
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 12 August 2022
Sec. Cardiovascular Therapeutics
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.909178
 Ye-Wei Yang1
Ye-Wei Yang1 Nian-Hua Deng2Kai-Jiang Tian2Lu-Shan Liu2
Nian-Hua Deng2Kai-Jiang Tian2Lu-Shan Liu2 Zuo Wang2
Zuo Wang2 Dang-Heng Wei2Hui-Ting Liu2
Dang-Heng Wei2Hui-Ting Liu2 Zhi-Sheng Jiang2*
Zhi-Sheng Jiang2*Hydrogen sulfide (H2S), a gas transmitter found in eukaryotic organisms, plays an essential role in several physiological processes. H2S is one of the three primary biological gas transmission signaling mediators, along with nitric oxide and carbon monoxide. Several animal and in vitro experiments have indicated that H2S can prevent coronary endothelial mesenchymal transition, reduce the expression of endothelial cell adhesion molecules, and stabilize intravascular plaques, suggesting its potential role in the treatment of atherosclerosis (AS). H2S donors are compounds that can release H2S under certain circumstances. Development of highly targeted H2S donors is a key imperative as these can allow for in-depth evaluation of the anti-atherosclerotic effects of exogenous H2S. More importantly, identification of an optimal H2S donor is critical for the creation of H2S anti-atherosclerotic prodrugs. In this review, we discuss a wide range of H2S donors with anti-AS potential along with their respective transport pathways and design-related limitations. We also discuss the utilization of nano-synthetic technologies to manufacture H2S donors. This innovative and effective design example sheds new light on the production of highly targeted H2S donors.
Hydrogen sulfide (H2S) is a colorless gas that smells like rotten eggs and has toxic effects at concentrations approaching 20 ppm. Initial research on H2S was largely conducted in the context of elimination of H2S waste gas in industrial operations and protection from dangerous gases in wartime (1). H2S poisoning is caused by the reaction of H2S with trivalent iron in oxidized cytochrome oxidase, which inhibits the function of cellular respiratory enzymes, resulting in cellular hypoxia (2). H2S can also inactivate glutathione by coupling with its sulfhydryl group, inducing cell death (3). However, it was only in 1996 that Abe and Kimura (4) published the findings of a seminal investigation on endogenous H2S generation and signaling. Consequently, over the next 20 years, the general perception of H2S shifted from that of a poisonous gas to a gas transmitter with potential for pharmacological therapy. Carbon monoxide and nitrogen oxide (5) are all similar gas transmitters. All gas transmitters have comparable qualities, such as solubility, free diffusion, and the need for certain enzymes and substrates for production. The characteristics of the three gas transmitters are summarized in Table 1.

Table 1. Comparison of common characteristics of the three gas transmitters.
H2S has numerous key regulatory effects in AS, including anti-oxidative stress, prevention of endothelial mesenchymal transition, reduction of foam cell production, and modulation of mitochondrial autophagy (6–8). Several studies have demonstrated the cardiovascular benefits of H2S in clinical settings. In a randomized controlled, double-blind trial involving 120 hypertensive patients, taurine supplementation significantly decreased the clinic and 24-h ambulatory blood pressure (9). Furthermore, changes in blood pressure were negatively correlated with both the plasma H2S and taurine levels in taurine-treated prehypertensive individuals. The potential underlying mechanism is that taurine up-regulates the expression of H2S synthase by inhibiting calcium influx in transient receptor potential channels, thereby reducing vascular reactivity. In addition, H2S prodrug SG1002 is already being investigated in Phase 1 clinical trials. David J. Polhemus' team conducted a small-sample non-randomized controlled clinical study to show that in vitro administration of the H2S prodrug, SG1002, can alleviate heart failure. SG1002 reduced the level of brain natriuretic peptide (BNP), and showed no apparent toxic side-effects (10). Thus, H2S is a novel cardiovascular disease drug worthy of further research and development.
H2S donors are compounds that can release H2S when particular trigger conditions are met. Given the features of the gas transmitter, developing an H2S donor with high targeting ability and stability during transportation is critical for studying the anti-AS properties of exogenous H2S. Moreover, development of suitable H2S donor would provide the foundation for future H2S anti-AS prodrug research and clinical therapy. In this article, we review the various kinds of donors with anti-AS potential and discuss the pros and cons of each donor design. We also propose the idea of preparing H2S donors using nanomolecule technology in combination with chemical synthesis technology. We anticipate that multidisciplinary collaboration will foster the translation of H2S anti-AS from basic research to clinical treatment in the future.
H2S has a molecular weight of 34.08, which is slightly greater than the molecular weight of water. It has a vapor pressure of 2,026.5 kPa/25.5°C, flash point of −50°C, melting point of −85.5°C, boiling point of −60.4°C, relative density of 1.19 (air = 1), and tipping point of 292°C. H2S is highly soluble in water; it is also soluble in alcohol, petroleum solvents, and crude oil. It may be hydrolyzed to hydrogen ions, sulfur-hydrogen ions, and sulfur ions in water or plasma using the reaction equations below. The first step: H2S+H2O=HS− +H3O+; the second step: HS− + H2O=S2−+H3O+. H2S is soluble up to 80 mmol/L at typical human body temperature and can be oxidized to sulfides, sulfates, persulfides, and sulfites (11).
AS is the primary cause of atherosclerotic heart disease, cerebral infarction, and peripheral vascular diseases. Persistent inflammation plays a key role in the pathogenesis of AS (12). The pathogenetic mechanism is complex and involves endothelial dysfunction, leukocyte adhesion and aggregation, lipid plaque deposition, smooth muscle cell proliferation, and extracellular matrix remodeling, among other factors (13). Of note, a large number of studies have demonstrated that H2S can protect the cardiovascular system against several elements involved in AS progression, such as by ameliorating myocardial fibrosis; inhibiting IL-1 and IL-18 production; inhibiting ICAM-1 expression in TNF-alpha-induced HUVECs via the NF-κB pathway; preventing lipid peroxidation; inhibiting intravascular thrombosis; protecting against apoptosis induced by oxidative stress via the SIRT1 pathway; inhibiting smooth muscle cell proliferation; and attenuating foam cell formation (Table 2 and Figure 1). These findings demonstrate the anti-AS potential of H2S.
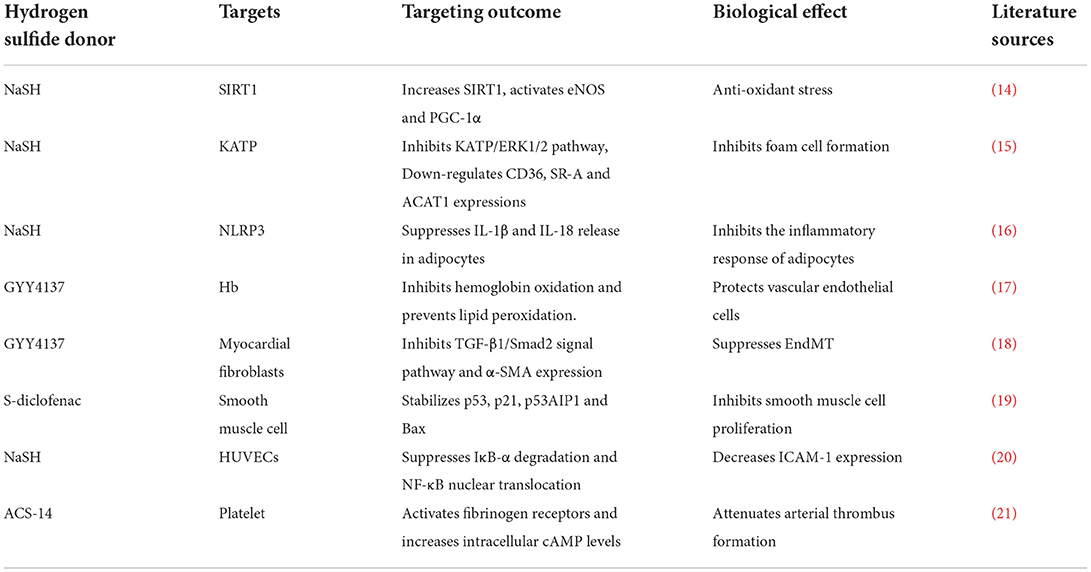
Table 2. Anti-AS effect of H2S from different mechanisms.
