 Anastasia Milusev
Anastasia Milusev Robert Rieben
Robert Rieben Nicoletta Sorvillo1*
Nicoletta Sorvillo1*- 1Department for BioMedical Research (DBMR), University of Bern, Bern, Switzerland
- 2Graduate School for Cellular and Biomedical Sciences (GCB), University of Bern, Bern, Switzerland
The physiological, anti-inflammatory, and anti-coagulant properties of endothelial cells (ECs) rely on a complex carbohydrate-rich layer covering the luminal surface of ECs, called the glycocalyx. In a range of cardiovascular disorders, glycocalyx shedding causes endothelial dysfunction and inflammation, underscoring the importance of glycocalyx preservation to avoid disease initiation and progression. In this review we discuss the physiological functions of the glycocalyx with particular focus on how loss of endothelial glycocalyx integrity is linked to cardiovascular risk factors, like hypertension, aging, diabetes and obesity, and contributes to the development of thrombo-inflammatory conditions. Finally, we consider the role of glycocalyx components in regulating inflammatory responses and discuss possible therapeutic interventions aiming at preserving or restoring the endothelial glycocalyx and therefore protecting against cardiovascular disease.
Introduction
The endothelial glycocalyx is a carbohydrate-rich layer of proteoglycans and glycosaminoglycans lining the luminal surface of endothelial cells (ECs) in all blood vessels, ranging from small capillaries (1) to large arteries and veins (2). The thickness and structure of the glycocalyx differs between vascular beds and is highly dependent on the shear stress applied to the EC surface (3, 4). The expression of glycocalyx components is regulated and maintained by physiological blood flow conditions, leading to an intact glycocalyx layer. The thickness of the glycocalyx ranges from a few nanometers up to one micrometer. However, the observed thickness depends largely on the investigation method and whether the measurement was performed in vivo or on fixed tissues (5–7). Interestingly, glycocalyx thickness correlates with glycocalyx integrity and function. In fact, reduced thickness has been observed in inflammatory disorders and is associated with vessel dysfunction (8).
The endothelial glycocalyx fulfills diverse functions, ranging from mechanotransduction, maintenance of vascular integrity and vascular tone (9), to supporting the production of nitric oxide (NO) as well as providing anti-inflammatory and anti-coagulant properties by interacting with plasma proteins and plasma cells (10, 11). Two important families of proteins that interact with the glycocalyx and help maintaining vascular hemostasis are complement regulatory proteins including factor H (fH) and C1 inhibitor (12), and regulatory proteins of the coagulation system such as antithrombin III (ATIII) (13). These proteins, like many other plasma proteins, interact with the glycocalyx using heparan sulfate (HS) binding domains (14). The specific interaction of these proteins with HS underlines the importance of the structure and integrity of the glycocalyx for EC function. In fact, alterations of the glycocalyx cause loss of protein binding and are associated with disease. During several thrombo-inflammatory conditions including atherosclerosis, myocardial infarction, stroke, and ischemia/reperfusion injury, but also during infections and sepsis, syndecans (a core protein) or HS are released from the endothelial surface and represent a readout of glycocalyx damage (15–17). Once released, glycocalyx components induce activation of dendritic cells causing secretion of pro-inflammatory cytokines (18). At the endothelial level, partial or total shedding of the glycocalyx leads to increased leukocyte rolling and adhesion (19–21), elevated vessel permeability (22, 23), impaired vascular tone (24), and coagulation (8). The essential role of the glycocalyx in maintaining vascular hemostasis underscores the importance of protecting or restoring the glycocalyx as a potential therapeutic intervention for many inflammatory disorders. In this review we discuss the physiological functions of the glycocalyx with particular focus on how alteration of its integrity plays a role downstream of multiple cardiovascular risk factors in the context of thrombo-inflammatory conditions. Finally, we explore how prevention of shedding or restoration of the glycocalyx integrity can protect against the development of inflammatory disorders and therefore represent a potential therapeutic target.
Biosynthesis And Composition Of The Endothelial Glycocalyx
The glycocalyx consists of different glycoproteins and proteoglycans. Glycoproteins contain oligosaccharide chains with terminal sialic acid residues (25) while proteoglycans are membrane-anchored core proteins covalently linked to negatively charged glycosaminoglycan (GAG) chains through tetrasaccharide bridges (26). The mechanical features of each GAG chain are determined by its specific sugar composition, a combination of disaccharides containing hexosamine (N-sulfated or N-acetylated) and uronic acid or galactose residues. This results in the formation of the five primary groups of GAGs: heparan sulfate, chondroitin sulfate, keratan sulfate, dermatan sulfate, or hyaluronan (8, 10, 26).
Both the protein and polysaccharide components of PGs are synthesized in the endoplasmic reticulum and Golgi apparatus, except for hyaluronan, which in turn is synthesized directly on the cell surface (27). Once decorated with a variety of different GAGs, PGs are transported from the Golgi apparatus to the cell surface where they embed into the plasma membrane. The majority of endothelial PGs are membrane anchored syndecans and glypicans, which can be associated with all five different combinations of the above-mentioned GAGs (26). The resulting large variety of PG composition makes the glycocalyx extremely complex and variable between vessels and could therefore contribute, in addition to heterogeneity given by vessel type and organ function, to the different behavior of ECs in cardiovascular disorders. The most common GAG side chain found on ECs is HS (28), which, together with chondroitin sulfate, is the main GAG linked to syndecan. As shown in Figure 1, HS is composed of repeated disaccharide units consisting of a uronic sugar, β-D-glucuronic acid (GlcA) or α-L-iduronic acid (IdoA), and an amino sugar, N-acetyl-α-D-glucosamine (GlcNAc) or N-sulfo-α-D-glucosamine (GlcNS) (29). HS synthesis is initiated by addition of GlcNAc to a carbohydrate linker attached to the PG core protein and is mediated by the exostosin-like (EXTL1-3) family of glycosyltransferases. The growing GAG chain is then extended by a HS-polymerase complex consisting of EXT1 and EXT2 that adds the monosaccharides uronic acid or D-glucosamine (Figure 1) (30, 31). HS is highly heterogenic and undergoes several steps of post-translational modifications, in which the transfer of sulfate groups to distinct positions within the GlcNAc monosaccharide region leads to N-, 2-O-, 3-O-, or 6-O-sulfation (Figure 1). These modifications are mediated by specific sulfotransferases: N-deacetylase/N-sulfotransferase (NDST) initiates N-sulfation already during synthesis of the growing GAG chain, HS 2-O-sulfotransferase (HS2ST) sulfates uronic acid during chain elongation, HS 3-O-sulfotransferase (HS3ST) and HS 6-O-sulfotransferase (HS6ST) finalizes the sulfation of HS by adding sulfate groups to the 3-O and 6-O position within GlcNAc (Figure 1). HS sulfation motifs are instrumental to the interaction with plasma proteins; fibroblast growth factor 2, for example, requires NS- and 2O-sulfation, and ATIII specifically recognizes 3O-sulfation (31–33). Binding of these proteins to the glycocalyx is essential for their activity (12, 34).
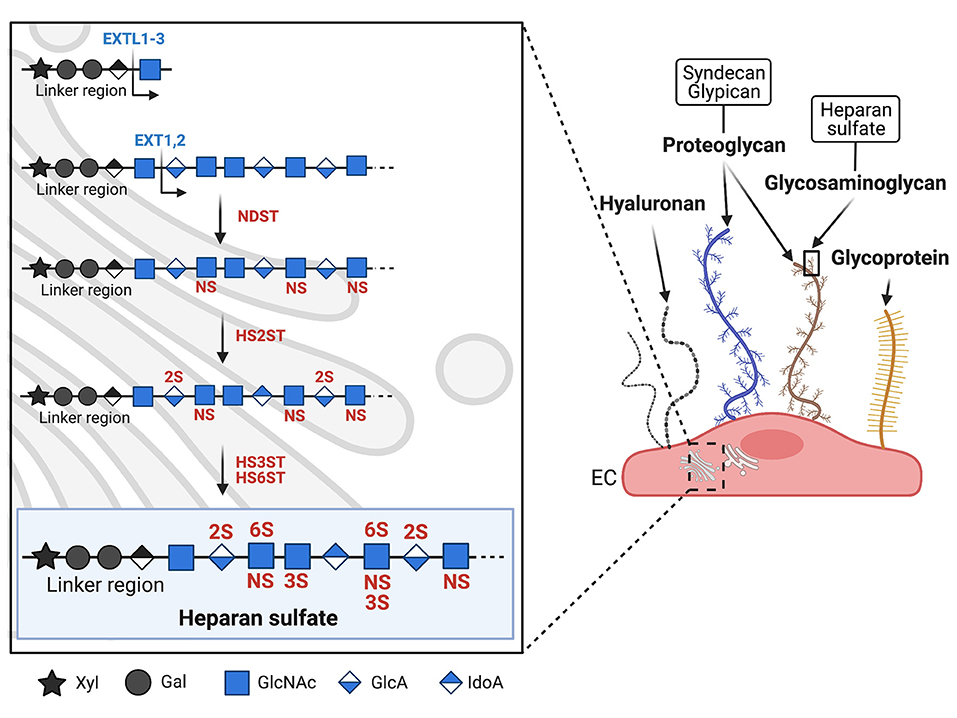
Figure 1. Biosynthesis and structure of the endothelial glycocalyx. Schematic representation of the major glycocalyx components covering the luminal surface of microvascular endothelial cells (EC). On the right panel, syndecan (blue) and glypican (brown) are shown as two examples of proteoglycans (PGs, see text chapter 'Biosynthesis and composition of the endothelial glycocalyx'). PGs carry long glycosaminoglycan (GAGs) side chains, while other glycoproteins (shown in yellow) carry shorter, unbranched carbohydrate side chains. The left panel shows the biosynthesis of heparan sulfate (HS), the major GAG expressed on EC. HS biosynthesis takes place in the Golgi apparatus and is mediated by different enzymes. Synthesis is initiated by the enzyme EXTL1-3 which adds the first sugar to the linker region. Chain elongation is performed by EXT1-2 which add GlcNAc and GlcA. Sulfotransferases then initiate HS sulfation, starting with NDST which sulfates GlcNAc at the N-acetyl position. HS2ST sulfates uronic acid, HS3ST and HS6ST finish sulfation by adding sulfate respectively to the 3-O and 6-O position of GlcNAc. Xyl, xylose; Gal, galactose; GlcNAc, N-acetylglucosamine; GlcA, glucuronic acid; IdoA, iduronic acid; NS, N-sulfation; 2S, 2-O sulfation; 3S, 3-O sulfation; 6S, 6-O sulfation; EXTL, exostosin-like glycosyltransferase; NDST, N-deacetylase/N-sulfotransferase; HS2ST, HS 2O-sulfotransferase; HS3ST, HS 3O-sulfotransferase; HS6ST, HS 6O-sulfotransferase.
The expression of the GAG modifying enzymes is tightly regulated and has been shown to change under inflammatory conditions. For instance, the expression of NDST1 in cultured human microvascular ECs was shown to increase after stimulation with interferon gamma (IFNγ) or tumor necrosis factor alpha (TNFα). This boosts the HS N-sulfation, leading to an enhanced interaction with the chemokine CCL5 and a rise in leukocyte adhesion (35). Elevated levels of TNFα have also been associated with a decrease in HS6ST expression. This alters the sulfation pattern of HS and consequently induces leukocyte rolling and adhesion (19). Furthermore, HS acts as a viral attachment factor, facilitating viral entry and infection. This is currently of great interest in the context of the global coronavirus disease-19 (COVID-19) pandemic, as SARS-CoV-2 requires HS as a recognition molecule for infection (36). In particular, the SARS-CoV-2 spike protein specifically binds N- and 6-O sulfated HS domains while other viruses such as herpes simplex virus require 3-O sulfation (37). Based on this, it is reasonable to speculate that changes in the sulfation pattern contribute to the different viral infection rates observed in the population. Further studies should assess whether modifications on HS play a role in EC infection.
Altogether, post-translational modifications of HS are largely responsible for changes in the interaction of plasma proteins, cells and virus with the endothelial glycocalyx. The differential regulation of HS modifying enzymes accounts for alterations in glycocalyx structure and function during pathological conditions.
Functions Of The Endothelial Glycocalyx Under Physiological Conditions
Initially thought to be a passive layer on the EC surface, it is now accepted that the glycocalyx serves multiple functions and actively takes part in regulating vascular hemostasis. The functions of the glycocalyx are mediated by both its mechanical and biochemical properties. Mechanical functions enclose the maintenance of vascular tone, mechanotransduction of extracellular signals, as well as preservation of vascular integrity, while biochemical functions are mediated by the interaction with plasma proteins and plasma cells (Figure 2). The vascular tone is mainly regulated by nitric oxide (NO), a vasodilator produced by ECs. When released abluminally, NO interacts with the adjacent vascular smooth muscle cells and activates soluble guanylate cyclase (sGC), which converts guanosine-triphosphate (GTP) to cyclical guanosine-monophosphate (cGMP). This leads to activation of protein kinase G and subsequently decreases intracellular calcium levels, causing relaxation of smooth muscle cells and vasodilation. A major regulator of NO production is vascular shear stress, which induces the expression of nitric oxide synthase (eNOS), the enzyme responsible for NO production within the endothelium (38, 39). On the other hand, removal of glycocalyx components has been shown to block the expression of NO and impair flow-mediated vasodilation (40–42). Consistent with this, eNOS can be upregulated only when HS structures are preserved, but not after shedding of HS by heparinase III treatment (43). Similarly, enzymatic shedding of chondroitin sulfate and sialic acids from cultured aortic ECs blocks shear-induced NO production (40). Overall, these reports indicate that the glycocalyx plays a significant role in shear-dependent NO release, but the mechanistic basis is still poorly explored. For instance, Boo et al. suggested that the increase in NO production is mediated by transmission of mechanosignals through membrane anchored PGs such as syndecan and glypican-1, located in membrane caveolae. The force applied to the glycocalyx components in the caveolae leads to phosphorylation of eNOS in a phosphoinositide-3-kinase and protein kinase A-dependent manner, thereby activating and increasing NO production (Figure 2A) (44). In line with this evidence, Ebong et al. showed that glypican-1 indeed plays a crucial role in shear-induced production of NO by phosphorylating eNOS, in such a way that silencing of glypican-1 abrogated eNOS activation (45).
