 Juexing Li
Juexing Li Lei Zhou
Lei Zhou Hui Gong
Hui Gong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 15 September 2022
Sec. General Cardiovascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.903902
Sodium-glucose cotransporter 2 inhibitors (SGLT2is) are newly emerging insulin-independent anti-hyperglycemic agents that work independently of β-cells. Quite a few large-scale clinical trials have proven the cardiovascular protective function of SGLT2is in both diabetic and non-diabetic patients. By searching all relevant terms related to our topics over the previous 3 years, including all the names of agents and their brands in PubMed, here we review the mechanisms underlying the improvement of heart failure. We also discuss the interaction of various mechanisms proposed by diverse works of literature, including corresponding and opposing viewpoints to support each subtopic. The regulation of diuresis, sodium excretion, weight loss, better blood pressure control, stimulation of hematocrit and erythropoietin, metabolism remodeling, protection from structural dysregulation, and other potential mechanisms of SGLT2i contributing to heart failure improvement have all been discussed in this manuscript. Although some remain debatable or even contradictory, those newly emerging agents hold great promise for the future in cardiology-related therapies, and more research needs to be conducted to confirm their functionality, particularly in metabolism, Na+-H+ exchange protein, and myeloid angiogenic cells.
Heart failure, a chronic pathological condition, is a universal prevailing disease, with more than 26 million sufferers (1). It has come to light that several factors are attributed to the heart failure process, including disorders of the structure, function, rhythm, and conductive systems (2). Such abnormal diseases place a tremendous cost on society and the economy. Thus, we should give sufficient emphasis to seeking more economical and effective treatment techniques for heart failure.
SGLT2is, a new class of oral anti-hyperglycemic agents, were required by the U.S. FDA to undergo cardiovascular safety testing prior to being officially marketed. The outcomes exceed expectations—SGLT2is have shown unexpected cardiorenal protection in both heart failure with reduced ejection fraction (HFrEF) and heart failure with preserved ejection fraction (HFpEF) in several clinical trials (3–7), which have been detailed in Table 1. As a result, SGLT2is are recommended as one of the fundamental treatments for heart failure in recent guidelines, particularly the 2022 AHA/ACC/HFSA one (8). Together with ACEI/ARNI, BB, and MRA, they are collectively called “new quadruple therapy.” It is noteworthy that SGLT2is can be applied across the entire stages of heart failure management, even if in the at-risk stage for heart failure.
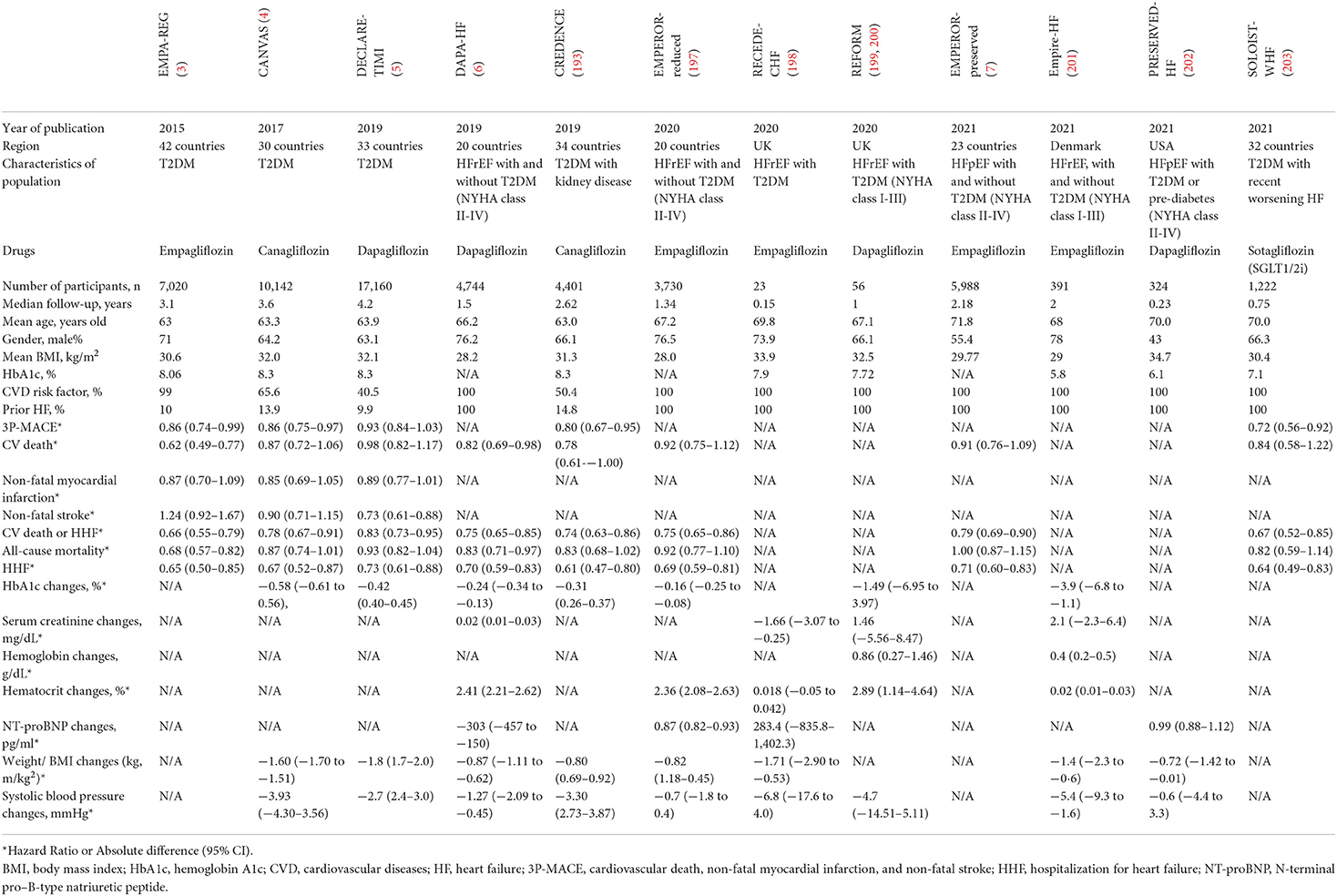
Table 1. Contribution of SGLT2i to heart failure in major clinical trials.
Those clinical trial results are groundbreaking. Thus, researchers have conducted quite a few studies concerning the cardioprotective function of SGLT2is. It has been thoroughly researched in the regulation of diuresis, sodium excretion, weight loss, blood pressure improvement, hematocrit and erythropoietin stimulation, metabolism remolding, protection from structural dysregulation, etc. Some of them have been sophisticatedly proven, but the in-depth mechanisms and some other recently emerging mechanism theories such as Na+-H+ exchange protein and myeloid angiogenic cells remain vague.
Thus, this manuscript reviews these newly emerging studies from the perspectives of natriuresis, weight loss, blood pressure reduction, etc., containing all the research that may be concerned with this topic during the past 3 years so as to identify the potential mechanisms and provide a full landscape of its benefits to patients with heart failure.
The maximum transport capacity (Tmax) of the proximal tubule in humans is 500 g of glucose per day under normal conditions. Thus, as blood circulates through, almost all glucose will be reabsorbed. When blood glucose levels are above a certain threshold, which implies they are higher than the renal tubules' capacity to reabsorb glucose, glucose starts to show up in the urine and is then expelled (9, 10).
The active transporters or symporters (SGLTs) and the facilitated transporters or uniporters (GLUTs) are two different types of glucose transporters that are known to contribute to glucose homeostasis (11). The SGLT family contributes to the active absorption of glucose/galactose in the site of the intestine as well as the reabsorption of glucose in the kidney (12). SGLT receptors are categorized into six types in human bodies (11). SGLT1/2 are mainly enriched in the kidney, while SGLT2, a symporter for Na+ and glucose, plays a considerable role during glucose reabsorption in the kidney (11). The new anti-hyperglycemic drug SGLT2i directly binds to the corresponding receptor to curb the reabsorption of glucose at the site of the proximal convoluted tubule, and the unabsorbed part is excreted from the body, thereby achieving the effect of blood sugar lowering (Figure 1, which was created with BioRender.com).
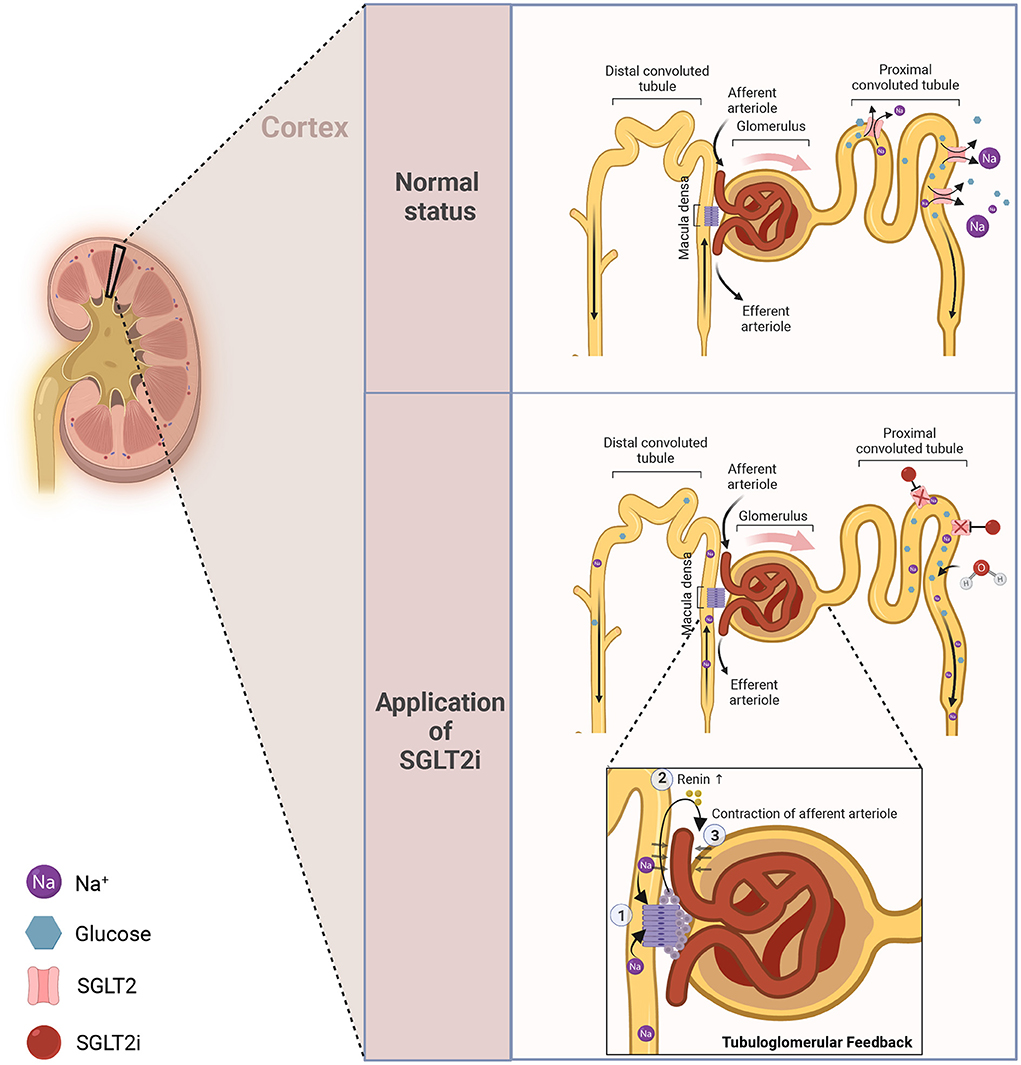
Figure 1. The anti-hyperglycemic mechanism of SGLT2i. In normal status, glucose and Na+ can be efficiently reabsorbed at the site of the proximal convoluted tubule to maintain glucose homeostasis. Once applied with SGLT2i, such reabsorption will be inhibited, thereby leading to diuresis and natriuresis.
