
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 13 June 2022
Sec. Cardio-Oncology
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.896792
This article is part of the Research Topic HF2Cancer: Exploring bidirectional interaction between cardiovascular diseases and cancer View all 18 articles
 Guoxia Zhang1†
Guoxia Zhang1† Chao Yuan2†
Chao Yuan2† Xin Su1†Jianzhen Zhang3Priyanka Gokulnath4Gururaja Vulugundam5Guoping Li4Xinyu Yang6
Xin Su1†Jianzhen Zhang3Priyanka Gokulnath4Gururaja Vulugundam5Guoping Li4Xinyu Yang6 Na An3
Na An3 Can Liu1
Can Liu1 Wanli Sun1
Wanli Sun1 Hengwen Chen1
Hengwen Chen1 Min Wu1Shipeng Sun1*
Min Wu1Shipeng Sun1* Yanwei Xing1*
Yanwei Xing1*Anthracyclines (ANTs) are a class of anticancer drugs widely used in oncology. However, the clinical application of ANTs is limited by their cardiotoxicity. The mechanisms underlying ANTs-induced cardiotoxicity (AIC) are complicated and involve oxidative stress, inflammation, topoisomerase 2β inhibition, pyroptosis, immunometabolism, autophagy, apoptosis, ferroptosis, etc. Ferroptosis is a new form of regulated cell death (RCD) proposed in 2012, characterized by iron-dependent accumulation of reactive oxygen species (ROS) and lipid peroxidation. An increasing number of studies have found that ferroptosis plays a vital role in the development of AIC. Therefore, we aimed to elaborate on ferroptosis in AIC, especially by doxorubicin (DOX). We first summarize the mechanisms of ferroptosis in terms of oxidation and anti-oxidation systems. Then, we discuss the mechanisms related to ferroptosis caused by DOX, particularly from the perspective of iron metabolism of cardiomyocytes. We also present our research on the prevention and treatment of AIC based on ferroptosis. Finally, we enumerate our views on the development of drugs targeting ferroptosis in this emerging field.
With the advancements in medical technology, while the survival time of cancer patients has been prolonged, cardiovascular toxicity has become one of the most severe complications of cancer treatment (1, 2). Studies have shown that cancer survivors are at an eight-times higher risk of developing cardiovascular disease (CVD) than the general population (3). Anthracyclines (ANTs) are a class of chemotherapy drugs commonly used in clinical practice that significantly improve the survival rate of patients. However, the use of ANTs is restricted due to their cardiotoxic effects (2, 4). The incidence of left ventricular dysfunction, which is up to 48%, is positively correlated with dose (2). In some cancer survivors, the death rate of CVDs even exceeds that of their primary cancers (5). Therefore, it is necessary to explore the mechanism of cardiotoxicity caused by cancer therapy. The mechanisms of ANTs-induced cardiotoxicity (AIC) involve oxidative stress, inflammation, topoisomerase 2β inhibition, pyroptosis, immunometabolism, autophagy, apoptosis, etc. (6–8). Besides, in recent years, more and more studies have shown that ferroptosis plays a vital role in AIC (9, 10). Inhibiting the ferroptosis of cardiomyocytes can reduce AIC, which may be a novel prevention and treatment strategy in cardio-oncology.
Ferroptosis is a new form of regulated cell death (RCD) different from apoptosis, necrosis, necroptosis, pyroptosis, and autophagy. It is characterized by iron overload and reactive oxygen species (ROS) accumulation, resulting in lipid peroxidation of cell membranes (11, 12). A few decades ago, it was demonstrated that glutamate could inhibit the uptake of cystine, leading to a decrease in glutathione (GSH) levels within cells, thereby causing oxidative death of cells, and termed this process as “oxytosis” (13, 14). We believe that doxorubicin (DOX) can induce ferroptosis in cardiomyocytes through the following mechanisms: firstly, by regulating iron homeostasis-related proteins and iron-responsive elements (IREs)/iron regulatory proteins (IRPs), leading to increased iron levels in cardiomyocytes; secondly, DOX can increase ROS, thereby causing cell membrane lipid peroxidation. The nuclear factor (erythroid-derived 2)-like 2 (Nrf2) signaling pathway plays an important role. As the central organelle for ROS generation and the site where iron accumulation may occur, mitochondria are crucial for developing doxorubicin-induced cardiomyopathy (DIC). Further, we summarize the current treatments to prevent and treat AIC by inhibiting the ferroptosis of cardiomyocytes. Finally, we provide our future perspectives on this emerging field.
Ferroptosis is an iron-dependent lipid peroxidation induced novel RCD, caused by redox imbalances between the oxidant and antioxidant systems. Antioxidant systems include the Cyst(e)ine-GSH-glutathione peroxidase 4 (GPX4) pathway, the ferroptosis suppressor protein 1 (FSP1)-coenzyme Q10 (CoQ10)-nicotinamide adenine dinucleotide phosphate (NADPH) pathway, the GTP cyclohydrolase-1 (GCH1)-tetrahydrobiopterin (BH4) pathway, etc. (15, 16). Intracellular iron overload is a necessary condition for ferroptosis. Therefore, lipid peroxidation is the most common cause of ferroptosis (15) (Figure 1).
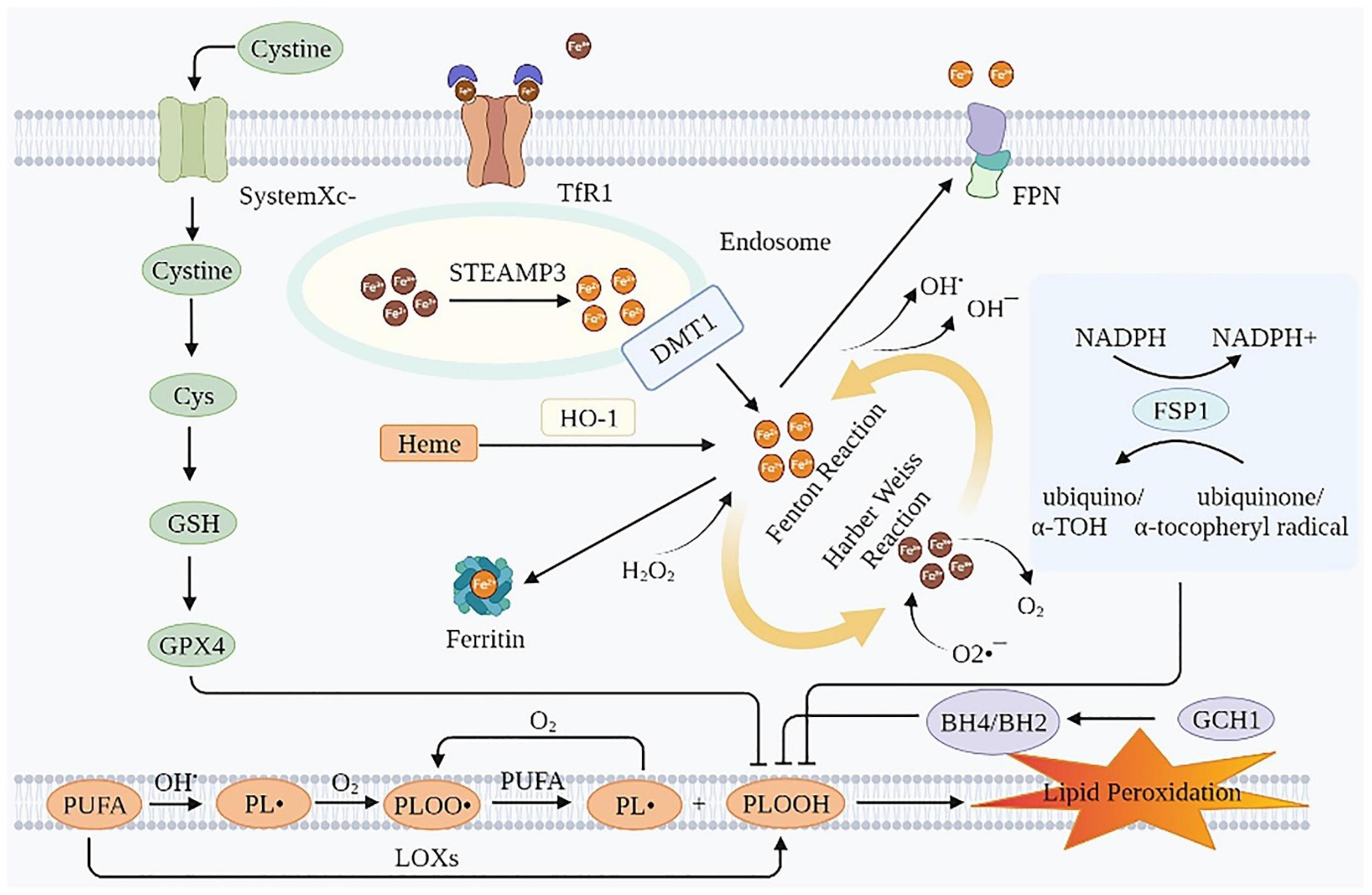
Figure 1. Mechanisms of ferroptosis. Ferroptosis is essentially an iron-dependent lipid peroxidation. Intracellular iron overload is a necessary condition for ferroptosis, and lipid peroxidation is the presentation form of ferroptosis. TfR1, transferrin receptor 1; FPN, ferroportin; Cys, cysteine; GSH, glutathione; GPX4, glutathione peroxidase 4; STEAP3, six-transmembrane epithelial antigen of the prostate 3; DMT1, divalent metal transporter 1; HO-1, heme oxygenase 1; NADPH, nicotinamide adenine dinucleotide phosphate; FSP1, ferroptosis suppressor protein 1; PUFA, polyunsaturated fatty acids; GCH1, GTP cyclohydrolase-1; BH4/BH2, tetrahydrobiopterin/dihydrobiopterin; LOXs, lipoxygenases; PL•, phospholipid radical; PLOO•, phospholipid peroxyl radical; PLOOH, phospholipid hydroperoxide; α-TOH, α-tocopherol.
Iron overload is a prerequisite of ferroptosis. The erastin-induced ferroptosis was inhibited by deferoxamine (DFO, an iron chelator), evidenced by increased cell viability and decreased lipid ROS production in HT-1080 cells. In contrast, the erastin-induced ferroptosis was triggered by incubation with three different exogenous iron supplements (11). In intestinal ischemia/reperfusion-induced acute lung injury model of C57BL/6 mice, the injection of Fe (15 mg/kg) aggravated lung injury and pulmonary edema, while the injection of ferrostatin-1 (Fer-1, 5 mg/kg) rescued this injury (17). Iron transport involves import, storage, and export (18, 19). Circulating iron exists in the form of ferric iron (Fe3+) by binding to transferrin (Tf). Fe3+ enters the endosome through membrane protein transferrin receptor 1 (TfR1). Then, Fe3+ is reduced to ferrous iron (Fe2+) by the iron reductase activity of the six-transmembrane epithelial antigen of the prostate 3. The divalent metal transporter 1 (DMT1, also known as SLC11A2) releases Fe2+ from the endosome into the cytoplasm. While part of the Fe2+ in the cytoplasm is stored as ferritin, part is oxidized to Fe3+ and transported outside the cell by the membrane protein iron transporter ferroportin (FPN, an iron efflux pump, also known as SLC11A3), and the rest is stored in the labile iron pool (LIP) of the cytoplasm or mitochondria (20). The iron in LIP spontaneously undergoes redox reactions, namely, Fenton and Harber Weiss reactions, to generate ROS, which in turn leads to lipid peroxidation (21). Moreover, iron and iron derivatives, such as heme or [Fe-S] clusters, also affect ferroptosis as they act on the active centers of ROS producing enzymes, such as lipoxygenase (LOX), cytochrome P450, NADPH oxidase and so on (22). Therefore, iron overload is essential for ferroptosis. Maintaining the LIP within a relatively narrow concentration range is crucial for preventing ferroptosis. Using DFO could prevent cell death caused by erastin and RSL3 (11, 23).
In this process, the core negative regulators of ferroptosis are ferritin heavy chain (FTH) (24–27) and FPN (28), and the core positive regulators of ferroptosis are Tf (29–31), TfR1 (24, 26, 31–33), and DMT1 (24, 34, 35). These iron homeostasis proteins involved in iron uptake, storage, utilization, and efflux from cells are regulated by IREs/IRPs (36, 37). In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced PD mice models, apoferritin inhibited ferroptosis by downregulating the iron importers DMT1 and FSP1, and conversely upregulating long-chain acyl-CoA synthetase 4 (38). The lipopolysaccharide then increased the expression of nuclear receptor co-activator 4, which directly interacted with ferritin and degraded ferritin in a ferritin phagocytosis-dependent manner. It then released a large amount of iron (39). In addition, heme oxygenase 1 (HO-1) mediates the release of free iron from heme, resulting in the accumulation of Fe2+ in LIP, which also exacerbates ferroptosis (40, 41).