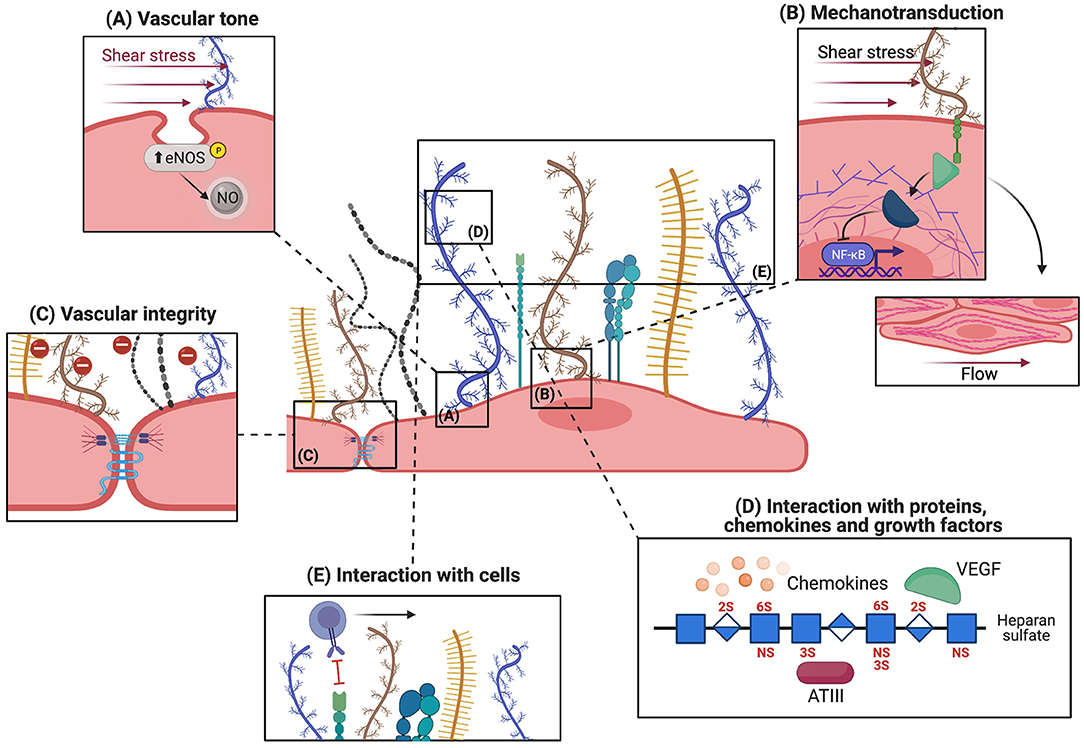
Figure 2. Physiological functions of the endothelial glycocalyx. The microvascular glycocalyx fulfills both mechanical and biochemical functions under physiological conditions, the five main functions are depicted here. (A) Sensing and transmission of shear stress by syndecans (shown in blue), located near membrane caveolae, increases eNOS activity and NO release. This assures vasodilatory functions. (B) Transmission of shear stress to the cytoskeleton via the cytoplasmic linker region (green) of syndecans leads to rearrangement of actin filaments and cell alignment in the direction of flow. Mechanotransduction also activates intracellular signaling molecules like Rho GTPases which regulate NFKB and MAPK. (C) The glycocalyx acts as a charge- and size-selective barrier to proteins. HS is important for maintaining intact junctions and vascular integrity. (D) Different molecules can bind to glycocalyx components: regulatory plasma proteins whose activity is potentiated by interaction with the glycocalyx, chemokines which are locally concentrated, and growth factors. (E) The intact glycocalyx shields off selectins and integrins expressed on EC thereby inhibiting leukocyte adhesion.
The coupling of an extracellular signal, such as shear stress, to intracellular changes like expression of eNOS as detailed above, illustrates how the glycocalyx plays a significant role in mechanotransduction. Along with this concept, syndecans interact using their cytoplasmatic tail with intracellular linker proteins dynamin, tubulin and actin, and transfer shear stress signals to the cytoskeleton leading to the alignment of ECs in the direction of flow (Figure 2B) (46, 47). This alignment can be clearly visualized by the rearrangement of actin microfilaments into a series of parallel elongated fibers along the axis of ECs (48). In line with this, knock-out of syndecan-4 is associated with alignment disruption both in vivo and in vitro (49). Besides transducing shear stress information to cytoplasmic components, syndecans also impact the organization of tight junction complexes, possibly affecting vascular permeability/integrity (50). In addition, mechanotransduction of shear stress regulates a range of intracellular pathways involved in control of thrombosis and fibrinolysis, such as thrombomodulin and tissue plasminogen, and inflammation (51). For example, laminar unidirectional shear stress activates Rho family GTPases, which results in the transcriptional activity of NFκB (Figure 2B). This pathway controls the expression of the NLRP3, pro-IL-1β, and pro-IL-18 genes, which participate in inflammasome assembly (51). Similarly, shear stress controls the activation of Ras-Mitogen-Activated Protein Kinases (MAPKs) in ECs, ultimately resulting in the activation of different transcription factors such as Krüppel-like factor 2 (KLF2), necessary for cell survival, and monocyte chemoattractant protein 1 (MCP1), that regulates migration and infiltration of monocytes. Finally, shear stress is known to modulate the composition of the endothelial glycocalyx itself. For instance, low shear (4 dyn/cm2) upregulates syndecan-1 expression, while high shear (10–14 dyn/cm2) increases the expression of syndecan-4 (52, 53). Altogether, the glycocalyx, and syndecans in particular, seem to play an essential role in mechanotransduction of vascular shear stress forces to preserve diverse aspects of vascular physiology.
Physically, ECs control migration of molecules and cells across the vessel wall and prevent vascular leakage by creating a solid EC monolayer in which cells are interconnected through tight and adherens junctions (Figure 2C). Tight junctions form a paracellular barrier that regulates cellular permeability as well as an intramembrane barrier that restrains exchanges between the luminal and basolateral cell membrane (54). Instead, adherens junctions maintain tissue structure and, by transduction of signals, modulate cell specification and cell growth (55, 56). Although endothelial junctions are the major players in maintaining vascular integrity, the luminal glycocalyx layer is also involved by acting as a charge-selective barrier that restrains transport of molecules into the intra- and subcellular space. In line with this concept, proteins that display a similar charge interact with the glycocalyx in a similar manner in vitro and have similar entry rates to the intra- and subcellular space, despite their difference in size (e.g., albumin and fibrinogen) (57). The charge-based selectivity of glycocalyx in vitro is also demonstrated by Vink et al., who show that migration to the abluminal side of ECs is slower for anionic than for neutral dextran (58). Although in vitro damage of the glycocalyx influences EC permeability to proteins (23), in vivo settings revealed that vascular permeability is regulated by both the glycocalyx composition and the structure of EC junctions in a coordinated fashion (59). In fact, removal of HS from the endothelial glycocalyx layer reduces the expression of the gap junction protein connexin 43 by 30%, causing an increase in vascular permeability (60). This exemplifies how both the glycocalyx and the endothelial junctions are essential in maintaining vascular integrity and regulating vascular permeability.
In addition to the so far mentioned functions, the glycocalyx is also able to directly interact with different plasma proteins and circulating cells, regulating inflammation and coagulation (Figure 2D). The sulfated GAG side chain HS is the major player for these interactions and has been shown to express binding sites for different growth factors, cytokines, and chemokines (61). Vice versa, HS binding domains are represented in a wide range of plasma proteins. These domains are characterized by clusters of one to three basic amino acids interspaced with one or two non-basic residues such as glycine and serine (62). Examples of growth factors binding to HS include fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF) (63), and granulocyte macrophage colony stimulating factor (GM-CSF) (64). Binding of these proteins to heparan sulfate proteoglycans (HSPGs) not only influences their stability, bioavailability and protects them from degradation, but also leads to an increase of their concentration on the cell surface, facilitating and amplifying intracellular signaling. For instance, IFNγ dimers and IL-8 bind to a small internal domain of HS rich in GlcA which leads to an increase in neutrophil migration (65–67). MCP1, Regulated on Activation Normal T-cell Expressed and Secreted (RANTES), and macrophage inflammatory peptides 1α and β (MIP-1α and MIP-1β, respectively) chemokines interact with the glycocalyx to generate the so called “chemokine-cloud,” a local concentration of chemokines within the glycocalyx layer (68). This facilitates leukocyte activation and amplifies pro-inflammatory signals (69).
Proteins that have binding sites for HS include ATIII (11, 13) which regulates hemostasis, superoxide and xanthine dismutase (SOD and XOD, respectively) (70, 71) that protects against oxidative stress, and complement fH (12) as well as C1 inhibitor which are involved in regulation of complement activation. The interaction of these proteins with HS potentiates their function. For example, interaction of fH with HSPGs enhances its capacity to bind and subsequently degrade C3b (72, 73). Given that HS is one of the main components shed during pathological conditions, it is more than plausible that the inflammatory and pro-coagulant conditions observed after glycocalyx shedding are due to loss of interaction of the glycocalyx with these regulatory plasma proteins. Analysis of such interactions would allow a better understanding of the pathophysiological mechanisms behind many inflammatory conditions.
In addition to interacting with plasma proteins the intact glycocalyx also acts as a barrier to leukocyte adhesion (Figure 2E) (21, 74). Rolling and adhesion of leukocytes to vessels is mediated by selectins and integrins expressed on activated ECs (75). The thickness of the intact glycocalyx, measuring up to one micrometer, exceeds the length of receptors involved in leukocyte adhesion which are only 20–40 nm long (76). Consistent with this, increased adhesion of leukocytes is observed only upon degradation of the glycocalyx (20).
On the other hand, HS has been shown to directly bind herpes simplex virus 1, human immunodeficiency virus 1 (HIV-1) (77, 78) and SARS-CoV-2, and facilitate entry into host epithelial cells and favor infection (79). Although HS is important for viral entry in epithelial cells, whether this is also true for ECs is still unknown.
In summary, an intact glycocalyx is essential to maintain vascular integrity and avoid coagulation dysregulations and leukocyte adhesion and subsequent transmigration into the surrounding tissue.
Endothelial Glycocalyx Damage And Its Link To Risk Factors Of Atherosclerosis
Atherosclerosis is a chronic inflammatory process that induces cholesterol plaque formation within the artery wall. Accumulation of oxidized lipoproteins in the vessel wall first occurs at sites of endothelial dysfunction, characterized by increased permeability, impaired cellular communication and vessel tone, complement activation, leukocyte adhesion, platelet aggregation and alteration of the glycocalyx (80–83). During atherosclerosis, EC activation induces local inflammation and promotes monocyte migration to the intima of the vessel and their subsequent differentiation into macrophages. Oxidized lipoproteins are then taken up by macrophages giving rise to foam cells and finally leading to plaque formation (84). In addition, vessel injury induces a pro-thrombotic and anti-fibrinolytic EC phenotype predisposing the atherosclerotic lesion site to thrombosis and reducing NO production and release (85). This in turn impairs endothelial-dependent vasodilation further allowing accumulation of lipoproteins (86). Tsiantoulas et al. demonstrated that the intact HSPGs structures protect against atherosclerotic plaque development by interacting with A PRoliferation-Inducing Ligand (APRIL). Binding of APRIL to HSPGs reduces retention of lipoproteins and migration of macrophages to the vessel intima thereby limiting plaque formation (87). In contrast, reduced glycocalyx thickness is linked to increased lipid retention and plaque formation (88).
Most atherosclerotic lesions develop in vessel areas subjected to disturbed or reduced flow like bifurcations, curvatures and branching points (89). Disturbed or reduced flow profiles are known to damage the endothelial glycocalyx through the activity of sheddases, proteolytic enzymes that cleave glycocalyx components releasing soluble PGs or GAGs (Figure 3) (90). For example, low shear stress activates hyaluronidase, which induces shedding of hyaluronan from the glycocalyx leading to a thin and unstable glycocalyx layer (91). In vivo studies have confirmed the presence of a thin and disrupted glycocalyx layer in regions of vessels presenting disturbed flow. In the same areas atherosclerotic plaques were found (88). Interestingly, in patients with coronary heart disease, a thin endothelial glycocalyx layer was associated with a plasmatic increase of hyaluronic acid and syndecan-1 (Figure 3) (92, 93). This confirms that shedding of the glycocalyx and vascular dysfunction are key elements in atherosclerosis. In line with these findings, Nagy et al. proved that treatment of apolipoprotein E–deficient mice with 4-methylumbelliferone, an inhibitor of hyaluronan synthesis, damages the endothelial glycocalyx causing atherosclerosis (94). Loss of glycocalyx compromises the endothelial barrier functions resulting in an increase of vascular permeability. This is followed by cholesterol accumulation and macrophage infiltration of the vessel wall, which eventually leads to atherosclerosis (88, 95, 96). Although it appears to correlate with the formation of atherosclerotic plaques, glycocalyx integrity alone is still not considered a risk factor for the development of cardiovascular diseases in clinical settings (97). Nevertheless, cardiovascular risk factors for atherosclerosis such as hypertension, aging, diabetes, and obesity are known to damage the endothelial glycocalyx, underlining the importance of this endothelial structure in cardiovascular disorders. For instance, high sodium levels, typical of a high salt diet, reduce HS content on human ECs (98) and are associated with syndecan-1 shedding (99). In hypertensive patients, loss of glycocalyx integrity and reduced thickness is associated with increased vascular stiffness, which is an independent predictor of cardiovascular risk caused by degeneration of the extracellular matrix of elastic arteries (Figure 3) (100–102). Vascular stiffness is initiated by high blood pressure as well as vascular aging and it negatively affects the endothelial glycocalyx (103). In agreement, ECs cultured on stiff matrices that mimic rigid aged vessels, express low levels of the endothelial glycocalyx components HS, glypican-1 and hyaluronan compared to ECs cultured on a soft matrix (104). Furthermore, low levels of glypican-1 are found in a mouse model of age-mediated vascular stiffness and are associated with endothelial dysfunction. Knocking out glypican-1 in young mice results in a vascular phenotype typical of advanced age, characterized by arterial stiffness and endothelial dysfunction (105). Although endothelial glycocalyx damage is linked to vascular stiffness, the molecular mechanisms behind have not been elucidated so far.
