 Shuangshuang Wang1
Shuangshuang Wang1 Siwang Hu2*
Siwang Hu2*- 1Department of Cardiology, The First People's Hospital of Wenling (The Affiliated Wenling Hospital of Wenzhou Medical University), Wenling, China
- 2The Orthopedic Center, The First People's Hospital of Wenling (The Affiliated Wenling Hospital of Wenzhou Medical University), Wenling, China
Vascular calcification (VC) is a common pathological change in many chronic diseases, such as diabetes and chronic kidney disease. It is mainly deposited in the intima and media of vessels in the form of hydroxyapatite. Recently, a lot of research has been performed to show that VC is associated with various cellular stresses, such as hyperphosphate, hyperglycemia and oxidative stress. Unfortunately, our understanding of the pathogenesis of calcification is far from comprehensive. Sirtuins belong to a family of class III highly conserved deacetylases that are involved in the regulation of biological and cellular processes including mitochondrial biogenesis, metabolism, oxidative stress, inflammatory response, DNA repair, etc. Numerous studies have shown that sirtuins might play protective roles in VC, and restoring the activity of sirtuins may be a potentially effective treatment for VC. However, the exact mechanism of their vascular protection remains unclear. Here, we reviewed the roles of sirtuins in the osteogenic transformation of vascular smooth muscle cells and the development of VC. We also elucidated the applications of sirtuins agonists for the treatment of VC.
Introduction
As the aging trend of the world population continues, vascular calcification (VC) is becoming a major global health problem. VC is characterized by the deposition of hydroxyapatite in the vessel wall, which is mainly divided into intima calcification and medial calcification (1, 2). The former is common in patients with atherosclerosis, while medial calcification often occurs in patients with chronic kidney disease (CKD), diabetes, and hypertension, which causes arterial remodeling and vascular stiffness (3). The recent study showed that abdominal aortic calcification was associated with increased risk of atherosclerotic vascular disease mortality and all-cause mortality (4). And the patients with macrocalcifications had a three- to four-fold risk of developing a fatal cardiovascular event (5). Unfortunately, due to the unclear mechanism of VC, there is no effective drug or method for preventing or reversing VC. Therefore, it is urgent to deeply explore the pathogenesis of VC and develop new strategies to alleviate the onset and progression of VC.
Sirtuins (SIRTs) comprise a family of highly conserved nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases, with seven homologous analogs in mammals, SIRT1-SIRT7 (6). The SIRT1-7 share a conserved 275-amino-acid catalytic core domain, but have different subcellular localization, among which SIRT1, SIRT6 and SIRT7 are mainly located in the nucleus, SIRT3, SIRT4 and SIRT5 in the mitochondria, and SIRT2 in the cytoplasm (7). SIRTs have been proven to be involved in the regulation of several pathophysiological processes via deacetylating various substrates, including cell metabolism, inflammation, oxidative stress, and aging (8–11). All these pathophysiological processes play crucial parts in VC. However, the mechanisms of different SIRTs homologs in VC are complicated, and the pathways vary from the homologs on account of their different localization in cells. In this review, we were dedicated to clarifying the role and exact mechanism of SIRTs during the occurrence and development of VC.
The Pathophysiology of Vascular Calcification
VC is a significant feature of vascular changes in various physiological or pathological states. Increasing evidence shows that aging is an important contributor to VC (12). In addition to aging, many other risk factors including CKD, diabetes, hypertension, and dyslipidemia, might participate in the progression of VC (13). Notably, CKD can significantly increase the risk of cardiovascular disease, especially VC (14). The patients with CKD are frequently exposed to various pro-calcification factors, such as phosphate, indolyl sulfate, advanced glycation end products, and pro-inflammatory cytokines (15, 16). High phosphorus due to mineral imbalance in CKD drives the occurrence of VC, and protein-bound uremic toxin could further aggravate VC (17, 18). Besides, studies revealed that postmenopausal women were also at a significantly increased risk of VC (19–21). In recently menopausal women, estrogen supplementation could slow the progression of coronary artery calcification (22). Originally, VC was thought to be a passive degenerative process that marked the aging of blood vessels. However, more and more studies have shown that VC is an active process regulated by multiple factors, including abnormal calcium and phosphate homeostasis, oxidative stress, inflammation, matrix remodeling, and so on (23). Therefore, VC is a common disease with complex regulatory mechanisms.
The vessel wall is composed of endothelial cells, vascular smooth muscle cells (VSMCs), fibroblasts, and pericytes (24). Normally, a monolayer of endothelial cells forms the innermost layer that acts as a barrier between the vessel lumen and the vessel wall in order to maintain the non-thrombotic surface and quiescence in the vascular wall. Under pathological conditions, endothelial cells also respond to different stimuli and exist in various activation states, including inflammatory, angiogenic, and osteogenic phenotypes (25–27). Growing evidence suggests that endothelial cells influence the development of VC in a variety of pathways, including transition to mesenchymal and osteoblastic lineages, secretion of calcific growth factors, disruption of the proteolytic activity of IELs, induction of endothelial alkaline phosphatase and inappropriate interactions with underlying cells (27).
As the most abundant cell type, VSMCs are mainly located in the tunica media with a high degree of plasticity and regulate the dilation and contraction of vessels (28). The senescence of VSMCs, the osteogenic transformation of VSMCs, the apoptosis of VSMCs, and the deposition of extracellular matrix have been confirmed as the reasons for VSMCs dysfunction and the development of VC (11). The transition of VSMCs from a contractile state to a secretory phenotype has been identified as a key event in VC, and the synthesized VSMCs undergoes further maladaptive osteogenic differentiation under continuous stimulation, ultimately leading to hydroxyapatite deposition in blood vessels (Figure 1) (29, 30). As VSMCs transition to an osteogenic phenotype upon specific stimuli, these cells could acquire the characteristics of osteoblasts (31). It is mainly manifested by increased expression of osteogenic markers such as ALP, BMP2, and RUNX2, while decreased expression of calcification-inhibitor protein (32). What's more, senescent VSMCs could also promote their osteogenic transformation (33). To sum up, VSMCs are involved in the process of VC from multiple perspectives.
