 Yongfei Song
Yongfei Song Zequn Zheng
Zequn Zheng Jiangfang Lian
Jiangfang Lian
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 13 May 2022
Sec. Cardiac Rhythmology
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.889519
From carrying potentially pathogenic genes to severe clinical phenotypes, the basic research in the inherited cardiac ion channel disease such as long QT syndrome (LQTS) has been a significant challenge in explaining gene-phenotype heterogeneity. These have opened up new pathways following the parallel development and successful application of stem cell and genome editing technologies. Stem cell-derived cardiomyocytes and subsequent genome editing have allowed researchers to introduce desired genes into cells in a dish to replicate the disease features of LQTS or replace causative genes to normalize the cellular phenotype. Importantly, this has made it possible to elucidate potential genetic modifiers contributing to clinical heterogeneity and hierarchically manage newly identified variants of uncertain significance (VUS) and more therapeutic options to be tested in vitro. In this paper, we focus on and summarize the recent advanced application of human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) combined with clustered regularly interspaced short palindromic repeats/CRISPR-associated system 9 (CRISPR/Cas9) in the interpretation for the gene-phenotype relationship of the common LQTS and presence challenges, increasing our understanding of the effects of mutations and the physiopathological mechanisms in the field of cardiac arrhythmias.
Long QT syndrome (LQTS) is an inherited cardiac channelopathy (ICCs) associated with prolonged cardiac repolarization caused by functional changes in cardiac ion channels. It is characterized by the prolongation of QT interval and fatal arrhythmias (1). Causative mutations with autosomal dominant inheritance are associated with congenital long QT syndrome in at least 1:2,000 live births, with type 1 LQTS (LQT1) to type 3 LQTS (LQT3) accounting for ~75% of clinically defined cases. KCNQ1, KCNH2, and SCN5A encode ion channels Kv7.1, Kv11.1, and Nav1.5 to cause LQT1, LQT2, and LQT3, respectively (2–5). Single nucleotide variants (SNVs) occurring in each gene to cause loss-of-function (LOF) (Kv7.1, Kv11.1) and gain-of-function (Nav1.5) to abnormal ionic currents in each channel are thought to underlie the pathogenesis of these ICCs (6).
The pillars of current therapy include the following: β-blockers (propranolol and nadolol), left cardiac sympathetic denervation, and the automatic implantable cardioverter-defibrillator (7). Various limitations, including drug intolerance, poor compliance, and high complications of invasive strategies, urgently call for safer and more effective drug administration (8). From correction of pathogenic phenotypes in the laboratory to clinical translation, an available treatment always requires robust disease models that ideally replicate the pathophysiological features of the patient without gross deviations.
Unfortunately, the search for pathogenesis and effective therapeutic strategies for ICC is limited mainly by the availability of disease models; while human primary cardiomyocytes would be an ideal option, their limited provenance, non-replication, and poor consistency hinder this choice. In desperation, animal models and various modified immortalized tool cells are always alternatives. Overexpression of mutant channels in heterologous expression systems, such as Xenopus oocytes, human embryonic kidney (HEK) cells, and Chinese hamster ovary (CHO) cells, has been used in most functional studies of specific mutations associated with LQTS. One significant limit of these models is that they lack critical cardiac ion channel macromolecular complex components that may be required to recapitulate the exact molecular and electrophysiological phenotype associated with the mutation (9, 10).
The rise of the human-induced pluripotent stem (hiPSC) technology and improvements in hiPSC directed differentiation schemes with CRISPR/Cas9 gene-editing tools have established powerful approaches for ICC-related research, human disease modeling, and drug development or screening (9). Cardiomyocytes generated by disease-specific hiPSC-induced differentiation called hiPSC-CMs are expected to retain a gene-specific clinical phenotype in vitro to better model disease (11). Furthermore, CRISPR/Cas9 enables us to create any variant we desire in hiPSC in a dish to generate isogenic cardiomyocyte lines. These isogenic hiPSC-CMs are generating new study ideas for SNV in LQTS, particularly for those classified as variations of unknown significance (VUS) that haven't been clinically defined and characterized in the lab, or even pathogenic variants to investigate the influence of modifier genes (7, 12, 13). More importantly, through the continuous development of improved CRISPR/Cas9, building on the successful modeling of hiPSC-CMs, we are able to use it to introduce or correct disease-causing mutations at a much smaller cost to the cell (14–16). In this paper, we will discuss how these technologies can be used to elucidate common LQTS-related gene phenotypes in a single dish and strategies for doing so, current achievements, and future challenges.
Before developing hiPSC-CMs, it was useful to use immortalized cell lines overexpressing specific ion channels or transgenic mouse models to replicate LQTS in vitro and in vivo (10). However, the limitations are pretty obvious. Firstly, these cells cannot replicate the physiological environment of primary cardiomyocytes, which could result in false positives or negatives. For example, to model LQT2, the expression of the KCNH2 gene encoding a potassium channel in HEK-293 cells alone seems less than ideal for its intracellular maturation and supracellular membrane function. The presentation of the hERG channel's full physiological state requires the auxiliary subunit minK to form a stable multimer, which is lacking in a heterologous expression system (17, 18). Co-expression of them in HEK-293 cells, on the other hand, requires consideration of the operation strategy of the researcher, such as differences in transfection ratios and their sensitivity to cardiotoxic drugs when present alone or together in a physiological context (19).
As for animal models, the implications of species differences in inconsistency in cardiac electrical activity need to be considered. Although ion channels are highly conserved in humans and mice, the substantial differences in gene expression profiles and physiology between species severely limit the validity of extrapolating human data from rodents. Such differences are more pronounced in mice, as evidenced by higher heart-rate differences and repolarization performance (20). And the low level of expression of channels mediating repolarizing currents limits the use of models in knockdown expression. Because of species differences in cardiac electrical function features, these different models do not accurately represent all aspects of the human disease (20).
hiPSC-CMs express a variety of ion currents essential for the heart, such as INa, ICaL, Ito, IKr, and IKs (21). Although still immature, mainly reflected by the low expression of IK1 and the high expression of the pacemaker current, they are important for forming resting potentials, notably for the action potential (AP) generation and profile (22, 23). The fetal-like myocardial function may impair the accurate modeling of adult LQTS. For example, low expression of IK1 makes hiPSC-CMs dependent on IKr to obtain the maximum diastolic potential (MDP), which is markedly depolarized with IKr blockers, negatively affecting LQT2 disease replication and screening for hERG channel toxicity (23–26). These deficiencies are not negligible for the study of LQTS. Multiple cues to promote the maturation of hiPSC-CMs, including prolonged culture time, biophysical stimulation, and 3D culture, also help to address this (23). 3D Cardiac tissue shows longer AP duration (APD), hyperpolarized resting membrane potential, and faster upward velocity, with sodium current density and upward velocity similar to human ventricular tissue (27).
Circumventing the ethical limitations of hESCs, reprogrammed pluripotent stem cells from somatic cells of LQTS patients are induced by four specific transcription factors (28–30). Transcription factors are mainly delivered into any cell type with proliferative potential by viral vector-based methods (retroviruses, lentiviruses, and inducible viruses) and non-viral vector methods (plasmids or linear DNA and transposons) (31). Initially, skin dermal fibroblasts were used, followed by non-invasive peripheral blood cells, including T and B cells, and even uroepithelial cells (32, 33). Retroviruses and lentiviruses, as delivery vectors, raise concerns about the risk of insertional mutations and even karyotypic abnormalities (34–36). The use of non-integrating viruses (e.g., Sendai virus), free vectors, or the formation of gene integration-free hiPSC via direct delivery of reprogramming factors (e.g., proteins or mRNA) has emerged as an excellent option (37–39) (Figure 1). Of course, each approach has its own set of benefits and drawbacks. And no reprogramming delivery mechanism has ever been modified without encountering severe restrictions or unintended consequences (40).
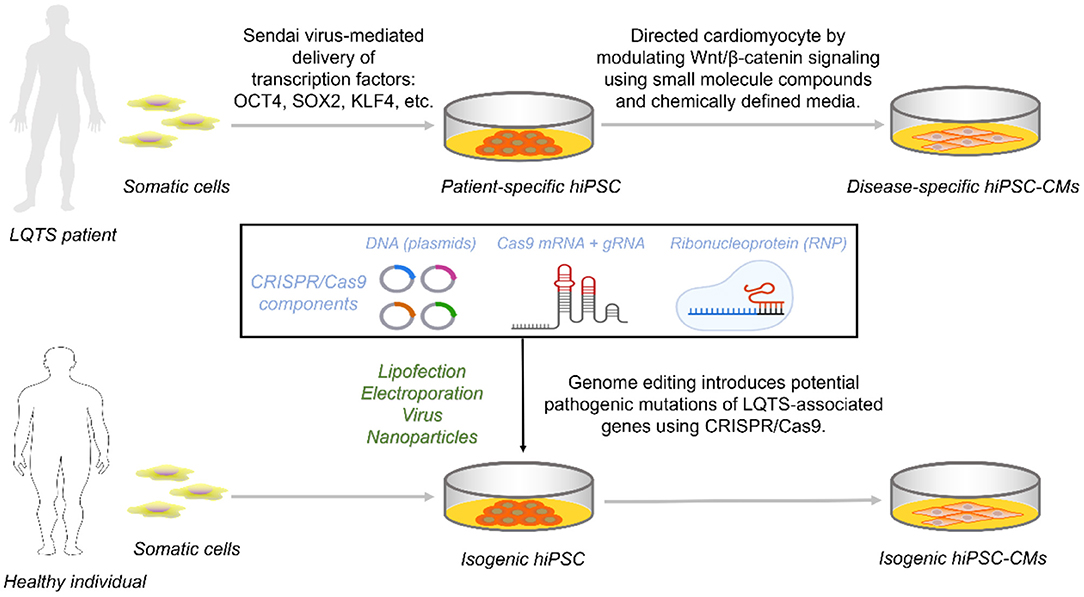
Figure 1. Access to disease-specific hiPSC-CMs from LQTS patients and isogenic cell lines by CRISPR/Cas9. Reprogramming somatic cells from healthy individuals or patients with transcription factors generates hiPSCs that are further targeted to inhibit Wnt/β-catenin capable of directed differentiation into hiPSC-CMs carrying the genetic information. For hiPSCs from healthy individuals, constructing an isogenic set as control requires the introduction of one of the three CRISPR/Cas9 components into the cell via a different vector.
The development of hiPSC-CMs is designed to mimic the human mesoderm and heart development process (41). Briefly, TGF-β activates the typical Wnt/β-catenin pathway to signal initiation and subsequent mesoderm formation and is inhibited late in the cardiac lineage (42). Chemically defined media containing Wnt inhibitors promote mesoderm formation capable of efficient formation of cardiomyocytes (43, 44) (Figure 1).
LQTS individual-derived hiPSCs are directionally differentiated and retain their genetic background to form disease-specific hiPSC-CMs, giving most experimental platforms a novel approach to ICCs research. LQT1 was the first channelopathy to be modeled using hiPSC-CM. This study derived hiPSCs from two LQT1 patients carrying the genetic heterozygous missense mutation KCNQ1-R190Q. LQT1 hiPSC-CMs summarize the distinctive features of LQT1, including a reduction in AP repolarisation currents IKs, leading to a prolongation of APD and a channel protein transport defect that exhibits a dominant-negative effect (45). Although it is not always possible to enroll LQTS patients in some laboratories, the development of CRISPR/Cas9 has alleviated this problem. Researchers can alter the genetic information of normal people or LQTS patients nearly at will by modifying the genome of hiPSC with CRISPR/Cas9 (46) (Figure 1). And the reproducibility of such genomic modifications in hiPSC is far superior to the manipulations performed in terminally differentiated hiPSC-CMs.
