 Dominga Iacobazzi
Dominga Iacobazzi Valeria Vincenza Alvino
Valeria Vincenza Alvino Massimo Caputo
Massimo Caputo Paolo Madeddu
Paolo Madeddu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 26 May 2022
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.892861
This article is part of the Research TopicCardiovascular Cell Senescence in Aging and DiseaseView all 5 articles
An increasing number of patients with congenital heart disease (CHD) survive into adulthood but develop long-term complications including heart failure (HF). Cellular senescence, classically defined as stable cell cycle arrest, is implicated in biological processes such as embryogenesis, wound healing, and aging. Senescent cells have a complex senescence-associated secretory phenotype (SASP), involving a range of pro-inflammatory factors with important paracrine and autocrine effects on cell and tissue biology. While senescence has been mainly considered as a cause of diseases in the adulthood, it may be also implicated in some of the poor outcomes seen in patients with complex CHD. We propose that patients with CHD suffer from multiple repeated stress from an early stage of the life, which wear out homeostatic mechanisms and cause premature cardiac aging, with this term referring to the time-related irreversible deterioration of the organ physiological functions and integrity. In this review article, we gathered evidence from the literature indicating that growing up with CHD leads to abnormal inflammatory response, loss of proteostasis, and precocious age in cardiac cells. Novel research on this topic may inspire new therapies preventing HF in adult CHD patients.
Congenital heart disease (CHD) is the most common type of birth defect, with a reported prevalence of 9 per 1,000 births. Heart failure (HF) represents an important cause of morbidity and mortality in patients with CHD (1–4). It occurs in ≈25% of adult CHD (ACHD) patients by the age of 30 and is not limited to severe cardiac defects (5–8). Individuals with lower-complexity malformations, which constitute the majority of ACHD, have a higher burden of adverse cardiovascular events relative to the general population, with hazard ratios ranging from 2 for coronary artery disease to 13 for HF (9). In the US, hospitalization for ACHD-related HF increased 91% from 1998 to 2011 and charges increased 258%, more than double that for non-ACHD HF.
Several factors may participate in the pathogenesis of ACHD-related HF, such as inherited architectural disorganization of myocytes and vascular cells, cardiac damage due to the surgical trauma, and insufficient protection during cardiopulmonary bypass, and the hemodynamic load to the heart from residual defects and failing grafts (10). Moreover, genetic and epigenetic factors play both distinct and additive roles in maladaptive myocardial remodeling (6). Investigation of mechanisms has mainly focused on canonical pathways, such as the renin-angiotensin-aldosterone system (RAS) and adrenergic system (11, 12). Nonetheless, trials using RAS inhibitors in ACHD-related HF failed to show any benefit on ventricular function (13, 14), thus conventional therapy is unsupported by clinical evidence (15). New efforts are urgently needed to unveil new druggable targets for prevention and treatment. Investigation in neonatal animal models could aid in this endeavor. Mice modeling monogenic causes of CHD recapitulate cardiac septation defects, heart valve malformations, and conotruncal heart defects, as well as more complex CHD (16, 17). More recently, patient-specific induced pluripotent stem cells (iPSCs) have been employed to study human CHD (18). In addition, large animal models simulating the corrective treatments employed in children are warranted to determine the positive and negative effects of cardiac surgery on post-natal heart maturation (19–22). Finally, computational growth models can help in tracking disease progression and stratifying patients with CHD (23, 24).
Accelerated cardiac cell senescence is a key determinant of the increased risk for HF in patients with CHD. Hence, identifying the causes of heart’s aging may improve the outcome of CHD patients.
We propose that growing up with CHD exposes the patient to multiple and repeated stress, related not only to the defect severity, but also to the damage from reconstructive and multiple cardiac operations, cardio-pulmonary bypass and cardioplegic arrest, and implantation of foreign graft material (Figure 1). Cardiac cells (especially stromal cells which have immunomodulatory roles) respond to early in life and subsequent repeated stress by activating the inflammasome. Stress-induced response pathways will attempt to reduce protein synthesis and increase the cellular capacity for protein folding and degradation. With unremitting stress, proteostasis will be overloaded, resulting in misfolded proteins accumulation in subcellular compartments, including the mitochondria where proteotoxic stress will incite ROS production. An increasing number of cells will become senescent in the CHD heart and will transfer proteostatic stress to neighboring cells through the secretion of inflammatory chemokines and misfolded peptides. This vicious cycle will lead to cardiomyocyte loss, microvascular rarefaction, and fibro-calcific interstitial remodeling of the heart, compromising myocardial perfusion and contractile function.
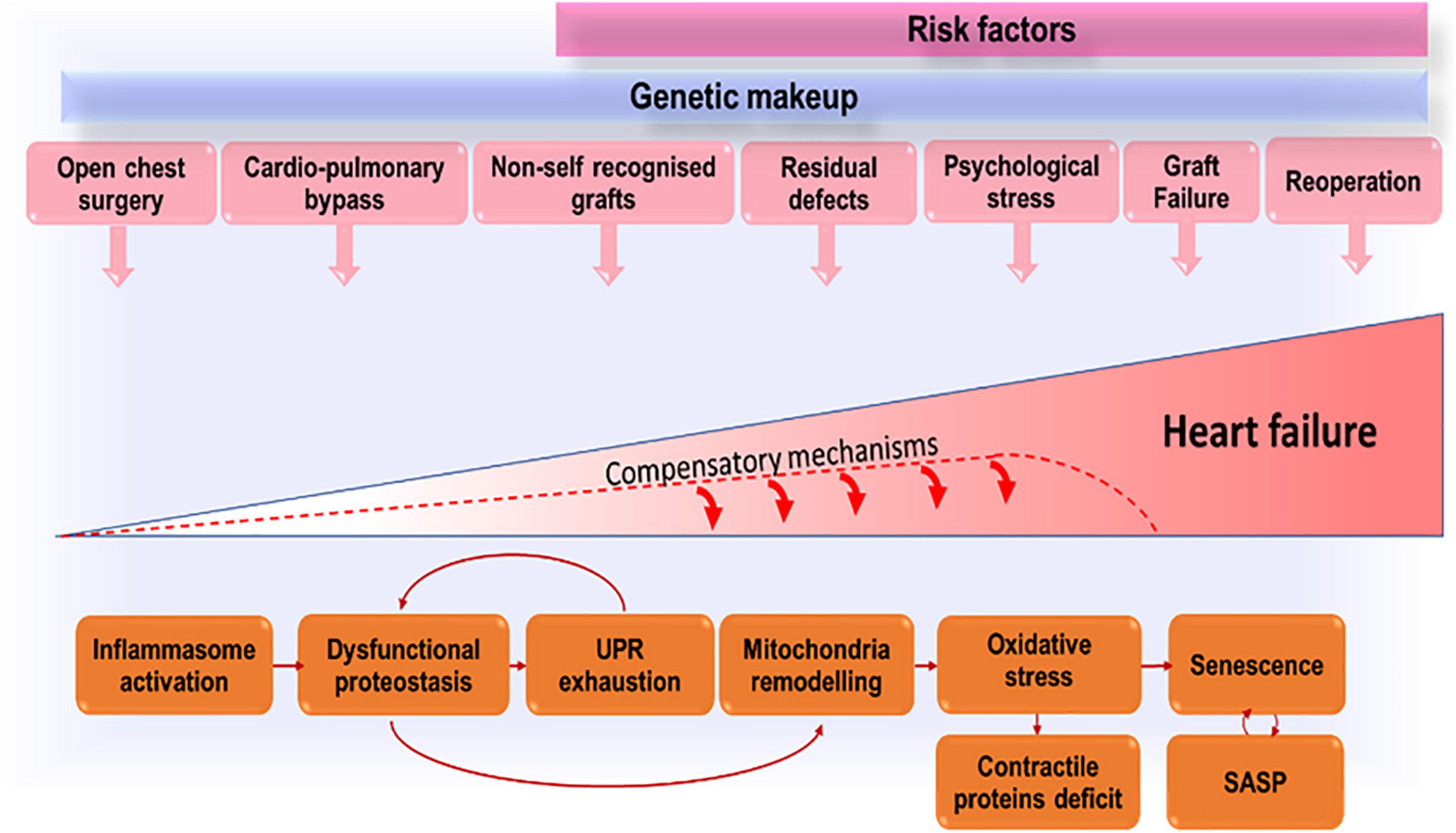
Figure 1. Accruing stressors that, by inducing premature cells senescence, lead to exhaustion of compensatory mechanisms and eventually heart failure.
The stress from the defect and surgical trauma is unavoidable. However, drugs able to modulate the inflammatory response and to improve proteostasis could halt cardiac deterioration in CHD patients, thus succeeding where conventional therapy has failed. Moreover, improvement in graft biocompatibility, e.g., cellularization prior to implantation, may reduce the inflammatory reaction to the prosthesis and the need for reinterventions, therefore delaying heart’s aging and failure.
In a definition proposed by Mitchell et al. in 1971, Congenital Heart Disease is referred to as “A gross structural abnormality of the heart or intrathoracic great vessels that is actually or potentially of functional significance” (25). Since then, more than forty different types of defects have been characterized, from the most common ventricular septal defects (VSD), to the severe Tetralogy of Fallot (26).
The classification of CHD based on the anatomic complexity of the defect, from mild to moderate to complex, and on the patients’ related risk of morbidity and mortality is the most commonly applied system (27). However, in a more simplified stratification, CHD are normally divided into cyanotic and acyanotic (28, 29). Tetralogy Of Fallot (TOF) and Transposition of Great Arteries (TGA) are the two most common cyanotic CHD conditions, characterized by a right to left shunt resulting in deoxygenated blood entering the oxygenated limb of the vascular circuit, thus causing an oxygen saturation level below 90%. On the other hand, the majority of septal defects such as Ventricular Septal Defect (VSD), Atrial Septal Defect (ASD) and Atrioventricular Septal Defect (AVSD) are classified as acyanotic CHD lesions, with oxygen saturation normally above 95%.
Furthermore, CHD malformations can occur as single lesions or in combination with other heart defects. Commonly diagnosed CHD single lesions are ASD, VSD, and pulmonary stenosis (PS), whereas complex or combination lesions include AVSD, TOF, and TGA (30). Within this yet complex scenario, the mostly unknown etiology of CHD adds a further level of complexity to the CHD condition.
Although numerous etiologic investigations have been conducted, only approximately 15% of cases of CHD can be attributable to a known cause (31). Indeed, the etiology of CHD remains in most cases unknown. Numerous theories have been put forward to explain the pathogenesis of CHD, the earliest of which dates back 1968, whereby the author conducted and extensive and systematic investigation into the potential factors underlying the development of CHD (32). While the genetic basis alone, determined by either gross chromosomal aberration or single mutant genes, was considered and tested by reviewing karyotypes of CHD patients in the literature as well as 104 personal cases, this hypothesis was rejected for lack of conclusive evidence. Conversely, the author supported the hypothesis of the so called “multifactorial inheritance,” which encompasses both genetic and environmental factors as known to participate in the etiology of CHD.
Although the gene-environment interaction could certainly affect the normal embryo-fetal development, separate environmental and genetic causes have been also identified (33, 34).
In 2007, the American Heart Association Congenital Cardiac Defects Committee conducted an extensive review on the contribution of genetics to the origin of CHD, with an insight on the available diagnostic genetic tools and their applications. Chromosome analysis and FISH technology were indicated as important tools to detect the chromosomal 22q11 deletion, which is associated with a series of CHD, including Tetralogy of Fallot, interrupted aortic arch type B, truncus arteriosus, cono-ventricular VSDs, and aortic arch anomalies (35, 36). Another common microdeletion, at chromosome 7q11.23, has been identified by FISH and associated to Williams-Beuren syndrome, an autosomal dominant disorder characterized by specific cardiovascular anomalies, including supravalvular aortic stenosis, often in conjunction with supravalvular pulmonary stenosis and peripheral pulmonary stenosis. The review also provides a list of single genes, whose mutation, thanks to the DNA Mutation Analysis technique, was found to be associated to a number of heart defects (i.e., GATA4 in ASD/VSD; CFC1 in TGA, FBN1 in Marfan syndrome, etc.) (33).
With the development of new genomic techniques, such as chromosomal microarray and next-generation sequencing (NGS), the discoveries of new gene mutations and numerous pathogenic copy number variants (CNVs) and have significantly advanced the understanding of causes of CHD (37). An updated summary of knowledge of the genetic contributions to the pathogenesis of CHD has been reported in 2018 by the same American Heart Association Congenital Cardiac Defects Committee that conducted the initial investigation on the genetic basis of heart defect in 2007 (38).
As more and more young patients with severe types of CHD are surviving into their adulthood, thanks to the advances in surgical strategies of intervention, ascertaining the genetic cause of the defect is an important determinant in providing more accurate prognosis for the CHD and outcomes for CHD–related interventions. Furthermore, determining whether there is an underlying genetic pattern to the heart defect can help evaluate associated extracardiac organ involvement and identify genetic reproductive risks for other family members.
Although genetic factors play an important role in the development of CHD, accounting for about the 20% of all defects, it has become increasingly evident that some CHD might be preventable through interventions addressed to reduce environmental exposure (39). Indeed, environmental factors are identified as the cause of approximately 5–10% of CHD, as estimated by the World Health Organization. Epigenetic changes during pregnancy can influence the development of child’s heart structure (40–42). Epigenetic mechanisms implicated in the pathogenesis of CHD include DNA methylation, histone modifications, higher-order chromatin structure, and the activity of non-coding RNA species reviewed in Lim et al. (43).
Epidemiological studies have suggested an association of CHD with various types of environmental factors to which pregnant women and their fetuses may be exposed, such as air pollutants, pesticides, solvents, metals, radiations, contaminants, and chemicals (44, 45). Lifestyle and socioeconomic determinant might constitute risk factors too (45, 46).
Among the main mechanism of action of exposure to chemical pollutant are DNA methylation, oxidative stress, inflammatory processes, and epigenetic mechanism (47–55).
It is conceivable that the impact of epigenetics is not restricted to the embryonic heart development, but the same responsible mechanisms could rather influence the aging clock of the congenitally defective heart.
Advances in cardiac surgery and postoperative care during the past few decades have led to more people with CHD living longer with a better quality of life. Despite the progresses in this field, it is estimated that death rates in the population from 20 to > 70 years of age may be twice to seven times higher for the ACHD population than for their peers (56).
Residual hemodynamic abnormalities and new sequelae, after childhood surgical repair, might manifest later on in life, adding on to superimposed acquired heart disease and to the comorbidity of the natural aging process of the heart. It is now widely acknowledged that age represents the largest risk factor for cardiovascular disease, as cardiac aging entails a series of pathophysiological changes, impairing the myocardium at structural, cellular, molecular, and functional levels (57). These changes are highly relevant to cardiomyocytes, because they are postmitotic cells and thus not replenished by proliferation. Therefore, diminished myocardial reserves, due to a physiologic age-associated disruption of cardiac homeostasis, leads to increased vulnerability to cardiovascular diseases that occurs with aging.