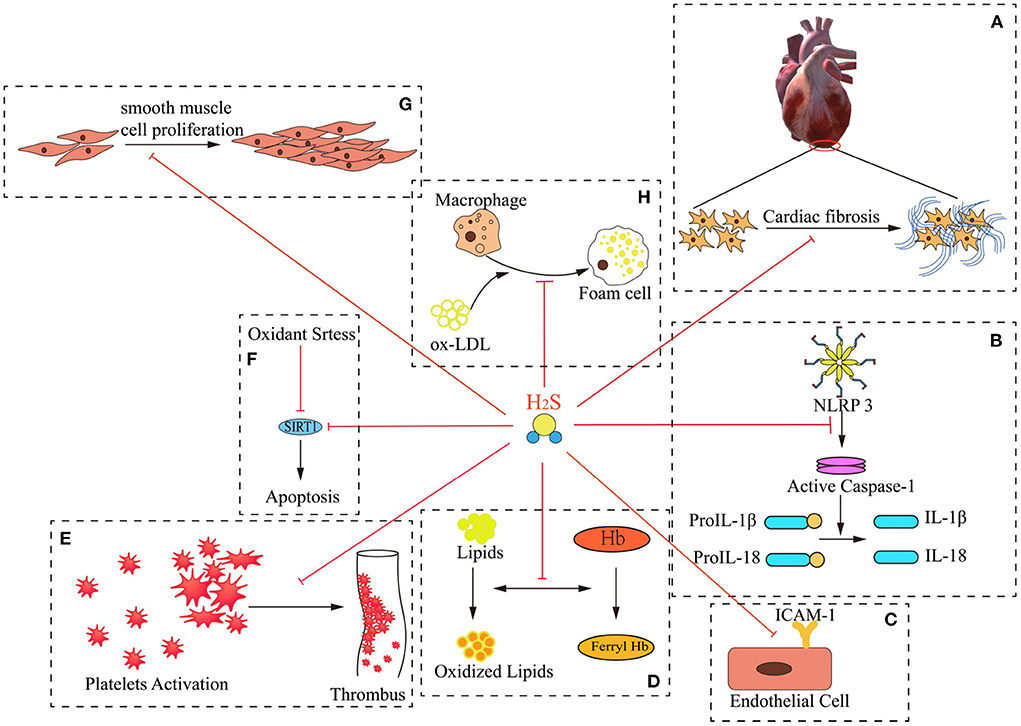
Figure 1. Mechanisms of H2S “multi-angle” inhibition of AS. H2S exerts numerous critical effects against the pathogenesis of atherogenesis. These include: (A) improving myocardial fibrosis; (B) inhibiting the production of IL-1β and IL-18; (C) inhibiting ICAM-1 expression in TNF-alpha-induced HUVECs via the NF-kappa B pathway; (D) preventing lipid peroxidation; (E) inhibiting intravascular thrombosis; (F) protecting against apoptosis under oxidative stress through SIRT1 pathway; (G) inhibiting smooth muscle cell proliferation; (H) attenuating foam cell formation.
Diffuse myocardial fibrosis is strongly related with prior cardiovascular events and may result in serious complications. Ambale-Venkatesh et al. (22) used contrast-enhanced cardiac magnetic resonance (CMR) to assess differences in myocardial fibrosis measured at the year-10 examination between participants with and without cardiovascular (CV) events accrued in a large population-based study over a 10-year follow-up period. The findings implied that the prevalence of CV events during the last decade was related with an increased risk of ischemic myocardial scarring on advanced gadolinium-enhanced imaging and greater diffuse interstitial fibrosis as measured by T1 imaging in a multiethnic free-living population.
The effect of H2S on fibrosis has previously been examined. Sheng et al. (23) examined human atrial fibroblasts using the BrdU test. The research demonstrated that NaHS at concentrations of 100, 300, and 500 μM inhibited the proliferation of atrial fibroblasts by 33.1 ± 4.2, 43.7 ± 3.1, and 58.4 ± 6.2% respectively. Additionally, they verified the combined inhibitory effects of H2S on BKCa and Ito currents in suppressing cellular growth using whole cell patch clamping. Cx43 is intimately linked to myocardial fibrosis, and lower Cx43 expression predisposes to collagen accumulation (24). By partly ligating the rat abdominal aorta, Huang et al. generated a rat hypertrophic cardiomyopathy model. They verified in this experiment that sodium hydride-treated rats had smaller LVMI, cardiomyocyte size and area, and CVF than control rats (25). Additionally, H2S has been shown to greatly boost CX43 expression in rat cardiomyocytes. These findings suggest that H2S may act as an anti-fibrosis agent via increasing CX43 expression.
Adipose tissue is regarded as an important endocrine organ and is known to be involved in regulating inflammation (26). Numerous experimental and epidemiological studies have implicated obesity-related adipose dysfunction as one of the main causes of endothelial dysfunction (27). Adipocyte dysfunction often leads to activation of NLRP3, and its activation leads to caspase-1 activation and the release of the inflammatory cytokines interleukin 1(IL-1) and interleukin 18 (IL-18) (28). These two inflammatory factors increase monocyte chemo-attractant protein-1 (MCP-1) and vascular cell adhesion molecule 1 (VCAM-1), thereby promoting leukocyte-endothelial cell adhesion, causing endothelial dysfunction. Endothelial cell dysfunction, as demonstrated by increased production of endothelial cell adhesion molecules and pro-inflammatory mediators, results in infiltration of monocytes in the subendothelial layer (29). Under the influence of macrophage colony-stimulating factor, these monocytes transform into macrophages and engulf non-biodegradable oxidized LDL cholesterol, ultimately transforming into foam cells, leading to the development of atheromatous plaques (30).
Exogenous H2S has been shown to inhibit high glucose-induced activation of the NLRP3 inflammasome in adipocytes. In the study by Hu et al., high glucose was found to induce the up-regulation of NLRP3 in adipocytes, which promoted the release of downstream molecules IL-1β and IL-18. The above phenomenon was found to be inhibited by exogenous sodium hydrosulfide (16). This suggests a potential anti-atherosclerotic effect of exogenous H2S in obese patients, especially those with adipose tissue dysfunction. However, further studies are required for in-depth characterization of the molecular mechanisms underlying the anti-inflammatory effect of H2S.
ICAM-1 plays an important role in immune and inflammatory responses, including atherosclerosis. These proteins are ligands for the leukocyte adhesion protein LFA-1 (integrin alpha-L/beta-2). During leukocyte trans-endothelial migration, ICAM1 engagement promotes the assembly of endothelial apical cups through activation of ARHGEF26/SGEF and RHOG, eventually leading to trans-endothelial migration of leukocytes (31). ICAM-1 in the blood has been recognized as a marker of vascular inflammation in atherosclerosis; it has been shown to predict cardiovascular risk and future cardiovascular disease (32).
The study of H2S and ICAM-1 was originally reported in the context of non-steroidal drug therapy. H2S was found to reduce the increase in ICAM-1 caused by non-steroidal anti-inflammatory drugs (33). Subsequent studies found that exogenous H2S can slow down the expression of ICAM-1 in the blood of apoE knockout mice. In addition, under the influence of exogenous sodium hydrosulfide, the size of arterial plaque also decreased (20). This phenomenon was attributed to the prevention of the activation of the nfkb signaling pathway by H2S.
In the atherosclerotic lesions, a pathological change called “infiltration of red blood cells” is usually found (34). Some of the damaged red blood cells are phagocytosed by macrophages and degraded in lysosomes. As a product of lysosomal digestion, iron ions are excreted by macrophages by exocytosis, inducing the oxidation of LDL to OxLDL. Subsequently, the macrophages phagocytose OxLDL (35). In addition, degraded red blood cells release hemoglobin (Hb), which can react with surrounding plaque lipids. This leads to the formation of different oxidatively modified hemoglobin species, such as metHb (Fe3+) and ferrylHb (Fe4+ =O2−) (36). Oxidized Hb species sensitize vascular endothelial cells to oxidant-mediated killing, suggesting that it is a potential causative factor in atherosclerosis.
Potor et al. found that H2S significantly reduces oxidation of Hb preventing the formation of ferrylHb derivatives. By inhibiting Hb-lipid interactions, sulfide lowers oxidized Hb-mediated induction of adhesion molecules in endothelium and disruption of endothelial integrity (37). Chemically, H2S is a reductant, and there is evidence that H2S can convert oxidized low-density lipoproteins to lipoalcohols (38). This heralds the potential of H2S in inhibiting hemoglobin oxidation and preventing lipid peroxidation.