The patch-clamp technique is the gold standard for measuring various AP parameters from hiPSC-CM. They chiefly include resting membrane potential (RMP), MDP, and APD at different repolarization percentage levels (i.e., APD20, APD50, and APD90) that correspond to specific ion current characteristics (21). Researchers have focused more on APD for LQTS since APD90 is most dependent on IKs and IKr (9). Using a dynamic clamp characterized by the real-time evaluation and injection of simulated membrane current, the injection of IK1 into hiPSC-CMs to achieve a close-to-physiological RMP minimizes the LQTS model's inaccuracy (47–50).
Compared to the patch-clamp, no other technique can isolate individual sodium or potassium channels. However, this low-throughput and relatively complex technique is insufficient for academic and industrial laboratories. Despite the lower resolution, a high-throughput multi-electrode array (MEA) can obtain ex vivo cardiac field potential durations (FPD) correlated with QT interval characteristics in ECG and aid in the analysis of normal or prolonged APD (51–57).
Abnormal electrical activity, such as decreased Ikr in LQT2 and Iks in LQT1, increased late sodium currents (INaL) in LQT3, prolonged APD and FPD, and the occurrences of after early depolarizations (EADs), can be detected in the laboratory using these techniques in disease-specific or CRISPR/Cas9 editing-based LQTS models (Table 1). Different LQTS-associated SNVs can be assessed for drug response in hiPSC-CM using high-throughput electrical measurements and simple gene editing (53, 54, 108, 109). Moreover, optical evaluations by mapping the hiPSC-CMs with genetically encoded voltage and fluorescent calcium indicators fulfill the potential for studying conduction and arrhythmogenesis (110, 111). They are progressively identifying intracellular calcium transient abnormalities as an important mechanism of abnormal phenotypic in LQTS, which could lead to new therapeutic options (8).
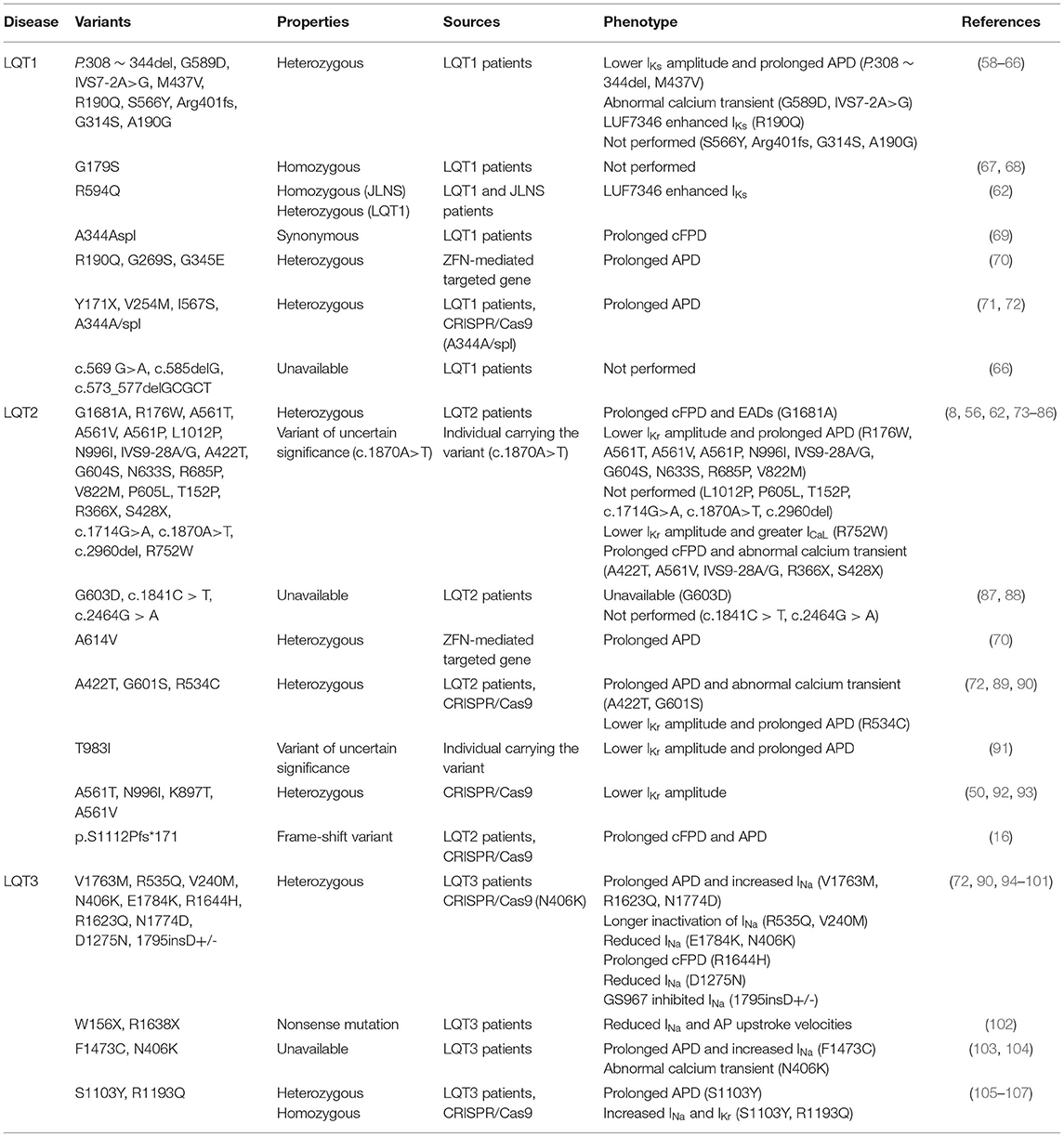
Table 1. Disease-specific and CRISPR/Cas9-edited LQTS-associated variants characterized in hiPSC-CMs.
Earlier, various single-gene modification efforts were performed in hiPSC using genome editing techniques such as zinc finger nuclease (ZFN) and transcription activator-like effector nuclease (TALEN) (112). ZFN-driven transgene addition of KCNQ1 and KCNH2 has successfully modeled the disease (70). Unlike ZFN and TALEN, which use interactions between amino acid residues and nucleotides to identify DNA target sites and induce double-strand breaks (DSBs), CRISPR/Cas9 uses absolutely specific nucleotide-nucleotide base pairing to make it a more efficient and precise gene-editing tool (113, 114). Genome editing based on CRISPR/Cas9 has undoubtedly become the primary means of modifying the expression of specific genes at the hiPSC. Off-target mutations in hiPSCs may be sufficiently low to be a non-issue in disease modeling and other applications (115).
The CRISPR/Cas9 system consists of two components, CRISPR and Cas9. Two repair mechanisms of Cas9-induced DNA DSBs, non-homologous end joining (NHEJ) or homologous directed repair (HDR), are utilized for the alteration of genomic DNA sequences to prevent or treat a disease: disruption of a gene, deletion of a specific genomic region, and correction of a gene (Figure 2). CRISPR can usually be designed and substituted as a single guide RNA (sgRNA or gRNA) at LQTS-associated gene target sites (116, 117). CRISPR/Cas9 cargoes can be of three types, namely (1) DNA plasmid encoding both the Cas9 protein and the guide RNA, (2) a combination of sgRNA and Cas9 mRNA, and (3) Cas9 protein with gRNA (ribonucleoprotein complex, RNP) (118–120) (Figure 1). Furthermore, both gene correction and gene addition require an exogenous single- or double-stranded DNA (ssDNA) template with homologous arms or a single-stranded DNA oligonucleotide (ssODN) (46, 113). For example, SNV for KCNH2, isogenic correction ssODN templates need to contain wild nucleotide sequences and CRISPR/Cas-blocking mutations, preferably in the PAM, to minimize undesirable re-editing (91, 121) (Figure 4).
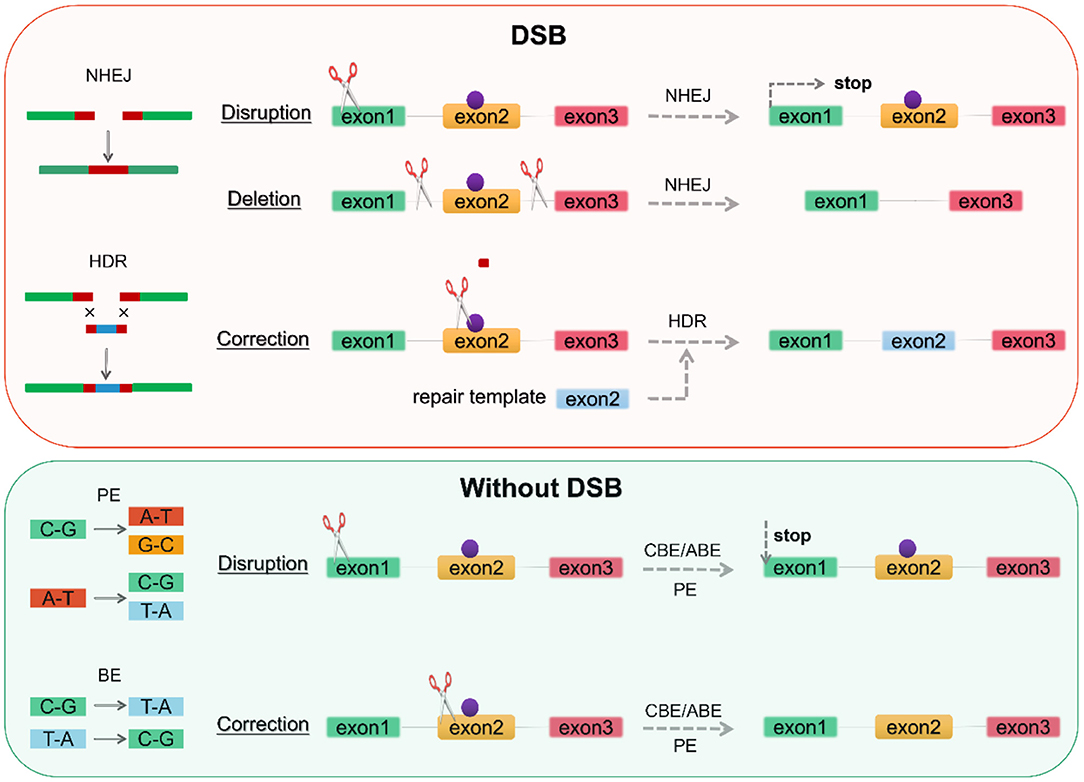
Figure 2. Genome editing according to repair mechanism. DSBs induce two repair mechanisms, NHEJ and HDR. NHEJ can generate an indel at the gene of interest-based on gene disruption resulting in premature translation stop to achieve knockout, while gene deletion requires the creation of two DSBs on both sides of the pathogenic mutation (purple point). BE and PE can perform base substitution without DSBs, with the former being limited to 8 base substitutions and the latter enabling arbitrary base substitutions. DSB, double-strand breaks; NHEJ, non-homologous end joining; HDR, homologous directed repair; BE, base editor; PE, prime editor. CBE, cytosine base editor; ABE, adenine base editor.