Fe2+ in LIP can spontaneously undergo redox reactions to produce ROS, including both Fenton and Harber-Weiss reactions. The chemical equations are as follows (42):
The electron transport system of mitochondria is the primary source of H2O2 and active oxygen (O2•–) (43). ROS, generated by the above mechanism, causes damage to the biomembrane in two ways. One is an enzyme-independent way, that is: Firstly, the hydroxyl radical (OH•) combines with polyunsaturated fatty acids (PUFA) on the biomembrane to generate the phospholipid radical (PL•). Secondly, PL• reacts with O2 generating a phospholipid peroxyl radical (PLOO•). Thirdly, PLOO• reacts with PUFA to generate phospholipid hydroperoxide (PLOOH) and PL•, which can react again with O2, forming a vicious circle. The other is the enzymatic way, that is, PUFA generates PLOOH under the action of LOXs. However, the detailed mechanism of PLOOH resulting in ferroptotic lipid peroxidative cell death remains obscure. Continued oxidation and consumption of PUFA may alter the structure of lipid pores, ultimately leading to compromised membrane integrity. In addition, PLOOH may be decomposed into active toxic aldehydes, such as 4-hydroxy-2-nonenal or malondialdehyde (MDA), causing cytotoxic effects (44). The hallmark of ferroptosis is the iron-dependent accumulation of lipid hydroperoxides to cell-lethal levels, especially peroxidized phosphatidylethanolamine (PEox). However, only a few studies detected and quantified these directly. In heart transplantation mice models, hydroperoxy-arachidonoyl-phosphatidylethanolamine (HOO-C20:4/C18:0-PE) was elevated and the resulting ferroptosis triggered early inflammation by recruiting neutrophils. In IRI mice models, the abundance of several hydroxyeicosatetraenoic acids (HETE) (such as 5-HETE, 11-HETE, 12-HETE, and 15-HETE) and epoxyeicosatrienoic acid species were increased (45). Besides, in RSL3-induced ferroptosis in H9C2 cardiomyocytes, via LC/MS, three significant species of hydroperoxy-PE were found to be up-regulated, namely, PE(36:4)-OOH, PE(38:4)-OOH, and PE(40:4)-OOH (46). Sparvero et al. used gas cluster ion beam secondary ion mass spectrometry imaging with a 70 keV (H2O) (n) (+) (n > 28000) cluster ion beam to visualize them at the single-cell and subcellular levels (47). The ferroptosis inhibitor, Fer-1 inhibited ferroptosis by reducing PLOOH.
The cyst(e)ine-GSH-GPX4 pathway is considered the canonical pathway for restricting ferroptosis. System Xc–, a heterodimeric 12-pass transmembrane cystine–glutamate anti-porter, consists of a xCT light chain (also known as SLC7A11) that mediates cystine transport specificity, and a 4F2 heavy chain (also known as SLC3A2) (48), plays an important role in this process. SLC7A11 transports extracellular cystine, which is rapidly reduced to cysteine (Cys) by an NADPH-consuming reduction process. Cys participates in the production of GSH, a fundamental component of GPX4 (49). GPX4 is the primary inhibitor of ferroptosis. It can prevent lipid peroxidation by reducing PLOOH to non-toxic phospholipid alcohols (49). Cardiac impairments were ameliorated in GPX4 Tg mice and exacerbated in GPX4 heterodeletion mice. In cultured cardiomyocytes, GPX4 overexpression prevented DOX-induced ferroptosis (50). Surprisingly, a recent study showed that in non-small-cell lung cancer cell lines, cystine starvation induces an unexpected accumulation of γ-glutamyl-peptides under the influence of glutamate-cysteine ligase catalytic subunit, which limits the accumulation of glutamate, thereby protecting against ferroptosis (51). In addition, methionine can be used as one of the sources of intracellular cystine through the trans-sulfuration pathway (49). GPX4 is a selenoenzyme and its biosynthesis relies on the co-translational incorporation of selenocysteine (49). Selenium augments GPX4 and other genes by enhancing adaptive transcription factors TFAP2c and Sp1 to protect the cells from ferroptosis (52).
The FSP1-CoQ10-NADPH pathway exists as a GPX4-independent one. FSP1 catalyzes the transformation of CoQ10 into ubiquinol, which is an excellent radical-trapping antioxidant in phospholipids and lipoproteins (53–55). Furthermore, the pathway can reduce oxidized α-tocopheryl radical to its non-radical form, increasing antioxidant capacity (56). The MDM2-MDMX complex is a negative regulator of FSP1. It changes the activity of PPARα, resulting in a decrease in the level of FSP1 protein and an increase in the level of CoQ10 (57). MiR-4443, whose target gene is METLL3, inhibited FSP1-mediated ferroptosis induced by cisplatin treatment in vitro and enhanced tumor growth in vivo (58).
The GTP-GCH1-BH4 pathway is also not dependent on GPX4. BH4/dihydrobiopterin synthesis by GCH1-expressing cells caused lipid remodeling, suppressing ferroptosis by selectively preventing depletion of phospholipids with two polyunsaturated fatty acyl tails (59). Using a co-culture model system, iNOS/NO (•) in M1 macrophages has been confirmed to inhibit the effect of NO (•) on epithelial cells by inhibiting phospholipid peroxidation, especially the generation of 15-HpETE-PE signal that promotes ferroptosis. It is an intercellular mechanism that distantly prevents the ferroptosis of epithelial cells stimulated by Pseudomonas aeruginosa (60).
Nuclear factor (erythroid-derived 2)-like 2 signaling is implicated in many molecular aspects of ferroptosis, including glutathione homeostasis, mitochondrial function, and lipid metabolism (61, 62). The Nrf2-Focad-Fak signaling pathway is closely related to ferroptosis caused by Cys deprivation. In non-small-cell lung carcinoma, brusatol (an Nrf2 inhibitor) was added based on ferroptosis inducer erastin or RSL3. The therapeutic effect based on ferroptosis was better than single treatment in vivo and in vitro (63). In immunocompetent mice and humanized mice, ZVI-NP, a dual-functional nanomedicine, enhanced the degradation of Nrf2 by GSK3/β-TrCP through AMP-activated protein kinase (AMPK)/rapamycin activation, leading to cancer-specific ferroptosis of lung cancer cells (64).
The antioxidant system of the heart is very complex. In addition to the above three major systems, the antioxidant system of the heart also includes some other elements that inhibit ferroptosis. O2•–, OH•, OH–, H2O2, PL•, PLOO•, PLOOH, ROS, etc. play important roles in the occurrence and development of ferroptosis. Superoxide dismutase (SOD) and superoxide reductases can reduce O2•– to H2O2. Catalase catalyzes H2O2 to water and O2. Water-soluble ascorbic acid (vitamin C), lipid-soluble vitamin E or α-tocopherol (α-TOH), and lipoic acid can reduce lipid hydroperoxide production and peroxyl radicals. Besides, ascorbic acid increases the vitamin E content by reducing vitamin E semiquinone (65). The thioredoxin system, consisting of the thioredoxin (Trx) and thioredoxin reductase (TrxR), is also an important antioxidant system (66). Ferroptocide causes an accumulation of lipid peroxidation by inhibiting this system, thereby inducing ferroptosis (67). TrxR and NADPH reduce the active site disulfide in Trx. Under the combination of Trx and TrxR, peroxides, including lipid hydroperoxides and H2O2 were observed to be reduced effectively. In addition, there are several crosstalks between these antioxidants. For example, the thioredoxin system promotes the regeneration of certain antioxidants. It reduces ascorbyl free radical to ascorbic acid and turns GSSG to GSH. The thioredoxin system increases the content of ascorbic acid by reducing dehydroascorbic acid (65).
Doxorubicin is one of the most cardiotoxic anticancer agents. Current studies on DIC based on ferroptosis mainly focus on DOX. DOX’s anti-cancer activity is primarily mediated by DNA intercalation and inhibition of the topoisomerase II enzyme in rapidly proliferating tumors. However, DOX causes cumulative and dose-dependent cardiotoxicity, resulting in increased mortality risks among cancer patients and thus limits its wide clinical applications (68). Ferroptosis is involved in DIC both in vivo and in vitro (50, 69–72). The survival rate of rats was markedly elevated with the ferroptosis inhibitor Fer-1 than with apoptosis inhibitor emricasan, necroptosis inhibitor necrostatin-1, and autophagy inhibitor 3-methyladenine (73, 74). Besides, compared with apoptosis-defective (Ripk3–/–) mice and necroptosis-defective (Mlkl–/–) mice, intraperitoneal injections of Fer-1 (20 mg/kg) followed by DOX in normal mice improved their survival rate remarkably (74). The mechanisms of DOX causing cardiac ferroptosis are as follows (Figure 2 and Table 1):
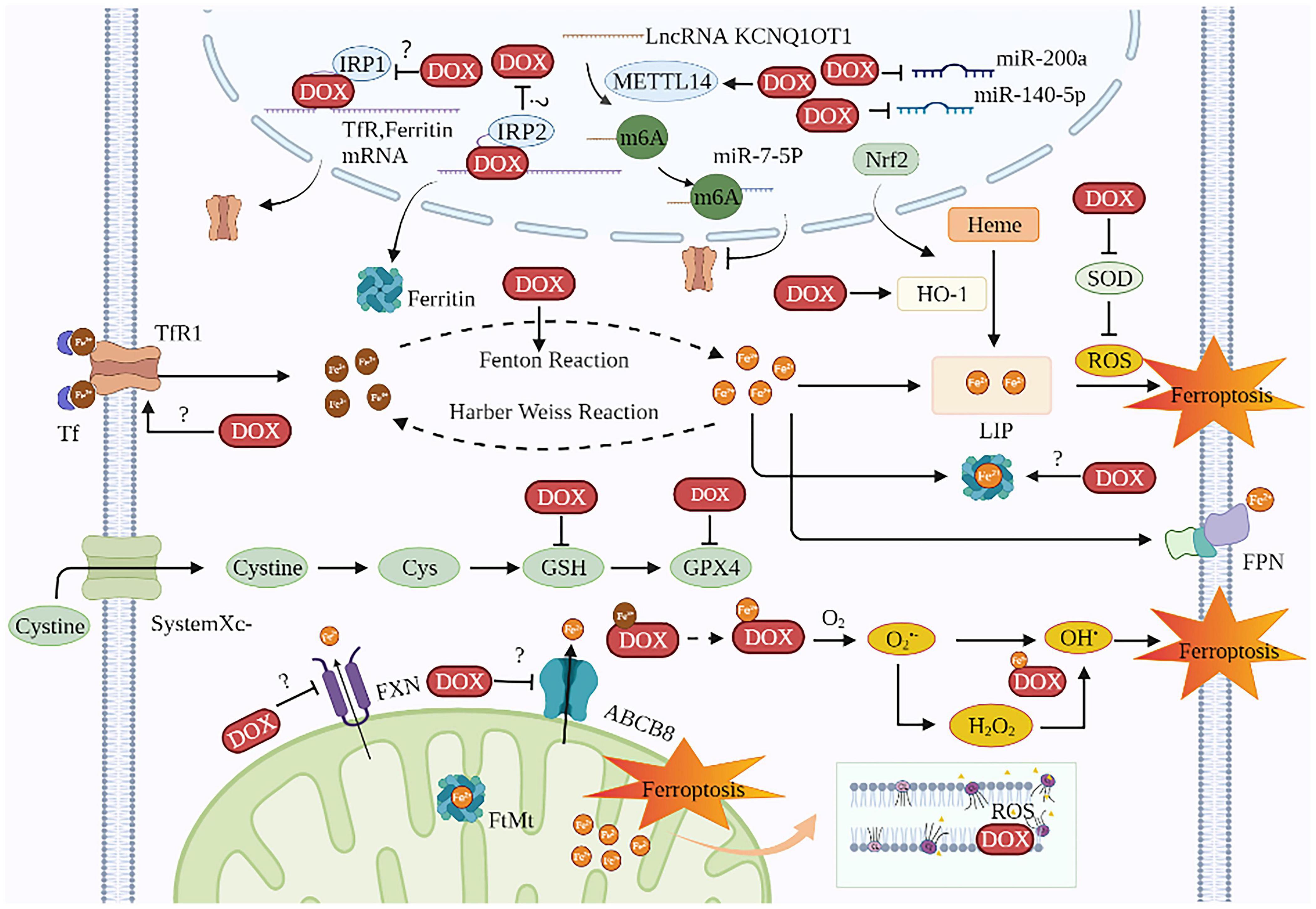
Figure 2. The mechanisms of DIC based on ferroptosis. DOX induces ferroptosis in cardiomyocytes involves two major mechanisms: one is to disrupt iron homeostasis and the other is to promote lipid peroxidation. The targets of DOX on iron disorder are Tf, ferritin, HO-1, FXN, ABCB8, IRE, IRP, and KCNQ1OT1m6A. The targets of DOX for lipid peroxidation are ROS, SOD, GPX4, and GSH. The site of iron death in cardiomyocytes is probably the mitochondria. TfR1, transferrin receptor 1; Tf, transferrin; DOX, doxorubicin; IRP, iron regulatory protein; METTL14, methyltransferase-like 14; Nrf2, nuclear factor (erythroid-derived 2)-like 2; LIP, labile iron pool; HO-1, heme oxygenase 1; ROS, reactive oxygen species; SOD, superoxide dismutase; Cys, cysteine; GSH, glutathione; GPX4, glutathione peroxidase 4; FPN, ferroportin; FXN, frataxin; ABCB8, ABC protein-B8; O2, oxygen; O2•–, active oxygen; FtMt, mitochondrial ferritin; CL, cardiolipin; OH•, hydroxyl radical.