The course and prognosis of heart failure are closely tied to sodium and water retention, which is responsible for 90% of heart failure hospitalizations (13). Water and sodium retention result from reduced circulation, decreased renal blood flow, and elevated aldosterone when the heart's pumping capacity is compromised (14). Furthermore, the blood detained in vein vessels will seep into the interstitial fluid under the pressure of capillaries, and those penetrative fluids remain there when the volume of the fluids exceeds the surrounding cells' abilities of absorption (remarkably, the fluid in lower limbs) (15).
In terms of its mode of action, SGLT2i is distinct from traditional diuretics. With the application of SGLT2i, glucose reabsorption is hindered at the site of renal tubules, and therefore the unabsorbed portion will flow into the distal nephron. However, water can still be reabsorbed unimpededly as usual during this period. Thus, the reduction of the osmotic gradient leads to a decrease in water reabsorption, termed osmotic diuresis (16). One of the critical factors in a significantly reduced rate of heart failure deterioration is a decrease in circulation volume, which is caused by the natriuretic action of SGLT2i. But the eyes-catching role of diuresis in the process of heart failure has been undermined by another large clinical trial which concludes that patients with a high propensity for fluid retention show no remarkably extra benefits compared to the control group under the application of empagliflozin (17). Beyond this diuresis function, an additional independent element during the progression of heart failure may be the improvement of pulmonary artery diastolic pressure (18).
On the other hand, prior research has shown that diabetes patients have higher sodium levels in their skin and muscles (19). Moreover, tissue sodium is closely linked to ventricular hypertrophy regardless of blood pressure and water-sodium retention conditions. It is reported that the application of dapagliflozin for 6 weeks can decrease tissue sodium in patients with T2DM (20) compared to control groups, thereby reducing the risk of left ventricular hypertrophy and chronic heart failure. In contrast, in an emergency scenario, increased glycosuria rather than natriuresis, with SGLT2i utilization, contributes to improving the patient's state (21).
Excessive adipose tissue exerts vital functions in the onset and progression of heart failure (22). Several studies have revealed that SGLT2i plays a crucial part in weight shedding during its pharmacological action in both types of diabetes (23–25), and the baseline body mass index will not impair the effectiveness of SGLT2i, contrary to what is known as the “obesity paradox” (26).
Attributed to the calories loss caused by the excretion of glucose (27) and enhanced hypothalamic insulin responsiveness (28), the application of SGLT2i can significantly reduce the weight of patients, leading to the reduction of total fat mass, subdermal fat, visceral fat, and liver fat content (29–33). What's more, it has been revealed that SGLT2i can reduce the size of adipocytes in the perivascular adipose tissue (34) and be capable of inducing mitochondrial biogenesis through the AMPK/SIRT1 pathway and β3-adrenoceptor-cAMP-PKA signaling pathway, thereby increasing the energy consumption of adipocytes directly in vivo (35–37). Some researchers believe that it is possible to achieve pounds shedding by inducing the beigeing of fat (38), while beigeing refers to a specific metabolism remolding of fat tissue (39). Additionally, a recent study reveals that the activated innervation of intra-adipose sympathetic induced by SGLT2i in mice with a high-calorie diet also contributes to such an increased energy consumption (37, 40). Fibroblast growth factor 21 (FGF21) is a coordinator for the SGLT2i-dependent decline in adiposity and the trigger of lipolysis during weight loss (41). This mediator can perform a role in the activation of the nervous system, thereby achieving the biological effect of white/brown adipose tissue and the induction of thermogenesis (40, 42).
This weight loss impact does not last indefinitely; it progressively reaches a plateau between 24 and 52 weeks. These phenomena may be explained by the fact that adipose tissue has an anti-lipolytic function, and the decline in leptin levels has boosted compensatory eating (43–45). Such increased intake is a higher calorie intake (46) with a constant proportion of nutrients as previously, although those intake calories are ultimately countered by glucose loss (28, 47). Figure 2, which was produced using BioRender.com, shows intricate mechanisms. Therefore, although SGLT2i cannot permanently treat obesity, lowering body weight and fat percentage may still positively affect the cardiovascular system.
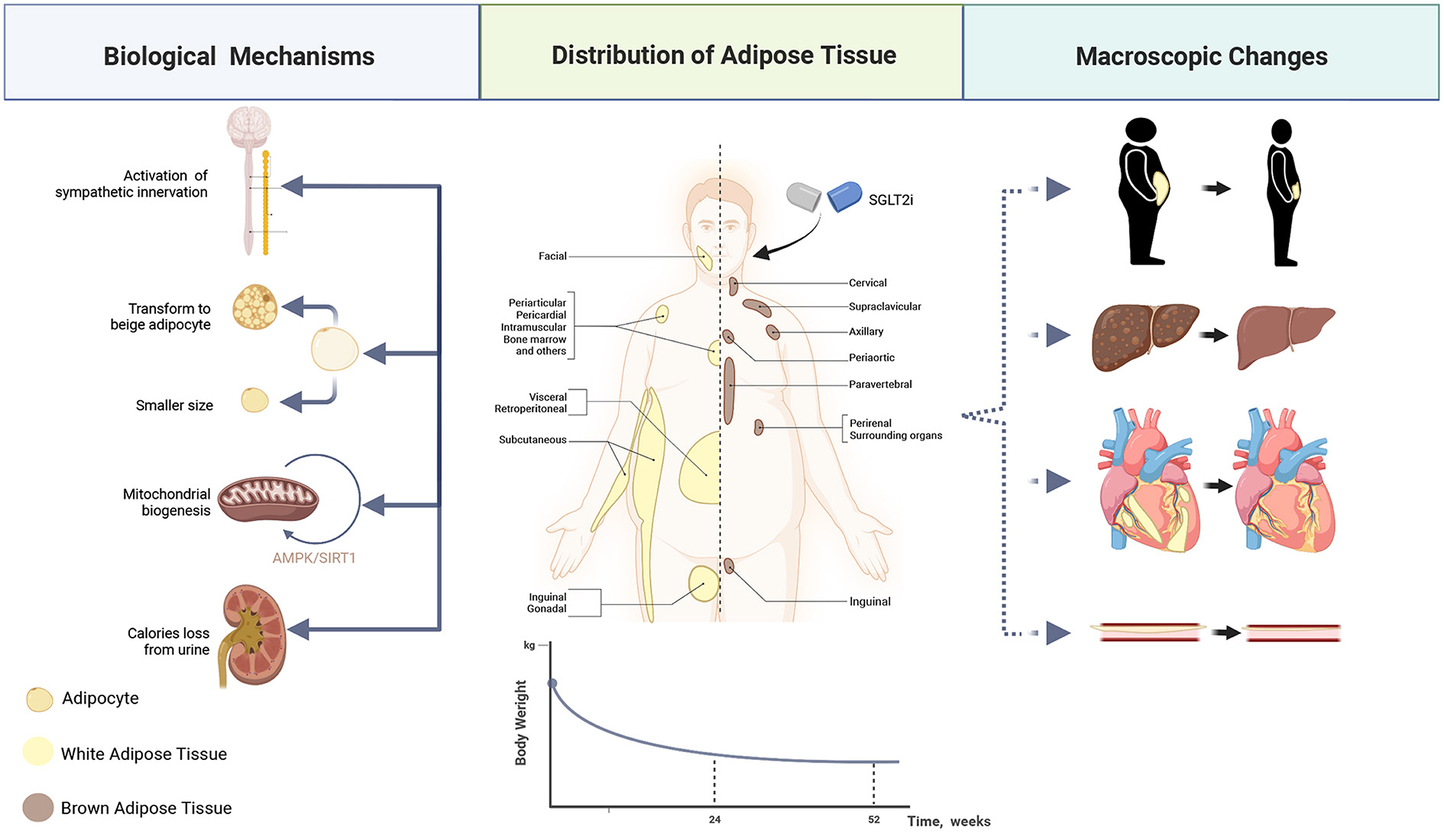
Figure 2. Mechanisms of weight loss. Full view of how SGLT2i affects adipocytes—how to reduce the size, and how to stimulate its transformation to beige adipocytes, as well as how it makes the body pounds shed directly via calories loss. Consequently, the subcutaneous, visceral, and perivascular fat content decreases under the treatment of SGLT2i.
The blood volume in the body is tightly connected to blood pressure levels. Nevertheless, it has been observed that volume changes and urinary sodium excretion, under the application of SGLT2i, do not significantly lower blood pressure (48), suggesting that additional processes may have been at work throughout this blood pressure-lowering process.
In clinical trials, the 24 h ambulatory blood pressure shows a slight reduction among people suffering from nocturnal hypertension, diabetes, or salt sensitivity with the application of SGLT2i, thereby lowering the incidence of heart failure and the tolls of cardiovascular mortality (49). Such an efficacy may be achieved by regulating renal HIF-1α, impairing inflammation and oxidative stress (50), regulating the function of the paraventricular nucleus of the hypothalamus (PVN) (51), and sGC pathways (52). A meta-analysis of 43 RCT studies reveals that under SGLT2i treatment, patients' systolic blood pressure (SBP) decreases by a mean of 2.46 mmHg and diastolic blood pressure (DBP) by a mean of 1.46 mmHg (25, 53), while another meta-analysis confirms that SGLT2i significantly reduced 24 h dynamic SBP and dynamic DBP, and such an effect is concluded as SGLT2i-like effect (54). Therefore, SGLT2i generates cardiovascular benefits by reducing blood volume, lowering blood pressure, and improving ventricular load through its diuretic and natriuretic effects. In the meantime, it also exerts cardiovascular effects by regulating the central nervous system and affecting PVN.
An endogenous glycoprotein hormone called erythropoietin (EPO) can promote erythropoiesis (More details on this concept have been fully explicated in the reviews by Lappin, T.R. and Koury, M.J.) (55, 56). EPO is mostly produced by the kidney and in minor quantities by the liver. At the same time, a hypoxic environment will trigger its production. Moreover, the values of hematocrit and hemoglobin within normal ranges are negatively correlated to the incidence or prognosis of cardiovascular diseases (57–60). EPO and hematocrit significantly rise after using empagliflozin for 1–6 months, causing a continuous rise in blood cells that peaks in 2–3 months (29, 61, 62), and this tendency is independent of the initial levels of anemia (63).
These changes in EPO and hematocrit can be elaborated as follows: (1) Decreased plasma volume and blood concentration induced by osmotic diuresis (64), but some hold the contradictory view (2) Increased erythropoiesis and hematocrit via inhibiting hepcidin and regulating other iron regulatory proteins (65), (3) The application of SGLT2i can reduce the reabsorption of glucose and reduce the expensed ATP in Na+/K+ pumps, thereby decreasing the metabolic load of the EPO-related cells and improving the status of hypoxia. To a certain extent, such an improvement of hypoxia restores myofibroblasts back to erythropoietin-producing fibroblasts, leading to enhanced hematopoietic function and increased hematocrit (64, 66, 67), (4) Inhibition of SGLT2 will induce the activity of HIF and SIRT1. The HIF-2α activation in the kidney and liver can promote the production of erythropoietin (68) and strengthen the activation of SIRT1, resulting in modification of macrophage polarization which promotes anti-inflammatory phenotypes and impairs myocardial inflammation (69). (5) In vivo, it has also been observed that dapagliflozin can increase endogenous antioxidant enzymes and regulate fibrosis markers in renal tissues, thereby reducing the oxidative stress of rat kidneys.
Incessant contraction of the heart requires enough energy to support. Previous studies (70) have concluded that when the heart goes through the process of failing, abnormalities in cardiac energy metabolism, particularly in advanced stages, will occur, often manifested as increased glucose intake (mainly on anaerobic glycolysis) (71–73), reduced fatty acid oxidation, and decreased ketone oxidation, coincident with severely impaired mitochondrial function, leading to an overall depleting ATP content (More on the metabolic profile of the failing heart can be found in the reviews by Stanley et al. (70). Thus, it is essential to shift the substrate preference to satisfy such a pathological energy demand. However, such an alternation comes with a price—patients may be trapped in a vicious circle that worsens their more advanced heart failure.