Provided that a donor template is available, DSB-mediated HDR repair mechanisms allow the introduction of targeted mutations associated with the LQTS phenotype or the correction of pathogenic mutations. Nevertheless, in addition to consideration of off-target, DSB leads to activation of the p53 pathway inducing DNA damage responses and cell cycle arrest (122, 123). And genome editing by CRISPR/Cas9-induced DSBs is generally less efficient in hiPSCs compared to 293T cells (124, 125). DNA base editors (BEs), including the cytosine base editor (CBE) and adenine base editor (ABE), have been proposed to perform precise nucleotide substitution without the need for a donor template or the introduction of DSBs (46, 126–128) (Figure 2). Prime editors (PEs) extend the limited editing window of BEs (C-G to T-A conversion for CBE and A-T to G-C conversion for ABE) to eight transition mutations (C→ A, C→ G, G→ C, G→ T, A→ C, A→ T, T→ A, and T→ G) (129–132) (Figure 2). PEs substantially expands the scope and capabilities of genome editing and, in principle, could correct up to 89% of known genetic variants associated with human diseases.
Clinical genetic testing has become the standard for diagnosing genetic variants in suspected monogenic disorders. However, these results are often found for VUS, for which they do not have sufficient evidence of pathogenicity. VUS is becoming a significant challenge in clinical genetics. The critical issue remains that the variable expression and incomplete penetrance between individuals with the same LQTS pathogenic mutation remain largely unexplained (7). As a result of the progressive use of genetic testing, the discovery of more rare variants, modifier genes with clear variants, and VUS, combined with the lack of experimental platforms, the focus has become on how to rapidly and reliably determine the functional significance of the genetics of variants.
In vitro hiPSC-CMs models of LQTS can be derived from disease-specific somatic cell differentiation and differentiation of CRISPR/Cas9-edited hiPSCs. The main reason for generating these hiPSC-CMs is to observe the effects of mutations on cell phenotypes, particularly genome editing using CRISPR/Cas9. It is worth considering that multiple genetic and epigenetic differences exist between cell lines and that reprogramming and genome editing are themselves the causes of epigenetic variation. Moreover, genetic background variation may confound disease characteristics, particularly for LQTS with incomplete penetrances (73). It requires the generation of appropriate control cell lines (isogenic cell lines) with the same genetic background (Figure 1). Isogenic hiPSCs provide reliable and effective model alternatives for human disease research (50, 73, 89, 91, 133).
For disease-specific differentiation of hiPSC, unrelated healthy individuals need to be recruited to generate hiPSC-CMs under the same conditions. CRISPR/Cas9-edited hiPSCs can then insert the same mutations into control hiPSC lines or introduce disease-causing variants in normal hiPSCs (Figure 1). Even comparing LQTS-specific to genome-edited isogenic cell lines is a strategy that could be considered. It could be an attempt to discern their superiority or inferiority in LQTS modeling by eliminating genetic background interference (89). In terms of these considerations, disease-specific hiPSC-CMs may have an advantage in overcoming the detrimental effects, such as p53 pathway-induced cell apoptosis associated with genome editing. Subsequently, CRISPR/Cas9 corrective mutations performed in the same genetic background from the same individuals to produce isogenic control sets would provide stronger evidence for the reversal of abnormal cell phenotypes than control sets from unrelated individuals. Unfortunately, sometimes, such disease-specific individual patients are not easily recruited, especially those with rare variants, which impedes the timely and effective management of those patients carrying what is defined as VUS after the genetic screening. Due to its high efficiency, CRISPR/Cas9 is also a powerful tool for establishing homogeneous controls when outlining the disease profile of LQTS.
In 1996, KCNQ1 was identified as the gene responsible for LQT1 (134). LOF mutations in KCNQ1 to reduce IKs are the most common cause of congenital LQTS, occurring in 40–50% of all patients, and are referred to as LQT1 (135) (Figure 3). Numerous models of LQT1 hiPSC-CMs carrying SNVs are summarized in Table 1.
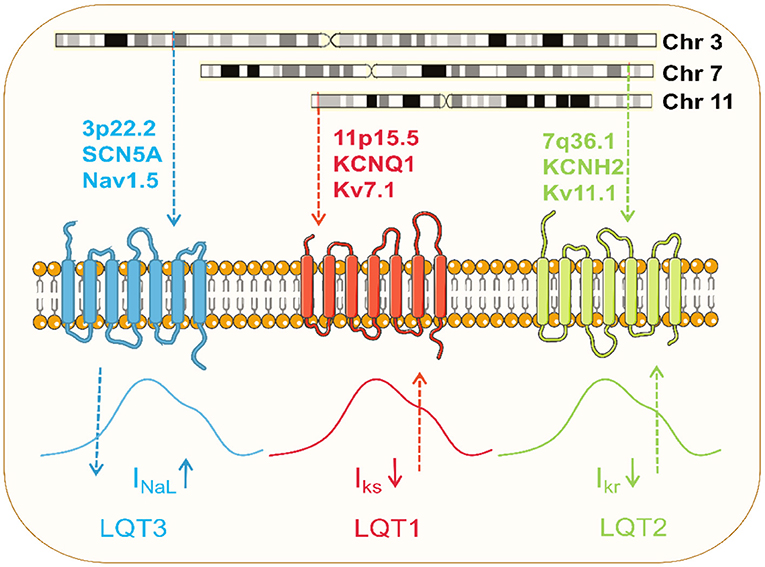
Figure 3. Pathophysiology of the common LQTS. Dominant mutations in KCNQ1 on chromosome 11 and KCNH2 on chromosome 7 result in loss of function of Kv7.1 and Kv11.1 potassium channels, respectively, and these channel currents play a significant role in myocardial repolarization. Pathogenic mutation in SCN5A on chromosome 3 causes gain of function of Nav1.5 channel, resulting in increased late sodium currents to prolong the action potential.
Before applying genome editing in hiPSC, controls were generated based on unrelated healthy individuals with hiPSC-CMs. The generation of isogenic cells is not only salutary for modeling but also for explaining clinical heterogeneity (13). At this point, the introduction of CRISPR/Cas9 can target not only SCN5A itself but also SCN5A modifiers, such as myotubularin-related protein 4 (MTMR4) (13). The impact of an indirect generation of wild or mutated modifiers on the phenotype of isogenic cell lines identifies the underlying regulation of SCN5A to produce diverse clinical manifestations. Their recognition can improve risk stratification and clinical management. And even though mutation correction is inefficient and unavailable for mature heart cells, the CRISPR/Cas9 system remains a powerful tool for validating its gene therapy potential by generating normal isogenic controls (14, 71).
LQT2 is the second most common LQTS and results from mutations in the KCNH2, also known as the hERG gene (136). Dominant mutations present a haplotype deficiency or dominant-negative effect, causing a partial or complete reduction in IKr current and prolonging APD (137) (Figure 3). Modeling hiPSC-CMs avoids the problem of changes in channel activity generated by the absence of auxiliary subunits. IKr has been reported in hiPSC-CM with a current density value comparable to native human myocardium (10). Like LQT1, several SNVs in LQT2-hiPSC-CMs have been developed (Table 1). Itzhaki et al. first established a model containing the KCNH2-A614V mutation using dermal fibroblasts from LQT2 patients. APD and FPD were significantly prolonged compared to healthy control models (138).
The isogenic set generated by CRISPR/Cas9 editing makes it convenient to replicate the newly identified LQTS variants in hiPSC-CMs. More recently, in a genetically elusive multigenerational LQTS pedigree, a frameshift variant (p.S1112Pfs*171) in patient-specific hiPSC-CMs was identified as a novel LQT2-causative variant to induce prolonged FPD, a result compared to CRISPR/Cas9-corrected isogenic control hiPSC-CMs (16). Besides, as hERG channels are the most common targets of drug-acquired LQTS, modeling using hiPSC-CMs combined with high-throughput assays of MEA offers great advantages in assessing multiple groups of cardiotoxic drugs (53, 54, 139). More importantly, the genetic background may influence individual sensitivity to these drugs (19, 55, 72, 73). Through introducing different variants in healthy wild-type hiPSC without any known disease-causing mutations, isogenic sets of hiPSC-CMs with distinct KCNH2 mutations differ functionally and in susceptibility to drug-induced arrhythmias (50, 72, 89, 91). Targeted editing of KCNH2 using CRISPR/Cas9 to generate hiPSC-CMs carrying SNPs supports hiPSC-CMs as solid candidates for evaluating the underlying severity of KCNH2 mutations, which could facilitate patient risk stratification.
LQT3 is caused exclusively by a gain-of-function mutation in the SCN5A gene that encodes for the alpha subunit of the Nav1.5 (94, 140). SCN5A mutations cause gain-of-function of Nav1.5 channels by impairing channel inactivation and accelerating recovery from inactivation, increasing INaL to counter repolarization and prolonging APD (103) (Figure 3; Table 1).
The primary question for simulations of the SCN5A mutation may be the relationship between genotype-phenotype (141). Mutations are associated with several genetically heterogeneous disorders, including Brugada syndrome (BrS), cardiac conduction disease (CCD), sick sinus syndrome (SSS), and others (142). Genotype-phenotype studies in large pedigrees have established that several single SCN5A mutations present with multiple clinical manifestations due to the various biophysical defects.
CRISPR/Cas9 generation knockdown of SCN5A to generate the Nav1.5 KO hiPSC lineage or the introduction of pathogenic mutations to build control models has great breakthroughs in analyzing the pathophysiological mechanisms of the phenotype (15, 105, 106). Similarly, drug arrhythmogenic susceptibility can be determined using genomically corrected SCN5A variant isogenic control cell lines (105). Even introducing CRISPR/Cas9 in hiPSCs makes it possible to compare the properties of different variants, a single variant in different cell models, or even differences between homozygous or heterozygous mutation (107, 143).
Modifier genes are genes at the same or other loci that affect the phenotype of the major gene (144). Non-synonymous coding variants common to the same major mutant gene and common variants in genes encoding ion channel regulators or auxiliary subunits can also act as gene modifiers (144). KCNE1 encoding the Kv7.1 auxiliary subunit (p.Asp85Asn, also known as D85N) is more susceptible to LQTS and may be associated with an increase in disease severity (145). These observations can be explained by impaired repolarisation currents (IKs, IKr) when this KCNE1 variant interacts with KCNQ1 or hERG channels. Non-coding elements required for mRNA stability and translation exist in the genes' 3′-untranslated region (3′-UTR). Variants in the 3′-UTR are only related to a prolonged QT interval in heterozygous KCNQ1 mutations, not in the general population (146, 147). Conversely, the SNVs of the gene may also be an independent modifier to confer protection against cardiac events in patients with LQTS (13, 148).
It is helpful to explain monogenic diseases' incomplete penetration and variable expression. Further examples include AKAP9 and NOS1AP variants (3, 149–151). Recently, two SNVs on the myotubularin-related protein 4 (MTMR4) gene were identified by whole-exome sequencing as potential contributors to the clinical phenotype of LQT1 patients (13). As an interactor of Nedd4L, the MTMR4 variant is thought to be responsible for the different clinical manifestations of members of the same family carrying the same mutation, KCNQ1-Y111C. It was discovered that hiPSC-CMs from asymptomatic patients showed increased degradation of KCNQ1 than hiPSC-CMs from symptomatic patients, who showed reduced degradation of KCNQ1 and hERG proteins due to reduced Nedd4L activity caused by the MTMR4 variant. And the correction of SNVs in MTMR4 by CRISPR/Cas9 unmasked the LQTS phenotype (13). While new modifier genes are proposed, it is worth testing whether such protective modifications are present in more monogenic diseases, as in the case of KCNQ1 polymorphism rs2074238 T-allele (148).
Given the above, a conclusion can also be drawn: if no disease phenotype is observed, this does not necessarily mean that the variant is benign. The disease phenotype may be masked if the mutation has low penetration and the control hiPSC line selected carries a protective genetic modifier. In such cases, it is essential to correct for these protective SNVs in the patient-derived hiPSC to demonstrate their effect on the disease phenotype.