Platelets play an important role in the pathogenesis of coronary thrombosis and atherogenesis. Abnormal activation of platelets contributes to atherothrombosis (39). Activation of platelets induces the release of chemokines, leading to the aggregation of leukocytes, followed by the progress of leukocyte and platelet adhesion, which is mainly mediated by P-selectin and ligand PSGL-1 (40). In addition, degranulation of platelets leads to a release of inflammatory cells molecules, including various chemokines, cytokines, lipids, and proteins (41–43). The recruitment of leukocytes to the thrombus is a complex process. Specifically, leukocytes act by binding to p-selectin on the surface of platelets (44), rolling on the endothelium, and finally adhering to activated platelets. As a next step, leukocytes undergo integrin-mediated changes in shape and cellular functions such as motility, migration, degranulation, or phagocytosis. Almost at the same time, leukocytes promote platelet aggregation and secretion (45), enhance the production of thrombin and tissue factor (46, 47), and play an important role in the stability of the thrombus.
Previous studies have confirmed that H2S could inhibit platelet activation and aggregation (48–50). In the study by Grambow et al., exogenous H2S treatment was found to reduce platelet and leukocyte aggregation, and this inhibition showed a significant correlation with the concentration of H2S. The team then used scanning electron microscopy to directly analyze the specific effects of H2S on platelet activity, and found that the activated platelets in the control group exhibited changes in shape with formation of pseudopodia, leading to the appearance of a thorn apple-like shape under microscope. Cells treated with GYY4137 did not exhibit the above morphological changes, but instead were characterized by “round platelet morphology” and “less pseudopodia formation” (51). This phenomenon may be related to the inhibitory effect of H2S on the extracellular action of platelet p-selectin. Currently, there is a lack of direct evidence that H2S inhibits platelet activation, such as the lack of necessary rescue experiments to demonstrate that H2S has a direct effect on platelet p-selectin. However, the available evidence suggests the antithrombotic potential of this gas molecule, providing a new avenue of antagonism toward atherosclerotic and thrombotic diseases.
Oxidative stress is recognized as a distinct factor in the pathogenesis of cardiovascular diseases. It is manifested specifically by an imbalance in the production and removal of oxygen free radicals in cells, with the production of some reactive oxygen species (ROS) such as O2−, OH−, ONOO−, and H2O2 (44). ROS can induce upregulation of endothelial cell adhesion molecules, proliferation and migration of vascular smooth muscle cells (VSMCs), platelet activation, lipid oxidation, and activation of matrix metalloproteinase, all of which can contribute to the advancement of atherosclerotic disease (52). The following mechanisms of the protective effect of H2S against oxidative stress have been identified: (1) it acts as a reducing agent to directly induce ROS scavenging in vivo (53); (2) protects proteins from oxygen radical attack by modulating the expression and activity of classic antioxidants, such as glutathione (GSH) and thioredoxin (Trx) (45); (3) regulates mitochondrial metabolism to limit ROS formation (46); (4) reduces ROS production by interacting with cytochrome c and providing electrons to the mitochondrial ATP synthesis mechanism, which can substitute oxygen in the ATP production mechanism (47).
Warnholtz et al. (54) established a hypertension model generated by AngII and found that AngII substantially enhanced superoxide anion generation in the aorta, which was reduced by NaHS therapy. Later, Hsin-Ying et al. discovered that exogenous H2S may suppress the IL-6-induced oxidative stress in rat vascular smooth muscle cells and decrease their ROS content (55). Notably, iNOS can sustainably produce NO under the continuous stimulation of ROS. Excessive NO release is seen as a potential risk factor for cardiovascular disease. H2S has been shown to reduce ROS release by IL-6 stimulation, thereby inhibiting the sustained increase in NO release by iNOS activation, ultimately delaying the phenotypic transformation of endothelial smooth muscle cells (55). This suggests that the anti-oxidative stress effect of H2S may be mediated via inhibition of the ROS-iNOS-NO pathway.
VSMCs are located in the medial layer of arteries and play an important role in the regulation of the vascular system (56). Physiologically, VSMCs possesses both systolic and diastolic phenotypes, regulating both vasoconstriction and relaxation. Under the stimulation of pathological conditions such as vascular injury, angiotensin II (Ang II), platelet-derived growth factor, insulin-like growth factor 1, ROS, and endothelin-1, the phenotype of cells undergoes a deconversion from a contractile phenotype to a synthetic phenotype, which ultimately leads to cell proliferation and migration (57–59). Proliferation of VSMCs is associated with various vascular diseases such as atherosclerosis, restenosis, and hypertension. In particular, this phenotypic switch plays an important role in atherosclerosis and plaque stability, and inhibition of vascular smooth muscle phenotype switch may be beneficial in advanced atherosclerosis (60). The myocardin serum response factor regulatory module, for example, is a critical component of phenotypic regulation because it permits the combinatorial interactions of activating and repressing signals and cofactors that operate on the majority of VSMC contractile genes. Compared to myocardin+/+ littermates, myocardin+/− mice on an ApoE−/− background showed increased atherosclerosis with greater concentration of macrophage or macrophage-like cells (61).
Earlier studies confirmed that the body transmitter H2S plays a broad role in the cardiovascular system. For instance, H2S was shown to induce a dose-dependent suppression of the proliferation of VSMCs through the MAPK pathway (62). With further advancement of research, H2S was found to inhibit the proliferation of VSMCs by regulating chromatin remodeling and target gene expression. Brg1 is the central catalytic subunit of the SWI/SNF apparatus (an ATP-dependent chromatin remodeling complex). Li et al. demonstrated that Brg1 plays an important role in the inhibition of VSMC proliferation induced by H2S by overexpressing and knocking out the Brg1 gene. The effect of H2S on Brg1 was confirmed by luciferase reporter assay and real-time quantitative PCR at the transcriptional level. Finally, they used chromatin immunoprecipitation experiments to confirm that H2S inhibited the recruitment of Brg1 to the Pcna, Ntf3, and Pdgfα promoters, thereby acting as anti-VSMC proliferation (63). The above studies suggest that H2S regulates the proliferation of smooth muscle cells through epigenetic modification. Because epigenetic changes may be temporary or reversible, this provides a new idea for the development of late-stage drug therapy.
Accumulation of foam cells is a hallmark pathological change in the development of atherosclerosis. Macrophages are an important source of membrane cell formation. Macrophages phagocytose cholesterol or oxidized LDL, and then esterify the above substances to extrude them from the cell. This is a normal compensatory phenomenon; however, disruption of this compensatory mechanism leads to excessive accumulation of lipids in cells, eventually leading to the formation of foam cells (64). Foam cell accumulation is involved in the development of atherosclerosis, such as the release of matrix-degrading enzymes, leading to plaque rupture or vascular occlusion (65).
Since macrophage phagocytosis of oxidized LDL is critical for foam cell formation, H2S has been reported to inactivate macrophage phagocytosis of LDL. This phenomenon can be reflected in the uptake of lipids by macrophages. In a study, the uptake rate of oxidized LDL was found to have significantly decreased in cells pretreated with sodium hydrosulfide (15). Further research found that the endogenous H2S inhibitor PPG could reverse the passivation effect of H2S and restore the efficiency of lipid uptake by macrophages. ACAT-1 is a kind of mitochondrially localized enzyme that catalyzes the reversible formation of acetoacetyl-CoA from two molecules of acetyl-CoA. Defects in this gene are associated with 3-ketothiolase deficiency (66). Currently, ACAT-1 is believed to be a key enzyme promoting the intracellular cholesterol accumulation within macrophages. In a study, NaHS was shown to reduce macrophage ACAT-1 expression and inhibit foam cell formation (15). This demonstrated that ACAT-1 may be the target of H2S to inhibit the transformation of macrophages into foam cells.
Although several studies have demonstrated the protective effect of H2S against atherosclerosis, its precise molecular mechanism is not clear. S-sulfhydration has recently been recognized as a major mechanism of the physiological effects of H2S, and several recent studies have explored H2S signaling by S-sulfhydration. S-sulfhydration modifies the functions of a specific protein by causing post-translational modifications. In a study, H2S was shown to increase keap1 protein thiolation, boost Nrf2 nuclear translocation, and limit production in endothelial cells, and this effect was abolished when Keapl was mutated at Cys151, but not Cys273, in endothelial cells (67). The enzymatic process involved in the synthesis of H2S in mammalian tissues includes cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS) (68). A study found that H2S donor treatment can lead to thiolation of C252, C255, C307, and C310 of CSE, promote its binding to L-hcy, and prevent hyperhomocysteinemia-induced atherosclerosis in mice (69).