Several environmental factors can contribute to accelerate the heart aging clock in ACHD people. Sedentary behavior and physical inactivity are among the leading modifiable risk factors worldwide for cardiovascular disease and all-cause mortality. Sedentary behavior is increasingly recognized as a risk factor for cardiovascular disease, independent of physical activity (58). The global childhood inactivity crisis is worrisome for the consequence on metabolic and cardiovascular disease (59). Children with CHD may be even less active than their healthy peers, although this evidence is matter of debate (60–63). In addition, very little is known about sedentary behavior in this population. A cross-sectional study with 316 children and adolescents with CHD seen in an outpatient clinic of a reference hospital showed the prevalence of excess weight was 26.9%, which is similar to the prevalence of that described in the literature for children without congenital disease (64).
Psychomotor development is influenced by factors such as low birth weight, cyanosis, hospitalizations, repeated examinations, physical constraints and consequently, school and social withdrawal. Children with CHD reportedly have a developmental delay (65). A larger study analyzing data from the 1997 to 2011 National Health Interview Survey showed that children with CHD are more likely to report worse health overall, to need more healthcare services, and to have other health conditions (e.g., autism, intellectual disability, or asthma), compared to children without CHD (66). The caregivers of children with CHD undergoing cardiac surgery are also under stress due to the uncertainty of the surgical outcome and the stressful experience of being admitted to an intensive care unit. The stress may influence the children perception of their state and amplify their frailty (67). Restriction in physical activity might be medically or parentally imposed. Furthermore, parents might overprotect their children in fear of sudden complications during physical exercise.
Growing to an adult with CHD exposes to an often traumatic transition from pediatric care to a different care model that lasts through a lifelong trajectory (68). Surprisingly, these concerns seem to be contradicted by a study that measured Quality of life (QoL) in patients with CHD (69). This observational case control study showed that patients with CHD felt their environment was healthier, had more opportunities for leisure and were happier with their access to healthcare system than controls. Except for the need for more medical treatment in patients with great CHD defects, no significant differences were seen in the World Health Organization QoL-BREF Index according to the anatomical complexity.
The recognition of an association of CHD with age-related comorbidity would strengthen the assumption of a more rapid senescence at the heart and systemic level. In geriatric ACHD patients, mortality is seemingly driven by acquired comorbid conditions. In a population-based cohort study, the most powerful predictors of mortality were dementia, gastrointestinal bleed, and chronic kidney disease (70). The increased risk for dementia, particularly early onset dementia, was confirmed by another cohort study using the medical registries and records from Danish hospitals between 1963 and 2012 (71). There was a 60% increased lifetime risk of dementia in individuals born with CHD compared to the public. In addition, even more alarming, there was a 160% higher chance of having dementia earlier than 65 years of age.
Coronary artery disease (CAD) is one of the most important causes for mortality in ACHD patients, and the consequences of an acute coronary event can be more fateful in a patient with a corrected congenital defect than in age-matched controls (72). The prevalence of CAD in a population of ACHD patients in the US was 6.8% (73). This cohort comprised ACHD patients who were symptom-free, one reason being that the heart might become denervated during an open-heart operation, as for example in patients whose transposition of the great arteries was corrected with arterial switch operation. The increased risk of CAD is not associated with a greater accrual of risk factors, with the only exception being arterial hypertension, whose prevalence is slightly higher and insurgence more precocious in patients with CHD (74). Moreover, a retrospective study suggested that metabolic syndrome was more common among adults with CHD than in the general population (75).
Interestingly, the literature indicates that patients with cyanotic CHD might be protected against atherosclerosis. This might be due to a combination of reduced atherosclerotic risk factors such as lower blood pressure, lower total cholesterol levels, higher bilirubin levels and lower thrombocyte levels (76–78). Another study argued that children and adolescents with cyanotic CHD have subclinical atherosclerosis as indicated by cross sectional assessment of carotid intima-media thickness (79).
Evidence of accelerated aging at the systemic level is supported by the observation that CHD patients suffer from sarcopenia, a condition characterized by progressive and generalized loss of skeletal muscle mass and strength (80, 81), osteoporosis (82), and have a twofold higher risk of developing cancer compared with healthy matched controls, with this risk being significantly higher among patients with CHD from the most recent birth cohort (83). The relative risk of cancer was high, regardless of whether patients had undergone surgical procedures or were exposed to diagnostic low dose ionizing radiation, which contradict the interpretation of another report from a different group (84). Despite these conflicting results, however, most research agrees that a senescent phenotype is observable in irradiated organs, in a process that starts with DNA damage induced by radiation, followed by G2 arrest and mitotic bypass. Ataxia telangiectasia-mutated protein, p53, and p21 are among the crucial mediators of the DNA damage response signaling network (85). The ionizing radiation-mediated cell senescence occurs in virtually all eukaryotic cells. Nonetheless, some cell types are more radioresistant than others. With regard to the heart, cardiomyocytes, which are terminally differentiated, quiescent cells, are highly radioresistant, whereas endothelial cell (EC) readily undergo senescence or permanent cell-cycle arrest after exposure to moderate dose of radiation (86, 87).
It was reported that activation of the insulin-like growth factor 1 (IGF1)-phosphtidylinositol-3-kinase (PI3K)-Akt/mechanistic target of mTOR pathway acts upstream of the p53-p21 pathway in EC senescence induced by radiation. Additionally, radiation-induced senescent EC exhibit decreased production of nitric oxide and elevated production of ROS, probably due in part to downregulation and/or upcoupling of endothelial nitric oxide synthase (eNOS) and upregulation of NADPH oxidases. The authors also report an increased expression of adhesion molecules and inflammatory cytokines, in addition to an inability to proliferate and form capillary-like structures in vitro, suggesting that EC senescence can lead to endothelial dysfunction by dysregulation of vasodilation and hemostasis, induction of oxidative stress and inflammation and inhibition of angiogenesis, which can potentially contribute to radiation-induced late effects leading to cardiovascular diseases.
Many of the age-related cardiac changes seem to be in common with the alterations underlying the development of the major cardiac pathologies, such as chronic HF, ventricular hypertrophy and atrial fibrillation. Highlighting the potential common pathways of heart aging and cardiac pathophysiology might provide an explanation of the age risk factor for these diseases and for the higher susceptibility of the ACHD population. The hallmark of cardiac aging is the functional impairment that manifests as systolic and diastolic dysfunction, as well as disturbance in the electrical activity of the heart, presenting with different types of arrhythmias (57, 88).
On a structural level, the most striking phenomenon seen with age is an increase in the thickness of the Left Ventricle (LV) wall because of increased cardiomyocyte size. LV hypertrophy is mostly seen as a compensatory response after the loss of cardiomyocytes with aging, and because of the increased afterload produced by large artery stiffening (89, 90). Similar anatomical changes, although not as prominent as in the left side, are observed in the right side of the heart, in conjunction with reduced end systolic and end diastolic volumes (ESV and EDV). Degenerative or calcific aortic valve disease is another common feature encountered among elderly patients, ranging from mild valve thickening without obstruction of blood flow, to severe calcification with impaired leaflet motion, or aortic stenosis (91).
At a cellular level, remodeling includes loss of cardiomyocytes, by either apoptosis/necrosis or autophagy, which induces a compensatory alterations of extracellular matrix (ECM) composition involving the synthesis of fibroblasts and the degradation of collagen through transforming growth factor-β (TGF-β) signaling (92). The resulting fibrotic status of the heart increases cardiac stiffness and reduces the cardiac compliance.
The above mentioned structural and functional changes are the direct consequences of the numerous changes that occur at a molecular level. Investigation of molecular mechanisms underlying cardiac aging has mainly focused on canonical pathways of neurohormonal signaling dysregulation, such as the renin-angiotensin-aldosterone system (RAAS) and adrenergic system (57, 88). Nonetheless, pharmacological intervention targeting these pathways did not show any benefit on ventricular function (13, 14).
Among the other molecular pathways associated with cardiac aging, the development of oxidative stress, because of excessive mitochondrial ROS generation and impaired antioxidant defense, has been extensively described. Elevated ROS levels in the aging myocardium lead to enhanced DNA and protein oxidation/nitration, activation of inflammatory response, apoptosis, and endoplasmic reticulum (ER) stress (93, 94). Moreover, the mitochondrial dysfunction, resulting from ROS-induced damage to mitochondrial DNA and proteins, impairs respiratory chain efficiency, leading to further ROS production in a vicious cycle that ultimately impacts on energy production and myocardial function (95). The excitation-contraction coupling also results affected by the oxidative damage, with the sarcoplasmic reticulum Ca2 + ATPase pump (SERCA) being a direct target of the ROS action, meaning a decreased pump activity and slow contraction in the aging heart (96).
Another important consequence of mitochondrial stress is the impairment of the proteostasis network, which plays a key role in safeguarding the functioning of the heart by maintaining proper cell function through adequate synthesis, folding, assembly, trafficking, function, and degradation of proteins (97, 98). Mitochondrial stress activates the mitochondrial unfolded protein response (UPRmito), which is regulated by activation of the transcription factor associated with stress 1 (ATFS1). The import of ATFS1 in the mitochondrion is prevented in stress situations, which results in ATFS1 being translocated to the nucleus instead, where it binds to the promotor region of genes involved in resolving mitochondrial stress (99). This derailed proteostatic network, in addition to other stress-responsive pathways [i.e., heat shock response (HSR), endoplasmic reticulum unfolded protein response (UPRER)], and the aberrant protein degradation/autophagy activity, are common denominator in age and multiple cardiac diseases culminating in HF, suggesting that proteostasis maintenance is an important aspect of healthy cardiac aging, and therefore susceptible to potential pharmacologic intervention (99, 100).
More recently, miRNAs have been proposed as novel key players of cellular senescence regulators (101, 102). Among the several miRNAs differently expressed in the old vs. young mouse hearts, miRNA-22 overexpression was shown to be involved in aging-related cardiac fibrosis, while the aging-induced expression of miR-34 was associated to contractile dysfunction during age (103, 104). Therefore, investigating potential aging-related miRNAs and targeting them via pharmacological modulation could represent a strategic means to combat cardiovascular aging in a clinical setting.
Lastly, the reduced regenerative capacity of cardiac stem cell must not be ignored, as increasing evidence suggests (105–107). Such decline may in part be responsible for the impaired myocardial repair in aged hearts and, thus, enhancing the function of endogenous cardiac stem cells might represent a new promising therapeutic strategy of intervention in the senescent myocardium.
It should be noted that heart aging is not a homogenous process, as the changes with age do not occur in everyone at the same pace. The rate of aging myocardium is influenced by multiple factors, including genetic features, lifestyle, environmental exposure, and most importantly comorbidity.
Deeper research aimed at preventing the injury and pathophysiological changes that predispose the young CHD heart to premature aging are warranted. Furthermore, identifying markers of early senescence in ACHD is crucial to guide the development of novel therapeutic interventions.
Early hemodynamic overload represents the obvious mechanism making the heart prone to premature aging (108). Right ventricular (RV) failure can develop in young patients as consequence of the pressure overload such as in TOF, in which the RV becomes the systemic ventricle, and in Pulmonary Artery Hypertension (PAH) associated with left to right shunt. An overloaded RV undergoes a remodeling phase characterized by mitochondrial dysfunction, ischemic stress, and ROS overproduction.
RV failure in young CHD can be prevented by focused interventions, such as by reversing PAH through surgical correction of aortopulmonary shunts. However, this reversibility potential is lost beyond a certain point in time, and late correction leads to irreversible damage to the pulmonary vasculature resulting in chronic PAH development (i.e., Eisenmenger’s syndrome) (109). An interesting study has demonstrated how irreversible PAH-CHD is associated with a switch from a proliferative to a senescent vascular phenotype (110). In a shunt-induced PAH rat model, the authors found a significantly reduced level of genes associated with DNA repair (i.e., Rad51) and increased levels of survivin, a protein associated with apoptosis resistance, in irreversible vs. reversible PAH animals.
Senescence markers, such as p16ink4A and p21cip1, and SASP were also upregulated in irreversible PAH compared to reversible PAH. The vascular profile of irreversible shunt-induced PAH in rats was further corroborated in human lung explant tissue from patients with irreversible PAH-CHD vs. controls, confirming markers of senescence (survivin, p16ink4A and p21cip1) in human PAH-CHD tissue, and higher vulnerability to senescence of PAH-CHD tissue derived endothelial cells in response to shear stress. These observations have led the authors to the recommendation that PAH-CHD should be repaired in early infancy when PAH is still in at a reversible stage.
In the next sections, we will examine the mechanisms and pathways that lead to a progressively faster transition to irreversible aging of the congenitally defective heart.
As previously reported, one of the main features of senescent cells is the secretion of SASPs, including soluble signaling factors, proteases, and insoluble proteins/extracellular matrix (ECM) components released into the surrounding cellular environment. Specifically, the secretome of senescent cardiomyocytes and vascular smooth muscle cells is rich in the inflammatory cytokines IL-1 and IL-6 and tumor necrosis factor a (TNF-α), inducing inflammation in the local surrounding cardiac microenvironment and systemically (111, 112).
Systemic chronic inflammation and immune activation are crucial determinant in the pathogenesis of various cardiovascular diseases and therefore regarded as potential therapeutic targets. Accumulating evidence indicates chronic inflammation and immune senescence have a pathogenic relevance in the progressive deterioration of heart function in patients with CHD (113).
Although the inflammatory syndrome observed in CHD patients is generally described as a consequence of hemodynamic changes or postoperative response after corrective heart surgery, innate factors such as the host’s genotype might determine the magnitude of their systemic inflammatory status and the individual inflammatory response profile to stress factors (114, 115). In a study analyzing the systemic inflammation in new-borns with CHD undergoing heart surgery, it was found that serum IL12p70, IL-6, IL-8, IL-10, and TNF-α levels were significantly higher in CHD patients than in normal cord blood. In a similar study, Mou et al. provided direct evidence of myocardial inflammatory activation in children with CHD not only after CPB but even before surgery, when elevated pre-operative TNF-α levels and nuclear translocation of NF-KB were detected (116).
Additionally, genetic syndromes associated with CHDs can indirectly contribute to altered inflammatory and immune responses. In DiGeorge Syndrome, the abnormal development of pharyngeal precursor structure for both the heart and thymus results in cardiac defects and humoral and T-cell deficiency (117). Children with Down Syndrome are at higher risk for CHD, infections, and immune cell malignancies (118). In Turner Syndrome, congenital defects of bicuspid aortic valve and aortic damage are often associated with autoimmune disease. This immune connection is highlighted by the inflammatory remodeling of the bicuspid valves, which undergo premature immune-mediated calcification, and a systemic pro-inflammatory state (119).