The chief role SGLT2i plays in the metabolism of a failing heart is to improve the overall cardiac ATP production through the increased consumption of fatty acids, ketone bodies, and amino acids, with a concomitant decrease in glucose utilization (73, 74). Quantitatively, it can increase the overall ATP content by ~30% (75). Concerning glucose metabolism, SGLT2i-treated heart failure models show a reduced intake of cardiac glucose and a decreased level of metabolism-related enzymes, mainly shifting the substrate preference to ketone bodies, free fatty acids (73). Also, SGLT2i improves glucose tolerance and insulin resistance via several mechanisms such as inflammasome suppression (76), while the plasma insulin has been down-regulated and the glucagon shows a rise (37, 41, 77).
In mitochondrial dysfunction models, the improved potency of fatty acid oxidation, the reduced aggregation of fatty acid intermediates, as well as the improved synthetic efficiency of mitochondrial bioenergy are attributed to the function of SGLT2i, thereby enhancing the oxidation of the fatty acids in cardiac mitochondria and, as a result, avoiding mitochondrial failure (36, 78–80). A similar trend has also been observed in clinical trials (74). From another perspective, both in vitro and in vivo, SGLT2i has been confirmed to act as an activator of the AMPK signaling pathway to regulate metabolism, ameliorate inflammation, and maintain mitochondrial homeostasis (81–83).
SGLT2i can upregulate the plasma ketone levels, in part, mediated by the reduced plasma glucose levels and decreased insulin levels (41), coincident with a climb in free fatty acids (84, 85). As a result of the ongoing disorder of glucose, the failing heart becomes more dependent on ketone bodies to serve as extra fuel (86, 87). It will simulate a pseudo fasting state and give priority to the utilization of ketone bodies (29, 80, 88, 89). Such an effect of ketone bodies production is directly induced without other factors' mediation (90). SGLT2i also upregulates ketogenic enzymes and transporters in livers, kidneys, and intestines, which, accordingly, increases the level of beta-hydroxybutyric acid in the bodies and tissues (91). However, this promising hypothesis remains debatable and controversial due to the absence of further cogent and robust evidence (92). Some find that the efficiency of ketone bodies oxidation remains approximately unchanged under the application of SGLT2i and believe that the increased production of ATP benefits, in part, from the extra supplement of ketone bodies (75). Nevertheless, some researchers observe the contradictory results that bioavailability and oxidation have been increased by SGLT2i (93). This vague issue requires more research to tackle with. And such an increase in ketone bodies may, in turn, bring about worries about diabetic ketoacidosis, which is worthy of caution during its application.
As both a cause and a result of cardiovascular diseases, inflammatory cytokines, such as interleukin-1 (IL-1), interleukin-6 (IL-6), tumor necrosis factor (TNF), galectin-3, and others, have a very significant positive link with the pathophysiology and development of these conditions (94). The inflammatory profile of heart failure has been well-illustrated in the reviews by Murphy et al. (95) and Adamo et al. (96). Inflammasomes were initially reported as a kind of complex for caspase-activator (97). Moreover, now, they are referred to as multiprotein signaling complexes, including NOD-like receptor 1 (NLRP1), NOD-like receptor 3 (NLRP3), NOD-like receptor 6 (NLRP6), absent in melanoma 2 (AIM2), etc., which regulate inflammatory processes as well as anti-pathogen defenses (98, 99), inducing an increase in pro-inflammatory cytokines (100, 101). Those factors have a close connection with impaired blood vessels (102), disordered mitochondrial function (103), elevated oxidative stress, deteriorated hypertension (104), progressed atherosclerosis (99, 105), and advanced myocardial damage (106), etc.
Inflammatory cytokines and inflammasomes can both be suppressed by SGLT2i. To be more specific, it has been confirmed in rodent models that the crucial inflammasome NLRP3, a complex of caspase-1, IL-1β, and IL-18 cytokine-triggering factors (76, 101, 107), can be suppressed under the application of SGLT2i (108–110), which is, in part, calcium-mediated (105, 109). Several studies have shown that the AMPK pathway can restrain the increase of inflammasomes (111), and treatment with SGLT2i can bring about a climb of AMPK phosphorylation in LPS-treated cardio-fibroblasts (69), thereby avoiding the consequently augmented inflammation.
From another perspective, β-hydroxybutyrate also plays a significant role in regulating inflammasomes and inflammatory cytokines. Oscillated levels of β-hydroxybutyrate and insulin go hand in hand with inflammation (or inflammasome) levels. It has been established that SGLT2i can increase the β-hydroxybutyrate levels (discussed in the previous section), and this climb acts as a defender against endoplasmic reticulum stress-related inflammasomes via the activation of AMPK (111). However, some researchers believe this regulation is not AMPK-dependent. They reveal that the suppressive function of β-hydroxybutyrate on inflammasomes has nothing to do with fasting-related mechanisms such as AMPK, ROS, or other factors. Instead, it works by reducing the apoptosis-associated speck-like protein containing CARD (ASC) oligomerization and speck formation (112, 113), and this can be mediated through the G-protein-coupled receptor 109a (Gpr109a)–NLRP3 pathway associated with the increased influx of extracellular calcium (105). Furthermore, β-hydroxybutyrate is able to regulate mitochondrial protein acetylation, lowering the levels of NLRP3 and inflammatory cytokines, thus achieving the benefits in HFpEF models (103).
The most logical explanation for how SGLT2i reduces inflammation seems to be that inflammation is closely associated with hyperglycemia (114–117), and these negative effects will be lessened as soon as the glucose level falls. Recently, more attention has been paid to the regulation of insulin, which is regarded universally as an anti-inflammatory factor in most cases (118–120). However, recent studies report that in some specific circumstances, insulin can trigger the pro-inflammatory pattern of macrophages, thereby contributing to a climb in IL-β mediated by activated inflammasome and overproduction of ROS (121, 122). Such a function of insulin has been validated in a clinical setting by other research conducted by La Grotta et al. (123). Likewise, given the glucose-lowering function of SGLT2i, the insulin shows a decrease along with the dropped levels of blood glucose correspondingly, contributing to the avoidance of progressed inflammation and increased inflammasome. Moreover, alleviated insulin resistance, which has been mentioned above, also brings benefits to inflammation reduction, because the insulin resistance status possesses strong links with pro-inflammatory and inflammatory states (124–126). So, insulin-related mechanisms can at least partially mediate SGLT2i's ability to suppress inflammation.
Oxidative stress is a biological process characterized by high levels of free radicals that is accompanied by detrimental consequences (127, 128). It is well-known that a high level of blood glucose can induce oxidative stress intracellularly (129, 130), and such a stress-related function is negatively correlated with the level of HbA1c (131). SGLT2i can significantly contribute to the reduction of oxidative stress through a variety of mechanisms besides blood sugar lowering in its capacity as an anti-hyperglycemic drug.
Both in vivo and in vitro, it is reported that SGLT2i can attenuate cell apoptosis induced by endoplasmic reticulum stress (132–136). This process may be mediated by the curbed TGF-β/Smad pathway (137, 138) and the activated Nrf2/ARE signaling (139). Furthermore, SGLT2i use can eliminate intracellular ROS, block ROS-activated signaling pathways, and improve the hyperglycemic state in this process as well (140, 141). It can also normalize the size and quantity of mitochondria by regulating autophagy and reducing mitochondrial abnormalities attributed to myocardial infarction (142). What's more, SGLT2i is able to suppress inflammatory factors and oxidative stress by inhibiting the NO-sGC-cGMP pathway and curbing the polymerization of PKGIa (141, 143). Therefore, the methods above, as well as SGLT2i-mediated suppression of endoplasmic reticulum stress-induced apoptosis, can support cardioprotective function in heart failure.
In patients with T2DM, hyperglycemia and hyperinsulinemia often lead to sympathetic hyperexcitation, which consequently results in aggravated hypertension and an increased occurrence of other cardiovascular diseases (144–146). Contrary to our expectations, the heart rate, under the effects of those mechanisms above, including diuresis, decreased blood volume, and decreased blood pressure, has not shown a significant increase, suggesting that SGLT2i can reduce the heart rate of patients with a heart rate faster than 70 bpm (147). It may have the potential effect of inhibiting sympathetic nerve excitation. This role can be described as a “mediator” of SGLT2i to produce cardiovascular benefits.
In vivo, dapagliflozin significantly improves mice's blood pressure and endothelial function via reduced excitability of the sympathetic nervous system and down-regulated IL-6 (148). Interestingly, such a function goes hand in hand with the natriuretic effect of SGLT2i rather than its sugar-lowering function (149). At the same time, it is well-known that SGLT2 receptors are mainly enriched in the tissues of the central nervous system involved in autonomous regulation, and SGLT2i may affect the central nervous system in an uncharted way, exerting a sympathetic inhibition effect (51). In clinical trials of those who suffer from acute myocardial infarction with T2DM, the application of SGLT2i for 24 weeks improves sympathetic hyperexcitability and parasympathetic nerve function through changes in hemodynamics, myocardial energy metabolism, and function of the hepatic vagus nerve (150). On the other hand, from the perspective of cardiorenal syndrome, SGLT2i can also indirectly suppress sympathetic hyperexcitability by improving renal function.
However, there are limited clinical studies on how SGLT2i affects the sympathetic nervous system in the human body, and further clinical studies are required to be conducted.
It has been reported that SGLT2i can prevent and delay heart remodeling (135, 151–153), thereby protecting against the deterioration of heart failure.
In various animal models, SGLT2i shows the ability to prevent atrial remodeling, electrical remodeling (154, 155), and endothelial dysfunction (156–158). SGLT2i is confirmed to be capable of delaying the progression of left ventricular concentric hypertrophy in aorta cells by reducing sympathetic nerve tension and inflammation in the aorta (159). At the same time, the left ventricular systolic and diastolic functions have been enhanced, and the stiffness of cardiomyocytes has been improved (160, 161). Additionally, SGLT2i directly affects the phenotype and function of human cardiac myofibroblasts by weakening their activities and regulating cell-mediated collagen remodeling. Also, it can regulate the PERK-eIF2α-CHOP axis and activate the SIRT1 pathway (135, 162), thereby improving cardiac remodeling and heart failure.
On the other hand, SGLT2i can also exert a cardioprotective effect on patients suffering from myocardial infarction, preventing heart failure after that occurrence. It can reduce the infarct area (163, 164) independently of the blood sugar state, improving the heart function, remodeling, and metabolic state after myocardial infarction, etc. (93, 165, 166). However, cardiomyocytes chiefly express SGLT1 and seldom express SGLT2. It has been reported that specific knockdown of SGLT1 can reduce the infarct size in mice through function with EGFR (167), and SGLT1 knockdown can also ameliorate cardiac fibrosis and pyroptosis in vivo (168, 169), suggesting the potential ability of SGLT1 to provide cardiovascular benefits. So, is there a possibility that SGLT1/2i will have more beneficial impacts compared with SGLT2i? The SOLOIST-WHF trial shows that SGLT1/2 brings great benefits to the patients (which has also been detailed in Table 1), but whether SGLT1/2i is superior to SGLT2i or not remains unclear. Moreover, what is worth mentioning is that dual inhibitors of SGLT1/2 have been reported to exacerbate cardiac dysfunction after myocardial infarction in rats (170), which contradicts the assumption and clinical trials. The underlying reason for such a phenomenon requires additional preclinical research.
It has been revealed that empagliflozin can curb the activity of NHE1 (Na+/H+ Exchange Protein 1) in the hearts of mice, rats, and rabbits, thereby reducing the concentration of sodium and calcium in cardiomyocytes (171–173). Also, it can function as a direct cardiac effect to inhibit the NHE of the isolated intact heart and delay the occurrence of ischemic contracture of the heart in the absence of insulin (174). Similarly, empagliflozin can reduce the calcium sensitivity of human myocardial myofilament (175), improving the diastolic dysfunction of the human myocardium and inhibiting NHE in human bodies. By and large, SGLT2i successfully reduces the amount of Na+ entering cells by inhibiting NHE1 and improves the contractile dysfunction in heart failure by normalizing the intracellular pH, thereby producing cardiovascular benefits. However, there are some opposite voices that the activity of cardiac NHE1 will not be influenced by empagliflozin or other SGLT2i, as under the application of therapeutic doses, empagliflozin shows no regulatory effect on the concentration of Na+ (176). They believe that the role of SGLT2i in heart failure should not be interpreted as being mediated by myocardial NHE1 or intracellular Na+. In general, the function of SGLT2i for NHE is controversial since the clinical research data is very scarce, and more evidence is still required.
