLR4/NF-κB/NLRP3 inflammasome signaling pathway to prevent myocardial pyroptosis caused by I/R injury (91).
In AS, miR-125a-5p mediated oxLDL-induced pyroptosis in vascular endothelial cells (VECs) by downregulating TET2 and increasing NF-κB activation, which activated NLRP3 and caspase-1, contributing to VECs and AS pyroptosis (92). TET2, as a DNA demethylase, protects VECs from the dysfunction caused by oxLDL in hydrogen sulfide (93). Furthermore, miR-125a-5p inhibition reduced ROS production by improving DNA methylation and mitochondrial function, as well as downregulating the expression of pyroptosis-related proteins, such as IL-1β, caspase-1, and NLRP3 (93). Peng et al. discovered that miR-30c-5p could inhibit NLRP3 inflammasome-mediated endothelial cell pyroptosis in AS by downregulating FOXO3 expression (94). In addition to oxLDL, shear stress played an important role in the onset and progression of AS, causing dramatic changes in signaling and gene expression in endothelial cells (ECs), regulating cell migration, proliferation, growth, and death (95). With the augmented expression of inflammasome-dependent pyroptosis in human umbilical vein endothelial cells (HUVECs), shear stress suppressed mechanosensitive miR-181b-5p expression, and miR-181-5p also inhibited signal transducer and transcriptional activation factor 3 (STAT3) gene expression by directly binding to its 3′UTR, leading to the downregulation of NLRP3 transcription and NLRP3 inflammasome-related pyroptosis (96). Notably, piceatannol inhibits pyroptosis in macrophages by upregulating Nrf2, which is known for its antioxidant and anti-inflammatory properties (97), via the miR-200a/Nrf2/GSDMD axis (81). Furthermore, miR-200a suppressed Nrf2 expression, resulting in increased levels of IL-18, IL-1β, and caspase-1.
miRNAs play an important role in cardiac hypertrophy (CH) and heart failure (HF). CH causes changes in cardiomyocytes such as calcium processing, metabolism, and gene expression, as well as cell death (apoptosis and autophagy), extracellular matrix (ECM) changes (fibrosis), and angiogenesis. HF is a crippling condition in which the heart fails to supply oxygen to the body. The heart initially responds to additional stress or heart damage in a compensatory manner, increasing its volume and mass to normalize wall stress and cardiovascular function at rest. Pathological CH refers to the typical enlargement of the heart (98). According to a recent study, MLK3 can promote pyroptosis of cardiomyocytes primarily via NFκB/NLRP3 signaling pathway, aggravating myocardial fibrosis in the early stages of chronic heart failure (CHF). The cardiac function of mice subjected to transverse aortic constriction (TAC) could then be significantly improved due to MLK3′s suppressive effect via upregulation of miR-351 expression (99). Similarly, Zhu et al. discovered that targeting IKKε to suppress pyroptosis could reduce cardiomyocyte hypertrophy caused by Ang II (100).
Furthermore, Fan et al. discovered that miR-599 inhibited pyroptosis caused by H2O2 by downregulating the expression of ASC as well as the downstream inflammatory factors IL-18 and IL-1β in oxidative stress-induced cardiac injury (101). Another group reported that overexpression of the lncRNA TUG1 alleviated NLRP3-dependent pyroptosis of cardiomyocytes by regulating miR-186-5p/XIAP axis in a study on coronary microembolization (CME)-induced myocardial damage.
Diabetic cardiomyopathy (DC), another common type of CVD, is defined as cardiac dysfunction in diabetic patients who do not have other cardiovascular diseases (102). Inflammation, oxidative stress, impaired immune regulation, improper activation of the renin-angiotensin-aldosterone system, abnormal subcellular components, and systemic metabolic disorders all contribute to heart stiffness/diastolic dysfunction, interstitial fibrosis of cardiac tissue, and posterior systolic dysfunction in diabetic cardiomyopathy (103). According to Jeyabal et al., miR-9 inhibition increased ELAV-like protein 1 (ELAVL1) expression as well as IL-1β and caspase-1 expression, whereas miR-9 mimic transfection decreased caspase-1, IL-1β, and ELAVL1 expression, which inhibited cardiomyocyte pyroptosis (104). Furthermore, upregulation of miR-30d in streptozotocin (STZ)-induced diabetic rats and high-glucose-treated cardiomyocytes aggravated pyroptosis by decreasing the concentrations of FOXO3a as well as its downstream protein like ARC and resulted in high expression of pro-pyroptosis factors or cytokines like IL-18, IL-1β, and caspase-1 (105). Furthermore, in STZ-induced diabetic cardiac fibrosis, miR-21–3p exacerbates pyroptosis of cardiomyocytes by suppressing AR expression via the NLRP3/caspase-1 pathways (106). miRNAs are also important in other CVDs. Sepsis-induced myocardial dysfunction (SIMD) is now thought to be the inherent systolic and diastolic myocardial dysfunction on the left and right sides of the heart caused by sepsis. (107, 108).
Recent research in SIMD found that the lncRNA ZFAS1, which is activated by the transcription factor SP1, reduces the expression of miR-590-3p, which regulates the AMPK/mTOR signal pathway, influencing NLRP3-dependent pyroptosis of cardiomyocytes and promoting SIMD (109). miRNAs also played a role in the pathogenesis of uremic cardiomyopathy (UC), another fatal cardiac disease. Diastolic insufficiency is a common feature of UC in patients with chronic kidney disease (CKD) who also have left ventricular hypertrophy (LVH) and myocardial fibrosis (110). Wang et al. discovered that macrophage-derived miR-155–containing exosomes aggravated pyroptosis and alterations (CH and cardiac tissue fibrosis) in UC by directly targeting FOXO3a (111). miRNAs also play important cellular roles in aortic dissection (AD). AD is a potentially fatal disease caused by the rupture of the aortic intima or bleeding of the aortic wall, which results in aortic wall detachment (112). Macrophages have dual anti-inflammatory and anti-inflammatory functions, play a critical role in aortic wall inflammation, and contribute to the development of AD and AD complications (113–116). A recent study found that miR-133a could negatively regulate the expression level of NLRP3 by specifically binding to its 3′UTR and that upregulating miR-133a inhibited pyroptosis induced by LPS in vSMCs (117). Similarly, in coronary artery disease (CAD), Wang et al. discovered that upregulation of miR-223 significantly reduced oxLDL-induced cell death, effectively eliminating NLRP3 inflammasome-mediated pyroptosis in human VECs (118).
lncRNAs
Similar to miRNAs, lncRNAs function in the regulation of pyroptosis in CVDs (119). In MI, lncRNA KLF3-AS1 in hMSC exosomes acted as a ceRNA to sponge miR-138-5p, which targeted SIRT1. Interfering with miR-138-5p increased the KLF3-AS1 expression while decreasing the level of IL-18, IL-1β, cleaved-caspase-1, ASC, cleaved-GSDMD, and NLRP3, indicating that NLRP3-mediated pyroptosis occurred via the miR-138-5p/SIRT1 axis in MI (120). Han et al. reported that overexpression of CYP1B1 abolished the suppressive effect of lncRNA H19 on pyroptosis, whereas H19 inhibited the activity of CYP1B1 promoters by targeting PBX3 to inhibit IL-18, IL-1β, ASC, and NLRP3 in hypoxic cardiomyocytes (121).
Furthermore, in AS, intragastric administration of melatonin reduced the expression of pyroptosis-related genes, such as cleaved-caspase-1, ASC, NLRP3, GSDMD-N termini, NF-κB/GSDMD cleaved-caspase-1, IL-1β, and IL-18, which significantly reduced atherosclerotic plaques in mouse aortas fed a high-fat diet via downregulating lncRNA MEG3, high expression of which prompted pyroptosis in HAECs (122). Song et al. (123) discovered that the lncRNA MALAT1 sponged miR-22 to promote NLRP3 inflammasome expression, causing pyroptosis in human ECs exposed to high glucose. Similarly, as explained in Han's study, pyroptosis was suppressed in HG-oxLDL-treated macrophages due to MALAT1 overexpression via the miR-23c/ELAVL1 axis (124). Anti-pyroptosis lncRNANEXN-AS1 was found to be upregulated in atorvastatin-treated HVECs. High lncRNANEXN-AS1 expression reduces the expression of pyroptosis-related genes (IL-18, IL-1β, GSDMD, caspase-1, and NLRP3) by upregulating NEXN (125).
Yang et al. also reported in DC that lncRNA sponged miR-214-3p by competing for binding sites and regulating gene expression. Silencing the lncRNA Kcnq1ot1 reduced pyroptosis and fibrosis in diabetic cardiomyopathy by targeting miR-214, which influenced the expression level of its downstream proteins (e.g., caspase-1, TGF-β1) (126, 127). Xu discovered that overexpression of the lncRNA GAS5 repressed miR-34b-3p expression while increasing aryl hydrocarbon receptor (AHR) expression, which inhibited NLRP3-dependent pyroptosis in DC by playing a role in miR-34b-3p/AHR signaling (128). In addition, in CME-induced myocardial damage, overexpression of lncRNA TUG1 alleviated pyroptosis of cardiomyocytes caused by the NLRP3 inflammasome via the miR-186-5p/XIAP axis (129).
circRNAs
So far, only a few circRNAs have been proven to play a critical role in CVDs. Yang et al. discovered in DC that the caspase-1-associated circRNA (CACR), hsa_circ_0076631, functioned as a ceRNA, sequestering miR-214-3p and thus alleviating its suppressive effect on caspase-1 expression. Pyroptosis in cardiomyocytes treated with high glucose could thus be significantly reduced (130).