Table 1. The main molecular mechanism of DOX-induced ferroptosis in cardiomyocytes.
The quinine moiety of DOX received electrons from NADPH oxidase and nitric oxide synthase (NOS) to become semiquinones, which was then accompanied by the ROS production such as O2•– and OH• generation (75). In addition, DOX can turn Fe3+ into Fe2+, thereby aggravating the Fenton reaction to produce more ROS (76). DOX can lead to downregulation of the antioxidant system. Several studies support the view that the levels of antioxidant substances (GPX4, SOD, and GSH) in DOX-treated rats and mice were significantly lower and the content of MDA was significantly higher than in control groups (50, 77, 78). One study suggested that ENPP2 overexpression enhances the expression levels of the ferroptosis-associated gene “GPX4” in H9C2 cells while FoxO4 regulates gene transcription negatively by the suppression of post-transcriptional coding mRNAs. In the H9C2 cells overexpressing ENPP2, DOX-induced increased Fe2+ activity, ROS and NOX4 production, while decreasing SLC7A11 and reversing GPX4 and FPN expression (79).
We know that ferroptosis is an iron-dependent lipid peroxidation process, and iron is essential in both the occurrence and development of this process. It is generally believed that excessive iron can aggravate DIC. More than 20 years ago, research indicated that iron overload aggravated DIC (80). DOX reduced the viability of H9C2 cardiomyocytes, while ferric ammonium citrate aggravated it in a concentration-dependent manner (81). Male Sprague Dawley rats fed with iron-rich chow showed significantly higher DOX cardiotoxicity, accompanied with a significant weight loss and severe myocyte injury as evidenced through electron microscopy and light microscopy. However, feeding an iron-rich meal alone did not result in any cardiotoxicity (82). Paradoxically, in cultured H9C2 cardiomyocytes and male C57BL/6 mice, researchers concluded that pretreatment with dextran-iron (125–1,000 μg/mL) in combination with DOX did not potentiate DIC and even prevented some aspects of it (83). Therefore, the regulation of DIC by iron is a complicated process, which may involve the balance between iron dosage, protection, and damage.
The HFE gene encodes HFE protein, which binds to TfR1 and facilitates the uptake of iron-bound to Tf. The elevation of iron concentration in the heart was much more accentuated in DOX-treated HFE–/– mice. Mutations in the HFE gene led to iron overload in cardiomyocytes, which increased the susceptibility of cardiomyocytes to ferroptosis and exacerbated DIC (84). One study concluded that the mutation status of HFE RS1799945 H63D could be used as one of the critical markers to identify patients at high risk for AIC (85). Among survivors of high-risk acute lymphoblastic leukemia in children, patients with the C282Y mutation of the HFE gene had a more severe DIC, which was reflected in higher levels of cardiac troponin-T, lower left ventricular quality and thickness, and worsened left ventricular function in echocardiography (86). These studies on the iron-regulated gene HFE support the view that iron plays a pivotal role in DIC.
Proteins associated with iron transport are considered significant indicators of cellular iron homeostasis and are mainly involved in cellular iron uptake (TfR1) and storage (ferritin). As for TfR1, most studies believe that DOX could elevate its expression (87–89). A study believed that TfR1 is critical for the DOX-induced increase in iron uptake. After incubation with the specific anti-TfR antibody (12 μg/ml), DOX-induced increase of 55Fe uptake in bovine aortic endothelial (BAEC) cells was reversed (88). The mechanism of increased TfR1 could be because DOX inhibited the expression of miR-7-5p by increasing the METTL14-mediated expression of KCNQ1OT1m6A, thus reducing the degradation of TfR1 (89). In AC16 cells, METTL14 knockdown, KCNQ1OT1 silencing, and miR-7-5p mimic attenuated the DOX-induced increase of Fe2+ and lipid ROS, and reduced DOX-induced decrease in mtDNA and MMP levels. In METTL14 shRNA mice models, DOX-induced increase in the levels of MDA and 4-HNE were also alleviated (89). However, one study suggested that TfR1 was reduced in heart lysates extracted from DOX-treated mice that received a single i.p. DOX dose (20 mg/kg) as observed in Western-blot and RT-PCR analysis (90). As for ferritin, DOX elevated it, and this change was accompanied by an increase in the level of iron bound to it (90). After being treated with 5 and 10 μM DOX for 24 h, the content of ferritin, especially ferritin heavy chain (FHC), in H9C2 cells increased, and was ROS-dependent. The increase in DOX-induced ferritin was reversed after clearing ROS using N-acetylcysteine (a known ROS scavenger) in H9C2 cells. Interestingly, the increased ferritin induced by DOX protected H9C2 cells from iron toxicity, as demonstrated by increased cell viability in 500, 750, and 1,000 μg/ml FAC measured by MTT assays (91). Similarly, ferritin mRNA and protein levels were also increased in an adult cell line of cardiomyocytes (HL-1) exposed to 5 μM DOX (92). In addition, elevated intracellular iron levels were also associated with high mobility group box 1 (HMGB1)-mediated heme degradation. Compared with the DOX group, the ferroptosis-related indexes (PTGS2, MDA, and 4-HNE) and cardiac injury-related indexes (Anp, Bnp, and Myh7) of the rat heart in the DOX + shHMGB1 group were significantly decreased (73). Therefore, the effect of DOX on iron homeostasis regulatory proteins is a complex process, possibly related to cell lines, drug dosage, and imbalances in protection and injury.
At present, many studies have shown that DOX can act on IRPs, but the results are numerous. Some studies believed that DOX irreversibly inactivated IRP1 and IRP2. This study believed that the secondary alcohol doxorubicinol (DOXol) and certain products of DOX metabolism, converted cytoplasmic aconitase to the cluster-free IRP1 by removing iron from its catalytic [Fe-S] clusters. This eventually produced a null protein, which could not adapt the levels of TfR1 and ferritin, and IRP2 was inactivated only by DOX related ROS (93). Some scholars thought H2O2 activated IRP1, leading to the upregulation of TfR1 and cellular iron accumulation, which was considered an important molecular mechanism in DIC (94). One study demonstrated that the IRP1 activity of BAEC cells increased in a dose-dependent manner after being treated with 0.5 μM DOX (88). However, one study found that even exposing DOX-sensitive GLC4 cells to 12.5 μM DOX for 24 h did not change IRP activity. It can be seen that the effect of DOX on cellular IRP activity may be related to cell line and cell resistance (95). However, Juliana et al. believed that the effect of DOX on cardiomyocyte IRP activity was closely related to DOX concentration and incubation time. After 6 h of DOX administration, total IRP1 did not change significantly, but active IRP1 and active IRP2 decreased in a concentration-dependent manner (1 μM DOX, 5 μM DOX, 10 μM DOX, and 20 μM DOX). IRP2, in particular, dropped by more than 50% at 20 μM DOX. After 24 h of administration, while the total IRP1 level decreased, the active IRP1 and active IRP2 did not significantly reduce but even increased. Interestingly, they thought that the free radical scavengers, DOXol, and cis-aconitate had little effect on IRP-RNA-binding activity in SK-Mel-28 melanoma cells and cardiomyocytes. The Fe and Cu complexes of anthracyclines altered iron metabolism in cardiomyocytes (96). Gianfranca suggested that DOX had differential effects on the two IPRs, as evidenced by reduced IRP2 activity and unchanged IRP1 activity. The effect of DOX on IRP2 resulted in an upregulation of ferritin gene expression and a decrease in TfR1 expression. Surprisingly, this change reduced the iron content in LIP and protected cardiomyocytes from ferroptosis induced by DOX (97). Furthermore, one study concluded that DOX at low concentrations (≈1 μM) activated IRP1 in cardiomyocytes, while at higher concentrations (>5 μM), it irreversibly inactivated IRP1 in BAEC cells (88). Besides, according to one study, DOX can directly interact with IREs. DOX intercalated double-stranded RNA by recognizing the IREs hairpins located in the 50-UTR of ferritin mRNAs, thereby changing the tertiary structure of the RNA drastically altering the effectiveness of the IREs/IRPs interaction (98). Anyway, DOX could modify the expression of genes involved in iron metabolism by inactivating IRPs binding to IREs (99).
Under normal physiological conditions, Nrf2 binds to Keap1 in the cytoplasm, and then Nrf2 is degraded by the ubiquitin-proteasome system. In the case of oxidative stress, Nrf2 dissociates from Keap1 and translocates to the nucleus, where it binds to promoter regions (AREs), activates the transcription of a series of downstream genes, and exerts physiological functions (100). Notably, many of the genes associated with ferroptosis are target genes for Nrf2, but DIC-related studies mainly focus on GPX4 and HO-1 genes. Nrf2 up-regulates the expression of GPX4 and has an anti-ferroptosis effect (101–103). Nrf2 can be methylated by the protein arginine methyltransferase 4 (PRMT4), leading to its nuclear restriction and consequently decreased GPX4 expression. PRMT4 aggravated the expression of ferroptosis markers (ROS, MDA, NCO4, and Fe2+) in the DOX-induced primary neonatal rat ventricular myocytes and C57BL/6 J mice cardiotoxicity models, and this influence can be mitigated by PRMT4 knockout (103). However, studies have also shown that Nrf2-mediated activation of HO-1 promotes ferroptosis. HO-1 mediates the release of Fe2+ from heme, which accumulates in cardiomyocytes and induces ferroptosis (74, 104). Through Nrf2+/+ mice, Nrf2–/–mice, Znpp (an HO-1 inhibitor), Hemin (an HO-1 activator), DOX has been proven to increase HO-1 by affecting Nrf2 and accelerating the degradation of heme, leading to an increase in non-heme iron and myocardial ferroptosis. An increase in intestinal iron absorption did not accompany this increase of iron content. In this study, DOX was confirmed to be able to upregulate hepatic Hamp1 mRNA to increase hepcidin and reduce FPN degradation (74). In addition, Nrf2 can be activated by deacetylation of SIRT1, and Fisetin activated Nrf2 by up-regulating the expression of SIRT1, leading to the up-regulation of HO-1, FTH1, TfR1, and FPN, and exerting an anti-ferroptosis effect. After being transfected with SIRT1 and Nrf2 siRNA, the anti-ferroptosis effect of Fisetin was abolished in H9C2 cells (105).
The morphological changes of ferroptosis under the electron microscope were mainly observed in the mitochondria (11). The metabolic activity of mitochondria drives ferroptosis. Mitochondria are the main source of cellular ROS. When electrons are transferred to O2, some escape from the ETC and react directly with O2 to form O2•–, a precursor to many other ROS such as OH• and H2O2 (106). Energy stress inhibits ferroptosis by activating AMPK, while AMPK inactivation promotes ferroptosis (107). ETC complex inhibitors can inhibit ferroptosis, indicating that mitochondria play an important role in ferroptosis, and possibly through the activation of AMPK (108). The mitochondrial TCA cycle is involved in ferroptosis induced by Cys deprivation, for which glutaminolysis is essential. However, mitochondria are not essential for ferroptosis induced by GPX4 inhibition (108).