Adipose tissues secrete a large amount of biologically active substances collectively known as adipokines, which have been fully described in the reviews by Lelis et al. (177) and Kim et al. (178). Adipokines are in a harmonious balance—adiponectin and SFPR5 can down-regulate the expression of many pro-inflammatory mediators and prevent a variety of obesity-related endocrine or cardiovascular diseases (179, 180). Simultaneously, leptin can stimulate inflammatory responses and up-regulate pro-inflammatory elements such as TNF- and IL-6, hastening the onset and progression of cardiovascular disease (181, 182). Leptin and adiponectin exert contrary effects on subclinical inflammation and insulin resistance. Under the pathological condition of obesity, hypertrophic lipocytes and immunocytes in adipose tissue will activate and accelerate the chronic pro-inflammatory response and regulate the secretion of adipokines and other regulatory factors, with a deterioration of cardiometabolic diseases (183). Meta-analysis shows that SGLT2i can effectively reduce circulating leptin levels, increase circulating adiponectin levels (184), and inhibit the excretion of IL-6 and TNF-α in animal aortas (137, 159, 185), thereby producing cardiac vascular protection and delaying the progression of heart failure. Furthermore, SGLT2i can also reduce the size of epicardial adipose tissue (186) in patients by improving systemic micro-inflammation (187) and alleviating adipose-related vascular diseases (188), benefiting heart failure patients.
Myeloid angiogenic cells are abbreviated to MAC, which means vascular endothelial progenitor cells, and play a vital role in the pathological process of atherosclerosis. Its quantity is negatively correlated with cardiovascular events, serving as a biomarker (189). The regulation of SGLT2i on such progenitor cells expression is controversial, and some studies confirm that treatment with SGLT2i can up-regulate the expression of vascular progenitor cells (190). However, some studies believe that SGLT2i does not increase circulating stem cells and endothelial progenitor cells, while such an increase of endothelial progenitor cells may be indirectly achieved through the improvement of blood glucose level, and SGLT2i has no direct effect on endothelial progenitor cells (191, 192). In conclusion, the relationship between SGLT2i and endothelial progenitor cells is an emerging direction, and the results of existing studies are inconsistent and controversial.
With several clinical trials finishing in succession, it has been approved that SGLT2i will bring quite a few renal benefits (3–5, 193, 194), and those renal benefits will, synchronously, favor the treatment of heart failure. The dominant mechanism for its renal-protection is attributed to the tubuloglomerular feedback change under the application of SGLT2i (which has also been shown in Figure 1). Briefly speaking, when applied with SGLT2i, the reabsorption of Na+ and glucose will be impaired. Then, the macula dense can detect such changes, activating the tubuloglomerular feedback and inducing the contraction of afferent arteriole to maintain homeostasis. As a result, some pathological states, such as high perfusion, high load, and high filtration of the glomerulus, will be relieved, leading to sound renal protection (195, 196). From another perspective, it has been discussed in previous sections that SGLT2i can also be capable of reducing the kidney's metabolic burden and mitigating oxidative stress or inflammation at that site in vivo. The improved renal function will, in turn, bring benefits to heart failure via increased EPO generation, etc., thereby creating a virtuous cycle for cardiorenal interaction.
With the advent of SGLT2i, a breakthrough in the treatment of cardiovascular diseases has appeared. At this stage, SGLT2i has been applied clinically and has been accepted in a variety of guidelines. By and large, SGLT2i achieves its cardiovascular outcomes and benefits patients with heart failure through combined effects of multiple ways (which has been visualized in Figure 3), which means it is hard to fully explain its rapid and comprehensive benefits only by one of those mechanisms. Nevertheless, some of those mechanisms mentioned above are still uncertain, while others are contradictory and controversial, like Na+-H+ exchange protein and myeloid angiogenic cells. Thus, further studies on cells, animals, and clinics ought to be supplemented. It is promising that in-depth research will further open up more paths and hold great promise for cardiovascular diseases, including heart failure, myocardial hypertrophy, and even treatments for comorbidity.
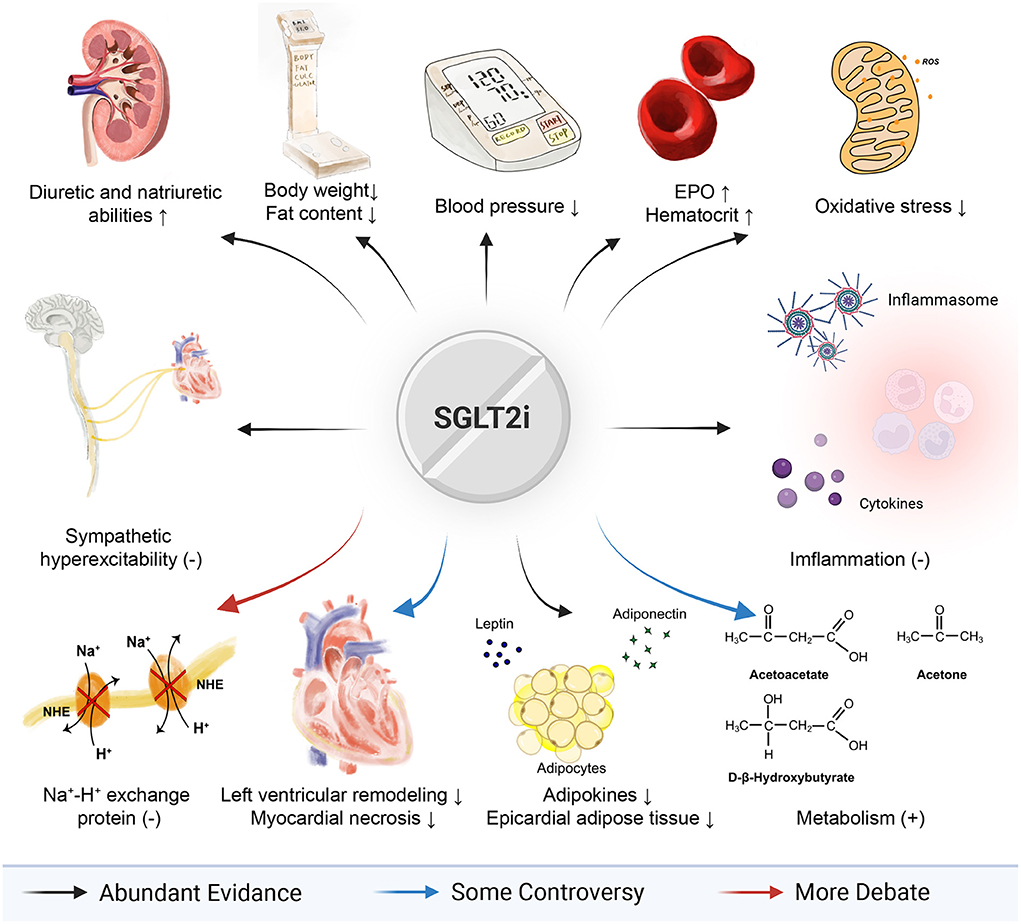
Figure 3. Full view of SGLT2i in heart failure.
JL was responsible for paper collecting and article writing. LZ was responsible for the revision of this article. HG took responsibility of ideas generation and suggestion making. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Natural Science Foundation of Shanghai (No. 19JC1415703).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Savarese G, Lund LH. Global public health burden of heart failure. Card Fail Rev. (2017) 3:7–11. doi: 10.15420/cfr.2016:25:2
2. McMurray JJ, Pfeffer MA. Heart failure. Lancet. (2005) 365:1877–89. doi: 10.1016/S0140-6736(05)66621-4
3. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
4. Neal B, Perkovic V, Mahaffey KW, De Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
5. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2019) 380:347–57. doi: 10.1056/NEJMoa1812389
6. McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. (2019) 381:1995–2008. doi: 10.1056/NEJMoa1911303
7. Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Böhm M, et al. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. (2021) 385:1451–61. doi: 10.1056/NEJMoa2107038
8. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. AHA/ACC/HFSA guideline for the management of heart failure: a report of the american college of cardiology/American heart association joint committee on clinical practice guidelines. Circulation. (2022) 145:e895–1032. doi: 10.1161/CIR.0000000000001073
9. Himsworth HP. The relation of glycosuria to glycaemia and the determination of the renal threshold for glucose. Biochem J. (1931) 25:1128–46. doi: 10.1042/bj0251128
10. Mackay RL. Observations on the renal threshold for glucose. Biochem J. (1927) 21:760–4. doi: 10.1042/bj0210760
11. Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. (2011) 91:733–94. doi: 10.1152/physrev.00055.2009
12. Han L, Qu Q, Aydin D, Panova O, Robertson MJ, Xu Y, et al. Structure and mechanism of the SGLT family of glucose transporters. Nature. (2022) 601:274–9. doi: 10.1038/s41586-021-04211-w
13. Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dais S, De Simone G, et al., Heart disease and stroke statistics−2010 update: a report from the American Heart Association. Circulation. (2010) 121:e46–215. doi: 10.1161/CIRCULATIONAHA.109.192667
14. Zannad F. Aldosterone and heart failure. Eur Heart J. (1995) 16:98–102. doi: 10.1093/eurheartj/16.suppl_N.98
15. Nijst P, Verbrugge FH, Grieten L, Dupont M, Steels P, Tang WHW, et al. The pathophysiological role of interstitial sodium in heart failure. J Am Coll Cardiol. (2015) 65:378–88. doi: 10.1016/j.jacc.2014.11.025
16. Bakris GL, Fonseca VA, Sharma K, Wright EM. Renal sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Kidney Int. (2009) 75:1272–7. doi: 10.1038/ki.2009.87
17. Packer M, Anker SD, Butler J, Filippatos G, Ferreira JP, Pocock SJ, et al. Empagliflozin in patients with heart failure, reduced ejection fraction, and volume overload: EMPEROR-reduced trial. J Am Coll Cardiol. (2021) 77:1381–92. doi: 10.1016/j.jacc.2021.01.033
18. Nassif ME, Qintar M, Windsor SL, Jermyn R, Shavelle DM, Tang F, et al. Empagliflozin effects on pulmonary artery pressure in patients with heart failure: results from the EMBRACE-HF trial. Circulation. (2021) 143:1673–86. doi: 10.1161/CIRCULATIONAHA.120.052503
19. Kannenkeril D, Karg MV, Bosch A, Ott C, Linz P, Nagel AM, et al. Tissue sodium content in patients with type 2 diabetes mellitus. J Diabetes Complications. (2019) 33:485–9. doi: 10.1016/j.jdiacomp.2019.04.006
20. Karg MV, Bosch A, Kannenkeril D, Striepe K, Ott C, Schneider MP, et al. SGLT-2-inhibition with dapagliflozin reduces tissue sodium content: a randomised controlled trial. Cardiovasc Diabetol. (2018) 17:5. doi: 10.1186/s12933-017-0654-z
21. Boorsma EM, Beusekamp JC, Ter Maaten JM, Figarska SM, Danser AHJ, van Veldhuisen DJ, et al. Effects of empagliflozin on renal sodium and glucose handling in patients with acute heart failure. Eur J Heart Fail. (2021) 23:68–78. doi: 10.1002/ejhf.2066
22. Haykowsky MJ, Nicklas BJ, Brubaker PH, Hundley WG, Brinkley TE, Upadhya B, et al. Regional adipose distribution and its relationship to exercise intolerance in older obese patients who have heart failure with preserved ejection fraction. JACC Heart Fail. (2018) 6:640–9. doi: 10.1016/j.jchf.2018.06.002
23. Palmer SC, Tendal B, Mustafa RA, Vandvik PO, Li S, Hao Q, et al. Sodium-glucose cotransporter protein-2 (SGLT-2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists for type 2 diabetes: systematic review and network meta-analysis of randomised controlled trials. BMJ. (2021) 372:m4573. doi: 10.1136/bmj.m4573
24. Tsapas A, Karagiannis T, Kakotrichi P, Avgerinos I, Mantsiou C, Tousinas G, et al. Comparative efficacy of glucose-lowering medications on body weight and blood pressure in patients with type 2 diabetes: a systematic review and network meta-analysis. Diabetes Obes Metab. (2021) 23:2116–24. doi: 10.1111/dom.14451
25. Inzucchi SE, Davies MJ, Khunti K, Trivedi P, George JT, Zwiener I, et al. Empagliflozin treatment effects across categories of baseline HbA1c, body weight and blood pressure as an add-on to metformin in patients with type 2 diabetes. Diabetes Obes Metab. (2021) 23:425–33. doi: 10.1111/dom.14234