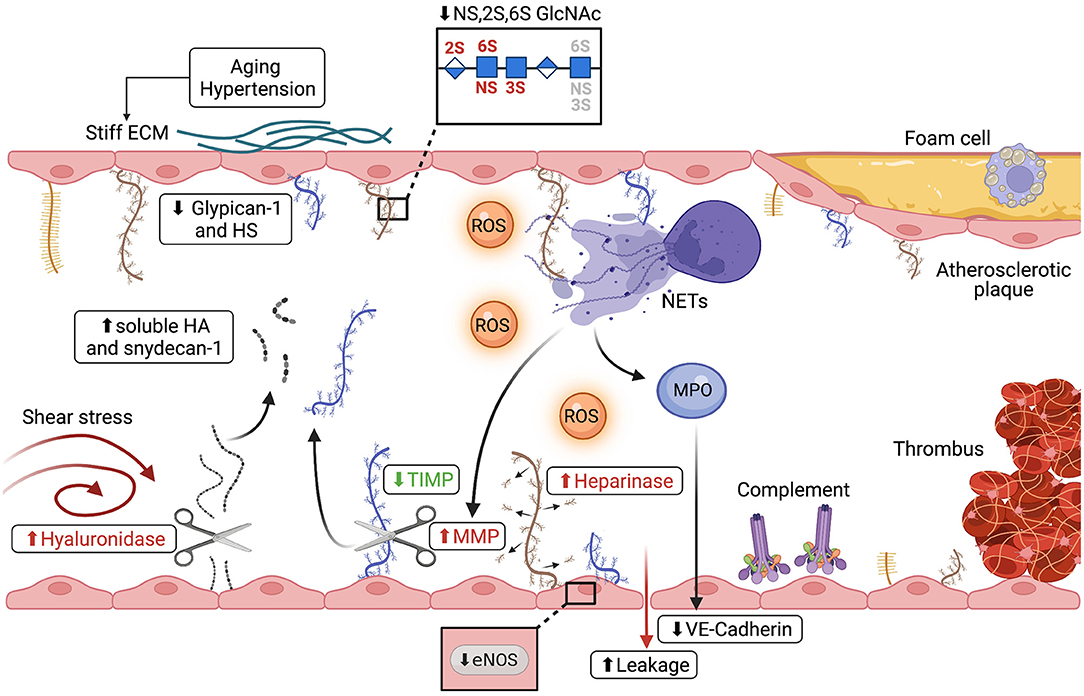
Figure 3. Alterations of the endothelial glycocalyx during thrombo-inflammatory conditions. Different mechanisms lead to shedding of the micro- and macrovascular glycocalyx in atherosclerosis and during ischemia/reperfusion injury (IRI). (I) Disturbed flow increases hyaluronidase expression leading to shedding of hyaluronan (shown in gray) and increased plasma syndecan-1 (shown in blue) and hyaluronic acid. (II) Hypertension, vascular stiffness and aging, all risk factors of cardiovascular disorders, cause thinning of the glycocalyx, evidenced by reduction of both glypican-1 and HS, as well as changes in the sulfation pattern of HS. (III) ROS (shown in orange), advanced glycation end products and sheddases all directly damage the endothelial glycocalyx and are linked to cardiovascular risk factors such as diabetes and obesity. (IV) Disruption of the glycocalyx is shown to be linked to reduced NO production and eNOS activity as well as activation of coagulation pathways and disturbed vasodilation. (V) Both complement deposition on the EC surface and neutrophil activation are associated with glycocalyx damage. Upon activation, neutrophils release neutrophil extracellular traps (NETs), decondensed chromatin decorated with neutrophil proteins. NETs can directly alter the glycocalyx or, through the release of MMPs and MPO (shown in purple), shed glycocalyx components and degrade junction proteins such as VE-Cadherin, leading to increased vascular leakage.
Vascular aging, an important cardiovascular risk factor associated with increased vessel stifness, is characterized by microvascular dysfunction, impaired perfusion and reduced capillary density (106). In mice, glycocalyx thickness and integrity are reduced with age (107, 108). A reduction of the endothelial glycocalyx thickness of ~30% was observed also in humans with a mean age of 60 years compared to 30-year-old individuals (109). In vitro, HS is abundantly expressed on the surface of low-passage human umbilical vein ECs compared to cells at higher passages (110). However, this result needs a word of caution, as the study setup cannot discriminate between cell aging and phenotypic drift. Interestingly, not only the amount of HS on the surface of ECs is influenced by aging but also the HS sulfation pattern can differ in old compared to young individuals (Figure 3). Human blood outgrowth cells isolated from old individuals present with a lower amount of tri-sulfated NS, 2S, 6S N-acetylglucosamine as compared to cells from young individuals (111). This suggests that aging alters the fine structure of HS, which could lead to reduced plasma protein interaction and therefore interfere with hemostasis (112).
Other risk factors of atherosclerosis in which alterations in the endothelial glycocalyx have been observed are diabetes and obesity. Thinning of the glycocalyx and endothelial stiffness were detected in the aorta of diabetic mice, whereby the damage to the glycocalyx was associated with endothelial stiffening and a reduction in endothelial NO production (113). Consistent with this, hyperglycemia results in glycocalyx shedding, as evidenced by the plasma increase of hyaluronan and syndecan-4, endothelial dysfunction, activation of coagulation pathways and reduction of eNOS activity (114–116). The three main factors reported to result in glycocalyx degradation in diabetes are reactive oxygen species (ROS), advanced glycation end products and sheddase activation. Both ROS and advanced glycation end products depolymerize hyaluronan, causing a damage in the glycocalyx integrity, while sheddases, like hyaluronidase, heparinase, metalloproteinase and neuraminidase, cleave glycocalyx components (Figure 3) (117). Notably, all these factors are increased in plasma of diabetic patients, which indicates a strong correlation between glycocalyx damage and insulin resistance (118–120).
Obesity is also known to induce endothelial dysfunction and, as for diabetes, it can manifest as loss of glycocalyx barrier properties and reduced NO availability (121, 122). The dilatory response of arteries to flow is impaired during obesity. This loss of flow-induced vasodilation is due to a decrease in NO production (123). Recent studies have shown that the activity of the flow sensitive K+ channel Kir, responsible for vasodilation, is highly dependent on HS. Heparinase treatment of freshly isolated ECs from obese mice reduces Kir activity. Interestingly, impairment of flow-mediated vasodilation in obesity differs between vascular beds. Endothelial dysfunction is observed in visceral adipose arteries but not in subcutaneous adipose arteries, where Kir activity, glycocalyx integrity and normal vasodilation are maintained (124). In contrast, in vivo data revealed that a thicker glycocalyx in the brain vasculature of obese mice is protective against inflammation during critical pathological conditions (125). This so called “obesity paradox” suggest that the glycocalyx might be structurally and functionally different in distinct organs, thus revealing an unexpected level of structural complexity.
Shedding Of Endothelial Glycocalyx During Ischemia/Reperfusion Injury
Ischemia/reperfusion injury (IRI) refers to the tissue damage sustained once blood flow is re-established after an ischemic period and can occur under different circumstances such as myocardial infarction with thrombotic vessel occlusion, heart surgery, transplantation, and ischemic stroke. Common to these circumstances are (I) tissue damage due to ROS produced upon reperfusion (81) (II) mitochondrial dysfunction with opening of the mitochondrial transition pore and loss of ATP production (126) (III) activation of the complement system (127, 128), and (IV) endothelial dysfunction (129). Glycocalyx shedding is recently emerging as an additional common event during IRI. Animal models of cardiac IRI have shown that the thickness of the glycocalyx is reduced as early as 5 min after reperfusion and that its shedding leads to decreased endothelium dependent vasodilation mediated by NO (130, 131). Early shedding of syndecan-1 and HS was also observed during reperfusion in patients undergoing cardiac surgery (132).
ROS are known to be key players in glycocalyx shedding/damage during IRI. Administration of the anti-oxidative reagent superoxide dismutase (SOD) maintains glycocalyx integrity and protects microvessels from IR damage (133). In agreement with this, patients with acute coronary syndrome, survivors of cardiac arrest or patients undergoing coronary bypass all present increased blood levels of circulating glycocalyx components such as syndecan-1 (92) and HS (15, 134). This has also been confirmed in ischemic stroke patients, where multiple soluble glycocalyx components, including 3 different GAGs (HS, keratan sulfate, chondroitin sulfate) and 3 different PGs (CD44, syndecan-2 and -3), were increased in the plasma of patients 1 week after the initial incident (135). Taken together, these studies indicate that mainly the core PG syndecan-1 and the GAG side chain HS are disrupted in IRI. In particular, HS shedding during IRI could explain some of the observed pathological vessel changes like increased permeability, complement activation, thrombosis and leukocyte infiltration toward the damaged tissue. In fact, one of the earliest inflammatory responses during cardiac IRI, next to endothelial dysfunction, is the activation of the complement system and the interaction of innate immune cells, such as neutrophils, with the vessel wall (136).
Different animal studies have shown the involvement of the complement system in IRI (137). Deposition of the complement components C3d and C5b-9 was seen in reperfused hearts of myocardial infarction patients (138) and associated with an increase in shedding of syndecan-1, a core protein of the endothelial glycocalyx (139, 140). Complement inhibition, by administration of a membrane-targeted molecule derived from complement receptor 1 (141) or by administration of dextran sulfate, was shown to be beneficial in experimental myocardial infarction as well as in cardiac transplantation (142). Dextran sulfate binding to the damaged myocardial blood vessels correlated with reduced vascular staining for HSPGs, suggesting a replenishment of the shed glycocalyx by dextran sulfate (143). Although complement deposition is correlated with loss of glycocalyx integrity, the exact link between the complement system and degradation of the glycocalyx in IRI is not very well-understood. On one hand, degradation of the glycocalyx might cause loss of interaction of complement regulatory proteins present in plasma such as fH or C1-inhibitor with the endothelial glycocalyx, resulting in increased complement deposition and tissue injury (144, 145). On the other hand, IRI might cause the expression of neoantigens on the EC surface (81). Binding of naturally occurring IgM antibodies to the neoantigens can then in turn lead to complement activation and tissue injury (146). The endothelial glycocalyx not only interacts with regulatory plasma proteins but also shields off cell surface adhesion molecules thereby limiting their interaction with immune cells. Shedding of the glycocalyx during both myocardial infarction and stroke has been shown to contribute to vascular edema as well as neutrophil and platelet adhesion to the vessel wall (147). Figure 3 illustrates how during neutrophil-mediated immune response, shedding of glycocalyx constituents can occur via enzymatic digestion by metalloproteases (MPOs) and hyaluronidase, or non-enzymatic degradation through oxidative stress. Other than degrading the glycocalyx, neutrophil enzymes such as MPOs, elastases, and cathepsins, can also cleave endothelial cell-cell junctions, in particular VE-Cadherin, leading to impaired junction integrity and vascular leakage (148). During myocardial infarction, stroke, and peripheral vascular disease, activated neutrophils also release neutrophil extracellular traps (NETs), web-like structures of decondensed chromatin covered with histones and cytoplasmic and granular proteins (149). It is well-accepted that the extracellular histones released during NET formation are highly cytotoxic for ECs and interact with the endothelial glycocalyx causing microvascular leakage and barrier dysfunction (150, 151). In fact, incubation of microvascular ECs in vitro with calf thymus histones results in EC death. Interestingly, this cytotoxicity is prevented by addition of negatively charged heparan sulfate tetra- or decasaccharides (152). Nucleosomes and histone 4 have been also detected in perfusates of isolated rat hearts subjected to ischemia reperfusion. Higher histone levels correlated to bigger infarct size (153).
Both neutrophil activation and complement deposition are considered key players in loss of endothelial glycocalyx integrity during IRI (Figure 3). In fact, knock-out of the complement receptor 5a in a mouse model of myocardial infarction resulted in reduction of neutrophil transmigration to post ischemic myocardium and diminished the expression of the matrix metalloproteinase 9, a sheddase of the endothelial glycocalyx (154). Both sheddases and sulfatases alter the endothelial glycocalyx by respectively removing entire GAG side chains and PGs or by changing the sulfation pattern of the GAG side chains. Many different sheddases are upregulated during IRI and are shown to be responsible for endothelial glycocalyx degradation. For instance, tryptase β (155), heparinase (156, 157) and atrial natriuretic peptide (ANP) (158) lead to an increase of soluble syndecan-1 (159). Similarly, the activity and release of matrix metalloproteinases (MMPs), which cleave entire PGs, is also upregulated during IRI. Cardiomyocytes and neutrophils have been identified as possible sources for MMPs during IRI (160, 161). Although ECs also produce MMPs, it is not clear whether MMPs of endothelial origin contribute to IRI (162). Shedding of syndecan under ischemic conditions is not only caused by upregulation of MMPs but is also due to downregulation of their natural inhibitors, the tissue inhibitors of metalloproteinases (TIMPs) (163–165). This indicates that IRI modulates not only the release of sheddases but also regulates their enzymatic activity. Other than syndecans, also removal of hyaluronan from the glycocalyx has been observed during ischemic stroke indicating that the sheddase hyaluronidase is involved in IRI (166, 167).