Doxorubicin can cause cell iron metabolism disorders, but there is much debate about where iron metabolic disorders occur. Some studies have suggested that the site where the DIC occurs is the mitochondria. Compared with cytoplasm, DOX and iron preferentially accumulated in mitochondria (109), especially in mitochondrial cardiolipin (110–112). After incubation with 10 μM DOX, the accumulation of DOX in the mitochondria of neonatal rat cardiomyocytes increased significantly compared with the cytoplasm, and DOX caused a significant increase in mitochondrial iron levels detected by 55Fe colorimetric measurement of mitochondrial non-heme iron (109). In the presence of Fe2+, DOX induced the activation of the mitochondrial permeability transition pore (113). Tadokoro et al. believed that ferroptosis was triggered by GPX4 deficiency in mitochondria for the following reasons: (1) DOX-induced lipid peroxidation occurred on mitochondria rather than other organelles; and (2) even though the cell viability was improved and lipid peroxidation indexes (MitoPeDPP, MDA) were reduced in both isolated neonatal rat ventricular cardiomyocytes cells transfected with Ad-cytoGPx4-FLAG and Ad-mitoGPx4-FLAG, electron microscopy revealed that mitochondrial GPX4 was almost exclusively localized to mitochondria during this process. In contrast, cytoplasmic GPX4 was transferred to mitochondria (50). Mitochondria-2,2,6,6-tetramethylpiperidin-N-oxyl, a mitochondrial superoxide scavenger, abolished DOX-induced lipid peroxidation and cardiac ferroptosis in DOX-treated mice models. In contrast, the non-mitochondrial targeted version only mildly rescued the DOX-induced effects (74). Dexrazoxane (DXZ) reduced iron in mitochondria. The poor impact of DFO in treating DIC compared with DXZ may be attributed to its inability to penetrate mitochondria and chelate iron in mitochondria specifically (109, 114). The most likely target of free radicals produced by ANTs through redox reactions was cardiolipin, a major phospholipid component of the inner mitochondrial membrane, known to be susceptible to peroxidative injury with abundant PUFA. Iron overload can aggravate the damage of ANTs to the mitochondria of cardiomyocytes (115).
The mechanism of DOX causing myocardial ferroptosis may be related to its effect on mitochondrial iron regulation-related proteins. Compared with wild-type mice, cardiotoxicity was more pronounced in FtMt–/– mice injected intraperitoneally with a single dose (15 mg/kg of body weight) of DOX, as manifested by higher mortality, more morphological changes (incomplete cristae, condensation, and fragmentation of most myofibril), more severe lipid peroxidation, and worse cardiac function (ATP and BNP) (116). In mice, neonatal cardiomyocytes, and H9C2 cardiomyoblasts, DOX led to the reduction of frataxin, a nuclear-encoded mitochondrial protein involved in maintaining mitochondrial iron homeostasis through the ubiquitin-proteasome system. In addition, the mitochondrial iron export protein ABC protein-B8 (ABCB8) is essential for maintaining mitochondrial iron homeostasis. The depletion of ABCB8 led to compromised systolic and diastolic functions, a significant accumulation of 55Fe in the mitochondria, and higher lipid peroxidation levels both in vivo and in vitro (87, 109, 117). The levels of ABCB8 in explanted hearts from patients with end-stage cardiomyopathy were significantly reduced (117). Intriguingly, one study concluded that there was no effect on ABCB8 in the myocardium of the DIC mice models, and the silencing of ABCB8 did not increase the iron content in cultured cardiomyocytes after 30 h of exposure to 2 μM DOX (50).
On the contrary, another study showed that DOX affected more cytosolic than mitochondrial iron metabolism in murine hearts and human HeLa cells, as manifested by alterations in proteins associated with cytoplasmic iron transport proteins (ferritin, TfR1, and hepcidin). In contrast, mitochondrial iron-related proteins (aconitase, succinate-dehydrogenase, and frataxin) appear to be unaffected (90). In addition, Kwok thought that the mechanism of DOX-induced cardiomyocyte ferroptosis was related to lysosomes. It may be that ANTs act on lysosomes to inhibit ferritin degradation, thereby affecting the iron-dependent life activities of cells. However, this study was conducted in SK cells, not cardiomyocytes, so the conclusion is debatable (118). Interestingly, some studies believed that DOX affected neither total cellular Fe content nor total cellular ferritin protein levels (119). Furthermore, the myocardial iron content was not statistically different between the saline and the DOX groups (82). In the presence of NADPH-cytochrome p450 reductase, ANTs underwent redox cycling to generate superoxide, which mediated a slow reductive release of iron from ferritin. More the cardiotoxic ANTs, more the rapid and extensive iron release it led to (120).
As mentioned earlier, iron promotes ROS generation through the Fenton and Harber-Weiss reactions (42, 121, 122). PUFA undergoes lipid peroxidation under the action of ROS, which in turn leads to ferroptosis. Furthermore, DOX can also be combined with Fe3+ to form the DOX-Fe3+ complex (123, 124), which generates the DOX-Fe2+ complex through both enzymatic or non-enzymatic reactions. And this DOX-Fe2+ complex reacts with oxygen to form O2•–, which is transformed into OH and H2O2 through disproportionation reaction (125). H2O2 can also react with DOX-Fe2+ complexes to generate OH• (126). Thus, under the action of the DOX-iron complex, OH and O2, PUFA undergoes lipid peroxidation. In addition, even without free Fe3+ and Fe2+, DOX can extract Fe3+ directly from ferritin to form DOX-Fe3+ complexes, resulting in lipid peroxidation (127). In this process, it is not the free Fe3+, free Fe2+, or DOX-Fe3+ iron complexes, but the DOX-Fe2+ complexes that induce ferroptosis in cardiomyocytes. The use of specific Fe2+ chelators like Mito-FerroGreen and bathophenanthroline effectively attenuate cardiomyocyte lipid peroxidation, measured using C11-BODIPY 581/591 and MDA (50, 124). It should be noted that lipid peroxidation is not the only pathological change brought about by ferroptosis. Oxidative stress, apoptosis, necrosis, and other forms of RCD share characteristics of lipid peroxidation. So, if we want to prove that ferroptosis is DIC’s mechanism, we must measure lipid peroxidation directly (56).
Evaluations performed before the initiation of anticancer therapy in patients without significant CVDs should be regarded as the primary prevention strategy (128). An appropriate cancer treatment and anti-cardiotoxicity prevention and treatment strategies should be selected after a comprehensive discussion by a multidisciplinary team of cardiovascular, oncology and hematology experts, especially to balance the effects of drugs after cancer treatment regimens and the risk of specific CVDs in all aspects (129). Commonly used drugs and their mechanism of action are described as follows (Figure 3 and Table 2):
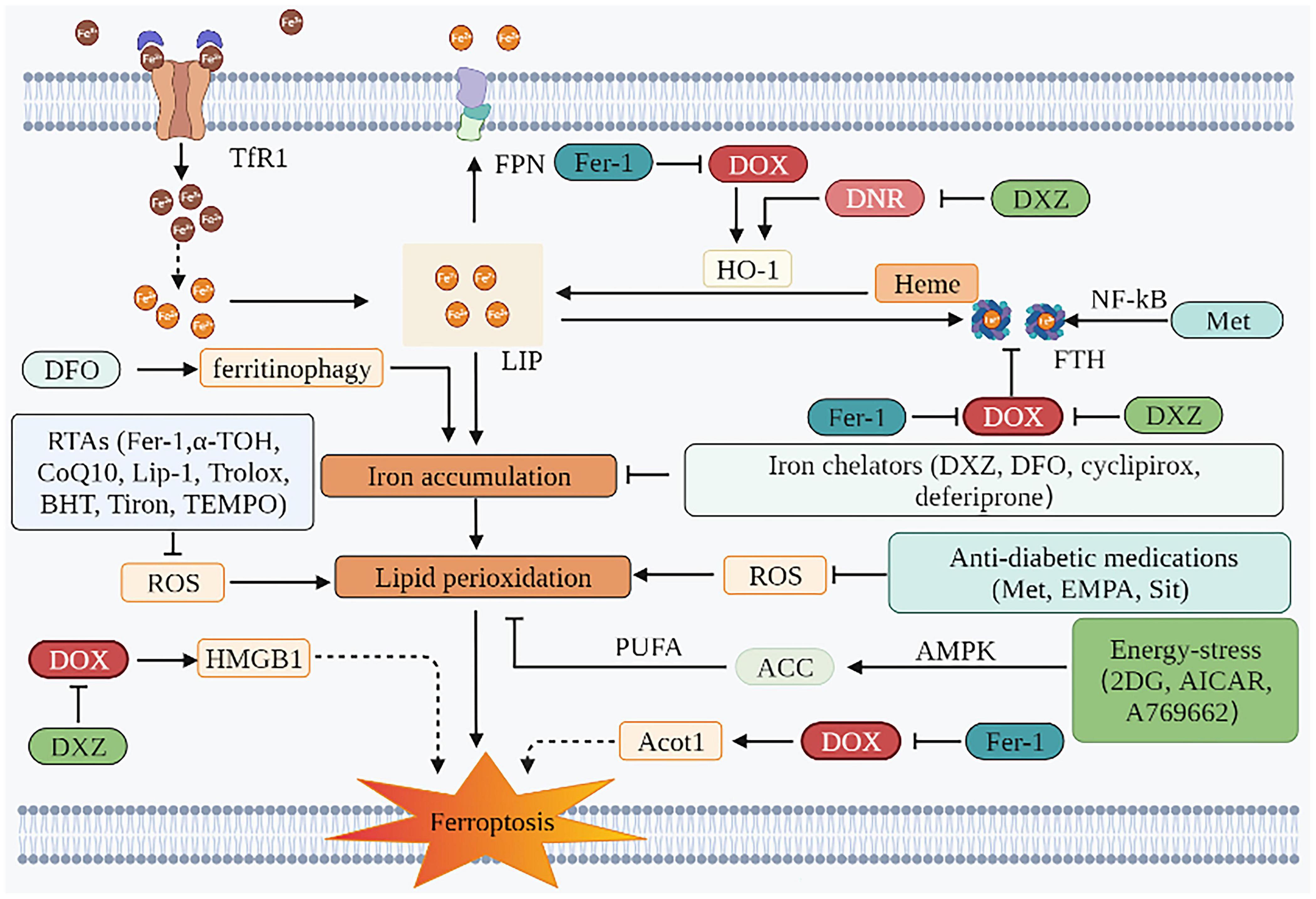
Figure 3. Prevention and SOD treatment of AIC based on ferroptosis. The mechanism of preventing ferroptosis of cardiomyocytes is mainly in two aspects. One is to inhibit iron accumulation, and the other is to inhibit lipid peroxidation. Iron chelators can play a role through the former. The effects of RTAs, anti-diabetic medications, and energy-stress inducers are mainly attributed to the latter. TfR1, transferrin receptor 1; FPN, ferroportin; DOX, doxorubicin; DNR, daunorubicin; LIP, labile iron pool; HO-1, heme oxygenase 1; ROS, reactive oxygen species; Fer-1, ferrostatin-1; DFO, deferoxamine; DXZ, dexrazoxane; PUFA, polyunsaturated fatty acids; ACC, acetyl-CoA carboxylase; AMPK, AMP-activated protein kinase; EMPA, empagliflozin; Sit, sitagliptin; 2DG, 2-deoxy-d-glucose; AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; HMGB1, high mobility group box 1; HO-1, heme oxygenase 1; FTH, ferritin heavy chain; NF-κB, nuclear factor-kappa B; α-TOH, α-tocopherol; CoQ10, coenzyme Q10; Lip-1, liproxstatin-1; BHT, butylated hydroxytoluene; Acot1, acyl-CoA thioesterase 1; TEMPO, 2,2,6,6-tetramethylpiperidin-N-oxyl; Met, metformin.
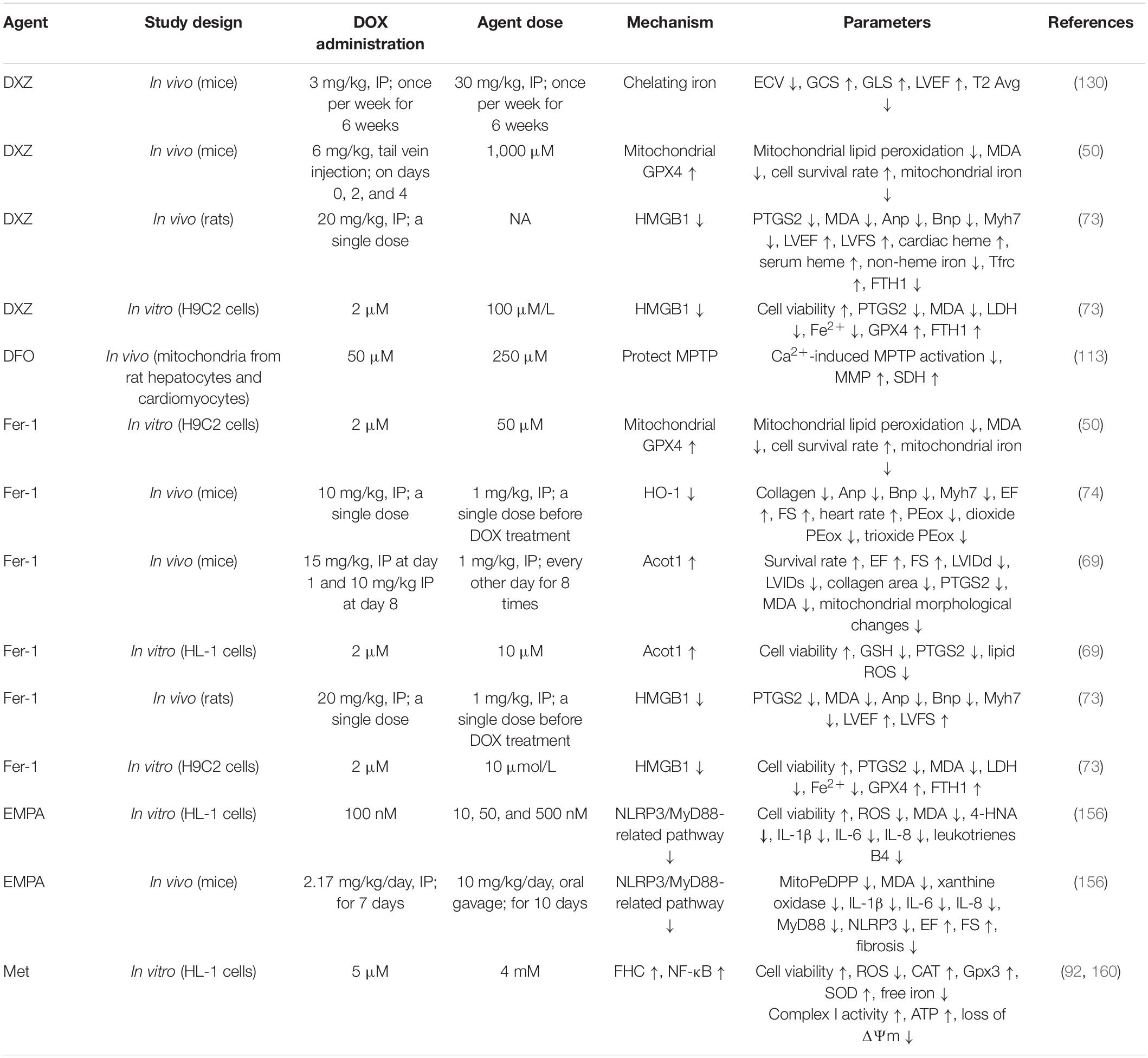
Table 2. The therapeutic strategies against ferroptosis in DIC.