26. Adamson C, Jhund PS, Docherty KF, Bělohlávek J, Chiang CE, Diez M, et al. Efficacy of dapagliflozin in heart failure with reduced ejection fraction according to body mass index. Eur J Heart Fail. (2021) 23:1662–72. doi: 10.1002/ejhf.2308
27. Matsuba I, Takihata M, Takai M, Maeda H, Kubota A, Iemitsu K, et al. Effects of 1-year treatment with canagliflozin on body composition and total body water in patients with type 2 diabetes. Diabetes Obes Metab. (2021) 23:2614–22. doi: 10.1111/dom.14508
28. Kullmann S, Hummel J, Wagner R, Dannecker C, Vosseler A, Fritsche L, et al. Empagliflozin improves insulin sensitivity of the hypothalamus in humans with prediabetes: a randomized, double-blind, placebo-controlled, phase 2 trial. Diabetes Care. (2022) 45:398–406. doi: 10.2337/dc21-1136
29. Gaborit B, Ancel P, Abdullah AE, Maurice F, Abdesselam I, Calen A, et al. Effect of empagliflozin on ectopic fat stores and myocardial energetics in type 2 diabetes: the EMPACEF study. Cardiovasc Diabetol. (2021) 20:57. doi: 10.1186/s12933-021-01237-2
30. Koshizaka M, Ishikawa K, Ishibashi R, Takahashi S, Sakamoto K, Yokoh H, et al. Comparison of visceral fat reduction by ipragliflozin and metformin in elderly type 2 diabetes patients: sub-analysis of a randomized-controlled study. Diabetes Ther. (2021) 12:183–96. doi: 10.1007/s13300-020-00949-0
31. Kahl S, Gancheva S, Straßburger K, Herder C, Machann J, Katsuyama H, et al. Empagliflozin effectively lowers liver fat content in well-controlled type 2 diabetes: a randomized, double-blind, phase 4, placebo-controlled trial. Diabetes Care. (2020) 43:298–305. doi: 10.2337/dc19-0641
32. Nasiri-Ansari N, Nikolopoulou C, Papoutsi K, Kyrou I, Mantzoros CS, Kyriakopoulos G, et al. Empagliflozin attenuates non-alcoholic fatty liver disease (NAFLD) in high fat diet fed ApoE (-/-) mice by activating autophagy and reducing ER stress and apoptosis. Int J Mol Sci. (2021) 22:818. doi: 10.3390/ijms22020818
33. Ghosh A, Dutta K, Bhatt SP, Gupta R, Tyagi K, Ansari IA, et al. Dapagliflozin improves body fat patterning, and hepatic and pancreatic fat in patients with type 2 diabetes in North India. J Clin Endocrinol Metab. (2022) 107:e2267–75. doi: 10.1210/clinem/dgac138
34. Mori Y, Terasaki M, Hiromura M, Saito T, Kushima H, Koshibu M, et al. Luseogliflozin attenuates neointimal hyperplasia after wire injury in high-fat diet-fed mice via inhibition of perivascular adipose tissue remodeling. Cardiovasc Diabetol. (2019) 18:143. doi: 10.1186/s12933-019-0947-5
35. Yang X, Liu Q, Li Y, Tang Q, Wu T, Chen L, et al. The diabetes medication canagliflozin promotes mitochondrial remodelling of adipocyte via the AMPK-Sirt1-Pgc-1α signalling pathway. Adipocyte. (2020) 9:484–94. doi: 10.1080/21623945.2020.1807850
36. Lee JY, Lee M, Lee JY, Bae J, Shin E, Lee YH, et al. Ipragliflozin, an SGLT2 inhibitor, ameliorates high-fat diet-induced metabolic changes by upregulating energy expenditure through activation of the AMPK/SIRT1 pathway. Diabetes Metab J. (2021) 2020:187. doi: 10.4093/dmj.2020.0187
37. Yang X, Liu Q, Li Y, Ding Y, Zhao Y, Tang Q, et al. Inhibition of the sodium-glucose co-transporter SGLT2 by canagliflozin ameliorates diet-induced obesity by increasing intra-adipose sympathetic innervation. Br J Pharmacol. (2021) 178:1756–71. doi: 10.1111/bph.15381
38. Matthews JR, Herat LY, Magno AL, Gorman S, Schlaich MP, Matthews VB, et al. SGLT2 inhibitor-induced sympathoexcitation in white adipose tissue: a novel mechanism for beiging. Biomedicines. (2020) 8:514. doi: 10.3390/biomedicines8110514
39. Li Y, Wang D, Ping X, Zhang Y, Zhang T, Wang L, et al. Local hyperthermia therapy induces browning of white fat and treats obesity. Cell. (2022) 185:949–66. doi: 10.1016/j.cell.2022.02.004
40. Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, et al. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab. (2014) 20:670–7. doi: 10.1016/j.cmet.2014.07.012
41. Osataphan S, Macchi C, Singhal G, Chimene-Weiss J, Sales V, Kozuka C, et al. SGLT2 inhibition reprograms systemic metabolism via FGF21-dependent and -independent mechanisms. JCI Insight. (2019) 4:123130. doi: 10.1172/jci.insight.123130
42. Douris N, Stevanovic DM, Fisher FM, Cisu TI, Chee MJ, Nguyen NL, et al. Central fibroblast growth factor 21 browns white fat via sympathetic action in male mice. Endocrinology. (2015) 156:2470–81. doi: 10.1210/en.2014-2001
43. Yoshida A, Matsubayashi Y, Nojima T, Suganami H, Abe T, Ishizawa M, et al. Attenuation of weight loss through improved antilipolytic effect in adipose tissue via the sglt2 inhibitor tofogliflozin. J Clin Endocrinol Metab. (2019) 104:3647–60. doi: 10.1210/jc.2018-02254
44. Ferrannini G, Hach T, Crowe S, Sanghvi A, Hall KD, Ferrannini E, et al. energy balance after sodium-glucose cotransporter 2 inhibition. Diabetes Care. (2015) 38:1730–5. doi: 10.2337/dc15-0355
45. Kosugi R, Nakatani E, Okamoto K, Aoshima S, Arai H, Inoue T, et al. Effects of sodium-glucose cotransporter 2 inhibitor (dapagliflozin) on food intake and plasma fibroblast growth factor 21 levels in type 2 diabetes patients. Endocr J. (2019) 66:677–82. doi: 10.1507/endocrj.EJ19-0013
46. Devenny JJ, Godonis HE, Harvey SJ, Rooney S, Cullen MJ, Pelleymounter MA, et al. Weight loss induced by chronic dapagliflozin treatment is attenuated by compensatory hyperphagia in diet-induced obese (DIO) rats. Obesity. (2012) 20:1645–52. doi: 10.1038/oby.2012.59
47. Matsuba I, Kanamori A, Takihata M, Takai M, Maeda H, Kubota A, et al. Canagliflozin increases calorie intake in type 2 diabetes without changing the energy ratio of the three macronutrients: CANA-K study. Diabetes Technol Ther. (2020) 22:228–34. doi: 10.1089/dia.2019.0372
48. Scholtes RA, Muskiet MHA, van Baar MJB, Hesp AC, Greasley PJ, Karlsson C, et al. Natriuretic effect of two weeks of dapagliflozin treatment in patients with type 2 diabetes and preserved kidney function during standardized sodium intake: results of the DAPASALT trial. Diabetes Care. (2021) 44:440–7. doi: 10.2337/dc20-2604
49. Kario K, Okada K, Kato M, Nishizawa M, Yoshida T, Asano T, et al. 24-hour blood pressure-lowering effect of an sglt-2 inhibitor in patients with diabetes and uncontrolled nocturnal hypertension: results from the randomized, placebo-controlled SACRA study. Circulation. (2018) 139:2089–97. doi: 10.1161/CIRCULATIONAHA.118.037076
50. Kim S, Jo CH, Kim GH. Effects of empagliflozin on nondiabetic salt-sensitive hypertension in uninephrectomized rats. Hypertens Res. (2019) 42:1905–15. doi: 10.1038/s41440-019-0326-3
51. Nguyen T, Wen S, Gong M, Yuan X, Xu D, Wang C, et al. Dapagliflozin activates neurons in the central nervous system and regulates cardiovascular activity by inhibiting SGLT-2 in mice. Diabetes Metab Syndr Obes. (2020) 13:2781–99. doi: 10.2147/DMSO.S258593
52. Reverte V, Rodriguez F, Oltra L, Moreno JM, Llinás MT, Shea CM, et al. SGLT2 inhibition potentiates the cardiovascular, renal, and metabolic effects of sGC stimulation in hypertensive rats with prolonged exposure to high-fat diet. Am J Physiol Heart Circ Physiol. (2022) 322:H523–36. doi: 10.1152/ajpheart.00386.2021
53. Mazidi M, Rezaie P, Gao HK, Kengne AP. Effect of sodium-glucose cotransport-2 inhibitors on blood pressure in people with type 2 diabetes mellitus: a systematic review and meta-analysis of 43 randomized control trials with 22 528 patients. J Am Heart Assoc. (2017) 6:e004007. doi: 10.1161/JAHA.116.004007
54. Baker WL, Buckley LF, Kelly MS, Bucheit JD, Parod ED, Brown R, et al. Effects of sodium-glucose cotransporter 2 inhibitors on 24-hour ambulatory blood pressure: a systematic review and meta-analysis. J Am Heart Assoc. (2017) 6:e005686 doi: 10.1161/JAHA.117.005686
55. Lappin TR, Lee FS. Update on mutations in the HIF: EPO pathway and their role in erythrocytosis. Blood Rev. (2019) 37:100590. doi: 10.1016/j.blre.2019.100590
56. Koury MJ. Erythropoietin: the story of hypoxia and a finely regulated hematopoietic hormone. Exp Hematol. (2005) 33:1263–70. doi: 10.1016/j.exphem.2005.06.031
57. Salisbury AC, Kosiborod M. Outcomes associated with anemia in patients with heart failure. Heart Fail Clin. (2010) 6:359–72. doi: 10.1016/j.hfc.2010.03.005
58. Aronson D, Suleiman M, Agmon Y, Suleiman A, Blich M, Kapeliovich M, et al. Changes in haemoglobin levels during hospital course and long-term outcome after acute myocardial infarction. Eur Heart J. (2007) 28:1289–96. doi: 10.1093/eurheartj/ehm013
59. Anand I, McMurray JJ, Whitmore J, Warren M, Pham A, McCamish MA, et al. Anemia and its relationship to clinical outcome in heart failure. Circulation. (2004) 110:149–54. doi: 10.1161/01.CIR.0000134279.79571.73
60. Kishimoto S, Maruhashi T, Kajikawa M, Matsui S, Hashimoto H, Takaeko Y, et al. Hematocrit, hemoglobin and red blood cells are associated with vascular function and vascular structure in men. Sci Rep. (2020) 10:11467. doi: 10.1038/s41598-020-68319-1
61. Mazer CD, Hare GMT, Connelly PW, Gilbert RE, Shehata N, Quan A, et al. Effect of empagliflozin on erythropoietin levels, iron stores, and red blood cell morphology in patients with type 2 diabetes mellitus and coronary artery disease. Circulation. (2020) 141:704–7. doi: 10.1161/CIRCULATIONAHA.119.044235
62. Aberle J, Menzen M, Schmid SM, Terkamp C, Jaeckel E, Rohwedder K, et al. Dapagliflozin effects on haematocrit, red blood cell count and reticulocytes in insulin-treated patients with type 2 diabetes. Sci Rep. (2020) 10:22396. doi: 10.1038/s41598-020-78734-z
63. Ferreira JP, Anker SD, Butler J, Filippatos G, Iwata T, Salsali A, et al. Impact of anaemia and the effect of empagliflozin in heart failure with reduced ejection fraction: findings from EMPEROR-Reduced. Eur J Heart Fail. (2021) 2021:ejhf.2409. doi: 10.1002/ejhf.2409
64. Thiele K, Rau M, Hartmann NK, Möllmann J, Jankowski J, Böhm M, et al. Effects of empagliflozin on erythropoiesis in patients with type 2 diabetes: data from a randomized, placebo-controlled study. Diabetes Obes Metab. (2021) 23:2814–8. doi: 10.1111/dom.14517