Finally, sulfatases can be upregulated during IRI. However, differently to sheddases, sulfatases seem to have a protective role. In a mouse model of myocardial infarction, the increase of sulfatase-1 and−2 was associated with a decrease in HS 6-O sulfation. This led to a reduction in the interaction of the glycocalyx with VEGF in the infarcted zone enhancing its bioavailability and increasing ischemic tissue repair (168). It is currently unknown whether upregulation or downregulation of sulfatases can have a detrimental effect on IRI.
Soluble Endothelial Glycocalyx Components As Regulators Of Inflammation
The endothelial glycocalyx regulates inflammation through different mechanisms: (I) it shields off integrins and other co-stimulatory molecules inhibiting binding of leukocytes to the endothelial surface, impeding antigen presentation and T cell activation, (II) it creates a local chemokine gradient that favors leukocyte activation (69, 169) (III) it regulates the activity of chemokines, cytokines and growth factors by protecting them from enzymatic degradation (68) and (IV) it binds regulatory proteins such as factor H or, in a soluble form, mannan-binding lectin serine protease 2 (MASP-2), thereby mediating complement inhibition (170).
Recent studies have shown that upon shedding of the glycocalyx, soluble HS propagates inflammation by directly interacting with toll-like receptors (TLRs). In vitro experiments proved that soluble HS fragments can stimulate the release of pro-inflammatory cytokines such as IL-1β, IL-6, IL-8, IL-10, and TNFα from human peripheral blood mononuclear cells (18). Additionally, both HS and hyaluronan induce dendritic cell maturation by TLR4 signaling (Figure 4A) (171). In agreement, mutation of TLR4 or incubation of dendritic cells with the TLR4 antagonist s-DPLA inhibited the upregulation of the co-stimulatory molecules CD86 and CD40 on the cell surface (172). The overall impact of soluble HS on vascular physiology remains largely debated. On one hand, stimulation of freshly isolated cardiac fibroblasts with soluble HS increases, through TLR4 signaling, the expression of ICAM-1 and VCAM-1 on the cell surface, favoring the adhesion of spleen mononuclear cells and bone marrow granulocytes. This promotes cardiac fibroblast differentiation into myofibroblasts. On the other hand, treatment of cardiac fibroblasts with HS was able to reduce alpha smooth muscle actin expression, impeding the differentiation into myofibroblast and protecting against a pro-fibrotic phenotype. This suggests that soluble HS can be both protective and deleterious (173).
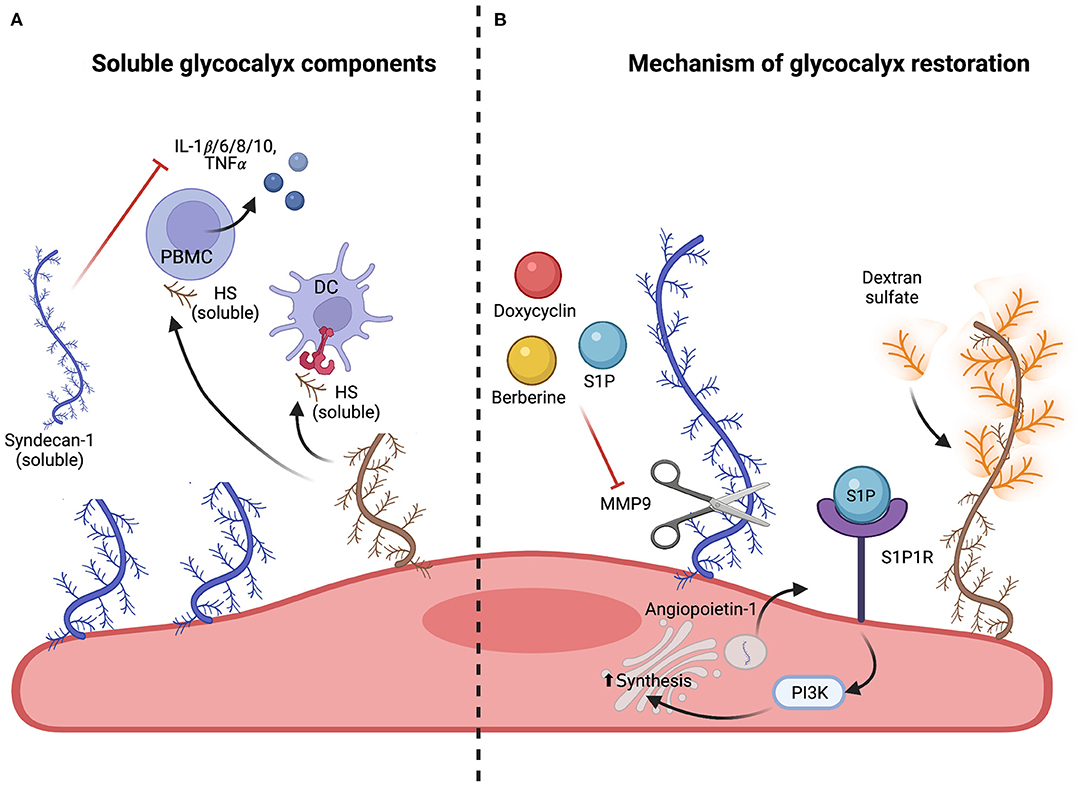
Figure 4. Soluble glycocalyx components and mechanism of glycocalyx restoration. (A) Effect of soluble glycocalyx components. Released soluble glycocalyx components such as HS (brown), syndecan-1 and −3 (blue) can propagate inflammatory responses by activating peripheral blood mononuclear cells (PBMCs) and dendritic cells (DCs) via TLR4 signaling. This leads to release of pro-inflammatory cytokines and DC maturation. However, soluble syndecan-1 (blue) has also been shown to reduce inflammation by directly inhibiting cytokine and chemokine release and blocking leukocyte adhesion. (B) Protection and restoration of the endothelial glycocalyx. Maintenance of glycocalyx integrity can be achieved by inhibition of sheddases. Doxycycline (shown in red), berberine (shown in yellow) or S1P (shown in blue) have been shown to directly inhibit matrix metalloproteinase 9 (MMP9) and therefore impede glycocalyx shedding. Regeneration of the glycocalyx can be achieved by upregulating the expression and extravasation of glycocalyx components such as syndecans with S1P and angiopoietin-1 or by replacing shed components such as HS with structurally similar agents such as dextran sulfate (shown in orange), a highly branched polysaccharide.
Although it is well-accepted that HS binds TLR4, the molecular mechanism of action of soluble HS is still not fully understood. Studies suggest that HS signaling through TLR4 activates a different NFκB pathway with respect to the response induced by the TLR4 ligand lipopolysaccharide (LPS). Indeed, TLR4 activation by soluble HS triggers a slower translocation of NFκB to the nucleus when compared to activation by LPS, and a more sustained increase of intracellular levels of calcium in macrophages (174). Whether this gives rise to different immune responses has not been yet investigated. Also, it is still unclear whether the main source of soluble HS derives from shedding of endothelial glycocalyx or from heparinase degradation of the extracellular matrix (172). Similarly, soluble syndecan-4 propagates the inflammatory response by upregulating ICAM1, VCAM1, IL-1β, and TNFα expression on cardiomyocytes increasing inflammatory cell recruitment (175).
Interestingly, soluble glycocalyx components not only propagate inflammation, but can also downregulate inflammatory signaling. For example, soluble HS fragments can directly inhibit cytokines such as IL-10 and interact with leukocytes, blocking their adhesion to the EC surface (Figure 4A) (176, 177). Addition of syndecan-1, which is rich in HS, was shown to directly inhibit the accumulation of CXC chemokines, thereby reducing neutrophil accumulation in various organs in a murine endotoxemia model (178). Along the same line of research, syndecan-1 was shown to block the expression of IL-1β, IL-6, and TNFα and the activity of pro-inflammatory chemokines CCL7, CCL11, and CCL17 (179, 180). Shed syndecan-3 also binds inflammatory chemokines like CCL2, CCL7, and CXCL8 and inhibits leukocyte migration in a mouse model of rheumatoid arthritis (181). Although syndecan-3 expression increases during myocardial infarction, its role in cardiovascular disorders is unknown (182).
Soluble components of the EG have the potential to both propagate and inhibit inflammation. The mechanism leading toward one direction rather than the other is still unknown and should be investigated.
Protection And Regeneration Of The Endothelial Glycocalyx
A damaged glycocalyx can regenerate over time. Although in vivo experiments have shown that 7 days are sufficient to reinstate the endothelial glycocalyx layer following enzymatic digestion, the mechanism behind and whether the restored glycocalyx is similar in its composition to the original has not been investigated so far (183). Nevertheless, various preventive and restorative approaches, aimed to re-establish the physiological glycocalyx function, have been suggested as potential therapies for several inflammatory disorders.
Different molecules can prevent endothelial glycocalyx degradation (Table 1). For example, angiopoietin-1 (184), hydrocortisone (189) ATIII (188), and SOD (133) maintain vascular integrity and prevent endothelial dysfunction during IRI by impeding the degradation of the glycocalyx components syndecan-1, HS and hyaluronic acid and have therefore been proposed as potential therapeutic candidates. However, how these molecules support endothelial glycocalyx integrity is still unclear. In Figure 4B, the two different proposed mechanisms of action are shown: angiopoietin-1 has a fast-acting effect in preventing glycocalyx shedding, suggesting that it protects the glycocalyx not by de novo synthesis of its components but rather by translocation of pre-formed GAG components from the Golgi apparatus to the cell surface. In fact, inhibiting the translocation of vesicles abolished the protective effect of angiopoietin-1. The second mechanism, proposed for hydrocortisone and ATIII, is a direct inhibition of sheddases which prevents glycocalyx degradation. One of the most abundant sheddases responsible for glycocalyx damage is the matrix-metalloproteinase-9 (MMP9). MMP9 belongs to a family of zinc-dependent endopeptidases implicated in both physiological and pathophysiological tissue remodeling. MMP9 is regulated transcriptionally by NFκB and once synthesized, its enzymatic activity is modulated by endogenous TIMPs (201). Berberine (187), doxycycline (185), and sphingosine-1 phosphate (S1P) (186) alleviate endothelial glycocalyx degradation by inhibiting MMP9 (Table 1, Figure 4B). Interestingly, S1P not only avoids glycocalyx shedding by directly interfering with MMP activity, but it also restores the glycocalyx once damaged. In fact, as depicted in Figure 4B, S1P interacts with its receptor S1P1R expressed on ECs and increases the synthesis of syndecan-1 and HS via intracellular phosphatidyl inositol-3 kinase (PI3K) signaling (60, 202).
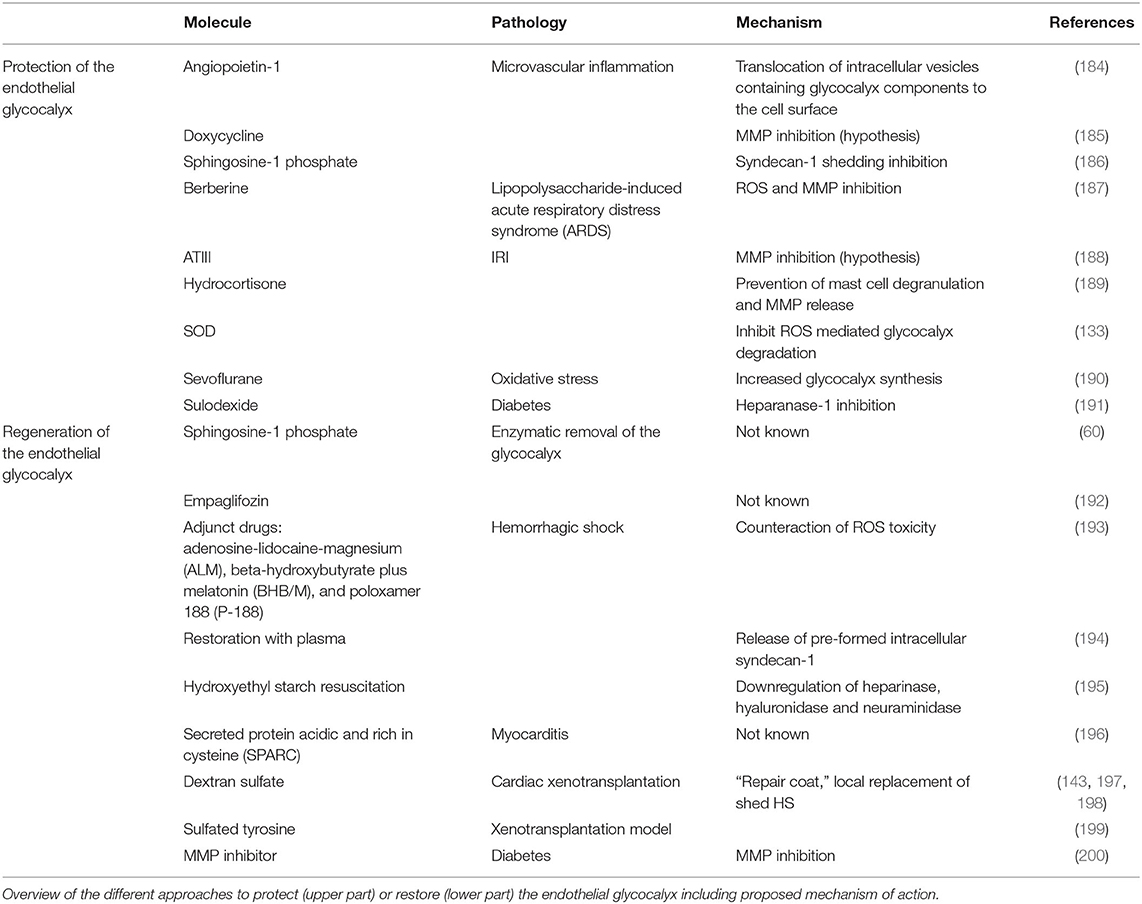
Table 1. Protection and regeneration of the endothelial glycocalyx.