Dexrazoxane is the only formally preventive drug approved by the FDA. For patients planning to receive high-dose of ANT therapy, DXZ is recommended (2). DXZ is traditionally known as an iron chelator. Under the action of the iron-ANT complex, the ring of DXZ was opened and hydrolyzed to ADR-925. This presumably exerted its cardioprotective effects by either binding freely or loosely to iron or iron complexed with DOX, thus preventing or reducing site-specific oxygen radical production that damages cellular components (130). In addition, DOX-induced cardiac ferroptosis in rats was observed to be mediated by the upregulation of HMGB1, and correspondingly ferroptosis was inhibited when shHMGB1 was used. DXZ reversed DOX-induced elevation of HMGB1. DXZ also modulated iron metabolism-related protein levels in cardiac myocytes and reversed the upregulation of HO-1 induced by daunorubicin. It therefore inhibited the conversion of heme iron to non-heme iron, which reduced the Fe2+ content in the LIP in cardiac myocytes (131). In addition, this study also showed that DXZ reversed the DOX-induced decrease of FTH1 protein in the H9C2 cells (73). However, this view has been greatly challenged. An increasing number of studies believe that DXZ plays a protective role in the heart mainly because it inhibited DOX-mediated damage of cardiomyocyte topoisomerase IIβ (132, 133). Also, in one study, the chelating metabolite ADR-925 therapy on neonatal ventricular myocytes receiving was neither able to mitigate AIC, nor did it significantly impact daunorubicin-induced mortality, blood congestion, and biochemical and functional markers of cardiac dysfunction in a chronic rabbit model in vivo (132). Although the mechanism of DXZ chelating iron in mitochondria does not depend on the topoisomerase IIβ pathway (109), the contribution of DXZ in regulating the cardioprotective effect against ferroptosis is not apparent. Besides, DXZ has been shown to inhibit DOX-induced cardiomyocyte necrosis and apoptosis through several alternate mechanisms (134–136). Therefore, the cardiomyocyte protective effect exerted by DXZ is not achieved only through the inhibition of ferroptosis as it is not a simple, specific ferroptosis inhibitor.
Although DXZ is generally considered to reduce AIC and is recommended as the only approved cardioprotective agent (137, 138), the clinical use of DXZ is encountering various challenges. Firstly, there are concerns that DXZ may increase the risk of acute myeloid leukemia and secondary solid tumors in children (139–141). Secondly, the effectiveness of DXZ is also being questioned. One study found that the preventive use of DXZ before high-dose DOX in eight sarcoma patients did not reduce their cardiotoxicity satisfactorily. There were six patients with high-sensitivity troponin T levels exceeding 10 ng/ml, four patients with LVEF that decreased by more than 5%, and three patients with global longitudinal peak systolic strain changed by more than 15% (142). Finally, the European Society of Cardiology only recommends DXZ for patients with advanced or metastatic breast cancer receiving cumulative doses of DOX over 300 mg/m2 (2). In spite of this, DIC development is not a dose-dependent response, and in our clinical work, we have found that some patients develop DIC when treated with small doses of anthracyclines.
Deferoxamine is a widely used iron chelator that can chelate excess intracellular iron, thereby reducing DOX-induced ferroptosis in cardiomyocytes. In addition, DFO can also be regarded as a protective agent for mitochondrial permeability transition pore, as it weakens the opening of calcium-dependent pores induced by iron and iron-DOX complexes and reduces the uptake of Fe2+ in mitochondria, thus protecting mitochondrial function (113). Intriguingly, DFO was demonstrated to aggravate ferroptosis by inducing ferritinophagy, leading to the accumulation of iron and ROS (10, 143, 144). The effect of DFO in protecting cardiomyocytes from DIC is still contentious. In this study, the cardioprotective benefit of DFO requires a strict dose, and a slight deviation from it would diminish this effect (145). Besides, DFO failed to reverse myocardial damage in a well-established spontaneously hypertensive rat models of chronic ANT cardiomyopathy (146). Moreover, DFO’s side effects, such as hypotension and renal insufficiency, limit its clinical application (147).
As a radical-trapping antioxidant (RTA), Fer-1 attenuates lipid peroxidation by decreasing erastin-induced accumulation of cytosolic and lipid ROS, consequently inhibiting ferroptosis (11). This is primarily due to its powerful chain-carrying peroxyl trapping ability (148). The experimental results have shown that Fer-1 could reduce the lipid peroxides labeled with MDA and MitoPeDPP (50). In addition, the mechanisms by which Fer-1 protects the heart from DOX-induced ferroptosis could be describes as follows: Fer-1 inhibited DOX-induced elevation of non-heme iron in cardiomyocytes by downregulating Nrf2/HO-1, thereby inhibiting ferroptosis (74). In an alternate way, DOX downregulated the Acyl-CoA thioesterase 1 gene, causing alterations in the composition of free fatty acids in mitochondrial membranes, particularly in the proportion of C22:6N3, leading to ferroptosis. Fer-1 inhibited DOX downregulation of this gene which reduced the sensitivity of cardiomyocytes to ferroptosis induced by DOX (69). Besides, Fer-1 increased the expression of FTH1 protein to increase iron in storage, resulting in the reduction of Fe2+ in LIP (73).
In addition to Fer-1, parallel experiments in GPX4 gene-deficient mouse embryonic fibroblast ferroptosis model and extracellular high concentration glutamate-induced ferroptosis model have shown that other RTAs (liproxstatin-1 and α-TOH) can also improve cell survival rate. In HEK-293 cells, the mechanism by which Fer-1, liproxstatin-1, and α-TOH inhibit lipid hydroperoxides and bring about ferroptosis was demonstrated to be achieved by capturing chain-carrying peroxyl radicals, rather than inhibiting LOXs or restoring GSH levels (148–150). However, in a striatal cell model, the mechanism of action of α-TOH against ferroptosis was proved to be associated with LOX. Its endogenous metabolite, α-tocopherol hydroquinone, inhibited 15-LOX activity by reducing its non-heme Fe3+ center to the inactive Fe2+, thereby inhibiting lipid peroxidation (151). Other antioxidants (Trolox, butylated hydroxytoluene, Tiron, and TEMPO) have less inhibitory effects on erastin-induced ferroptosis than Fer-1 and this could be attributed to Fer-1 containing an aromatic amine (11). Another RTA, CoQ10, was shown to inhibit ferroptosis by inhibiting the propagation of lipid peroxides. Inhibition of CoQ10 synthesis with 4-chlorobenzoic acid or knockout of COQ2, an enzyme required for CoQ10 synthesis, can increase the sensitivity of cells to RSL3-induced ferroptosis in vitro (152, 153). Other iron chelators such as ciclopirox and deferiprone also alleviate iron-dependent lipid peroxidation by depriving iron (12).
Although many experiments have demonstrated that ferroptosis could be attenuated by inhibiting lipid peroxidation and iron accumulation, studies using DOX-induced cardiomyopathy models are scarce, and the efficiency and safety of these drugs against ferroptosis are still questionable (56).
According to recent research, DIC was also associated with insulin signaling imbalance and cardiac insulin resistance (154, 155). Metformin, empagliflozin, and sitagliptin have been shown to reduce ferroptosis triggering lipid peroxidation in the mitochondria and cytoplasm of cardiomyocytes (156–158). Metformin and sitagliptin have been shown to attenuate myocardial lipid peroxidation in rats by reversing the DOX-induced decrease in GSH (159). In cardiomyocytes (HL-1 cell line) exposed to DOX and C57Bl/6 mice treated with DOX, empagliflozin reduced lipid peroxidation levels, decreased cardiomyocyte fibrosis, inhibited cardiomyocyte inflammation, increased ejection fraction percentage (% EF) and fractional shortening percentage (% FS), improved cardiac function, and protected cardiomyocytes from ferroptosis (156). Metformin also activated nuclear factor-kappa B, thereby increasing FTH and reducing iron accumulation in LIP, thus protecting adult mouse cardiomyocytes from DIC (92, 160).
In immortalized mouse embryonic fibroblasts (MEFs), the energy-stress inducers (2-deoxy-D-glucose, 5-aminoimidazole-4-carboxamide ribonucleotide, and A769662) were demonstrated to activate AMPK. This further inhibited acetyl-CoA carboxylase, and in turn palmitic acid (C16:0), thereby reducing the synthesis of PUFA, all of which suppressed ferroptosis (107).
In conclusion, anti-DIC therapy based on these ferroptosis triggering mechanisms mainly include two aspects: iron chelation and antioxidant treatment. Notably, human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) have a great potential in predicting patient susceptibility to DIC. Furthermore, the human-derived DOX cardiomyocyte injury model established by hiPSC-CMs overcomes the species differences of current research models and can be accurately used to understand the mechanism of ferroptosis in DIC (161).
In summary, ferroptosis has been demonstrated by several studies to mediate the occurrence of AIC. In fact, DOX can increase the ROS content and affect iron metabolism in cardiomyocytes by acting on iron homeostasis regulatory proteins, such as, IREs/IRPs, and Nrf2/HO-1, resulting in the accumulation of lipid peroxides, thereby inducing ferroptosis. Mitochondria are the main organelle for inducing ferroptosis in cardiomyocytes. With further research, inhibition of ferroptosis could act as an effective strategy in both prevention and treatment of AIC. This would require screening for potential drugs that inhibit ferroptosis forcefully in cardiomyocytes or developing novel ferroptosis inhibitors and will surely benefit cancer patients with heart diseases as well as patients with high cardiovascular risk stratification.
YX and SS designed this study. GZ wrote the first draft of this manuscript and created the figures. CY and XS searched the literature. JZ and XY participated in discussions and improved pictures related to the manuscript. PG, GV, GL, NA, CL, WS, HC, and MW critically revised and approved the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the CACMS Innovation Fund (Grant No. CI2021A00919), the National Natural Science Foundation of China (Grant No. 82174349), and the National Key R&D Program of China (Grant Nos. 2018YFC1704901 and 2018YFC1704900).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Biorender (http://biorender.com) for providing an easy tool to make figures.
ANT, anthracyclines; RCD, regulated cell death; ROS, reactive oxygen species; PUFA, polyunsaturated fatty acids; GSH, glutathione; GPX4, glutathione peroxidase 4; FSP1, ferroptosis suppressor protein 1; CoQ10, coenzyme Q10; NADPH, nicotinamide adenine dinucleotide phosphate; GCH1, GTP cyclohydrolase-1; BH4, tetrahydrobiopterin; AIC, anthracycline-induced cardiotoxicity; DIC, doxorubicin-induced cardiomyopathy; DOX, doxorubicin; CVDs, cardiovascular diseases; DFO, deferoxamine; Fe3+, ferric iron; Tf, transferrin; TfR1, transferrin receptor 1; Fe2+, ferrous iron; DMT1, divalent metal transporter 1; FPN, ferroportin; LIP, labile iron pool; LOX, lipoxygenase; FTH, ferritin heavy chain; IRP, iron regulatory protein; IREs, iron-responsive elements; HO-1, heme oxygenase 1; O2•–, active oxygen; OH•, hydroxyl radical; PL•, phospholipid radical; PLOO•, phospholipid peroxyl radical; PLOOH, phospholipid hydroperoxide; MDA, malondialdehyde; Cys, cysteine; Nrf2, nuclear factor (erythroid-derived 2)-like 2; AMPK, AMP-activated protein kinase; HMGB1, high mobility group box 1; DXZ, dexrazoxane; ABCB8, ABC protein-B8; RTA, radical-trapping antioxidant; α-TOH, α-tocopherol; Fer-1, ferrostatin-1; SOD, superoxide dismutase; Trx, thioredoxin; TrxR, thioredoxin reductase; HETE, hydroxyeicosatetraenoic acid; PE, phosphatidylethanolamine; PRMT4, protein arginine methyltransferase 4.