There is a reciprocal augmentation between hemodynamic alterations (myocardial injury, hypoxia, edema, and hypoperfusion) and systemic and local myocardial inflammatory response mediated by circulating immune cells and cardiac macrophages (115, 120). Changes in the cytokine profile are acknowledged as part of a compensatory response to the hemodynamic stress in a variety of CHD, including septal defects and shunts. Cytokine response to mechanical stress encompasses high levels of necrosis factor (TNF)-a and acute-phase reactants (121, 122), which is intended to facilitate a compensatory structural remodeling (123).
Hypoxia induces the transcription of several inflammation-promoting genes, such as NF-κB and TLRs, via the alpha subunit of the hypoxia-inducible transcription factor (HIF-1α). Inflammatory cells and cytokines have been shown to be activated in the pulmonary microvasculature of patients with communications between cardiac chambers or the great arteries leading to increased pulmonary blood flow and pressure. A differential pattern of cytokines was described in children with high pulmonary vascular resistance compared to children with high pulmonary blood flow, with the first group showing higher levels of Macrophage migration inhibitory factor (MIF) and Interleukin 16 (124).
Reversal of flow through a septal defect in Eisenmenger syndrome is associated with a systemic proinflammatory response, with increased C-reactive protein (CRP) and Interferon (IFN)-γ levels (125). Elevation of hsCRP is considered an independent predictor of cardiovascular morbidity and all-cause mortality in patients with CHD (126). Furthermore, in children with coarctation of the aorta, increased inflammatory and apoptotic mediators, including IL-6, IL-10, TNF-a, and soluble Fas (sFas) may contribute to vascular disease as these children enter the ACHD stage (127).
Hypoperfusion and congestion of the bowel facilitate the translocation of bacterial endotoxins from the gut lumen into blood circulation (116, 120). Abnormalities in the inflammatory response and immaturity of the immune system predispose to infection by common pathogens, such as respiratory syncytial virus, and to increased risk of developing bronchopneumonia and sepsis. The readers are invited to consult an extensive review of the immune and inflammatory markers associated with CHD from Singampalli et al. (113). Most common features include reduced granulocyte activity against bacterial infections, T and B lymphocyte levels, naïve T-cell production and T-Cell Receptor Excision Circles (TREC) levels IgA and IgG levels, and complement levels, as well as increased suppressor T-cell function.
Aging is a risk factor for cardiac surgery. The other way round, the trauma of cardiac surgery could accelerate the biological clock of the heart. ACHD patients undergo repeated corrective surgical procedures, cardiopulmonary bypass, and general anesthesia, which can influence the organ homeostasis in different manners.
Open-heart surgery will leave a highly noticeable scar along the area where the incision was made. These scars are the superficial equivalent of heart scars. Surgical correction of the cardiac defect requires cutting, suturing, and inserting a graft or a stent in the attempt to restore the physiologic anatomy and perfusion. Unfortunately, reparative corrections may need to be performed in subsequent stages to allow maturation of the infant or must be repeated because of graft failure. Recurrent damage can trigger an endless cycle of inflammatory responses, scarring, and repair. Cardiac scars are dynamic living structures. Surgical trauma, like other forms of cardiac injury, is followed by tissue necrosis, neutrophil infiltration, and macrophage-driven clearance of cellular debris. Infiltration of circulating and local macrophages occurs as early as a few hours post-injury. Next, the production of extracellular matrix proteins by fibroblasts creates the structural basis for a stabilized fibrillar scar. This whole process can take several weeks. The mature scar contains a spectrum of cells, is metabolically dynamic, and subjected to further spontaneous evolution, i.e., expansion and extension. Contractile properties of the scar rely on the presence of inflammatory cell, which persist in cardiac scars for many years following injury (128).
The healing of a surgically corrected cardiac defect is also dependent on the biocompatibility and integration of prosthetic grafts. Bioprosthetic xenografts, allografts, and synthetic materials have traditionally been used for correction of cardiac defects. However, none of these grafts has the capability to growth in accordance to the child’s growth. Prosthetic conduits and patches used for correction of cardiac defects are non-viable and are recognized as a foreign material by the host’s immune system (129). The consequent inflammatory reaction leads to fibrocalcific remodeling similar to that seen in atherosclerotic lesions (130).
Surgical access for corrective or palliative operations often involves the partial or complete removal of the thymus (131). Thymectomized CHD patients reportedly develop cancer, autoimmune diseases, and atopic diseases and have a higher risk for bacterial and viral infections compared with those without thymectomy (132). This increased risk has been attributed immunologic exhaustion, after early thymectomy (133, 134). A reduction in total and naïve CD4 + T helper and CD8 + cytotoxic T cell compartments reportedly persisted for 2 decades after thymectomy (135, 136). Furthermore, these patients have a shorter T cell telomere length compared to controls and showed a diminished T cell receptor repertoire with signs of oligoclonality as well as augmented memory T cell levels (136, 137). These are typical features of immune senescence as seen in the adaptive immune deficiency typical of the older general population.
Most neonates with congenital heart defects require surgery on cardiopulmonary bypass (CPB) in early infancy and multiple subsequent surgeries to correct residual defects or failing grafts. For instance, the hypoplastic left heart syndrome (HLHS), one of the most critical forms of CHD, requires an early palliative intervention, the Norwood operation, which is associated with a high mortality risk. Although these multiple corrections are necessary to prevent the development of HF, they are not free from complications and adverse effects.
There is a consensus that a significant proportion of morbidity is related to the non-physiologically nature of total CPB, which activates an inflammatory response that closely resembles the systemic inflammatory response syndrome (SIRS), a condition characterized by cardiovascular and pulmonary dysfunction, coagulopathy, and multisystem organ dysfunction/failure (138). The reintroduction of high oxygen level following CPB exposes to reoxygenation injury is considered the primary trigger of the adverse inflammatory response (139, 140). Heterogeneity in the magnitude of the inflammatory response to reoxygenation may be attributed to genetic predisposition and severity of the cardiac defect. For instance, cyanotic CHD children suffering from chronic hypoxia are more prone to the reoxygenation injury during and after CPB, due to the reduced antioxidant reserve capacity in hypoxic conditions (141, 142).
Seminal transcriptomic and proteomic studies have attempted to dissect the complexity of the inflammatory pathways (143, 144). A recent investigation explored the whole blood transcriptome profile in neonates with HLHS before and after their Norwood operation, attempting to unveil an association with the development of the post-operative cardiac output syndrome (LCOS). There were distinctive differences in gene expression between patients with or without LCOS both in pre-operative (14 genes) and post-operative samples (8 genes). Moreover, those who developed LCOS showed larger differences between post-operative and pre-operative samples. Pathway analysis revealed differential regulation of inflammatory pathways (IL signaling, PDGF, NOTCH1, NGF, GPCR) and metabolic pathways (heme metabolism, oxidative phosphorylation, protein metabolism including amino acid and derivatives, fatty acid metabolism, TCA cycle and respiratory electron transport chain). The authors concluded that transcriptome profiling could provide diagnostic hints and potential therapeutic targets to improve outcomes in this high-risk population (114).
Moreover, CPB predisposes to postoperative infections, which impact on the postoperative rehabilitation of children with CHD. This predisposition has been attributed to suppression of the Toll-like receptor (TLR)-mediated signal transduction pathway, which compromises the inflammatory response (145). The same group showed that CPB suppresses the expression of the nucleotide binding and oligomerization domain (NOD)-like receptors signaling-mediated inflammatory response in pediatric CHD patients (146). NOD1 and NOD2 belong to the large class of Pattern recognition receptors which act as sensors of different ligands of bacterial origin. They both recruit and interact with the adaptor protein receptor-interacting protein kinase 2 (RIP2) and thereby activate the NF-κB pathway and mitogen-activated protein kinase (MAPK) pathway, which eventually induces the production of inflammatory cytokines, such as TNF-α and IL-6. Interestingly, peripheral blood leukocytes from CHD patients release TNF-α and IL-6 after stimulation with a NOD agonist, but this response is reduced after CPB (146). Altogether, these findings indicate that the injury from reperfusion causes a dysregulation of the innate immune response, resulting in susceptibility to infection and protracted low-grade inflammation.
Anti-inflammatory treatment, immunomodulatory drugs, and refinement of surgery to avoid or limit the need for thymectomy should be considered to reduce the damage caused by chronic inflammation and immune senescence. Translational work involving scar-modifying treatments has mainly focused on myocardial infarction (MI), with the main aims being to develop interventions that will steer scar properties toward compaction, mechanical strength, and electrical integration. Besides conventional drug treatments, more recent attempts to influence fibroblast activation involve approaches modulating the production of extracellular matrix protein, blocking inflammatory mediators, and interfering with TGF-β or Smad3 signaling (147–149). The effectiveness and safety of such interventions in children with CHD remains unknown. More advanced is the translational attempt to improve the biocompatibility of prosthetic grafts by tissue engineering, which, by combining the use of autologous/allogenic cells and biocompatible scaffolds, holds the promise to create cardiovascular grafts with the potential to remodel, repair, and grow alongside the children’s growth. In particular, the immune-favorable and immune-privileged status of stem/progenitor cells has made these cell types the ideal candidates for correction of congenital pathologies through a tissue engineering approach (150–153). Moreover, pharmacological modulation of altered pathways could be applied to reduce autologous stem cells senescence ex vivo, in order to enhance their reparative ability upon implantation (154). The readers are directed to recent reviews and preclinical studies on this topic (19, 20, 133, 155, 156).
In the last decade, different strategies have been proposed with the aim of avoiding reoxygenation injury during and after CPB. A study comparing propofol combined with low dose fentanyl and midazolam showed that the former is superior in reducing inflammation and oxidase stress and in improving post-operation recovery in children with CHD undergoing cardiac surgery (157). The use of controlled reoxygenation using a partial pressure of oxygen in arterial blood (PaO2), similar to the patient’s preoperative oxygen saturation, has shown to decrease the markers of organ damage (troponin I), stress (cortisol), inflammation (IL-6, IL-8, IL10) and oxidative stress (8-isoprostane) in single-ventricle and TOF patients undergoing cardiac surgery (158, 159). Other studies in adults and small randomized controlled trials in children have suggested some benefits (i.e., reduction of oxidative stress injury) in keeping the blood at normal body temperature throughout surgery (“normothermia”) as opposed to the standard hypotermic procedure, whereby the blood is cooled down during the operation (160, 161).
Senescent vascular cells can drive an irreversible state of vasoconstriction through a paracrine inflammatory mechanism. Therefore, their eradication could be an effective modality to halt the progression of PAH. Human pulmonary endothelial cells of patients with PAH are more vulnerable to senescence than controls in response to shear stress and the senolytic ABT263 reportedly induced apoptosis in senescent, but not in normal, endothelial cells. Interestingly, the senolytic treatment reversed the hemodynamic and structural changes associated with severe PAH in a rodent model (110). The pros and cons of senolytic therapy as a treatment of cardiovascular disease has been recently reviewed (162), although evidence supporting a benefit in children with CHD is lacking. The possible side effects on the young heart are also unknown.
Vitamin supplementation has been considered as a possible avenue for the primary prevention and treatment of cardiac defects. Peri-conceptional multivitamin use was associated with a reduced risk for CHD, particularly outflow tract defects and ventricular septal defects, but no risk reduction was evident when multivitamin use was started after the first month of pregnancy (163). Later studies focusing on folic acid showed that lower dietary folate intake during pregnancy was associated with increased risk (164). Moreover, maternal folate supplementation was significantly associated with a decreased risk of CHD, with this effect being possibly ascribed to anti-apoptotic protection of neural crest cells which participate in the formation of the truncus arteriosus and its division into the pulmonary artery and aorta (165, 166). However, another large study conducted in cohorts from Denmark and Norway found no association between individual-level of maternal folic acid supplementation and offspring heart defects. The authors concluded that, although most likely not harmful, the preventive effect of maternal folic acid supplementation indicated by time trend analyses should be questioned, at least in regions with sufficient intake of dietary folate (167).
Several studies reported that up to 90% of children with CHD undergoing corrective surgery have post-operative serum vitamin D concentrations below 50 nmol/L (168–170). Importantly, there was statistically significant direct correlation between serum vitamin D concentrations and post-operative cardiovascular dysfunction (171). The abrupt decline in vitamin D concentrations during surgery coincided with the initiation of cardiopulmonary bypass (170, 171). Usual low-dose daily supplementation is inadequate to maintain post-operative vitamin D concentrations above 50 nmol/L in the CHD population (172).
As previously mentioned, the oxidative stress has been identified as one of the major contributors to the cardiovascular aging process. Elevated oxidative stress in the senescent myocardium has several consequences such as enhanced protein oxidation/nitration, activation of inflammatory response, proteostasis impairment, mitochondrial dysfunction and endoplasmic reticulum (ER) stress (88).
The ER plays key roles in the synthesis, folding, and translocation of secretory and membrane proteins and in calcium homeostasis and lipid biosynthesis (173). A variety of environmental stresses, including nutrient deprivation, hypoxia, calcium depletion, and cytotoxic drug administration, can cause the accumulation of unfolded proteins in the ER, thereby inducing the ER stress response. In the developing heart, an excessive or too prolonged activation of the UPR may reduce expression of essential proteins, adversely affect cell function, and eventually lead to cell death.
Interestingly, a recent study showed that exposure of mouse embryos to short-term gestational hypoxia induced the UPR in cardiac progenitor cells and caused common types of heart defect in about 50% of newborns (174). One of the early UPR responses was PERK-dependent attenuation of protein synthesis, which was expected to result in the reduction in embryo growth rate. However, in mouse embryos exposed to hypoxia, the translation block was only affecting the levels of specific proteins that are immediately and continuously available for cardiomyocyte maturation. In particular, gestational stress resulted in reduced FGFR1 translation, which profoundly reduced FGF signaling in cardiac progenitor cells of the second heart field, whereas other receptor tyrosine kinases characterized by rapid recycling were not affected (174).
In the above mouse model, the induction of the UPR was reversible but FGF-1 signaling was persistently abrogated (174). However, it is tempting to speculate that persistent hypoxia after birth may cause a progressive exhaustion of the UPR, leading to accumulation of unfolded proteins in the ER and mitochondria. A study from Jian et al. examined this possibility on myocardial samples from patients with cyanotic congenital cardiac defects (175). They found that the upregulation of HIF-1α in the hypoxic myocardium was associated with a marked induction of the chaperone GRP78/Bip in cardiomyocytes, suggesting an activation of the UPR. Then, they assessed the activity of the key signaling pathways of UPR, ATF6α, and PERK, and found that, at variance with the mouse embryo hypoxia model, ATF6α was significantly upregulated whereas ERK signaling was not significantly different between the cyanotic and acyanotic groups. This was interpreted as a compensatory mechanism, as pharmacological inhibition or silencing of ATF6α promoted cardiomyocyte apoptosis following long-lasting in vitro hypoxia or hypoxia/re-oxygenation (175).