Protein sulfhydrylation triggered by H2S causes proteins to have various effects. These findings shed light on the specific molecular mechanism of the anti-antherosclerotic effect of H2S. Thus, H2S- induced protein sulfhydrylation is a potential promising topic for future research on this subject.
Hydrolysis-triggered, intestinal flora metabolism-triggered, biothiol-triggered, esterase-triggered, pH-triggered, and other mechanisms are used by the current H2S donors with anti-AS properties. The pharmacokinetic features of the donors are determined by the various triggering mechanisms, which are critical for their continued development as prodrugs.
Many inorganic sulfide salts have been utilized to synthesize H2S donors, including Na2S, NaHS, and CaS. Water usually produces these inorganic salts, such as Na2S9H2O, which are often used in laboratories. Dissolution of inorganic sulfate in water gradually leads to a state of chemical equilibrium between S2, HS, and H2S. The specific reaction formula is as follows: NaHS + H2O NaOH + H2S↑ (70). The benefit is that H2S is rapidly produced after hydrolysis and no by-products are formed during the chemical reaction, essentially eliminating bias in experimental findings caused by by-product activity. This is why it holds tremendous promise for research on use of exogenous H2S in the treatment of AS. Unfortunately, there are several major obstacles to developing inorganic sulfide salts as anti-AS prodrugs. First, it is difficult to determine the exact concentration of inorganic sulfate salts in solution due to the variety of purity levels used; secondly, inorganic sulfate salts are extremely volatile in aqueous solution following H2S hydrolysis, making it difficult to maintain the active ingredient during the configuration of the drug in clinical practice when intravenous drip treatment is used. Change of mode of administration to intravenous injection to reduce the impact of volatility of H2S may lead to an inordinately high concentration of H2S at the injection site, which is liable to lead to adverse effects. Finally, the produced H2S is rapidly diluted, making it difficult to sustain a reasonably extended period of time at the lesion. Due to the aforementioned deficiencies, the use of inorganic sulfide salt donors has been largely confined to animal experiments or cytohistological research.
Allicin is a naturally occurring compound in common foods such as onions and garlic. After digestion and absorption, allicin in certain foods can slowly produce H2S (71). Although allicin is a natural compound with a high safety profile and widespread availability, its decomposition products (DADS, DAS, and DATS) have low water solubility, slow rate of production of H2S, and a low pharmacokinetic profile, which frequently results in a lengthy test period. Second, interaction of allicin in the body leads to production of several by-products, making it impossible to identify the specific chemical responsible for anti-AS effects. These issues restrict the research on allicin as an H2S donor.
Moore's team at the National University of Singapore published a report on Morpholinol thiosphosphonate, a water-soluble, long-acting H2S donor chemical, in 2008 (72). H2S produced by hydrolysis of this molecule is quite moderate when compared to inorganic sulfides. GYY4137 hydrolysis produces a peak H2S release of 10 min, while sodium hydrosulfide hydrolysis produces a peak H2S release of just 10 s. GYY4137 is progressively gaining favor among researchers because it seems to compensate for the unstable H2S emission during hydrolysis of inorganic sulfates. In a study by Qiu et al., diabetic model rats who were administered GYY4137 before myocardial ischemia-reperfusion injury showed smaller infarcts, reduced apoptosis, and lower oxidative stress than the control group, implying that the protective effect of GYY4137 against myocardial ischemia-reperfusion injury is linked to p-Akt and nuclear Nrf2 protein (73). In addition, Zheng et al. employed GYY4137 to show that H2S molecules regulate the PI3K/Akt/TLR4 signaling pathway to stabilize vascular plaques (74). As research continues, the atherosclerotic potential of GYY4137 is increasingly being unraveled, such as prevention of vascular inflammation and oxidative stress (75), protection against myocardial fibrosis (18), and attenuation of adverse remodeling (76).
Many researchers theorize GYY4137 as a beneficial research tool; however it does have some disadvantages. First, the end-product of GYY4137 is often sold as a dichloromethane complex, and one of the metabolites of dichloromethane is CO, another gaseous molecular signal with biological effects comparable to H2S. As a result, determining whether CO is involved in the biological effects of GYY3137 is challenging. Second, since the rate of hydrolysis of GYY4137 is exceptionally slow, greater dosages may be necessary to attain therapeutic H2S concentrations. This would increase the risk of adverse effects due to high doses of GYY4137. To the best of our knowledge, no extensive pharmacokinetic studies of GYY4147 have been conducted nor is there a reliable way to determine the quantity of H2S generated by hydrolysis of this compound (77). Because of the aforementioned flaws, GYY4137 has a limited role in the development of H2S prodrugs.
NOSH-aspirin, is a synthetic derivative of 1,2-dithio-3-thione and aspirin with a nitrate moiety. It was developed as a potential substitute for the commonly used anti-platelet medication aspirin (78). Currently, NOSH-aspirin research is being largely conducted in the context of pancreatic cancer, colon cancer, neurodegeneration, antiplatelet, and anti-inflammatory, analgesic, and antipyretic (79). Despite the paucity of research on the development of this molecule as an anti-AS agent, its unique drug metabolism properties appear to hold a lot of promise. NOSH-aspirin can be broken down in the body into three different chemicals that have different physiological effects: H2S, nitric oxide, and aspirin (80). In clinical practice, aspirin is regarded the first-line anti-atherosclerotic drug, while the cardiovascular protective properties of nitric oxide (a gaseous molecular signal), have already been well-established (81, 82). In addition to the combination of H2S and nitric oxide, which functions as a stomach mucosal prostaglandin mimic and effectively suppresses the gastrointestinal adverse effects induced by aspirin, H2S may further boost the anti-atherosclerotic activity of aspirin (83). Although NOSH-aspirin is a promising H2S donor, its anti-atherosclerotic capabilities have not been well-investigated. Because all of the metabolites have anti-atherosclerotic properties, suitable compounds must be developed to assess the individual effects of each of the products, and further research is expected in the future.
Thiol-triggered H2S donors are non-hydrolyzation-triggered H2S donors and were one of the first documented synthetic H2S donors. N-(Benzoylthio)benzamides are representative thiol-triggered H2S donors. The donor design is based on the instability of the S-N bond, which when broken releases H2S. The S-N bond is protected by an acyl group, which acts as a switch by regulating the chemical reaction of the acyl group, allowing the S-N bond to be exposed again and thus indirectly controlling the release of H2S (84). In addition, Acyl Perthiol Donors were also designed to be a mercaptan-triggered H2S donor. Acyl perthiol donors (RC(O)-S-SR) were first synthesized by Xian, where the R group is derived from penicillamine (85). The donor was synthesized from thiobenzoic acid derivatives and n-benzoyl cysteine methyl ester. Briefly, C- and N-protected penicillamine was first treated with 2,2 -dibenzothioazolyl disulfide to provide a penicillamine-benzothioazolyl disulfide intermediate, and then treated with corresponding thioacids to furnish the desired penicillamine-based donors. Subsequent experiments demonstrated that the donor could significantly reduce the area of myocardial ischemia-reperfusion injury, and the cytotoxicity test showed lower cytotoxicity of the donor compared to that of sodium hydride. Dithioperoxyanhydrides is another thiol-triggered donor of H2S. It is prepared in a single reaction step involving thiobenzoic acid and methoxycarbonylsulfenyl chloride (CH3OC(O)SCl) with fair overall yields. In vivo experiments confirmed that Dithioperoxyanhydrides can also induce total vasorelaxation of isolated rat aortic rings pre-contracted with phenylephrine (86). Other types of thiol-triggered H2S donors include Arylthioamides and S-aroylthiooximes. Studies have also demonstrated the potential for clinical application of these two kinds of donors. Arylthioamides were shown to strongly abolish the noradrenaline-induced vasoconstriction in isolated rat aortic rings and to hyperpolarize the membranes of human vascular smooth muscle cells in a concentration-dependent manner (87). In a study by Jeffrey et al., S-aroylthiooximes were found to significantly reduce the survival of HCT116 colon cancer cells relative to Na2S, GYY4137, and a small molecule SATO (88), indicating that this donor may inhibit smooth muscle proliferation and migration in atherosclerotic diseases.