As with LQT1, modifier genes explain the clinical genotype-phenotype inconsistency of LQT2. It is important to investigate this issue by examining modifier genes, introducing them to hiPSC-CMs, and characterizing their function. KCNH2-K897T is the most commonly reported genetic modifier of KCNH2, which impairs the repolarisation reserve associated with LQT1 and LQT2 mutations, and exacerbates LQTS (144, 152–155). For example, K897T with the KCNH2 mutation (A1116V) exacerbates the reduction in IKr caused by the latter to produce symptoms, whereas there are no symptoms when A1116V is present alone (154).
Conversely, some protective mutations like KCNH2-R1135H maintain IKr channel function (156). Isogenic hiPSC-CMs lines with K897T in cis and trans were produced using CRISPR/Cas9 to see if the linkage phase of the K897T polymorphism to the major KCNH2 polymorphisms A561T and N996I caused changes in IKr (92). The findings show that the common polymorphism KCNH2-K897T has a different effect on LQT2-causing KCNH2 mutations depending on whether it is present in cis or trans (92). Similarly, the correction of K897T variants with CRISPR/Cas9 would be valuable in determining whether this common polymorphism plays a protective or aggravating role in QTc prolongation and the corresponding effect on disease severity. Genome sequencing provides more evidence of genetic modifiers to explain genotype-phenotype discordance (74, 157, 158). In the LQT2 lineage, genomics identified two modifier genes, KCNK17 and REM2, to elucidate the contributors to variable expressivity of KCNH2-R752W mutation (74). CRISPR/Cas9 editing to correct a REM2 variant reversed the enhanced ICaL and prolonged APD observed in hiPSC-CMs from severely affected individuals (74).
Another study of interest is the only one to date to characterize KCNH2-T983I (KCNH22948C>T) VUS in LQTS using hiPSC-CMs combined with CRISPR/Cas9 editing (91) (Figure 4). Determining the pathogenicity of a VUS is challenging due to the lack of suitable model systems and accessible technologies (12). Approximately 40% of variants are designated as still not classifiable as pathogenic or benign (159). Multiple family members carrying the same mutation may have different QT intervals and clinical presentations. Moreover, in the era of next-generation sequencing, the interpretation of the increasing number of identified unknown variants will pose an even greater therapeutic challenge. Thus, Garg et al.'s research was groundbreaking, demonstrating that genome editing in hiPSC could be a helpful tool for determining the pathogenicity of VUS in cardiac channelopathies (91). They generated hiPSC-CMs from peripheral blood mononuclear cells carrying the novel VUS mutation KCNH2-T983I. VUS hiPSC-CMs exhibited significantly prolonged APD, reduced IKr, and abnormal calcium transients compared to a healthy control line. Further correction of VUS hiPSC-CMs with CRISPR/Cas9 resulted in a normal phenotype, and the introduction of VUS in healthy hiPSC-CMs recapitulated the hallmark features of LQTS disease (91) (Figure 4). These attempts were able to categorize and manage VUS in a timely and effective manner without the development of SCD.
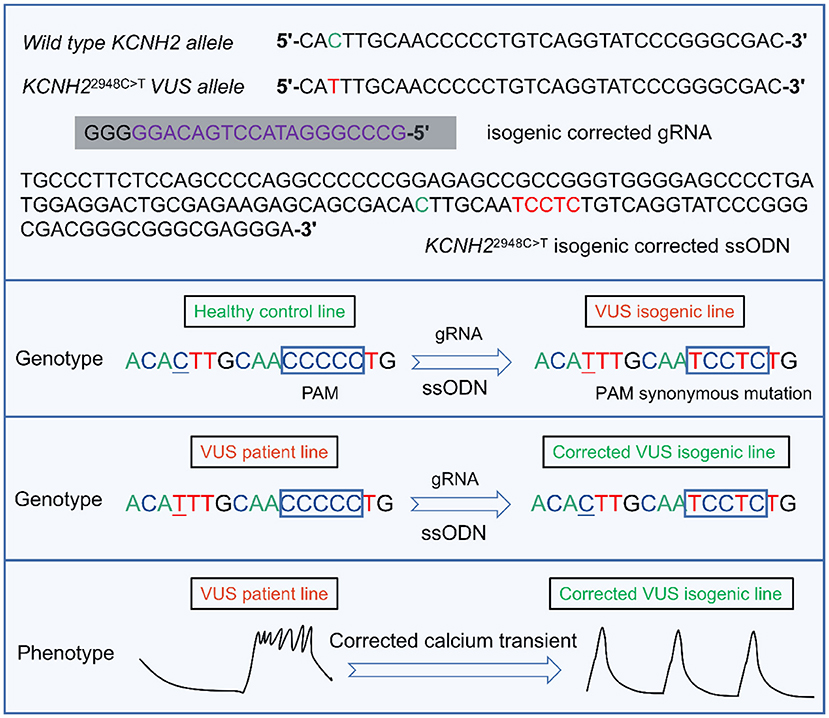
Figure 4. A representative example of the targeted design of isogenic corrected gRNA or ssODN for the locus where KCNH22948C>T VUS is located, enabling correction of the point mutation (91). gRNA is designed by targeting the upstream of the 5'-NGG (PAM) of the KCNH2 VUS locus. By introducing ssODN containing homologous arms, the VUS isogenic line can be generated in a healthy control cell line. Similarly, the VUS line can also be corrected to enable a phenotype of abnormal calcium transient to be reversed. VUS, variants of uncertain significance. PAM, protospacer adjacent motif. gRNA, guide RNA. ssODN, single-stranded DNA oligonucleotide.
Identifying these variants can guide genotype-specific management or facilitate rapid screening of potentially high-risk relatives. Although distinguishing pathogenic from benign is a significant challenge, primarily when classified as VUS, this new approach still represents a significant advance in precision medicine for the management of LQTS disease (157).
A modifier gene can also explain the phenotype caused by the SNVs in SCN5A. SCN3B encoding the Nav1.5 β-subunit amplifies the reduced sodium current generated by mutation SCN5A-E1784K and thus masks the Brs phenotype (95). Compared to the almost 100% penetration of the typical ΔKPQ variant initially described, the R1193Q is close to zero (107, 160). The prevalence of this variant in an unaffected population (6.1% in the East Asian allele) strongly suggests that it cannot play an essential role in disease manifestion (161).
The underlying mechanisms of SCN5A ΔKPQ and R1193Q-inducible variants were assessed by designing gRNAs to introduce R1193Q into SCN5A exon 19 of hiPSC from healthy individuals (107). The study suggests that the low penetration rate of the R1193Q mutation involves PI3Kα-mediated changes in PIP3-modulated INaL. Unlike ΔKPQ, the sensitivity of R1193Q to PIP3 means distinct pathologies need to be considered when interpreting the severity of the excess late current functional defect in Nav1.5 (107). The observation of late current in an in vitro setting does not necessarily translate into a highly pathogenic LQT3 phenotype but rather depends on the underlying mechanisms. Nav1.5 channels differ in hiPSC-CMs of LQTS, or Brs may rely on different Tbx5 variants, such that LQT3 may be due to the failure of the Tbx5-D111Y mutation to repress CAMK2D and SPTBN4, which significantly enhances INaL (162).
Attempts at effective management strategies in disease-specific hiPSC-CMs are valuable. The corrective effect of applying CRISPR/Cas9 against the SNVs of KCNQ1 was first demonstrated in a hiPSC-CMs carrying a double mutation (c. 605-2A>G and c. 815G>A). Splicing mutations causing exon skipping lead to significantly longer APD90 and EADs; designing gRNAs to target these SNVs can correct aberrant cellular phenotypes (14). Nevertheless, unable to rescue two mutations simultaneously by constructing a CRISPR/Cas9 system containing two guide RNAs. In LQT1, KCNQ1 variants often exhibit dominant-negative effects (163–167). Therefore, substitution alone, such as silencing the mutant allele using RNA interference (RNAi) without affecting the wild allele, is not sufficient; such a strategy requires that the effect of silencing exceeds that of wild-type expression, and the best outcome is to present a haplotype deficiency. Attempts to replace the mutant allele are feasible, and targeting SNV in CRISPR/Cas9 to design a segment of ssODN as a template for repair is feasible. The recently reported strategy of a dual-component suppression-and-replacement (SupRep) KCNQ1 gene therapy targeting two variants of KCNQ1 is also a clever optional strategy (71). Unfortunately, all of these tools face the problem of how to push data to the living body for the next validation step.
As previously stated, allele-specific RNAi was developed with targeted mutant KD to eliminate dominant-negative interference, assuming that the remaining WT allele would provide enough function to alleviate the disease phenotype despite haploinsufficiency. In previous studies of LQTS, allele-specific small interfering RNAs rescued the hERG current in heterologous expression systems through specific KD of the dominant-negative missense variants KCNH2-E637K (168). Such experiments in hiPSC-CMs were also victorious against KCNH2-G1681A (75). However, the end of the haplotype deficiency generated by dominant-negative effect RNAi appears promising. However, its implementation is still limited, such as the difficulty of designing each RNAi targeting SNVs and the fact that designing separate RNAi targeting specific individual variants is impractical when multiple variants are present simultaneously (71).
The advent of BEs and PEs seems to have made the correction of SNVs easier. But CRISPR/Cas9 still has a high probability of off-targeting associated with spatially inhomogeneous tolerances for pairwise mismatches in sgRNA-DNA heteroduplexes. They are still some way from being studied and applied in vivo (169). The most significant breakthrough in LQT2 treatment came with identifying the chemical chaperone lumacaftor, a drug approved by the FDA for the treatment of cystic fibrosis (8). Lumacaftor corrected two mutations, KCNH2-IVS9-28A/G and -A561V, representing trafficking defects in patient-specific hiPSC-CMs (8). Subsequent clinical translations showed good therapeutic effects in LQT2 patients (170). Targeting more KCNH2 variants, lumacaftor had opposite effects in different cellular models (76, 171–173). They suggest that the dominance of the hiPSC-CMs model is responsible for the role of lumacaftor, which further delineates the differences between hiPSC and heterologous expression systems. Currently, a total of 160 KCNH2 mutations representing protein trafficking defects are presented in the Milan database tested using Orkambi (lumacaftor plus ivacaftor) (7). This is another grand attempt by researchers to use the hiPSC-based platform to move toward precision medicine.
After a decade of development, from the initial use of hiPSC to building a disease model to understand the clinical heterogeneity of the disease today combined with CRISPR/Cas9, the researchers have made a great deal of work. To achieve this, a large number of issues including consistently available in vitro research models, avoidance of ethical requirements and immune rejection, safety and efficiency of the reprogramming process, feasibility and efficiency of targeted differentiation, characterization of successful myocardial differentiation, off-target and cell safety of gene editing, quality control of epigenetics, and ex vivo and in vitro dissemination of the strategy, are being progressively addressed.
From somatic cells to hiPSC-CMs, the development of genome editing gives us more options in elucidating diseases. Experts in every field are working tirelessly to achieve the goal of precision medicine. In the case of a monogenic ICC such as LQTS, genome sequencing of individuals, and ongoing genome-wide association studies, identifies many possible variants that could be involved but need to be validated.
We need to figure out why people with the same SNV have varied clinical features, but we also need to test potential therapeutic options. Meanwhile, because there is a lack of VUS knowledge compared to typical dominant mutations, it is even more crucial to open a window of opportunity for timely intervention for patients carrying VUS or rare variants. Finally, achieving precision medicine may require collaboration from experts in clinical cardiology, electrophysiology, stem cells, genomics, and even pharmacology.