The regulation of UPR under hypoxia may involve a group of hypoxia-microRNAs. For instance, miR-199a-5p is reportedly downregulated in hypoxic myocardial samples from children with CHD (139). This is mediated by the activation of IL-6/IL-11/STAT3 signaling. Binding of pSTAT3 to the promoter region of miR-199a-2 gene, more significantly under hypoxic conditions, reduced the expression of pri-miR-199a-2, a precursor of miR-199a-5p, thereby activating its UPR targets, GRP78 and ATF6 (176, 177).
Recent findings indicate that cardiac aging is accompanied by a gradual derailment of proteostasis, which predisposes the heart to the development of age-related cardiac diseases, including atrial fibrillation (99).
Unfolded proteins can accumulate in cardiomyocytes due to genetic mutations, such as in desmin-related cardiomyopathy, but also as the consequence of cellular stress that damages nascent or aging proteins, or normal wear and tear. When terminally misfolded proteins exceed the UPR capacity, they are targeted for degradation through the ubiquitin–proteasome or autophagy–lysosomal pathways. Eventually, accumulation of misfolded proteins in aggresomes occurs when protein misfolding is not fixed. It is well acknowledged that the functionality of these regulatory proteostasis checkpoints is damaged during aging, with senescent cells becoming unable to maintain proteostasis and causing the heart to suffer from age-associated proteinopathies (178).
Ubiquitin plays a key role in the degradation of unfolded or misfolded proteins by targeting ubiquitin chains in the ubiquitin–proteasome or autophagy system. Failure to eliminate the abnormal protein aggregates causes the cells to move to apoptosis and death. SUMOylation is an essential component of the ubiquitination proteasome system. SUMO (Small ubiquitin-like modifier) conjugation pathway is abundantly expressed in the heart and implicated in cardiovascular development via modifying transcription factors necessary for normal cardiovascular development (179). A balanced post-translational SUMO conjugation-deconjugation pathway is essential for normal cardiac development. In fact, both hetero- and homo-zygous SUMO-1 knockout mice exhibited ventricular defects with high mortality rates, which were rescued by cardiac re-expression of the SUMO-1 transgene (179). Moreover, transgenic mice with cardiac-specific expression of SENP2, a SUMO-specific protease that deconjugates sumoylated proteins, resulted in premature death of mice due to atrial and/or ventricular septal defects. Surviving mice showed growth retardation, and developed cardiomyopathy with impaired cardiac function with aging (180).
Lysosomes are one of the main components of the autophagy pathway, which is crucial for maintaining cardiomyocyte morphology and function by regulating organelle turnover and cardiac remodeling. Alterations of mitochondrial function can impair processes such as cardiomyocyte differentiation, response to ischemic events, apoptosis, and autophagy (181). Mitophagy, the degradation of mitochondria by the autophagy machinery, is dysfunctional in primary human derived from subject with Down Syndrome and this is associated with low Parkin and p62 levels and delayed PINK1 activation (182). Moreover, mTOR hyperactivity and reduced ATG proteins involved in autophagosome formation contribute to downregulation of mitophagy (182). Organelle dysfunction could contribute to accelerated aging in patients with Down Syndrome-related CHD (183).
Therapies specifically targeting the ER stress and proteostasis in heart disease are at experimental stage. Some pharmacological agents used in clinical settings such as angiotensin II type 1 receptor antagonists and the antidiabetic agent pioglitazone may inhibit UPR pathways. Pharmaceutical AMPK activators reduce cardiac ER stress and prevents the progression of HF (173). Additional drugs include small-molecule activators of the heat shock response, such as geranylgeranylacetone, currently used in Japan for treatment of ulcers, and UPRER activators, such as the ATF6 pathway activator compound (184). The rationale for the use of these compounds is to activate stress response pathways to enhance cytoprotection against protein misfolding and aggregation. However, strategies to reduce protein synthesis could negatively interfere with regenerative processes. In contrast, the use of chemical chaperones such as 4-phenylbutyrate or agents that reduce misfolded protein concentrations may represent a more effective modality in CHD, where the UPR is exhausted and protein misfolding accrues slowly over time. In these conditions, activation of autophagy (e.g., rapamycin or spermidine), pharmacological proteasome induction, and interfering RNA–based therapeutics represent additional promising options (185). On the other hand, inhibition of autophagy might represent a target for treatment of some cardiovascular diseases which are not caused by the overexpression of a mutant protein, and are rather associated to excessive activation of autophagy, like myocardial infarction, and atrial fibrillation (99). Nonetheless, inhibitors of autophagy, such as bafilomycin, can cause cellular toxicity (186).
Moreover, longevity genes and proteins could have a significant impact in delaying age-related cardiac dysfunction (187). In this respect, we discovered that a genetic polymorphism of the BPIFB4 gene is associated with exceptional longevity. The related mutant protein forms a complex with 14-3-3 and HSP90, activating the prosurvival eNOS—Akt signaling. Supplementation of the longevity genes significantly improved cardiac function in different models of age-related cardiac disease (188–191) and reverse immune senescence (192). It would be interesting to determine the expression of BPIFB4 in CHD and, if this is found downregulated as in elderly cardiovascular patients (193), to assess the benefit of supplementation on cardiac cells from CHD patients and CHD models.
Personalized interventions are urgently needed for the treatment of rare cardiac diseases characterized by an alteration in protein disposal. For instance, the Koolen-de Vries syndrome (KdVS) is a rare monogenic disorder characterized by intellectual disability, heart failure, hypotonia, and congenital malformations. Mechanistic study shows that KANSL1 modulates autophagosome-lysosome fusion for cargo degradation via transcriptional regulation of autophagosomal gene, STX17. Kansl1 ± mice have an impaired autophagic clearance of damaged mitochondria and tend to accumulate ROS which in turn cause defective neuronal and cardiac functions. An FDA-approved drug, 13-cis retinoic acid, can reverse these mitophagic defects and neurobehavioral abnormalities in Kansl1 ± mice by promoting autophagosome-lysosome fusion. Hence, KANSL1 could represent a possible therapeutic target to treat autophagy and KdVS (194).
With the heart being a highly metabolic organ that is reliant on the maintenance of cellular-energetic homeostasis, mitochondria are important determinants of cellular longevity. It is, therefore, not surprising that defects in mitochondrial bioenergetics have been related to normal cardiac aging (195–197). Many factors contribute to the age-related reduced energetic capacity of the cardiac mitochondria including increased ROS, mutation and deletions in the mitochondrial genome, and dysregulation of proteostasis and mitochondrial biogenesis throughout the organism lifespan as well as in the embryonic development (95, 198).
Accruing evidence suggests that mitochondria maturation and processing not only serve as the powerhouse enabling the heart to beat but also play a critical role in regulating embryonic and neonatal heart development in mammals (199). The normal assembly of respiratory chain components, i.e., the oxidative phosphorylation (OXPHOS) complexes, in embryonic hearts is paramount to prepare the heart to undergo the fetal to neonatal metabolic shift -from anaerobic glycolysis to aerobic oxidative phosphorylation- after birth (199). Several studies have reported how defective OXPHOS in embryonic hearts leads to severe heart defects and embryonic or neonatal lethality in mouse models and human patients (200–202).
Additionally, a mismatch between mitochondrial programming and the substrate availability was proposed as the major cause for the juvenile cardiomyopathy described in mice with cardiac ectopic expression of MFN2 AA (203). Another in vivo study showed that cardiac ablation of both peroxisome proliferator-activated receptor gamma coactivator 1-α (Ppargc1α) and peroxisome proliferator-activated receptor gamma coactivator 1-β (Ppargc1β), nuclear transcriptional coactivators required for mitochondrial biogenesis, caused mitochondrial dysfunction and late gestational defects in cardiac function and cardiomyocytes maturation (204).
Strong evidence indicates that mitochondrial dysfunction, in particular impaired mitochondrial biogenesis (highlighted by decreased mitochondrial mass and DNA content), participates in the development of HF (205–207). Myocardial ischemia, reperfusion, and chronic overload significantly damage the mitochondrial structure and function, severely compromising cellular viability. A recent study showed reduced mtDNA replication and depletion of mtDNA together with pathological changes of mitochondrial ultrastructure occurs early during the development of RV hypertrophy, preceding the clinical manifestation of HF (208). The reasons for these alterations remain unknown but the authors suggested that “these findings provide an important basis for the future development of mitochondrial oriented clinical and surgical treatment of patients with overloaded RV caused by CHD” (208).
As previously described in the mechanisms of cardiac aging, mitochondrial dysfunction leads to elevate ROS production also in fetal heart, an event that triggers the DNA damage pathway that blocks cardiomyocyte proliferation (199). A study conducted on TOF patients with RV hypertrophy showed an increase of 4-HNE on mitochondrial function and structure. This bioproduct of lipid peroxidation, a form of oxidative stress in aging heart, altered the energy production in the mitochondrion of RV leading to its failure and cardiomyocyte dysfunction (209). An in vivo genetic model of mitochondrial dysfunction has been achieved by inactivating the mitochondrial transcription factor A (tfam), which resulted in elevated ROS production and activated DNA damage response, thus causing cardiomyocyte cell cycle arrest and ultimately lethal cardiomyopathy (210). The authors further showed that inhibition of ROS or the DNA damage response pathway rescued the cell proliferation defect observed in cultured fetal cardiomyocytes in which Tfam was deleted, making these pathways potential targets of intervention to prevent cardiac aberration caused by some forms of mitochondrial dysfunction.
Genetic factors, like mtDNA mutations, might as well underlie the mitochondrial stress typical of some heart disease, as we have previously described in the etiology of CHD (211–213). A Chinese study conducted a systematic screening assessing mtDNA mutations among coronary heart disease patients (214). One three-generation family presenting evidence of coronary heart disease maternally inherited was identified, with 6/24 analyzed adults in this family exhibiting coronary heart disease of vary severity. The sequencing of the mitochondrial genomes of these individuals showed a tRNAThr 15910C > T mutation of the M7b’c haplogroup, which is predicted to destabilize the strongly conserved (24C-10G) base-pairing, thereby disrupting tRNAThr functionality. Interestingly, only limited changes in mitochondrial functionality were observed, suggesting that 15910C > T mutation is relatively mild and may by itself be insufficient to produce a clinical phenotype, nonetheless it represents an inherited risk factor for the development of coronary heart disease. This finding points out how the diagnostic process of mitochondrial disease of the heart can be very challenging, especially in CHD patients, as it goes asymptomatic in the early stages, and later it manifests itself as cardiomyopathy, HF, arrhythmia and so on (215–217).
Identifying early markers of mitochondrial dysfunction would be beneficial to prevent the later manifestations of cardiac disorders in CHD population. It has been suggested that mitochondrial respiration in peripheral blood mononuclear cells (PBMCs) may have utility for predicting HF risk in some CHD, as elevated PBMCs respiration was found to be correlated with HF in post-Fontan SV-CHD young patients (218).
Current treatments are not effective in preserving mitochondrial structure and function and therefore accruing mitochondria dysfunction through repeated operations could rapidly lead to energetic deficits in cardiomyocytes of CHD patients. A novel therapy based on mitochondrial transplantation may become a game-changer in this clinical field. Autologous mitochondria isolated from the patient’s own body and then directly injected into the ischemic myocardium significantly ameliorated ischemia/reperfusion injury and enhanced myocardial cellular viability and post-ischemic functional recovery (219). The authors reported that improvement in energy generation were associated with upregulation cardioprotective cytokines. The initial uncontrolled clinical application in humans was performed in pediatric patients suffering from myocardial ischemia reperfusion injury following coronary artery occlusion and revascularization (220). In a subsequent pilot study, mitochondrial transplantation enhanced ventricular strain in patients requiring postcardiotomy extracorporeal membrane oxygenation for severe refractory cardiogenic shock after ischemia-reperfusion injury (221).
The successful diagnosis, advances in imaging, catheter-based interventions, surgery, and clinical management of CHD have yielded a dramatic change in age distribution of the CHD population. The improvement in survival has increased the number of ACHD and related complications. Comparing ACHD group with the general population, the mortality of ACHD is drastically high (56). Results from the CONCOR study in Netherlands, showed that ∼ 0.6% of ACHD patients died because of the presence of late complications for cardiac causes. The 26% died for chronic HF and 19% died for unknown sudden death. Among the 23% who died for non-cardiovascular causes, 14% died for pneumonia, and 9% died for malignant diseases (222). Growing up with CHD implore clinicians and researchers to gain a better understanding of aging, as this will be the keystone to how this rapidly evolving group of patients will be planned and monitored (223). There is an urgent need to standardize a clinical-educational program in the transition phase between adolescents and adults to allow young patients to become more skilled about their disease and reducing the probability of delaying CHD care (diagnosis, complication assessment, therapy) in adulthood (224). Patients must have more awareness of their CHD conditions and take the responsibility of their decisions of care, such as, major interventions or transplantation (225). American College of Cardiology (ACC) and American Heart Association (AHA) member committee in 2018 have provided clinical guidelines with all the recommendations for the management of adult patients with CHD (226). This guideline is a complete revision of previous 2008 ACC/AHA guidelines and focuses only on the care of ACHD. This document was set up because of the successful diagnosis and treatment strategies employed in babies and children with this disease which have led to a high prevalence of the adult population. ACHD management requires specific needs, and cardiologists must be specifically trained for their care in highly specialized ACHD centers. Recently European Society of Cardiology (ESC) working group published a position paper with the recommendation for the transcatheter intervention in ACHD highlighting the issues of its allocation on national scale and how to train the surgeons to reach a standard recognition as ACHD interventionists (227). This process is running at slow pace because it will require infrastructures and expertise of different cardiologists for each procedure enabling the training of the future ACHD interventionists. In addition, stem cell therapy, immunosuppressive therapy for cardiac transplantation, mechanical pumps, artificial hearts and new machines using algorithms to estimate prognosis and guide therapies in ACHD patients will be one of the new interesting clinical research in the field of CHD (225, 228).
Beyond clinical and educational research program and specialized ACHD centers, several measures of aging underpinning ACHD will need to be studied. Scientific data on functional and biological markers of aging (telomere length, markers of inflammation, epigenetic clock) in CHD is hugely needed. For instance, exercise and diet could be potential strategies for maintaining the telomere. In addition, epigenetic modifications such as, DNA methylation, histone modifications, non-coding RNA, can be chemically reversibile and used as target for rejuvenation strategies (223, 229). Once aging is investigated over the entire life course and the predictor factors causing accelerated senescence in ACHD are characterized, a targeted therapy could be developed and applied to these patients. Furthermore, it is paramount to secure more resources which welcome patients in the field of CHD management and ensure a complete consciousness and responsibility of conducting a healthy life (225).
The increase in lifespan of CHD patients poses new challenges to the National Health Systems and the society, but also provides an opportunity to increase translational efforts to design an holistic approach for the prevention and cure of age-related diseases. As for older people, it is vital to prolong the period ACHD patients enjoy a healthy life. Further refinement of surgical approaches including the merging of minimally invasive surgery and tissue engineering will limit the need of repeated operations. Anti-aging treatments targeting inflammatory pathways, and homeostatic response to stress merit consideration. Senolytic drugs provide a promising means to interrupt the vicious cycle of cell senescence and inflammation. Finally, genetic interventions transferring longevity genes and proteins could confer cardiovascular protection as shown in preclinical models of aging.