The “trimethyl lock” lactonisation reaction, which entails cleavage of a phenolic ester by esterase, followed by spatial repulsion of the three methyl groups triggering lactonisation and the release of the drug from the adjacent carbonyl group, is the basis for the enzyme-triggered H2S donor design (89).
Enzymes are tissue- and substrate-specific active proteins that are found in all living organisms (90). Sofia-Iris Bibli demonstrated greater CSE expression in endothelial cells near carotid plaques than in endothelial cells in superior mesenteric arteries (91). We hypothesize that by combining the aforementioned qualities, enzyme-triggered H2S donors may be created to allow for the optimal concentration of H2S in the causative lesion, laying the groundwork for targeted drug therapy. Despite the paucity of research on enzyme-triggered H2S donors, enzyme-triggered design should be a trend in future pro-sulfide donor design.
Based on the poor targeting of many of the current mainstream H2S donors, Xian's team used intramolecular cyclisation to activate phosphorothioates to design a new pH-regulated release of H2S donor JK; JK series H2S donors have great potential for development in cardiovascular disease research, especially in the field of myocardial ischemia reperfusion injury. Due to myocardial ischemia or hypoperfusion, tissue hypoxia leads to accumulation of lactic acid, reducing the pH level in the responsible lesion and nearby tissues, which provides a basis for targeted therapy for this type of donor to release H2S (92). In addition, due to the donor's special mechanism of triggering the release of H2S, the protective effect of donors on organs in the context of gastric diseases is also well-reflected. In the study by Yang et al., JK donor significantly alleviated gastric mucosal damage, a side-effect of NSAIDs (93). Intragastric pre-administration of JK-1 was found to mitigate the side effects of NSAIDs, such as inflammatory cell infiltration, increased IL-6 and TNF-α release, and oxidative loss. Increase in CBS and CSE levels is a well-known requirement for in vitro osteogenic differentiation. JK has also been shown to promote osteogenic differentiation in vitro (94). In conclusion, JK has shown potential therapeutic value in other therapeutic fields besides cardiovascular diseases. These findings indicate that this compound is a H2S donor that is worthy of further research and development.
In addition to the types of H2S donors described above, there are other types of H2S donors, such as “UV-triggered H2S donors” and “carbonyl H2S donors” (95, 96). However, these donors have poor potential for the development of anti-AS pre-drugs. For example, UV-triggered H2S donors have weak penetration due to the short wavelength of UV light, making it impossible to trigger the donor drug deep in the tissue. Carbonyl H2S donors have poor water solubility and poor targeting. As a result, these donors are mostly used in in vitro assays.
It has been more than 20 years since the H2S donor was successfully developed (97). Despite considerable breakthroughs in the research on H2S donors in the last two decades, targeting of the donor is still a key issue to be addressed. Clinical use of H2S as an anti-AS therapy depends on the design of a well-targeted donor and its delivery and release in the vicinity of the target lesion.
Nanomaterials were initially used to improve the targeting of antitumor drugs. The targeting of nanomaterials is mainly reflected in two aspects—active targeting and passive targeting. First, the alteration of ligands on the surface of nanoshells and the binding of ligands to receptors to provide precisely focused treatment is referred to as active targeting. Second, the diameter of the nanoparticles is more easily absorbed in some lesions due to passive targeting because of inadequate capillary endothelial connection in the microcirculation at the lesion site (e.g., in tumor tissue or inflammatory tissue). Due to the presence of inflammatory lesions near the arterial plaque lesions, the capillary endothelial junctions in the tissue are incomplete, and this forms the basis for passive targeting of nanomaterials. This implies that in normal tissues, drugs modified by nanomaterials are not easily absorbed but accumulate adjacent to the inflamed tissues, thereby achieving targeted therapy. Willem's team created unique multimodality HDL-mimicking nanoparticles by incorporating gold, iron oxide, or quantum dot nanocrystals for computed tomography, magnetic resonance, and fluorescence imaging, respectively (98), It is generally known that macrophage infiltration around arterial plaques is a specific pathophysiological change in AS. The team cleverly used this principle to design a plaque-targeted imaging drug. Moreover, a study by Wang et al. demonstrated good biocompatibility of macrophage membrane functionalized biomimetic nanoparticles: the biomimetic particles were rapamycin-loaded poly(lactic-co-glycolic acid) copolymers made from macrophage membrane coating. Such nanoparticles can be efficiently targeted and accumulated in atherosclerotic lesions in vivo. After a 4-week treatment plan, MM/RAPNPs were found to have significantly delayed AS progression (99).
Similar principles can be used to design vascular-targeted H2S donors based on the above research. Theoretically, this design idea offers three advantages: First, because surface proteins on the macrophage membrane provide good active targeting of H2S donors, especially α4β1, this receptor can tightly bind to vascular cell adhesion molecule-1 (VCAM-1) on the surface of endothelial cells, which is highly expressed in the inflamed endothelium. Second, nanotechnology-modified H2S donors are also passively targeted and can accumulate around inflammatory tissues near vascular plaques to achieve ideal therapeutic concentrations. The last point is also the most critical; since nanomaterials are foreign bodies, they often induce the activation of innate immunity. The above situation can be significantly avoided by using macrophage membranes as biomimetic coatings of nanomaterials (100, 101). This technology is poised to gradually mature in the near future.
In addition to drug targeting, several other important questions remain unanswered in the field of H2S donors, which require multidisciplinary collaboration involving chemistry, pharmacology, and biomaterials science. Firstly, there is considerable variability in the therapeutic concentration window for H2S molecules used to correct different pathophysiological states and even different stages of the same pathophysiological state. Therefore, clarifying the therapeutic window of H2S for specific targets is a crucial imperative. Secondly, as H2S is highly volatile and rapidly metabolized, the relationship between the effective dose of the drug and the donor dose is not necessarily a simple linear relationship. Therefore, it is crucial to define the release rate of the H2S for the development of H2S donors. In addition, whether the by-products or the donor itself can produce biological effects after the triggering of the H2S donor and bias the experimental results is also an important issue to be addressed. As the field of H2S research continues to evolve, these critical issues must be addressed in order to progress toward clinical treatment. Future multidisciplinary collaborations involving the disciplines of nanotechnology, chemical synthesis, drug metabolism, and biology may eventually make clinical pathways for H2S treatment possible.