YS and ZZ: conceptualization, data curation, writing—original draft, software, and resources. JL: conceptualization, writing—review and editing, and supervision. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of Ningbo [Grant Number 2021J296] and the Natural Science Foundation of the Zhejiang Province [Grant Numbers LQQ20H160001 and LY21H020001].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.889519/full#supplementary-material
1. Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. (2008) 51:2291–300. doi: 10.1016/j.jacc.2008.02.068
2. Wallace E, Howard L, Liu M, O'Brien T, Ward D, Shen S, et al. Long QT syndrome: genetics and future perspective. Pediatr Cardiol. (2019) 40:1419–30. doi: 10.1007/s00246-019-02151-x
3. Lahrouchi N, Tadros R, Crotti L, Mizusawa Y, Postema PG, Beekman L, et al. Transethnic genome-wide association study provides insights in the genetic architecture and heritability of long QT syndrome. Circulation. (2020) 142:324–38. doi: 10.1161/CIRCULATIONAHA.120.045956
4. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. (2009) 120:1761–7. doi: 10.1161/CIRCULATIONAHA.109.863209
5. Tester DJ, Ackerman MJ. Genetics of long QT syndrome. Methodist DeBakey Cardiovasc J. (2014) 10:29–33. doi: 10.14797/mdcj-10-1-29
6. Skinner JR, Winbo A, Abrams D, Vohra J, Wilde AA. Channelopathies that lead to sudden cardiac death: clinical and genetic aspects. Heart Lung Circ. (2019) 28:22–30. doi: 10.1016/j.hlc.2018.09.007
7. Wu JC, Garg P, Yoshida Y, Yamanaka S, Gepstein L, Hulot JS, et al. Towards precision medicine with human iPSCS for cardiac channelopathies. Circ Res. (2019) 125:653–8. doi: 10.1161/CIRCRESAHA.119.315209
8. Mehta A, Ramachandra CJA, Singh P, Chitre A, Lua CH, Mura M, et al. Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur Heart J. (2018) 39:1446–55. doi: 10.1093/eurheartj/ehx394
9. Garg P, Garg V, Shrestha R, Sanguinetti MC, Kamp TJ, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes as models for cardiac channelopathies: a primer for non-electrophysiologists. Circ Res. (2018) 123:224–43. doi: 10.1161/CIRCRESAHA.118.311209
10. Hoekstra M, Mummery CL, Wilde AA, Bezzina CR, Verkerk AO. Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front Physiol. (2012) 3:346. doi: 10.3389/fphys.2012.00346
11. Musunuru K, Sheikh F, Gupta RM, Houser SR, Maher KO, Milan DJ, et al. Induced pluripotent stem cells for cardiovascular disease modeling and precision medicine: a scientific statement from the American Heart Association. Circ Genom Precis Med. (2018) 11:e000043. doi: 10.1161/HCG.0000000000000043
12. Guo H, Liu L, Nishiga M, Cong L, Wu JC. Deciphering pathogenicity of variants of uncertain significance with CRISPR-edited iPSCs. Trends Genet. (2021) 37:1109–23. doi: 10.1016/j.tig.2021.08.009
13. Lee YK, Sala L, Mura M, Rocchetti M, Pedrazzini M, Ran X, et al. MTMR4 SNVs modulate ion channel degradation and clinical severity in congenital long QT syndrome: insights in the mechanism of action of protective modifier genes. Cardiovasc Res. (2021) 117:767–79. doi: 10.1093/cvr/cvaa019
14. Wang Z, Wang L, Liu W, Hu D, Gao Y, Ge Q, et al. Pathogenic mechanism and gene correction for LQTS-causing double mutations in KCNQ1 using a pluripotent stem cell model. Stem Cell Res. (2019) 38:101483. doi: 10.1016/j.scr.2019.101483
15. Pierre M, Djemai M, Poulin H, Chahine M. Na(V)1.5 knockout in iPSCs: a novel approach to study Na(V)1.5 variants in a human cardiomyocyte environment. Sci Rep. (2021) 11:17168. doi: 10.1038/s41598-021-96474-6
16. Tobert KE, Tester DJ, Zhou W, Haglund-Turnquist CM, Giudicessi JR, Ackerman MJ. Genome sequencing in a genetically elusive multigenerational long QT syndrome pedigree identifies a novel LQT2-causative deeply intronic KCNH2 variant. Heart Rhythm. (2022). S1547-5271(22)00121-7. doi: 10.1016/j.hrthm.2022.02.004
17. McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW, et al. A minK-HERG complex regulates the cardiac potassium current I(Kr). Nature. (1997) 388:289–92. doi: 10.1038/40882
18. Abbott GW. Control of biophysical and pharmacological properties of potassium channels by ancillary subunits. Handb Exp Pharmacol. (2021) 267:445–80. doi: 10.1007/164_2021_512
19. Shah RR. Drug-induced QT interval prolongation–regulatory guidance and perspectives on hERG channel studies. Novartis Found Symp. (2005) 266:251–85. doi: 10.1002/047002142x.ch19
20. Salama G, London B. Mouse models of long QT syndrome. J Physiol. (2007) 578(Pt 1):43–53. doi: 10.1113/jphysiol.2006.118745
21. Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, et al. High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circul Physiol. (2011) 301:H2006–17. doi: 10.1152/ajpheart.00694.2011
22. Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. (2015) 117:80–8. doi: 10.1161/CIRCRESAHA.117.305365
23. Ahmed RE, Anzai T, Chanthra N, Uosaki H. A brief review of current maturation methods for human induced pluripotent stem cells-derived cardiomyocytes. Front Cell Dev Biol. (2020) 8:178. doi: 10.3389/fcell.2020.00178
24. Veerman CC, Kosmidis G, Mummery CL, Casini S, Verkerk AO, Bellin M. Immaturity of human stem-cell-derived cardiomyocytes in culture: fatal flaw or soluble problem? Stem Cells Dev. (2015) 24:1035–52. doi: 10.1089/scd.2014.0533
25. Jiang Y, Park P, Hong SM, Ban K. Maturation of cardiomyocytes derived from human pluripotent stem cells: current strategies and limitations. Mol Cells. (2018) 41:613–21. doi: 10.14348/molcells.2018.0143
26. Denning C, Borgdorff V, Crutchley J, Firth KS, George V, Kalra S, et al. Cardiomyocytes from human pluripotent stem cells: from laboratory curiosity to industrial biomedical platform. Biochim Biophy Acta. (2016) 1863(7 Pt B):1728–48. doi: 10.1016/j.bbamcr.2015.10.014
27. Campostrini G, Windt LM, van Meer BJ, Bellin M, Mummery CL. Cardiac tissues from stem cells: new routes to maturation and cardiac regeneration. Circ Res. (2021) 128:775–801. doi: 10.1161/CIRCRESAHA.121.318183
28. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
29. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. (2007) 131:861–72. doi: 10.1016/j.cell.2007.11.019
30. Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. (2009) 458:771–5. doi: 10.1038/nature07864
31. Bekhite MM, Schulze PC. Human induced pluripotent stem cell as a disease modeling and drug development platform-A cardiac perspective. Cells. (2021) 10:3483. doi: 10.3390/cells10123483
32. Okita K, Yamakawa T, Matsumura Y, Sato Y, Amano N, Watanabe A, et al. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells. (2013) 31:458–66. doi: 10.1002/stem.1293
33. Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, et al. Generation of human induced pluripotent stem cells from urine samples. Nat Prot. (2012) 7:2080–9. doi: 10.1038/nprot.2012.115
34. Higuchi A, Ling QD, Kumar SS, Munusamy MA, Alarfaj AA, Chang Y, et al. Generation of pluripotent stem cells without the use of genetic material. Lab Invest. (2015) 95:26–42. doi: 10.1038/labinvest.2014.132
35. Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. (2008) 118:3143–50. doi: 10.1172/JCI35798
36. Liu X, Li C, Zheng K, Zhao X, Xu X, Yang A, et al. Chromosomal aberration arises during somatic reprogramming to pluripotent stem cells. Cell Div. (2020) 15:12. doi: 10.1186/s13008-020-00068-z
37. Gähwiler EKN, Motta SE, Martin M, Nugraha B, Hoerstrup SP, Emmert MY. Human iPSCs and genome editing technologies for precision cardiovascular tissue engineering. Front Cell Dev Biol. (2021) 9:639699. doi: 10.3389/fcell.2021.639699
38. Jia F, Wilson KD, Sun N, Gupta DM, Huang M, Li Z, et al. A nonviral minicircle vector for deriving human iPS cells. Nat Methods. (2010) 7:197–9. doi: 10.1038/nmeth.1426
39. Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. (2009) 4:472–6. doi: 10.1016/j.stem.2009.05.005
40. Al Abbar A, Ngai SC, Nograles N, Alhaji SY, Abdullah S. Induced pluripotent stem cells: reprogramming platforms and applications in cell replacement therapy. Biores Open Access. (2020) 9:121–36. doi: 10.1089/biores.2019.0046
41. MacGrogan D, Münch J, de la Pompa JL. Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat Rev Cardiol. (2018) 15:685–704. doi: 10.1038/s41569-018-0100-2
42. Wang G, Jacquet L, Karamariti E, Xu Q. Origin and differentiation of vascular smooth muscle cells. J Physiol. (2015) 593:3013–30. doi: 10.1113/JP270033
43. Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. (2014) 11:855–60. doi: 10.1038/nmeth.2999
44. Minami I, Yamada K, Otsuji TG, Yamamoto T, Shen Y, Otsuka S, et al. A small molecule that promotes cardiac differentiation of human pluripotent stem cells under defined, cytokine- and xeno-free conditions. Cell Rep. (2012) 2:1448–60. doi: 10.1016/j.celrep.2012.09.015
45. Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. (2010) 363:1397–409. doi: 10.1056/NEJMoa0908679
46. Nishiga M, Qi LS, Wu JC. Therapeutic genome editing in cardiovascular diseases. Adv Drug Delivery Rev. (2021) 168:147–57. doi: 10.1016/j.addr.2020.02.003
47. Verkerk AO, Veerman CC, Zegers JG, Mengarelli I, Bezzina CR, Wilders R. Patch-clamp recording from human induced pluripotent stem cell-derived cardiomyocytes: improving action potential characteristics through dynamic clamp. Int J Mol Sci. (2017) 18:1873. doi: 10.3390/ijms18091873
48. Wilders R. Dynamic clamp: a powerful tool in cardiac electrophysiology. J Physiol. (2006) 576(Pt 2):349–59. doi: 10.1113/jphysiol.2006.115840
49. Meijer van Putten RME, Mengarelli I, Guan K, Zegers JG, van Ginneken ACG, Verkerk AO, et al. Ion channelopathies in human induced pluripotent stem cell derived cardiomyocytes: a dynamic clamp study with virtual IK1. Front Physiol. (2015) 6:7. doi: 10.3389/fphys.2015.00007