In summary, ncRNAs are demonstrated to play an important role in CVDs through the regulation of pyroptosis, as shown in Table 2 and Figure 3.
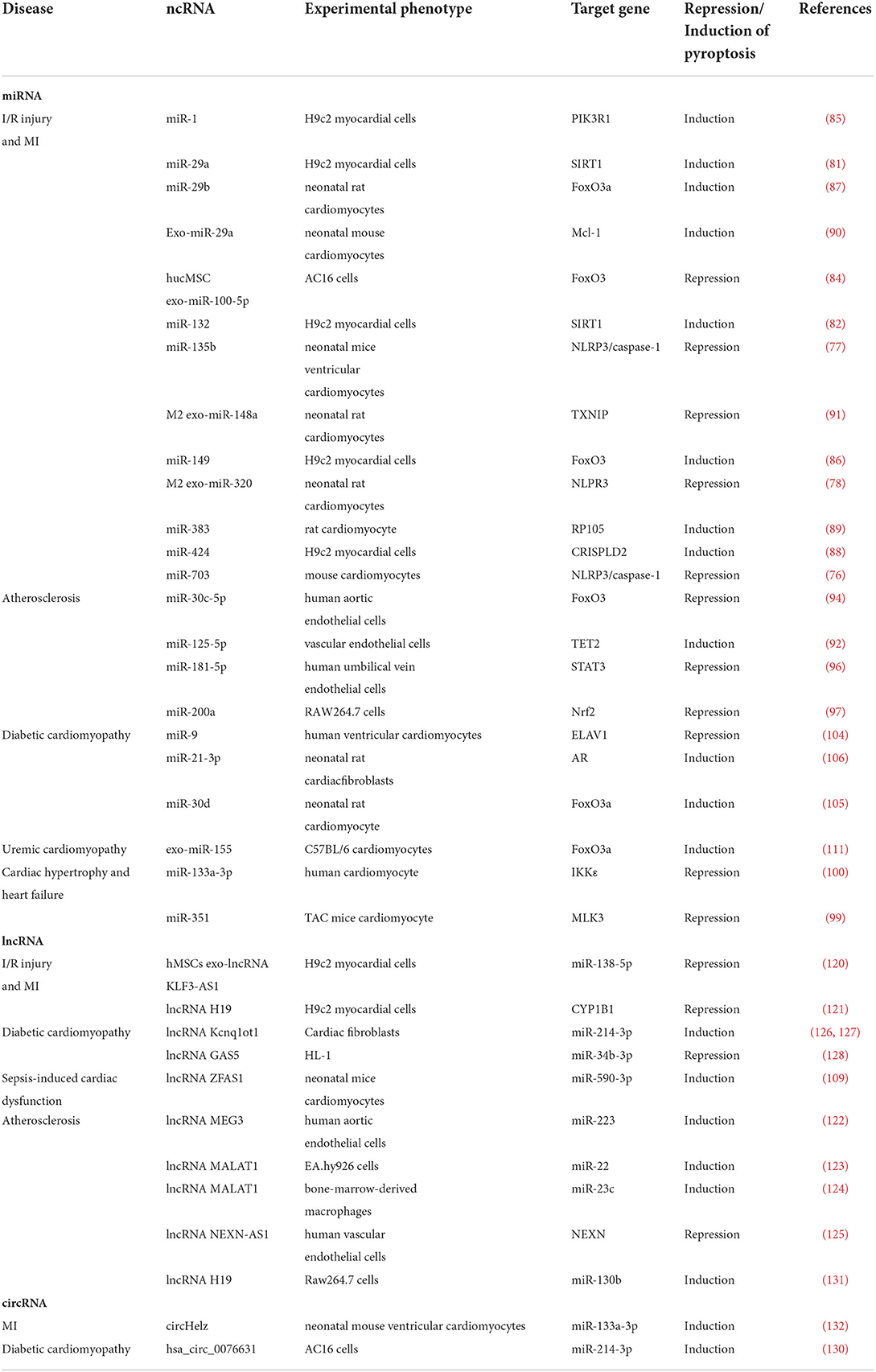
Table 2. Non-coding RNAs regulate pyroptosis in various types of CVDs.
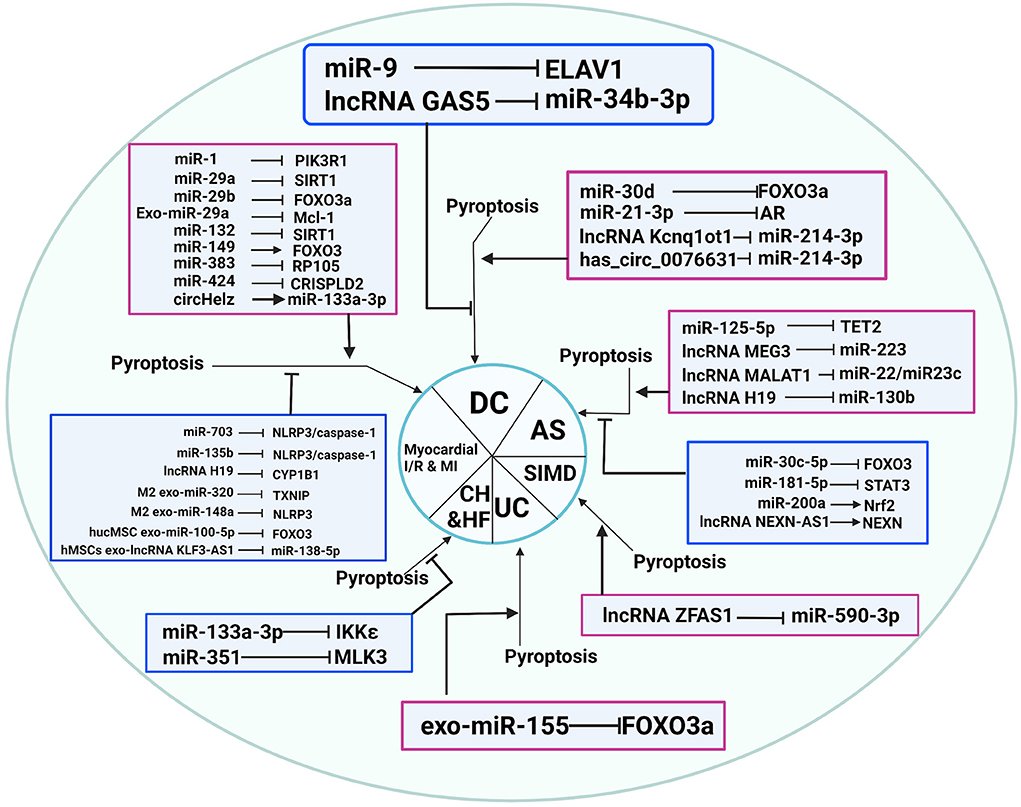
Figure 3. Non-coding RNAs play an essential role in CVDs through pyroptosis regulation. AS, atherosclerosis; CH, cardiac hypertrophy; DC, diabetic cardiomyopathy; HF, heart failure; I/R, ischemia-reperfusion; SIMD, sepsis-induced myocardial dysfunction; UC: uremic cardiomyopathy.
How to detect pyroptosis
The mechanism of pyroptosis involves three major signaling pathways, all activating downstream GSDMD and E, which leads to the release of IL-1 and IL-18. Accordingly, corresponding markers can be employed for detecting pyroptosis, such as the cleavage of GSDM D and E, release of IL-1β and −18, or caspase-1, −3, −4, −5, −8, and −11. Immunological techniques like western blotting can be applied as the most effective methods to detect or monitor pyroptosis owing to little involvement in transcription and translation during the modulation of pyroptosis (133).
Ferroptosis in CVDs
Ferroptosis signaling pathway
Ferroptosis is an oxidation iron-dependent cell death that differs from apoptosis, necroptosis, pyroptosis, and other types of cell death (134). Mechanically, low-glutathione levels induce the downregulation of glutathione peroxidase 4 (GPX4) and intracellular iron (mostly Fe2+) accumulation, which improves the level of lipid-based ROS, which activates the high level of expression of unsaturated fatty acids on the cell membrane, ultimately resulting in liposome peroxidation and cell death (135). The ferroptosis pathway involves the ferroptosis induced by inhibition of the cystine-glutamate transport receptor (systemXc-), ferroptosis induced by P53, and ferroptosis driven by GPX4 (136). Regardless of the different upstream pathways, the final effects all lead to the downregulation of glutathione peroxidase (GPXs), which reduces cells' antioxidant capacity and promotes the reaction of lipid peroxidation, resulting in ferroptosis (137). Ferroptosis inducers possess two types: one contains sulfoximine, sulfasalazine, erastin, etc. This kind of inducer suppresses the function of system Xc- and decreases the level of glutathione (GSH), which results in cell redox imbalance; another kind of inducer contains DPI13, RSL3, DPI7, DPI12, DPI10, etc. Synthetic compounds can suppress GPX4 and increase the level of peroxide in cardiomyocytes. Finally, due to the unnatural metabolism of iron ions in cardiomyocytes, the high level of ROS results in ferroptosis (138). The specific molecular signaling mechanism of ferroptosis is demonstrated in Figure 1.