Z-SJ, Y-WY, and N-HD: manuscript conceptualization. Y-WY and N-HD: writing original manuscript draft. K-JT, L-SL, and ZW: literature search and articles acquisition. D-HW and H-TL: figures drawing. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (91839103 to Z-SJ), and Postgraduate Scientific Research Innovation Project of Hunan Province (CX20210958 to N-HD).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Powell CR, Dillon KM, Matson JB. A review of hydrogen sulfide (HS) donors: Chemistry and potential therapeutic applications. Biochem Pharmacol. (2018) 149:110–23. doi: 10.1016/j.bcp.2017.11.014
2. Malone Rubright SL, Pearce LL, Peterson J. Environmental toxicology of hydrogen sulfide. Nitric Oxide. (2017) 71:1–13. doi: 10.1016/j.niox.2017.09.011
3. Ng PC, Hendry-Hofer TB, Witeof AE, Brenner M, Mahon SB, Boss GR, et al. Hydrogen sulfide toxicity: mechanism of action, clinical presentation, and countermeasure development. J Med Toxicol. (2019) 15:287–94. doi: 10.1007/s13181-019-00710-5
4. Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. (1996) 16:1066–71. doi: 10.1523/JNEUROSCI.16-03-01066.1996
5. Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. (2002) 16:1792–8. doi: 10.1096/fj.02-0211hyp
6. Chen DB, Feng L, Hodges JK, Lechuga TJ, Zhang H. Human trophoblast-derived hydrogen sulfide stimulates placental artery endothelial cell angiogenesis. Biol Reprod. (2017) 97:478–89. doi: 10.1093/biolre/iox105
7. Zhang D, Wang X, Chen S, Chen S, Yu W, Liu X, et al. Endogenous hydrogen sulfide sulfhydrates IKKβ at cysteine 179 to control pulmonary artery endothelial cell inflammation. Clin Sci. (2019) 133:2045–59. doi: 10.1042/CS20190514
8. Liu HT, Zhou ZX, Ren Z, Yang S, Liu LS, Wang Z, et al. EndMT: potential target of H2S against atherosclerosis. Curr Med Chem. (2021) 28:3666–80. doi: 10.2174/0929867327999201116194634
9. Qianqian S, Bin W, Yingsha L, Fang P, Weijie X. Taurine supplementation lowers blood pressure and improves vascular function in prehypertension: randomized, double-blind, placebo-controlled study. Hypertension. (2016) 67:541–9. doi: 10.1161/HYPERTENSIONAHA.115.06624
10. Polhemus DJ, Li Z, Pattillo CB, Gojon G, Gojon G, Giordano T, et al. A novel hydrogen sulfide prodrug, SG1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc Ther. (2015) 33:216–26. doi: 10.1111/1755-5922.12128
11. Cuevasanta E, Denicola A, Alvarez B, Moller MN. Solubility and permeation of hydrogen sulfide in lipid membranes. PLoS ONE. (2012) 7:e34562. doi: 10.1371/journal.pone.0034562
12. von Scheidt M, Zhao Y, de Aguiar Vallim TQ, Che N, Wierer M, Seldin MM, et al. The transcription factor MAFF regulates an atherosclerosis relevant network connecting inflammation and cholesterol metabolism. Circulation. (2021) 143:1809–23. doi: 10.1161/CIRCULATIONAHA.120.050186
13. Afonso MS, Sharma M, Schlegel MP, van Solingen C, Koelwyn GJ, Shanley LC, et al. miR-33 silencing reprograms the immune cell landscape in atherosclerotic plaques. Circ Res. (2021) 128:1122–38. doi: 10.1161/CIRCRESAHA.120.317914
14. Wu D, Hu Q, Liu X, Pan L, Xiong Q, Zhu YZ. Hydrogen sulfide protects against apoptosis under oxidative stress through SIRT1 pathway in H9c2 cardiomyocytes. Nitric Oxide. (2015) 46:204–12. doi: 10.1016/j.niox.2014.11.006
15. Zhao ZZ, Wang Z, Li GH, Wang R, Tan JM, Cao X, et al. Hydrogen sulfide inhibits macrophage-derived foam cell formation. Exp Biol Med. (2011) 236:169–76. doi: 10.1258/ebm.2010.010308
16. Hu TX, Zhang NN, Ruan Y, Tan QY, Wang J. Hydrogen sulfide modulates high glucose-induced NLRP3 inflammasome activation in 3T3-L1 adipocytes. Exp Ther Med. (2020) 19:771–6. doi: 10.3892/etm.2019.8242
17. Potor L, Nagy P, Méhes G, Hendrik Z, Jeney V, Petho D, et al. Hydrogen sulfide abrogates hemoglobin-lipid interaction in atherosclerotic lesion. Oxid Med Cell Longev. (2018) 2018:3812568.
18. Meng G, Zhu J, Xiao Y, Huang Z, Zhang Y, Tang X, et al. Hydrogen sulfide donor GYY4137 protects against myocardial fibrosis. Oxid Med Cell Longev. (2015) 2015:691070. doi: 10.1155/2015/691070
19. Baskar R, Sparatore A, Del Soldato P, Moore PK. Effect of S-diclofenac, a novel hydrogen sulfide releasing derivative inhibit rat vascular smooth muscle cell proliferation. Eur J Pharmacol. (2008) 594:1–8. doi: 10.1016/j.ejphar.2008.07.029
20. Wang Y, Xia Z, Jin H, Wei H, Du J. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. (2009) 29:173–9. doi: 10.1161/ATVBAHA.108.179333
21. Emerson M. Hydrogen sulfide and platelets: a possible role in thrombosis. Handb Exp Pharmacol. (2015) 230:153–62. doi: 10.1007/978-3-319-18144-8_7
22. Ambale-Venkatesh B, Liu CY, Liu YC, Donekal S, Ohyama Y, Sharma RK, et al. Association of myocardial fibrosis and cardiovascular events: the multi-ethnic study of atherosclerosis. Eur Heart J Cardiovasc Imaging. (2019) 20:168–76. doi: 10.1093/ehjci/jey140
23. Sheng J, Shim W, Wei H, Lim SY, Liew R, Lim TS, et al. Hydrogen sulphide suppresses human atrial fibroblast proliferation and transformation to myofibroblasts. J Cell Mol Med. (2013) 17:1345–54. doi: 10.1111/jcmm.12114
24. Valls-Lacalle L, Negre-Pujol C, Rodriguez C, Varona S, Valera-Canellas A, Consegal M, et al. Opposite effects of moderate and extreme Cx43 deficiency in conditional Cx43-deficient mice on angiotensin II-induced cardiac fibrosis. Cells. (2019) 8:1299. doi: 10.3390/cells8101299
25. Huang J, Wang D, Zheng J, Huang X, Jin H. Hydrogen sulfide attenuates cardiac hypertrophy and fibrosis induced by abdominal aortic coarctation in rats. Mol Med Rep. (2012) 5:923–8. doi: 10.3892/mmr.2012.748
26. Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Mol Cell Endocrinol. (2010) 316:129–39. doi: 10.1016/j.mce.2009.08.018
27. Whitlock G, Lewington S, Sherliker P, Clarke R, Emberson J, Halsey J, et al. Body-mass index and cause-specific mortality in 900000 adults: collaborative analyses of 57 prospective studies. Lancet. (2009) 373:1083–96. doi: 10.1016/S0140-6736(09)60318-4
28. Kursawe R, Dixit VD, Scherer PE, Santoro N, Narayan D, Gordillo R, et al. Role of the inflammasome in the low storage capacity of the abdominal subcutaneous adipose tissue in obese adolescents. Diabetes. (2016) 65:610–8. doi: 10.2337/db15-1478
29. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. (2019) 124:315–27. doi: 10.1161/CIRCRESAHA.118.313591
30. Kasikara C, Doran AC, Cai B, Tabas I. The role of non-resolving inflammation in atherosclerosis. J Clin Invest. (2018) 128:2713–23. doi: 10.1172/JCI97950
31. van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, García-Mata R, et al. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol. (2007) 178:1279–93. doi: 10.1083/jcb.200612053
32. Gholami M, Amoli M, Sharifi F. Letter to the Editor: Comments on “Association between the ICAM-1 gene polymorphism and coronary heart disease risk: a meta-analysis”. Biosci Rep. (2019) 39. doi: 10.1042/BSR20190554
33. Fiorucci S, Antonelli E, Distrutti E, Rizzo G, Mencarelli A, Orlandi S, et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. (2005) 129:1210–24. doi: 10.1053/j.gastro.2005.07.060
34. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis a report from the committee on vascular lesions of the council on arteriosclerosis, American heart association. Circulation. (1995) 92:1355. doi: 10.1161/01.CIR.92.5.1355