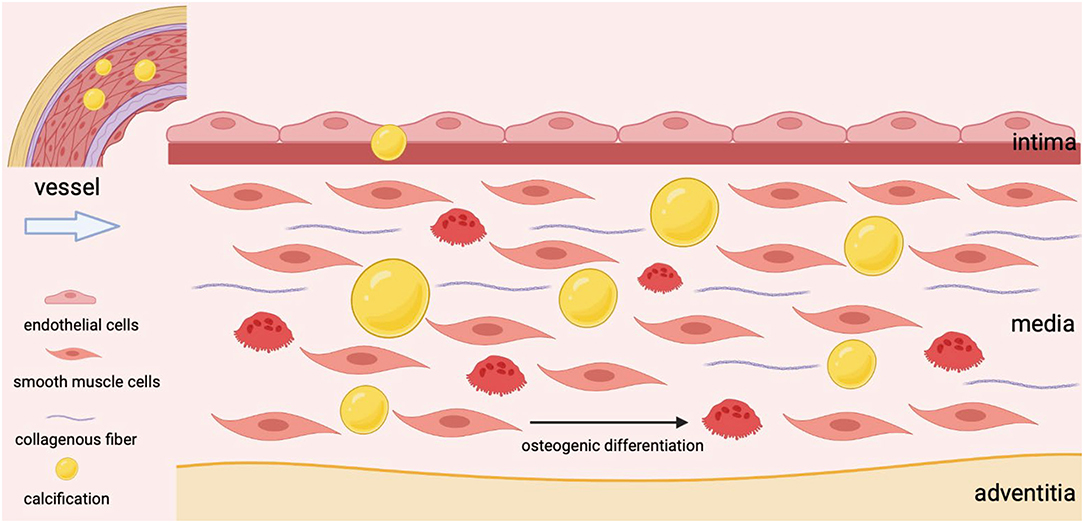
Figure 1. The osteogenic differentiation of VSMCs during vascular calcification.
Sirtuins and Vascular Calcification
As classical deacetylases, SIRTs catalyze the deacetylation of proteins mainly by breaking the bond between NAD+ and nicotinamide ribosomes, and then participate in the regulation of various processes (34, 35). For example, SIRT1 might mediate cellular senescence by regulating p53 and p65 acetylation (33). And the decreased vascular SIRT1 expression was associated with age-related arteriosclerosis (36). In recent years, the molecular mechanism of SIRTs in the phenotypic changes of VSMCs has been extensively studied in VC. In the following, we mainly focused on the roles of SIRTs in VSMCs osteogenic differentiation and their potential as therapeutic targets for VC.
SIRT1 and Vascular Calcification
SIRT1 is the well-studied family member of NAD+-dependent deacetylase, which could protect against cardiovascular diseases (37, 38). In Japanese hemodialysis patients, SIRT1 polymorphisms (rs7069102 and rs2273773) were confirmed to be associated with coronary artery calcification (39). In-vitro cultured aortas from SIRT1−/− mice exhibited accelerated medial calcification upon phosphate stimulation (40). A transgenic mouse model was used to demonstrate that lifetime overexpression of SIRT1 might ameliorate aortic stiffness and prevent aortic calcification with advancing age (41). Therefore, SIRT1 plays an essential role in VC.
The Molecular Mechanism of SIRT1 in VC
Existing evidence from cell and animal models indicated that SIRT1 was involved in VC through a variety of signaling pathways (Figure 2). Most importantly, SIRT1 could prevent arterial calcification via inhibition of VSMCs osteogenic differentiation (42). And SIRT1 could serve as a positive regulator of the osteoblast transcription factor, RUNX2 (43). Under high glucose conditions, the downregulation of SIRT1 expression promoted acetylation of the RUNX2 promoter region, thereby increasing the osteogenic differentiation of VSMCs (44). On the other hand, low expression of SIRT1 fails to deacetylate β-Catenin and HMGB1, thus promoting their translocation to the nucleus, and promoting the development of VC by activating Wnt signaling (45). The lncRNA HOTAIR can upregulate SIRT1 expression via miR-126/Klotho axis, and then attenuate VSMCs calcification and VC by inhibiting Wnt/β-catenin pathway (46). Additionally, SIRT1 downregulation may promote NBS1 acetylation and inhibit ATM activation, thereby inhibiting MRN complex formation, and ultimately leading to failure of DNA repair (47). And increased SIRT1 could promote MRN activation, leading to increased DNA repair and cell survival, ultimately attenuating DNA damage-induced VSMCs osteogenic differentiation and VC in diabetes (48).
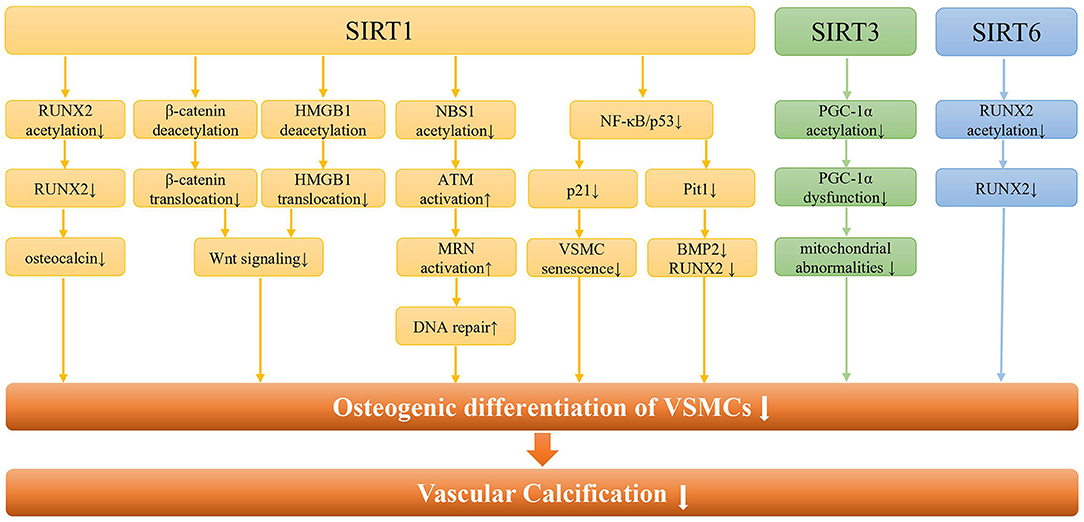
Figure 2. The molecular pathways of SIRTs in vascular calcification. Regarding SIRT1, it can regulate the osteogenic differentiation of VSMCs and vascular calcification through the RUNX2/osteocalcin pathway, the Wnt signaling pathway, the DNA repair pathway, and the NF-κB pathway. In addition, SIRT1 can alleviate the osteogenic differentiation of VSMCs by regulating the senescence of VSMCs through NF-κB/p53/p21 pathway. As for SIRT3, it is mainly involved in the osteogenic differentiation of VSMCs by regulating mitochondrial function through PGC-1a. Besides, SIRT6 regulates vascular calcification through RUNX2 deacetylation in CKD.
Furthermore, phosphate-induced VSMCs calcification may be associated with both premature and replicative cellular senescence (42). With increasing age, the SIRT1 expression in human VSMCs was reduced, which led to reducing migratory and proliferative capacity, as well as induction of cellular senescence (49). Han et al. reported that upregulated SIRT1 expression could retard IL-1β-induced VSMCs senescence via NF-κB/p53/p21 pathways (50). In addition, high glucose and high phosphate can synergistically inhibit SIRT1 expression, increase p65 acetylation, and activate NF-κB signaling pathway, thereby increasing osteogenic transformation, cellular senescence, and ROS levels in VSMCs, resulting in VC (51). The upregulated miR-34a could promote senescence in VSMCs and ultimately trigger VC occurrence and progression (52, 53). And miR-34a directly downregulated Axl and SIRT1 to induce cellular senescence and promote VSMC mineralization and VC (54). Therefore, cell senescence might synergize with downregulation of SIRT1 to cause the osteogenic transformation of VSMCs, which ultimately leads to the occurrence of calcification.