Contributions of ferroptosis to the pathophysiology of CVDs
Ferroptosis is also considered a crucial part of the pathophysiology of diverse CVDs.
During myocardial I/R injury, large amounts of degraded ferritin lead to the release of tremendous amount of iron into the coronary arteries. Subsequently, excessive iron in coronary arteries can impair cardiac function and exacerbate myocardial damage (139, 140).
In the early and middle stages of MI, the expression of Gpx4 is inhibited, but the content of ROS increases, thus causing the ferroptosis of myocardial cells and the deterioration of cardiac function (141).
Iron homeostasis plays an indispensable role in maintaining myocardial function. Either iron deficiency or iron overload can worsen cardiac function in HF (142).
ncRNAs regulate ferroptosis in CVDs
Song et al. reported in MI that exosomes derived from HUCB-MSCs could carry miR-23a-3p to protect cardiomyocytes against I/R-induced ferroptosis by downregulating divalent metal transporter 1 (DMT1) expression, thereby attenuating myocardial injury in AMI mice (143). Another relevant study found that the expression of miR-30d decreased in cardiomyocytes after MI, which could inhibit autophagy by binding to autophagy-related protein 5 (ATG5). Furthermore, autophagy after MI might also promote ferroptosis (144).
miR-214 has been reported to mediate iron translocation into cells in I/R injury by modulating transferrin receptor 1 protein (TFR1) via binding to 3′UTR of TFR1. Furthermore, overexpression of PVT1 or silencing of miR-214 significantly reduced the effects of Fer-1 on ferroptosis in vitro (145).
Zheng et al. discovered in HF that miR-224-5p could bind to the 3′UTR region of ferritin heavy chain 1 (FTH1) and functioned as a downstream target of circSnx12, which regulated miR-224-5p expression. Furthermore, silencing circSnx12 or overexpression of miR-224-5p could cause cardiac cell death by lowering FTH1 expression and directly regulating iron overload in cardiomyocytes (146).
As indicated above, ncRNAs are involved in CVDs by influencing the occurrence of ferroptosis, as shown in Table 3 and Figure 2.
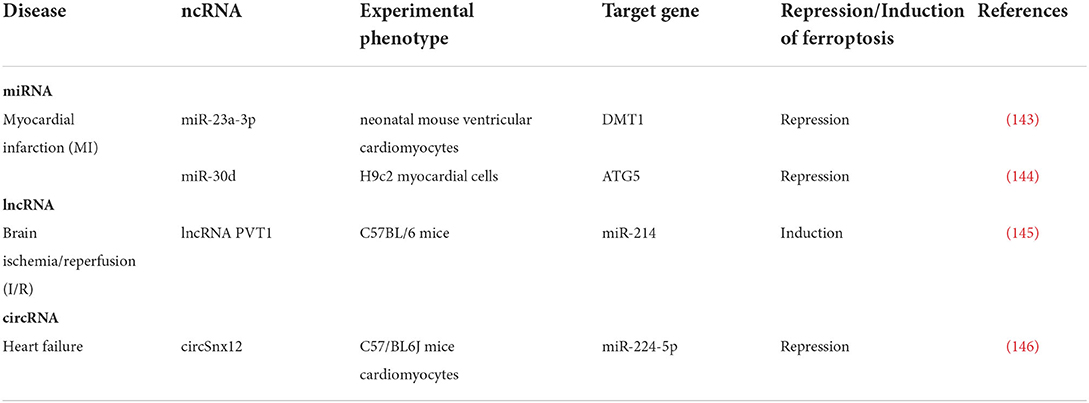
Table 3. Non-coding RNAs regulate ferroptosis in various types of CVDs.
How to detect ferroptosis
Ferroptosis is driven by the lethal accumulation of lipid peroxides in plasma membranes. Therefore, the determination of lipid peroxide level plays a key role in analyzing ferroptosis in biological samples. Recently, a newly discovered technique using BODIPY-C11 probe and flow cytometry to detect ferroptosis has been developed, which can assay cellular lipid peroxide levels in live cells with hypersensitivity and high accuracy (147).
ncRNAs as therapeutic targets in CVDs
Many ncRNAs have been identified as drug targets for treating CVDs in recent years, indicating the great potential of using ncRNAs as therapeutic targets in CVDs by influencing the manner of cell death. Recently, we found that piperine treatment can attenuate pyroptosis via downregulating the expression of miR-383 in the rat I/R injury model (148). In the rat model of MI, for example, the β blocker propranolol can reduce miR-1 expression by inhibiting the beta-adrenoceptor–cAMP–protein kinase A (PKA) signaling pathway and suppressing the expression of transcriptional factor serum response factor (SRF) (149), thus the propranolol may improve MI via downregulating miR-1 then inhibiting pyroptosis in cardiomyocytes (85). Another study found that Tanshinone IIA, an active component of Chinese medicine Salvia miltiorrhiza, could suppress the level of miR-1 in rats 3 months after MI (150). Likewise, in Laura's study, she discovered that Trimethylamine n-Oxide (TMAO), which is metabolized from Choline, L-carnitine, and betaine, can upregulate miR-30c-5p in HEPG-2, THP-1, mouse liver organoids, and primary human macrophages (151). Then, miR-30c-5p is associated with pyroptosis repression, which can lead to AS (94). Schisandrin A has been studied for its protective role in chronic heart failure via the upregulation of miR-155, which represses the necroptosis of cardiomyocytes (152). Moreover, we found that nicorandil combined with trimetazidine treatment can attenuate the progress of CHD via upregulating the miR-223-3p (153). Additionally, a study found that anti-miR132 can potentially play a role in heart failure treatment, which induces pyroptosis (148). In Wang's study, MIR-17-5P silencing improves myocardial tissue microcirculation by reducing the rate of apoptosis and repairing vascular damage, thereby protecting cardiac function after acute myocardial infarction (154). Moreover, prostaglandin E1 protects cardiomyocytes against hypoxia-reperfusion-induced injury via the miR-21-5p/FASLG axis (155). As a result, the studies described above demonstrate the infinite possibilities of targeting ncRNAs for treating CVDs, and more relevant studies will be published soon, contributing to the development of CVD targeted therapy.
Current limitations and future prospects
Clinical implication and limitation of ncRNAs in necroptosis, pyroptosis, and ferroptosis of CVDs
Significant progress has been made in the field of ncRNA over the last few decades, particularly in terms of its nature, structure, and function. Part of the research findings has recently been successfully implemented in clinical applications. Except as a therapeutic target, one of its most useful applications in CVDs is as a biomarker. For instance, a study verified that lncRNA MALAT1 could serve as biomarkers with independent predictive properties for diagnosing, severity, and prognosis of patients with sepsis (156). Because biomarkers are an essential tool for CVD screening, diagnosis, and prognosis, using good warning indicators can help doctors identify high-risk cardiovascular diseases as soon as possible, which is important for lowering death rates and improving prognosis. As a result, their clinical application is steadily increasing. Currently, the proliferation of biomedical technologies has broadened the range of new blood-derived biomarkers (157). Many researchers have affirmed that numerous circulating miRNAs are significantly upregulated or downregulated in various CVDs (158, 159). Recent research showed that overexpression of miRNA-195, miRNA-23a, miRNA-24, miRNA-23b, and other miRNAs could lead to cardiomyocyte hypertrophy in vitro (159).
When compared to traditional biomarkers, ncRNAs have four distinct advantages as follows: (1) abnormal ncRNA expression is linked to the onset and progression of diseases. (2) ncRNA expression is found in cells, tissues, and organs and has high sensitivity and specificity for diagnosing certain diseases. (3) Furthermore, due to their ability to secrete into blood compartments and other body fluids, ncRNAs are simple to acquire and detect. (4) Finally, ncRNAs are relatively stable and capable of transmitting signals locally or over long distances due to exosome transport (157, 160). Therefore, an increasing body of evidence supports the key role of ncRNA in the pathologic mechanism and development of CVDs. Accordingly, regardless of clinical or preclinical studies, both of them are striving to evaluate the diagnostic and prognostic potency of ncRNA and appraise the effectiveness of the treatment based on ncRNAs for CVDs. For instance, the usual biochemical markers for AMI include MB, CK-MB, cTnI, and cTnT (161). However, their clinical application effect in the early detection of AMI is not ideal and satisfactory. According to Pan et al., the concentrations of cTnI and CK-MB in the AMI group were significantly higher than in the control group, peaking 12 h after the disease onset. The expression level of miR-130 was also strongly upregulated in the AMI class, reaching a peak 6 h after the occurrence of AMI in the AMI class, compared to the administer classification. Circulating miR-130 levels peaked earlier than cTnI and CK-MB, implying that miR-130 levels can improve the diagnostic accuracy of cTnI and CK-MB and provide useful clues for the early detection of AMI (162).