PM was responsible as corresponding author for the final writing and the data presented. All authors read, approved the final manuscript, conceived, and designed the review article.
This study was supported by the grants from the Sir Jules Thorn Charitable Trust (MC and PM), the Enid Linder Foundation (MC), the National Institute for Health Research (NIHR) Bristol Biomedical Research Centre in Cardiovascular Medicine (MC and PM), the British Heart Foundation (no. PG/18/38/33707) grant entitled “Microfabrication and in vivo testing of small-diameter tissue-engineered vascular grafts” (PM), and the Medical Research Council (no. MR/N027086/1) grant entitled “In vitro and in vivo preclinical testing of pericyte-engineered grafts for correction of congenital heart defects” (PM and MC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rossano JW, Shaddy RE. Heart failure in children: etiology and treatment. J Pediatr. (2014) 165:228–33. doi: 10.1016/j.jpeds.2014.04.055
2. Massin MM, Astadicko I, Dessy H. Epidemiology of heart failure in a tertiary pediatric center. Clin Cardiol. (2008) 31:388–91. doi: 10.1002/clc.20262
3. Sommers C, Nagel BH, Neudorf U, Schmaltz AA. [Congestive heart failure in childhood. An epidemiologic study]. Herz. (2005) 30:652–62. doi: 10.1007/s00059-005-2596-6
4. Hayabuchi Y, Mori K, Kitagawa T, Sakata M, Kagami S. Polytetrafluoroethylene graft calcification in patients with surgically repaired congenital heart disease: evaluation using multidetector-row computed tomography. Am Heart J. (2007) 153:806.e1–8. doi: 10.1016/j.ahj.2007.01.035
5. Konstam MA, Kiernan MS, Bernstein D, Bozkurt B, Jacob M, Kapur NK, et al. Evaluation and management of right-sided heart failure: a scientific statement from the American heart association. Circulation. (2018) 137:e578–622. doi: 10.1161/CIR.0000000000000560
6. Hinton RB, Ware SM. Heart failure in pediatric patients with congenital heart disease. Circ Res. (2017) 120:978–94.
7. Best KE, Rankin J. Long-term survival of individuals born with congenital heart disease: a systematic review and meta-analysis. J Am Heart Assoc. (2016) 5:e002846.
8. Norozi K, Wessel A, Alpers V, Arnhold JO, Geyer S, Zoege M, et al. Incidence and risk distribution of heart failure in adolescents and adults with congenital heart disease after cardiac surgery. Am J Cardiol. (2006) 97:1238–43. doi: 10.1016/j.amjcard.2005.10.065
9. Saha P, Potiny P, Rigdon J, Morello M, Tcheandjieu C, Romfh A, et al. Substantial cardiovascular morbidity in adults with lower-complexity congenital heart disease. Circulation. (2019) 139:1889–99. doi: 10.1161/CIRCULATIONAHA.118.037064
10. Budts W, Roos-Hesselink J, Radle-Hurst T, Eicken A, McDonagh TA, Lambrinou E, et al. Treatment of heart failure in adult congenital heart disease: a position paper of the working group of grown-up congenital heart disease and the heart failure association of the European society of cardiology. Eur Heart J. (2016) 37:1419–27. doi: 10.1093/eurheartj/ehv741
11. Andersen S, Andersen A, Nielsen-Kudsk JE. The renin-angiotensin-aldosterone-system and right heart failure in congenital heart disease. Int J Cardiol Heart Vasc. (2016) 11:59–65. doi: 10.1016/j.ijcha.2016.03.013
12. Miyamoto SD, Stauffer BL, Nakano S, Sobus R, Nunley K, Nelson P, et al. Beta-adrenergic adaptation in paediatric idiopathic dilated cardiomyopathy. Eur Heart J. (2014) 35:33–41. doi: 10.1093/eurheartj/ehs229
13. van der Bom T, Winter MM, Bouma BJ, Groenink M, Vliegen HW, Pieper PG, et al. Effect of valsartan on systemic right ventricular function: a double-blind, randomized, placebo-controlled pilot trial. Circulation. (2013) 127:322–30. doi: 10.1161/CIRCULATIONAHA.112.135392
14. Hsu DT, Zak V, Mahony L, Sleeper LA, Atz AM, Levine JC, et al. Enalapril in infants with single ventricle: results of a multicenter randomized trial. Circulation. (2010) 122:333–40. doi: 10.1161/circulationaha.109.927988
15. Stout KK, Daniels CJ, Aboulhosn JA, Bozkurt B, Broberg CS, Colman JM, et al. 2018 AHA/ACC guideline for the management of adults with congenital heart disease: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Circulation. (2019) 139:e698–800.
16. Majumdar U, Yasuhara J, Garg V. In vivo and in vitro genetic models of congenital heart disease. Cold Spring Harb Perspect Biol. (2021) 13:a036764. doi: 10.1101/cshperspect.a036764
17. Broberg M, Hastbacka J, Helle E. From stem cells to populations-using hiPSC, next-generation sequencing, and GWAS to explore the genetic and molecular mechanisms of congenital heart defects. Genes (Basel). (2021) 12:921. doi: 10.3390/genes12060921
18. Mercer EJ, Evans T. Congenital heart disease in a dish: progress toward understanding patient-specific mutations. J Thorac Dis. (2017) 9:E510–3. doi: 10.21037/jtd.2017.03.178
19. Alvino VV, Thomas AC, Ghorbel MT, Rapetto F, Narayan SA, Kilcooley M, et al. Reconstruction of the swine pulmonary artery using a graft engineered with syngeneic cardiac pericytes. Front Bioeng Biotechnol. (2021) 9:715717. doi: 10.3389/fbioe.2021.715717
20. Iacobazzi D, Rapetto F, Albertario A, Swim MM, Narayan S, Skeffington K, et al. Wharton’s jelly-mesenchymal stem cell-engineered conduit for pediatric translation in heart defect. Tissue Eng Part A. (2021) 27:201–13. doi: 10.1089/ten.TEA.2020.0088
21. Ghorbel MT, Jia H, Swim MM, Iacobazzi D, Albertario A, Zebele C, et al. Reconstruction of the pulmonary artery by a novel biodegradable conduit engineered with perinatal stem cell-derived vascular smooth muscle cells enables physiological vascular growth in a large animal model of congenital heart disease. Biomaterials. (2019) 217:119284. doi: 10.1016/j.biomaterials.2019.119284
22. Albertario A, Swim MM, Ahmed EM, Iacobazzi D, Yeong M, Madeddu P, et al. Successful reconstruction of the right ventricular outflow tract by implantation of thymus stem cell engineered graft in growing swine. JACC Basic Transl Sci. (2019) 4:364–84. doi: 10.1016/j.jacbts.2019.02.001
23. Sophocleous F, Bone A, Shearn AIU, Forte MNV, Bruse JL, Caputo M, et al. Feasibility of a longitudinal statistical atlas model to study aortic growth in congenital heart disease. Comput Biol Med. (2022) 144:105326. doi: 10.1016/j.compbiomed.2022.105326
24. Sandrini C, Lombardi C, Shearn AIU, Ordonez MV, Caputo M, Presti F, et al. Three-dimensional printing of fetal models of congenital heart disease derived from microfocus computed tomography: a case series. Front Pediatr. (2019) 7:567. doi: 10.3389/fped.2019.00567
25. Mitchell SC, Korones SB, Berendes HW. Congenital heart disease in 56,109 births. Incidence and natural history. Circulation. (1971) 43:323–32. doi: 10.1161/01.cir.43.3.323
26. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. (2002) 39:1890–900.
27. Warnes CA, Liberthson R, Danielson GK, Dore A, Harris L, Hoffman JI, et al. Task force 1: the changing profile of congenital heart disease in adult life. J Am Coll Cardiol. (2001) 37:1170–5. doi: 10.1016/s0735-1097(01)01272-4
28. Rohit M, Shrivastava S. Acyanotic and cyanotic congenital heart diseases. Indian J Pediatr. (2018) 85:454–60. doi: 10.1007/s12098-017-2454-6
29. Ossa Galvis MM, Bhakta RT, Tarmahomed A, Mendez MD. Cyanotic Heart Disease. Treasure Island, FL: StatPearls (2022).
30. Thiene G, Frescura C. Anatomical and pathophysiological classification of congenital heart disease. Cardiovasc Pathol. (2010) 19:259–74. doi: 10.1016/j.carpath.2010.02.006
31. van der Bom T, Zomer AC, Zwinderman AH, Meijboom FJ, Bouma BJ, Mulder BJ. The changing epidemiology of congenital heart disease. Nat Rev Cardiol. (2011) 8:50–60.
32. Nora JJ. Multifactorial inheritance hypothesis for the etiology of congenital heart diseases. The genetic-environmental interaction. Circulation. (1968) 38:604–17. doi: 10.1161/01.cir.38.3.604
33. Pierpont ME, Basson CT, Benson DW Jr, Gelb BD, Giglia TM, Goldmuntz E, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American heart association congenital cardiac defects committee, council on cardiovascular disease in the young: endorsed by the American academy of pediatrics. Circulation. (2007) 115:3015–38. doi: 10.1161/CIRCULATIONAHA.106.183056
34. Jenkins KJ, Correa A, Feinstein JA, Botto L, Britt AE, Daniels SR, et al. Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American heart association council on cardiovascular disease in the young: endorsed by the American academy of pediatrics. Circulation. (2007) 115:2995–3014. doi: 10.1161/CIRCULATIONAHA.106.183216
35. Marino B, Digilio MC, Toscano A, Anaclerio S, Giannotti A, Feltri C, et al. Anatomic patterns of conotruncal defects associated with deletion 22q11. Genet Med. (2001) 3:45–8. doi: 10.1097/00125817-200101000-00010
36. Goldmuntz E, Clark BJ, Mitchell LE, Jawad AF, Cuneo BF, Reed L, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol. (1998) 32:492–8. doi: 10.1016/s0735-1097(98)00259-9
37. Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. (2017) 49:1593–601. doi: 10.1038/ng.3970
38. Pierpont ME, Brueckner M, Chung WK, Garg V, Lacro RV, McGuire AL, et al. Genetic basis for congenital heart disease: revisited: a scientific statement from the American heart association. Circulation. (2018) 138:e653–711.
39. Hobbs CA, Cleves MA, Simmons CJ. Genetic epidemiology and congenital malformations: from the chromosome to the crib. Arch Pediatr Adolesc Med. (2002) 156:315–20. doi: 10.1001/archpedi.156.4.315
40. Zaidi S, Brueckner M. Genetics and genomics of congenital heart disease. Circ Res. (2017) 120:923–40. doi: 10.1161/CIRCRESAHA.116.309140
41. Moore-Morris T, van Vliet PP, Andelfinger G, Puceat M. Role of epigenetics in cardiac development and congenital diseases. Physiol Rev. (2018) 98:2453–75. doi: 10.1152/physrev.00048.2017
42. Cao J, Wu Q, Huang Y, Wang L, Su Z, Ye H. The role of DNA methylation in syndromic and non-syndromic congenital heart disease. Clin Epigenetics. (2021) 13:93. doi: 10.1186/s13148-021-01077-7
43. Lim TB, Foo SYR, Chen CK. The role of epigenetics in congenital heart disease. Genes (Basel). (2021) 12:390. doi: 10.3390/genes12030390
44. Castilla EE, Lopez-Camelo JS, Campana H, Rittler M. Epidemiological methods to assess the correlation between industrial contaminants and rates of congenital anomalies. Mutat Res. (2001) 489:123–45. doi: 10.1016/s1383-5742(01)00067-9
45. Shi M, Wehby GL, Murray JC. Review on genetic variants and maternal smoking in the etiology of oral clefts and other birth defects. Birth Defects Res C Embryo Today. (2008) 84:16–29. doi: 10.1002/bdrc.20117
46. Miranda ML, Maxson P, Edwards S. Environmental contributions to disparities in pregnancy outcomes. Epidemiol Rev. (2009) 31:67–83. doi: 10.1093/epirev/mxp011
47. Taylor K, Elhakeem A, Thorbjornsrud Nader JL, Yang TC, Isaevska E, Richiardi L, et al. Effect of maternal prepregnancy/early-pregnancy body mass index and pregnancy smoking and alcohol on congenital heart diseases: a parental negative control study. J Am Heart Assoc. (2021) 10:e020051. doi: 10.1161/JAHA.120.020051
48. Taylor K, Ferreira DLS, West J, Yang T, Caputo M, Lawlor DA. Differences in pregnancy metabolic profiles and their determinants between white European and south Asian women: findings from the born in Bradford cohort. Metabolites. (2019) 9:190. doi: 10.3390/metabo9090190
49. Giannakoulas G, Mouratoglou SA, Gatzoulis MA, Karvounis H. Blood biomarkers and their potential role in pulmonary arterial hypertension associated with congenital heart disease. a systematic review. Int J Cardiol. (2014) 174:618–23. doi: 10.1016/j.ijcard.2014.04.156
50. Mostafavi N, Vlaanderen J, Chadeau-Hyam M, Beelen R, Modig L, Palli D, et al. Inflammatory markers in relation to long-term air pollution. Environ Int. (2015) 81:1–7. doi: 10.1016/j.envint.2015.04.003
51. Pirinccioglu AG, Alyan O, Kizil G, Kangin M, Beyazit N. Evaluation of oxidative stress in children with congenital heart defects. Pediatr Int. (2012) 54:94–8. doi: 10.1111/j.1442-200X.2011.03478.x
52. Stein RA. Epigenetics and environmental exposures. J Epidemiol Community Health. (2012) 66:8–13.
53. Tang D, Li TY, Chow JC, Kulkarni SU, Watson JG, Ho SS, et al. Air pollution effects on fetal and child development: a cohort comparison in China. Environ Pollut. (2014) 185:90–6. doi: 10.1016/j.envpol.2013.10.019
54. Yuan Y, Jin L, Wang L, Li Z, Zhang L, Zhu H, et al. Levels of PAH-DNA adducts in placental tissue and the risk of fetal neural tube defects in a Chinese population. Reprod Toxicol. (2013) 37:70–5. doi: 10.1016/j.reprotox.2013.01.008
55. Zhang QJ, Liu ZP. Histone methylations in heart development, congenital and adult heart diseases. Epigenomics. (2015) 7:321–30. doi: 10.2217/epi.14.60
56. Verheugt CL, Uiterwaal CS, van der Velde ET, Meijboom FJ, Pieper PG, van Dijk AP, et al. Mortality in adult congenital heart disease. Eur Heart J. (2010) 31:1220–9.