Different studies have shown that also adjunct drugs (193), restoration with plasma (194), heparin (203, 204), sulodexide (191), and hydroxyethyl starch downregulate heparinase, hyaluronidase and neuraminidase (195) during inflammation or sepsis and therefore prevent endothelial dysfunction by preserving glycocalyx integrity (Table 1). Whether these approaches are beneficial also for cardiovascular disorders is not known. Also, the respective mechanism of action is still unclear. Dextran sulfate, a highly branched polysaccharide resembling HS, has also been recommended as a potential EC protectant. It was suggested that dextran sulfate creates a “repair coat” that mimics the missing endothelial glycocalyx layer and thereby inhibits complement deposition during acute vascular rejection (Table 1, Figure 4B) (197, 198, 205). Further studies are needed to confirm such a hypothesis.
Upregulation of glycocalyx synthesis genes could be a secondary approach for restoration of the endothelial glycocalyx during disease. However, little is known on the beneficial effects of induction of glycocalyx gene expression in damaged cells. It was shown that the anesthetic sevoflurane increases sialyltransferase expression during oxidative stress thereby promoting endothelial glycocalyx restoration and vasodilation (Table 1) (190). This suggests that other interventions leading to an increase of expression of glycocalyx synthesis genes could be helpful and open new avenues of research in the field of glycocalyx restoration.
Although shedding of the endothelial glycocalyx has been related to various cardiovascular and inflammatory conditions, a clear understanding of the mechanisms involved in glycocalyx shedding is still missing. Nonetheless, glycocalyx disruption occurs already early during disease development, suggesting that glycocalyx components could be potentially important, early biomarkers of disease. Given that prevention of glycocalyx shedding can be achieved, recognizing glycocalyx damage and inhibiting shedding might be beneficial in slowing down or even preventing certain cardiovascular conditions. Further research should focus on understanding the molecular mechanisms behind glycocalyx injury and uncover new therapeutic options for the protection or restoration of the endothelial glycocalyx in cardiovascular disorders.
Author Contributions
AM wrote the manuscript. NS and RR revised the manuscript. NS supervised the review. All authors approved the submitted version.
Funding
This study was supported by Swiss National Science Foundation, grant no. 310030_182264.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
All figures have been created using Biorender. We thank Emanuele Pignatti for his critical inputs.
References
1. Okada H, Takemura G, Suzuki K, Oda K, Takada C, Hotta Y, et al. Three-dimensional ultrastructure of capillary endothelial glycocalyx under normal and experimental endotoxemic conditions. Critical Care. (2017) 21:261. doi: 10.1186/s13054-017-1841-8
2. Megens RTA, Reitsma S, Schiffers PHM, Hilgers RHP, Mey JGRD, Slaaf DW, et al. Two-photon microscopy of vital murine elastic and muscular arteries. JVR. (2007) 44:87–98. doi: 10.1159/000098259
3. Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. (2005) 6:902–10. doi: 10.1038/ni1233
4. Zeng Y, Tarbell JM. The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS ONE. (2014) 9:e86249. doi: 10.1371/journal.pone.0086249
5. Cerny V, Astapenko D, Burkovskiy I, Hyspler R, Ticha A, Trevors MA, et al. Glycocalyx in vivo measurement. Clin Hemorheol Microcirc. (2017) 67:499–503. doi: 10.3233/CH-179235
6. Rovas A, Lukasz A-H, Vink H, Urban M, Sackarnd J, Pavenstädt H, et al. Bedside analysis of the sublingual microvascular glycocalyx in the emergency room and intensive care unit - the GlycoNurse study. Scand J Trauma Resusc Emerg Med. (2018) 26:16. doi: 10.1186/s13049-018-0483-4
7. Haymet AB, Bartnikowski N, Wood ES, Vallely MP, McBride A, Yacoub S, et al. Studying the endothelial glycocalyx in vitro: what is missing? Front Cardiovasc Med. (2021) 8:647086. doi: 10.3389/fcvm.2021.647086
8. Reitsma S, Slaaf DW, Vink H, Zandvoort MAMJ, Egbrink MGA. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch Eur J Physiol. (2007) 454:345–59. doi: 10.1007/s00424-007-0212-8
9. Cosgun ZC, Fels B, Kusche-Vihrog K. Nanomechanics of the endothelial glycocalyx: from structure to function. Am J Pathol. (2020) 190:732–41. doi: 10.1016/j.ajpath.2019.07.021
10. Jackson RL, Busch SJ, Cardin AD. Glycosaminoglycans: molecular properties, protein interactions, and role in physiological processes. Physiol Rev. (1991) 71:59. doi: 10.1152/physrev.1991.71.2.481
11. Stern D, Nawroth P, Marcum J, Handley D, Kisiel W, Rosenberg R, et al. Interaction of antithrombin III with bovine aortic segments. Role of heparin in binding and enhanced anticoagulant activity. J Clin Invest. (1985) 75:272–9. doi: 10.1172/JCI111685
12. Meri S, Pangburn MK. Regulation of alternative pathway complement activation by glycosaminoglycans: specificity of the polyanion binding site on factor H. Biochem Biophys Res Commun. (1994) 198:52–9. doi: 10.1006/bbrc.1994.1008
13. Lindahl U, Backstr G, Thunberg L, Ledert IG. Evidence for a 3- O-sulfated D-glucosamine residue in the antithrombin-binding sequence of heparin. Proc Natl Acad Sci USA. (1980) 5:6551. doi: 10.1073/pnas.77.11.6551
14. Mulloy B, Linhardt RJ. Order out of complexity – protein structures that interact with heparin. Curr Opin Struct Biol. (2001) 11:623–8. doi: 10.1016/S0959-440X(00)00257-8
15. Grundmann S, Fink K, Rabadzhieva L, Bourgeois N, Schwab T, Moser M, et al. Perturbation of the endothelial glycocalyx in post cardiac arrest syndrome. Resuscitation. (2012) 83:715–20. doi: 10.1016/j.resuscitation.2012.01.028
16. Ostrowski SR, Pedersen SH, Jensen JS, Mogelvang R, Johansson PI. Acute myocardial infarction is associated with endothelial glycocalyx and cell damage and a parallel increase in circulating catecholamines. Critical Care. (2013) 17:R32. doi: 10.1186/cc12532
17. Goligorsky MS, Sun D. Glycocalyx in endotoxemia and sepsis. Am J Pathol. (2020) 190:791–8. doi: 10.1016/j.ajpath.2019.06.017
18. Goodall KJ, Poon IKH, Phipps S, Hulett MD. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS ONE. (2014) 9:e109596. doi: 10.1371/journal.pone.0109596
19. Rops AL, van den Hoven MJ, Baselmans MM, Lensen JF, Wijnhoven TJ, van den Heuvel LP, et al. Heparan sulfate domains on cultured activated glomerular endothelial cells mediate leukocyte trafficking. Kidney Int. (2008) 73:52–62. doi: 10.1038/sj.ki.5002573
20. Constantinescu Alina A, Hans V, Spaan Jos AE. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface arteriosclerosis, thrombosis, and vascular. Biology. (2003) 23:1541–7. doi: 10.1161/01.ATV.0000085630.24353.3D
21. Mulivor AW, Lipowsky HH. Role of glycocalyx in leukocyte-endothelial cell adhesion. Am J Physiol Heart Circ Physiol. (2002) 283:10. doi: 10.1152/ajpheart.00117.2002
22. Chelazzi C, Villa G, Mancinelli P, De Gaudio A, Adembri C. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit Care. (2015) 19:26. doi: 10.1186/s13054-015-0741-z
23. Betteridge KB, Arkill KP, Neal CR, Harper SJ, Foster RR, Satchell SC, et al. Sialic acids regulate microvessel permeability, revealed by novel in vivo studies of endothelial glycocalyx structure and function. J Physiol. (2017) 595:5015–35. doi: 10.1113/JP274167
24. Yen W, Cai B, Yang J, Zhang L, Zeng M, Tarbell JM, et al. Endothelial surface glycocalyx can regulate flow-induced nitric oxide production in microvessels in vivo. PLoS ONE. (2015) 10:e0117133. doi: 10.1371/journal.pone.0117133
25. Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. (2007) 9:121–67. doi: 10.1146/annurev.bioeng.9.060906.151959
26. Lindahl U, Couchman J, Kimata K, Esko JD. Proteoglycans and sulfated glycosaminoglycans. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, et al., editors, Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press (2015). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK453033/ (accessed June 11, 2021).
27. Möckl L. The emerging role of the mammalian glycocalyx in functional membrane organization and immune system regulation. Front Cell Dev Biol. (2020) 8:253. doi: 10.3389/fcell.2020.00253
28. Ihrcke NS, Wrenshall LE, Lindman BJ, Platt JL. Role of heparan sulfate in immune system-blood vessel interactions. Immunol Today. (1993) 14:500–5. doi: 10.1016/0167-5699(93)90265-M
29. Rabenstein DL. Heparin and heparan sulfate: structure and function. Nat Prod Rep. (2002) 19:312–31. doi: 10.1039/b100916h
30. Kreuger J, Kjellén L. Heparan sulfate biosynthesis: regulation and variability. J Histochem Cytochem. (2012) 2012:22155412464972. doi: 10.1369/0022155412464972
31. Lamanna WC, Kalus I, Padva M, Baldwin RJ, Merry CLR, Dierks T. The heparanome—the enigma of encoding and decoding heparan sulfate sulfation. J Biotechnol. (2007) 129:290–307. doi: 10.1016/j.jbiotec.2007.01.022
32. Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. (2002) 71:435–71. doi: 10.1146/annurev.biochem.71.110601.135458
33. Liu J, O'Leary T. Heparan sulfate (glucosamine) 3-O-sulfotransferase 1-6 (HS3ST1-6). In: Taniguchi N, Honke K, Fukuda M, Narimatsu H, Yamaguchi Y, Angata T, editors, Handbook of Glycosyltransferases and Related Genes. Tokyo: Springer Japan (2014). p. 1081–9. doi: 10.1007/978-4-431-54240-7_117
34. Wuillemin WA, te Velthuis H, Lubbers YTP, de Ruig CP, Eldering EF, Hack CE. Potentiation of C1 inhibitor by glycosaminoglycans: dextran sulfate species are effective inhibitors of in vitro complement activation in plasma. J. Immunol. Res. 159:1953–60.