However, several huge challenges must be overcome before the clinical applications of ncRNAs in necroptosis, pyroptosis, and ferroptosis of CVDs: (1) low expression in body fluids: First, the relative abundance of specific lncRNAs may pose a problem. Because, despite the fact that ncRNAs are present in both peripheral blood and whole blood samples, most peripheral blood miRNAs may originate primarily from well-vascularized tissues (163). Low expression of ncRNAs in body fluids makes it difficult to detect their specific biological functions (164). Thus, more powerful techniques are urgently needed to identify ncRNAs that modulate the necroptosis, pyroptosis, and ferroptosis signaling pathways and uncover their exact mechanisms. (2) maintaining the effectiveness and stability: Indeed, it is unclear whether the link between lncRNAs and miRNAs can serve as a potentially powerful therapeutic target. Because low-level lncRNAs bind to highly expressed miRNAs may not cause significant effects (165). In addition, maintaining the stability of ncRNAs is still a major difficulty (166). (3) The complexity of the regulatory mechanism: Additionally, ncRNAs have complex functions in necroptosis, pyroptosis, and ferroptosis of CVDs, influencing a wide range of pathways and biological functions, revealing the unexpected complexity of the clinical applications of ncRNAs in necroptosis, pyroptosis, and ferroptosis of CVDs. Therefore, there is an urgent need to comprehensively identify the distinct pathways, and these functional mechanisms and their applications are waiting for further research. (4) Plasma storage conditions and sample handling: Some researchers hypothesized that long-term storage and improper sample processing could affect the level of some miRNA in plasma, thereby influencing diagnostic results (167–169). (5) The high cost: Finally, the cost may be another considerable blockage since the manufacturing process of ncRNAs is relatively complicated, like controlling RNase activity, reverse transcription-polymerase (PCR), RNA purification, etc. (167, 170).
Future prospects of ncRNAs in necroptosis, pyroptosis, and ferroptosis of CVDs
In recent years, with the rapid development of quantification and standardization methodologies, the prevalence of ncRNAs as biomarkers has increased due to their disease specificity, accessibility, stability, and other distinguishing features, making ncRNAs valuable as biomarkers (171, 172). Considering the close link between ncRNAs and necroptosis, pyroptosis, and ferroptosis, it holds promise in developing ncRNA-based methods to monitor and interfere with necroptosis, pyroptosis, and ferroptosis in CVDs. Furthermore, the tissue-specific expression patterns of ncRNAs provide a unique possibility to regulate tissue-specific necroptosis, pyroptosis, and ferroptosis in CVDs. With more in-depth research, more necroptosis, pyroptosis, and ferroptosis-related ncRNAs will be discovered, and their underlying mechanisms of the necroptosis, pyroptosis, and ferroptosis pathways will increasingly be deciphered as well. But the fact is that methodological and biological characteristics currently limit the high specificity and sensitivity of ncRNA detection. (e.g., a short sequence of miRNAs, susceptibility to degradation, and kinetics) (173, 174). Consequently, further research is urgently needed to elucidate the relationship between the expression levels of ncRNAs in plasma and tissues. In addition, the quantities, storage conditions, sample handling, and kinetics of circulating ncRNAs should be improved, and the cost of ncRNAs should be cut down. Therefore, altering ncRNAs in vivo by novel therapeutics holds promise in developing a novel therapeutic approach to diagnosing and treating necroptosis, pyroptosis, and ferroptosis-related CVDs. Further investigations will continue to strive for these. The future trend and development direction in this field will construct a novel ncRNAs detection strategy with higher sensitivity and specificity (175).
Conclusion
CVDs are the leading cause of death from nearly all diseases worldwide. Cell death has long been recognized as necessary for our tissues and bodies to maintain their typical morphological character and physiological function, but it has recently been identified as the primary cause of clinical diseases and severe pathological injuries. Recently, it has been reported that new types of PCD, such as pyroptosis, necroptosis, and ferroptosis, play a different role in the progression of CVDs. In this review, we summarized and analyzed the pathway of ncRNAs involved in necroptosis, pyroptosis, and ferroptosis and their impact on CVD pathogenesis, as shown in Figures 2, 3. Thus, revealing the role of ncRNA-regulated programmed cell death in CVDs will identify novel diagnostic markers and potential therapeutic targets in the clinical treatment of CVDs.
Author contributions
PY and JZ: conceptualization, methodology, funding acquisition, and project administration. YC and YZ: writing original draft and formal analysis. ZL: visualization and supervision. PX: software and investigation. AS and XC: data curation, validation, writing—review and editing and resources. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Natural Science Foundation in Jiangxi Province, China (Grant Nos. 202002BAB216022, 20192ACBL21037, and 202004BCJL23049), the National Natural Science Foundation of China (Nos. 82160371 and 82100869), and National Clinical Research Center for Geriatrics-JiangXi Branch Center (2021ZDG02001).
Acknowledgments
The graphical abstracts were created with BioRender software (BioRender.com).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mensah GA, Roth GA, Fuster V. The global burden of cardiovascular diseases and risk factors: 2020 and beyond. J Am Coll Cardiol. (2019) 74:2529–32. doi: 10.1016/j.jacc.2019.10.009
2. Yusuf S, Wood D, Ralston J, Reddy KS. CardioPulse: prevention of global cardiovascular disease. Eur Heart J. (2016) 37:505–7. Available online at: https://pubmed.ncbi.nlm.nih.gov/27458620/
3. Wu S, Zhang S, Wu X, Zhou X. m(6)A RNA methylation in cardiovascular diseases. Molecular therapy: J Am Soc Gene Therapy. (2020) 28:2111–9. doi: 10.1016/j.ymthe.2020.08.010
4. Huang Y. The novel regulatory role of lncRNA-miRNA-mRNA axis in cardiovascular diseases. J Cell Mol Med. (2018) 22:5768–75. doi: 10.1111/jcmm.13866
5. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. (2019) 99:1765–817. doi: 10.1152/physrev.00022.2018
6. Karunakaran D, Geoffrion M, Wei L, Gan W, Richards L, Shangari P, et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. (2016) 2:e1600224. doi: 10.1126/sciadv.1600224
7. Wu Q, Kaji N, Yasui T, Rahong S, Yanagida T, Kanai M, et al. A millisecond micro-RNA separation technique by a hybrid structure of nanopillars and nanoslits. Sci Rep. (2017) 7:43877. doi: 10.1038/srep43877
8. Hombach S, Kretz M. Non-coding RNAs: classification, biology and functioning. Adv Exp Med Biol. (2016) 937:3–17. doi: 10.1007/978-3-319-42059-2_1
9. Kalayinia S, Arjmand F, Maleki M, Malakootian M, Singh CP. MicroRNAs: roles in cardiovascular development and disease. Cardiovasc Pathol. (2021) 50:107296. doi: 10.1016/j.carpath.2020.107296
10. Statello L, Guo CJ, Chen LL, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. (2021) 22:96–118. doi: 10.1038/s41580-020-00315-9
11. Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. (2013) 495:333–8. doi: 10.1038/nature11928
12. Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. (2011) 12:99–110. doi: 10.1038/nrg2936
13. Dong Y, Liu C, Zhao Y, Ponnusamy M, Li P, Wang K. Role of noncoding RNAs in regulation of cardiac cell death and cardiovascular diseases. CMLS. (2018) 75:291–300. doi: 10.1007/s00018-017-2640-8
14. Xu C, Jia Z, Cao X, Wang S, Wang J, An L. Hsa_circ_0007059 promotes apoptosis and inflammation in cardiomyocytes during ischemia by targeting microRNA-378 and microRNA-383. Cell Cycle (Georgetown, Tex). (2022) 21:1003–19. doi: 10.1080/15384101.2022.2040122