35. Yuan XM Li W, Olsson AG, Brunk UT. Iron in human atheroma and LDL oxidation by macrophages following erythrophagocytosis. Atherosclerosis. (1996) 124:61–73. doi: 10.1016/0021-9150(96)05817-0
36. Jeney V, Balla G, Balla J. Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front Physiol. (2014) 5:379. doi: 10.3389/fphys.2014.00379
37. László P, Péter N, Gábor M, Zoltán H, Viktória J, Dávid P, et al. Hydrogen sulfide abrogates hemoglobin-lipid interaction in atherosclerotic lesion. Oxid Med Cell Longev. (2018) 2018:1–16. doi: 10.1155/2018/3812568
38. Muellner MK, Schreier SM, Laggner H, Hermann M, Esterbauer H, Exner M, et al. Hydrogen sulfide destroys lipid hydroperoxides in oxidized LDL. Biochem J. (2009) 420:277–81. doi: 10.1042/BJ20082421
39. Heber S, Fischer B, Sallaberger-Lehner M, Hausharter M, Volf I. Effects of high-intensity interval training on platelet function in cardiac rehabilitation: a randomised controlled trial. Heart. (2020) 106:69–79. doi: 10.1136/heartjnl-2019-315130
40. Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. (2003) 9:61. doi: 10.1038/nm810
41. Piccardoni P, Sideri R, Manarini S, Piccoli A, Evangelista V. Platelet/polymorphonuclear leukocyte adhesion: a new role for SRC kinases in Mac-1 adhesive function triggered by P-selectin. Blood. (2001) 98:108–16. doi: 10.1182/blood.V98.1.108
42. Simon DI, Chen Z, Xu H, Li CQ, Dong J, Mcintire LV, et al. Platelet glycoprotein Ibα is a counterreceptor for the leukocyte integrin mac-1 (Cd11b/Cd18). J Exp Med. (2000) 192:193. doi: 10.1084/jem.192.2.193
43. Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. (2007) 25:619–47. doi: 10.1146/annurev.immunol.25.022106.141618
44. Forman HJ, Zhang H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov. (2021) 20:689–709. doi: 10.1038/s41573-021-00233-1
45. Xie ZZ, Liu Y, Bian JS. Hydrogen sulfide and cellular redox homeostasis. Oxid Med Cell Longev. (2016) 2016:6043038. doi: 10.1155/2016/6043038
46. Liu N, Wu J, Zhang L, Gao Z, Sun Y, Yu M, et al. Hydrogen Sulphide modulating mitochondrial morphology to promote mitophagy in endothelial cells under high-glucose and high-palmitate. J Cell Mol Med. (2017) 21:3190–203. doi: 10.1111/jcmm.13223
47. Yong R, Searcy DG. Sulfide oxidation coupled to ATP synthesis in chicken liver mitochondria. Comp Biochem Physiol B Biochem Mol Biol. (2001) 129:129–37. doi: 10.1016/S1096-4959(01)00309-8
48. Grambow E, Mueller-Graf F, Delyagina E, Frank M, Kuhla A, Vollmar B. Effect of the hydrogen sulfide donor GYY4137 on platelet activation and microvascular thrombus formation in mice. Platelets. (2014) 25:166. doi: 10.3109/09537104.2013.786823
49. Lin G, Cheng C, Sparatore A, Zhang H, Wang C. Hydrogen sulfide inhibits human platelet aggregation in vitro in part by interfering gap junction channels: effects of ACS14, a hydrogen sulfide-releasing aspirin. Heart Lung Circ. (2015) 24:77–85. doi: 10.1016/j.hlc.2014.05.019
50. Zhang H, Lauver DA, Hollenberg P. CYP-independent inhibition of platelet aggregation in rabbits by a mixed disulfide conjugate of clopidogrel. Thromb Haemost. (2014) 111:1304–11. doi: 10.1160/th14-04-0388
51. Grambow E, Leppin C, Leppin K, Kundt G, Vollmar B. The effects of hydrogen sulfide on platelet–leukocyte aggregation and microvascular thrombolysis. Platelets. (2017) 28:509–17. doi: 10.1080/09537104.2016.1235693
52. Zhang H, Bai Z, Zhu L, Liang Y, Fan X, Li J, et al. Hydrogen sulfide donors: therapeutic potential in anti-atherosclerosis. Eur J Med Chem. (2020) 205:112665. doi: 10.1016/j.ejmech.2020.112665
53. Wedmann R, Bertlein S, Macinkovic I, Boltz S, Miljkovic JL, Munoz LE, et al. Working with “H2S”: facts and apparent artifacts. Nitric Oxide. (2014) 41:85–96. doi: 10.1016/j.niox.2014.06.003
54. Warnholtz A, Nickenig G, Schulz E, Macharzina R, Bräsen JH, Skatchkov M, et al. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation. (1999) 99:2027–33. doi: 10.1161/01.CIR.99.15.2027
55. Lu HY, Hsu HL, Li CH, Li SJ, Lin SJ, Shih CM, et al. Hydrogen sulfide attenuates aortic remodeling in aortic dissection associating with moderated inflammation and oxidative stress through a NO-dependent pathway. Antioxidants. (2021). 10:682. doi: 10.3390/antiox10050682
56. Jaminon A, Reesink K, Kroon A, Schurgers L. The role of vascular smooth muscle cells in arterial remodeling: focus on calcification-related processes. Int J Mol Sci. (2019) 20:5694. doi: 10.3390/ijms20225694
57. Huynh D, Jin Y, Myung CS, Heo KS. Inhibition of p90RSK is critical to abolish Angiotensin II-induced rat aortic smooth muscle cell proliferation and migration. Biochem Biophys Res Commun. (2019) 523:267–73. doi: 10.1016/j.bbrc.2019.12.053
58. Planas-Rigol E, Terrades-Garcia N, Corbera-Bellalta M, Lozano E, Alba MA, Segarra M, et al. Endothelin-1 promotes vascular smooth muscle cell migration across the artery wall: a mechanism contributing to vascular remodelling and intimal hyperplasia in giant-cell arteritis. Ann Rheum Dis. (2017) 76:1624–34. doi: 10.1136/annrheumdis-2016-210792
59. Shi J, Yang Y, Cheng A, Xu G, He F. Metabolism of vascular smooth muscle cells in vascular diseases. AJP Heart Circ Physiol. (2020) 319:H613–31. doi: 10.1152/ajpheart.00220.2020
60. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
61. Ackers-Johnson M, Talasila A, Sage AP, Long X, Sinha S. Myocardin regulates vascular smooth muscle cell inflammatory activation and disease. Arterioscler Thromb Vasc Biol. (2015) 35:817. doi: 10.1161/ATVBAHA.114.305218
62. Du J, Yan H, Cheung Y, Geng B, Jiang H, Chen X, et al. The possible role of hydrogen sulfide as a smooth muscle cell proliferation inhibitor in rat cultured cells. Heart Vessels. (2004) 19:75–80. doi: 10.1007/s00380-003-0743-7
63. Li L, Liu D, Bu D, Chen S, Wu J, Tang C. Brg1-dependent epigenetic control of vascular smooth muscle cell proliferation by hydrogen sulfide. Biochim Biophys Acta. (2013) 1833:1347–55. doi: 10.1016/j.bbamcr.2013.03.002
64. Bobryshev YV, Ivanova EA, Chistiakov DA, Nikiforov NG, Orekhov AN. Macrophages and their role in atherosclerosis: pathophysiology and transcriptome analysis. BioMed Res Int. (2016) 2016:9582430. doi: 10.1155/2016/9582430
65. Maguire EM, Pearce SWA, Xiao Q. Foam cell formation: a new target for fighting atherosclerosis and cardiovascular disease. Vascul Pharmacol. (2019) 112:14–71. doi: 10.1016/j.vph.2018.08.002
66. Hans CP, Zerfaoui M, Naura AS, Catling A, Boulares AH. Differential effects of PARP inhibition on vascular cell survival and ACAT-1 expression favouring atherosclerotic plaque stability. Cardiovasc Res. (2008) 78:429–39. doi: 10.1093/cvr/cvn018
67. Xie L, Yue G, Wen M, Shuang Z, Wan W, Yan M, et al. Hydrogen sulfide induces keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes. (2017) 10:3171. doi: 10.2337/db16-0020
68. Wen YD, Hong W, Zhu YZ. The drug developments of hydrogen sulfide on cardiovascular disease. Oxid Med Cell Longev. (2018) 2018:1–21. doi: 10.1155/2018/4010395
69. Fan J, Zheng F, Li S, Cui C, Jiang S, Zhang J, et al. Hydrogen sulfide lowers hyperhomocysteinemia dependent on cystathionine γ lyase S-sulfhydration in ApoE-knockout atherosclerotic mice. Br J Pharmacol. (2019) 176:3180–92. doi: 10.1111/bph.14719
70. Lu X, Li H, Wang S. Hydrogen sulfide protects against uremic accelerated atherosclerosis via nPKCδ/Akt signal pathway. Front Mol Biosci. (2021). doi: 10.3389/fmolb.2020.615816