57. Steenman M, Lande G. Cardiac aging and heart disease in humans. Biophys Rev. (2017) 9:131–7. doi: 10.1007/s12551-017-0255-9
58. Saunders TJ, Chaput JP, Tremblay MS. Sedentary behaviour as an emerging risk factor for cardiometabolic diseases in children and youth. Can J Diabetes. (2014) 38:53–61. doi: 10.1016/j.jcjd.2013.08.266
59. Sallis JF, Bull F, Guthold R, Heath GW, Inoue S, Kelly P, et al. Progress in physical activity over the Olympic quadrennium. Lancet. (2016) 388:1325–36. doi: 10.1016/S0140-6736(16)30581-5
60. McCrindle BW, Williams RV, Mital S, Clark BJ, Russell JL, Klein G, et al. Physical activity levels in children and adolescents are reduced after the Fontan procedure, independent of exercise capacity, and are associated with lower perceived general health. Arch Dis Child. (2007) 92:509–14. doi: 10.1136/adc.2006.105239
61. Massin MM, Hovels-Gurich HH, Gerard P, Seghaye MC. Physical activity patterns of children after neonatal arterial switch operation. Ann Thorac Surg. (2006) 81:665–70. doi: 10.1016/j.athoracsur.2005.07.034
62. Stone N, Obeid J, Dillenburg R, Milenkovic J, MacDonald MJ, Timmons BW. Objectively measured physical activity levels of young children with congenital heart disease. Cardiol Young. (2015) 25:520–5. doi: 10.1017/S1047951114000298
63. Kao CC, Chang PC, Chiu CW, Wu LP, Tsai JC. Physical activity levels of school-age children with congenital heart disease in Taiwan. Appl Nurs Res. (2009) 22:191–7. doi: 10.1016/j.apnr.2007.12.002
64. Barbiero SM, D’Azevedo Sica C, Schuh DS, Cesa CC, de Oliveira Petkowicz R, Pellanda LC. Overweight and obesity in children with congenital heart disease: combination of risks for the future? BMC Pediatr. (2014) 14:271. doi: 10.1186/1471-2431-14-271
65. Mari MA, Cascudo MM, Alchieri JC. Congenital heart disease and impacts on child development. Braz J Cardiovasc Surg. (2016) 31:31–7. doi: 10.5935/1678-9741.20160001
66. Razzaghi H, Oster M, Reefhuis J. Long-term outcomes in children with congenital heart disease: national health interview survey. J Pediatr. (2015) 166:119–24. doi: 10.1016/j.jpeds.2014.09.006
67. Ni ZH, Lv HT, Ding S, Yao WY. Home care experience and nursing needs of caregivers of children undergoing congenital heart disease operations: a qualitative descriptive study. PLoS One. (2019) 14:e0213154. doi: 10.1371/journal.pone.0213154
68. Brida M, Gatzoulis MA. Adult congenital heart disease: the quest to address the simple and the complex. JACC Case Rep. (2021) 3:353–5. doi: 10.1016/j.jaccas.2021.01.001
69. Martinez-Quintana E, Estupinan-Leon H, Rojas-Brito AB, Deniz-Deniz L, Barreto-Martin A, Rodriguez-Gonzalez F. Evaluation of quality of life in patients with congenital heart disease: an observational case control study. Am J Cardiovasc Dis. (2021) 11:73–9.
70. Afilalo J, Therrien J, Pilote L, Ionescu-Ittu R, Martucci G, Marelli AJ. Geriatric congenital heart disease: burden of disease and predictors of mortality. J Am Coll Cardiol. (2011) 58:1509–15. doi: 10.1016/j.jacc.2011.06.041
71. Bagge CN, Henderson VW, Laursen HB, Adelborg K, Olsen M, Madsen NL. Risk of dementia in adults with congenital heart disease: population-based cohort study. Circulation. (2018) 137:1912–20. doi: 10.1161/circulationaha.117.029686
72. Jokinen E. Coronary artery disease in patients with congenital heart defects. J Intern Med. (2020) 288:383–9. doi: 10.1111/joim.13080
73. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke statistics-2018 update: a report from the American heart association. Circulation. (2018) 137:e67–492.
74. Moons P, Van Deyk K, Dedroog D, Troost E, Budts W. Prevalence of cardiovascular risk factors in adults with congenital heart disease. Eur J Cardiovasc Prev Rehabil. (2006) 13:612–6. doi: 10.1097/01.hjr.0000197472.81694.2b
75. Deen JF, Krieger EV, Slee AE, Arslan A, Arterburn D, Stout KK, et al. Metabolic syndrome in adults with congenital heart disease. J Am Heart Assoc. (2016) 5:e001132.
76. Duffels MG, Mulder KM, Trip MD, de Groot E, Gort J, van Dijk AP, et al. Atherosclerosis in patients with cyanotic congenital heart disease. Circ J. (2010) 74:1436–41.
77. Perloff JK. Cyanotic congenital heart disease the coronary arterial circulation. Curr Cardiol Rev. (2012) 8:1–5. doi: 10.2174/157340312801215836
78. Tarp JB, Jensen AS, Engstrom T, Holstein-Rathlou NH, Sondergaard L. Cyanotic congenital heart disease and atherosclerosis. Heart. (2017) 103:897–900. doi: 10.1136/heartjnl-2016-311012
79. Honicky M, Souza JN, Cardoso SM, de Carlos Back I, Vieira FGK, de Fragas Hinnig P, et al. Dietary patterns are associated with central adiposity and carotid intima-media thickness in children and adolescents with congenital heart disease. Eur J Nutr. (2021) 60:4295–306. doi: 10.1007/s00394-021-02586-0
80. Shiina Y, Matsumoto N, Okamura D, Takahashi Y, Kijima Y, Fukuda T, et al. Sarcopenia in adults with congenital heart disease: nutritional status, dietary intake, and resistance training. J Cardiol. (2019) 74:84–9. doi: 10.1016/j.jjcc.2019.01.003
81. Sandberg C, Johansson K, Christersson C, Hlebowicz J, Thilen U, Johansson B. Sarcopenia is common in adults with complex congenital heart disease. Int J Cardiol. (2019) 296:57–62. doi: 10.1016/j.ijcard.2019.06.011
82. Sandberg C, Johansson K, Christersson C, Hlebowicz J, Thilen U, Johansson B. Low bone mineral density in adults with complex congenital heart disease. Int J Cardiol. (2020) 319:62–6. doi: 10.1016/j.ijcard.2020.06.053
83. Mandalenakis Z, Karazisi C, Skoglund K, Rosengren A, Lappas G, Eriksson P, et al. Risk of cancer among children and young adults with congenital heart disease compared with healthy controls. JAMA Netw Open. (2019) 2:e196762. doi: 10.1001/jamanetworkopen.2019.6762
84. Cohen S, Liu A, Gurvitz M, Guo L, Therrien J, Laprise C, et al. Exposure to low-dose ionizing radiation from cardiac procedures and malignancy risk in adults with congenital heart disease. Circulation. (2018) 137:1334–45. doi: 10.1161/circulationaha.117.029138
85. Li M, You L, Xue J, Lu Y. Ionizing radiation-induced cellular senescence in normal, non-transformed cells and the involved DNA damage response: a mini review. Front Pharmacol. (2018) 9:522. doi: 10.3389/fphar.2018.00522
86. Wang Y, Boerma M, Zhou D. Ionizing radiation-induced endothelial cell senescence and cardiovascular diseases. Radiat Res. (2016) 186:153–61. doi: 10.1667/RR14445.1
87. Azimzadeh O, Sievert W, Sarioglu H, Merl-Pham J, Yentrapalli R, Bakshi MV, et al. Integrative proteomics and targeted transcriptomics analyses in cardiac endothelial cells unravel mechanisms of long-term radiation-induced vascular dysfunction. J Proteome Res. (2015) 14:1203–19. doi: 10.1021/pr501141b
88. Nakou ES, Parthenakis FI, Kallergis EM, Marketou ME, Nakos KS, Vardas PE. Healthy aging and myocardium: a complicated process with various effects in cardiac structure and physiology. Int J Cardiol. (2016) 209:167–75. doi: 10.1016/j.ijcard.2016.02.039
89. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, et al. Evidence for cardiomyocyte renewal in humans. Science. (2009) 324:98–102. doi: 10.1126/science.1164680
90. Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res. (1991) 68:1560–8. doi: 10.1161/01.res.68.6.1560
91. Iung B, Baron G, Butchart EG, Delahaye F, Gohlke-Barwolf C, Levang OW, et al. A prospective survey of patients with valvular heart disease in Europe: the Euro heart survey on valvular heart disease. Eur Heart J. (2003) 24:1231–43. doi: 10.1016/s0195-668x(03)00201-x
92. Boyle AJ, Shih H, Hwang J, Ye J, Lee B, Zhang Y, et al. Cardiomyopathy of aging in the mammalian heart is characterized by myocardial hypertrophy, fibrosis and a predisposition towards cardiomyocyte apoptosis and autophagy. Exp Gerontol. (2011) 46:549–59. doi: 10.1016/j.exger.2011.02.010
93. Bachschmid MM, Schildknecht S, Matsui R, Zee R, Haeussler D, Cohen RA, et al. Vascular aging: chronic oxidative stress and impairment of redox signaling-consequences for vascular homeostasis and disease. Ann Med. (2013) 45:17–36. doi: 10.3109/07853890.2011.645498
94. Wu J, Xia S, Kalionis B, Wan W, Sun T. The role of oxidative stress and inflammation in cardiovascular aging. Biomed Res Int. (2014) 2014:615312.
95. Tocchi A, Quarles EK, Basisty N, Gitari L, Rabinovitch PS. Mitochondrial dysfunction in cardiac aging. Biochim Biophys Acta. (2015) 1847:1424–33.
96. Babusikova E, Lehotsky J, Dobrota D, Racay P, Kaplan P. Age-associated changes in Ca(2+)-ATPase and oxidative damage in sarcoplasmic reticulum of rat heart. Physiol Res. (2012) 61:453–60. doi: 10.33549/physiolres.932320
97. Diaz-Villanueva JF, Diaz-Molina R, Garcia-Gonzalez V. Protein folding and mechanisms of proteostasis. Int J Mol Sci. (2015) 16:17193–230. doi: 10.3390/ijms160817193
98. Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem. (2015) 84:435–64. doi: 10.1146/annurev-biochem-060614-033955
99. Wiersma M, Henning RH, Brundel BJ. Derailed proteostasis as a determinant of cardiac aging. Can J Cardiol. (2016) 32:1166.e11–20. doi: 10.1016/j.cjca.2016.03.005
100. Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, et al. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res. (2012) 93:79–88. doi: 10.1093/cvr/cvr274
101. Menghini R, Stohr R, Federici M. MicroRNAs in vascular aging and atherosclerosis. Ageing Res Rev. (2014) 17:68–78. doi: 10.1016/j.arr.2014.03.005
102. Zhang X, Azhar G, Wei JY. The expression of microRNA and microRNA clusters in the aging heart. PLoS One. (2012) 7:e34688. doi: 10.1371/journal.pone.0034688
103. Jazbutyte V, Fiedler J, Kneitz S, Galuppo P, Just A, Holzmann A, et al. MicroRNA-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age (Dordr). (2013) 35:747–62. doi: 10.1007/s11357-012-9407-9
104. Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, et al. MicroRNA-34a regulates cardiac ageing and function. Nature. (2013) 495:107–10. doi: 10.1038/nature11919
105. Anversa P, Rota M, Urbanek K, Hosoda T, Sonnenblick EH, Leri A, et al. Myocardial aging–a stem cell problem. Basic Res Cardiol. (2005) 100:482–93. doi: 10.1007/s00395-005-0554-3
106. Ballard VL, Edelberg JM. Stem cells and the regeneration of the aging cardiovascular system. Circ Res. (2007) 100:1116–27. doi: 10.1161/01.RES.0000261964.19115.e3
107. Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. (2004) 94:514–24. doi: 10.1161/01.RES.0000117306.10142.50
108. Iacobazzi D, Suleiman MS, Ghorbel M, George SJ, Caputo M, Tulloh RM. Cellular and molecular basis of RV hypertrophy in congenital heart disease. Heart. (2016) 102:12–7. doi: 10.1136/heartjnl-2015-308348
109. van der Feen DE, Bartelds B, de Boer RA, Berger RMF. Pulmonary arterial hypertension in congenital heart disease: translational opportunities to study the reversibility of pulmonary vascular disease. Eur Heart J. (2017) 38:2034–41. doi: 10.1093/eurheartj/ehx034
110. van der Feen DE, Bossers GPL, Hagdorn QAJ, Moonen JR, Kurakula K, Szulcek R, et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci Transl Med. (2020) 12:eaaw4974. doi: 10.1126/scitranslmed.aaw4974
111. Chen MS, Lee RT, Garbern JC. Senescence mechanisms and targets in the heart. Cardiovasc Res. (2022) 118:1173–87. doi: 10.1093/cvr/cvab161
112. Cui S, Xue L, Yang F, Dai S, Han Z, Liu K, et al. Postinfarction hearts are protected by premature senescent cardiomyocytes via GATA 4-dependent CCN 1 secretion. J Am Heart Assoc. (2018) 7:e009111. doi: 10.1161/JAHA.118.009111
113. Singampalli KL, Jui E, Shani K, Ning Y, Connell JP, Birla RK, et al. Congenital heart disease: an immunological perspective. Front Cardiovasc Med. (2021) 8:701375. doi: 10.3389/fcvm.2021.701375
114. Jain PN, Robertson M, Lasa JJ, Shekerdemian L, Guffey D, Zhang Y, et al. Altered metabolic and inflammatory transcriptomics after cardiac surgery in neonates with congenital heart disease. Sci Rep. (2021) 11:4965. doi: 10.1038/s41598-021-83882-x
115. Pironkova RP, Giamelli J, Seiden H, Parnell VA, Gruber D, Sison CP, et al. Brain injury with systemic inflammation in newborns with congenital heart disease undergoing heart surgery. Exp Ther Med. (2017) 14:228–38. doi: 10.3892/etm.2017.4493
116. Mou SS, Haudek SB, Lequier L, Pena O, Leonard S, Nikaidoh H, et al. Myocardial inflammatory activation in children with congenital heart disease. Crit Care Med. (2002) 30:827–32. doi: 10.1097/00003246-200204000-00018
117. Soshnick SH, Joseph T, Bennett NJ. Humoral immunodeficiency and immune globulin replacement therapy (IGRT) usage in digeorge syndrome. J Clin Immunol. (2021) 41:1208–12. doi: 10.1007/s10875-021-01012-8
118. Ram G, Chinen J. Infections and immunodeficiency in down syndrome. Clin Exp Immunol. (2011) 164:9–16. doi: 10.1111/j.1365-2249.2011.04335.x