15. Zhe-Wei S, Li-Sha G, Yue-Chun L. The role of necroptosis in cardiovascular disease. Front Pharmacol. (2018) 9:721. doi: 10.3389/fphar.2018.00721
16. Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of pyroptosis in cardiovascular disease. Cell Prolif. (2019) 52:e12563. doi: 10.1111/cpr.12563
17. Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. (2021) 11:3052–9. doi: 10.7150/thno.54113
18. Jusic A, Devaux Y. Mitochondrial noncoding RNA-regulatory network in cardiovascular disease. Basic Res Cardiol. (2020) 115:23. doi: 10.1007/s00395-020-0783-5
19. Qi T, Chen Y, Li H, Pei Y, Woo SL, Guo X, et al. A role for PFKFB3/iPFK2 in metformin suppression of adipocyte inflammatory responses. J Mol Endocrinol. (2017) 59:49–59. doi: 10.1530/JME-17-0066
20. Jaé N, Dimmeler S. Noncoding RNAs in vascular diseases. Circ Res. (2020) 126:1127–45. doi: 10.1161/CIRCRESAHA.119.315938
21. Gomes CPC, Schroen B, Kuster GM, Robinson EL, Ford K, Squire IB, et al. Regulatory RNAs in heart failure. Circulation. (2020) 141:313–28. doi: 10.1161/CIRCULATIONAHA.119.042474
22. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. (2014) 15:135–47. doi: 10.1038/nrm3737
23. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. (2003) 114:181–90. doi: 10.1016/S0092-8674(03)00521-X
24. Han J, Zhong CQ, Zhang DW. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol. (2011) 12:1143–9. doi: 10.1038/ni.2159
25. Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. (2011) 471:363–7. doi: 10.1038/nature09852
26. Moquin DM, McQuade T, Chan FK. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE. (2013) 8:e76841. doi: 10.1371/journal.pone.0076841
27. Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. (2010) 327:1135–9. doi: 10.1126/science.1182364
28. Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. (2013) 20:1381–92. doi: 10.1038/cdd.2013.94
29. Legarda-Addison D, Hase H, O'Donnell MA, Ting AT. NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell Death Differ. (2009) 16:1279–88. doi: 10.1038/cdd.2009.41
30. Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, et al. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J. (2011) 433:447–57. doi: 10.1042/BJ20101738
31. Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y. Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. (2013) 5:70–8. doi: 10.1016/j.celrep.2013.08.044
32. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. (2014) 16:55–65. doi: 10.1038/ncb2883
33. Meng L, Jin W, Wang Y, Huang H, Li J, Zhang C. RIP3-dependent necrosis induced inflammation exacerbates atherosclerosis. Biochem Biophys Res Commun. (2016) 473:497–502. doi: 10.1016/j.bbrc.2016.03.059
34. Horvath C, Young M, Jarabicova I, Kindernay L, Ferenczyova K, Ravingerova T, et al. Inhibition of cardiac RIP3 mitigates early reperfusion injury and calcium-induced mitochondrial swelling without altering necroptotic signalling. Int J Mol Sci. (2021) 22:57983. doi: 10.3390/ijms22157983
35. Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res. (2014) 103:206–16. doi: 10.1093/cvr/cvu146
36. Oerlemans MI, Liu J, Arslan F, den Ouden K, van Middelaar BJ, Doevendans PA, et al. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol. (2012) 107:270. doi: 10.1007/s00395-012-0270-8
37. Su Z, Yang Z, Xu Y, Chen Y, Yu Q. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget. (2015) 6:8474–90. doi: 10.18632/oncotarget.3523
38. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. (2011) 17:1391–401. doi: 10.1038/nm.2507
39. Yan HF, Tuo QZ, Yin QZ, Lei P. The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zoological Res. (2020) 41:220–30. doi: 10.24272/j.issn.2095-8137.2020.042
40. Qin D, Wang X, Li Y, Yang L, Wang R, Peng J, et al. MicroRNA-223-5p and−3p Cooperatively suppress necroptosis in ischemic/reperfused hearts. J Biol Chem. (2016) 291:20247–59. doi: 10.1074/jbc.M116.732735
41. Tan H, Qi J, Fan BY, Zhang J, Su FF, Wang HT. MicroRNA-24-3p Attenuates Myocardial Ischemia/Reperfusion Injury by Suppressing RIPK1 Expression in Mice. Int J Exper Cell Physiol, Biochem Pharmacol. (2018) 51:46–62. doi: 10.1159/000495161
42. Saleh M, Ambrose JA. Understanding myocardial infarction. F1000Research. (2018) 7:15096. doi: 10.12688/f1000research.15096.1
43. Zhou X, Zhang P, Wang Q, Ji N, Xia S, Ding Y, et al. Metformin ameliorates experimental diabetic periodontitis independently of mammalian target of rapamycin (mTOR) inhibition by reducing NIMA-related kinase 7(Nek7) expression. J Periodontol. (2019). doi: 10.1002/jper.10311
44. Zaafan MA, Abdelhamid AM. The cardioprotective effect of microRNA-103 inhibitor against isoprenaline-induced myocardial infarction in mice through targeting FADD/RIPK pathway. Eur Rev Med Pharmacol Sci. (2021) 25:837–44. doi: 10.26355/eurrev_202101_24648
45. Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. (2011) 35:572–82. doi: 10.1016/j.immuni.2011.08.014
46. Liu J, van Mil A, Vrijsen K, Zhao J, Gao L, Metz CH, et al. MicroRNA-155 prevents necrotic cell death in human cardiomyocyte progenitor cells via targeting RIP1. J Cell Mol Med. (2011) 15:1474–82. doi: 10.1111/j.1582-4934.2010.01104.x
47. Bäck M, Yurdagul A Jr, Tabas I, Öörni K, Kovanen PT Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nature Rev Cardiol. (2019) 16:389–406. doi: 10.1038/s41569-019-0169-2
48. Karshovska E, Wei Y, Subramanian P, Mohibullah R, Geißler C, Baatsch I, et al. HIF-1α (Hypoxia-Inducible Factor-1α) promotes macrophage necroptosis by regulating miR-210 and miR-383. Arterioscler Thromb Vasc Biol. (2020) 40:583–96. doi: 10.1161/ATVBAHA.119.313290
49. Yang T, Cao C, Yang J, Liu T, Lei XG, Zhang Z, et al. miR-200a-5p regulates myocardial necroptosis induced by Se deficiency via targeting RNF11. Redox Biol. (2018) 15:159–69. doi: 10.1016/j.redox.2017.11.025
50. Jiang L, Liu XQ, Ma Q, Yang Q, Gao L, Li HD, et al. hsa-miR-500a-3P alleviates kidney injury by targeting MLKL-mediated necroptosis in renal epithelial cells. FASEB. (2019) 33:3523–35. doi: 10.1096/fj.201801711R
51. Huang G, Xue J, Sun X, Wang J, Yu LL. Necroptosis in 3-chloro-1, 2-propanediol (3-MCPD)-dipalmitate-induced acute kidney injury in vivo and its repression by miR-223-3p. Toxicology. (2018) 406:33-43. doi: 10.1016/j.tox.2018.05.015
52. Gu C, Hou C, Zhang S. miR-425-5p improves inflammation and septic liver damage through negatively regulating the RIP1-mediated necroptosis. Inflamm Res. (2020) 69:299–308. doi: 10.1007/s00011-020-01321-5
53. Wang K, Liu F, Liu CY, An T, Zhang J, Zhou LY, et al. The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ. (2016) 23:1394–405. doi: 10.1038/cdd.2016.28
54. He S, Huang S, Shen Z. Biomarkers for the detection of necroptosis. CMLS. (2016) 73:2177–81. doi: 10.1007/s00018-016-2192-3
55. Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, et al. Activation of necroptosis in multiple sclerosis. Cell Rep. (2015) 10:1836–49. doi: 10.1016/j.celrep.2015.02.051
56. Jia C, Chen H, Zhang J, Zhou K, Zhuge Y, Niu C, et al. Role of pyroptosis in cardiovascular diseases. Int Immunopharmacol. (2019) 67:311–8. doi: 10.1016/j.intimp.2018.12.028
57. Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol. (2016) 26:R568–r72. doi: 10.1016/j.cub.2016.02.019
58. Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. (2000) 38:31–40. doi: 10.1046/j.1365-2958.2000.02103.x
59. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
60. Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, et al. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell. (2020) 180:941–55.e20. doi: 10.1016/j.cell.2020.02.002
61. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
62. de Torre-Minguela C, Mesa Del Castillo P, Pelegrín P. The NLRP3 and pyrin inflammasomes: implications in the pathophysiology of autoinflammatory diseases. Front Immunol. (2017) 8:43. doi: 10.3389/fimmu.2017.00043
63. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
64. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. (2013) 341:1246–9. doi: 10.1126/science.1240248
65. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
66. Zeng CY Li CG, Shu JX, Xu LH, Ouyang DY, Mai FY, et al. ATP induces caspase-3/gasdermin E-mediated pyroptosis in NLRP3 pathway-blocked murine macrophages. Apoptosis. (2019) 24:703–17. doi: 10.1007/s10495-019-01551-x
67. Sarhan J, Liu BC, Muendlein HI Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A. (2018) 115:E10888–e97. doi: 10.1073/pnas.1809548115
68. Popov SV, Maslov LN, Naryzhnaya NV, Mukhomezyanov AV, Krylatov AV, Tsibulnikov SY, et al. The Role of Pyroptosis in Ischemic and Reperfusion Injury of the Heart. J Cardiovasc Pharmacol Ther. (2021) 26:562–74. doi: 10.1177/10742484211027405
69. Wu X, Zhang H, Qi W, Zhang Y, Li J, Li Z, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. (2018) 9:171. doi: 10.1038/s41419-017-0257-3
70. Li G, Xing W, Zhang M, Geng F, Yang H, Zhang H, et al. Antifibrotic cardioprotection of berberine via downregulating myocardial IGF-1 receptor-regulated MMP-2/MMP-9 expression in diabetic rats. Am J Physiol Heart Circ Physiol. (2018) 315:H802–h13. doi: 10.1152/ajpheart.00093.2018
71. Bai Y, Sun X, Chu Q, Li A, Qin Y, Li Y, et al. Caspase-1 regulate AngII-induced cardiomyocyte hypertrophy via upregulation of IL-1β. Bioscience Rep. (2018) 38:1438. doi: 10.1042/BSR20171438
72. Luo B, Huang F, Liu Y, Liang Y, Wei Z, Ke H, et al. NLRP3 inflammasome as a molecular marker in diabetic cardiomyopathy. Front Physiol. (2017) 8:519. doi: 10.3389/fphys.2017.00519
73. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. (2018) 141:1202–7. doi: 10.1016/j.jaci.2017.08.034
74. Gao J, Chen X, Wei P, Wang Y, Li P, Shao K. Regulation of pyroptosis in cardiovascular pathologies: Role of noncoding RNAs. Molecular Therapy Nucl Acids. (2021) 25:220–36. doi: 10.1016/j.omtn.2021.05.016
75. Wang Y, Liu X, Shi H, Yu Y, Yu Y, Li M, et al. NLRP3 inflammasome, an immune-inflammatory target in pathogenesis and treatment of cardiovascular diseases. Clin Transl Med. (2020) 10:91–106. doi: 10.1002/ctm2.13
76. Wei X, Peng H, Deng M, Feng Z, Peng C, Yang D. MiR-703 protects against hypoxia/reoxygenation-induced cardiomyocyte injury via inhibiting the NLRP3/caspase-1-mediated pyroptosis. J Bioenerg Biomembr. (2020) 52:155–64. doi: 10.1007/s10863-020-09832-w
77. Li A, Yu Y, Ding X, Qin Y, Jiang Y, Wang X, et al. MiR-135b protects cardiomyocytes from infarction through restraining the NLRP3/caspase-1/IL-1β pathway. Int J Cardiol. (2020) 307:137–45. doi: 10.1016/j.ijcard.2019.09.055
78. Tang J, Jin L, Liu Y, Li L, Ma Y, Lu L, et al. Exosomes derived from mesenchymal stem cells protect the myocardium against ischemia/reperfusion injury through inhibiting pyroptosis. Drug Des Devel Ther. (2020) 14:3765–75. doi: 10.2147/DDDT.S239546
79. Huang K, Gao X, Wei W. The crosstalk between Sirt1 and Keap1/Nrf2/ARE anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-β1 expressions in rat glomerular mesangial cells. Exp Cell Res. (2017) 361:63–72. doi: 10.1016/j.yexcr.2017.09.042
80. Shah SA, Khan M, Jo MH, Jo MG, Amin FU, Kim MO. Melatonin stimulates the SIRT1/Nrf2 signaling pathway counteracting lipopolysaccharide (LPS)-induced oxidative stress to rescue postnatal rat brain. CNS Neurosci Ther. (2017) 23:33–44. doi: 10.1111/cns.12588
81. Ding S, Liu D, Wang L, Wang G, Zhu Y. Inhibiting MicroRNA-29a protects myocardial ischemia-reperfusion injury by targeting SIRT1 and suppressing oxidative stress and NLRP3-mediated pyroptosis pathway. J Pharmacol Exp Ther. (2020) 372:128–35. doi: 10.1124/jpet.119.256982
82. Zhou Y, Li KS, Liu L, Li SL. MicroRNA132 promotes oxidative stress induced pyroptosis by targeting sirtuin 1 in myocardial ischaemia-reperfusion injury. Int J Mol Med. (2020) 45:1942–50. doi: 10.3892/ijmm.2020.4557
83. Sengupta A, Molkentin JD, Paik JH, DePinho RA, Yutzey KE. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J Biol Chem. (2011) 286:7468–78. doi: 10.1074/jbc.M110.179242
84. Liang C, Liu Y, Xu H, Huang J, Shen Y, Chen F, et al. Exosomes of human umbilical cord mscs protect against hypoxia/reoxygenation-induced pyroptosis of cardiomyocytes via the miRNA-100-5p/FOXO3/NLRP3 Pathway. Front Bioeng Biotechnol. (2020) 8:615850. doi: 10.3389/fbioe.2020.615850
85. Yao RD Li HL, Liu Y, Sun LT. MiRNA-1 promotes pyroptosis of cardiomyocytes and release of inflammatory factors by downregulating the expression level of PIK3R1 through the FoxO3a pathway. Eur Rev Med Pharmacol Sci. (2020) 24:11243–50.
86. Lin J, Lin H, Ma C, Dong F, Hu Y, Li H. MiR-149 Aggravates Pyroptosis in Myocardial Ischemia-Reperfusion Damage via Silencing FoxO3. Med Sci Monit. (2019) 25:8733–43. doi: 10.12659/MSM.918410
87. Zhong Y, Li YP, Yin YQ, Hu BL, Gao H. Dexmedetomidine inhibits pyroptosis by down-regulating miR-29b in myocardial ischemia reperfusion injury in rats. Int Immunopharmacol. (2020) 86:106768. doi: 10.1016/j.intimp.2020.106768
88. Lou Y, Wang S, Qu J, Zheng J, Jiang W, Lin Z, et al. miR-424 promotes cardiac ischemia/reperfusion injury by direct targeting of CRISPLD2 and regulating cardiomyocyte pyroptosis. Int J Clin Exp Pathol. (2018) 11:3222–35.
89. Guo X, Hu S, Liu JJ, Huang L, Zhong P, Fan ZX, et al. Piperine protects against pyroptosis in myocardial ischaemia/reperfusion injury by regulating the miR-383/RP105/AKT signalling pathway. J Cell Mol Med. (2021) 25:244–58. doi: 10.1111/jcmm.15953
90. Wang Y, Qiu Z, Yuan J, Li C, Zhao R, Liu W, et al. Hypoxia-reoxygenation induces macrophage polarization and causes the release of exosomal miR-29a to mediate cardiomyocyte pyroptosis. In Vitro Cell Dev Biol Anim. (2021) 57:30–41. doi: 10.1007/s11626-020-00524-8
91. Dai Y, Wang S, Chang S, Ren D, Shali S, Li C, et al. M2 macrophage-derived exosomes carry microRNA-148a to alleviate myocardial ischemia/reperfusion injury via inhibiting TXNIP and the TLR4/NF-κB/NLRP3 inflammasome signaling pathway. J Mol Cell Cardiol. (2020) 142:65–79. doi: 10.1016/j.yjmcc.2020.02.007
92. Zhaolin Z, Jiaojiao C, Peng W, Yami L, Tingting Z, Jun T, et al. OxLDL induces vascular endothelial cell pyroptosis through miR-125a-5p/TET2 pathway. J Cell Physiol. (2019) 234:7475–91. doi: 10.1002/jcp.27509
93. Peng J, Tang ZH, Ren Z, He B, Zeng Y, Liu LS, et al. TET2 Protects against oxLDL-Induced HUVEC Dysfunction by Upregulating the CSE/H(2)S System. Front Pharmacol. (2017) 8:486. doi: 10.3389/fphar.2017.00486
94. Li P, Zhong X, Li J, Liu H, Ma X, He R, et al. MicroRNA-30c-5p inhibits NLRP3 inflammasome-mediated endothelial cell pyroptosis through FOXO3 down-regulation in atherosclerosis. Biochem Biophys Res Commun. (2018) 503:2833–40. doi: 10.1016/j.bbrc.2018.08.049
95. Sakamoto N, Ueki Y, Oi M, Kiuchi T, Sato M. Fluid shear stress suppresses ICAM-1-mediated transendothelial migration of leukocytes in coculture model. Biochem Biophys Res Commun. (2018) 502:403–8. doi: 10.1016/j.bbrc.2018.05.182
96. Xu X, Yang Y, Wang G, Yin Y, Han S, Zheng D, et al. Low shear stress regulates vascular endothelial cell pyroptosis through miR-181b-5p/STAT-3 axis. J Cell Physiol. (2021) 236:318–27. doi: 10.1002/jcp.29844
97. Mu Z, Zhang H, Lei P. Piceatannol inhibits pyroptosis and suppresses oxLDL-induced lipid storage in macrophages by regulating miR-200a/Nrf2/GSDMD axis. Biosci Rep. (2020) 40:1366. doi: 10.1042/BSR20201366
98. Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. (2015) 89:1401–38. doi: 10.1007/s00204-015-1477-x
99. Wang J, Deng B, Liu Q, Huang Y, Chen W, Li J, et al. Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis. (2020) 11:574. doi: 10.1038/s41419-020-02777-3
100. Zhu YF, Wang R, Chen W, Cao YD Li LP, Chen X. miR-133a-3p attenuates cardiomyocyte hypertrophy through inhibiting pyroptosis activation by targeting IKKε. Acta Histochem. (2021) 123:151653. doi: 10.1016/j.acthis.2020.151653