65. Ghanim H, Abuaysheh S, Hejna J, Green K, Batra M, Makdissi A, et al. Dapagliflozin suppresses hepcidin and increases erythropoiesis. J Clin Endocrinol Metab. (2020) 105:e1056–63. doi: 10.1210/clinem/dgaa057
66. Sano M. and Goto S. Possible mechanism of hematocrit elevation by sodium glucose cotransporter 2 inhibitors and associated beneficial renal and cardiovascular effects. Circulation. (2019) 139:1985–7. doi: 10.1161/CIRCULATIONAHA.118.038881
67. Sano M, Takei M, Shiraishi Y, Suzuki Y. Increased hematocrit during sodium-glucose cotransporter 2 inhibitor therapy indicates recovery of tubulointerstitial function in diabetic kidneys. J Clin Med Res. (2016) 8:844–7. doi: 10.14740/jocmr2760w
68. Packer M. Critical examination of mechanisms underlying the reduction in heart failure events with SGLT2 inhibitors: identification of a molecular link between their actions to stimulate erythrocytosis and to alleviate cellular stress. Cardiovasc Res. (2021) 117:74–84. doi: 10.1093/cvr/cvaa064
69. Ye Y, Bajaj M, Yang HC, Perez-Polo JR, Birnbaum Y. SGLT-2 inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Cardiovasc Drugs Ther. (2017) 31:119–32. doi: 10.1007/s10557-017-6725-2
70. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. (2005) 85:1093–129. doi: 10.1152/physrev.00006.2004
71. Allard MF, Schönekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. (1994) 267:H742–50. doi: 10.1152/ajpheart.1994.267.2.H742
72. Kagaya Y, Kanno Y, Takeyama D, Ishide N, Maruyama Y, Takahashi T, et al. Effects of long-term pressure overload on regional myocardial glucose and free fatty acid uptake in rats. A quantitative autoradiographic study. Circulation. (1990) 81:1353–61. doi: 10.1161/01.CIR.81.4.1353
73. Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, Picatoste B, et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol. (2019) 73:1931–44. doi: 10.1016/j.jacc.2019.01.056
74. Op DEN, Kamp YJM, De Ligt M, Dautzenberg B, Kornips E, Esterline R, et al. Effects of the SGLT2 inhibitor dapagliflozin on energy metabolism in patients with type 2 diabetes: a randomized, double-blind crossover trial. Diabetes Care. (2021) 44:1334–43. doi: 10.2337/dc20-2887
75. Verma S, Rawat S, Ho KL, Wagg CS, Zhang L, Teoh H, et al. Empagliflozin increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC Basic Transl Sci. (2018) 3:575–87. doi: 10.1016/j.jacbts.2018.07.006
76. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. (2011) 17:179–88. doi: 10.1038/nm.2279
77. Swe MT, Thongnak L, Jaikumkao K, Pongchaidecha A, Chatsudthipong V, Lungkaphin A, et al. Dapagliflozin attenuates renal gluconeogenic enzyme expression in obese rats. J Endocrinol. (2020) 245:193–205. doi: 10.1530/JOE-19-0480
78. Makrecka-Kuka M, Korzh S, Videja M, Vilks K, Cirule H, Kuka J, et al. Empagliflozin protects cardiac mitochondrial fatty acid metabolism in a mouse model of diet-induced lipid overload. Cardiovasc Drugs Ther. (2020) 34:791–7. doi: 10.1007/s10557-020-06989-9
79. Trang NN, Chung CC, Lee TW, Cheng WL, Kao YH, Huang SY, et al. Empagliflozin and liraglutide differentially modulate cardiac metabolism in diabetic cardiomyopathy in rats. Int J Mol Sci. (2021) 22:1177. doi: 10.3390/ijms22031177
80. Wallenius K, Kroon T, Hagstedt T, Löfgren L, Sörhede-Winzell M, Boucher J, et al. The SGLT2 inhibitor dapagliflozin promotes systemic FFA mobilization, enhances hepatic β-oxidation, and induces ketosis. J Lipid Res. (2022) 63:100176. doi: 10.1016/j.jlr.2022.100176
81. Koyani CN, Plastira I, Sourij H, Hallström S, Schmidt A, Rainer PP, et al. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol Res. (2020) 158:104870. doi: 10.1016/j.phrs.2020.104870
82. Sun X, Han F, Lu Q, Li X, Ren D, Zhang J, et al. Empagliflozin ameliorates obesity-related cardiac dysfunction by regulating sestrin2-mediated AMPK-mTOR signaling and redox homeostasis in high-fat diet-induced obese mice. Diabetes. (2020) 69:1292–305. doi: 10.2337/db19-0991
83. Liu X, Xu C, Xu L, Li X, Sun H, Xue M, et al. Empagliflozin improves diabetic renal tubular injury by alleviating mitochondrial fission via AMPK/SP1/PGAM5 pathway. Metabolism. (2020) 111:154334. doi: 10.1016/j.metabol.2020.154334
84. Daniele G, Xiong J, Solis-Herrera C, Merovci A, Eldor R, Tripathy D, et al. Dapagliflozin enhances fat oxidation and ketone production in patients with type 2 diabetes. Diabetes Care. (2016) 39:2036–41. doi: 10.2337/dc15-2688
85. Merovci A, Solis-Herrera C, Daniele G, Eldor R, Fiorentino TV, Tripathy D, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. (2014) 124:509–14. doi: 10.1172/JCI70704
86. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, et al. The failing heart relies on ketone bodies as a fuel. Circulation. (2016) 133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355
87. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, et al, Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. (2016) 133:706–16. doi: 10.1161/CIRCULATIONAHA.115.017545
88. Garcia-Ropero A, Santos-Gallego CG, Zafar MU, Badimon JJ. Metabolism of the failing heart and the impact of SGLT2 inhibitors. Expert Opin Drug Metab Toxicol. (2019) 15:275–85. doi: 10.1080/17425255.2019.1588886
89. Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME trial: a “thrifty substrate” hypothesis. Diabetes Care. (2016) 39:1108–14. doi: 10.2337/dc16-0330
90. Capozzi ME, Coch RW, Koech J, Astapova II, Wait JB, Encisco SE, et al. The limited role of glucagon for ketogenesis during fasting or in response to SGLT2 inhibition. Diabetes. (2020) 69:882–92. doi: 10.2337/db19-1216
91. Kim JH, Lee M, Kim SH, Kim SR, Lee BW, Kang ES, et al. Sodium-glucose cotransporter 2 inhibitors regulate ketone body metabolism via inter-organ crosstalk. Diabetes Obes Metab. (2019) 21:801–11. doi: 10.1111/dom.13577
92. Lopaschuk GD, Verma S. Empagliflozin's fuel hypothesis: not so soon. Cell Metab. (2016) 24:200–2. doi: 10.1016/j.cmet.2016.07.018
93. Yurista SR, Silljé HHW, Oberdorf-Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, et al. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail. (2019) 21:862–73. doi: 10.1002/ejhf.1473
94. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
95. Murphy SP, Kakkar R, McCarthy CP, Januzzi JL Jr. Inflammation in heart failure: JACC state-of-the-art review. J Am Coll Cardiol. (2020) 75:1324–40. doi: 10.1016/j.jacc.2020.01.014
96. Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. (2020) 17:269–85. doi: 10.1038/s41569-019-0315-x
97. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
98. Deets KA, Vance RE. Inflammasomes and adaptive immune responses. Nat Immunol. (2021) 22:412–22. doi: 10.1038/s41590-021-00869-6
99. Schunk SJ, Kleber ME, März W, Pang S, Zewinger S, Triem S, et al. Genetically determined NLRP3 inflammasome activation associates with systemic inflammation and cardiovascular mortality. Eur Heart J. (2021) 42:1742–56. doi: 10.1093/eurheartj/ehab107
100. Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity. (2013) 39:432–41. doi: 10.1016/j.immuni.2013.08.037
101. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. (2013) 13:397–411. doi: 10.1038/nri3452
102. Bruder-Nascimento T, Ferreira NS, Zanotto CZ, Ramalho F, Pequeno IO, Olivon VC, et al. NLRP3 inflammasome mediates aldosterone-induced vascular damage. Circulation. (2016) 134:1866–80. doi: 10.1161/CIRCULATIONAHA.116.024369
103. Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, et al. Targeting mitochondria-inflammation circuit by β-hydroxybutyrate mitigates HFpEF. Circ Res. (2021) 128:232–45. doi: 10.1161/CIRCRESAHA.120.317933
104. Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B, et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med. (2017) 23:174–84. doi: 10.1038/nm.4267
105. Zhang SJ, Li ZH, Zhang YD, Chen J, Li Y, Wu FQ, et al. Ketone body 3-hydroxybutyrate ameliorates atherosclerosis via receptor gpr109a-mediated calcium influx. Adv Sci. (2021) 8:2003410. doi: 10.1002/advs.202003410
106. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. (2018) 15:203–14. doi: 10.1038/nrcardio.2017.161
107. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. (2010) 11:897–904. doi: 10.1038/ni.1935
108. Philippaert K, Kalyaanamoorthy S, Fatehi M, Long W, Soni S, Byrne NJ, et al. Cardiac late sodium channel current is a molecular target for the sodium/glucose cotransporter 2 inhibitor empagliflozin. Circulation. (2021) 143:2188–204. doi: 10.1161/CIRCULATIONAHA.121.053350
109. Byrne NJ, Matsumura N, Maayah ZH, Ferdaoussi M, Takahara S, Darwesh AM, et al. Empagliflozin blunts worsening cardiac dysfunction associated with reduced NLRP3 (nucleotide-binding domain-like receptor protein 3) inflammasome activation in heart failure. Circ Heart Fail. (2020) 13:e006277. doi: 10.1161/CIRCHEARTFAILURE.119.006277
110. Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. (2020) 11:2127. doi: 10.1038/s41467-020-15983-6
111. Bae HR, Kim DH, Park MH, Lee B, Kim MJ, Lee EK, et al. β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget. (2016) 7:66444–54. doi: 10.18632/oncotarget.12119
112. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. (2011) 12:408–15. doi: 10.1038/ni.2022
113. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. (2015) 21:263–9. doi: 10.1038/nm.3804
114. Forbes JM, Cooper ME, Thallas V, Burns WC, Thomas MC, Brammar GC, et al. Reduction of the accumulation of advanced glycation end products by ACE inhibition in experimental diabetic nephropathy. Diabetes. (2002) 51:3274–82. doi: 10.2337/diabetes.51.11.3274
115. Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol. (2009) 20:742–52. doi: 10.1681/ASN.2008050514
116. Kang R, Tang D, Lotze MT, Zeh HJ. RAGE regulates autophagy and apoptosis following oxidative injury. Autophagy. (2011) 7:442–4. doi: 10.4161/auto.7.4.14681
117. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. (2000) 404:787–90. doi: 10.1038/35008121
118. Dandona P, Aljada A, Mohanty P. The anti-inflammatory and potential anti-atherogenic effect of insulin: a new paradigm. Diabetologia. (2002) 45:924–30. doi: 10.1007/s00125-001-0766-5