71. Tocmo R, Wu Y, Liang D, Fogliano V, Huang D. Boiling enriches the linear polysulfides and the hydrogen sulfide-releasing activity of garlic. Food Chem. (2017) 221:1867–73. doi: 10.1016/j.foodchem.2016.10.076
72. Szabo C, Papapetropoulos A. International union of basic and clinical pharmacology. CII: pharmacological modulation of HS Levels: HS donors and HS biosynthesis inhibitors. Pharmacol Rev. (2017) 69:497–564. doi: 10.1124/pr.117.014050
73. Qiu Y, Wu Y, Meng M, Luo M, Zhao H, Sun H, et al. GYY4137 protects against myocardial ischemia/reperfusion injury via activation of the PHLPP-1/Akt/Nrf2 signaling pathway in diabetic mice. J Surg Res. (2018) 225:29–39. doi: 10.1016/j.jss.2017.12.030
74. Zheng Y, Lv P, Huang J, Ke J, Yan J. GYY4137 exhibits anti-atherosclerosis effect in apolipoprotein E (-/-) mice via PI3K/Akt and TLR4 signalling. Clin Exp Pharmacol Physiol. (2020) 47:1231–9. doi: 10.1111/1440-1681.13298
75. Liu Z, Han Y, Li L, Lu H, Meng G, Li X, et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E(-/-) mice. Br J Pharmacol. (2013) 169:1795–809. doi: 10.1111/bph.12246
76. Lilyanna S, Peh MT, Liew OW, Wang P, Moore PK, Richards AM, et al. GYY4137 attenuates remodeling, preserves cardiac function and modulates the natriuretic peptide response to ischemia. J Mol Cell Cardiol. (2015) 87:27–37. doi: 10.1016/j.yjmcc.2015.07.028
77. Mitsuhashi H, Yamashita S, Ikeuchi H, Kuroiwa T, Kaneko Y, Hiromura K, et al. Oxidative stress-dependent conversion of hydrogen sulfide to sulfite by activated neutrophils. Shock. (2005) 24:529–34. doi: 10.1097/01.shk.0000183393.83272.de
78. Kodela R, Chattopadhyay M, Velázquez-Martínez CA, Kashfi K. NOSH-aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid has enhanced chemo-preventive properties compared to aspirin, is gastrointestinal safe with all the classic therapeutic indications. Biochem Pharmacol. (2015) 98:564–72. doi: 10.1016/j.bcp.2015.09.014
79. Chattopadhyay M, Kodela R, Santiago G, Le TTC, Nath N, Kashfi K. NOSH-aspirin (NBS-1120) inhibits pancreatic cancer cell growth in a xenograft mouse model: Modulation of FoxM1, p53, NF-κB, iNOS, caspase-3 and ROS. Biochem Pharmacol. (2020) 176:113857. doi: 10.1016/j.bcp.2020.113857
80. Antoniou C, Xenofontos R, Chatzimichail G, Christou A, Kashfi K, Fotopoulos V. Medicago sativaexploring the potential of nitric oxide and hydrogen sulfide (NOSH)-releasing synthetic compounds as novel priming agents against drought stress in plants. Biomolecules. (2020) 10:120. doi: 10.3390/biom10010120
81. Fasano S, Iacono D, Riccardi A, Ciccia F, Valentini G. The role of aspirin in the primary prevention of accelerated atherosclerosis in systemic autoimmune rheumatic diseases. Rheumatology. (2020) 59:3593–602. doi: 10.1093/rheumatology/keaa335
82. Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. (2017) 120:713–35. doi: 10.1161/CIRCRESAHA.116.309326
83. Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn't the stomach digest itself? Physiol Rev. (2008) 88:1547–65. doi: 10.1152/physrev.00004.2008
84. Zhao Y, Wang H, Xian M. Cysteine-activated hydrogen sulfide (H2S) donors. J Am Chem Soc. (2011) 133:15–7. doi: 10.1021/ja1085723
85. Zhao Y, Bhushan S, Yang C, Otsuka H, Stein JD, Pacheco A, et al. Controllable hydrogen sulfide donors and their activity against myocardial ischemia-reperfusion injury. ACS Chem Biol. (2013) 8:1283–90. doi: 10.1021/cb400090d
86. Roger T, Raynaud F, Bouillaud F, Ransy C, Simonet S, Crespo C, et al. New biologically active hydrogen sulfide donors. Chembiochem. (2013) 14:2268–71. doi: 10.1002/cbic.201300552
87. Martelli A, Testai L, Citi V, Marino A, Pugliesi I, Barresi E, et al. Arylthioamides as H2S Donors: l-Cysteine-Activated Releasing Properties and Vascular Effects in vitro and in vivo. ACS Med Chem Lett. (2013) 4:904–8. doi: 10.1021/ml400239a
88. Foster JC, Radzinski SC, Zou X, Finkielstein CV, Matson JB. H2S-releasing polymer micelles for studying selective cell toxicity. Mol Pharm. (2017) 14:1300–6. doi: 10.1021/acs.molpharmaceut.6b01117
89. Borchardt RT, Cohen LA. Stereopopulation control. 3. Facilitation of intramolecular conjugate addition of the carboxyl group. J Am Chem Soc. (1972) 94:9175–82. doi: 10.1021/ja00781a031
90. Glanz VY, Myasoedova VA, Grechko AV, Orekhov AN. Trans-sialidase associated with atherosclerosis: defining the identity of a key enzyme involved in the pathology. Curr Drug Targets. (2019) 20:938–41. doi: 10.2174/1389450120666190308111619
91. Bibli SI, Hu J, Leisegang MS, Wittig J, Zukunft S, Kapasakalidi A, et al. Shear stress regulates cystathionine γ lyase expression to preserve endothelial redox balance and reduce membrane lipid peroxidation. Redox Biol. (2020) 28:101379. doi: 10.1016/j.redox.2019.101379
92. Shen Y, Liu X, Shi J, Wu X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int J Biol Macromol. (2019) 125:496–502. doi: 10.1016/j.ijbiomac.2018.11.190
93. Yang CT, Lai ZZ, Zheng ZH, Kang JM, Xian M, Wang RY, et al. A novel pH-controlled hydrogen sulfide donor protects gastric mucosa from aspirin-induced injury. J Cell Mol Med. (2017) 21:2441–51. doi: 10.1111/jcmm.13166
94. Al-Bishari AM, Yie KHR, Al-Baadani MA, Al-Shaaobi BA, Zhou Z, Fang K, et al. JK-2 loaded electrospun membrane for promoting bone regeneration. Mater Sci Eng C Mater Biol Appl. (2021) 130:112471. doi: 10.1016/j.msec.2021.112471
95. Devarie-Baez NO, Bagdon PE, Peng B, Zhao Y, Park CM, Xian M. Light-induced hydrogen sulfide release from “caged” gem-dithiols. Org Lett. (2013) 15:2786–9. doi: 10.1021/ol401118k
96. Steiger AK, Marcatti M, Szabo C, Szczesny B, Pluth MD. Inhibition of Mitochondrial Bioenergetics by Esterase-Triggered COS/HS Donors. ACS Chem Biol. (2017) 12:2117–23. doi: 10.1021/acschembio.7b00279
97. Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of HS as a novel endogenous gaseous K(ATP) channel opener. EMBO J. (2001) 20:6008–16. doi: 10.1093/emboj/20.21.6008
98. Cormode DP, Skajaa T, van Schooneveld MM, Koole R, Jarzyna P, Lobatto ME, et al. Nanocrystal core high-density lipoproteins: a multimodality contrast agent platform. Nano Lett. (2008) 8:3715–23. doi: 10.1021/nl801958b
99. Wang Y, Zhang K, Li T, Maruf A, Qin X, Luo L, et al. Macrophage membrane functionalized biomimetic nanoparticles for targeted anti-atherosclerosis applications. Theranostics. (2021) 11:164–80. doi: 10.7150/thno.47841
100. Gao C, Huang Q, Liu C, Kwong CHT, Yue L, Wan JB, et al. Treatment of atherosclerosis by macrophage-biomimetic nanoparticles via targeted pharmacotherapy and sequestration of proinflammatory cytokines. Nat Commun. (2020) 11:2622. doi: 10.1038/s41467-020-16439-7
Keywords: hydrogen sulfide, donor, atherosclerotic, nanotechnology, drug delivery and targeting
Citation: Yang Y-W, Deng N-H, Tian K-J, Liu L-S, Wang Z, Wei D-H, Liu H-T and Jiang Z-S (2022) Development of hydrogen sulfide donors for anti-atherosclerosis therapeutics research: Challenges and future priorities. Front. Cardiovasc. Med. 9:909178. doi: 10.3389/fcvm.2022.909178
Received: 31 March 2022; Accepted: 28 July 2022;
Published: 12 August 2022.
Edited by:
Masuko Ushio-Fukai, Augusta University, United StatesReviewed by:
Guangdong Yang, Laurentian University, CanadaCopyright © 2022 Yang, Deng, Tian, Liu, Wang, Wei, Liu and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi-Sheng Jiang, enNqaWFuZzIwMDVAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.