1. Curigliano G, Lenihan D, Fradley M, Ganatra S, Barac A, Blaes A, et al. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann Oncol. (2020) 31:171–90. doi: 10.1016/j.annonc.2019.10.023
2. Zamorano JL, Lancellotti P, Rodriguez Muñoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 Esc position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the esc committee for practice guidelines: the task force for cancer treatments and cardiovascular toxicity of the European society of cardiology (Esc). Eur Heart J. (2016) 37:2768–801. doi: 10.1093/eurheartj/ehw211
3. Cardinale D, Colombo A, Lamantia G, Colombo N, Civelli M, De Giacomi G, et al. Cardio-oncology: a new medical issue. Ecancermedicalscience. (2008) 2:126. doi: 10.3332/ecancer.2008.126
4. Curigliano G, Cardinale D, Dent S, Criscitiello C, Aseyev O, Lenihan D, et al. Cardiotoxicity of anticancer treatments: epidemiology, detection, and management. CA A Cancer J Clin. (2016) 66:309–25. doi: 10.3322/caac.21341
5. Narayan V, Ky B. Common cardiovascular complications of cancer therapy: epidemiology, risk prediction, and prevention. Annu Rev Med. (2018) 69:97–111. doi: 10.1146/annurev-med-041316-090622
6. Rocca C, Pasqua T, Cerra MC, Angelone T. Cardiac damage in anthracyclines therapy: focus on oxidative stress and inflammation. Antioxid Redox Signal. (2020) 32:1081–97. doi: 10.1089/ars.2020.8016
7. Kitakata H, Endo J, Ikura H, Moriyama H, Shirakawa K, Katsumata Y, et al. Therapeutic targets for dox-induced cardiomyopathy: role of apoptosis Vs. ferroptosis. Int J Mol Sci. (2022) 23:1414. doi: 10.3390/ijms23031414
8. Vejpongsa P, Yeh ET. Prevention of anthracycline-induced cardiotoxicity: challenges and opportunities. J Am Coll Cardiol. (2014) 64:938–45. doi: 10.1016/j.jacc.2014.06.1167
9. Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. (2021) 11:3052–9. doi: 10.7150/thno.54113
10. Zhai Z, Zou P, Liu F, Xia Z, Li J. Ferroptosis is a potential novel diagnostic and therapeutic target for patients with cardiomyopathy. Front Cell Dev Biol. (2021) 9:649045. doi: 10.3389/fcell.2021.649045
11. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
12. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
13. Tan S, Schubert D, Maher P. Oxytosis: a novel form of programmed cell death. Curr Topics Med Chem. (2001) 1:497–506. doi: 10.2174/1568026013394741
14. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. (1989) 2:1547–58. doi: 10.1016/0896-6273(89)90043-3
15. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. (2020) 32:920–37. doi: 10.1016/j.cmet.2020.10.011
16. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. (2021) 18:280–96. doi: 10.1038/s41571-020-00462-0
17. Qiang Z, Dong H, Xia Y, Chai D, Hu R, Jiang H. Nrf2 and STAT3 alleviates ferroptosis-mediated IIR-ALI by regulating SLC7a11. Oxid Med Cell Longev. (2020) 2020:5146982. doi: 10.1155/2020/5146982
18. Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. (2021) 6:49. doi: 10.1038/s41392-020-00428-9
19. Qin Y, Guo T, Wang Z, Zhao Y. The role of iron in doxorubicin-induced cardiotoxicity: recent advances and implication for drug delivery. J Mater Chem B. (2021) 9:4793–803. doi: 10.1039/d1tb00551k
20. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. (2016) 23:369–79. doi: 10.1038/cdd.2015.158
21. Gonciarz RL, Collisson EA, Renslo AR. Ferrous iron-dependent pharmacology. Trends Pharmacol Sci. (2021) 42:7–18. doi: 10.1016/j.tips.2020.11.003
22. Doll S, Conrad M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life. (2017) 69:423–34. doi: 10.1002/iub.1616
23. Stamenkovic A, Pierce GN, Ravandi A. Phospholipid oxidation products in ferroptotic myocardial cell death. Am J Physiol Heart Circ Physiol. (2019) 317:H156–63. doi: 10.1152/ajpheart.00076.2019
24. Zeng X, An H, Yu F, Wang K, Zheng L, Zhou W, et al. Benefits of iron chelators in the treatment of Parkinson’s disease. Neurochem Res. (2021) 46:1239–51. doi: 10.1007/s11064-021-03262-9
25. Li G, Li X, Dong J, Han Y. Electroacupuncture ameliorates cerebral ischemic injury by inhibiting ferroptosis. Front Neurol. (2021) 12:619043. doi: 10.3389/fneur.2021.619043
26. Guo H, Zhu L, Tang P, Chen D, Li Y, Li J, et al. Carthamin yellow improves cerebral ischemia-reperfusion injury by attenuating inflammation and ferroptosis in rats. Int J Mol Med. (2021) 47:52. doi: 10.3892/ijmm.2021.4885
27. Tsai Y, Xia C, Sun Z. The inhibitory effect of 6-gingerol on ubiquitin-specific peptidase 14 enhances autophagy-dependent ferroptosis and anti-tumor in vivo and in vitro. Front Pharmacol. (2020) 11:598555. doi: 10.3389/fphar.2020.598555
28. Song Z, Xiang X, Li J, Deng J, Fang Z, Zhang L, et al. Ruscogenin induces ferroptosis in pancreatic cancer cells. Oncol Rep. (2020) 43:516–24. doi: 10.3892/or.2019.7425
29. Zhu S, Yu Q, Huo C, Li Y, He L, Ran B, et al. Ferroptosis: a novel mechanism of artemisinin and its derivatives in cancer therapy. Curr Med Chem. (2021) 28:329–45. doi: 10.2174/0929867327666200121124404
30. Xie Y, Chen G. Dioscin induces ferroptosis and synergistic cytotoxicity with chemotherapeutics in melanoma cells. Biochem Biophys Res Commun. (2021) 557:213–20. doi: 10.1016/j.bbrc.2021.04.024
31. Ikeda Y, Hamano H, Horinouchi Y, Miyamoto L, Hirayama T, Nagasawa H, et al. Role of ferroptosis in cisplatin-induced acute nephrotoxicity in mice. J Trace Elem Med Biol. (2021) 67:126798. doi: 10.1016/j.jtemb.2021.126798
32. Zhang Q, Yi H, Yao H, Lu L, He G, Wu M, et al. Artemisinin derivatives inhibit non-small cell lung cancer cells through induction of ros-dependent apoptosis/ferroptosis. J Cancer. (2021) 12:4075–85. doi: 10.7150/jca.57054
33. Lu Y, Yang Q, Su Y, Ji Y, Li G, Yang X, et al. Mycn mediates TFRC-dependent ferroptosis and reveals vulnerabilities in neuroblastoma. Cell Death Dis. (2021) 12:511. doi: 10.1038/s41419-021-03790-w
34. Li B, Jiang Y, Wang T, He X, Ma L, Li B, et al. Effect of atrazine on accumulation of iron via the iron transport proteins in the midbrain of SD rats. Sci Total Environ. (2021) 780:146666. doi: 10.1016/j.scitotenv.2021.146666
35. Qiu L, Ge L, Hu Q. Dexmedetomidine protects SK-N-SH nerve cells from oxidative injury by maintaining iron homeostasis. Biol Pharm Bull. (2020) 43:424–31. doi: 10.1248/bpb.b19-00711
36. Núñez MT, Hidalgo C. Noxious iron-calcium connections in neurodegeneration. Front Neurosci. (2019) 13:48. doi: 10.3389/fnins.2019.00048
37. Wang H, Liu C, Zhao Y, Gao G. Mitochondria regulation in ferroptosis. Eur J Cell Biol. (2020) 99:151058. doi: 10.1016/j.ejcb.2019.151058
38. Song LM, Xiao ZX, Zhang N, Yu XQ, Cui W, Xie JX, et al. Apoferritin improves motor deficits in MPTP-treated mice by regulating brain iron metabolism and ferroptosis. iScience. (2021) 24:102431. doi: 10.1016/j.isci.2021.102431
39. Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. (2020) 160:303–18. doi: 10.1016/j.freeradbiomed.2020.08.009
40. Fernández-Mendívil C, Luengo E, Trigo-Alonso P, García-Magro N, Negredo P, López MG. Protective role of microglial HO-1 blockade in aging: implication of iron metabolism. Redox Biol. (2021) 38:101789. doi: 10.1016/j.redox.2020.101789
41. Menon AV, Liu J, Tsai HP, Zeng L, Yang S, Asnani A, et al. Excess hemeupregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice withsickle cell disease. Blood. (2022) 139:936–41. doi: 10.1182/blood.2020008455
42. Ai Y, Yan B, Wang X. The oxidoreductases POR and CYB5R1 catalyze lipid peroxidation to execute ferroptosis. Mol Cell Oncol. (2021) 8:1881393. doi: 10.1080/23723556.2021.1881393
43. Indo HP, Yen HC, Nakanishi I, Matsumoto K, Tamura M, Nagano Y, et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J Clin Biochem Nutr. (2015) 56:1–7. doi: 10.3164/jcbn.14-42
44. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. (2019) 35:830–49. doi: 10.1016/j.ccell.2019.04.002
45. Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Investig. (2019) 129:2293–304. doi: 10.1172/jci126428
46. Jang S, Chapa-Dubocq XR, Tyurina YY, St Croix CM, Kapralov AA, Tyurin VA, et al. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. (2021) 45:102021. doi: 10.1016/j.redox.2021.102021
47. Sparvero LJ, Tian H, Amoscato AA, Sun WY, Anthonymuthu TS, Tyurina YY, et al. Direct mapping of phospholipid ferroptotic death signals in cells and tissues by gas cluster ion beam secondary ion mass spectrometry (GCIB-SIMS). Angew Chem Int Ed Engl. (2021) 60:11784–8. doi: 10.1002/anie.202102001
48. Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. (2016) 15:348–66. doi: 10.1038/nrd.2015.6
49. Lei G, Mao C, Yan Y, Zhuang L, Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell. (2021) 12:836–57. doi: 10.1007/s13238-021-00841-y
50. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. (2020) 5:e132747. doi: 10.1172/jci.insight.132747
51. Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. (2021) 33:174–89.e7. doi: 10.1016/j.cmet.2020.12.007
52. Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. (2019) 177:1262–79.e25. doi: 10.1016/j.cell.2019.03.032
53. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
54. Hadian K. Ferroptosis suppressor protein 1 (FSP1) and coenzyme Q(10) cooperatively suppress ferroptosis. Biochemistry. (2020) 59:637–8. doi: 10.1021/acs.biochem.0c00030
55. Dai E, Zhang W, Cong D, Kang R, Wang J, Tang D. Aifm2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun. (2020) 523:966–71. doi: 10.1016/j.bbrc.2020.01.066
56. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
57. Venkatesh D, O’Brien NA, Zandkarimi F, Tong DR, Stokes ME, Dunn DE, et al. MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev. (2020) 34:526–43. doi: 10.1101/gad.334219.119
58. Song Z, Jia G, Ma P, Cang S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. (2021) 276:119399. doi: 10.1016/j.lfs.2021.119399
59. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. Gtp cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
60. Dar HH, Anthonymuthu TS, Ponomareva LA, Souryavong AB, Shurin GV, Kapralov AO, et al. A new thiol-independent mechanism of epithelial host defense against Pseudomonas Aeruginosa: iNOS/NO() sabotage of theft-ferroptosis. Redox Biol. (2021) 45:102045. doi: 10.1016/j.redox.2021.102045
61. La Rosa P, Petrillo S, Turchi R, Berardinelli F, Schirinzi T, Vasco G, et al. The NRF2 induction prevents ferroptosis in friedreich’s ataxia. Redox Biol. (2021) 38:101791. doi: 10.1016/j.redox.2020.101791
62. Lovatt M, Adnan K, Kocaba V, Dirisamer M, Peh GSL, Mehta JS. Peroxiredoxin-1 regulates lipid peroxidation in corneal endothelial cells. Redox Biol. (2020) 30:101417. doi: 10.1016/j.redox.2019.101417
63. Liu P, Wu D, Duan J, Xiao H, Zhou Y, Zhao L, et al. NRF2 regulates the sensitivity of human NSCLC cells to cystine deprivation-induced ferroptosis via FOCAD-FAK signaling pathway. Redox Biol. (2020) 37:101702. doi: 10.1016/j.redox.2020.101702
64. Hsieh CH, Hsieh HC, Shih FS, Wang PW, Yang LX, Shieh DB, et al. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics. (2021) 11:7072–91. doi: 10.7150/thno.57803
65. Nordberg J, Arnér ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. (2001) 31:1287–312. doi: 10.1016/s0891-5849(01)00724-9
66. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. (2021) 31:107–25. doi: 10.1038/s41422-020-00441-1
67. Llabani E, Hicklin RW, Lee HY, Motika SE, Crawford LA, Weerapana E, et al. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat Chem. (2019) 11:521–32. doi: 10.1038/s41557-019-0261-6
68. Rawat PS, Jaiswal A, Khurana A, Bhatti JS, Navik U. Doxorubicin-induced cardiotoxicity: an update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed Pharmacother. (2021) 139:111708. doi: 10.1016/j.biopha.2021.111708
69. Liu Y, Zeng L, Yang Y, Chen C, Wang D, Wang H. Acyl-coa thioesterase 1 prevents cardiomyocytes from doxorubicin-induced ferroptosis via shaping the lipid composition. Cell Death Dis. (2020) 11:756. doi: 10.1038/s41419-020-02948-2
70. Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. (2000) 60:1789–92.