119. Chaudhuri A, Janicke D, Wilson MF, Tripathy D, Garg R, Bandyopadhyay A, et al. Anti-inflammatory and profibrinolytic effect of insulin in acute ST-segment-elevation myocardial infarction. Circulation. (2004) 109:849–54. doi: 10.1161/01.CIR.0000116762.77804.FC
120. Aljada A, Ghanim H, Saadeh R, Dandona P. Insulin inhibits NFkappaB and MCP-1 expression in human aortic endothelial cells. J Clin Endocrinol Metab. (2001) 86:450–3. doi: 10.1210/jcem.86.1.7278
121. Dror E, Dalmas E, Meier DT, Wueest S, Thévenet J, Thienel C, et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol. (2017) 18:283–92. doi: 10.1038/ni.3659
122. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. (2010) 11:136–40. doi: 10.1038/ni.1831
123. La Grotta R, de Candia P, Olivieri F, Matacchione G, Giuliani A, Rippo MR, et al. Anti-inflammatory effect of SGLT-2 inhibitors via uric acid and insulin. Cell Mol Life Sci. (2022) 79:273. doi: 10.1007/s00018-022-04289-z
124. Donath MY, Størling J, Maedler K, Mandrup-Poulsen T. Inflammatory mediators and islet beta-cell failure: a link between type 1 and type 2 diabetes. J Mol Med. (2003) 81:455–70. doi: 10.1007/s00109-003-0450-y
125. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. (2006) 116:1793–801. doi: 10.1172/JCI29069
126. Shimobayashi M, Albert V, Woelnerhanssen B, Frei IC, Weissenberger D, Meyer-Gerspach AC, et al. Insulin resistance causes inflammation in adipose tissue. J Clin Invest. (2018) 128:1538–50. doi: 10.1172/JCI96139
127. Ing DJ, Zang J, Dzau VJ, Webster KA, Bishopric NH. Modulation of cytokine-induced cardiac myocyte apoptosis by nitric oxide, Bak, and Bcl-x. Circ Res. (1999) 84:21–33. doi: 10.1161/01.RES.84.1.21
128. Cheng XS, Shimokawa H, Momii H, Oyama J, Fukuyama N, Egashira K, et al. Role of superoxide anion in the pathogenesis of cytokine-induced myocardial dysfunction in dogs in vivo. Cardiovasc Res. (1999) 42:651–9. doi: 10.1016/S0008-6363(98)00317-4
129. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. (2001) 414:813–20. doi: 10.1038/414813a
130. Sottero B, Gargiulo S, Russo I, Barale C, Poli G, Cavalot F, et al. Postprandial dysmetabolism and oxidative stress in type 2 diabetes: pathogenetic mechanisms and therapeutic strategies. Med Res Rev. (2015) 35:968–1031. doi: 10.1002/med.21349
131. Steven S, Frenis K, Oelze M, Kalinovic S, Kuntic M, Bayo Jimenez MT, et al. Vascular inflammation and oxidative stress: major triggers for cardiovascular disease. Oxid Med Cell Longev. (2019) 2019:7092151. doi: 10.1155/2019/7092151
132. Shibusawa R, Yamada E, Okada S, Nakajima Y, Bastie CC, Maeshima A, et al. Dapagliflozin rescues endoplasmic reticulum stress-mediated cell death. Sci Rep. (2019) 9:9887. doi: 10.1038/s41598-019-46402-6
133. Shih JY, Lin YW, Fisch S, Cheng JT, Kang NW, Hong CS, et al. Dapagliflozin suppresses ER stress and improves subclinical myocardial function in diabetes: from bedside to bench. Diabetes. (2020) 70:262–7. doi: 10.2337/db20-0840
134. Lahnwong S, Palee S, Apaijai N, Sriwichaiin S, Kerdphoo S, Jaiwongkam T, et al. Acute dapagliflozin administration exerts cardioprotective effects in rats with cardiac ischemia/reperfusion injury. Cardiovasc Diabetol. (2020) 19:91. doi: 10.1186/s12933-020-01066-9
135. Ren FF, Xie ZY, Jiang YN, Guan X, Chen QY, Lai TF, et al. Dapagliflozin attenuates pressure overload-induced myocardial remodeling in mice via activating SIRT1 and inhibiting endoplasmic reticulum stress. Acta Pharmacol Sin. (2021) 43:1721–32. doi: 10.1038/s41401-021-00805-2
136. Olgar Y, Tuncay E, Degirmenci S, Billur D, Dhingra R, Kirshenbaum L, et al. Ageing-associated increase in SGLT2 disrupts mitochondrial/sarcoplasmic reticulum Ca(2+) homeostasis and promotes cardiac dysfunction. J Cell Mol Med. (2020) 24:8567–78. doi: 10.1111/jcmm.15483
137. Shentu Y, Li Y, Xie S, Jiang H, Sun S, Lin R, et al. Empagliflozin, a sodium glucose cotransporter-2 inhibitor, ameliorates peritoneal fibrosis via suppressing TGF-β/Smad signaling. Int Immunopharmacol. (2021) 93:107374. doi: 10.1016/j.intimp.2021.107374
138. Tian J, Zhang M, Suo M, Liu D, Wang X, Liu M, et al. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J Cell Mol Med. (2021) 25:7642–59. doi: 10.1111/jcmm.16601
139. Li C, Zhang J, Xue M, Li X, Han F, Liu X, et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol. (2019) 18:15. doi: 10.1186/s12933-019-0816-2
140. El-Daly M, Pulakazhi Venu VK, Saifeddine M, Mihara K, Kang S, Fedak PWM, et al. Hyperglycaemic impairment of PAR2-mediated vasodilation: prevention by inhibition of aortic endothelial sodium-glucose-co-Transporter-2 and minimizing oxidative stress. Vascul Pharmacol. (2018) 109:56–71. doi: 10.1016/j.vph.2018.06.006
141. Satoh T, Wang L, Espinosa-Diez C, Wang B, Hahn SA, Noda K, et al. Metabolic syndrome mediates ROS-miR-193b-NFYA-dependent downregulation of soluble guanylate cyclase and contributes to exercise-induced pulmonary hypertension in heart failure with preserved ejection fraction. Circulation. (2021) 144:615–37. doi: 10.1161/CIRCULATIONAHA.121.053889
142. Mizuno M, Kuno A, Yano T, Miki T, Oshima H, Sato T, et al. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol Rep. (2018) 6:e13741. doi: 10.14814/phy2.13741
143. Kolijn D, Pabel S, Tian Y, Lódi M, Herwig M, Carrizzo A, et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc Res. (2020) 12:cvaa123. doi: 10.1093/cvr/cvaa123
144. Huggett RJ, Scott EM, Gilbey SG, Stoker JB, Mackintosh AF, Mary DA, et al. Impact of type 2 diabetes mellitus on sympathetic neural mechanisms in hypertension. Circulation. (2003) 108:3097–101. doi: 10.1161/01.CIR.0000103123.66264.FE
145. Guyenet PG, Stornetta RL, Souza G, Abbott SBG, Brooks VL. Neuronal networks in hypertension: recent advances. Hypertension. (2020) 76:300–11. doi: 10.1161/HYPERTENSIONAHA.120.14521
146. Brunner-La Rocca HP, Esler MD, Jennings GL, Kaye DM. Effect of cardiac sympathetic nervous activity on mode of death in congestive heart failure. Eur Heart J. (2001) 22:1136–43. doi: 10.1053/euhj.2000.2407
147. Sano M, Chen S, Imazeki H, Ochiai H, Seino Y. Changes in heart rate in patients with type 2 diabetes mellitus after treatment with luseogliflozin: subanalysis of placebo-controlled, double-blind clinical trials. J Diabetes Investig. (2018) 9:638–41. doi: 10.1111/jdi.12726
148. Herat LY, Magno AL, Rudnicka C, Hricova J, Carnagarin R, Ward NC, et al. SGLT2 inhibitor-induced sympathoinhibition: a novel mechanism for cardiorenal protection. JACC Basic Transl Sci. (2020) 5:169–79. doi: 10.1016/j.jacbts.2019.11.007
149. Gueguen C, Burke SL, Barzel B, Eikelis N, Watson AMD, Jha JC, et al. Empagliflozin modulates renal sympathetic and heart rate baroreflexes in a rabbit model of diabetes. Diabetologia. (2020) 63:1424–34. doi: 10.1007/s00125-020-05145-0
150. Shimizu W, Kubota Y, Hoshika Y, Mozawa K, Tara S, Tokita Y, et al. Effects of empagliflozin versus placebo on cardiac sympathetic activity in acute myocardial infarction patients with type 2 diabetes mellitus: the EMBODY trial. Cardiovasc Diabetol. (2020) 19:148. doi: 10.21203/rs.3.rs-35207/v2
151. Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Garcia-Ropero A, Ishikawa K, Watanabe S, et al. Empagliflozin ameliorates diastolic dysfunction and left ventricular fibrosis/stiffness in nondiabetic heart failure: a multimodality study. JACC Cardiovasc Imaging. (2021) 14:393–407. doi: 10.1016/j.jcmg.2020.07.042
152. Santos-Gallego CG, Vargas-Delgado AP, Requena-Ibanez JA, Garcia-Ropero A, Mancini D, Pinney S, et al. Randomized trial of empagliflozin in nondiabetic patients with heart failure and reduced ejection fraction. J Am Coll Cardiol. (2021) 77:243–55. doi: 10.1016/j.jacc.2020.11.008
153. Jiang K, Xu Y, Wang D, Chen F, Tu Z, Qian J, et al. Cardioprotective mechanism of SGLT2 inhibitor against myocardial infarction is through reduction of autosis. Protein Cell. (2021) 13:336–59. doi: 10.1007/s13238-020-00809-4
154. Nishinarita R, Niwano S, Niwano H, Nakamura H, Saito D, Sato T, et al. Canagliflozin suppresses atrial remodeling in a canine atrial fibrillation model. J Am Heart Assoc. (2021) 10:e017483. doi: 10.1161/JAHA.119.017483
155. Azam MA, Chakraborty P, Si D, Du B, Massé S, Lai PFH, et al. Anti-arrhythmic and inotropic effects of empagliflozin following myocardial ischemia. Life Sci. (2021) 276:119440. doi: 10.1016/j.lfs.2021.119440
156. Shao Q, Meng L, Lee S, Tse G, Gong M, Zhang Z, et al. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc Diabetol. (2019) 18:165. doi: 10.1186/s12933-019-0964-4
157. Park SH, Farooq MA, Gaertner S, Bruckert C, Qureshi AW, Lee HH, et al. Empagliflozin improved systolic blood pressure, endothelial dysfunction and heart remodeling in the metabolic syndrome ZSF1 rat. Cardiovasc Diabetol. (2020) 19:19. doi: 10.1186/s12933-020-00997-7
158. Correale M, Mazzeo P, Mallardi A, Leopizzi A, Tricarico L, Fortunato M, et al. Switch to SGLT2 inhibitors and improved endothelial function in diabetic patients with chronic heart failure. Cardiovasc Drugs Ther. (2021) 14:1–8. doi: 10.1007/s10557-021-07254-3
159. Zhang N, Feng B, Ma X, Sun K, Xu G, Zhou Y, et al. Dapagliflozin improves left ventricular remodeling and aorta sympathetic tone in a pig model of heart failure with preserved ejection fraction. Cardiovasc Diabetol. (2019) 18:107. doi: 10.1186/s12933-019-0914-1
160. Withaar C, Meems LMG, Markousis-Mavrogenis G, Boogerd CJ, Silljé HHW, Schouten EM, et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc Res. (2021) 117:2108–24. doi: 10.1093/cvr/cvaa256
161. Wei R, Wang W, Pan Q, Guo L. Effects of SGLT-2 inhibitors on vascular endothelial function and arterial stiffness in subjects with type 2 diabetes: a systematic review and meta-analysis of randomized controlled trials. Front Endocrinol. (2022) 13:826604. doi: 10.3389/fendo.2022.826604
162. Kang S, Verma S, Hassanabad AF, Teng G, Belke DD, Dundas JA, et al. Direct effects of empagliflozin on extracellular matrix remodelling in human cardiac myofibroblasts: novel translational clues to explain EMPA-REG OUTCOME results. Can J Cardiol. (2020) 36:543–53. doi: 10.1016/j.cjca.2019.08.033