50. Brandão KO, van den Brink L, Miller DC, Grandela C, van Meer BJ, Mol MPH, et al. Isogenic sets of hiPSC-CMs harboring distinct KCNH2 Mutations differ functionally and in susceptibility to drug-induced arrhythmias. Stem Cell Rep. (2020) 15:1127–39. doi: 10.1016/j.stemcr.2020.10.005
51. Egashira T, Yuasa S, Suzuki T, Aizawa Y, Yamakawa H, Matsuhashi T, et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc Res. (2012) 95:419–29. doi: 10.1093/cvr/cvs206
52. Nozaki Y, Honda Y, Tsujimoto S, Watanabe H, Kunimatsu T, Funabashi H. Availability of human induced pluripotent stem cell-derived cardiomyocytes in assessment of drug potential for QT prolongation. Toxicol Appl Pharmacol. (2014) 278:72–7. doi: 10.1016/j.taap.2014.04.007
53. Gilchrist KH, Lewis GF, Gay EA, Sellgren KL, Grego S. High-throughput cardiac safety evaluation and multi-parameter arrhythmia profiling of cardiomyocytes using microelectrode arrays. Toxicol Appl Pharmacol. (2015) 288:249–57. doi: 10.1016/j.taap.2015.07.024
54. Blinova K, Stohlman J, Vicente J, Chan D, Johannesen L, Hortigon-Vinagre MP, et al. Comprehensive translational assessment of human-induced pluripotent stem cell derived cardiomyocytes for evaluating drug-induced arrhythmias. Toxicol Sci. (2017) 155:234–47. doi: 10.1093/toxsci/kfw200
55. Yamamoto W, Asakura K, Ando H, Taniguchi T, Ojima A, Uda T, et al. Electrophysiological characteristics of human ipsc-derived cardiomyocytes for the assessment of drug-induced proarrhythmic potential. PLoS ONE. (2016) 11:e0167348. doi: 10.1371/journal.pone.0167348
56. Lahti AL, Kujala VJ, Chapman H, Koivisto AP, Pekkanen-Mattila M, Kerkelä E, et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Models Mech. (2012) 5:220–30. doi: 10.1242/dmm.008409
57. Chang Y, Li YN, Bai R, Wu F, Ma S, Saleem A, et al. hERG-deficient human embryonic stem cell-derived cardiomyocytes for modelling QT prolongation. Stem Cell Res Ther. (2021) 12:278. doi: 10.1186/s13287-021-02346-1
58. Kiviaho AL, Ahola A, Larsson K, Penttinen K, Swan H, Pekkanen-Mattila M, et al. Distinct electrophysiological and mechanical beating phenotypes of long QT syndrome type 1-specific cardiomyocytes carrying different mutations. International journal of cardiology Heart Vasculat. (2015) 8:19–31. doi: 10.1016/j.ijcha.2015.04.008
59. Ma D, Wei H, Lu J, Huang D, Liu Z, Loh LJ, et al. Characterization of a novel KCNQ1 mutation for type 1 long QT syndrome and assessment of the therapeutic potential of a novel IKs activator using patient-specific induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res Ther. (2015) 6:39. doi: 10.1186/s13287-015-0027-z
60. Kuusela J, Larsson K, Shah D, Prajapati C, Aalto-Setälä K. Low extracellular potassium prolongs repolarization and evokes early afterdepolarization in human induced pluripotent stem cell-derived cardiomyocytes. Biol Open. (2017) 6:777–84. doi: 10.1242/bio.024216
61. Sogo T, Morikawa K, Kurata Y, Li P, Ichinose T, Yuasa S, et al. Electrophysiological properties of iPS cell-derived cardiomyocytes from a patient with long QT syndrome type 1 harboring the novel mutation M437V of KCNQ1. Regen Ther. (2016) 4:9–17. doi: 10.1016/j.reth.2015.12.001
62. Sala L, Yu Z, Ward-van Oostwaard D, van Veldhoven JP, Moretti A, Laugwitz KL, et al. A new hERG allosteric modulator rescues genetic and drug-induced long-QT syndrome phenotypes in cardiomyocytes from isogenic pairs of patient induced pluripotent stem cells. EMBO Mol Med. (2016) 8:1065–81. doi: 10.15252/emmm.201606260
63. Ge N, Liu M, Krawczyk J, McInerney V, Galvin J, Shen S, et al. Generation of eight human induced pluripotent stem cell (iPSC) lines from familial Long QT Syndrome type 1 (LQT1) patients carrying KCNQ1 c.1697C>A mutation (NUIGi005-A, NUIGi005-B, NUIGi005-C, NUIGi006-A, NUIGi006-B, NUIGi006-C, NUIGi007-A, and NUIGi007-B). Stem Cell Res. (2019) 39:101502. doi: 10.1016/j.scr.2019.101502
64. Ge N, Liu M, Ding Y, Krawczyk J, McInerney V, Galvin J, et al. Generation and characterization of twelve human induced pluripotent stem cell (iPSC) lines from four familial long QT syndrome type 1 (LQT1) patients carrying KCNQ1 c.1201dupC mutation. Stem Cell Res. (2019) 41:101650. doi: 10.1016/j.scr.2019.101650
65. Lavra L, Magi F, Ulivieri A, Morgante A, Paulis M, Sala L, et al. Generation and characterization of the human induced pluripotent stem cell (hiPSC) line NCUFi001-A from a patient carrying KCNQ1 G314S mutation. Stem Cell Res. (2021) 54:102418. doi: 10.1016/j.scr.2021.102418
66. Lee Y, Park H, Kyung Koo S, Kim JH. Establishment of a human induced pluripotent stem cell line, KSCBi015-A, from a long QT syndrome type 1 patient harboring a KCNQ1 mutation. Stem Cell Res. (2021) 56:102521. doi: 10.1016/j.scr.2021.102521
67. Mura M, Ginevrino M, Zappatore R, Pisano F, Boni M, Castelletti S, et al. Generation of the human induced pluripotent stem cell (hiPSC) line PSMi003-A from a patient affected by an autosomal recessive form of Long QT Syndrome type 1. Stem Cell Res. (2018) 29:170–3. doi: 10.1016/j.scr.2018.04.003
68. Mura M, Bastaroli F, Corli M, Ginevrino M, Calabrò F, Boni M, et al. Generation of the human induced pluripotent stem cell (hiPSC) line PSMi006-A from a patient affected by an autosomal recessive form of long QT syndrome type 1. Stem Cell Res. (2020) 42:101658. doi: 10.1016/j.scr.2019.101658
69. Wuriyanghai Y, Makiyama T, Sasaki K, Kamakura T, Yamamoto Y, Hayano M, et al. Complex aberrant splicing in the induced pluripotent stem cell-derived cardiomyocytes from a patient with long QT syndrome carrying KCNQ1-A344Aspl mutation. Heart Rhythm. (2018) 15:1566–74. doi: 10.1016/j.hrthm.2018.05.028
70. Wang Y, Liang P, Lan F, Wu H, Lisowski L, Gu M, et al. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J Am Coll Cardiol. (2014) 64:451–9. doi: 10.1016/j.jacc.2014.04.057
71. Dotzler SM, Kim CSJ, Gendron WAC, Zhou W, Ye D, Bos JM, et al. Suppression-replacement KCNQ1 gene therapy for type 1 long QT syndrome. Circulation. (2021) 143:1411–25. doi: 10.1161/CIRCULATIONAHA.120.051836
72. Yoshinaga D, Baba S, Makiyama T, Shibata H, Hirata T, Akagi K, et al. Phenotype-based high-throughput classification of long QT syndrome subtypes using human induced pluripotent stem cells. Stem Cell Rep. (2019) 13:394–404. doi: 10.1016/j.stemcr.2019.06.007
73. Bellin M, Casini S, Davis RP, D'Aniello C, Haas J, Ward-van Oostwaard D, et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. (2013) 32:3161–75. doi: 10.1038/emboj.2013.240
74. Chai S, Wan X, Ramirez-Navarro A, Tesar PJ, Kaufman ES, Ficker E, et al. Physiological genomics identifies genetic modifiers of long QT syndrome type 2 severity. J Clin Invest. (2018) 128:1043–56. doi: 10.1172/JCI94996
75. Matsa E, Dixon JE, Medway C, Georgiou O, Patel MJ, Morgan K, et al. Allele-specific RNA interference rescues the long-QT syndrome phenotype in human-induced pluripotency stem cell cardiomyocytes. Eur Heart J. (2014) 35:1078–87. doi: 10.1093/eurheartj/eht067
76. O'Hare BJ, John Kim CS, Hamrick SK, Ye D, Tester DJ, Ackerman MJ. Promise and potential peril with lumacaftor for the trafficking defective type 2 long-QT syndrome-causative variants, p.G604S, p.N633S, and p.R685P, using patient-specific re-engineered cardiomyocytes. Circ Genom Precis Med. (2020) 13:466–75. doi: 10.1161/CIRCGEN.120.002950
77. Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A, et al. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. European Heart J. (2011) 32:952–62. doi: 10.1093/eurheartj/ehr073
78. Mehta A, Sequiera GL, Ramachandra CJ, Sudibyo Y, Chung Y, Sheng J, et al. Re-trafficking of hERG reverses long QT syndrome 2 phenotype in human iPS-derived cardiomyocytes. Cardiovasc Res. (2014) 102:497–506. doi: 10.1093/cvr/cvu060
79. Jouni M, Si-Tayeb K, Es-Salah-Lamoureux Z, Latypova X, Champon B, Caillaud A, et al. Toward personalized medicine: using cardiomyocytes differentiated from urine-derived pluripotent stem cells to recapitulate electrophysiological characteristics of type 2 long QT syndrome. J Am Heart Assoc. (2015) 4:e002159. doi: 10.1161/JAHA.115.002159
80. Fatima A, Ivanyuk D, Herms S, Heilmann-Heimbach S, O'Shea O, Chapman C, et al. Generation of human induced pluripotent stem cell line from a patient with a long QT syndrome type 2. Stem Cell Res. (2016) 16:304–7. doi: 10.1016/j.scr.2015.12.039
81. Mura M, Mehta A, Ramachandra CJ, Zappatore R, Pisano F, Ciuffreda MC, et al. The KCNH2-IVS9–28A/G mutation causes aberrant isoform expression and hERG trafficking defect in cardiomyocytes derived from patients affected by Long QT Syndrome type 2. Int J Cardiol. (2017) 240:367–71. doi: 10.1016/j.ijcard.2017.04.038
82. Perry MD, Ng CA, Mangala MM, Ng TYM, Hines AD, Liang W, et al. Pharmacological activation of IKr in models of long QT Type 2 risks overcorrection of repolarization. Cardiovasc Res. (2020) 116:1434–45. doi: 10.1093/cvr/cvz247
83. Mondéjar-Parreño G, Jahng JWS, Belbachir N, Wu BC, Zhang X, Perez MV, et al. Generation of three heterozygous KCNH2 mutation-carrying human induced pluripotent stem cell lines for modeling LQT2 syndrome. Stem Cell Res. (2021) 54:102402. doi: 10.1016/j.scr.2021.102402
84. Wang T, Zhou Y, Zhou R, Huang W, Wang J, Li H, et al. Generation of induced pluripotent stem cells (iPSCs) from a Chinese infant (XACHi015-A) with type 2 Long QT syndrome carrying the heterozygous mutation c.1814C>T(p.P605L) in KCNH2. Stem Cell Res. (2021) 56:102509. doi: 10.1016/j.scr.2021.102509
85. Lee Y, Koo SK, Kim JH. Establishment of a human-induced pluripotent stem cell line, KSCBi014-A, from a long QT syndrome type 2 patient harboring a KCNH2 mutation. Stem Cell Res. (2021) 57:102570. doi: 10.1016/j.scr.2021.102570
86. Ge N, Liu M, Krawczyk J, McInerney V, Ward D, Shen S, et al. Generation and characterization of an induced pluripotent stem cell (iPSC) line (NUIGi003-A) from a long QT syndrome type 2 (LQT2) patient harbouring the KCNH2 c.2464G>A pathogenic variant. Stem Cell Res. (2020) 49:101997. doi: 10.1016/j.scr.2020.101997