All the above highlights that SIRT1 serves as a key factor in both senescence and osteogenic differentiation of VSMCs, which exist a synergistic role in VC. It provides new insights and rationale for future development of specific therapeutic targets with potent SIRT1 analogs.
SIRT1 as a Potential Therapeutic Target for VC
Based on the above evidence, SIRT1 is an important target in preventing the occurrence and development of VC. Strategies to enhance the activity of SIRT1 may be an effective mean to prevent and improve VC. In this regard, several SIRT1 activators have been synthesized to activate SIRT1, such as SRT1720, SRT2014 and SRT1460 (11). SRT1720 could protect VSMCs from calcification by activating SIRT1 in either osteogenesis or hyperglycemia condition (44, 51). Besides, some natural drugs have been shown to act as activators of SIRT1, including resveratrol, spermidine, terpene-4-ol, etc. (Table 1).
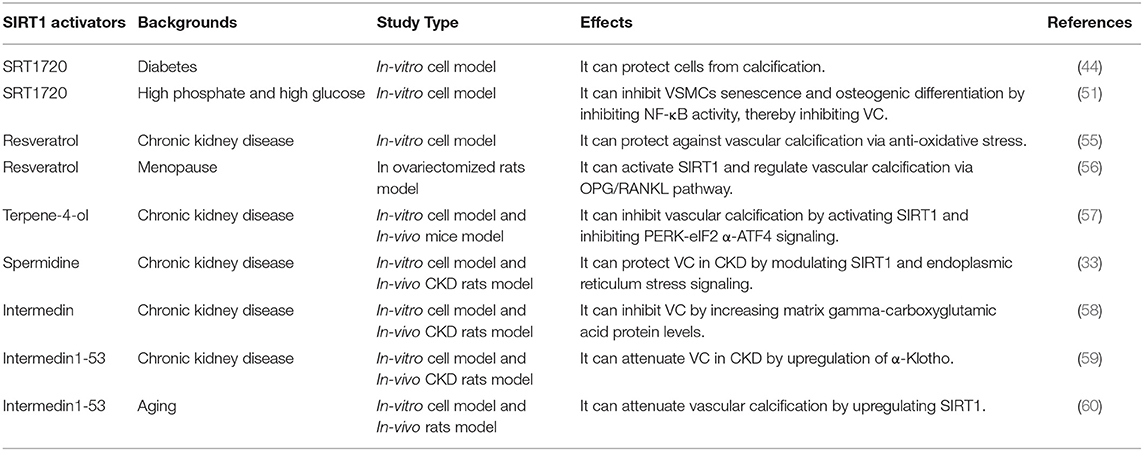
Table 1. The applications of SIRT1 activators in vascular calcification.
Resveratrol (3,5,4'-trihydroxy-trans-stilbene), known as phytoestrogens in grapes, peanuts, and legumes, is a scavenger for free radicals (61). It has been reported that resveratrol could inhibit the oxidative stress-induced proliferation of VSMCs by activating SIRT1 signaling pathway, thereby improving cardiovascular diseases (61). Zhang et al. found that resveratrol could effectively improve VC in patients with end-stage renal disease, and the mechanism may be to inhibit the oxidative damage of VSMCs by regulating SIRT1 and Nrf2 (55). On the other hand, since resveratrol has a similar structure to estrogen, it can bind to estrogen receptors to produce similar effects. Resveratrol could reduce the expression levels of ALP, RUNX2 and increase the expression levels of OPG in the aortas of ovariectomized rats (56). In addition, the authors speculated that resveratrol might play key roles in regulating aortic calcification through SIRT1 activation. Therefore, resveratrol may be a potential drug for VC.
Terpene-4-ol is a common component in plant essential oils, which has anti-inflammatory and anti-tumor effects (62, 63). It has been reported to reduce calcium deposition in VSMCs and CKD mouse arteries and inhibit the phenotype switching of VSMCs (57). Mechanistically, terpinen-4-ol exerts anti-calcification effects by activating SIRT1 to deacetylate PERK and then inhibiting the endoplasmic reticulum stress pathway–PERK-eIF2α-ATF4 signaling pathway. Moreover, endoplasmic reticulum stress increases the expression of ATF4, which could bind to the RUNX2 promoter, affecting VSMC calcification (64, 65). Taken together, terpinen-4-ol could be a promising therapeutic agent for CKD-related VC.
The natural polyamine spermidine, a novel autophagy inducer and longevity elixir, could extend lifespan and reduce oxidative stress (66, 67). The previous study showed that spermidine exerted an anti-aging effect to reduce arterial sclerosis by increasing NO bioavailability, reducing oxidative stress, modifying structural factors, and enhancing autophagy (68). In addition, spermidine could improve cardiomyocyte aging by activating mitochondrial biogenesis and function through SIRT1-mediated deacetylation of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) (69). Spermidine has been shown to protect VC in CKD by modulating SIRT1 and endoplasmic reticulum stress signaling (33). Moreover, spermidine was confirmed with the low toxicity and strong efficacy, suggesting it is a promising VC therapy in clinical application (70).
Intermedin (IMD) is a paracrine/endocrine peptide belonging to the calcitonin gene-related peptide superfamily, and IMD1-53 may be the main active fragment of IMD (71). IMD1-53 could improve vascular function by increasing endothelial nitric oxide synthase activity and inhibiting oxidative stress (72, 73). Studies have shown that IMD1-53 may attenuate VC in CKD by upregulating α-Klotho, and IMD could alleviate vitamin D3 plus nicotine (VDN)-induced VC by increasing the level of matrix gamma-carboxyglutamic acid protein (58, 59). Chen et al. found that IMD1-53 upregulated SIRT1 to protect senescence-associated VSMC calcification through PI3K/Akt, AMPK and cAMP/PKA signaling (60). In general, IMD1-53 has protective effects on VC caused by different causes through various pathways.
Overall, both natural and synthetic substances can relieve VC by improving SIRT1 activity. There is still a need to further explore efficient and safe drugs. Of course, diet and exercise also play an important role in vascular health. For example, calorie restriction can promote eNOS activity and SIRT1 expression, thereby reducing atherosclerosis development (74). Therefore, the therapeutic effects and mechanisms of diet and exercise in VC are worthy of study.