35. Carter NM. Endothelial inflammation: the role of differential expression of N-deacetylase/N-sulphotransferase enzymes in alteration of the immunological properties of heparan sulphate. J Cell Sci. (2003) 116:3591–600. doi: 10.1242/jcs.00662
36. Wadowski PP, Jilma B, Kopp CW, Ertl S, Gremmel T, Koppensteiner R. Glycocalyx as possible limiting factor in COVID-19. Front Immunol. (2021) 12:607306. doi: 10.3389/fimmu.2021.607306
37. De Pasquale V, Quiccione MS, Tafuri S, Avallone L, Pavone LM. Heparan sulfate proteoglycans in viral infection and treatment: a special focus on SARS-CoV-2. Int J Mol Sci. (2021) 22:6574. doi: 10.3390/ijms22126574
38. Uematsu M, Ohara Y, Navas JP, Nishida K, Murphy TJ, Alexander RW, et al. Regulation of endothelial cell nitric oxide synthase mRNA expression by shear stress. Am J Physiol Cell Physiol. (1995) 269:C1371–8. doi: 10.1152/ajpcell.1995.269.6.C1371
39. Ranjan V, Xiao Z, Diamond SL. Constitutive NOS expression in cultured endothelial cells is elevated by fluid shear stress. Am J Physiol. (1995) 269:H550–5. doi: 10.1152/ajpheart.1995.269.2.H550
40. Pahakis MY, Kosky JR, Dull RO, Tarbell JM. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem Biophys Res Commun. (2007) 355:228–33. doi: 10.1016/j.bbrc.2007.01.137
41. Cho JM, Ly K, Ly S, Park S-K, Babu PVA, Balagurunathan K, et al. Procedures to evaluate the role of heparan sulfate on the reactivity of resistance and conductance arteries ex vivo. Methods Mol Biol. (2022) 2303:495–511. doi: 10.1007/978-1-0716-1398-6_40
42. Dragovich MA, Chester D, Fu BM, Wu C, Xu Y, Goligorsky MS, et al. Mechanotransduction of the endothelial glycocalyx mediates nitric oxide production through activation of TRP channels. Am J Physiol. (2016) 311:C846–53. doi: 10.1152/ajpcell.00288.2015
43. Florian Jeffry A, Kosky Jason R, Ainslie K, Pang Z, Dull Randal O, Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circulat Res. (2003) 93:e136–42. doi: 10.1161/01.RES.0000101744.47866.D5
44. Boo YC, Hwang J, Sykes M, Michell BJ, Kemp BE, Lum H, et al. Shear stress stimulates phosphorylation of eNOS at Ser635 by a protein kinase A-dependent mechanism. Am J Physiol. (2002) 283:H1819–28. doi: 10.1152/ajpheart.00214.2002
45. Ebong EE, Spray DC, Tarbell JM. The endothelial glycocalyx: its structure and role in eNOS mechano-activation. In: Proceedings of the 2010 IEEE 36th Annual Northeast Bioengineering Conference (NEBEC). (2010). p. 1–2. doi: 10.1109/NEBC.2010.5458171
46. Fu BM, Tarbell JM. Mechano-sensing and transduction by endothelial surface glycocalyx: composition, structure, and function: Mechano-sensing and transduction by endothelial surface glycocalyx. WIREs Syst Biol Med. (2013) 5:381–90. doi: 10.1002/wsbm.1211
47. Rapraeger AC. Syndecan-regulated receptor signaling. J Cell Biol. (2000) 149:995–8. doi: 10.1083/jcb.149.5.995
48. Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. (1995) 42:519. doi: 10.1152/physrev.1995.75.3.519
49. Baeyens N, Mulligan-Kehoe MJ, Corti F, Simon DD, Ross TD, Rhodes JM, et al. Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc Natl Acad Sci USA. (2014) 111:17308–13. doi: 10.1073/pnas.1413725111
50. Thi MM, Tarbell JM, Weinbaum S, Spray DC. The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a “bumper-car” model. Proc Natl Acad Sci USA. (2004) 101:16483–8. doi: 10.1073/pnas.0407474101
51. Li Y-SJ, Haga JH, Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J Biomech. (2005) 38:1949–71. doi: 10.1016/j.jbiomech.2004.09.030
52. Liu J-X, Yan Z-P, Zhang Y-Y, Wu J, Liu X-H, Zeng Y. Hemodynamic shear stress regulates the transcriptional expression of heparan sulfate proteoglycans in human umbilical vein endothelial cell. Cell Mol Biol. (2016) 62:8. doi: 10.14715/cmb/2016.62.8.5
53. Voyvodic PL, Min D, Liu R, Williams E, Chitalia V, Dunn AK, et al. Loss of syndecan-1 induces a pro-inflammatory phenotype in endothelial cells with a dysregulated response to atheroprotective flow. J Biol Chem. (2014) 289:9547–59. doi: 10.1074/jbc.M113.541573
54. Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol. (2016) 17:564–80. doi: 10.1038/nrm.2016.80
55. Harris TJC, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol. (2010) 11:502–14. doi: 10.1038/nrm2927
56. Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. (2004) 84:869–901. doi: 10.1152/physrev.00035.2003
57. van Haaren PMA, VanBavel E, Vink H, Spaan JAE. Localization of the permeability barrier to solutes in isolated arteries by confocal microscopy. Am J Physiol Heart Circ Physiol. (2003) 285:H2848–2856. doi: 10.1152/ajpheart.00117.2003
58. Vink H, Duling BR. Capillary endothelial surface layer selectively reduces plasma solute distribution volume. Am J Physiol. (2000) 278:H285–9. doi: 10.1152/ajpheart.2000.278.1.H285
59. Rehm M, Zahler S, Lötsch M, Welsch U, Conzen P, Jacob M, et al. Endothelial glycocalyx as an additional barrier determining extravasation of 6% hydroxyethyl starch or 5% albumin solutions in the coronary vascular bed. Anesthesiology. (2004) 100:1211–23. doi: 10.1097/00000542-200405000-00025
60. Mensah SA, Cheng MJ, Homayoni H, Plouffe BD, Coury AJ, Ebong EE. Regeneration of glycocalyx by heparan sulfate and sphingosine 1-phosphate restores inter-endothelial communication. PLoS ONE. (2017) 12:e0186116. doi: 10.1371/journal.pone.0186116
61. Tumova S, Woods A, Couchman JR. Heparan sulfate proteoglycans on the cell surface: versatile coordinators of cellular functions. Int J Biochem Cell Biol. (2000) 32:269–88. doi: 10.1016/S1357-2725(99)00116-8
62. Muñoz EM, Linhardt RJ. Heparin-binding domains in vascular biology. Arterioscler Thromb Vasc Biol. (2004) 24:1549–57. doi: 10.1161/01.ATV.0000137189.22999.3f
63. Hu Z, Cano I, D'Amore PA. Update on the role of the endothelial glycocalyx in angiogenesis and vascular inflammation. Front Cell Dev Biol. (2021) 9:734276. doi: 10.3389/fcell.2021.734276
64. Ruoslahti E, Yamaguchi Y. Proteoglycans as modulators of growth factor activities. Cell Press. (1991) 64:867–9. doi: 10.1016/0092-8674(91)90308-L
65. Lortat-Jacob H, Turnbull JE, Grimaud JA. Molecular organization of the interferon gamma-binding domain in heparan sulphate. Biochem J. (1995) 310:497–505. doi: 10.1042/bj3100497
66. Frevert CW, Kinsella MG, Vathanaprida C, Goodman RB, Baskin DG, Proudfoot A, et al. Binding of interleukin-8 to heparan sulfate and chondroitin sulfate in lung tissue. Am J Respir Cell Mol Biol. (2003) 28:464–72. doi: 10.1165/rcmb.2002-0084OC
67. Rot A. Endothelial cell binding of NAP-1/IL-8: role in neutrophil emigration. In: Lindley IJD, Westwick J, Kunkel S, editors, The Chemokines: Biology of the Inflammatory Peptide Supergene Family II. Advances in Experimental Medicine and Biology. Boston, MA: Springer US (1993). p. 208. doi: 10.1007/978-1-4615-2952-1_54
68. Kuschert GSV, Coulin F, Power CA, Proudfoot AEI, Hubbard RE, Hoogewerf AJ, et al. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry. (1999) 38:12959–68. doi: 10.1021/bi990711d
69. Graham GJ, Handel TM, Proudfoot AEI. Leukocyte adhesion: reconceptualizing chemokine presentation by glycosaminoglycans. Trends Immunol. (2019) 40:472–81. doi: 10.1016/j.it.2019.03.009
70. Karlsson K, Lindahl U, Marklund SL. Binding of human extracellular superoxide dismutase C to sulphated glycosaminoglycans. Biochem J. (1988) 256:29–33. doi: 10.1042/bj2560029
71. Adachi T, Fukushima T, Usami Y, Hirano K. Binding of human xanthine oxidase to sulphated glycosaminoglycans on the endothelial-cell surface. Biochem J. (1993) 289:523–7. doi: 10.1042/bj2890523
72. Kajander T, Lehtinen MJ, Hyvärinen S, Bhattacharjee A, Leung E, Isenman DE, et al. Dual interaction of factor H with C3d and glycosaminoglycans in host–nonhost discrimination by complement. Proc Natl Acad Sci USA. (2011) 108:2897–902. doi: 10.1073/pnas.1017087108
73. Morgan HP, Schmidt CQ, Guariento M, Blaum BS, Gillespie D, Herbert AP, et al. Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol. (2011) 18:463–70. doi: 10.1038/nsmb.2018
74. Celie J WAM, Beelen RHJ, van den Born J. Heparan sulfate proteoglycans in extravasation: assisting leukocyte guidance. Front Biosci. (2009) 14:4932. doi: 10.2741/3578
75. Granger DN, Senchenkova E. Leukocyte–Endothelial Cell Adhesion. Morgan & Claypool Life Sciences (2010). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK53380/ (accessed February 19, 2022).
76. Springer TA. Adhesion receptors of the immune system. Nature. (1990) 346:425–34. doi: 10.1038/346425a0
77. Koehler M, Delguste M, Sieben C, Gillet L, Alsteens D. Initial step of virus entry: virion binding to cell-surface glycans. Ann Rev Virol. (2020) 7:143–65. doi: 10.1146/annurev-virology-122019-070025
78. Sato S, Ouellet M, St-Pierre C, Tremblay MJ. Glycans, galectins, and HIV-1 infection. Ann N Y Acad Sci. (2012) 1253:133–48. doi: 10.1111/j.1749-6632.2012.06475.x
79. Cagno V, Tseligka ED, Jones ST, Tapparel C. Heparan sulfate proteoglycans and viral attachment: true receptors or adaptation bias? Viruses. (2019) 11:596. doi: 10.3390/v11070596
80. Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
81. Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat Med. (2011) 17:1391–401. doi: 10.1038/nm.2507
82. Abassi Z, Armaly Z, Heyman SN. Glycocalyx degradation in ischemia-reperfusion injury. Am J Pathol. (2020) 8:19. doi: 10.1016/j.ajpath.2019.08.019
83. Zhang X, Sun D, Song JW, Zullo J, Lipphardt M, Coneh-Gould L, et al. Endothelial cell dysfunction and glycocalyx - a vicious circle. Matrix Biol. (2018) 71–2:421–31. doi: 10.1016/j.matbio.2018.01.026
84. Insull W. The pathology of atherosclerosis: plaque development and plaque responses to medical treatment. Am J Med. (2009) 122:S3–14. doi: 10.1016/j.amjmed.2008.10.013
85. Harding IC, Mitra R, Mensah SA, Herman IM, Ebong EE. Pro-atherosclerotic disturbed flow disrupts caveolin-1 expression, localization, and function via glycocalyx degradation. J Transl Med. (2018) 16:2. doi: 10.1186/s12967-018-1721-2
86. Xu S, Bendeck M, Gotlieb AI. Vascular pathobiology. In: Maximilian Buja L, Butany J, editors. Cardiovascular Pathology. 4th ed. Academic Press (2016). p. iv. doi: 10.1016/B978-0-12-420219-1.09995-X
87. Tsiantoulas D, Eslami M, Obermayer G, Clement M, Smeets D, Mayer FJ, et al. APRIL limits atherosclerosis by binding to heparan sulfate proteoglycans. Nature. (2021) 597:92–6. doi: 10.1038/s41586-021-03818-3
88. Cancel LM, Ebong EE, Mensah S, Hirschberg C, Tarbell JM. Endothelial glycocalyx, apoptosis and inflammation in an atherosclerotic mouse model. Atherosclerosis. (2016) 252:136–46. doi: 10.1016/j.atherosclerosis.2016.07.930
89. Gimbrone MA, García-Cardeña G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc Pathol. (2013) 22:9–15. doi: 10.1016/j.carpath.2012.06.006
90. Cooper S, McDonald K, Burkat D, Leask RL. Stenosis hemodynamics disrupt the endothelial cell glycocalyx by MMP activity creating a proinflammatory environment. Ann Biomed Eng. (2017) 45:2234–43. doi: 10.1007/s10439-017-1846-0
91. Zhang J, Kong X, Wang Z, Gao X, Ge Z, Gu Y, et al. AMP-activated protein kinase regulates glycocalyx impairment and macrophage recruitment in response to low shear stress. FASEB J. (2019) 33:7202–12. doi: 10.1096/fj.201801869RRR
92. Miranda CH, de Carvalh Borges M, Schmidt A, Marin-Neto JA, Pazin-Filho A. Evaluation of the endothelial glycocalyx damage in patients with acute coronary syndrome. Atherosclerosis. (2016) 247:184–8. doi: 10.1016/j.atherosclerosis.2016.02.023
93. Xue X-J, Jiang Y, Chen L, Chen S-L. Relationship between the endothelial glycocalyx and the extent of coronary atherosclerosis. Microcirculation. (2018) 25:e12504. doi: 10.1111/micc.12504
94. Nagy N, Till F, Ariane MB, Katharina R, Michael B, Holger J, et al. Inhibition of hyaluronan synthesis accelerates murine atherosclerosis. Circulation. (2010) 122:2313–22. doi: 10.1161/CIRCULATIONAHA.110.972653