101. Fan X, Zhan E, Yao Y, Zhang R, Sun Y, Tian X. MiR-599 Protects cardiomyocytes against oxidative stress-induced pyroptosis. Biomed Res Int. (2021) 2021:3287053. doi: 10.1155/2021/3287053
102. Paolillo S, Marsico F, Prastaro M, Renga F, Esposito L, De Martino F, et al. Diabetic cardiomyopathy: definition, diagnosis, and therapeutic implications. Heart Fail Clin. (2019) 15:341–7. doi: 10.1016/j.hfc.2019.02.003
103. Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia. (2018) 61:21–8. doi: 10.1007/s00125-017-4390-4
104. Jeyabal P, Thandavarayan RA, Joladarashi D, Suresh Babu S, Krishnamurthy S, Bhimaraj A, et al. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem Biophys Res Commun. (2016) 471:423–9. doi: 10.1016/j.bbrc.2016.02.065
105. Li X, Du N, Zhang Q, Li J, Chen X, Liu X, et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. (2014) 5:e1479. doi: 10.1038/cddis.2014.430
106. Shi P, Zhao XD, Shi KH, Ding XS, Tao H. MiR-21-3p triggers cardiac fibroblasts pyroptosis in diabetic cardiac fibrosis via inhibiting androgen receptor. Exp Cell Res. (2021) 399:112464. doi: 10.1016/j.yexcr.2020.112464
107. Zaky A, Deem S, Bendjelid K, Treggiari MM. Characterization of cardiac dysfunction in sepsis: an ongoing challenge. Shock (Augusta, Ga). (2014) 41:12–24. doi: 10.1097/SHK.0000000000000065
108. Antonucci E, Fiaccadori E, Donadello K, Taccone FS, Franchi F, Scolletta S. Myocardial depression in sepsis: from pathogenesis to clinical manifestations and treatment. J Crit Care. (2014) 29:500–11. doi: 10.1016/j.jcrc.2014.03.028
109. Liu JJ Li Y, Yang MS, Chen R, Cen CQ. SP1-induced ZFAS1 aggravates sepsis-induced cardiac dysfunction via miR-590-3p/NLRP3-mediated autophagy and pyroptosis. Arch Biochem Biophys. (2020) 695:108611. doi: 10.1016/j.abb.2020.108611
110. Wang X, Shapiro JI. Evolving concepts in the pathogenesis of uraemic cardiomyopathy. Nature Rev Nephrol. (2019) 15:159–75. doi: 10.1038/s41581-018-0101-8
111. Wang B, Wang ZM Ji JL, Gan W, Zhang A, Shi HJ, et al. Macrophage-Derived Exosomal Mir-155 regulating cardiomyocyte pyroptosis and hypertrophy in uremic cardiomyopathy. JACC Basic Trans Sci. (2020) 5:148–66. doi: 10.1016/j.jacbts.2019.10.011
112. Nienaber CA, Clough RE, Sakalihasan N, Suzuki T, Gibbs R, Mussa F, et al. Aortic dissection. Nature Rev Dis Primers. (2016) 2:16053. doi: 10.1038/nrdp.2016.53
113. Andreata F, Syvannarath V, Clement M, Delbosc S, Guedj K, Fornasa G, et al. Macrophage CD31 signaling in dissecting aortic aneurysm. J Am Coll Cardiol. (2018) 72:45–57. doi: 10.1016/j.jacc.2018.04.047
114. Visonà SD, de Boer OJ, Mackaaij C, de Boer HH, Pertiwi KR, de Winter RW, et al. Immunophenotypic analysis of the chronological events of tissue repair in aortic medial dissections. Cardiovasc Pathol. (2018) 34:9–14. doi: 10.1016/j.carpath.2018.01.009
115. Xu Y, Ye J, Wang M, Wang Y, Ji Q, Huang Y, et al. Increased interleukin-11 levels in thoracic aorta and plasma from patients with acute thoracic aortic dissection. Clin Chim Acta. (2018) 481:193–9. doi: 10.1016/j.cca.2018.03.014
116. Ye J, Wang M, Jiang H, Ji Q, Huang Y, Liu J, et al. Increased levels of interleukin-22 in thoracic aorta and plasma from patients with acute thoracic aortic dissection. Clin Chim Acta. (2018) 486:395–401. doi: 10.1016/j.cca.2017.10.033
117. Duan H, Zhang X, Song R, Liu T, Zhang Y, Yu A. Upregulation of miR-133a by adiponectin inhibits pyroptosis pathway and rescues acute aortic dissection. Acta Biochim Biophys Sin. (2020) 52:988–97. doi: 10.1093/abbs/gmaa078
118. Wang X, Li X, Wu Y, Song Y. Upregulation of miR-223 abrogates NLRP3 inflammasome-mediated pyroptosis to attenuate oxidized low-density lipoprotein (ox-LDL)-induced cell death in human vascular endothelial cells (ECs). In Vitro Cell Dev Biol Anim. (2020) 56:670–9. doi: 10.1007/s11626-020-00496-9
119. He D, Zheng J, Hu J, Chen J, Wei X. Long non-coding RNAs and pyroptosis. Clin Chim Acta. (2020) 504:201–8. doi: 10.1016/j.cca.2019.11.035
120. Mao Q, Liang XL, Zhang CL, Pang YH, Lu YX. LncRNA KLF3-AS1 in human mesenchymal stem cell-derived exosomes ameliorates pyroptosis of cardiomyocytes and myocardial infarction through miR-138-5p/Sirt1 axis. Stem Cell Res Ther. (2019) 10:393. doi: 10.1186/s13287-019-1522-4
121. Han Y, Dong B, Chen M, Yao C. LncRNA H19 suppresses pyroptosis of cardiomyocytes to attenuate myocardial infarction in a PBX3/CYP1B1-dependent manner. Mol Cell Biochem. (2021) 476:1387–400. doi: 10.1007/s11010-020-03998-y
122. Zhang Y, Liu X, Bai X, Lin Y, Li Z, Fu J, et al. Melatonin prevents endothelial cell pyroptosis via regulation of long noncoding RNA MEG3/miR-223/NLRP3 axis. J Pineal Res. (2018) 64:12449. doi: 10.1111/jpi.12449
123. Song Y, Yang L, Guo R, Lu N, Shi Y, Wang X. Long noncoding RNA MALAT1 promotes high glucose-induced human endothelial cells pyroptosis by affecting NLRP3 expression through competitively binding miR-22. Biochem Biophys Res Commun. (2019) 509:359–66. doi: 10.1016/j.bbrc.2018.12.139
124. Han Y, Qiu H, Pei X, Fan Y, Tian H, Geng J. Low-dose sinapic acid abates the pyroptosis of macrophages by downregulation of lncRNA-MALAT1 in rats with diabetic atherosclerosis. J Cardiovasc Pharmacol. (2018) 71:104–12. doi: 10.1097/FJC.0000000000000550
125. Wu LM, Wu SG, Chen F, Wu Q, Wu CM, Kang CM, et al. Atorvastatin inhibits pyroptosis through the lncRNA NEXN-AS1/NEXN pathway in human vascular endothelial cells. Atherosclerosis. (2020) 293:26–34. doi: 10.1016/j.atherosclerosis.2019.11.033
126. Yang F, Qin Y, Lv J, Wang Y, Che H, Chen X, et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis. (2018) 9:1000. doi: 10.1038/s41419-018-1029-4
127. Yang F, Qin Y, Wang Y, Li A, Lv J, Sun X, et al. LncRNA KCNQ1OT1 mediates pyroptosis in diabetic cardiomyopathy. Cell Physiol Biochem. (2018) 50:1230–44. doi: 10.1159/000494576
128. Xu Y, Fang H, Xu Q, Xu C, Yang L, Huang C. LncRNA GAS5 inhibits NLRP3 inflammasome activation-mediated pyroptosis in diabetic cardiomyopathy by targeting miR-34b-3p/AHR. Cell Cycle. (2020) 19:3054–65. doi: 10.1080/15384101.2020.1831245
129. Zhou Y, Li T, Chen Z, Huang J, Qin Z, Li L. Overexpression of lncRNA TUG1 Alleviates NLRP3 inflammasome-mediated cardiomyocyte pyroptosis through targeting the miR-186-5p/XIAP axis in coronary microembolization-induced myocardial damage. Front Immunol. (2021) 12:637598. doi: 10.3389/fimmu.2021.637598
130. Yang F, Li A, Qin Y, Che H, Wang Y, Lv J, et al. A novel circular RNA mediates pyroptosis of diabetic cardiomyopathy by functioning as a competing endogenous RNA. Mol Ther Nucleic Acids. (2019) 17:636–43. doi: 10.1016/j.omtn.2019.06.026
131. Han Y, Ma J, Wang J, Wang L. Silencing of H19 inhibits the adipogenesis and inflammation response in ox-LDL-treated Raw264 7 cells by up-regulating miR-130b. Mol Immunol. (2018) 93:107–14. doi: 10.1016/j.molimm.2017.11.017
132. Bian Y, Pang P, Li X, Yu S, Wang X, Liu K, et al. CircHelz activates NLRP3 inflammasome to promote myocardial injury by sponging miR-133a-3p in mouse ischemic heart. J Mol Cell Cardiol. (2021) 158:128–39. doi: 10.1016/j.yjmcc.2021.05.010
133. Feng Y, Huang X. Methodology for comprehensive detection of pyroptosis. Methods Mol Biol. (2021) 2255:149–57. doi: 10.1007/978-1-0716-1162-3_13
134. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
135. Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. (2016) 26:165–76. doi: 10.1016/j.tcb.2015.10.014
136. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by Gpx4 is required to prevent hydroperoxide-induced ferroptosis. Cell. (2018) 172:409–22.e21. doi: 10.1016/j.cell.2017.11.048
137. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. (2019) 572:402–6. doi: 10.1038/s41586-019-1426-6
138. Hong X, Roh W, Sullivan RJ, Wong KHK, Wittner BS, Guo H, et al. The lipogenic regulator SREBP2 induces transferrin in circulating melanoma cells and suppresses ferroptosis. Cancer Discov. (2021) 11:678–95. doi: 10.1158/2159-8290.CD-19-1500