SIRT3 and Vascular Calcification
As a mitochondrial sirtuin with the highest deacetylase activity, SIRT3 could regulate the acetylation levels of several mitochondrial proteins, thereby regulating mitochondrial energy metabolism, redox balance and mitochondrial dynamics (75). Mitochondria are the main energy source for maintaining cardiac function, and mitochondrial dysfunction regulated by SIRT3 can lead to the progression of various cardiac diseases, including hypertension, coronary atherosclerosis, and heart failure (76). Feng et al. found that SIRT3 expression was decreased in β GP-induced calcification of VSMCs and in arteries of CKD rats and patients (77). Downregulation of SIRT3 might induce an increase in the acetylation of PGC-1α, leading to PGC-1α dysfunction, which also compromised mitochondrial adenosine triphosphate (ATP) production and morphology damage, ultimately triggering VSMCs phenotypic transition and calcium deposition. Furthermore, deletion of soluble epoxide hydrolase was reported to stabilize and increase SIRT3, thereby ameliorating mitochondrial abnormalities and VC in CKD or high phosphate conditions (78). Currently, berberine, honokiol, and resveratrol have been confirmed to target SIRT3 to correct pathological condition. However, no drug targeting SIRT3 has been used in the treatment of VC, which should be investigated in the future.
SIRT6 and Vascular Calcification
SIRT6, not only a chromatin-associated deacetylase but also a mono-ADP-ribosyltransferase, has been recognized to serve a pivotal role in a variety of biological processes, including DNA damage repair, cellular metabolism, etc (79). Previous studies showed that the downregulation of SIRT6 was involved in kidney injury, atherosclerosis, and cholesterol accumulation (79, 80). SIRT6 can attenuate CKD fibrosis by blocking the expression of β-catenin target genes through deacetylation (81). In addition, decreased SIRT6 was associated with an increased risk of VC in CKD patients (82). And SIRT6 could inhibit VSMCs osteogenic differentiation and attenuate VC through deacetylating RUNX2 and promoting its ubiquitination and subsequent degradation. Moreover, bone marrow mesenchymal stem cell exosomes could inhibit hyperphosphate-induced aortic calcification via the SIRT6-HMGB1 deacetylation pathway (83). In short, SIRT6 acts as a protective regulator and a potential therapeutic target for VC.
Discussion and Conclusion
Acetylation and deacetylation of proteins regulate a variety of cellular biological processes, including cell proliferation, gene transcription, apoptosis, protein stability, and mitochondrial metabolism, which are closely related to cancers and cardiovascular diseases (76, 84). Accumulating studies showed that the deacetylase family SIRTs were essential for regulating several aspects of VC. In this review, we mainly introduced the regulatory roles of SIRTs in the osteogenic differentiation of VSMCs. The downregulation of both SIRT1 and SIRT6 could promote RUNX2 acetylation, thereby enabling osteogenic transformation of VSMCs. Also, SIRT1 can alleviate VC by regulating Wnt signaling pathway and DNA repair pathway. And the mitochondrial sirtuin SIRT3 could maintain mitochondrial function and inhibit VC by regulating mitochondrial protein acetylation levels. It suggests that SIRTs may form complex regulatory networks through multiple pathways to participate in VC.
SIRT2, the major cytoplasmic deacetylase, is downregulated in aging as well as cardiovascular diseases, including cardiac hypertrophy and heart failure (85). Specially, SIRT2 can protect vascular endothelial cells by inhibiting oxidative stress and regulating apoptosis (86). SIRT4 and SIRT5 are two other mitochondrial sirtuins, which have been reported to be regulators of cellular metabolism (87). For instance, mitochondrial SIRT3, 4 and 5 may affect cell metabolism and ROS homeostasis, ultimately regulating cardiovascular health (88). Interestingly, on the one hand, SIRT4 could attenuate inflammatory damage to human umbilical vein endothelial cells (89, 90). On the other hand, SIRT4 might increase ROS levels under Ang II-induced pathological stimulation and promote hypertrophic growth, fibrosis, and cardiac dysfunction (91). Therefore, the role and mechanism of SIRT4 are complex and must be explored in different states. SIRT7 mainly resides in the nucleolus and is involved in the regulation of overall genome stability, aging, and cell metabolism (92). Current evidence suggests that the SIRTs play important regulatory roles in VC. In addition to SIRT1, SIRT3 and SIRT6 mentioned above, the roles of other SIRTs family members in VC should also be explored to gain a more objective understanding of the SIRTs family.
Recently, miR-34a and lncRNA HOTAIR /miR-126/Klotho axis have been recognized to serve as upstream pathways of SIRT1. And SIRT1 could be involved in the regulation of VC through several downstream pathways, including the RUNX2/osteocalcin pathway, Wnt signaling pathway, DNA repair pathway, and NF-κB pathway. Moreover, myocardin, a myogenic coactivator responsible for the VSMC contractile phenotype, could interact directly with SIRT1. And maintenance of myocardin and SIRT1 and the normal contractile phenotype of VSMCs may be a protective mechanism of VC (93, 94). Interestingly, SIRT6 is the deacetylation target of SIRT1, and the two could work together to enhance DNA damage repair (95). The potential interaction between sirtuins and other molecules in VC should be validated in the future.
Oxidative stress plays an important role in VC in both CKD and diabetes. The elevated reactive oxygen species production in VSMCs can increase the expression of RUNX2, which is involved in the osteogenic transition of VSMCs and the occurrence of VC (31, 96). Though different SIRTs are in different locations, evidence suggests that SIRT1, SIRT3, and SIRT6 can all be involved in the regulation of oxidative stress. And the SIRT1 agonists have antioxidant effects during the treatment of VC, including resveratrol, spermidine, terpene-4-ol, and IMD. All these highlight the important regulatory role of SIRT1 and oxidative stress in VC. It also provides a theoretical basis for future randomized controlled trial studies of SIRT1 agonists in VC, thereby improving the current situation where no drugs are available.
Most previous studies have shown that SIRT1 levels of patients with advanced coronary disease and diabetic kidney disease decreased compared with healthy subjects (97, 98). As for uremic patients undergoing hemodialysis, the concentration of SIRT1 increased and was positively correlated with dialysis time (93). Angelika et al. found that the serum SIRT1 concentration in the CKD group was higher than that in the control group, and serum levels of SIRT1 tend to increase with the progression of CKD (99). This may be inconsistent with our above-mentioned tone, and the reasons for this need to be further explored in the future.
In conclusion, SIRTs play key roles in vascular protection by regulating multiple signaling pathways. SIRTs might be potential therapeutic targets to inhibit VSMC osteogenic differentiation and alleviate VC.
Author Contributions
SH contributes to the conception, design, and final approval of the submitted version. SW contributes to completing the table and writing the paper. Both authors contributed to the article and approved the submitted version.