95. Vink H, Constantinescu AA, Spaan JA. Oxidized lipoproteins degrade the endothelial surface layer : implications for platelet-endothelial cell adhesion. Circulation. (2000) 101:1500–2. doi: 10.1161/01.CIR.101.13.1500
96. Reitsma S, Egbrink M, Heijnen V, Megens R, Engels W, Vink H, et al. Endothelial glycocalyx thickness and platelet-vessel wall interactions during atherogenesis. Thromb Haemost. (2011) 106:939–46. doi: 10.1160/TH11-02-0133
97. Valerio L, Peters RJ, Zwinderman AH, Pinto-Sietsma S-J. Sublingual endothelial glycocalyx and atherosclerosis. A cross-sectional study. PLoS ONE. (2019) 14:e0213097. doi: 10.1371/journal.pone.0213097
98. Schierke F, Wyrwoll MJ, Wisdorf M, Niedzielski L, Maase M, Ruck T, et al. Nanomechanics of the endothelial glycocalyx contribute to Na+-induced vascular inflammation. Sci Rep. (2017) 7:46476. doi: 10.1038/srep46476
99. Koch J, Idzerda NMA, Ettema EM, Kuipers J, Dam W, van den Born J, et al. An acute rise of plasma Na + concentration associates with syndecan-1 shedding during hemodialysis. Am J Physiol. (2020) 319:F171–7. doi: 10.1152/ajprenal.00005.2020
100. Triantafyllidi H, Benas D, Vlachos S, Vlastos D, Pavlidis G, Schoinas A, et al. HDL cholesterol levels and endothelial glycocalyx integrity in treated hypertensive patients. J Clin Hypertens. (2018) 20:1615–23. doi: 10.1111/jch.13404
101. Ikonomidis I, Voumvourakis A, Makavos G, Triantafyllidi H, Pavlidis G, Katogiannis K, et al. Association of impaired endothelial glycocalyx with arterial stiffness, coronary microcirculatory dysfunction, and abnormal myocardial deformation in untreated hypertensives. J Clin Hypertens. (2018) 20:672–9. doi: 10.1111/jch.13236
103. Palombo C, Kozakova M. Arterial stiffness, atherosclerosis and cardiovascular risk: pathophysiologic mechanisms and emerging clinical indications. Vascul Pharmacol. (2016) 77:1–7. doi: 10.1016/j.vph.2015.11.083
104. Mahmoud M, Cancel L, Tarbell JM. Matrix stiffness affects glycocalyx expression in cultured endothelial cells. Front Cell Dev Biol. (2021) 9::731666. doi: 10.3389/fcell.2021.731666
105. Mahmoud M, Mayer M, Cancel LM, Bartosch AM, Mathews R, Tarbell JM. The glycocalyx core protein Glypican 1 protects vessel wall endothelial cells from stiffness-mediated dysfunction and disease. Cardiovasc Res. (2021) 117:1592–605. doi: 10.1093/cvr/cvaa201
106. Laina A, Stellos K, Stamatelopoulos K. Vascular ageing: underlying mechanisms and clinical implications. Exp Gerontol. (2018) 109:16–30. doi: 10.1016/j.exger.2017.06.007
107. Carge MJ, Liberati DM, Diebel LN. A biomimetic shock model on the effect of endothelial aging on vascular barrier properties. J Trauma Acute Care Surg. (2021) 91:849–55. doi: 10.1097/TA.0000000000003207
108. Morgan RG, Walker AE, Trott DW, Machin DR, Henson GD, Reihl KD, et al. Induced Trf2 deletion leads to aging vascular phenotype in mice associated with arterial telomere uncapping, senescence signaling, and oxidative stress. J Mol Cell Cardiol. (2019) 127:74–82. doi: 10.1016/j.yjmcc.2018.11.014
109. Machin DR, Bloom SI, Campbell RA, Phuong TTT, Gates PE, Lesniewski LA, et al. Advanced age results in a diminished endothelial glycocalyx. Am J Physiol. (2018) 315:H531–9. doi: 10.1152/ajpheart.00104.2018
110. Cheung TM, Yan JB, Fu JJ, Huang J, Yuan F, Truskey GA. Endothelial cell senescence increases traction forces due to age-associated changes in the glycocalyx and SIRT1. Cel Mol Bioeng. (2015) 8:63–75. doi: 10.1007/s12195-014-0371-6
111. Williamson KA, Hamilton A, Reynolds JA, Sipos P, Crocker I, Stringer SE, et al. Age-related impairment of endothelial progenitor cell migration correlates with structural alterations of heparan sulfate proteoglycans. Aging Cell. (2013) 12:139–47. doi: 10.1111/acel.12031
112. Machin DR, Phuong TTT, Donato AJ. The role of the endothelial glycocalyx in advanced age and cardiovascular disease. Curr Opin Pharmacol. (2019) 45:66–71. doi: 10.1016/j.coph.2019.04.011
113. Targosz-Korecka M, Jaglarz M, Malek-Zietek KE, Gregorius A, Zakrzewska A, Sitek B, et al. AFM-based detection of glycocalyx degradation and endothelial stiffening in the db/db mouse model of diabetes. Sci Rep. (2017) 7:15951. doi: 10.1038/s41598-017-16179-7
114. Li R, Xie J, Wu H, Li G, Chen J, Chen Q, et al. Syndecan-4 shedding impairs macrovascular angiogenesis in diabetes mellitus. Biochem Biophys Res Commun. (2016) 112:474: doi: 10.1016/j.bbrc.2016.03.112
115. Nieuwdorp M, van Haeften TW, Gouverneur MCLG, Mooij HL, van Lieshout MHP, Levi M, et al. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. (2006) 55:480–6. doi: 10.2337/diabetes.55.02.06.db05-1103
116. Lopez-Quintero SV, Cancel LM, Pierides A, Antonetti D, Spray DC, Tarbell JM. High glucose attenuates shear-induced changes in endothelial hydraulic conductivity by degrading the glycocalyx. PLoS ONE. (2013) 8:e78954. doi: 10.1371/journal.pone.0078954
117. Li Z, Wu N, Wang J, Zhang Q. Roles of endovascular calyx related enzymes in endothelial dysfunction and diabetic vascular complications. Front Pharmacol. (2020) 11::590614. doi: 10.3389/fphar.2020.590614
118. Kennett EC, Davies MJ. Glycosaminoglycans are fragmented by hydroxyl, carbonate, and nitrogen dioxide radicals in a site-selective manner: implications for peroxynitrite-mediated damage at sites of inflammation. Free Radic Biol Med. (2009) 47:389–400. doi: 10.1016/j.freeradbiomed.2009.05.002
119. Katsumura C, Sugiyama T, Nakamura K, Obayashi H, Hasegawa G, Oku H, et al. Effects of advanced glycation end products on hyaluronan photolysis: a new mechanism of diabetic vitreopathy. ORE. (2004) 36:327–31. doi: 10.1159/000081635
120. Nieuwdorp M, Holleman F, de Groot E, Vink H, Gort J, Kontush A, et al. Perturbation of hyaluronan metabolism predisposes patients with type 1 diabetes mellitus to atherosclerosis. Diabetologia. (2007) 50:1288–93. doi: 10.1007/s00125-007-0666-4
121. Eskens BJM, Leurgans TM, Vink H, Van Teeffelen JWGE. Early impairment of skeletal muscle endothelial glycocalyx barrier properties in diet-induced obesity in mice. Physiol Rep. (2014) 2:e00194. doi: 10.1002/phy2.194
122. Williams IL, Wheatcroft SB, Shah AM, Kearney MT. Obesity, atherosclerosis and the vascular endothelium: mechanisms of reduced nitric oxide bioavailability in obese humans. Int J Obes. (2002) 26:754–64. doi: 10.1038/sj.ijo.0801995
123. Toda N, Okamura T. Obesity impairs vasodilatation and blood flow increase mediated by endothelial nitric oxide: an overview. J Clin Pharmacol. (2013) 53:1228–39. doi: 10.1002/jcph.179
124. Ahn SJ, Le Master E, Lee JC, Phillips SA, Levitan I, Fancher IS. Differential effects of obesity on visceral versus subcutaneous adipose arteries: role of shear-activated Kir21 and alterations to the glycocalyx. Am J Physiol. (2022) 322:H156–66. doi: 10.1152/ajpheart.00399.2021
125. Mitsuda S, Uzawa K, Sawa M, Ando T, Yoshikawa T, Miyao H, et al. Vascular endothelial glycocalyx plays a role in the obesity paradox according to intravital observation. Front Cardiovasc Med. (2021) 8:727888. doi: 10.3389/fcvm.2021.727888
126. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Chapter six - cell biology of ischemia/reperfusion injury. In: KW Jeon, editor, International Review of Cell and Molecular Biology. Cambridge, MA: Academic Press (2012). p. 229–317 doi: 10.1016/B978-0-12-394309-5.00006-7
127. Banz Y, Rieben R. Role of complement and perspectives for intervention in ischemia-reperfusion damage. Ann Med. (2012) 44:205–17. doi: 10.3109/07853890.2010.535556
128. Kilgore KS, Friedrichs GS, Homeister JW, Lucchesi BR. The complement system in myocardial ischaemia/reperfusion injury. Cardiovasc Res. (1994) 28:437–44. doi: 10.1093/cvr/28.4.437
129. Carden DL, Granger DN. Pathophysiology of ischaemia–reperfusion injury. J Pathol. (2000) 190:255–66. doi: 10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6
130. Platts SH, Linden J, Duling BR. Rapid modification of the glycocalyx caused by ischemia-reperfusion is inhibited by adenosine A2A receptor activation. Am J Physiol. (2003) 284:H2360–7. doi: 10.1152/ajpheart.00899.2002
131. Beresewicz A, Czarnowska E, Maczewski M. Ischemic preconditioning and superoxide dismutase protect against endothelial dysfunction and endothelium glycocalyx disruption in the postischemic guinea-pig hearts. Mol Cell Biochem. (1998) 186:87–97. doi: 10.1023/A:1006867214448
132. Passov A, Schramko A, Salminen U-S, Aittomäki J, Andersson S, Pesonen E. Endothelial glycocalyx during early reperfusion in patients undergoing cardiac surgery. PLoS ONE. (2021) 16:e0251747. doi: 10.1371/journal.pone.0251747
133. Rubio-Gayosso I, Platts SH, Duling BR. Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am J Physiol. (2006) 290:H2247–56. doi: 10.1152/ajpheart.00796.2005
134. Rehm M, Dirk B, Frank C, Peter C, Manfred T, Matthias J, et al. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation. (2007) 116:1896–906. doi: 10.1161/CIRCULATIONAHA.106.684852
135. DellaValle B, Henrik H, Flemming F, Iversen Helle K, Jørgen R, Casper H. Multiple soluble components of the glycocalyx are increased in patient plasma after ischemic stroke. Stroke. (2019) 50:2948–51. doi: 10.1161/STROKEAHA.119.025953
136. Morgan BP. The complement system: an overview. In: BP Morgan, editor, Complement Methods and Protocols. Methods in Molecular Biology. Totowa, NJ: Humana Press (2000). p. 1–13 doi: 10.1385/1-59259-056-X:1
137. Diepenhorst GMP, van Gulik TM, Hack CE. Complement-mediated ischemia-reperfusion injury: lessons learned from animal and clinical studies. Ann Surg. (2009) 249:889–99. doi: 10.1097/SLA.0b013e3181a38f45
138. Nijmeijer R, Krijnen PaJ, Assink J, Klaarenbeek MaR, Lagrand WK, Veerhuis R, et al. C-reactive protein and complement depositions in human infarcted myocardium are more extensive in patients with reinfarction or upon treatment with reperfusion. Eur J Clin Investig. (2004) 34:803–10. doi: 10.1111/j.1365-2362.2004.01425.x
139. Chaban V, Nakstad ER, Stær-Jensen H, Schjalm C, Seljeflot I, Vaage J, et al. Complement activation is associated with poor outcome after out-of-hospital cardiac arrest. Resuscitation. (2021) 166:129–36. doi: 10.1016/j.resuscitation.2021.05.038
140. Bro-Jeppesen J, Johansson PI, Hassager C, Wanscher M, Ostrowski SR, Bjerre M, et al. Endothelial activation/injury and associations with severity of post-cardiac arrest syndrome and mortality after out-of-hospital cardiac arrest. Resuscitation. (2016) 107:71–9. doi: 10.1016/j.resuscitation.2016.08.006
141. Banz Y, Hess OM, Robson SC, Csizmadia E, Mettler D, Meier P, et al. Attenuation of myocardial reperfusion injury in pigs by Mirococept, a membrane-targeted complement inhibitor derived from human CR1. Cardiovasc Res. (2007) 76:482–93. doi: 10.1016/j.cardiores.2007.07.016
142. Atkinson C, He S, Morris K, Qiao F, Casey S, Goddard M, et al. Targeted complement inhibitors protect against posttransplant cardiac ischemia and reperfusion injury and reveal an important role for the alternative pathway of complement activation. J Immunol. (2010) 185:7007–13. doi: 10.4049/jimmunol.1001504
143. Banz Y, Hess OM, Robson SC, Mettler D, Meier P, Haeberli A, et al. Locally targeted cytoprotection with dextran sulfate attenuates experimental porcine myocardial ischaemia/reperfusion injury. Eur Heart J. (2005) 26:2334–43. doi: 10.1093/eurheartj/ehi421
144. Langford-Smith A, Day AJ, Bishop PN, Clark SJ. Complementing the sugar code: role of GAGs and sialic acid in complement regulation. Front Immunol. (2015) 6:25. doi: 10.3389/fimmu.2015.00025
145. Wuillemin WA, Velthuis H, Lubbers YT, Ruig CP de, Eldering E, Hack CE. Potentiation of C1 inhibitor by glycosaminoglycans: dextran sulfate species are effective inhibitors of in vitro complement activation in plasma. J Immunol. (1997) 159:1953–60.
146. Busche MN, Pavlov V, Takahashi K, Stahl GL. Myocardial ischemia and reperfusion injury is dependent on both IgM and mannose-binding lectin. Am J Physiol. (2009) 297:H1853–9. doi: 10.1152/ajpheart.00049.2009
147. Chappell D, Heindl B, Jacob M, Annecke T, Chen C, Rehm M, et al. Sevoflurane reduces leukocyte and platelet adhesion after ischemia-reperfusion by protecting the endothelial glycocalyx. Anesthesiology. (2011) 115:483–91. doi: 10.1097/ALN.0b013e3182289988
148. Ma Y, Yang X, Chatterjee V, Meegan JE, Beard RS, Yuan SY. Role of neutrophil extracellular traps and vesicles in regulating vascular endothelial permeability. Front Immunol. (2019) 10:1037. doi: 10.3389/fimmu.2019.01037
149. Sorvillo N, Cherpokova D, Martinod K, Wagner DD. Extracellular DNA NET-works with dire consequences for health. Circ Res. (2019) 125:470–88. doi: 10.1161/CIRCRESAHA.119.314581
150. Meegan JE, Yang X, Beard RS, Jannaway M, Chatterjee V, Taylor-Clark TE, et al. Citrullinated histone 3 causes endothelial barrier dysfunction. Biochem Biophys Res Commun. (2018) 503:1498–502. doi: 10.1016/j.bbrc.2018.07.069
151. Freeman CG, Parish CR, Knox KJ, Blackmore JL, Lobov SA, King DW, et al. The accumulation of circulating histones on heparan sulphate in the capillary glycocalyx of the lungs. Biomaterials. (2013) 34:5670–6. doi: 10.1016/j.biomaterials.2013.03.091
152. Zhang Y, Haeger SM, Yang Y, Dailey KL, Ford JA, Schmidt EP. Circulating heparan sulfate fragments attenuate histone-induced lung injury independently of histone binding. Shock. (2017) 48:666–73. doi: 10.1097/SHK.0000000000000907
153. Shah M, He Z, Rauf A, Beikoghli Kalkhoran S, Heiestad CM, Stensløkken K-O, et al. Extracellular histones are a target in myocardial ischaemia–reperfusion injury. Cardiovasc Res. (2021) 2021:cvab139. doi: 10.1093/cvr/cvab139
154. Mueller M, Herzog C, Larmann J, Schmitz M, Hilfiker-Kleiner D, Gessner JE, et al. The receptor for activated complement factor 5 (C5aR) conveys myocardial ischemic damage by mediating neutrophil transmigration. Immunobiology. (2013) 218:1131–8. doi: 10.1016/j.imbio.2013.03.006
155. Annecke T, Fischer J, Hartmann H, Tschoep J, Rehm M, Conzen P, et al. Shedding of the coronary endothelial glycocalyx: effects of hypoxia/reoxygenation vs. ischaemia/reperfusion. Br J Anaesth. (2011) 107:679–86. doi: 10.1093/bja/aer269
156. Becker BF, Jacob M, Leipert S, Salmon AHJ, Chappell D. Degradation of the endothelial glycocalyx in clinical settings: searching for the sheddases. Br J Clin Pharmacol. (2015) 80:389–402. doi: 10.1111/bcp.12629
157. Lee S-J, Lee C, Kang S, Park I, Kim YH, Kim SK, et al. Angiopoietin-2 exacerbates cardiac hypoxia and inflammation after myocardial infarction. J Clin Invest. (2018) 128:5018–33. doi: 10.1172/JCI99659
158. Bruegger D, Jacob M, Rehm M, Loetsch M, Welsch U, Conzen P, et al. Atrial natriuretic peptide induces shedding of endothelial glycocalyx in coronary vascular bed of guinea pig hearts. Am J Physiol. (2005) 289:H1993–9. doi: 10.1152/ajpheart.00218.2005
159. Mulivor AW, Lipowsky HH. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am J Physiol. (2004) 286:H1672–80. doi: 10.1152/ajpheart.00832.2003
160. Romanic AM, Harrison SM, Bao W, Burns-Kurtis CL, Pickering S, Gu J, et al. Myocardial protection from ischemia/reperfusion injury by targeted deletion of matrix metalloproteinase-9. Cardiovasc Res. (2002) 54:549–58. doi: 10.1016/S0008-6363(02)00254-7
161. Lalu MM, Pasini E, Schulze CJ, Ferrari-Vivaldi M, Ferrari-Vivaldi G, Bachetti T, et al. Ischaemia–reperfusion injury activates matrix metalloproteinases in the human heart. Eur Heart J. (2005) 26:27–35. doi: 10.1093/eurheartj/ehi007
162. Taraboletti G, D'Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol. (2002) 160:673–80. doi: 10.1016/S0002-9440(10)64887-0
163. Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. (2011) 41:271–90. doi: 10.1007/s00726-010-0689-x
164. Cheung PY, Sawicki G, Wozniak M, Wang W, Radomski MW, Schulz R. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation. (2000) 101:1833–9. doi: 10.1161/01.CIR.101.15.1833
165. Ali MM, Mahmoud AM, Le Master E, Levitan I, Phillips SA. Role of matrix metalloproteinases and histone deacetylase in oxidative stress-induced degradation of the endothelial glycocalyx. Am J Physiol Heart Circ Physiol. (2019) 316:H647–63. doi: 10.1152/ajpheart.00090.2018
166. Ko K, Suzuki T, Ishikawa R, Hattori N, Ito R, Umehara K, et al. Ischemic stroke disrupts the endothelial glycocalyx through activation of proHPSE via acrolein exposure. J Biol Chem. (2020) 295:18614–24. doi: 10.1074/jbc.RA120.015105
167. Al'Qteishat A, Gaffney J, Krupinski J, Rubio F, West D, Kumar S, et al. Changes in hyaluronan production and metabolism following ischaemic stroke in man. Brain. (2006) 129:2158–76. doi: 10.1093/brain/awl139
168. Korf-Klingebiel M, Reboll Marc R, Grote K, Schleiner H, Wang Y, Wu X, et al. Heparan sulfate–editing extracellular sulfatases enhance VEGF bioavailability for ischemic heart repair. Circ Res. (2019) 125:787–801. doi: 10.1161/CIRCRESAHA.119.315023
169. Hoogewerf AJ, Kuschert GSV, Proudfoot AEI, Borlat F, Clark-Lewis I, Power CA, et al. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry. (1997) 36:13570–8. doi: 10.1021/bi971125s
170. Talsma DT, Poppelaars F, Dam W, Meter-Arkema AH, Vivès RR, Gál P, et al. MASP-2 is a heparin-binding protease; identification of blocking oligosaccharides. Front Immunol. (2020) 11:732. doi: 10.3389/fimmu.2020.00732
171. Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. (2002) 195:99–111. doi: 10.1084/jem.20001858
172. Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by toll-like receptor 4. J Immunol. (2002) 168:5233–9. doi: 10.4049/jimmunol.168.10.5233
173. Olivares-Silva F, Landaeta R, Aránguiz P, Bolivar S, Humeres C, Anfossi R, et al. Heparan sulfate potentiates leukocyte adhesion on cardiac fibroblast by enhancing Vcam-1 and Icam-1 expression. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:831–42. doi: 10.1016/j.bbadis.2017.12.002
174. Collins LE, Troeberg L. Heparan sulfate as a regulator of inflammation and immunity. J Leukoc Biol. (2019) 105:81–92. doi: 10.1002/JLB.3RU0618-246R
175. Strand ME, Aronsen JM, Braathen B, Sjaastad I, Kvaløy H, Tønnessen T, et al. Shedding of syndecan-4 promotes immune cell recruitment and mitigates cardiac dysfunction after lipopolysaccharide challenge in mice. J Mol Cell Cardiol. (2015) 88:133–44. doi: 10.1016/j.yjmcc.2015.10.003
176. Salek-Ardakani S, Arrand JR, Shaw D, Mackett M. Heparin and heparan sulfate bind interleukin-10 and modulate its activity. Blood. (2000) 96:1879–88. doi: 10.1182/blood.V96.5.1879
177. Rops ALWMM, van der Vlag J, Lensen JFM, Wijnhoven TJM, van den Heuvel LPWJ, van Kuppevelt TH, et al. Heparan sulfate proteoglycans in glomerular inflammation. Kidney Int. (2004) 65:768–85. doi: 10.1111/j.1523-1755.2004.00451.x
178. Hayashida K, Parks WC, Park PW. Syndecan-1 shedding facilitates the resolution of neutrophilic inflammation by removing sequestered CXC chemokines. Blood. (2009) 114:3033–43. doi: 10.1182/blood-2009-02-204966
179. Zhang C, Guo F, Chang M, Zhou Z, Yi L, Gao C, et al. Exosome-delivered syndecan-1 rescues acute lung injury via a FAK/p190RhoGAP/RhoA/ROCK/NF-κB signaling axis and glycocalyx enhancement. Exp Cell Res. (2019) 384:111596. doi: 10.1016/j.yexcr.2019.111596
180. Xu J, Park PW, Kheradmand F, Corry DB. Endogenous attenuation of allergic lung inflammation by syndecan-1. J Immunol. (2005) 174:5758–65. doi: 10.4049/jimmunol.174.9.5758
181. Eustace AD, McNaughton EF, King S, Kehoe O, Kungl A, Mattey D, et al. Soluble syndecan-3 binds chemokines, reduces leukocyte migration in vitro and ameliorates disease severity in models of rheumatoid arthritis. Arthritis Res Ther. (2019) 21:172. doi: 10.1186/s13075-019-1939-2
182. Finsen AV, Woldbaek PR Li J, Wu J, Lyberg T, Tønnessen T, Christensen G. Increased syndecan expression following myocardial infarction indicates a role in cardiac remodeling. Physiol Genomics. (2004) 16:301–8. doi: 10.1152/physiolgenomics.00144.2002
183. Potter DR, Jiang J, Damiano ER. The recovery time course of the endothelial cell glycocalyx in vivo and its implications in vitro. Circ Res. (2009) 104:1318–25. doi: 10.1161/CIRCRESAHA.108.191585
184. Salmon AHJ, Neal CR, Sage LM, Glass CA, Harper SJ, Bates DO. Angiopoietin-1 alters microvascular permeability coefficients in vivo via modification of endothelial glycocalyx. Cardiovasc Res. (2009) 83:24–33. doi: 10.1093/cvr/cvp093
185. Mulivor AW, Lipowsky HH. Inhibition of glycan shedding and leukocyte-endothelial adhesion in postcapillary venules by suppression of matrixmetalloprotease activity with doxycycline. Microcirculation. (2009) 16:657–66. doi: 10.3109/10739680903133714
186. Zeng Y, Adamson RH, Curry F-RE, Tarbell JM. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am J Physiol. (2013) 306:H363–72. doi: 10.1152/ajpheart.00687.2013
187. Huang L, Zhang X, Ma X, Zhang D, Li D, Feng J, et al. Berberine alleviates endothelial glycocalyx degradation and promotes glycocalyx restoration in LPS-induced ARDS. Int Immunopharmacol. (2018) 65:96–107. doi: 10.1016/j.intimp.2018.10.001
188. Chappell D, Jacob M, Hofmann-Kiefer K, Rehm M, Welsch U, Conzen P, et al. Antithrombin reduces shedding of the endothelial glycocalyx following ischaemia/reperfusion. Cardiovasc Res. (2009) 83:388–96. doi: 10.1093/cvr/cvp097
189. Chappell D, Jacob M, Hofmann-Kiefer K, Bruegger D, Rehm M, Conzen P, et al. Hydrocortisone preserves the vascular barrier by protecting the endothelial glycocalyx. Anesthesiology. (2007) 107:776–84. doi: 10.1097/01.anes.0000286984.39328.96
190. Kazuma S, Tokinaga Y, Kimizuka M, Azumaguchi R, Hamada K, Yamakage M. Sevoflurane promotes regeneration of the endothelial glycocalyx by upregulating sialyltransferase. J Surg Res. (2019) 241:40–7. doi: 10.1016/j.jss.2019.03.018
191. Masola V, Onisto M, Zaza G, Lupo A, Gambaro G. A new mechanism of action of sulodexide in diabetic nephropathy: inhibits heparanase-1 and prevents FGF-2-induced renal epithelial-mesenchymal transition. J Transl Med. (2012) 10:213. doi: 10.1186/1479-5876-10-213
192. Cooper S, Teoh H, Campeau MA, Verma S, Leask RL. Empagliflozin restores the integrity of the endothelial glycocalyx in vitro. Mol Cell Biochem. (2019) 459:121–30. doi: 10.1007/s11010-019-03555-2
193. Torres Filho IP, Torres LN, Salgado C, Dubick MA. Novel adjunct drugs reverse endothelial glycocalyx damage after hemorrhagic shock in rats. Shock. (2017) 48:583–9. doi: 10.1097/SHK.0000000000000895
194. Kozar RA, Peng Z, Zhang R, Holcomb JB, Pati S, Park P, et al. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth Analg. (2011) 112:1289–95. doi: 10.1213/ANE.0b013e318210385c
195. Zhao H, Zhu Y, Zhang J, Wu Y, Xiang X, Zhang Z, et al. The beneficial effect of HES on vascular permeability and its relationship with endothelial glycocalyx and intercellular junction after hemorrhagic shock. Front Pharmacol. (2020) 11:597. doi: 10.3389/fphar.2020.00597
196. Rienks M, Carai P, van Teeffelen J, Eskens B, Verhesen W, Hemmeryckx B, et al. SPARC preserves endothelial glycocalyx integrity, and protects against adverse cardiac inflammation and injury during viral myocarditis. Matrix Biol. (2018) 74:21–34. doi: 10.1016/j.matbio.2018.04.015
197. Laumonier T, Walpen AJ, Maurus CF, Mohacsi PJ, Matozan KM, Korchagina EY, et al. Dextran sulfate acts as an endothelial cell protectant and inhibits human complement and natural killer cell-mediated cytotoxicity against porcine cells. Transplantation. (2003) 76:838. doi: 10.1097/01.TP.0000078898.28399.0A
198. Laumonier T, Mohacsi PJ, Matozan KM, Banz Y, Haeberli A, Korchagina EY, et al. Endothelial cell protection by dextran sulfate: a novel strategy to prevent acute vascular rejection in xenotransplantation. Am J Transplant. (2004) 4:181–7. doi: 10.1046/j.1600-6143.2003.00306.x
199. Laumonier T, Walpen AJ, Matozan KM, Korchagina EY, Bovin NV, Haeberli A, et al. Multimeric tyrosine sulfate acts as an endothelial cell protectant and prevents complement activation in xenotransplantation models. Xenotransplantation. (2004) 11:262–8. doi: 10.1111/j.1399-3089.2004.00125.x
200. Ramnath RD, Butler MJ, Newman G, Desideri S, Russell A, Lay AC, et al. Blocking matrix metalloproteinase-mediated syndecan-4 shedding restores the endothelial glycocalyx and glomerular filtration barrier function in early diabetic kidney disease. Kidney Int. (2020) 97:951–65. doi: 10.1016/j.kint.2019.09.035
201. Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase-9: many shades of function in cardiovascular disease. Physiology. (2013) 28:391–403. doi: 10.1152/physiol.00029.2013
202. Zeng Y, Liu X-H, Tarbell J, Fu B. Sphingosine 1-phosphate induced synthesis of glycocalyx on endothelial cells. Exp Cell Res. (2015) 339:90–5. doi: 10.1016/j.yexcr.2015.08.013
203. Lipowsky HH, Lescanic A. Inhibition of inflammation induced shedding of the endothelial glycocalyx with low molecular weight heparin. Microvasc Res. (2017) 112:72–8. doi: 10.1016/j.mvr.2017.03.007
204. Huang X, Han S, Liu X, Wang T, Xu H, Xia B, et al. Both UFH and NAH alleviate shedding of endothelial glycocalyx and coagulopathy in LPS-induced sepsis. Exp Ther Med. (2020) 19:913–22. doi: 10.3892/etm.2019.8285
Keywords: glycocalyx, endothelial cell (EC), cardiovascular risk factor, ischemia/reperfusion injury, inflammation, therapeutic target, heparan sulfate (HS), atherosclerosis
Citation: Milusev A, Rieben R and Sorvillo N (2022) The Endothelial Glycocalyx: A Possible Therapeutic Target in Cardiovascular Disorders. Front. Cardiovasc. Med. 9:897087. doi: 10.3389/fcvm.2022.897087
Received: 15 March 2022; Accepted: 21 April 2022;
Published: 13 May 2022.
Edited by:
Hideshi Okada, Gifu University, JapanReviewed by:
David Astapenko, University Hospital Hradec Kralove, CzechiaKristina Kusche-Vihrog, University of Lübeck, Germany
Copyright © 2022 Milusev, Rieben and Sorvillo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicoletta Sorvillo, bmljb2xldHRhLnNvcnZpbGxvQGRibXIudW5pYmUuY2g=