71. Li M, Sala V, De Santis MC, Cimino J, Cappello P, Pianca N, et al. Phosphoinositide 3-kinase gamma inhibition protects from anthracycline cardiotoxicity and reduces tumor growth. Circulation. (2018) 138:696–711. doi: 10.1161/circulationaha.117.030352
72. Singla DK, Johnson TA, Tavakoli Dargani Z. Exosome treatment enhances anti-inflammatory M2 macrophages and reduces inflammation-induced pyroptosis in doxorubicin-induced cardiomyopathy. Cells. (2019) 8:1224. doi: 10.3390/cells8101224
73. Zhang H, Wang Z, Liu Z, Du K, Lu X. Protective effects of dexazoxane on rat ferroptosis in doxorubicin-induced cardiomyopathy through regulating HMGB1. Front Cardiovasc Med. (2021) 8:685434. doi: 10.3389/fcvm.2021.685434
74. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA. (2019) 116:2672–80. doi: 10.1073/pnas.1821022116
75. Finn NA, Findley HW, Kemp MLA. Switching mechanism in doxorubicin bioactivation can be exploited to control doxorubicin toxicity. PLoS Comput Biol. (2011) 7:e1002151. doi: 10.1371/journal.pcbi.1002151
76. Lewandowski M, Gwozdzinski K. Nitroxides as antioxidants and anticancer drugs. Int J Mol Sci. (2017) 18:2490. doi: 10.3390/ijms18112490
77. Zhao L, Qi Y, Xu L, Tao X, Han X, Yin L, et al. Microrna-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. (2018) 15:284–96. doi: 10.1016/j.redox.2017.12.013
78. Sunitha MC, Dhanyakrishnan R, PrakashKumar B, Nevin KG. p-Coumaric acid mediated protection of H9c2 cells from doxorubicin-induced cardiotoxicity: involvement of augmented Nrf2 and autophagy. Biomed Pharmacother. (2018) 102:823–32. doi: 10.1016/j.biopha.2018.03.089
79. He L, Yang Y, Chen J, Zou P, Li J. Transcriptional activation of Enpp2 by Foxo4 protects cardiomyocytes from doxorubicin-induced toxicity. Mol Med Rep. (2021) 24:668. doi: 10.3892/mmr.2021.12307
80. Hershko C, Link G, Tzahor M, Kaltwasser JP, Athias P, Grynberg A, et al. Anthracycline toxicity is potentiated by iron and inhibited by deferoxamine: studies in rat heart cells in culture. J Lab Clin Med. (1993) 122:245–51.
81. Xu X, Sutak R, Richardson DR. Iron chelation by clinically relevant anthracyclines: alteration in expression of iron-regulated genes and atypical changes in intracellular iron distribution and trafficking. Mol Pharmacol. (2008) 73:833–44. doi: 10.1124/mol.107.041335
82. Panjrath GS, Patel V, Valdiviezo CI, Narula N, Narula J, Jain D. Potentiation of doxorubicin cardiotoxicity by iron loading in a rodent model. J Am Coll Cardiol. (2007) 49:2457–64. doi: 10.1016/j.jacc.2007.02.060
83. Guenancia C, Li N, Hachet O, Rigal E, Cottin Y, Dutartre P, et al. Paradoxically, iron overload does not potentiate doxorubicin-induced cardiotoxicity in vitro in cardiomyocytes and in vivo in mice. Toxicol Appl Pharmacol. (2015) 284:152–62. doi: 10.1016/j.taap.2015.02.015
84. Miranda CJ, Makui H, Soares RJ, Bilodeau M, Mui J, Vali H, et al. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood. (2003) 102:2574–80. doi: 10.1182/blood-2003-03-0869
85. Vaitiekus D, Muckiene G, Vaitiekiene A, Sereikaite L, Inciuraite R, Insodaite R, et al. Hfe gene variants’ impact on anthracycline-based chemotherapy-induced subclinical cardiotoxicity. Cardiovasc Toxicol. (2021) 21:59–66. doi: 10.1007/s12012-020-09595-1
86. Lipshultz SE, Lipsitz SR, Kutok JL, Miller TL, Colan SD, Neuberg DS, et al. Impact of hemochromatosis gene mutations on cardiac status in doxorubicin-treated survivors of childhood high-risk leukemia. Cancer. (2013) 119:3555–62. doi: 10.1002/cncr.28256
87. Mouli S, Nanayakkara G, AlAlasmari A, Eldoumani H, Fu X, Berlin A, et al. The role of frataxin in doxorubicin-mediated cardiac hypertrophy. Am J Physiol Heart Circ Physiol. (2015) 309:H844–59. doi: 10.1152/ajpheart.00182.2015
88. Kotamraju S, Chitambar CR, Kalivendi SV, Joseph J, Kalyanaraman B. Transferrin receptor-dependent iron uptake is responsible for doxorubicin-mediated apoptosis in endothelial cells: role of oxidant-induced iron signaling in apoptosis. J Biol Chem. (2002) 277:17179–87. doi: 10.1074/jbc.M111604200
89. Zhuang S, Ma Y, Zeng Y, Lu C, Yang F, Jiang N, et al. Mettl14 promotes doxorubicin-induced cardiomyocyte ferroptosis by regulating the KCNQ1OT1-mir-7-5p-TFRC axis. Cell Biol Toxicol. (2021). doi: 10.1007/s10565-021-09660-7 [Epub ahead of print].
90. Cocco E, Porrini V, Derosas M, Nardi V, Biasiotto G, Maccarinelli F, et al. Protective effect of mitochondrial ferritin on cytosolic iron dysregulation induced by doxorubicin in hela cells. Mol Biol Rep. (2013) 40:6757–64. doi: 10.1007/s11033-013-2792-z
91. Corna G, Santambrogio P, Minotti G, Cairo G. Doxorubicin paradoxically protects cardiomyocytes against iron-mediated toxicity: role of reactive oxygen species and ferritin. J Biol Chem. (2004) 279:13738–45. doi: 10.1074/jbc.M310106200
92. Asensio-López MC, Sánchez-Más J, Pascual-Figal DA, Abenza S, Pérez-Martínez MT, Valdés M, et al. Involvement of ferritin heavy chain in the preventive effect of metformin against doxorubicin-induced cardiotoxicity. Free Radic Biol Med. (2013) 57:188–200. doi: 10.1016/j.freeradbiomed.2012.09.009
93. Minotti G, Ronchi R, Salvatorelli E, Menna P, Cairo G. Doxorubicin irreversibly inactivates iron regulatory proteins 1 and 2 in cardiomyocytes: evidence for distinct metabolic pathways and implications for iron-mediated cardiotoxicity of antitumor therapy. Cancer Res. (2001) 61:8422–8.
94. Mueller S. Iron regulatory protein 1 as a sensor of reactive oxygen species. Biofactors. (2005) 24:171–81. doi: 10.1002/biof.5520240121
95. Brazzolotto X, Andriollo M, Guiraud P, Favier A, Moulis JM. Interactions between doxorubicin and the human iron regulatory system. Biochim Biophys Acta. (2003) 1593:209–18. doi: 10.1016/s0167-4889(02)00391-9
96. Kwok JC, Richardson DR. Unexpected anthracycline-mediated alterations in iron-regulatory protein-RNA-binding activity: the iron and copper complexes of anthracyclines decrease RNA-binding activity. Mol Pharmacol. (2002) 62:888–900. doi: 10.1124/mol.62.4.888
97. Corna G, Galy B, Hentze MW, Cairo G. IRP1-independent alterations of cardiac iron metabolism in doxorubicin-treated mice. J Mol Med. (2006) 84:551–60. doi: 10.1007/s00109-006-0068-y
98. Canzoneri JC, Oyelere AK. Interaction of anthracyclines with iron responsive element mRNAs. Nucleic Acids Res. (2008) 36:6825–34. doi: 10.1093/nar/gkn774
99. Christidi E, Brunham LR. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. (2021) 12:339. doi: 10.1038/s41419-021-03614-x
100. Dodson M, Castro-Portuguez R, Zhang DD. Nrf2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
101. Luo LF, Guan P, Qin LY, Wang JX, Wang N, Ji ES. Astragaloside IV inhibits adriamycin-induced cardiac ferroptosis by enhancing Nrf2 signaling. Mol Cell Biochem. (2021) 476:2603–11. doi: 10.1007/s11010-021-04112-6
102. Lu Z, Liu Z, Fang B. Propofol protects cardiomyocytes from doxorubicin-induced toxic injury by activating the nuclear factor erythroid 2-related factor 2/glutathione peroxidase 4 signaling pathways. Bioengineered. (2022) 13:9145–55. doi: 10.1080/21655979.2022.2036895
103. Wang Y, Yan S, Liu X, Deng F, Wang P, Yang L, et al. Prmt4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/Gpx4 pathway. Cell Death Differ. (2022). doi: 10.1038/s41418-022-00990-5 [Epub ahead of print].
104. Hou K, Shen J, Yan J, Zhai C, Zhang J, Pan JA, et al. Loss of Trim21 alleviates cardiotoxicity by suppressing ferroptosis induced by the chemotherapeutic agent doxorubicin. EBioMedicine. (2021) 69:103456. doi: 10.1016/j.ebiom.2021.103456
105. Li D, Liu X, Pi W, Zhang Y, Yu L, Xu C, et al. Fisetin attenuates doxorubicin-induced cardiomyopathy in vivo and in vitro by inhibiting ferroptosis through Sirt1/Nrf2 signaling pathway activation. Front Pharmacol. (2021) 12:808480. doi: 10.3389/fphar.2021.808480
106. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol. (2021) 220:e202105043. doi: 10.1083/jcb.202105043
107. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK Activation Inhibits Ferroptosis. Nat Cell Biol. (2020) 22:225–34. doi: 10.1038/s41556-020-0461-8
108. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. (2019) 73:354–63.e3. doi: 10.1016/j.molcel.2018.10.042
109. Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Investig. (2014) 124:617–30. doi: 10.1172/jci72931
110. Fox CA, Ryan RO. Dye binding assay reveals doxorubicin preference for DNA versus cardiolipin. Anal Biochem. (2020) 594:113617. doi: 10.1016/j.ab.2020.113617
111. Moulin M, Solgadi A, Veksler V, Garnier A, Ventura-Clapier R, Chaminade P. Sex-specific cardiac cardiolipin remodelling after doxorubicin treatment. Biol Sex Differ. (2015) 6:20. doi: 10.1186/s13293-015-0039-5
112. Pereira GC, Pereira SP, Tavares LC, Carvalho FS, Magalhães-Novais S, Barbosa IA, et al. Cardiac cytochrome C and cardiolipin depletion during anthracycline-induced chronic depression of mitochondrial function. Mitochondrion. (2016) 30:95–104. doi: 10.1016/j.mito.2016.07.005
113. Fedotcheva TA, Fedotcheva NI. Protectors of the mitochondrial permeability transition pore activated by iron and doxorubicin. Curr Cancer Drug Targets. (2021) 21:514–25. doi: 10.2174/1568009621999210120192558
114. Ihnat PM, Vennerstrom JL, Robinson DH. Synthesis and solution properties of deferoxamine amides. J Pharm Sci. (2000) 89:1525–36. doi: 10.1002/1520-6017(200012)89:123.0.co;2-t
115. Link G, Tirosh R, Pinson A, Hershko C. Role of iron in the potentiation of anthracycline cardiotoxicity: identification of heart cell mitochondria as a major site of iron-anthracycline interaction. J Lab Clin Med. (1996) 127:272–8. doi: 10.1016/s0022-2143(96)90095-5
116. Maccarinelli F, Gammella E, Asperti M, Regoni M, Biasiotto G, Turco E, et al. Mice lacking mitochondrial ferritin are more sensitive to doxorubicin-mediated cardiotoxicity. J Mol Med. (2014) 92:859–69. doi: 10.1007/s00109-014-1147-0
117. Ichikawa Y, Bayeva M, Ghanefar M, Potini V, Sun L, Mutharasan RK, et al. Disruption of ATP-binding cassette b8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc Natl Acad Sci USA. (2012) 109:4152–7. doi: 10.1073/pnas.1119338109
118. Kwok JC, Richardson DR. Examination of the mechanism(S) involved in doxorubicin-mediated iron accumulation in ferritin: studies using metabolic inhibitors, protein synthesis inhibitors, and lysosomotropic agents. Mol Pharmacol. (2004) 65:181–95. doi: 10.1124/mol.65.1.181
119. Kwok JC, Richardson DR. Anthracyclines induce accumulation of iron in ferritin in myocardial and neoplastic cells: inhibition of the ferritin iron mobilization pathway. Mol Pharmacol. (2003) 63:849–61. doi: 10.1124/mol.63.4.849
120. Thomas CE, Aust SD. Release of iron from ferritin by cardiotoxic anthracycline antibiotics. Arch Biochem Biophys. (1986) 248:684–9. doi: 10.1016/0003-9861(86)90523-0
121. Kaźmierczak-Barańska J, Boguszewska K, Adamus-Grabicka A, Karwowski BT. Two faces of vitamin C-antioxidative and pro-oxidative agent. Nutrients. (2020) 12:1501. doi: 10.3390/nu12051501
122. Koppenol WH. The haber-weiss cycle–70 years later. Redox Rep. (2001) 6:229–34. doi: 10.1179/135100001101536373
123. Sugioka K, Nakano M. Mechanism of phospholipid peroxidation induced by ferric ion-ADP-adriamycin-co-ordination complex. Biochim Biophys Acta. (1982) 713:333–43.