Funding
This research was supported by the grant from National Natural Science Foundation of China (81900441), Natural Science Foundation of Zhejiang Province (LQ19H020002), Zhejiang Provincial Program for Medicine and Health (2022KY446), Social Development Science and Technology Foundation of Taizhou (21ywb115, 21ywb118, and 20ywb143), and Social Development Science and Technology Foundation of Wenling (2021S00197, 2020S0180083, and 2021S00156).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wang S, Hu S, Wang J, Liu Y, Zhao R, Tong M, et al. Conditioned medium from bone marrow-derived mesenchymal stem cells inhibits vascular calcification through blockade of the BMP2-Smad1/5/8 signaling pathway. Stem Cell Res Ther. (2018) 9:160. doi: 10.1186/s13287-018-0894-1
2. Li T, Yu H, Zhang D, Feng T, Miao M, Li J, et al. Matrix vesicles as a therapeutic target for vascular calcification. Front Cell Dev Biol. (2022) 10:825622. doi: 10.3389/fcell.2022.825622
3. Li W, Su SA, Chen J, Ma H, Xiang M. Emerging roles of fibroblasts in cardiovascular calcification. J Cell Mol Med. (2021) 25:1808–16. doi: 10.1111/jcmm.16150
4. Teh R, Prince RL, Sim M, Schousboe JT, Raymond WD, Szulc P, et al. Abdominal aortic calcification, cardiac troponin I and atherosclerotic vascular disease mortality in older women. Heart. (2021). doi: 10.1136/heartjnl-2021-319879
5. Vengrenyuk Y, Carlier S, Xanthos S, Cardoso L, Ganatos P, Virmani R, et al. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc Natl Acad Sci U S A. (2006) 103:14678–83. doi: 10.1073/pnas.0606310103
6. Li L, Chen Z, Fu W, Cai S, Zeng Z. Emerging evidence concerning the role of sirtuins in sepsis. Crit Care Res Pract. (2018) 2018:5489571. doi: 10.1155/2018/5489571
7. Gandhirajan A, Roychowdhury S, Vachharajani V. Sirtuins and sepsis: cross talk between redox and epigenetic pathways. Antioxidants 11. (2021). doi: 10.3390/antiox11010003
8. Kupis W, Palyga J, Tomal E, Niewiadomska E. The role of sirtuins in cellular homeostasis. J Physiol Biochem. (2016) 72:371–80. doi: 10.1007/s13105-016-0492-6
9. Singh CK, Chhabra G, Ndiaye MA, Garcia-Peterson LM, Mack NJ, Ahmad N. The role of sirtuins in antioxidant and redox signaling. Antioxid Redox Signal. (2018) 28:643–61. doi: 10.1089/ars.2017.7290
10. Dai H, Sinclair DA, Ellis JL, Steegborn C. Sirtuin activators and inhibitors: promises, achievements, and challenges. Pharmacol Ther. (2018) 188:140–54. doi: 10.1016/j.pharmthera.2018.03.004
11. Lu CL, Liao MT, Hou YC, Fang YW, Zheng CM, Liu WC, et al. Sirtuin-1 and Its Relevance in Vascular Calcification. Int J Mol Sci, 21. (2020). doi: 10.3390/ijms21051593
12. Pescatore LA, Gamarra LF, Liberman M. Multifaceted mechanisms of vascular calcification in aging. Arterioscler Thromb Vasc Biol. (2019) 39:1307–16. doi: 10.1161/ATVBAHA.118.311576
13. Raucci A, Macri F, Castiglione S, Badi I, Vinci MC, Zuccolo E. MicroRNA-34a: the bad guy in age-related vascular diseases. Cell Mol Life Sci. (2021) 78:7355–78. doi: 10.1007/s00018-021-03979-4
14. Go S, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. (2004) 351:1296–305. doi: 10.1056/NEJMoa041031
15. Henaut L, Mary A, Chillon JM, Kamel S, Massy ZA. The impact of uremic toxins on vascular smooth muscle cell function. Toxins. (2018) 10:218. doi: 10.3390/toxins10060218
16. Jin D, Lin L, Xie Y, Jia M, Qiu H, Xun K. NRF2-suppressed vascular calcification by regulating the antioxidant pathway in chronic kidney disease. FASEB J. (2022) 36:e22098. doi: 10.1096/fj.202100625RR
17. Chen Y, Zhao X, Wu H. Arterial stiffness: a focus on vascular calcification and its link to bone mineralization. Arterioscler Thromb Vasc Biol. (2020) 40:1078–93. doi: 10.1161/ATVBAHA.120.313131
18. Hung SC, Kuo KL, Wu CC, Tarng DC. Indoxyl sulfate: a novel cardiovascular risk factor in chronic kidney disease. J Am Heart Assoc. (2017) 6:e005022. doi: 10.1161/JAHA.116.005022
19. Lampropoulos E, Kalamara P, Konsta M, Papaioannou I, Papadima E, Antoniou Z, et al. Osteoporosis and vascular calcification in postmenopausal women: a cross-sectional study. Climacteric. (2016) 19:303–7. doi: 10.3109/13697137.2016.1164134
20. El Khoudary SR, Aggarwal B, Beckie TM, Hodis HN, Johnson AE, Langer RD, et al. American Heart Association Prevention Science Committee of the Council on, Prevention. Menopause transition and cardiovascular disease risk: implications for timing of early prevention: a scientific statement from the American Heart Association. Circulation. (2020) 142:e506–32. doi: 10.1161/CIR.0000000000000912
21. El Khoudary SR, Greendale G, Crawford SL, Avis NE, Brooks MM, Thurston RC. The menopause transition and women's health at midlife: a progress report from the Study of Women's Health Across the Nation (SWAN). Menopause. (2019) 26:1213–27. doi: 10.1097/GME.0000000000001424
22. El Khoudary SR, Zhao Q, Venugopal V, Manson JE, Brooks MM, Santoro N, et al. Effects of hormone therapy on heart fat and coronary artery calcification progression: secondary analysis from the KEEPS trial. J Am Heart Assoc. (2019) 8:e012763. doi: 10.1161/JAHA.119.012763
23. Cannata-Andia JB, Carrillo-Lopez N, Messina OD, Hamdy NAT, Panizo S, Ferrari SL, et al. On Behalf of the International Osteoporosis Foundation Iof Working Group On, D. Cardiovascular pathophysiology of vascular calcification and bone loss: linked disorders of ageing? Nutrients. (2021) 13:3835. doi: 10.3390/nu13113835
24. Mazurek R, Dave JM, Chandran RR, Misra A, Sheikh AQ, Greif DM. Vascular cells in blood vessel wall development and disease. Adv Pharmacol. (2017) 78:323–50. doi: 10.1016/bs.apha.2016.08.001
25. Gimbrone MA Jr., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36 doi: 10.1161/CIRCRESAHA.115.306301
26. Eelen G, Treps L, Li X, Carmeliet P. Basic and therapeutic aspects of angiogenesis updated. Circ Res. (2020) 127:310–29. doi: 10.1161/CIRCRESAHA.120.316851
27. Zhang L, Yao J, Yao Y, Bostrom KI. Contributions of the Endothelium to Vascular Calcification. Front Cell Dev Biol. (2021) 9:620882. doi: 10.3389/fcell.2021.620882
28. Schurgers LJ, Akbulut AC, Kaczor DM, Halder M, Koenen RR, Kramann R. Initiation and Propagation of Vascular Calcification Is Regulated by a Concert of Platelet- and Smooth Muscle Cell-Derived Extracellular Vesicles. Front Cardiovasc Med. (2018) 5:36. doi: 10.3389/fcvm.2018.00036
29. Shroff RC, Shanahan CM. The vascular biology of calcification. Semin Dial. (2007) 20:103–9. doi: 10.1111/j.1525-139X.2007.00255.x
30. Kurabayashi M. [Vascular Calcification - Pathological Mechanism and Clinical Application -. Role of vascular smooth muscle cells in vascular calcification]. Clin Calcium. (2015) 25:661–9.