87. Okata S, Yuasa S, Yamane T, Furukawa T, Fukuda K. The generation of induced pluripotent stem cells from a patient with KCNH2 G603D, without LQT2 disease associated symptom. J Med Dental Sci. (2013) 60:17–22. doi: 10.17504/protocols.io.tvjen4n
88. Liu M, Ge N, Zhang J, Yang M, Yang F, Krawczyk J, et al. Derivation and characterization of two human induced pluripotent stem cell lines (NUIGi004-A) and (NUIGi012-A) from two patients with LQT2 disease. Stem Cell Res. (2021) 56:102555. doi: 10.1016/j.scr.2021.102555
89. Mesquita FCP, Arantes PC, Kasai-Brunswick TH, Araujo DS, Gubert F, Monnerat G, et al. R534C mutation in hERG causes a trafficking defect in iPSC-derived cardiomyocytes from patients with type 2 long QT syndrome. Sci Rep. (2019) 9:19203. doi: 10.1038/s41598-019-55837-w
90. Spencer CI, Baba S, Nakamura K, Hua EA, Sears MA, Fu CC, et al. Calcium transients closely reflect prolonged action potentials in iPSC models of inherited cardiac arrhythmia. Stem Cell Rep. (2014) 3:269–81. doi: 10.1016/j.stemcr.2014.06.003
91. Garg P, Oikonomopoulos A, Chen H, Li Y, Lam CK, Sallam K, et al. Genome editing of induced pluripotent stem cells to decipher cardiac channelopathy variant. J Am Coll Cardiol. (2018) 72:62–75. doi: 10.1016/j.jacc.2018.04.041
92. van den Brink L, Brandão KO, Yiangou L, Blanch-Asensio A, Mol MPH, Mummery CL, et al. The linkage phase of the polymorphism KCNH2-K897T influences the electrophysiological phenotype in hiPSC models of LQT2. Front Physiol. (2021) 12:755642. doi: 10.3389/fphys.2021.755642
93. Jiang Q, Li K, Lu WJ, Li S, Chen X, Liu XJ, et al. Identification of small-molecule ion channel modulators in C. elegans channelopathy models. Nat Commun. (2018) 9:3941. doi: 10.1038/s41467-018-06514-5
94. Fatima A, Kaifeng S, Dittmann S, Xu G, Gupta MK, Linke M, et al. The disease-specific phenotype in cardiomyocytes derived from induced pluripotent stem cells of two long QT syndrome type 3 patients. PLoS ONE. (2013) 8:e83005. doi: 10.1371/journal.pone.0083005
95. Okata S, Yuasa S, Suzuki T, Ito S, Makita N, Yoshida T, et al. Embryonic type Na(+) channel β-subunit, SCN3B masks the disease phenotype of Brugada syndrome. Sci Rep. (2016) 6:34198. doi: 10.1038/srep34198
96. Ma D, Wei H, Zhao Y, Lu J, Li G, Sahib NB, et al. Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int J Cardiol. (2013) 168:5277–86. doi: 10.1016/j.ijcard.2013.08.015
97. Malan D, Zhang M, Stallmeyer B, Muller J, Fleischmann BK, Schulze-Bahr E, et al. Human iPS cell model of type 3 long QT syndrome recapitulates drug-based phenotype correction. Basic Res Cardiol. (2016) 111:14. doi: 10.1007/s00395-016-0530-0
98. Hirose S, Makiyama T, Melgari D, Yamamoto Y, Wuriyanghai Y, Yokoi F, et al. Propranolol attenuates late sodium current in a long QT syndrome type 3-human induced pluripotent stem cell model. Front Cell Dev Biol. (2020) 8:761. doi: 10.3389/fcell.2020.00761
99. Hayama E, Furutani Y, Kawaguchi N, Seki A, Nagashima Y, Okita K, et al. Induced pluripotent stem cell-derived cardiomyocytes with SCN5A R1623Q mutation associated with severe long QT syndrome in fetuses and neonates recapitulates pathophysiological phenotypes. Biology. (2021) 10:1062. doi: 10.3390/biology10101062
100. Hayano M, Makiyama T, Kamakura T, Watanabe H, Sasaki K, Funakoshi S, et al. Development of a patient-derived induced pluripotent stem cell model for the investigation of SCN5A-D1275N-related cardiac sodium channelopathy. Circ J. (2017) 81:1783–1791. doi: 10.1253/circj.CJ-17-0064
101. Portero V, Casini S, Hoekstra M, Verkerk AO, Mengarelli I, Belardinelli L, et al. Anti-arrhythmic potential of the late sodium current inhibitor GS-458967 in murine Scn5a-1798insD+/- and human SCN5A-1795insD+/- iPSC-derived cardiomyocytes. Cardiovasc Res. (2017) 113:829–38. doi: 10.1093/cvr/cvx077
102. Kosmidis G, Veerman CC, Casini S, Verkerk AO, van de Pas S, Bellin M, et al. Readthrough-promoting drugs gentamicin and PTC124 Fail to rescue Nav1.5 function of human-induced pluripotent stem cell-derived cardiomyocytes carrying nonsense mutations in the sodium channel gene SCN5A. Circ Arrhythmia Electrophysiol. (2016) 9:e004227. doi: 10.1161/CIRCEP.116.004227
103. McKeithan WL, Feyen DAM, Bruyneel AAN, Okolotowicz KJ, Ryan DA, Sampson KJ, et al. Reengineering an antiarrhythmic drug using patient hiPSC cardiomyocytes to improve therapeutic potential and reduce toxicity. Cell Stem Cell. (2020) 27:813–21.e16. doi: 10.1016/j.stem.2020.08.003
104. Cashman JR, Ryan D, McKeithan WL, Okolotowicz K, Gomez-Galeno J, Johnson M, et al. Antiarrhythmic hit to lead refinement in a dish using patient-derived iPSC cardiomyocytes. J Med Chem. (2021) 64:5384–403. doi: 10.1021/acs.jmedchem.0c01545
105. Hamrick SK, John Kim CS, Tester DJ, Giudicessi JR, Ackerman MJ. Patient-specific, re-engineered cardiomyocyte model confirms the circumstance-dependent arrhythmia risk associated with the African-specific common SCN5A polymorphism p.S1103Y: Implications for the increased sudden deaths observed in black individuals during the COVID-19 pandemic. Heart rhythm. (2022) 19:822–7. doi: 10.1016/j.hrthm.2021.12.029
106. Wada Y, Yang T, Shaffer CM, Daniel LL, Glazer AM, Davogustto GE, et al. Common ancestry-specific ion channel variants predispose to drug-induced arrhythmias. Circulation. (2022) 145:299–308. doi: 10.1161/CIRCULATIONAHA.121.054883
107. Kroncke BM, Yang T, Roden DM. Multiple mechanisms underlie increased cardiac late sodium current. Heart Rhythm. (2019) 16:1091–7. doi: 10.1016/j.hrthm.2019.01.018
108. Terrenoire C, Wang K, Tung KWC, Chung WK, Pass RH, Lu JT, et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol. (2013) 141:61–72. doi: 10.1085/jgp.201210899
109. Limpitikul WB, Dick IE, Tester DJ, Boczek NJ, Limphong P, Yang W, et al. A Precision medicine approach to the rescue of function on malignant calmodulinopathic long-QT syndrome. Circ Res. (2017) 120:39–48. doi: 10.1161/CIRCRESAHA.116.309283
110. Shaheen N, Shiti A, Huber I, Shinnawi R, Arbel G, Gepstein A, et al. Human induced pluripotent stem cell-derived cardiac cell sheets expressing genetically encoded voltage indicator for pharmacological and arrhythmia studies. Stem Cell Rep. (2018) 10:1879–94. doi: 10.1016/j.stemcr.2018.04.006
111. Shinnawi R, Huber I, Maizels L, Shaheen N, Gepstein A, Arbel G, et al. Monitoring human-induced pluripotent stem cell-derived cardiomyocytes with genetically encoded calcium and voltage fluorescent reporters. Stem Cell Rep. (2015) 5:582–96. doi: 10.1016/j.stemcr.2015.08.009
112. Hotta A, Yamanaka S. From genomics to gene therapy: induced pluripotent stem cells meet genome editing. Annu Rev Genet. (2015) 49:47–70. doi: 10.1146/annurev-genet-112414-054926
113. Vermersch E, Jouve C, Hulot JS. CRISPR/Cas9 gene-editing strategies in cardiovascular cells. Cardiovasc Res. (2020) 116:894–907. doi: 10.1093/cvr/cvz250
114. Tamura R, Toda M. Historic overview of genetic engineering technologies for human gene therapy. Neurol Medico Chirurgica. (2020) 60:483–91. doi: 10.2176/nmc.ra.2020-0049
115. Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. (2014) 15:27–30. doi: 10.1016/j.stem.2014.07.009
116. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. (2012) 337:816–21. doi: 10.1126/science.1225829
117. Chylinski K, Le Rhun A, Charpentier E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. (2013) 10:726–37. doi: 10.4161/rna.24321
118. Lino CA, Harper JC, Carney JP, Timlin JA. Delivering CRISPR: a review of the challenges and approaches. Drug Delivery. (2018) 25:1234–57. doi: 10.1080/10717544.2018.1474964
119. González Castro N, Bjelic J, Malhotra G, Huang C, Alsaffar SH. Comparison of the feasibility, efficiency, and safety of genome editing technologies. Int J Mol Sci. (2021) 22:10355. doi: 10.3390/ijms221910355
120. Manjón AG, Linder S, Teunissen H, Friskes A, Zwart W, de Wit E, et al. Unexpected gene activation following CRISPR-Cas9-mediated genome editing. EMBO Rep. (2022) 23:e53902. doi: 10.15252/embr.202153902
121. Wei H, Zhang XH, Clift C, Yamaguchi N, Morad M. CRISPR/Cas9 gene editing of RyR2 in human stem cell-derived cardiomyocytes provides a novel approach in investigating dysfunctional Ca(2+) signaling. Cell Calcium. (2018) 73:104–11. doi: 10.1016/j.ceca.2018.04.009
122. Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. (2018) 24:939–46. doi: 10.1038/s41591-018-0050-6
123. Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. (2018) 24:927–30. doi: 10.1038/s41591-018-0049-z
124. Kim KT, Park JC, Jang HK, Lee H, Park S, Kim J, et al. Safe scarless cassette-free selection of genome-edited human pluripotent stem cells using temporary drug resistance. Biomaterials. (2020) 262:120295. doi: 10.1016/j.biomaterials.2020.120295
125. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. (2013) 339:823–6. doi: 10.1126/science.1232033
126. Habib O, Habib G, Hwang GH, Bae S. Comprehensive analysis of prime editing outcomes in human embryonic stem cells. Nucleic Acids Res. (2022) 50:1187–97. doi: 10.1093/nar/gkab1295
127. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. (2019) 576:149–57. doi: 10.1038/s41586-019-1711-4
128. Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nature reviews Genetics. (2018) 19:770–88. doi: 10.1038/s41576-018-0059-1
129. Zhou C, Sun Y, Yan R, Liu Y, Zuo E, Gu C, et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. (2019) 571:275–8. doi: 10.1038/s41586-019-1314-0
130. Zhang X, Zhu B, Chen L, Xie L, Yu W, Wang Y, et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat Biotechnol. (2020) 38:856–60. doi: 10.1038/s41587-020-0527-y