119. Schoepp M, Hannah-Shmouni F, Matta J, Ghanem AM, Hanover JA, Abd-Elmoniem KZ, et al. Coronary calcification in adults with turner syndrome. Genet Med. (2018) 20:664–8. doi: 10.1038/gim.2017.149
120. Sharma R, Bolger AP, Li W, Davlouros PA, Volk HD, Poole-Wilson PA, et al. Elevated circulating levels of inflammatory cytokines and bacterial endotoxin in adults with congenital heart disease. Am J Cardiol. (2003) 92:188–93. doi: 10.1016/s0002-9149(03)00536-8
121. Buchhorn R, Wessel A, Hulpke-Wette M, Bursch J, Werdan K, Loppnow H. Endogenous nitric oxide and soluble tumor necrosis factor receptor levels are enhanced in infants with congenital heart disease. Crit Care Med. (2001) 29:2208–10. doi: 10.1097/00003246-200111000-00026
122. Zhang X, Wang K, Yang Q, Wang J, Xuan C, Liu XC, et al. Acute phase proteins altered in the plasma of patients with congenital ventricular septal defect. Proteomics Clin Appl. (2015) 9:1087–96. doi: 10.1002/prca.201400166
123. Rouatbi H, Farhat N, Heying R, Gerard A, Vazquez-Jimenez JF, Seghaye MC. Right atrial myocardial remodeling in children with atrial septal defect involves inflammation, growth, fibrosis, and apoptosis. Front Pediatr. (2020) 8:40. doi: 10.3389/fped.2020.00040
124. Zorzanelli L, Maeda NY, Clave MM, Aiello VD, Rabinovitch M, Lopes AA. Serum cytokines in young pediatric patients with congenital cardiac shunts and altered pulmonary hemodynamics. Mediators Inflamm. (2016) 2016:7672048. doi: 10.1155/2016/7672048
125. Ramakrishnan S, Kukreti BB, Ramakrishnan L, Salahuddin S, Pendharkar A, Karthikeyan G, et al. Inflammatory markers are elevated in Eisenmenger syndrome. Pediatr Cardiol. (2013) 34:1791–6. doi: 10.1007/s00246-013-0715-3
126. Opotowsky AR, Valente AM, Alshawabkeh L, Cheng S, Bradley A, Rimm EB, et al. Prospective cohort study of C-reactive protein as a predictor of clinical events in adults with congenital heart disease: results of the Boston adult congenital heart disease biobank. Eur Heart J. (2018) 39:3253–61. doi: 10.1093/eurheartj/ehy362
127. Moutafi AC, Alissafi T, Chryssanthopoulos S, Thanopoulos V, Tousoulis D, Stefanadis C, et al. Neurohormones, cytokines, and aortic function in children with repaired coarctation of the aorta. Int J Cardiol. (2014) 172:e26–7. doi: 10.1016/j.ijcard.2013.12.091
128. Rog-Zielinska EA, Norris RA, Kohl P, Markwald R. The living scar–cardiac fibroblasts and the injured heart. Trends Mol Med. (2016) 22:99–114. doi: 10.1016/j.molmed.2015.12.006
129. Hibino N, Mejias D, Pietris N, Dean E, Yi T, Best C, et al. The innate immune system contributes to tissue-engineered vascular graft performance. FASEB J. (2015) 29:2431–8. doi: 10.1096/fj.14-268334
130. Salaun E, Clavel MA, Rodes-Cabau J, Pibarot P. Bioprosthetic aortic valve durability in the era of transcatheter aortic valve implantation. Heart. (2018) 104:1323–32. doi: 10.1136/heartjnl-2017-311582
131. Elder RW, George RP, McCabe NM, Rodriguez FH III, Book WM, Mahle WT, et al. Immunologic aging in adults with congenital heart disease: does infant sternotomy matter? Pediatr Cardiol. (2015) 36:1411–6. doi: 10.1007/s00246-015-1174-9
132. Roosen J, Oosterlinck W, Meyns B. Routine thymectomy in congenital cardiac surgery changes adaptive immunity without clinical relevance. Interact Cardiovasc Thorac Surg. (2015) 20:101–6. doi: 10.1093/icvts/ivu343
133. Boyd R, Parisi F, Kalfa D. State of the art: tissue engineering in congenital heart surgery. Semin Thorac Cardiovasc Surg. (2019) 31:807–17. doi: 10.1053/j.semtcvs.2019.05.023
134. Wienecke LM, Cohen S, Bauersachs J, Mebazaa A, Chousterman BG. Immunity and inflammation: the neglected key players in congenital heart disease? Heart Fail Rev. (2021). [Epub ahead of print]. doi: 10.1007/s10741-021-10187-6
135. Sauce D, Larsen M, Fastenackels S, Duperrier A, Keller M, Grubeck-Loebenstein B, et al. Evidence of premature immune aging in patients thymectomized during early childhood. J Clin Invest. (2009) 119:3070–8. doi: 10.1172/JCI39269
136. Gudmundsdottir J, Oskarsdottir S, Skogberg G, Lindgren S, Lundberg V, Berglund M, et al. Early thymectomy leads to premature immunologic ageing: an 18-year follow-up. J Allergy Clin Immunol. (2016) 138:1439–43.e10. doi: 10.1016/j.jaci.2016.05.014
137. Silva SL, Albuquerque A, Amaral AJ, Li QZ, Mota C, Cheynier R, et al. Autoimmunity and allergy control in adults submitted to complete thymectomy early in infancy. PLoS One. (2017) 12:e0180385. doi: 10.1371/journal.pone.0180385
138. Bronicki RA, Hall M. Cardiopulmonary bypass-induced inflammatory response: pathophysiology and treatment. Pediatr Crit Care Med. (2016) 17:S272–8. doi: 10.1097/PCC.0000000000000759
139. Modi P, Imura H, Caputo M, Pawade A, Parry A, Angelini GD, et al. Cardiopulmonary bypass-induced myocardial reoxygenation injury in pediatric patients with cyanosis. J Thorac Cardiovasc Surg. (2002) 124:1035–6. doi: 10.1067/mtc.2002.122536
140. del Nido PJ, Benson LN, Mickle DA, Kielmanowicz S, Coles JG, Wilson GJ. Impaired left ventricular postischemic function and metabolism in chronic right ventricular hypertrophy. Circulation. (1987) 76:V168–73.
141. Dhaliwal H, Kirshenbaum LA, Randhawa AK, Singal PK. Correlation between antioxidant changes during hypoxia and recovery on reoxygenation. Am J Physiol. (1991) 261:H632–8. doi: 10.1152/ajpheart.1991.261.3.H632
142. Ihnken K, Morita K, Buckberg GD, Sherman MP, Ignarro LJ, Young HH. Studies of hypoxemic/reoxygenation injury: with aortic clamping. XIII. Interaction between oxygen tension and cardioplegic composition in limiting nitric oxide production and oxidant damage. J Thorac Cardiovasc Surg. (1995) 110:1274–86. doi: 10.1016/s0022-5223(95)70014-5
143. Tomic V, Russwurm S, Moller E, Claus RA, Blaess M, Brunkhorst F, et al. Transcriptomic and proteomic patterns of systemic inflammation in on-pump and off-pump coronary artery bypass grafting. Circulation. (2005) 112:2912–20. doi: 10.1161/circulationaha.104.531152
144. Liangos O, Domhan S, Schwager C, Zeier M, Huber PE, Addabbo F, et al. Whole blood transcriptomics in cardiac surgery identifies a gene regulatory network connecting ischemia reperfusion with systemic inflammation. PLoS One. (2010) 5:e13658. doi: 10.1371/journal.pone.0013658
145. Li YP, Huang J, Huang SG, Xu YG, Xu YY, Liao JY, et al. The compromised inflammatory response to bacterial components after pediatric cardiac surgery is associated with cardiopulmonary bypass-suppressed Toll-like receptor signal transduction pathways. J Crit Care. (2014) 29:312.e7–13. doi: 10.1016/j.jcrc.2013.10.008
146. Yang Q, Liao J, Huang J, Li YP, Huang S, Zhou H, et al. Cardiopulmonary bypass down-regulates NOD signaling and inflammatory response in children with congenital heart disease. PLoS One. (2016) 11:e0162179. doi: 10.1371/journal.pone.0162179
147. Avolio E, Mangialardi G, Slater SC, Alvino VV, Gu Y, Cathery W, et al. Secreted protein acidic and cysteine rich matricellular protein is enriched in the bioactive fraction of the human vascular pericyte secretome. Antioxid Redox Signal. (2021) 34:1151–64. doi: 10.1089/ars.2019.7969
148. Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. (2007) 74:184–95. doi: 10.1016/j.cardiores.2006.10.002
149. Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia commonwealth university anakinra remodeling trial [VCU-ART] pilot study). Am J Cardiol. (2010) 105:1371–7.e1. doi: 10.1016/j.amjcard.2009.12.059
150. Ichiryu N, Fairchild PJ. Immune privilege of stem cells. Methods Mol Biol. (2013) 1029:1–16. doi: 10.1007/978-1-62703-478-4_1
151. Liu X, Li W, Fu X, Xu Y. The immunogenicity and immune tolerance of pluripotent stem cell derivatives. Front Immunol. (2017) 8:645. doi: 10.3389/fimmu.2017.00645
152. Weiss ARR, Dahlke MH. Immunomodulation by mesenchymal stem cells (MSCs): mechanisms of action of living, apoptotic, and dead MSCs. Front Immunol. (2019) 10:1191. doi: 10.3389/fimmu.2019.01191
153. Avolio E, Rodriguez-Arabaolaza I, Spencer HL, Riu F, Mangialardi G, Slater SC, et al. Expansion and characterization of neonatal cardiac pericytes provides a novel cellular option for tissue engineering in congenital heart disease. J Am Heart Assoc. (2015) 4:e002043. doi: 10.1161/JAHA.115.002043
154. Avolio E, Gianfranceschi G, Cesselli D, Caragnano A, Athanasakis E, Katare R, et al. Ex vivo molecular rejuvenation improves the therapeutic activity of senescent human cardiac stem cells in a mouse model of myocardial infarction. Stem Cells. (2014) 32:2373–85. doi: 10.1002/stem.1728
155. Kawai Y, Tohyama S, Arai K, Tamura T, Soma Y, Fukuda K, et al. Scaffold-free tubular engineered heart tissue from human induced pluripotent stem cells using bio-3D printing technology in vivo. Front Cardiovasc Med. (2021) 8:806215. doi: 10.3389/fcvm.2021.806215
156. Rapetto F, Taliotis D, Chen Q, Ttofi I, Iacobazzi D, Madeddu P, et al. Allogeneic mesenchymal stromal cell injection to alleviate ischemic heart failure following arterial switch operation. JACC Case Rep. (2021) 3:724–7. doi: 10.1016/j.jaccas.2021.02.039
157. Xia WF, Liu Y, Zhou QS, Tang QZ, Zou HD. Comparison of the effects of propofol and midazolam on inflammation and oxidase stress in children with congenital heart disease undergoing cardiac surgery. Yonsei Med J. (2011) 52:326–32. doi: 10.3349/ymj.2011.52.2.326
158. Caputo M, Mokhtari A, Miceli A, Ghorbel MT, Angelini GD, Parry AJ, et al. Controlled reoxygenation during cardiopulmonary bypass decreases markers of organ damage, inflammation, and oxidative stress in single-ventricle patients undergoing pediatric heart surgery. J Thorac Cardiovasc Surg. (2014) 148:792–801.e8. doi: 10.1016/j.jtcvs.2014.06.001
159. Ghorbel MT, Mokhtari A, Sheikh M, Angelini GD, Caputo M. Controlled reoxygenation cardiopulmonary bypass is associated with reduced transcriptomic changes in cyanotic tetralogy of Fallot patients undergoing surgery. Physiol Genomics. (2012) 44:1098–106. doi: 10.1152/physiolgenomics.00072.2012
160. Caputo M, Bays S, Rogers CA, Pawade A, Parry AJ, Suleiman S, et al. Randomized comparison between normothermic and hypothermic cardiopulmonary bypass in pediatric open-heart surgery. Ann Thorac Surg. (2005) 80:982–8. doi: 10.1016/j.athoracsur.2005.03.062
161. Durandy Y, Hulin S. Intermittent warm blood cardioplegia in the surgical treatment of congenital heart disease: clinical experience with 1400 cases. J Thorac Cardiovasc Surg. (2007) 133:241–6. doi: 10.1016/j.jtcvs.2006.10.004
162. Owens WA, Walaszczyk A, Spyridopoulos I, Dookun E, Richardson GD. Senescence and senolytics in cardiovascular disease: promise and potential pitfalls. Mech Ageing Dev. (2021) 198:111540. doi: 10.1016/j.mad.2021.111540
163. Botto LD, Mulinare J, Erickson JD. Occurrence of congenital heart defects in relation to maternal mulitivitamin use. Am J Epidemiol. (2000) 151:878–84. doi: 10.1093/oxfordjournals.aje.a010291
164. Mao B, Qiu J, Zhao N, Shao Y, Dai W, He X, et al. Maternal folic acid supplementation and dietary folate intake and congenital heart defects. PLoS One. (2017) 12:e0187996. doi: 10.1371/journal.pone.0187996
165. Feng Y, Wang S, Chen R, Tong X, Wu Z, Mo X. Maternal folic acid supplementation and the risk of congenital heart defects in offspring: a meta-analysis of epidemiological observational studies. Sci Rep. (2015) 5:8506. doi: 10.1038/srep08506
166. Ionescu-Ittu R, Marelli AJ, Mackie AS, Pilote L. Prevalence of severe congenital heart disease after folic acid fortification of grain products: time trend analysis in Quebec, Canada. BMJ. (2009) 338:b1673. doi: 10.1136/bmj.b1673
167. Oyen N, Olsen SF, Basit S, Leirgul E, Strom M, Carstensen L, et al. Association between maternal folic acid supplementation and congenital heart defects in offspring in birth cohorts from Denmark and Norway. J Am Heart Assoc. (2019) 8:e011615. doi: 10.1161/JAHA.118.011615
168. McNally JD, O’Hearn K, Fergusson DA, Lougheed J, Doherty DR, Maharajh G, et al. Prevention of post-cardiac surgery vitamin D deficiency in children with congenital heart disease: a pilot feasibility dose evaluation randomized controlled trial. Pilot Feasibility Stud. (2020) 6:159. doi: 10.1186/s40814-020-00700-3
169. Graham EM, Taylor SN, Zyblewski SC, Wolf B, Bradley SM, Hollis BW, et al. Vitamin D status in neonates undergoing cardiac operations: relationship to cardiopulmonary bypass and association with outcomes. J Pediatr. (2013) 162:823–6. doi: 10.1016/j.jpeds.2012.10.013
170. McNally JD, Menon K, Chakraborty P, Fisher L, Williams KA, Al-Dirbashi OY, et al. Impact of anesthesia and surgery for congenital heart disease on the vitamin D status of infants and children: a prospective longitudinal study. Anesthesiology. (2013) 119:71–80. doi: 10.1097/ALN.0b013e31828ce817
171. Dohain AM, Almogati J, Al-Radi OO, Elassal AA, Zaher ZF, Fatani TH, et al. Serum vitamin D status following pediatric cardiac surgery and association with clinical outcome. Eur J Pediatr. (2020) 179:635–43. doi: 10.1007/s00431-019-03538-x
172. Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. (2011) 96:53–8. doi: 10.1210/jc.2010-2704
173. Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. (2010) 107:1071–82. doi: 10.1161/CIRCRESAHA.110.227819
174. Shi H, O’Reilly VC, Moreau JL, Bewes TR, Yam MX, Chapman BE, et al. Gestational stress induces the unfolded protein response, resulting in heart defects. Development. (2016) 143:2561–72. doi: 10.1242/dev.136820
175. Jian Z, Li JB, Ma RY, Chen L, Wang XF, Xiao YB. Pivotal role of activating transcription factor 6alpha in myocardial adaptation to chronic hypoxia. Int J Biochem Cell Biol. (2012) 44:972–9. doi: 10.1016/j.biocel.2012.03.004
176. Zhou Y, Pang B, Xiao Y, Zhou S, He B, Zhang F, et al. The protective microRNA-199a-5p-mediated unfolded protein response in hypoxic cardiomyocytes is regulated by STAT3 pathway. J Physiol Biochem. (2019) 75:73–81. doi: 10.1007/s13105-018-0657-6
177. Zhou Y, Jia WK, Jian Z, Zhao L, Liu CC, Wang Y, et al. Downregulation of microRNA199a5p protects cardiomyocytes in cyanotic congenital heart disease by attenuating endoplasmic reticulum stress. Mol Med Rep. (2017) 16:2992–3000. doi: 10.3892/mmr.2017.6934
178. Henning RH, Brundel B. Proteostasis in cardiac health and disease. Nat Rev Cardiol. (2017) 14:637–53. doi: 10.1038/nrcardio.2017.89
179. Wang J, Chen L, Wen S, Zhu H, Yu W, Moskowitz IP, et al. Defective sumoylation pathway directs congenital heart disease. Birth Defects Res A Clin Mol Teratol. (2011) 91:468–76. doi: 10.1002/bdra.20816
180. Kim EY, Chen L, Ma Y, Yu W, Chang J, Moskowitz IP, et al. Enhanced desumoylation in murine hearts by overexpressed SENP2 leads to congenital heart defects and cardiac dysfunction. J Mol Cell Cardiol. (2012) 52:638–49. doi: 10.1016/j.yjmcc.2011.11.011
181. Ong SB, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res. (2010) 88:16–29. doi: 10.1093/cvr/cvq237
182. Bordi M, Darji S, Sato Y, Mellen M, Berg MJ, Kumar A, et al. mTOR hyperactivation in Down syndrome underlies deficits in autophagy induction, autophagosome formation, and mitophagy. Cell Death Dis. (2019) 10:563. doi: 10.1038/s41419-019-1752-5
183. Venegas-Zamora L, Bravo-Acuna F, Sigcho F, Gomez W, Bustamante-Salazar J, Pedrozo Z, et al. New molecular and organelle alterations linked to down syndrome heart disease. Front Genet. (2021) 12:792231. doi: 10.3389/fgene.2021.792231
184. Blackwood EA, Azizi K, Thuerauf DJ, Paxman RJ, Plate L, Kelly JW, et al. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat Commun. (2019) 10:187. doi: 10.1038/s41467-018-08129-2
185. Hofmann C, Katus HA, Doroudgar S. Protein misfolding in cardiac disease. Circulation. (2019) 139:2085–8. doi: 10.1161/circulationaha.118.037417
186. Drose S, Altendorf K. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J Exp Biol. (1997) 200:1–8. doi: 10.1242/jeb.200.1.1
187. Magenta A, Lorde R, Syed SB, Capogrossi MC, Puca A, Madeddu P. Molecular therapies delaying cardiovascular aging: disease- or health-oriented approaches. Vasc Biol. (2020) 2:R45–58. doi: 10.1530/VB-19-0029
188. Villa F, Carrizzo A, Spinelli CC, Ferrario A, Malovini A, Maciag A, et al. Genetic analysis reveals a longevity-associated protein modulating endothelial function and angiogenesis. Circ Res. (2015) 117:333–45. doi: 10.1161/CIRCRESAHA.117.305875
189. Puca AA, Carrizzo A, Spinelli C, Damato A, Ambrosio M, Villa F, et al. Single systemic transfer of a human gene associated with exceptional longevity halts the progression of atherosclerosis and inflammation in ApoE knockout mice through a CXCR4-mediated mechanism. Eur Heart J. (2020) 41:2487–97. doi: 10.1093/eurheartj/ehz459
190. Malavolta M, Dato S, Villa F, Rango F, Iannone F, Ferrario A, et al. LAV-BPIFB4 associates with reduced frailty in humans and its transfer prevents frailty progression in old mice. Aging (Albany NY). (2019) 11:6555–68. doi: 10.18632/aging.102209
191. Dang Z, Avolio E, Thomas AC, Faulkner A, Beltrami AP, Cervellin C, et al. Transfer of a human gene variant associated with exceptional longevity improves cardiac function in obese type 2 diabetic mice through induction of the SDF-1/CXCR4 signalling pathway. Eur J Heart Fail. (2020) 22:1568–81. doi: 10.1002/ejhf.1840
192. Ciaglia E, Lopardo V, Montella F, Carrizzo A, Di Pietro P, Malavolta M, et al. Transfer of the longevity-associated variant of BPIFB4 gene rejuvenates immune system and vasculature by a reduction of CD38(+) macrophages and NAD(+) decline. Cell Death Dis. (2022) 13:86. doi: 10.1038/s41419-022-04535-z
193. Villa F, Malovini A, Carrizzo A, Spinelli CC, Ferrario A, Maciag A, et al. Serum BPIFB4 levels classify health status in long-living individuals. Immun Ageing. (2015) 12:27. doi: 10.1186/s12979-015-0054-8
194. Li T, Lu D, Yao C, Li T, Dong H, Li Z, et al. Kansl1 haploinsufficiency impairs autophagosome-lysosome fusion and links autophagic dysfunction with Koolen-de Vries syndrome in mice. Nat Commun. (2022) 13:931.
196. Tatarkova Z, Kuka S, Racay P, Lehotsky J, Dobrota D, Mistuna D, et al. Effects of aging on activities of mitochondrial electron transport chain complexes and oxidative damage in rat heart. Physiol Res. (2011) 60:281–9. doi: 10.33549/physiolres.932019
197. Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. (1994) 91:10771–8. doi: 10.1073/pnas.91.23.10771
198. Lopez-Lluch G, Irusta PM, Navas P, de Cabo R. Mitochondrial biogenesis and healthy aging. Exp Gerontol. (2008) 43:813–9. doi: 10.1016/j.exger.2008.06.014
199. Zhao Q, Sun Q, Zhou L, Liu K, Jiao K. Complex regulation of mitochondrial function during cardiac development. J Am Heart Assoc. (2019) 8:e012731. doi: 10.1161/JAHA.119.012731
200. Ingraham CA, Burwell LS, Skalska J, Brookes PS, Howell RL, Sheu SS, et al. NDUFS4: creation of a mouse model mimicking a complex I disorder. Mitochondrion. (2009) 9:204–10. doi: 10.1016/j.mito.2009.02.001
201. Mohammed S, Bahitham W, Chan A, Chiu B, Bamforth F, Sergi C. Mitochondrial DNA related cardiomyopathies. Front Biosci (Elite Ed). (2012) 4:1706–16. doi: 10.2741/491
202. Schiff M, de Baulny HO, Lombes A. Neonatal cardiomyopathies and metabolic crises due to oxidative phosphorylation defects. Semin Fetal Neonatal Med. (2011) 16:216–21. doi: 10.1016/j.siny.2011.04.002
203. Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW II. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. (2015) 350:aad2459. doi: 10.1126/science.aad2459
204. Lai L, Leone T, Zechner C, Schaeffer P, Kelly S, Flanagan D, et al. Transcriptional coactivators PGC-1 alpha and PGC-I beta control overlapping programs required for cardiac mitochondrial maturation. Circ Res. (2008) 103:E56–56.
205. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. (2007) 356:1140–51. doi: 10.1056/NEJMra063052
206. Jullig M, Hickey AJ, Chai CC, Skea GL, Middleditch MJ, Costa S, et al. Is the failing heart out of fuel or a worn engine running rich? A study of mitochondria in old spontaneously hypertensive rats. Proteomics. (2008) 8:2556–72. doi: 10.1002/pmic.200700977
207. Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. (2010) 106:1541–8. doi: 10.1161/CIRCRESAHA.109.212753
208. Karamanlidis G, Bautista-Hernandez V, Fynn-Thompson F, Del Nido P, Tian R. Impaired mitochondrial biogenesis precedes heart failure in right ventricular hypertrophy in congenital heart disease. Circ Heart Fail. (2011) 4:707–13. doi: 10.1161/circheartfailure.111.961474
209. Hwang HV, Sandeep N, Paige SL, Ranjbarvaziri S, Hu DQ, Zhao M, et al. 4HNE impairs myocardial bioenergetics in congenital heart disease-induced right ventricular failure. Circulation. (2020) 142:1667–83. doi: 10.1161/CIRCULATIONAHA.120.045470
210. Zhang D, Li Y, Heims-Waldron D, Bezzerides V, Guatimosim S, Guo Y, et al. Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ Res. (2018) 122:74–87. doi: 10.1161/CIRCRESAHA.117.311349
211. Matam K, Shaik NA, Aggarwal S, Diwale S, Banaganapalli B, Al-Aama JY, et al. Evidence for the presence of somatic mitochondrial DNA mutations in right atrial appendage tissues of coronary artery disease patients. Mol Genet Genomics. (2014) 289:533–40. doi: 10.1007/s00438-014-0828-2
212. Mitrofanov KY, Zhelankin AV, Shiganova GM, Sazonova MA, Bobryshev YV, Postnov AY, et al. Analysis of mitochondrial DNA heteroplasmic mutations A1555G, C3256T, T3336C, capital ES, Cyrillic5178capital A, Cyrillic, G12315A, G13513A, G14459A, G14846capital A, Cyrillic and G15059A in CHD patients with the history of myocardial infarction. Exp Mol Pathol. (2016) 100:87–91. doi: 10.1016/j.yexmp.2015.12.003
213. Sobenin IA, Sazonova MA, Ivanova MM, Zhelankin AV, Myasoedova VA, Postnov AY, et al. Mutation C3256T of mitochondrial genome in white blood cells: novel genetic marker of atherosclerosis and coronary heart disease. PLoS One. (2012) 7:e46573. doi: 10.1371/journal.pone.0046573
214. Zhang ZX, Liu MY, He JS, Zhang XT, Chen YH, Li H. Maternally inherited coronary heart disease is associated with a novel mitochondrial tRNA mutation. BMC Cardiovasc Disord. (2019) 19:293. doi: 10.1186/s12872-019-01284-4
215. Finsterer J, Stollberger C. Cardiac manifestations of mitochondrial disorders. Eur J Heart Fail. (2010) 12:637. doi: 10.1093/eurjhf/hfq046
216. Meyers DE, Basha HI, Koenig MK. Cardiac manifestations of mitochondrial disorders. Tex Heart Inst J. (2013) 40:635–6.
217. Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. (2017) 14:238–50. doi: 10.1038/nrcardio.2016.203
218. Xu XX, Lin JHI, Bais AS, Reynolds MJ, Tan TT, Gabriel GC, et al. Mitochondrial respiration defects in single-ventricle congenital heart disease. Front Cardiovasc Med. (2021) 8:734388. doi: 10.3389/fcvm.2021.734388
219. Emani SM, McCully JD. Mitochondrial transplantation: applications for pediatric patients with congenital heart disease. Transl Pediatr. (2018) 7:169–75. doi: 10.21037/tp.2018.02.02
220. Emani SM, Piekarski BL, Harrild D, Del Nido PJ, McCully JD. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J Thorac Cardiovasc Surg. (2017) 154:286–9. doi: 10.1016/j.jtcvs.2017.02.018
221. Guariento A, Piekarski BL, Doulamis IP, Blitzer D, Ferraro AM, Harrild DM, et al. Autologous mitochondrial transplantation for cardiogenic shock in pediatric patients following ischemia-reperfusion injury. J Thorac Cardiovasc Surg. (2021) 162:992–1001. doi: 10.1016/j.jtcvs.2020.10.151
222. Triedman JK, Newburger JW. Trends in congenital heart disease: the next decade. Circulation. (2016) 133:2716–33. doi: 10.1161/CIRCULATIONAHA.116.023544
223. Moons P, Marelli A. Born to age: when adult congenital heart disease converges with geroscience. JACC Adv. (2022) 1:100012. doi: 10.1016/j.jacadv.2022.100012
224. Mackie AS, Rempel GR, Kovacs AH, Kaufman M, Rankin KN, Jelen A, et al. Transition intervention for adolescents with congenital heart disease. J Am Coll Cardiol. (2018) 71:1768–77.
225. Brida M, Gatzoulis MA. Adult congenital heart disease: past, present and future. Acta Paediatr. (2019) 108:1757–64. doi: 10.1111/apa.14921
226. Stout KK, Daniels CJ, Aboulhosn JA, Bozkurt B, Broberg CS, Colman JM, et al. 2018 AHA/ACC Guideline for the management of adults with congenital heart disease: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. J Am Coll Cardiol. (2019) 73:e81–192.
227. Chessa M, Baumgartner H, Michel-Behnke I, Berger F, Budts W, Eicken A, et al. ESC working group position paper: transcatheter adult congenital heart disease interventions: organization of care - recommendations from a joint working group of the European society of cardiology (ESC), European association of pediatric and congenital cardiology (AEPC), and the European association of percutaneous cardiac intervention (EAPCI). Eur Heart J. (2019) 40:1043–8. doi: 10.1093/eurheartj/ehy676
228. Diller GP, Kempny A, Babu-Narayan SV, Henrichs M, Brida M, Uebing A, et al. Machine learning algorithms estimating prognosis and guiding therapy in adult congenital heart disease: data from a single tertiary centre including 10 019 patients. Eur Heart J. (2019) 40:1069–77. doi: 10.1093/eurheartj/ehy915
Keywords: aging, congenital defect, proteostasis, inflammation, surgery, extracorporeal bypass
Citation: Iacobazzi D, Alvino VV, Caputo M and Madeddu P (2022) Accelerated Cardiac Aging in Patients With Congenital Heart Disease. Front. Cardiovasc. Med. 9:892861. doi: 10.3389/fcvm.2022.892861
Received: 09 March 2022; Accepted: 11 April 2022;
Published: 26 May 2022.
Edited by:
Antonio Paolo Beltrami, University of Udine, ItalyReviewed by:
Cecilia Vecoli, National Research Council (CNR), ItalyCopyright © 2022 Iacobazzi, Alvino, Caputo and Madeddu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Madeddu, cGFvbG8ubWFkZWRkdUBicmlzdG9sLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.