31. Jaminon, Reesink K, Kroon A, Schurgers L. The role of vascular smooth muscle cells in arterial remodeling: focus on calcification-related processes. Int J Mol Sci. (2019) 20:5694. doi: 10.3390/ijms20225694
32. Iyemere VP, Proudfoot D, Weissberg PL, Shanahan CM. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J Intern Med. (2006) 260:192–210. doi: 10.1111/j.1365-2796.2006.01692.x
33. Liu X, Chen A, Liang Q, Yang X, Dong Q, Fu M, et al. Spermidine inhibits vascular calcification in chronic kidney disease through modulation of SIRT1 signaling pathway. Aging Cell. (2021) 20:e13377. doi: 10.1111/acel.13377
34. Wang Y, He J, Liao M, Hu M, Li W, Ouyang H, et al. An overview of Sirtuins as potential therapeutic target: Structure, function and modulators. Eur J Med Chem. (2019) 161:48–77. doi: 10.1016/j.ejmech.2018.10.028
35. D'Onofrio N, Servillo L, Balestrieri ML. SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antioxid Redox Signal. (2018) 28:711–32. doi: 10.1089/ars.2017.7178
36. Donato J, Walker AE, Magerko KA, Bramwell RC, Black AD, Henson GD, et al. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell. (2013) 12:772–83. doi: 10.1111/acel.12103
37. Planavila A, Iglesias R, Giralt M, Villarroya F. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res. (2011) 90:276–84. doi: 10.1093/cvr/cvq376
38. Guarente L, Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med. (2011) 364:2235–44. doi: 10.1056/NEJMra1100831
39. Shimoyama Y, Mitsuda Y, Tsuruta Y, Suzuki K, Hamajima N, Niwa T, et al. 1 gene polymorphisms are associated with cholesterol metabolism and coronary artery calcification in Japanese hemodialysis patients. J Ren Nutr. (2012) 22:114–9. doi: 10.1053/j.jrn.2011.10.025
40. Akiyoshi T, Ota H, Iijima K, Son BK, Kahyo T, Setou M, et al. A novel organ culture model of aorta for vascular calcification. Atherosclerosis. (2016) 244:51–8. doi: 10.1016/j.atherosclerosis.2015.11.005
41. Machin R, Auduong Y, Gogulamudi VR, Liu Y, Islam MT, Lesniewski LA, et al. Lifelong SIRT-1 overexpression attenuates large artery stiffening with advancing age. Aging. (2020) 12:11314–24. doi: 10.18632/aging.103322
42. Takemura, Iijima K, Ota H, Son BK, Ito Y, Ogawa S, et al. Sirtuin 1 retards hyperphosphatemia-induced calcification of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2011) 31:2054–62. doi: 10.1161/ATVBAHA.110.216739
43. Zainabadi K, Liu CJ, Guarente L. SIRT1 is a positive regulator of the master osteoblast transcription factor, RUNX2. PLoS ONE. (2017) 12:e0178520. doi: 10.1371/journal.pone.0178520
44. Bartoli-Leonard F, Wilkinson FL, Schiro A, Inglott FS, Alexander MY, Weston R. Suppression of SIRT1 in diabetic conditions induces osteogenic differentiation of human vascular smooth muscle cells via RUNX2 signalling. Sci Rep. (2019) 9:878. doi: 10.1038/s41598-018-37027-2
45. Bartoli-Leonard F, Wilkinson FL, Langford-Smith AWW, Alexander MY, Weston R. The Interplay of SIRT1 and Wnt Signaling in Vascular Calcification. Front Cardiovasc Med. (2018) 5:183. doi: 10.3389/fcvm.2018.00183
46. Chen Y, Huang C, Zhu SY, Zou HC, Xu CY, Chen YX. Overexpression of HOTAIR attenuates Pi-induced vascular calcification by inhibiting Wnt/beta-catenin through regulating miR-126/Klotho/SIRT1 axis. Mol Cell Biochem. (2021) 476:3551–61. doi: 10.1007/s11010-021-04164-8
47. Yuan Z, Seto EA. functional link between SIRT1 deacetylase and NBS1 in DNA damage response. Cell Cycle. (2007) 6:2869–71. doi: 10.4161/cc.6.23.5026
48. Bartoli-Leonard F, Wilkinson FL, Schiro A, Serracino Inglott F, Alexander MY, Weston R. Loss of SIRT1 in diabetes accelerates DNA damage-induced vascular calcification. Cardiovasc Res. (2021) 117:836–49. doi: 10.1093/cvr/cvaa134
49. Thompson M, Wagner R, Rzucidlo EM. Age-related loss of SirT1 expression results in dysregulated human vascular smooth muscle cell function. Am J Physiol Heart Circ Physiol. (2014) 307:H533–41. doi: 10.1152/ajpheart.00871.2013
50. Han L, Zhang Y, Zhang M, Guo L, Wang J, Zeng F, et al. Interleukin-1beta-induced senescence promotes osteoblastic transition of vascular smooth muscle cells. Kidney Blood Press Res. (2020) 45:314–30. doi: 10.1159/000504298
51. Zhang M, Li T, Tu Z, Zhang Y, Wang X, Zang D, et al. Both high glucose and phosphate overload promote senescence-associated calcification of vascular muscle cells. Int Urol Nephrol. (2022). doi: 10.1007/s11255-022-03195-4. [Epub ahead of print].