131. Kim HS, Jeong YK, Hur JK, Kim JS, Bae S. Adenine base editors catalyze cytosine conversions in human cells. Nat Biotechnol. (2019) 37:1145–8. doi: 10.1038/s41587-019-0254-4
132. Lee S, Ding N, Sun Y, Yuan T, Li J, Yuan Q, et al. Single C-to-T substitution using engineered APOBEC3G-nCas9 base editors with minimum genome- and transcriptome-wide off-target effects. Sci Adv. (2020) 6:eaba1773. doi: 10.1126/sciadv.aba1773
133. Chavali NV, Kryshtal DO, Parikh SS, Wang L, Glazer AM, Blackwell DJ, et al. Patient-independent human induced pluripotent stem cell model: a new tool for rapid determination of genetic variant pathogenicity in long QT syndrome. Heart rhythm. (2019) 16:1686–95. doi: 10.1016/j.hrthm.2019.04.031
135. Shimizu W, Horie M. Phenotypic manifestations of mutations in genes encoding subunits of cardiac potassium channels. Circ Res. (2011) 109:97–109. doi: 10.1161/CIRCRESAHA.110.224600
136. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. (1995) 80:795–803. doi: 10.1016/0092-8674(95)90358-5
137. Kagan A, Yu Z, Fishman GI, McDonald TV. The dominant negative LQT2 mutation A561V reduces wild-type HERG expression. J Biol Chem. (2000) 275:11241–8. doi: 10.1074/jbc.275.15.11241
138. Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. (2011) 471:225–9. doi: 10.1038/nature09747
139. Navarrete EG, Liang P, Lan F, Sanchez-Freire V, Simmons C, Gong T, et al. Screening drug-induced arrhythmia [corrected] using human induced pluripotent stem cell-derived cardiomyocytes and low-impedance microelectrode arrays. Circulation. (2013) 128(11 Suppl. 1):S3–13. doi: 10.1161/CIRCULATIONAHA.112.000570
140. Napolitano C, Rivolta I, Priori SG. Cardiac sodium channel diseases. Clin Chem Lab Med. (2003) 41:439–44. doi: 10.1515/CCLM.2003.066
141. Davis RP, Casini S, van den Berg CW, Hoekstra M, Remme CA, Dambrot C, et al. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation. (2012) 125:3079–91. doi: 10.1161/CIRCULATIONAHA.111.066092
142. Remme CA, Wilde AA, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med. (2008) 18:78–87. doi: 10.1016/j.tcm.2008.01.002
143. de la Roche J, Angsutararux P, Kempf H, Janan M, Bolesani E, Thiemann S, et al. Comparing human iPSC-cardiomyocytes versus HEK293T cells unveils disease-causing effects of Brugada mutation A735V of Na(V)1.5 sodium channels. Sci Rep. (2019) 9:11173. doi: 10.1038/s41598-019-47632-4
144. Schwartz PJ, Crotti L, George AL Jr. Modifier genes for sudden cardiac death. Eur Heart J. (2018) 39:3925–31. doi: 10.1093/eurheartj/ehy502
145. Lahtinen AM, Marjamaa A, Swan H, Kontula K. KCNE1 D85N polymorphism–a sex-specific modifier in type 1 long QT syndrome? BMC Med Genet. (2011) 12:11. doi: 10.1186/1471-2350-12-11
146. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, et al. Variants in the 3′ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur Heart J. (2012) 33:714–23. doi: 10.1093/eurheartj/ehr473
147. Crotti L, Lahtinen AM, Spazzolini C, Mastantuono E, Monti MC, Morassutto C, et al. Genetic modifiers for the long-QT syndrome: how important is the role of variants in the 3′ untranslated region of KCNQ1? Circ Cardiovasc Genet. (2016) 9:330–9. doi: 10.1161/CIRCGENETICS.116.001419
148. Duchatelet S, Crotti L, Peat RA, Denjoy I, Itoh H, Berthet M, et al. Identification of a KCNQ1 polymorphism acting as a protective modifier against arrhythmic risk in long-QT syndrome. Circ Cardiovasc Genet. (2013) 6:354–61. doi: 10.1161/CIRCGENETICS.113.000023
149. de Villiers CP, van der Merwe L, Crotti L, Goosen A, George AL Jr, Schwartz PJ, et al. AKAP9 is a genetic modifier of congenital long-QT syndrome type 1. Circ Cardiovasc Genet. (2014) 7:599–606. doi: 10.1161/CIRCGENETICS.113.000580
150. Earle N, Yeo Han D, Pilbrow A, Crawford J, Smith W, Shelling AN, et al. Single nucleotide polymorphisms in arrhythmia genes modify the risk of cardiac events and sudden death in long QT syndrome. Heart Rhythm. (2014) 11:76–82. doi: 10.1016/j.hrthm.2013.10.005
151. Kolder I, Tanck MWT, Postema PG, Barc J, Sinner MF, Zumhagen S, et al. Analysis for genetic modifiers of disease severity in patients with long-QT syndrome type 2. Circ Cardiovasc Genet. (2015) 8:447–56. doi: 10.1161/CIRCGENETICS.114.000785
152. Paavonen KJ, Chapman H, Laitinen PJ, Fodstad H, Piippo K, Swan H, et al. Functional characterization of the common amino acid 897 polymorphism of the cardiac potassium channel KCNH2 (HERG). Cardiovasc Res. (2003) 59:603–11. doi: 10.1016/S0008-6363(03)00458-9
153. Anson BD, Ackerman MJ, Tester DJ, Will ML, Delisle BP, Anderson CL, et al. Molecular and functional characterization of common polymorphisms in HERG (KCNH2) potassium channels. Am J Physiol Heart Circ Physiol. (2004) 286:H2434–41. doi: 10.1152/ajpheart.00891.2003
154. Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. (2005) 112:1251–8. doi: 10.1161/CIRCULATIONAHA.105.549071
155. Santori M, Gil R, Blanco-Verea A, Riuró H, Díaz-Castro Ó, López-Abel B, et al. Sudden infant death as the most severe phenotype caused by genetic modulation in a family with atrial fibrillation. Forensic Sci Int Genet. (2019) 43:102159. doi: 10.1016/j.fsigen.2019.102159
156. Itoh H, Sakaguchi T, Ashihara T, Ding WG, Nagaoka I, Oka Y, et al. A novel KCNH2 mutation as a modifier for short QT interval. Int J Cardiol. (2009) 137:83–5. doi: 10.1016/j.ijcard.2008.05.050
157. van den Brink L, Grandela C, Mummery CL, Davis RP. Inherited cardiac diseases, pluripotent stem cells, and genome editing combined-the past, present, and future. Stem Cells. (2020) 38:174–86. doi: 10.1002/stem.3110
158. Caballero R, Utrilla RG, Amorós I, Matamoros M, Pérez-Hernández M, Tinaquero D, et al. Tbx20 controls the expression of the KCNH2 gene and of hERG channels. Proc Natl Acad Sci USA. (2017) 114:E416–25. doi: 10.1073/pnas.1612383114
159. Federici G, Soddu S. Variants of uncertain significance in the era of high-throughput genome sequencing: a lesson from breast and ovary cancers. J Exp Clin Cancer Res. (2020) 39:46. doi: 10.1186/s13046-020-01554-6
160. Wang DW, Yazawa K, George AL Jr, Bennett PB. Characterization of human cardiac Na+ channel mutations in the congenital long QT syndrome. Proc Natl Acad Sci USA. (1996) 93:13200–5. doi: 10.1073/pnas.93.23.13200
161. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
162. Nieto-Marín P, Tinaquero D, Utrilla RG, Cebrián J, González-Guerra A, Crespo-García T, et al. Tbx5 variants disrupt Nav1.5 function differently in patients diagnosed with Brugada or Long QT Syndrome. Cardiovasc Res. (2022) 118:1046–60. doi: 10.1093/cvr/cvab045
163. Mohammad-Panah R, Demolombe S, Neyroud N, Guicheney P, Kyndt F, van den Hoff M, et al. Mutations in a dominant-negative isoform correlate with phenotype in inherited cardiac arrhythmias. Am J Hum Genet. (1999) 64:1015–23. doi: 10.1086/302346
164. Synková I, Bébarová M, Andršová I, Chmelikova L, Švecová O, Hošek J, et al. Long-QT founder variant T309I-Kv7.1 with dominant negative pattern may predispose delayed afterdepolarizations under β-adrenergic stimulation. Sci Rep. (2021) 11:3573. doi: 10.1038/s41598-021-81670-1
165. Aizawa Y, Ueda K, Scornik F, Cordeiro JM, Wu Y, Desai M, et al. A novel mutation in KCNQ1 associated with a potent dominant negative effect as the basis for the LQT1 form of the long QT syndrome. J Cardiovasc Electrophysiol. (2007) 18:972–7. doi: 10.1111/j.1540-8167.2007.00889.x
166. Thomas D, Wimmer AB, Karle CA, Licka M, Alter M, Khalil M, et al. Dominant-negative I(Ks) suppression by KCNQ1-deltaF339 potassium channels linked to Romano-Ward syndrome. Cardiovasc Res. (2005) 67:487–97. doi: 10.1016/j.cardiores.2005.05.003
167. Li W, Du R, Wang QF, Tian L, Yang JG, Song ZF. The G314S KCNQ1 mutation exerts a dominant-negative effect on expression of KCNQ1 channels in oocytes. Biochem Biophys Res Commun. (2009) 383:206–9. doi: 10.1016/j.bbrc.2009.03.160
168. Lu X, Yang X, Huang X, Huang C, Sun HH, Jin L, et al. RNA interference targeting E637K mutation rescues hERG channel currents and restores its kinetic properties. Heart Rhythm. (2013) 10:128–36. doi: 10.1016/j.hrthm.2012.09.124
169. Satomura A, Nishioka R, Mori H, Sato K, Kuroda K, Ueda M. Precise genome-wide base editing by the CRISPR Nickase system in yeast. Sci Rep. (2017) 7:2095. doi: 10.1038/s41598-017-02013-7
170. Schwartz PJ, Gnecchi M, Dagradi F, Castelletti S, Parati G, Spazzolini C, et al. From patient-specific induced pluripotent stem cells to clinical translation in long QT syndrome Type 2. Eur Heart J. (2019) 40:1832–6. doi: 10.1093/eurheartj/ehz023
171. Al-Moubarak E, Zhang Y, Dempsey CE, Zhang H, Harmer SC, Hancox JC. Serine mutation of a conserved threonine in the hERG K channel S6-pore region leads to loss-of-function through trafficking impairment. Biochem Biophys Res Commun. (2020) 526:1085–91. doi: 10.1016/j.bbrc.2020.04.003
172. Harwood KH, McQuade RM, Jarnicki A, Schneider-Futschik EK. Anti-inflammatory influences of cystic fibrosis transmembrane conductance regulator drugs on lung inflammation in cystic fibrosis. Int J Mol Sci. (2021) 22:7606. doi: 10.3390/ijms22147606
Keywords: LQTS, hiPSC, hiPSC-CMs, CRISPR/Cas9, modifier genes, variants of uncertain significance (VUS)
Citation: Song Y, Zheng Z and Lian J (2022) Deciphering Common Long QT Syndrome Using CRISPR/Cas9 in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Front. Cardiovasc. Med. 9:889519. doi: 10.3389/fcvm.2022.889519
Received: 04 March 2022; Accepted: 22 April 2022;
Published: 13 May 2022.
Edited by:
Daniel M. Johnson, The Open University, United KingdomReviewed by:
Giulia Campostrini, Leiden University Medical Center, NetherlandsCopyright © 2022 Song, Zheng and Lian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiangfang Lian, aGptcGluQDE2My5jb20=; Yongfei Song, c29uZ3lvbmdmZWkxQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.