124. Miura T, Muraoka S, Ogiso T. Lipid peroxidation of rat erythrocyte membrane induced by adriamycin-Fe3+. Pharmacol Toxicol. (1991) 69:296–300. doi: 10.1111/bcpt.1991.69.4.296
125. Keizer HG, Pinedo HM, Schuurhuis GJ, Joenje H. Doxorubicin (adriamycin): a critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol Ther. (1990) 47:219–31. doi: 10.1016/0163-7258(90)90088-j
126. Simùnek T, Stérba M, Popelová O, Adamcová M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep. (2009) 61:154–71. doi: 10.1016/s1734-1140(09)70018-0
127. Miura T, Muraoka S, Ogiso T. Adriamycin-induced lipid peroxidation of erythrocyte membranes in the presence of ferritin and the inhibitory effect of ceruloplasmin. Biol Pharm Bull. (1993) 16:664–7. doi: 10.1248/bpb.16.664
128. Zhang Y, Liu Y, Xia Y. Dedicated to cardio-oncology. Eur Heart J. (2020) 41:907–9. doi: 10.1093/eurheartj/ehaa064
129. Lyon AR, Dent S, Stanway S, Earl H, Brezden-Masley C, Cohen-Solal A, et al. Baseline cardiovascular risk assessment in cancer patients scheduled to receive cardiotoxic cancer therapies: a position statement and new risk assessment tools from the cardio-oncology study group of the heart failure association of the European society of cardiology in collaboration with the international cardio-oncology society. Eur J Heart Fail. (2020) 22:1945–60. doi: 10.1002/ejhf.1920
130. Noel CV, Rainusso N, Robertson M, Romero J, Masand P, Coarfa C, et al. Early detection of myocardial changes with and without dexrazoxane using serial magnetic resonance imaging in a pre-clinical mouse model. Cardio Oncol. (2021) 7:23. doi: 10.1186/s40959-021-00109-8
131. Jirkovskı E, Lenèová-Popelová O, Hroch M, Adamcová M, Mazurová Y, Vávrová J, et al. Early and delayed cardioprotective intervention with dexrazoxane each show different potential for prevention of chronic anthracycline cardiotoxicity in rabbits. Toxicology. (2013) 311:191–204. doi: 10.1016/j.tox.2013.06.012
132. Jirkovskı E, Jirkovská A, Bavloviè-Piskáèková H, Skalická V, Pokorná Z, Karabanovich G, et al. Clinically translatable prevention of anthracycline cardiotoxicity by dexrazoxane is mediated by topoisomerase II beta and not metal chelation. Circ Heart Fail. (2021) 14:e008209. doi: 10.1161/circheartfailure.120.008209
133. Hasinoff BB, Patel D, Wu XA. Qsar study that compares the ability of bisdioxopiperazine analogs of the doxorubicin cardioprotective agent dexrazoxane (ICRF-187) to protect myocytes with DNA topoisomerase II inhibition. Toxicol Appl Pharmacol. (2020) 399:115038. doi: 10.1016/j.taap.2020.115038
134. Yu X, Ruan Y, Shen T, Qiu Q, Yan M, Sun S, et al. Dexrazoxane protects cardiomyocyte from doxorubicin-induced apoptosis by modulating mIR-17-5p. Biomed Res Int. (2020) 2020:5107193. doi: 10.1155/2020/5107193
135. Yu JL, Jin Y, Cao XY, Gu HH. Dexmedetomidine alleviates doxorubicin cardiotoxicity by inhibiting mitochondrial reactive oxygen species generation. Hum Cell. (2020) 33:47–56. doi: 10.1007/s13577-019-00282-0
136. Popelová O, Sterba M, Hasková P, Simùnek T, Hroch M, Guncová I, et al. Dexrazoxane-afforded protection against chronic anthracycline cardiotoxicity in vivo: effective rescue of cardiomyocytes from apoptotic cell death. Br J Cancer. (2009) 101:792–802. doi: 10.1038/sj.bjc.6605192
137. Basch E, Prestrud AA, Hesketh PJ, Kris MG, Feyer PC, Somerfield MR, et al. Antiemetics: American society of clinical oncology clinical practice guideline update. J Clin Oncol. (2011) 29:4189–98. doi: 10.1200/jco.2010.34.4614
138. Wengström Y, Margulies A. European oncology nursing society extravasation guidelines. Eur J Oncol Nurs. (2008) 12:357–61. doi: 10.1016/j.ejon.2008.07.003
139. Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS, et al. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin’s disease. J Clin Oncol. (2007) 25:493–500. doi: 10.1200/jco.2005.02.3879
140. Shaikh F, Dupuis LL, Alexander S, Gupta A, Mertens L, Nathan PC. Cardioprotection and second malignant neoplasms associated with dexrazoxane in children receiving anthracycline chemotherapy: a systematic review and meta-analysis. J Natl Cancer Inst. (2016) 108:djv357. doi: 10.1093/jnci/djv357
141. Seif AE, Walker DM, Li Y, Huang YS, Kavcic M, Torp K, et al. Dexrazoxane exposure and risk of secondary acute myeloid leukemia in pediatric oncology patients. Pediatr Blood Cancer. (2015) 62:704–9. doi: 10.1002/pbc.25043
142. Li J, Chang HM, Banchs J, Araujo DM, Hassan SA, Wagar EA, et al. Detection of subclinical cardiotoxicity in sarcoma patients receiving continuous doxorubicin infusion or pre-treatment with dexrazoxane before bolus doxorubicin. Cardio Oncol. (2020) 6:1. doi: 10.1186/s40959-019-0056-3
143. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ III, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
144. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. (2014) 509:105–9. doi: 10.1038/nature13148
145. Voest EE, van Acker SA, van der Vijgh WJ, van Asbeck BS, Bast A. Comparison of different iron chelators as protective agents against acute doxorubicin-induced cardiotoxicity. J Mol Cell Cardiol. (1994) 26:1179–85. doi: 10.1006/jmcc.1994.1136
146. Herman EH, Zhang J, Ferrans VJ. Comparison of the protective effects of desferrioxamine and ICRF-187 against doxorubicin-induced toxicity in spontaneously hypertensive rats. Cancer Chemother Pharmacol. (1994) 35:93–100. doi: 10.1007/bf00686629
147. Bentur Y, McGuigan M, Koren G. Deferoxamine (desferrioxamine). New toxicities for an old drug. Drug Saf. (1991) 6:37–46. doi: 10.2165/00002018-199106010-00004
148. Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. (2017) 3:232–43. doi: 10.1021/acscentsci.7b00028
149. Saito Y. Diverse cytoprotective actions of vitamin E isoforms- role as peroxyl radical scavengers and complementary functions with selenoproteins. Free Radic Biol Med. (2021) 175:121–9. doi: 10.1016/j.freeradbiomed.2021.08.234
150. Saito Y, Yoshida Y, Akazawa T, Takahashi K, Niki E. Cell death caused by selenium deficiency and protective effect of antioxidants. J Biol Chem. (2003) 278:39428–34. doi: 10.1074/jbc.M305542200
151. Hinman A, Holst CR, Latham JC, Bruegger JJ, Ulas G, McCusker KP, et al. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS One. (2018) 13:e0201369. doi: 10.1371/journal.pone.0201369
152. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
153. Santoro MM. The antioxidant role of non-mitochondrial CoQ10: mystery solved! Cell Metab. (2020) 31:13–5. doi: 10.1016/j.cmet.2019.12.007
154. Russo M, Della Sala A, Tocchetti CG, Porporato PE, Ghigo A. Metabolic aspects of anthracycline cardiotoxicity. Curr Treat Opt Oncol. (2021) 22:18. doi: 10.1007/s11864-020-00812-1
155. Ajzashokouhi AH, Bostan HB, Jomezadeh V, Hayes AW, Karimi GA. Review on the cardioprotective mechanisms of metformin against doxorubicin. Hum Exp Toxicol. (2020) 39:237–48. doi: 10.1177/0960327119888277
156. Quagliariello V, De Laurentiis M, Rea D, Barbieri A, Monti MG, Carbone A, et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol. (2021) 20:150. doi: 10.1186/s12933-021-01346-y
157. Chen J, Zhang S, Pan G, Lin L, Liu D, Liu Z, et al. Modulatory effect of metformin on cardiotoxicity induced by doxorubicin via the MAPK and AMPK pathways. Life Sci. (2020) 249:117498. doi: 10.1016/j.lfs.2020.117498
158. Ashour AE, Sayed-Ahmed MM, Abd-Allah AR, Korashy HM, Maayah ZH, Alkhalidi H, et al. Metformin rescues the myocardium from doxorubicin-induced energy starvation and mitochondrial damage in rats. Oxid Med Cell Longev. (2012) 2012:434195. doi: 10.1155/2012/434195
159. Sheta A, Elsakkar M, Hamza M, Solaiman A. Effect of metformin and sitagliptin on doxorubicin-induced cardiotoxicity in adult male albino rats. Hum Exp Toxicol. (2016) 35:1227–39. doi: 10.1177/0960327115627685
160. Asensio-Lopez MC, Sanchez-Mas J, Pascual-Figal DA, de Torre C, Valdes M, Lax A. Ferritin heavy chain as main mediator of preventive effect of metformin against mitochondrial damage induced by doxorubicin in cardiomyocytes. Free Radic Biol Med. (2014) 67:19–29. doi: 10.1016/j.freeradbiomed.2013.11.003
Keywords: ferroptosis, doxorubicin, iron, treatment, mechanism, cardiotoxicity
Citation: Zhang G, Yuan C, Su X, Zhang J, Gokulnath P, Vulugundam G, Li G, Yang X, An N, Liu C, Sun W, Chen H, Wu M, Sun S and Xing Y (2022) Relevance of Ferroptosis to Cardiotoxicity Caused by Anthracyclines: Mechanisms to Target Treatments. Front. Cardiovasc. Med. 9:896792. doi: 10.3389/fcvm.2022.896792
Received: 15 March 2022; Accepted: 24 May 2022;
Published: 13 June 2022.
Edited by:
Canan G. Nebigil, INSERM U1260 Nanomedicine Régénératrice (RNM), FranceReviewed by:
Sabzali Javadov, University of Puerto Rico, Puerto RicoCopyright © 2022 Zhang, Yuan, Su, Zhang, Gokulnath, Vulugundam, Li, Yang, An, Liu, Sun, Chen, Wu, Sun and Xing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shipeng Sun, c2hpcGVuZ3N1bkBnbWFpbC5jb20=; Yanwei Xing, eGluZ3lhbndlaTEyMzQ1QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.