52. Badi I, Burba I, Ruggeri C, Zeni F, Bertolotti M, Scopece A, et al. MicroRNA-34a induces vascular smooth muscle cells senescence by SIRT1 downregulation and promotes the expression of age-associated pro-inflammatory secretory factors. J Gerontol A Biol Sci Med Sci. (2015) 70:1304–11. doi: 10.1093/gerona/glu180
53. Zuccolo E, Badi I, Scavello F, Gambuzza I, Mancinelli L, Macri F, et al. The microRNA-34a-induced Senescence-Associated Secretory Phenotype (SASP) favors vascular smooth muscle cells calcification. Int J Mol Sci. (2020) 21:4454. doi: 10.3390/ijms21124454
54. Badi I, Mancinelli L, Polizzotto A, Ferri D, Zeni F, Burba I, et al. miR-34a promotes vascular smooth muscle cell calcification by downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Arterioscler Thromb Vasc Biol. (2018) 38, 2079–90. doi: 10.1161/ATVBAHA.118.311298
55. Zhang P, Li Y, Du Y, Li G, Wang L, Zhou F. Resveratrol ameliorated vascular calcification by regulating Sirt-1 and Nrf2. Transplant Proc. (2016) 48:3378–86. doi: 10.1016/j.transproceed.2016.10.023
56. Hammad SK, Eissa RG, Shaheen MA, Younis NN. Resveratrol Ameliorates Aortic Calcification in Ovariectomized Rats via SIRT1 Signaling. Curr Issues Mol Biol. (2021) 43:1057–71. doi: 10.3390/cimb43020075
57. Zhang Y, He L, Tu M, Huang M, Chen Y, Pan D, et al. The ameliorative effect of terpinen-4-ol on ER stress-induced vascular calcification depends on SIRT1-mediated regulation of PERK acetylation. Pharmacol Res. (2021) 170:105629. doi: 10.1016/j.phrs.2021.105629
58. Cai Y, Xu MJ, Teng X, Zhou YB, Chen L, Zhu Y, et al. Intermedin inhibits vascular calcification by increasing the level of matrix gamma-carboxyglutamic acid protein. Cardiovasc Res. (2010) 85:864–73. doi: 10.1093/cvr/cvp366
59. Chang JR, Guo J, Wang Y, Hou YL, Lu WW, Zhang JS, et al. Intermedin1-53 attenuates vascular calcification in rats with chronic kidney disease by upregulation of alpha-Klotho. Kidney Int. (2016) 89:586–600. doi: 10.1016/j.kint.2015.12.029
60. Chen Y, Zhang LS, Ren JL, Zhang YR, Wu N, Jia MZ, et al. Intermedin1-53 attenuates aging-associated vascular calcification in rats by upregulating sirtuin 1. Aging. (2020) 12:5651–74. doi: 10.18632/aging.102934
61. Madrigal-Perez LA, Ramos-Gomez M. Resveratrol inhibition of cellular respiration: new paradigm for an old mechanism. Int J Mol Sci. (2016) 17:368. doi: 10.3390/ijms17030368
62. Ning, Xu L, Zhao Q, Zhang YY, Shen CQ. The Protective Effects of Terpinen-4-ol on LPS-Induced Acute Lung Injury via Activating PPAR-gamma. J Inflamm. (2018) 41:2012-2017. doi: 10.1007/s10753-018-0844-1
63. Nakayama, Murata S, Ito H, Iwasaki K, Villareal MO, Zheng YW, et al. Terpinen-4-ol inhibits colorectal cancer growth via reactive oxygen species. Oncol Lett. (2017) 14:201524. doi: 10.3892/ol.2017.6370
64. Masuda, Ting TC, Levi M, Saunders SJ, Miyazaki-Anzai S, Miyazaki M. Activating transcription factor 4 regulates stearate-induced vascular calcification. J Lipid Res. (2012) 53:1543–52. doi: 10.1194/jlr.M025981
65. Zhang K, Wang M, Li Y, Li C, Tang S, Qu X, Feng N, Wu Y. The PERK-EIF2alpha-ATF4 signaling branch regulates osteoblast differentiation and proliferation by PTH. Am J Physiol Endocrinol Metab. (2019) 316:E590–E604. doi: 10.1152/ajpendo.00371.2018
66. Madeo F, Eisenberg T, Buttner S, Ruckenstuhl C, Kroemer G. Spermidine: a novel autophagy inducer and longevity elixir. Autophagy. (2010) 6:160–2. doi: 10.4161/auto.6.1.10600
67. Minois N, Carmona-Gutierrez D, Bauer MA, Rockenfeller P, Eisenberg T, Brandhorst S, et al. Spermidine promotes stress resistance in Drosophila melanogaster through autophagy-dependent and -independent pathways. Cell Death Dis. (2012) 3:e401. doi: 10.1038/cddis.2012.139
68. LaRocca TJ, Gioscia-Ryan RA, Hearon CM. Jr., Seals DR. The autophagy enhancer spermidine reverses arterial aging. Mech Ageing Dev. (2013) 134:314–20. doi: 10.1016/j.mad.2013.04.004
69. Wang J, Li S, Wang J, Wu F, Chen Y, Zhang H, et al. Spermidine alleviates cardiac aging by improving mitochondrial biogenesis and function. Aging. (2020) 12:650–71. doi: 10.18632/aging.102647
70. Madeo F, Eisenberg T, Pietrocola F, Kroemer G, Spermidine in health and disease. Science. (2018) 359:eaan2788. doi: 10.1126/science.aan2788
71. Ni X, Zhang J, Tang C, Qi Y. Intermedin/adrenomedullin2: an autocrine/paracrine factor in vascular homeostasis and disease. Sci China Life Sci. (2014) 57:781–9. doi: 10.1007/s11427-014-4701-7
72. Yang JH, Pan CS, Jia YX, Zhang J, Zhao J, Pang YZ., et al. Intermedin1-53 activates L-arginine/nitric oxide synthase/nitric oxide pathway in rat aortas. Biochem Biophys Res Commun. (2006) 341:567–72. doi: 10.1016/j.bbrc.2006.01.010
73. Lu WW, Jia LX, Ni XQ, Zhao L, Chang JR, Zhang JS., et al. Intermedin1-53 Attenuates Abdominal Aortic Aneurysm by Inhibiting Oxidative Stress. Arterioscler Thromb Vasc Biol. (2016) 36:2176–90.
74. Perry CA, Gadde KM. The Role of Calorie Restriction in the Prevention of Cardiovascular Disease. Curr Atheroscler Rep. (2022) 24:235–42. doi: 10.1007/s11883-022-00999-8
75. Yang W, Nagasawa K, Munch C, Xu Y, Satterstrom K, Jeong S, et al. Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell. (2016) 167:985–1000 e21 doi: 10.1016/j.cell.2016.10.016
76. Cao M, Zhao Q, Sun X, Qian H, Lyu S, Chen R, et al. Sirtuin 3: emerging therapeutic target for cardiovascular diseases. Free Radic Biol Med. (2022) 180:63-74. doi: 10.1016/j.freeradbiomed.2022.01.005
77. Feng H, Wang JY, Yu B, Cong X, Zhang WG, Li L, et al. Peroxisome Proliferator-Activated Receptor-gamma Coactivator-1alpha Inhibits Vascular Calcification Through Sirtuin 3-Mediated Reduction of Mitochondrial Oxidative Stress. Antioxid Redox Signal. (2019) 31:75–91. doi: 10.1089/ars.2018.7620
78. He W, Huang J, Liu Y, Xie C, Zhang K, Zhu X, et al. Deletion of soluble epoxide hydrolase suppressed chronic kidney disease-related vascular calcification by restoring Sirtuin 3 expression. Cell Death Dis. (2021) 12:992. doi: 10.1038/s41419-021-04283-6
79. Liu Z, Wang J, Huang X, Li Z, Liu P. Deletion Deletion of sirtuin 6 accelerates endothelial dysfunction and atherosclerosis in apolipoprotein E-deficient mice. Transl Res. (2016) 172:18–29 e2. doi: 10.1016/j.trsl.2016.02.005
80. Yang Q, Hu J, Yang Y, Chen Z, Feng J, Zhu Z, et al. Sirt6 deficiency aggravates angiotensin II-induced cholesterol accumulation and injury in podocytes. Theranostics. (2020) 10:7465–79. doi: 10.7150/thno.45003
81. Cai J, Liu Z, Huang X, Shu S, Hu X, Zheng M, et al. The deacetylase sirtuin 6 protects against kidney fibrosis by epigenetically blocking beta-catenin target gene expression. Kidney Int. (2020) 97:106–18. doi: 10.1016/j.kint.2019.08.028
82. Li W, Feng W, Su X, Luo D, Li Z, Zhou Y., et al. SIRT6 protects vascular smooth muscle cells from osteogenic transdifferentiation via Runx2 in chronic kidney disease. J Clin Invest. (2022) 132:e150051. doi: 10.1172/JCI150051
83. Wei W, Guo X, Gu L, Jia J, Yang M, Yuan W, et al. Bone marrow mesenchymal stem cell exosomes suppress phosphate-induced aortic calcification via SIRT6-HMGB1 deacetylation. Stem Cell Res Ther. (2021) 12:235. doi: 10.1186/s13287-021-02307-8
84. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. (2019) 20:156–74. doi: 10.1038/s41580-018-0081-3
85. Taneja A, Ravi V, Hong JY, Lin H, Sundaresan NR. Emerging roles of Sirtuin 2 in cardiovascular diseases. FASEB J. (2021)35:e21841. doi: 10.1096/fj.202100490R
86. Zhang W, Liu D, Ren J, Zhou P, Han X. Overexpression of Sirtuin2 prevents high glucose-induced vascular endothelial cell injury by regulating the p53 and NF-kappaB signaling pathways. Biotechnol Lett. (2018) 40:271–8. doi: 10.1007/s10529-017-2487-y
87. Taurone S, De Ponte C, Rotili D, De Santis E, Mai A, Fiorentino F, et al. Biochemical Functions and Clinical Characterizations of the Sirtuins in Diabetes-Induced Retinal Pathologies. Int J Mol Sci. 23 (2022). doi: 10.3390/ijms23074048
88. Zullo A, Guida R, Sciarrillo R, Mancini FP. Redox homeostasis in cardiovascular disease: the role of mitochondrial sirtuins. Front Endocrinol. (2022) 13:858330. doi: 10.3389/fendo.2022.858330
89. Tao Y, Yu S, Chao M, Wang Y, Xiong J, Lai H. SIRT4 suppresses the PI3K/Akt/NFkappaB signaling pathway and attenuates HUVEC injury induced by oxLDL. Mol Med Rep. (2019) 19:4973–9. doi: 10.3892/mmr.2019.10161
90. Tao Y, Huang C, Huang Y, Hong L, Wang H, Zhou Z, et al. SIRT4 Suppresses inflammatory responses in human umbilical vein endothelial cells. Cardiovasc Toxicol. (2015) 15:217–23. doi: 10.1007/s12012-014-9287-6
91. Luo YX, Tang X, An XZ, Xie XM, Chen XF, Zhao X, et al. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur Heart J. (2017) 38:1389–98. doi: 10.1093/eurheartj/ehw138
92. Kumari P, Tarighi S, Braun T, Ianni A. SIRT7 acts as a guardian of cellular integrity by controlling nucleolar and extra-nucleolar functions. Genes. (2021) 12:1361. doi: 10.3390/genes12091361
93. Zbroch E, Bazyluk A, Malyszko J, Koc-Zorawska E, Rydzewska-Rosolowska A, Kakareko K, et al. The serum concentration of anti-aging proteins, sirtuin1 and alphaklotho in patients with end-stage kidney disease on maintenance hemodialysis. Clin Interv Aging. (2020) 15:387–93. doi: 10.2147/CIA.S236980
94. Sharma A, Gautam V, Costantini S, Paladino A, Colonna G. Interactomic and pharmacological insights on human sirt-1. Front Pharmacol. (2012) 3:40. doi: 10.3389/fphar.2012.00040
95. Meng F, Qian M, Peng B, Peng L, Wang X, Zheng K, et al. Synergy between SIRT1 and SIRT6 helps recognize DNA breaks and potentiates the DNA damage response and repair in humans and mice. Elife. (2020) 9:e55828. doi: 10.7554/eLife.55828.sa2
96. Zhao M, Xu MJ, Cai Y, Zhao G, Guan Y, Kong W, et al. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. (2011) 79:1071–9. doi: 10.1038/ki.2011.18
97. Doulamis P, Tzani AI, Konstantopoulos PS, Samanidis G, Georgiopoulos G, Toutouzas KP, et al. A sirtuin 1/MMP2 prognostic index for myocardial infarction in patients with advanced coronary artery disease. Int J Cardiol. (2017) 230:447–53. doi: 10.1016/j.ijcard.2016.12.086
98. Shao Y, Ren H, Lv C, Ma X, Wu C, Wang Q. Changes of serum Mir-217 and the correlation with the severity in type 2 diabetes patients with different stages of diabetic kidney disease. Endocrine. (2017) 55:130–8. doi: 10.1007/s12020-016-1069-4
Keywords: sirtuins, vascular calcification, therapy, VSMCs, osteogenic differentiation
Citation: Wang S and Hu S (2022) The Role of Sirtuins in Osteogenic Differentiation of Vascular Smooth Muscle Cells and Vascular Calcification. Front. Cardiovasc. Med. 9:894692. doi: 10.3389/fcvm.2022.894692
Received: 12 March 2022; Accepted: 04 May 2022;
Published: 01 June 2022.
Edited by:
Emiel Van Der Vorst, Institute for Molecular Cardiovascular Research (IMCAR), GermanyReviewed by:
Nhat Tu Le, Houston Methodist Research Institute, United StatesAnna Dikalova, Vanderbilt University Medical Center, United States
Copyright © 2022 Wang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siwang Hu, c2l3YW5nX2gmI3gwMDA0MDt3bXUuZWR1LmNu