163. Patorno E, Htoo PT, Glynn RJ, Schneeweiss S, Wexler DJ, Pawar A, et al. Sodium-glucose cotransporter-2 inhibitors versus glucagon-like peptide-1 receptor agonists and the risk for cardiovascular outcomes in routine care patients with diabetes across categories of cardiovascular disease. Ann Intern Med. (2021) 174:1528–41. doi: 10.7326/M21-0893
164. Sayour AA, Celeng C, Oláh A, Ruppert M, Merkely B, Radovits T, et al. Sodium-glucose cotransporter 2 inhibitors reduce myocardial infarct size in preclinical animal models of myocardial ischaemia-reperfusion injury: a meta-analysis. Diabetologia. (2021) 64:737–48. doi: 10.1007/s00125-020-05359-2
165. Filion KB, Lix LM, Yu OH, Dell'Aniello S, Douros A, Shah B R, et al, Sodium glucose cotransporter 2 inhibitors and risk of major adverse cardiovascular events: multi-database retrospective cohort study. BMJ. (2020) 370:m3342. doi: 10.1136/bmj.m3342
166. McGuire DK, Shih WJ, Cosentino F, Charbonnel B, Cherney DZI, Dagogo-Jack S, et al. Association of SGLT2 inhibitors with cardiovascular and kidney outcomes in patients with type 2 diabetes: a meta-analysis. J Am Med Assoc Cardiol. (2021) 6:148–58. doi: 10.1001/jamacardio.2020.4511
167. Li Z, Agrawal V, Ramratnam M, Sharma RK, D'Auria S, Sincoular A, et al. Cardiac sodium-dependent glucose cotransporter 1 is a novel mediator of ischaemia/reperfusion injury. Cardiovasc Res. (2019) 115:1646–58. doi: 10.1093/cvr/cvz037
168. Lin H, Guan L, Meng L, Uzui H, Guo H. SGLT1 knockdown attenuates cardiac fibroblast activation in diabetic cardiac fibrosis. Front Pharmacol. (2021) 12:700366. doi: 10.3389/fphar.2021.700366
169. Sun Z, Chai Q, Zhang Z, Lu D, Meng Z, Wu W, et al. Inhibition of SGLT1 protects against glycemic variability-induced cardiac damage and pyroptosis of cardiomyocytes in diabetic mice. Life Sci. (2021) 271:119116. doi: 10.1016/j.lfs.2021.119116
170. Connelly KA, Zhang Y, Desjardins JF, Thai K, Gilbert RE. Dual inhibition of sodium-glucose linked cotransporters 1 and 2 exacerbates cardiac dysfunction following experimental myocardial infarction. Cardiovasc Diabetol. (2018) 17:99. doi: 10.1186/s12933-018-0741-9
171. Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet JWT, Koeman A, et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia. (2018) 61:722–6. doi: 10.1007/s00125-017-4509-7
172. Baartscheer A, Schumacher CA, Wüst RC, Fiolet JW, Stienen GJ, Coronel R, et al. Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia. (2017) 60:568–73. doi: 10.1007/s00125-016-4134-x
173. Lin K, Yang N, Luo W, Qian JF, Zhu WW, Ye SJ, et al. Direct cardio-protection of Dapagliflozin against obesity-related cardiomyopathy via NHE1/MAPK signaling. Acta Pharmacol Sin. (2022) 8:1–12. doi: 10.1038/s41401-022-00885-8
174. Uthman L, Nederlof R, Eerbeek O, Baartscheer A, Schumacher C, Buchholtz N, et al. Delayed ischaemic contracture onset by empagliflozin associates with NHE1 inhibition and is dependent on insulin in isolated mouse hearts. Cardiovasc Res. (2019) 115:1533–45. doi: 10.1093/cvr/cvz004
175. Trum M, Riechel J, Lebek S, Pabel S, Sossalla ST, Hirt S, et al. Empagliflozin inhibits Na(+) /H(+) exchanger activity in human atrial cardiomyocytes. ESC Heart Fail. (2020) 7:4429–37. doi: 10.1002/ehf2.13024
176. Chung YJ, Park KC, Tokar S, Eykyn TR, Fuller W, Pavlovic D, et al. Off-target effects of SGLT2 blockers: empagliflozin does not inhibit Na+/H+ exchanger-1 or lower [Na+]i in the heart. Cardiovasc Res. (2020) 2020:cvaa323. doi: 10.1093/cvr/cvaa323
177. Lelis DF, Freitas DF, Machado AS, Crespo TS, Santos SHS. Angiotensin-(1-7), adipokines and inflammation. Metabolism. (2019) 95:36–45. doi: 10.1016/j.metabol.2019.03.006
178. Kim JE, Kim JS, Jo MJ, Cho E, Ahn SY, Kwon YJ, et al. The roles and associated mechanisms of adipokines in development of metabolic syndrome. Molecules. (2022) 27:33. doi: 10.3390/molecules27020334
179. Zhao S, Kusminski CM, Scherer PE. Adiponectin, leptin and cardiovascular disorders. Circ Res. (2021) 128:136–49. doi: 10.1161/CIRCRESAHA.120.314458
180. Choi HM, Doss HM, Kim KS. Multifaceted physiological roles of adiponectin in inflammation and diseases. Int J Mol Sci. (2020) 21:1219. doi: 10.3390/ijms21041219
181. Ouchi N, Parker J, Lugus J, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. (2011) 11:85–97. doi: 10.1038/nri2921
182. López-Jaramillo P, Gómez-Arbeláez D, López-López J, López-López C, Martínez-Ortega J, Gómez-Rodríguez A, et al. The role of leptin/adiponectin ratio in metabolic syndrome and diabetes. Horm Mol Biol Clin Investig. (2014) 18:37–45. doi: 10.1515/hmbci-2013-0053
183. Scheja L, Heeren J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat Rev Endocrinol. (2019) 15:507–24. doi: 10.1038/s41574-019-0230-6
184. Wu P, Wen W, Li J, Xu J, Zhao M, Chen H, et al. Systematic review and meta-analysis of randomized controlled trials on the effect of SGLT2 inhibitor on blood leptin and adiponectin level in patients with type 2 diabetes. Horm Metab Res. (2019) 51:487–94. doi: 10.1055/a-0958-2441
185. Sardu C, Massetti M, Testa N, Martino LD, Castellano G, Turriziani F, et al. Effects of sodium-glucose transporter 2 inhibitors (SGLT2-I) in patients with ischemic heart disease (IHD) treated by coronary artery bypass grafting via miecc: inflammatory burden, and clinical outcomes at 5 years of follow-up. Front Pharmacol. (2021) 12:777083. doi: 10.3389/fphar.2021.777083
186. Requena-Ibáñez JA, Santos-Gallego CG, Rodriguez-Cordero A, Vargas-Delgado AP, Mancini D, Sartori S, et al. Mechanistic insights of empagliflozin in nondiabetic patients with HFrEF: from the EMPA-TROPISM study. JACC Heart Fail. (2021) 9:578–89. doi: 10.1016/j.jchf.2021.04.014
187. Bouchi R, Terashima M, Sasahara Y, Asakawa M, Fukuda T, Takeuchi T, et al. Luseogliflozin reduces epicardial fat accumulation in patients with type 2 diabetes: a pilot study. Cardiovasc Diabetol. (2017) 16:32. doi: 10.1186/s12933-017-0516-8
188. De Stefano A, Tesauro M, Di Daniele N, Vizioli G, Schinzari F, Cardillo C, et al. Mechanisms of SGLT2 (sodium-glucose transporter type 2) inhibition-induced relaxation in arteries from human visceral adipose tissue. Hypertension. (2021) 77:729–38. doi: 10.1161/HYPERTENSIONAHA.120.16466
189. Spigoni V, Fantuzzi F, Carubbi C, Pozzi G, Masselli E, Gobbi G, et al. Sodium-glucose cotransporter 2 inhibitors antagonize lipotoxicity in human myeloid angiogenic cells and ADP-dependent activation in human platelets: potential relevance to prevention of cardiovascular events. Cardiovasc Diabetol. (2020) 19:46. doi: 10.1186/s12933-020-01016-5
190. Hess DA, Terenzi DC, Trac JZ, Quan A, Mason T, Al-Omran M, et al. SGLT2 Inhibition with empagliflozin increases circulating provascular progenitor cells in people with type 2 diabetes mellitus. Cell Metab. (2019) 30:609–13. doi: 10.1016/j.cmet.2019.08.015
191. Bonora BM, Cappellari R, Albiero M, Avogaro A, Fadini GP. Effects of SGLT2 inhibitors on circulating stem and progenitor cells in patients with type 2 diabetes. J Clin Endocrinol Metab. (2018) 103:3773–82. doi: 10.1210/jc.2018-00824
192. Fadini GP. SGLT-2 inhibitors and circulating progenitor cells in diabetes. Cell Metab. (2020) 31:883. doi: 10.1016/j.cmet.2020.04.002
193. Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. (2019) 380:2295–306. doi: 10.1056/NEJMoa1811744
194. Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, et al. Dapagliflozin in patients with chronic kidney disease. N Engl J Med. (2020) 383:1436–46. doi: 10.1056/NEJMoa2024816
195. Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation. (2014) 129:587–97. doi: 10.1161/CIRCULATIONAHA.113.005081
196. Vallon V, Thomson SC. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat Rev Nephrol. (2020) 16:317–36. doi: 10.1038/s41581-020-0256-y
197. Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. (2020) 383:1413–24. doi: 10.1056/NEJMoa2022190
198. Mordi NA, Mordi IR, Singh JS, McCrimmon RJ, Struthers AD, Lang CC, et al. Renal and cardiovascular effects of sglt2 inhibition in combination with loop diuretics in patients with type 2 diabetes and chronic heart failure: the RECEDE-CHF trial. Circulation. (2020) 142:1713–24. doi: 10.1161/CIRCULATIONAHA.120.048739
199. Singh JSS, Mordi IR, Vickneson K, Fathi A, Donnan PT, Mohan M, et al. Dapagliflozin versus placebo on left ventricular remodeling in patients with diabetes and heart failure: the REFORM trial. Diabetes Care. (2020) 43:1356–9. doi: 10.2337/dc19-2187
200. Singh JS, Fathi A, Vickneson K, Mordi I, Mohan M, Houston JG, et al. Research into the effect Of SGLT2 inhibition on left ventricular remodelling in patients with heart failure and diabetes mellitus (REFORM) trial rationale and design. Cardiovasc Diabetol. (2016) 15:97. doi: 10.1186/s12933-016-0419-0
201. Jensen J, Omar M, Kistorp C, Tuxen C, Gustafsson I, Køber L, et al. Effects of empagliflozin on estimated extracellular volume, estimated plasma volume, and measured glomerular filtration rate in patients with heart failure (Empire HF Renal): a prespecified substudy of a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. (2021) 9:106–16. doi: 10.1016/S2213-8587(20)30382-X
202. Nassif ME, Windsor SL, Borlaug BA, Kitzman DW, Shah SJ, Tang F, et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: a multicenter randomized trial. Nat Med. (2021) 27:1954–60. doi: 10.1038/s41591-021-01536-x
Keywords: heart failure, mechanisms, metabolism, sodium-glucose cotransporter 2 inhibitors (SGLT2is), diabetes
Citation: Li J, Zhou L and Gong H (2022) New insights and advances of sodium-glucose cotransporter 2 inhibitors in heart failure. Front. Cardiovasc. Med. 9:903902. doi: 10.3389/fcvm.2022.903902
Received: 24 March 2022; Accepted: 15 August 2022;
Published: 15 September 2022.
Edited by:
Gary David Lopaschuk, University of Alberta, CanadaReviewed by:
Kim Lan Ho, University of Alberta, CanadaCopyright © 2022 Li, Zhou and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Gong, bGl5dWFuaG4yMDIxQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.