139. Chevion M, Jiang Y, Har-El R, Berenshtein E, Uretzky G, Kitrossky N. Copper and iron are mobilized following myocardial ischemia: possible predictive criteria for tissue injury. Proc Natl Acad Sci U S A. (1993) 90:1102–6. doi: 10.1073/pnas.90.3.1102
140. Berenshtein E, Vaisman B, Goldberg-Langerman C, Kitrossky N, Konijn AM, Chevion M. Roles of ferritin and iron in ischemic preconditioning of the heart. Mol Cell Biochem. (2002) 234–235:283–92. doi: 10.1023/A:1015923202082
141. Park TJ, Park JH, Lee GS, Lee JY, Shin JH, Kim MW, et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. (2019) 10:835. doi: 10.1038/s41419-019-2061-8
142. Ganz T. Systemic iron homeostasis. Physiol Rev. (2013) 93:1721–41. doi: 10.1152/physrev.00008.2013
143. Song Y, Wang B, Zhu X, Hu J, Sun J, Xuan J, et al. Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol Toxicol. (2021) 37:51–64. doi: 10.1007/s10565-020-09530-8
144. Tang S, Wang Y, Ma T, Lu S, Huang K, Li Q, et al. MiR-30d inhibits cardiomyocytes autophagy promoting ferroptosis after myocardial infarction. Panminerva Med. (2020). doi: 10.23736/S0031-0808.20.03979-8
145. Lu J, Xu F, Lu H. LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci. (2020) 260:118305. doi: 10.1016/j.lfs.2020.118305
146. Zheng H, Shi L, Tong C, Liu Y, Hou M. circSnx12 Is Involved in Ferroptosis During Heart Failure by Targeting miR-224-5p. Front Cardiovasc Med. (2021) 8:656093. doi: 10.3389/fcvm.2021.656093
147. Martinez AM, Kim A, Yang WS. Detection of Ferroptosis by BODIPY™ 581/591 C11. Methods Mol Biol. (2020) 2108:125–30. doi: 10.1007/978-1-0716-0247-8_11
148. Foinquinos A, Batkai S, Genschel C, Viereck J, Rump S, Gyöngyösi M, et al. Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat Commun. (2020) 11:633. doi: 10.1038/s41467-020-14349-2
149. Lu Y, Zhang Y, Shan H, Pan Z, Li X, Li B, et al. MicroRNA-1 downregulation by propranolol in a rat model of myocardial infarction: a new mechanism for ischaemic cardioprotection. Cardiovasc Res. (2009) 84:434–41. doi: 10.1093/cvr/cvp232
150. Shan H, Li X, Pan Z, Zhang L, Cai B, Zhang Y, et al. Tanshinone IIA protects against sudden cardiac death induced by lethal arrhythmias via repression of microRNA-1. Br J Pharmacol. (2009) 158:1227–35. doi: 10.1111/j.1476-5381.2009.00377.x
151. Díez-Ricote L, Ruiz-Valderrey P, Micó V, Blanco-Rojo R, Tomé-Carneiro J, Dávalos A, et al. Trimethylamine n-Oxide (TMAO) modulates the expression of cardiovascular disease-related micrornas and their targets. Int J Mol Sci. (2021) 22:11145. doi: 10.3390/ijms222011145
152. Gao L, Li T, Li S, Song Z, Chang Y, Yuan L. Schisandrin A protects against isoproterenol-induced chronic heart failure via miR-155. Mol Med Rep. (2022) 25:12540. doi: 10.3892/mmr.2021.12540
153. Wu Y, Fan Y, Huang N, Zhang S, Zhang H, Liu X, et al. Effect of nicorandil combined with trimetazidine on miR-223-3p and NRF2 expression in patients with coronary heart disease. Am J Transl Res. (2021) 13:4804–11. Available online at: https://pubmed.ncbi.nlm.nih.gov/34150061/
154. Yang S, Fan T, Hu Q, Xu W, Yang J, Xu C, et al. Downregulation of microRNA-17-5p improves cardiac function after myocardial infarction via attenuation of apoptosis in endothelial cells. Mol Genet Genomics. (2018) 293:883–94. doi: 10.1007/s00438-018-1426-5
155. Tang M, Pan H, Zheng Z, Guo Y, Peng J, Yang J, et al. Prostaglandin E1 protects cardiomyocytes against hypoxia-reperfusion induced injury via the miR-21-5p/FASLG axis. Biosci Rep. (2019) 39:190597. doi: 10.1042/BSR20190597
156. Chen J, He Y, Zhou L, Deng Y, Si L. Long noncoding RNA MALAT1 serves as an independent predictive biomarker for the diagnosis, severity and prognosis of patients with sepsis. Mol Med Rep. (2020) 21:1365–73. doi: 10.3892/mmr.2020.10923
157. Kim SH, Wei C, Hoffmann U, Borggrefe M, Akin I, Behnes M. Advantages and limitations of currentbiomarker research: from experimental research to clinical application. Curr Pharm Biotechnol. (2017) 18:445–55. doi: 10.2174/1389201018666170601091205
158. Voellenkle C, van Rooij J, Cappuzzello C, Greco S, Arcelli D, Di Vito L, et al. MicroRNA signatures in peripheral blood mononuclear cells of chronic heart failure patients. Physiol Genomics. (2010) 42:420–6. doi: 10.1152/physiolgenomics.00211.2009
159. van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. (2006) 103:18255–60. doi: 10.1073/pnas.0608791103
160. van den Berg NWE, Kawasaki M, Berger WR, Neefs J, Meulendijks E, Tijsen AJ, et al. MicroRNAs in atrial fibrillation: from expression signatures to functional implications. Cardiovasc Drugs Ther. (2017) 31:345–65. doi: 10.1007/s10557-017-6736-z
161. Danese E, Montagnana M. An historical approach to the diagnostic biomarkers of acute coronary syndrome. Ann Transl Med. (2016) 4:194. doi: 10.21037/atm.2016.05.19
162. Pan X, He Y, Chen Z, Yan G, Ma G. Circulating miR-130 is a potential bio signature for early prognosis of acute myocardial infarction. J Thorac Dis. (2020) 12:7320–5. doi: 10.21037/jtd-20-3207
163. Sunderland N, Skroblin P, Barwari T, Huntley RP, Lu R, Joshi A, et al. MicroRNA biomarkers and platelet reactivity: the clot thickens. Circ Res. (2017) 120:418–35. doi: 10.1161/CIRCRESAHA.116.309303
164. Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, et al. Mass-spectrometry-based draft of the human proteome. Nature. (2014) 509:582–7. doi: 10.1038/nature13319
165. Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell. (2014) 54:766–76. doi: 10.1016/j.molcel.2014.03.045
166. Yu SY, Tang L, Zhou SH. Long Noncoding RNAs: New Players in Ischaemia-Reperfusion Injury. Heart Lung Circ. (2018) 27:322–32. doi: 10.1016/j.hlc.2017.09.011
167. Glinge C, Clauss S, Boddum K, Jabbari R, Jabbari J, Risgaard B, et al. Stability of Circulating Blood-Based MicroRNAs - Pre-Analytic Methodological Considerations. PLoS ONE. (2017) 12:e0167969. doi: 10.1371/journal.pone.0167969
168. Borges DP, Cunha-Neto E, Bocchi EA, Rigaud VOC. Impact of Delayed Whole Blood Processing Time on Plasma Levels of miR- 1 and miR-423-5p up to 24 Hours. MicroRNA (Shariqah, United Arab Emirates). (2018) 7:115–9. doi: 10.2174/2211536607666180322093119
169. Matias-Garcia PR, Wilson R, Mussack V, Reischl E, Waldenberger M, Gieger C, et al. Impact of long-term storage and freeze-thawing on eight circulating microRNAs in plasma samples. PLoS ONE. (2020) 15:e0227648. doi: 10.1371/journal.pone.0227648
170. Peirson SN, Butler JN. RNA extraction from mammalian tissues. Methods Mol Bio l(Clifton, NJ). (2007) 362:315–27. doi: 10.1007/978-1-59745-257-1_22
171. Pogribny IP. MicroRNAs as biomarkers for clinical studies. Exp Biol Med (Maywood). (2018) 243:283–90. doi: 10.1177/1535370217731291
172. Kreth S, Hübner M, Hinske LC. MicroRNAs as clinical biomarkers and therapeutic tools in perioperative medicine. Anesth Analg. (2018) 126:670–81. doi: 10.1213/ANE.0000000000002444
173. Rao X, Di Leva G, Li M, Fang F, Devlin C, Hartman-Frey C, et al. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene. (2011) 30:1082–97. doi: 10.1038/onc.2010.487
174. Zhao Y, Chen F, Li Q, Wang L, Fan C. Isothermal Amplification of Nucleic Acids. Chem Rev. (2015) 115:12491–545. doi: 10.1021/acs.chemrev.5b00428
Keywords: non-coding RNAs, necroptosis, pyroptosis, ferroptosis, cardiovascular diseases, immunology
Citation: Cai Y, Zhou Y, Li Z, Xia P, ChenFu X, Shi A, Zhang J and Yu P (2022) Non-coding RNAs in necroptosis, pyroptosis, and ferroptosis in cardiovascular diseases. Front. Cardiovasc. Med. 9:909716. doi: 10.3389/fcvm.2022.909716
Received: 10 May 2022; Accepted: 04 July 2022;
Published: 04 August 2022.
Edited by:
Dan Meng, Fudan University, ChinaCopyright © 2022 Cai, Zhou, Li, Xia, ChenFu, Shi, Zhang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Yu, eXU4MjIwMTgyJiN4MDAwNDA7MTYzLmNvbQ==; Jing Zhang, emhhbmdqaW5nNjY2ZG9jJiN4MDAwNDA7MTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship